 Ana Drole Torkar1
Ana Drole Torkar1 Magdalena Avbelj Stefanija1
Magdalena Avbelj Stefanija1 Sara Bertok1
Sara Bertok1 Katarina Trebušak Podkrajšek2,3
Katarina Trebušak Podkrajšek2,3 Maruša Debeljak2Branislava Stirn Kranjc3,4
Maruša Debeljak2Branislava Stirn Kranjc3,4 Tadej Battelino1,3
Tadej Battelino1,3 Primož Kotnik1,3*
Primož Kotnik1,3*- 1University Children’s Hospital, Department of Endocrinology, Diabetes and Metabolism, Ljubljana University Medical Centre, Ljubljana, Slovenia
- 2University Children’s Hospital, Unit of Special Laboratory Diagnostics, Ljubljana University Medical Centre, Ljubljana, Slovenia
- 3Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
- 4University Eye Hospital, Ljubljana University Medical Centre, Ljubljana, Slovenia
A Caucasian girl with consanguineous parents presented with early severe obesity and retinal dystrophy. A novel, homozygous gene truncating variant (c.1897C>T) in the INPP5E gene confirmed the diagnosis of MORMS (OMIM #610156). A novel clinical finding in the presented syndrome is progressive cone-rod type retinal dystrophy diagnosed at the age of four months that progressed in the 1st decade of life. Severe obesity, insulin resistance with hyperinsulinism, and impaired glucose tolerance developed alongside other components of the metabolic syndrome - dyslipidemia, arterial hypertension, and obstructive hypopnea in sleep. At the age of 14 years, primary amenorrhea persists. The patient is managed by regular nutritional advice, metformin, antihypertensive medication, and non-invasive respiratory support during sleep. Differential diagnosis of this rare entity is discussed in extend.
Introduction
Monogenic obesity is rare, it presents, early and is severe, with minimal environmental influence on the phenotype (1). Characterization of genes involved in the development of monogenic obesity also contributed to the unraveling of mechanisms leading to polygenetic/common obesity and could contribute to the development of novel targeted therapeutic interventions (2).
Gene encoding inositol polyphosphate-5-phosphatase E (INPP5E) is located on chromosome 9q34. It is widely expressed in humans, especially in the brain (3, 4). INPP5E belongs to the 5-ptase family of enzymes and is involved in regulating diverse cellular processes, namely synaptic vesicle recycling, insulin signaling, and embryonic development (5–7). Loss of INPP5E function leads to decreased cilia stability and ciliopathy (5–9) as it plays an essential role in the primary cilium by controlling ciliary growth factor and phosphoinositide 3-kinase (PI3K) signaling and stability (2, 3, 5, 6, 10). Primary cilia play a critical role in the development and functioning of several cell types, including retinal photoreceptors, neurons, kidney tubules, and bile ducts (11, 12). Pathogenic variants in the INPP5E gene are linked to two partly overlapping but clinically distinct autosomal recessive ciliopathies: Joubert syndrome 1 (JBTS1; OMIM #213300) characterized by the malformation of the brainstem and agenesis or hypoplasia of the cerebellar vermis that causes abnormal respiratory pattern, nystagmus, hypotonia, ataxia, and developmental delay (6, 13); and a syndrome of static moderate mental disability, truncal obesity, congenital non-progressive retinal dystrophy, and micropenis in males (MORMS; OMIM #610156) (13).
In the case of MORMS, a nonsense mutation is located in the C-terminal domain of the gene, resulting in a truncated protein, affecting ciliary localization and stabilization of ciliary structures (5, 14).
MORMS has been, to our knowledge, so far described in a single complex consanguineous Pakistani kindred (13). We present a novel case of MORM syndrome in a Caucasian girl with consanguineous parents, including several novel clinical features.
Case Presentation
Family Medical History
Girl’s maternal and paternal grand grandfathers were brothers (Figure 1). The father has arterial hypertension, and the mother is overweight and had dermatitis and cholelithiasis. The girl has an older brother, who is also overweight, without visual impairment, but has not been available for further clinical or genetic evaluations. There is no data on intellectual disability, extreme obesity, or impaired vision in the extended family.
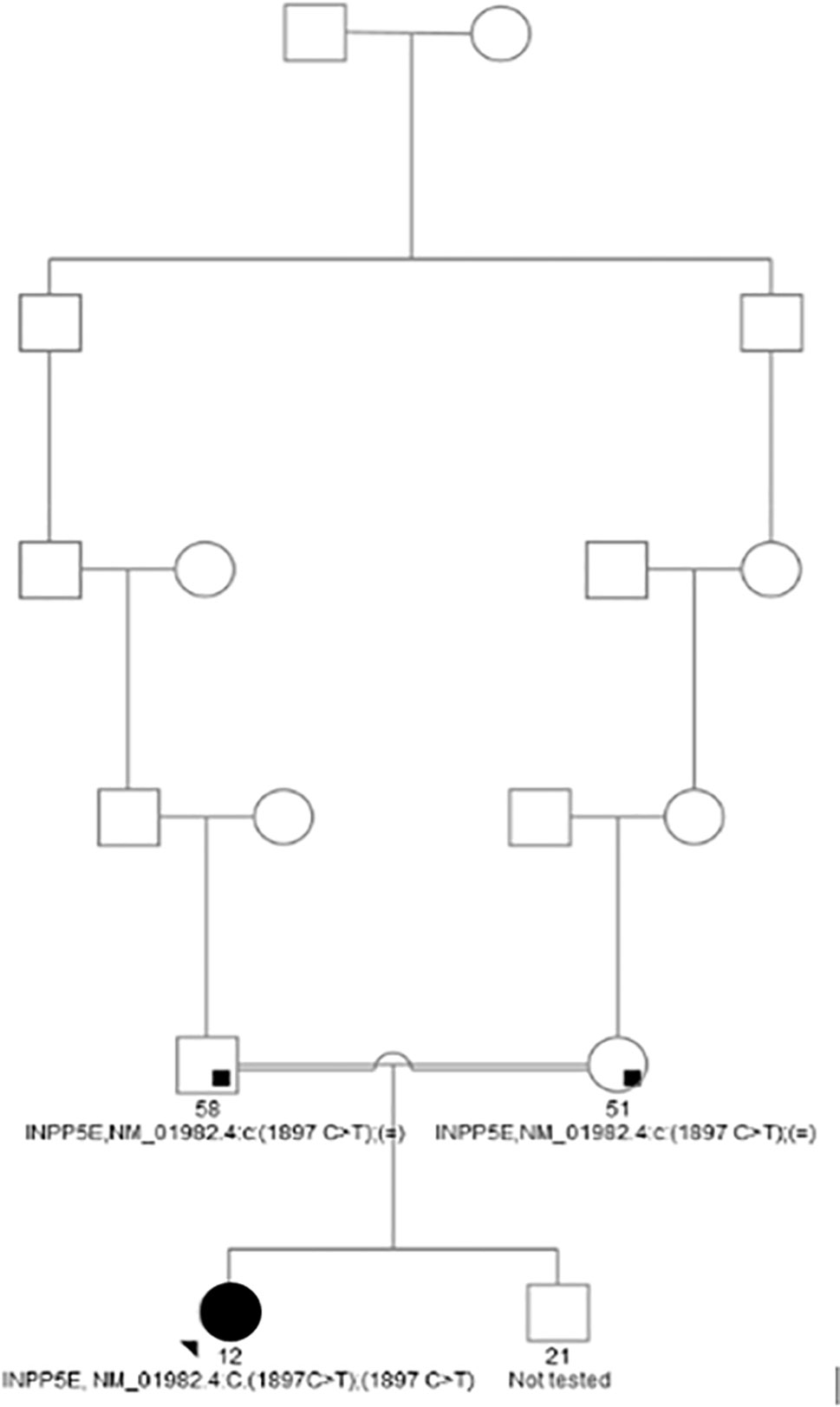
Figure 1 Family tree of the proband.
Clinical Observations and Investigations
The girl was born at term following mother’s uneventful second pregnancy, with a birth weight of 4.23 kg (SDS-W 1.8) and a birth length of 51 cm (SDS-L 1.5). There were no complications in the neonatal period.
Retinal Dystrophy of Cone-Rod Type
The first presenting sign of MORMS was horizontal nystagmus of low frequency and amplitude at the age of four months. At the age of two years, visual acuity was normal for age, but decreased to moderate myopic astigmatism by the age of ten years. The diagnosis of retinal dystrophy was confirmed at the age of five years, three years after some discrete pigmentary changes of the retina were first noticed. Retinal appearance showed progressive retinal dystrophy with optic disc pallor, attenuated retinal vessels, loss of foveal reflex and retinal pigment mottling in the posterior pole, and the retinal periphery. Electrophysiological examination (scotopic photopic electroretinography) confirmed cone-rod type retinal dystrophy. An uneven foveal reflex with a hyper-fluorescent ring around the macula was seen on fundus auto-fluorescence retina angiograph (HRA, Spectralis Heidelberg). 3D Optic Coherence Topography (Topcon 1000) analysis determined reduced retinal thickness with irregularities, predominantly of the outer retinal layer (Figure 2). Orthophoria, reduced visual acuity, and color vision with concentrically narrowed visual fields became evident. No signs of cataract developed in the 1st decade.
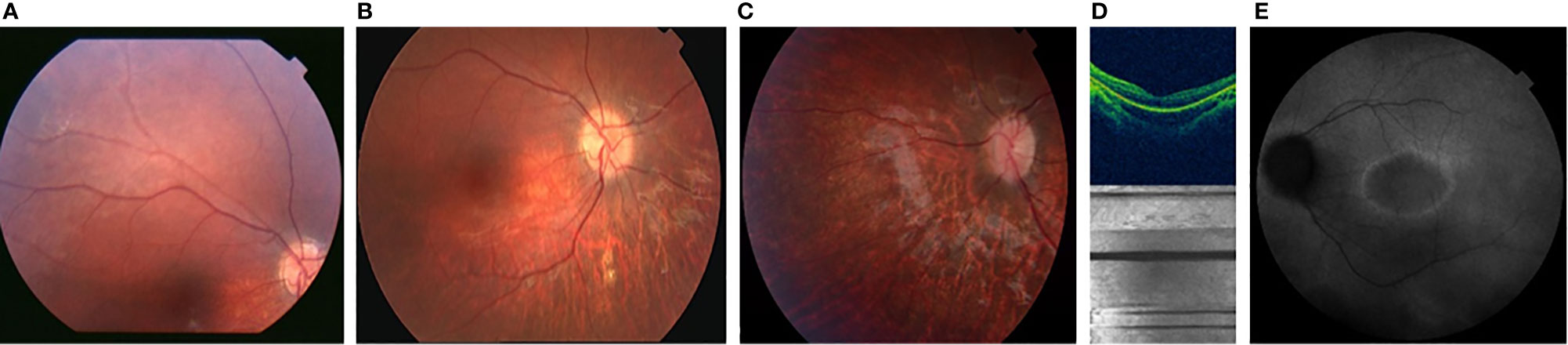
Figure 2 Color fundus photographs [(A) at the age of 2 yrs.], [(B) at the age of 6 yrs. [(C) at the age of 7 yrs.] showing the progression of the optic disc pallor with retinal arteriolar narrowing and pigment mottling; OCT image [(D) at the age of 7 years, showing thinner retina predominantly the outer retinal layer with irregularities] AF [(E) at the age of 10 years, showing hyper-fluorescent ring around the macula with irregular fluorescence of the posterior pole].
Obesity and Metabolic Syndrome Evaluation
Development of obesity, the second hallmark of MORMS, was noticed when a substantial weight gain from the age of six months (7.5 kg, SDS-W 0.2) to 12 months (14 kg, SDS-W 3.5) and 12 to 18 months (20.5 kg, SDS-W 5.2) was determined and pronounced food-seeking behavior was described. She was first presented to the pediatric endocrinologist at the age of two years. At the first visit, marked hyperinsulinism with normoglycemia and dyslipidemia were already determined (Table 1). Therapy with metformin was started at the age of five years. Obesity and severe hyperphagia progressed. Impaired glucose tolerance was determined at the age of ten years (Table 1).
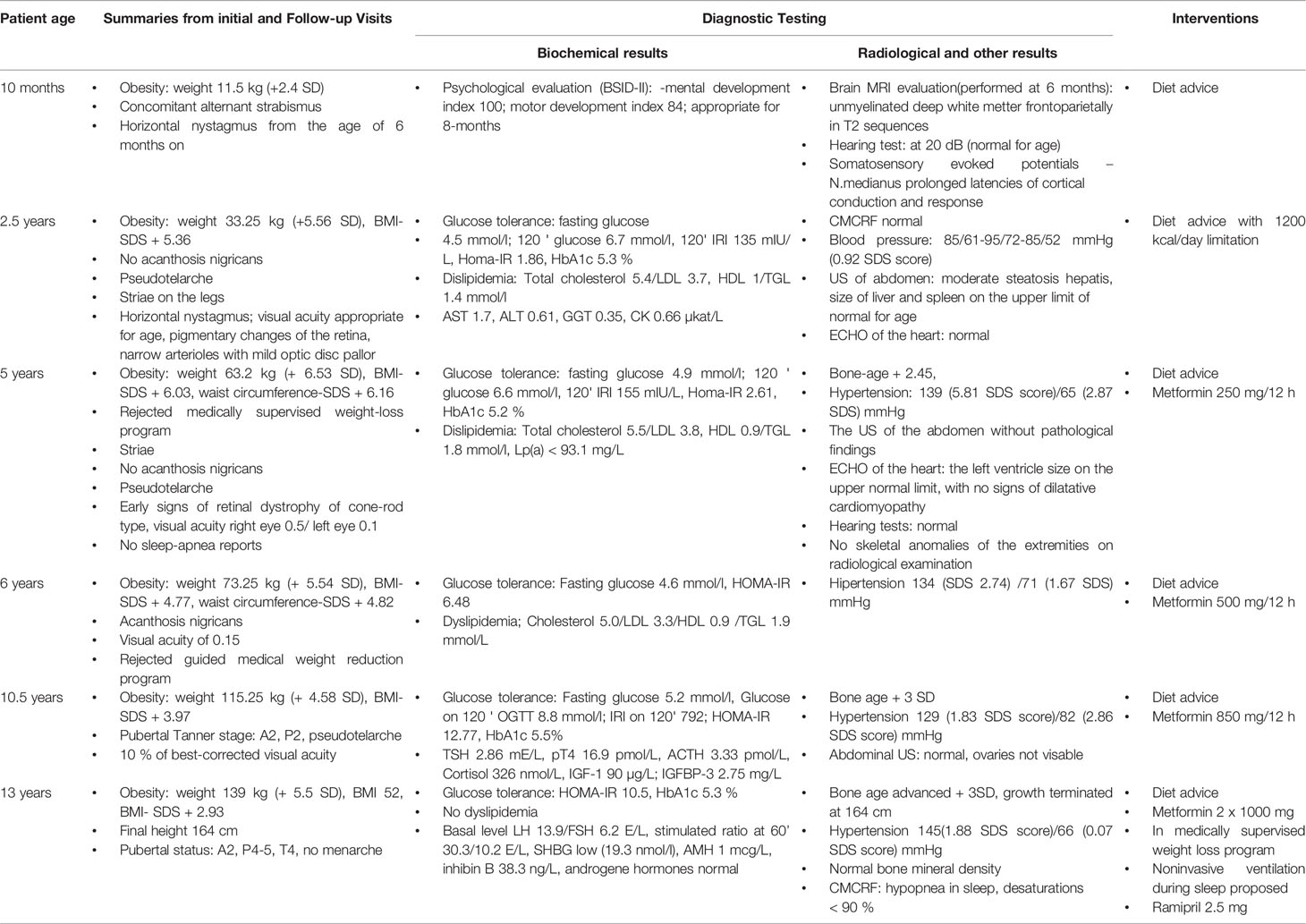
Table 1 Timeline of clinical findings, Diagnostic tests, and Interventions.
Blood pressure was elevated already at five years; anti-hypertensive medication was introduced at 13 years of age. Simultaneously, low basal oxygen saturation and hypopnea with marked desaturations during sleep were recorded, and non-invasive respiratory support with continuous positive airway pressure (CPAP) during sleep was proposed.
Neurodevelopmental History
Gross motor and speech developmental milestones were reached at an appropriate age. On the Verbal Comprehension Wechsler intelligence scale for children (WISC III), performed at the age of 13 years, she showed below-average abilities, especially in understanding common concepts and social situations in the subtests evaluating abstract thinking and on working memory testing. In the first year of life, delayed myelination pattern, with no visible malformations, was determined by magnetic resonance imaging.
Growth and Puberty
As illustrated in Table 1 and Figure 3, the girl was tall since the second year of life. Her bone age was advanced since the age of five years. IGF-1 and IGF-BP3 levels were normal (Table 1). Adrenarche started at the age of ten years. Aged 13 years, she attained her final height of 164 cm, 2 cm below her target height based on parental heights. At that time, pubertal staging, according to Tanner, was A2, T4, P3, and she did not have menarche. She had LH/FSH levels in the normal range for age and pubertal status.
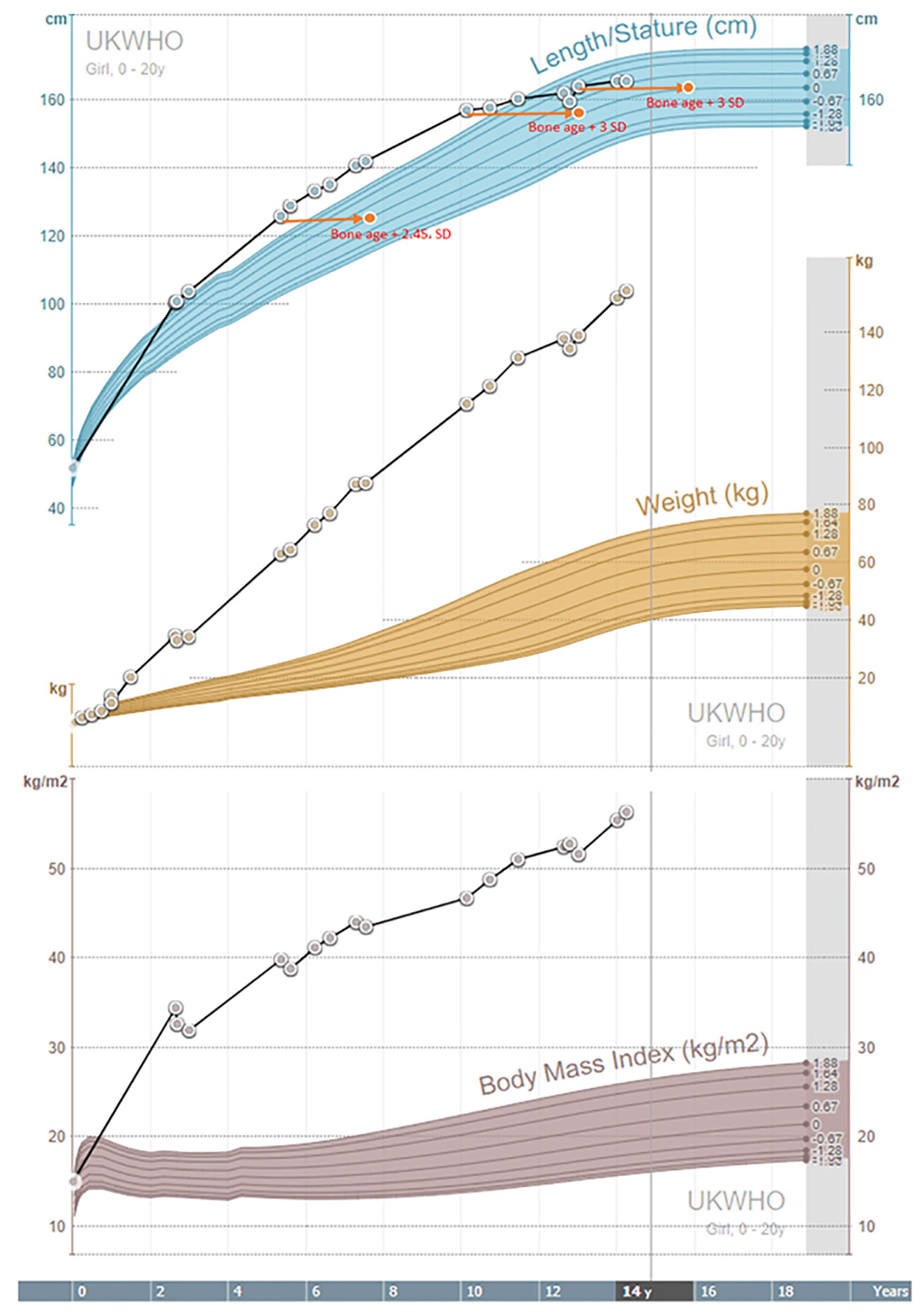
Figure 3 Growth charts.
Genetic Analysis
Informed consent for genetic testing was obtained from the parents. Genomic DNA was isolated from whole peripheral blood, following standard methods. No mutations in the ALMS1 gene were found on analysis by PCR and sequencing of both DNA strands of the entire coding region and the highly conserved exon-intron splice junctions.
Next-Generation Sequencing (NGS) was performed using MiSeq desktop sequencer (Illumina, San Diego, USA). The regions of interest were enriched using TruSightOne library enrichment kit (Illumina, San Diego, USA) and targeted analysis of genes causative of cone-rod type of retinal dystrophy (list of genes in the Appendix), with no causative mutations found. The selection of candidate causative variants was extended using PhenIX software (http://compbio.charite.de/PhenIX). 41,5% horizontal coverage of analyzed regions was reached, and 95,7% of target regions of the selected genetic panel were sequenced sufficiently.
In the gene INPP5E, a homozygous gene variant c.1897C>T causes a change from glutamine in amino acidic position 633 to stop codon and premature termination of translation (NP_063945.2:p.Gln633Ter). The variant is, to our knowledge, not yet described in The Human Gene Mutation Database, nor present in healthy population studies. Parents, who are consanguineous but healthy, were proved to be carriers of the variant p.Gln633Ter in a heterozygous form. The brother declined all testing.
Discussion
Further Phenotypic Characterization of MORMS
MORMS is a very rare autosomal recessive ciliopathy that has been, to our knowledge, so far reported in a single pedigree (13). We contribute the first description of MORMS in a Caucasian pedigree. The most prominent phenotypic features of the presented case were very early retinal dystrophy and early and severe obesity with the development of components of the metabolic syndrome.
Perinatal history of previously described individuals with MORMS (13) and our patient was uneventful; no congenital anomalies were observed. Affected individuals with MORMS in the described Pakistani kindred (13) had poor night vision evident in the first year of life; by the age of three, reduced visual acuity was apparent, but no further visual loss was noticed after that. Unlike in our patient, no nystagmus was evident. In previously described patients ophthalmologic examination of the fundi showed a mildly atrophic retina with thinned blood vessels, without increased pigmentation or optic atrophy. In our patient a complete ophthalmological evaluation was done in the first year of life. The diagnosis of retinal dystrophy was made on the grounds of both clinical and electrophysiological criteria. This is the first description of the retinal dystrophy associated with MORMS being of cone-rode type, similar to other ciliopathies (Table 2). Importantly, visual loss in our patient was progressive in the 1st decade of life (Figure 2), which should be considered in prognosis and management decisions of other MORMS patients.

Table 2 Phenotype comparison between MORM, Bardet-Biedl, Cohen, Alstörm, and Joubert 1 syndrome (OMIM database).
In the previously described subjects with MORMS, truncal obesity developed by the age of five years, while our patient was obese already at the age of nine months. No acanthosis or insulin resistance was reported in the previously described kindred (13). In our patient, hyperinsulinism was diagnosed at the age of 2.5 years, and impaired glucose tolerance developed at ten years. She developed obesity-related arterial hypertension and obstructive hypopnea in sleep at the age of 13 years.
All previously described individuals with MORMS had delayed language acquisition, while our patient did not. Like all other patients from the described kindred (13), a static mild to moderate intellectual disability with typical motor milestones acquisition and no disturbance of muscular tone were present. There is no description of pubertal development in affected females in Pakistani kindred (13) apart from the information that none have had offspring. Pubertal development started spontaneously and timely in our patient at the age of ten years. At the age of 13 years, while still in the premenarchal phase, puberty was assessed biochemically. Levels of gonadotropins were within the normal levels, with LH/FSH ratio of 3:1, no clinical or biochemical indicators of hyperandrogenism were found. AMH was below average according to her age and pubertal stage (15). Previously described individuals with MORMS (13) had normal growth parameters. Our patient was growing above the parental growth channel in the pre-pubertal phase and had advanced bone age (Table 1 and Figure 3), most probably due to severe childhood obesity and hyperinsulinism (16). However, she reached her final height at 13 years, when closed growth plates were determined, with the final height 2 cm below her target height.
The previously described kindred’s (13) nonsense mutation INPP5E gene was located at position 627 (13). A novel nonsense gene variant located in the near vicinity at position 633 was identified in our patient and results in premature termination of mRNA translation and impaired function of the INPP5E protein, as determined by in silico methods. Therefore, both pedigrees with MORMS had a truncating gene variant with a similar genetic location in the terminal exon of INPP5E, resulting in the omission of terminal amino acids and loss of the C-terminal transmembrane CaaX domain. The p.Gln627Ter variant in the Pakistani pedigree changes the ciliary localization of the affected cells, while the 5-phosphatase activity of the protein is retained (5). The protein variant in our patient was only six amino acids longer; therefore, similar functional consequences could be expected, reflected by similarities in clinical presentation and advocates for a significant correlation between genotype and phenotype.
Differential Diagnosis of MORMS
Childhood-onset obesity is a consistent feature of monogenic obesity syndromes. It is helpful to categorize obesity syndromes as those with dysmorphisms or developmental delay and those without these features (17). Ciliopathies are clinically and genetically heterogeneous group of monogenic obesity causes, where the common denominator is early-onset obesity in association with retinal dystrophy and developmental delay. The presented patient had moderate developmental delay and retinal dystrophy but no significant dysmorphisms.
Our first clinical differential diagnosis was Alström syndrome (Table 2), characterized by cone-rod retinal dystrophy, cardiomyopathy, and type 2 diabetes mellitus (18); however, a mutation of the ALMS1 gene was not found in our patient.
MORMS is also phenotypically and genetically distinct from the Bardet-Biedl syndrome (BBS; OMIM #209900) and Cohen syndrome (COH1; OMIM # 216550) by the age of onset and progression of the visual impairment, and the lack of several characteristics, including characteristic facies, skin or gingival infection, microcephaly, ‘mottled retina’ and polydactyly (19, 20) (Table 2).
Joubert syndrome and related disorders (JBTS) are clinically and genetically heterogeneous ciliopathies that share peculiar midbrain-hindbrain malformation named the ‘molar tooth sign’. JBTS1 shares the affected gene INPP5E with MORMS (10). It combines neurological signs with variable multi-organ involvement - the retina, kidneys, liver, and skeleton. The cardinal features are hypotonia that evolves to ataxia and developmental delay, altered respiratory pattern in the neonatal period, and abnormal ocular movements. The development of language skills is typically delayed, also mild to severe intellectual disability is common. Retinal dystrophy has a progressive course with variably conserved vision in JBTS (10). Our patient displayed some clinical characteristics of JBTS: intellectual disability, nystagmus, and progressive retinal dystrophy. Brain magnetic resonance imaging did not show pathological changes; she lacked hypotonia, ataxia, and respiratory pattern disorder and did not present any renal, hepatic, or skeletal involvement to fit the diagnostic criteria (Table 2).
MORMS also shares several features with common metabolic syndrome, namely obesity, hyperinsulinemia, and hypertriglyceridemia.
The assessment of severe obesity in children and adults should include screening for potentially treatable endocrine and neurological conditions and genetic diagnosis, also for genetic counseling. Clinical evaluation needs to address complications of severe obesity (21) regardless of the underlying cause.
In conclusion, a Caucasian girl with MORMS due to a novel INPP5E truncating variant is presented and compared to the only so far described MORMS pedigree. The main novel clinical finding is a progressive course of cone-rod type retinal dystrophy in the first decade of life, which is essential for prognosis and management decisions. Severe obesity develops in the first year of life, leading to the early development of prediabetes, dyslipidemia, and arterial hypertension. Delay in language acquisition is not necessarily present in association with below-average cognitive abilities.
Data Availability Statement
The data analyzed in this study is subject to the following licenses/restrictions: Unidentifiable human data. Requests to access these datasets should be directed to cHJpbW96LmtvdG5pa0BtZi51bmktbGouc2k=.
Ethics Statement
The patients’ parents gave their written informed consent for the publication of the report.
Author Contributions
The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted. ADT: Data Curation, Writing – Original Draft and Editing. MAS: Conceptualization, Clinical management of the patient, Data interpretation, Manuscript Editing. SB: Clinical and Genetic diagnostic management. KTP: Genetic analysis and diagnosis confirmation. MD: Genetic analysis, diagnosis confirmation. BSK: Ophthalmological evaluation, diagnostics, and management of the patient, Editing of the Manuscript. TB: Conceptualization, Resources, Supervision. PK: Conceptualization, Writing – Review and Editing, Supervision. All authors contributed to the article and approved the submitted version
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the family and our young patient for allowing us to learn and share their story.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.581134/full#supplementary-material
References
1. van der Klaauw AA, Farooqi IS. The Hunger Genes: Pathways to Obesity. Cell (2015) 161(1):119–32. doi: 10.1016/j.cell.2015.03.008
2. Kühnen P, Clément K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin Deficiency Treated With a Melanocortin-4 Receptor Agonist. N Engl J Med (2016) 375(3):240–6. doi: 10.1056/NEJMoa1512693
3. Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS, et al. The Role of the Inositol Polyphosphate 5-Phosphatases in Cellular Function and Human Disease. Biochem J (2009) 419(1):29–49. doi: 10.1042/BJ20081673
4. Kong AM, Speed CJ, O’Malley CJ, Layton MJ, Meehan T, Loveland KL, et al. Cloning and Characterisation of a 72-kDa Inositol-Polyphosphate 5-Phosphatase Localised to the Golgi Network. J Biol Chem (2000) 275(31):24052–64. doi: 10.1074/jbc.M000874200
5. Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, et al. INPP5E Mutations Cause Primary Cilium Signaling Defects, Ciliary Instability and Ciliopathies in Human and Mouse. Nat Genet (2009) 41(9):1027. doi: 10.1038/ng.427
6. Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, et al. Mutations in INPP5E, Encoding Inositol Polyphosphate-5-Phosphatase E, Link Phosphatidyl Inositol Signaling to the Ciliopathies. Nat Genet (2009) 41(9):1032. doi: 10.1038/ng.423
7. Travaglini L, Brancati F, Silhavy J, Iannicelli M, Nickerson E, Elkhartoufi N, et al. Phenotypic Spectrum and Prevalence of INPP5E Mutations in Joubert Syndrome and Related Disorders. Eur J Hum Genet (2013) 21(10):1074–8. doi: 10.1038/ejhg.2012.305
8. Plotnikova OV, Seo S, Cottle DL, Conduit S, Hakim S, Dyson JM, et al. INPP5E Interacts With AURKA, Linking Phosphoinositide Signalling to Primary Cilium Stability. J Cell Sci (2015) 128(2):364–72. doi: 10.1242/jcs.161323
9. Lancaster MA, Gleeson JG. The Primary Cilium as a Cellular Signalling Center: Lessons From Disease. Curr Opin Genet Dev (2009) 19:220–9. doi: 10.1016/j.gde.2009.04.008
10. Chávez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Cell (2015) 34(3):338–50. doi: 10.1016/j.devcel.2015.06.016
11. Badano JL, Mitsuma N, Beales PL, Katsanis N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu Rev Genomics Hum Genet (2006) 7:125–48. doi: 10.1146/annurev.genom.7.080505.115610
12. Conduit SE, Dyson JM, Mitchell CA. Inositol Polyphosphate 5-Phosphatases; New Players in the Regulation of Cilia and Ciliopathies. FEBS Lett (2012) 586(18):2846–57. doi: 10.1016/j.febslet.2012.07.037
13. Hampshire DJ, Ayub M, Springell K, Roberts E, Jafri H, Rashid Y, et al. MORM Syndrome (Mental Retardation, Truncal Obesity, Retinal Dystrophy and Micropenis), A New Autosomal Recessive Disorder, Links to 9q34. Eur J Hum Genet (2006) 14(5):543. doi: 10.1038/sj.ejhg.5201577
14. Farooqi IS, O’Rahilly S. Genetic Obesity Syndromes. In: The Genetics of Obesity. New York, NY: Springer (2014). p. 23–32.
15. Bhide P, Pundir J, Homburg R, Acharya G. Biomarkers of Ovarian Reserve in Childhood and Adolescence: A Systematic Review. Acta Obstet Gynecol Scand (2019) 98(5):563–72. doi: 10.1111/aogs.13574
16. Fintini D, Cianfarani S, Cofini M, Andreoletti A, Ubertini GM, Cappa M, et al. The Bones of Children With Obesity. Front Endocrinol (2020) 11(200):1–16. doi: 10.3389/fendo.2020.00200
17. Russell-Eggitt IM, Clayton PT, Coffey R, Kriss A, Taylor DS, Taylor JF. Alström Syndrome: Report of 22 Cases and Literature Review. Ophthalmology (1998) 105(7):1274–80. doi: 10.1016/S0161-6420(98)97033-6
18. Kivitie-Kallio S, Norio R. Cohen Syndrome: Essential Features, Natural History, and Heterogeneity. Am J Med Genet (2001) 102:125–35. doi: 10.1002/1096-8628(20010801)102:2<125::AID-AJMG1439>3.0.CO;2-0
19. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New Criteria for Improved Diagnosis of Bardet –Biedl Syndrome: Results of a Population Survey. J Med Genet (1999) 36:437–46.
20. El Chehadeh S, Aral B, Gigot N, Thauvin-Robinet C, Donzel A, Delrue MA, et al. Search for the Best Indicators for the Presence of a VPS13B Gene Mutation and Confirmation of Diagnostic Criteria in a Series of 34 Patients Genotyped for Suspected Cohen Syndrome. J Med Genet (2010) 47(8):549–53. doi: 10.1136/jmg.2009.075028
Keywords: MORMS, INPP5E gene, monogenic obesity, retinal dystrophy, case report
Citation: Drole Torkar A, Avbelj Stefanija M, Bertok S, Trebušak Podkrajšek K, Debeljak M, Stirn Kranjc B, Battelino T and Kotnik P (2021) Novel Insights Into Monogenic Obesity Syndrome Due to INPP5E Gene Variant: A Case Report of a Female Patient. Front. Endocrinol. 12:581134. doi: 10.3389/fendo.2021.581134
Received: 14 July 2020; Accepted: 01 June 2021;
Published: 15 June 2021.
Edited by:
Verónica Mericq, University of Chile, ChileReviewed by:
Anita Morandi, Integrated University Hospital Verona, ItalyOrit Pinhas-Hamiel, Edmond and Lily Safra Children’s Hospital, Israel
Copyright © 2021 Drole Torkar, Avbelj Stefanija, Bertok, Trebušak Podkrajšek, Debeljak, Stirn Kranjc, Battelino and Kotnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Primož Kotnik, cHJpbW96LmtvdG5pa0BtZi51bmktbGouc2k=