 Rossella Cannarella
Rossella Cannarella Rosita A. Condorelli
Rosita A. Condorelli Federica Barbagallo
Federica Barbagallo Sandro La Vignera
Sandro La Vignera Aldo E. Calogero
Aldo E. Calogero
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 22 February 2021
Sec. Endocrinology of Aging
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.554078
This article is part of the Research TopicReproductive Aging in Men and WomenView all 8 articles
Benign prostate hyperplasia (BPH), one of the most common diseases in older men, adversely affects quality-of-life due to the presence of low urinary tract symptoms (LUTS). Numerous data support the presence of an association between BPH-related LUTS (BPH-LUTS) and metabolic syndrome (MetS). Whether hormonal changes occurring in MetS play a role in the pathogenesis of BPH-LUTS is a debated issue. Therefore, this article aimed to systematically review the impact of hormonal changes that occur during aging on the prostate, including the role of sex hormones, insulin-like growth factor 1, thyroid hormones, and insulin. The possible explanatory mechanisms of the association between BPH-LUTS and MetS are also discussed. In particular, the presence of a male polycystic ovarian syndrome (PCOS)-equivalent may represent a possible hypothesis to support this link. Male PCOS-equivalent has been defined as an endocrine syndrome with a metabolic background, which predisposes to the development of type II diabetes mellitus, cardiovascular diseases, prostate cancer, BPH and prostatitis in old age. Its early identification would help prevent the onset of these long-term complications.
Prostate diseases, usually diagnosed in the elderly, mainly include benign prostatic hyperplasia (BPH) and prostate cancer (PCa). BPH is defined as an increased prostate volume, largely due to the cellular proliferation occurring in the transition zone, namely the portion of the prostatic tissue that surrounds the urethra. BPH is an age-dependent disease which can obstruct the prostatic urethra. Its prevalence varies between 5% and 10% in men aged 40, but reaches 80% in men aged 70–80 (1). PCa represents the second cause of mortality for oncologic diseases in Western countries (2). Although ethnicity also influences the epidemiology of PCa (3), increasing age is a widely-accepted risk factor for the development of this malignant tumor. Accordingly, it is more frequently diagnosed in aging men, whereas it is quite uncommon before the age of 45 (4).
Hormonal changes occurring in aging may play a role in the pathogenesis of prostate diseases. These include changes of sexual hormones, insulin-like growth factor 1 (IGF1), thyroid hormones, cortisol and insulin. Importantly, hyperinsulinemia seems to impact on prostate tissue and is associated with prostate inflammation and hyperplasia. However, the mechanisms of this association are not well-recognized yet. This review aims to provide comprehensive and updated insights into the endocrinology of the aging prostate, including possible pathogenic mechanisms linking prostate to endocrinological and metabolic health.
Prostate diseases and androgen decline represent some of the most common conditions in the aging male. Several large cross-sectional studies have shown an approximate 1%–2% annual decline in testosterone (T) levels in men during aging (5, 6). In middle-age men, the combination of low T and hypogonadism-related symptoms is known as “late-onset hypogonadism (LOH).” LOH has an estimated prevalence of 2.1% (7), but it reaches 15% when only biochemical criteria are used for its diagnosis (8). However, the presence of various diseases plays a crucial role in the decrease of T production during aging. These include obesity, type 2 diabetes mellitus and metabolic syndrome (MetS), which can also lead to an estrogen increase (9). Considering the high prevalence of these diseases in elderly men, it is important to understand the role of sex hormones, and, therefore, of their age-related change, in the development and the treatment for prostate disorders.
Androgens are essential for the development and the growth of the male genital system, including the prostate. The effects of androgens on the prostate are mainly mediated by the androgen receptor (AR), which is expressed in both prostatic stromal and epithelial cells (10). T, produced by Leydig cells of the testes, is the main circulating androgen in men and it is converted to its more potent metabolite, the 5α-dihydro-testosterone (DHT) by 5α-reductase whose type 2 is the isoform mostly expressed in the prostate tissue (11). Other androgens include adrenally-derived dehydroepiandrosterone (DHEA), androstenedione and 5α-androstenedione, which can be converted to sex steroids with higher potency and act on the prostate.
During development, androgens stimulate differentiation and proliferation of both the epithelial and the stromal compartments of the gland (12). Prostate differentiation and growth start at the 10–12th week of gestation (13). The increase of T that occurs shortly after birth and then, in the period known as “mini-puberty,” has a crucial role in prostate development (13). After mini-puberty, T levels and prostate volume decrease and prostate growth is quiescent until puberty (14). During puberty, a second phase of prostate growth starts and the prostate size increases from about 10 g at early puberty to almost 20 g around the age of twenty (15). In middle age, a third phase of prostate proliferation begins and continues up to the elderly (15). However, this third phase is different from the first two phases of prostate growth. First, the proliferation involves only one of the three anatomical areas of the prostate, the transitional zone surrounding the urethra, while the entire gland grows during the first two phases. Second, an age-related decline in serum T levels begins in middle age. Therefore, although androgens are responsible for prostate growth in the first two phases, the role of androgens is the object of great debate for middle-aged and elderly men (10). Indeed, a clear association between higher circulating T levels and benign prostatic hyperplasia (BPH) has never been demonstrated (16).
LUTS and BPH have long been considered a relative contraindication to testosterone replacement therapy (TRT). However, recent studies have contradicted the classic idea that androgens inevitably stimulate prostate growth (17). According to the saturation model, the prostate is sensitive to changes in androgen levels when they occur in a severe low range, but this sensitivity is lost for T levels corresponding to mild hypogonadism or eugonadal. Indeed, prostate ARs become saturated at relatively low T levels and thus, the gland becomes unresponsive to further increases in T levels (18). Experimental and clinical studies suggest that the saturation point for human prostate tissue probably occurs in the very low range of T levels, but a precise cutoff value has not been established; it could vary among men (17). According to this model, the association between T and prostate-specific antigen (PSA) is represented by a sigmoid curve. T levels of 8 nmol/L corresponds to the plateau value below which small changes in T levels cause greater variations in PSA (19). As reported in recent reviews (12, 17), studies evaluating the link between TRT and BPH/LUTS have mostly shown that TRT does not have any effect on BPH symptoms [measured by the International Prostate Symptom Score (IPSS)] and occasionally the IPSS score may decrease (20–22). A recent meta-analysis did not show a worsening of LUTS severity in hypogonadal patients treated with T vs. placebo (23). The safety of TRT in patients with severe LUTS remains to be established and the presence of LUTS of moderate-intensity (IPSS ≤19) is not a contraindication to TRT (23).
However, although current evidence suggests that higher T levels are not involved in BPH development, T seems to have a role in this pathogenic process by stimulating inflammation within prostate tissue. Inflammation has a crucial role in the pathogenesis of BPH and low, rather than high, T may stimulate prostate inflammation (12). Nevertheless, an anti-inflammatory effect of T in the prostate has also been described. In an experimental study using human prostatic stromal cell cultures, the treatment in-vitro with different pro-inflammatory stimuli, such as tumor necrosis factor α (TNF-α) or lipopolysaccharide (LPS), or co-culturing with activated CD4β lymphocytes, significantly enhanced the secretion of several cytokines and growth factors (24). The pre-treatment of BPH stromal cells with DHT inhibited the secretion of several inflammatory/growth factors and the proliferation of activated CD4β lymphocytes in a dose-dependent manner (24). The same authors developed a rabbit model of MetS, by feeding New Zealand male rabbits with a high-fat diet (HFD) for 12 weeks (25). Rabbits with MetS developed glucose intolerance, dyslipidemia, hypertension, increased visceral fat accumulation, hypogonadotropic hypogonadism, hyperestrogenism, and prostate inflammation. TRT has been shown to protect rabbits from prostatic hypoxia, fibrosis, and inflammation induced by the high-fat diet that appears to play a role in the development and even progression of BPH/LUTS (25). Interestingly, they found that MetS severity was associated with an increase of AR and estrogen receptor α (ERα), but not of estrogen receptor β (ERβ) gene expression within the prostate (10). This suggests that the prostate could become more sensitive to sex hormone changes that occur during MetS. Indeed, TRT not only corrects the low T levels and the rise of estrogen levels which are typical of MetS, but it also normalizes the majority of MetS-induced prostate alterations (25).
Accordingly, in human BPH stromal cells, AR activation by DHT inhibited TNFα, LPS, or CD4 (+) T cell-induced secretion of inflammatory/growth factors, including interleukin (IL)-6, IL-8, and basic fibroblast growth factor (bFGF), by blocking the nuclear translocation of the nuclear factor kappa-B (NF- κB) (24). Moreover, HFD prostates had increased phosphodiesterase type 5 (PDE5) expression. This was associated with a higher expression of cyclooxygenase 2 (COX2) and TNFα among inflammatory genes and of TGFβ, Rho-associated coiled-coil-containing protein kinase 2 (ROCK2) and α spinal muscular atrophy (αSMA) among those genes specifically involved in fibrosis and myofibroblast activation. Interestingly, HFD-induced PDE5 overexpression was counteracted by T treatment (26). Therefore, recent evidence suggests that the treatment of hypogonadism, which is frequently associated with MetS, not only does not seem to be dangerous for prostate health, but could even prevent the inflammatory process of this gland (12).
Even in inflammation of the prostate caused by Escherichia coli, the most frequent bacterial infection due to the reflux of infected urine into the prostate ducts (ascending urethral infection), T seems to play a protective role. Ho and colleagues have shown that T not only inhibits the invasion and colonization of the uropathogenic Escherichia coli (UPEC), but has also reduced the levels of pro-inflammatory cytokines (IL-1β, IL-6, and IL- 8) induced by UPEC in a dose-dependent manner. In particular, T plays an anti-inflammatory role in LPS-induced prostate cell inflammation by down-regulating Janus kinases, signal transducer and activator of transcription proteins (JAK/STAT1) signaling pathway (JAK/STAT1) (27).
The concern that T promotes the development of PCa has been the object of great debate since the discovery of PCa dependency on androgen in the 1940s thanks to the pioneering studies of Huggins and Hodges, who found that castration resulted in regression of metastatic PCa (28). However, according to the saturation model, androgen-dependency of prostate growth is evident only in hypogonadism (18). Thus, the relationship between T and the risk of PCa remains poorly understood. Several studies have shown an increased risk of PCa in men with hypogonadism (29). This hypothesis was firstly reported in 1996 by Morgentaler and colleagues, who found a higher prevalence of biopsy-detectable PCa in men with low total or free testosterone levels (30) and it was supported by subsequent studies (31–34). In contrast, studies assessing the relationship between high endogenous T levels and the development of PCa are less clear (29). However, there is evidence that previous T levels may influence Gleason scores, clinical outcomes and recurrence (29).
More controversial remains the safety of TRT in patients with a history of PCa after treatment. T could cause the growth of subclinical cancers that are frequently hidden in the prostate of older men (35). Aside, metastases of PCa are hormone-dependent, therefore T should not be administered to these patients (35). However, some clinicians have suggested considering TRT for patients with a history of organ-confined PCa if they have undergone radical prostatectomy, have undetectable PSA for at least two years and low-grade PCa (Gleason score <7). The possible risks and benefits of TRT should be discussed with the patient and TRT should be followed up with appropriate monitoring by experienced physicians (36).
Also, although adrenal androgens have weak androgenic effects, 11-oxy-androgens of adrenal origin can be metabolized in peripheral tissues to potent androgens which might have a role in the development of PCa. Moreover, emerging evidence suggests the role of microbiome components in the development of PCa that are only beginning to be understood. Recent studies have shown that gut bacteria are capable of metabolizing C21 glucocorticoids to 11-oxygenated C19 androgens via an enzyme known as steroid-17,20-desmolase (desAB) encoded by the desAB gene. A cortisol-inducible operon (desABCD) was previously identified in Clostridium scindens ATCC 35704 encoding enzyme involved in anaerobic side-chain cleavage. This operon also encodes a transketolase (desAB) which have steroid-17,20-desmolase activity (37). Interestingly, the compounds cleaved by steroid-17,20-desmolase, 11-ketoandrostenedione (11 KT), and 1,4-androstadiene-3,11,17- trione (AT) were found to stimulate proliferation of PCa cells (LNCaP), to a greater extent than dihydrotestosterone. Further researches are needed to fully understand the mechanism through which these compounds stimulate cell proliferation and the potential clinical role in the development and treatment of prostate cancer (38).
Prostate is commonly considered a target of androgens, but also estrogens can play an important role in prostate growth and differentiation. In fact, both ERα and ERβ are expressed in the prostate. ERα is mainly located in the stromal cells and its activation regulates the growth of both stromal and epithelial prostatic cells via paracrine mediators like stromal bFGF, epidermal growth factor (EGF), and IGF1 (39, 40). In contrast, ERβ is located in epithelial cells and their activation modulates the estrogenic signals in the prostate (41).
17ß-Estradiol (E2) is considered the most potent estrogen in men and it mainly originates from aromatization of T in fat and muscle, whereas about 20% is secreted by Leydig cells (42). In aging men, paralleling the decrease of T levels, the ratio of estrogens to androgens shows an important increase (43). There are several endogenous and exogenous estrogens which may play an important role in prostate. Endogenous estrogens include estrone (E1), which is considered to have minimal influence within the prostate, and estriol (E3), the main estrogen of pregnancy, which is present in minimal concentrations in men. However, E2 can be a potent inducer of prostatic proliferation (44). Local steroids with ER agonist activity include also 5α-androstane-3β, 17β-diol (3βAdiol), and 7α-hydroxy-DHEA (7HD). The effects of these sex steroids are not fully understood but they seem to influence prostate hyperplasia (44). Exogenous estrogens include therapeutic drugs, phytoestrogens, and endocrine disruptors. ERs have a high affinity for environmental estrogens such as bisphenol A (BPA), phthalates, pesticides, etc. In rodent studies, developmental BPA, DES, or E2 exposure affects prostate epigenome and thus causes increased prostate susceptibility to dysplasia and hormonal carcinogenesis with aging (45).
Estrogen hormone action in the prostate depends not only on the types of estrogens but most of all, from the type of ER. Specifically, ERα activation is associated with prostate hyperplasia, inflammation, and dysplasia (46). However, the main stimulation of inflammation in BPH by estrogens seem to be mediated by the membrane ER G protein-coupled receptor 30 (GPR30) or G protein-coupled ER (GPER), which are also expressed in prostate stromal cells (47). On the contrary, ERβ inhibits proliferation and its knockout results in prostate hyperplasia (48). The prostatic hyperplasia observed in ERβ knockout mice is attributed to the unopposed action to ERα, suggesting that the ratio ERα/ERβ is an important factor in estrogen-induced proliferation (44).
Recent studies suggest that not only low T levels, but also an increase of estrogens may favor BPH/LUTS progression (10). Marmorston and colleagues first reported that the E2/T ratio in 24-h urinary collections was elevated in men with BPH compared to normal controls (49). Other epidemiologic studies have found an association between BPH and higher serum estrogen levels or estrogen/androgen ratio (50, 51). As previously reported, prostate inflammation could be amplified and maintained by metabolic alteration occurring in conditions such as MetS. Vignozzi and colleagues showed that HFD rabbits had higher E2 to T ratio and lower urinary tract fibrosis, which improved with TRT (25). Recent evidence has also shown that leptin, a hormone produced by adipocytes, induces proliferative effects in prostate cells. This effect may be partially mediated by the direct effect of leptin on estrogen metabolism, as leptin induces aromatase expression (52).
Therefore, ERα, as a key mediator, is also a potential therapeutic target in BPH. The block of conversion of androgens to estrogens by aromatase inhibitors seems to prevent prostate hyperplasia (12). Similar to aromatase inhibitors, selective estrogen receptor modulators (SERMs) have shown anti-proliferative effects on prostate tissue (53).
Evidence suggests that ERα mediates the harmful effects of estrogen not only promoting BPH/LUTS, but also prostate carcinogenesis. In the early 1980s, Noble showed the estrogen-dependence of PCa in a rat model (54). In aromatase knockout (KO) mice, high T levels only lead to prostate hypertrophy and hyperplasia, whereas high E2 and low T levels induced also premalignant lesions (55). Indeed, prostatic intraepithelial neoplasia does not occur in ERαKO mice and SERMs that bind and inhibit ERα prevent PCa progression in mice and men (56–58). Epidemiological studies have confirmed the role of estrogens on PCa (59). African-American men, who have high serum E2 levels, present an increased risk of developing PCa (60). Indeed, ERα expression is significantly associated with high Gleason score and poor survival in PCa patients (61), whereas the expression of ERβ seems decreased or lost in PCa samples (62). Therefore, preclinical and clinical results suggest that ERβ agonists may be useful in PCa therapy, especially in the early stage (59).
Since the discovery of estrogen dependence of PCa, numerous clinical trials were conducted and molecular and functional effects of antiestrogen treatment in PCa have been conducted. Androgen deprivation therapy (ADT) is the first line of treatment for PCa. Nonetheless, ADT frequently induces resistance and PCa can progress toward an androgen-independent form, known as castration-resistant PCa, characterized by worse prognosis. For this reason, alternative approaches to androgen ablation have been investigated to prevent the progression of PCa. Currently, molecular networks of estrogenic signaling via ERα, ERβ, and GPR30 in PCa have not been fully understood but new compounds, whose efficacy has been successfully tested in preclinical and clinical models of PC, opened the way for novel therapeutic strategies for treating prostatic diseases (41, 59).
In conclusion, the above-mentioned evidence suggests a role for age-related changes in sex hormones; in particular, hypogonadism and the increase in estrogen levels related to obesity play a pivotal role in the pathogenesis of age-related prostatic diseases.
Several studies indicate that IGF1 declines with age (63). Interestingly, IGF1 seems to have an important role in the pathogenesis of metabolic syndrome. Indeed, several studies, both in-vitro and in-vivo have shown the association between low levels of IGF1 and altered lipid metabolism, cardiovascular disease and diabetes (64). Nevertheless, increased levels of IGF1 have also been reported in patients with diabetes mellitus (65).
A large body of evidence shows that the growth hormone (GH)- IGF1 endocrine axis has a pivotal role in the growth and development of prostate in normal physiology as in pathological conditions (66–69). IGF1 is secreted mainly by the liver under GH stimulation, but it is also expressed locally within the stromal and epithelial cells of the prostate, where it can act in autocrine/paracrine manner (70). The binding of IGF1 to the IGF1 receptor (IGF1R) or the insulin receptor (IR) in the prostate gland, activates the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway and RAF/MAPK pathway, which promote cell survival and proliferation (70).
Some studies have shown a greater prevalence of prostate enlargement and BPH in patients with acromegaly (67, 69). Kumar and colleagues documented structural changes in acromegalic patients by prostatic biopsy (71). They found that patients with acromegaly have higher IPSS, an increased rate of prostate enlargement on rectal exploration and ultrasound, higher PSA levels and obstructive pattern on uroflowmetry, and structural prostate changes, regardless of their age, disease activity, or gonadal status (71). These results suggest that the hyper-activation of the GH/IGF1 axis plays an important role in the pathogenesis of BPH in patients with acromegaly. This is further supported by the decrease of prostate volume in patients with inactive disease (for more than 24 months) (71).
Also, there is ample evidence that IGF1 and the signal transduction network that it regulates have important roles in the development of tumors (72). Epidemiological studies have reported that high circulating levels of IGF1 are associated with an increased risk of PCa, particularly advanced disease (70, 73). Experimental studies have shown that the binding of IGF1 to both the IGF1R and IR promotes mitogen signaling events, increases cell proliferation and inhibits apoptosis (72). Both IGF1R and IR are overexpressed in PCa tissue (74). Moreover, the expression of the components of the GH, insulin, and IGF1 axes can be finely modulated in the prostate by environmental factors such as the diet. As previously described, it was well documented that obesity promotes structural changes in the prostate (12). Prostate of obese mice presents altered mRNA expression levels of GH receptor (GHR) and glucose transporter 4 (GLUT4) and an up-regulation in IGF-binding protein 3 (Igfbp3) expression, which might have pathophysiological implications (75). Emerging evidence suggests a direct regulation of IGF/insulin signaling to the transmembrane serine protease 2 (TMPRSS2)/V-ETS avian erythroblastosis E26 oncogene homolog (ERG) gene fusion, one of the main somatic events in PCa (76). It has been hypothesized that ERG-positive tumors may be more sensitive to IGF/insulin signaling, which could promote PCa progression (70). Furthermore, another possible mechanism is the activation of AR. IGF1 signaling stimulates ARs by attenuating forkhead box-containing protein O subfamily (Foxo1) inhibition (77). Recent researches have highlighted the involvement of not only IGF1 signaling but also of GH itself. GH is responsible for the activation of transcription 5 (STAT5) protein which is involved in the development of several tumors including PCa (78). GHRH antagonists have been shown to decrease directly prostate volume without involving the androgen pathway (79).
Neoplastic, cardiovascular and respiratory disorders are common causes of mortality and morbidity in patients with acromegaly. Nonetheless the higher prevalence of “prostate structural changes” and a possibly increased risk of PCa in patients with acromegaly, no guidelines recommend screening for prostatic disorders. Therefore, a prostatic evaluation may be useful in the work-up of all male patients with acromegaly (71). Actual evidence supports the possible involvement of GH/IGF1 in the pathogenesis of BPH and PCa also in the general population and therapeutic agents targeting the IGF1R may be beneficial in the treatment of prostate diseases.
Finally, the increase of IGF1 seems to play a role in the pathogenesis of BPH and PCa, since it stimulates prostate cell proliferation. Notably, this hormone usually declines with age, although metabolic abnormalities and acromegaly leads to IGF1 increase. Therefore, IGF1 levels should be assessed in patients with prostatic diseases, when diabetes (or other metabolic alterations) or acromegalic features concomitantly occur.
Thyroid hormones (THs) are involved in cellular growth, metabolism and differentiation. Their effects are mainly mediated by triiodothyronine (T3) that, by binding the nuclear TH receptors (TRs), activates TH response elements (TREs) in the promoter of TH target genes. However, non-classical or non-genomic effects of TH have also been described (80).
The prevalence of thyroid dysfunction increases in the elderly. Recent data from observational studies suggest that serum thyroid-stimulating hormone (TSH) levels increase in older people. The US National Health and Nutritional Examination Survey (NHANES) III study showed that serum TSH concentrations, as well as serum thyroid peroxidase (TPOAb) and thyroglobulin (TgAb) antibodies, increased with age in both men and women (81). Hyperthyroidism is less common than hypothyroidism in the elderly and mainly caused by autonomously functioning thyroid nodules (82).
TRs are strongly expressed in the human prostate (83) and several studies have investigated the role of THs in the development of prostate diseases. Epidemiological studies have found that men with BPH or PCa have significantly increased serum T3 levels compared with euthyroid men (84, 85). Accordingly, Eldhose and colleagues found significantly increased levels of free T3 (FT3) and free thyroxine (FT4) and decreased levels of TSH in patients with BPH compared with controls. They also found that FT3 correlated positively and TSH negatively with prostate volume (86). It has also been shown that patients with hypothyroidism have a decreased risk of developing PCa compared to euthyroid men (87). A recent meta-analysis has shown that hyperthyroidism was associated with higher risks of PCa (pooled risk ratio: 1.35, 95%CI: 1.05-1.74) compared with euthyroidism (88). Experimental studies have shown that THs increase PCa cell proliferation in-vitro (89). Specifically, Hsieh and Juang found that T3 can enhance LNCaP cell proliferation and that this effect is cell-specific. LNCaP cells are androgen-sensitive and well-differentiated cells, derived from metastatic lymph nodes of a PCa patient (90). Other studies also indicated that T3 has an important role in the regulation of growth and differentiation of LNCaP cells and PSA expression (91–93). Specifically, a direct effect of T3 on PSA expression was described. Zhu and Young identified a functional TRE in the PSA promoter region, suggesting that T3 regulates PSA expression at the transcription level (93). Also, it has been described that T3 upregulates the proliferation of LNCaP cells through downregulation of the B-cell translocation gene 2 (BTG2), a gene involved in cell-cycle regulation, through the TRE pathway (94). Experimental studies have also shown that THs promote carcinogenesis by inducing angiogenesis (95). Non-classical mechanisms of THs may be also responsible for the effects on the prostate. The binding of TH to plasma membrane receptor integrin avb3 might activate various pro-carcinogenic pathways, including protein inhibitor 3 kinases (PI-3 K) and mitogen activated-protein kinases 1/2 (MAPK 1/2), thereby increasing cell proliferation and angiogenesis (80).
In conclusion, although a relationship between TH and prostate seems clear, the exact mechanisms by which they act need to be further investigated and targeting TH actions might become an alternative adjuvant therapy against PCa proliferation. Due to the age-related rise in TSH levels and the negative correlation between serum TSH levels and prostate volume (81), the role of this hormone in the pathogenesis of prostate diseases in the aging male seems unlikely.
BPH is considered an age-dependent disease, whose etiology is currently poorly understood. Several studies address to insulin-resistance and hyperinsulinemia, which are components of MetS, a role in the pathogenesis of BPH and LUTS. Accordingly, hyperinsulinemia has been shown to enhance prostatic epithelial cell proliferation in-vitro (96) and, conversely, hypoinsulinemia decreases prostate volume (97). In line with these findings, patients with serum insulin levels >13 mU/l have a greater prostate volume and annual BPH growth rate compared with those with insulin levels <7 mU/l (98).
Data from the second Nord-Trondelag Health Study, carried out in 21,694 patients, revealed a significantly higher risk for LUTS in patients with diabetes mellitus than in non-diabetic men (99). Also, the risk for developing LUTS has been esteemed twice in patients with diabetes compared with non-diabetic men (100). Accordingly, insulin-resistance is an independent predictor of severe LUTS development (101). This may be attributed to the autonomic nervous system hyperactivity, mainly involving the α-adrenergic pathway, which is closely associated with hyperinsulinemia and is involved in the pathogenesis of LUTS (102).
A recent retrospective study carried out in about 900 patients reported that, after correction for age, insulin levels and insulin-resistance are significantly associated with prostate volume. Interestingly, MetS predicted BPH/LUTS clinical progression (103), thus pointing to the additional role of MetS, other than insulin, in BPH/LUTS. A systematic review with meta-analysis performed on 8,476 participants, including 5,554 (30.1%) with and 12,922 (69.9%) without MetS showed a significantly higher prostate volume in patients compared to controls (104).
The National Cholesterol Education Program adult treatment panel III (2005 revision) defines MetS as the presence of three or more criteria among the following: i) abdominal obesity (waist circumference >102 cm), ii) hypertriglyceridemia (>150 mg/dl) or medications, iii) low high-density lipoprotein (HDL) cholesterol (<40 mg/dl) or medications, iv) hypertension (>130/85 mmHg) or medication, v) high fasting glucose (>110 mg/dl) or medication (105). The evidence supports an association between BPH and each of the MetS components (102).
In greater detail, a BMI >35 Kg/m2 has been associated with a 3.5-fold higher risk of developing an increased prostate volume (>40 ml) (106) and with a 1.2-fold higher risk of developing LUTS compared to those with a BMI <25 kg/m2 (107). Data obtained in 21,694 patients have confirmed these findings by showing that BMI is a predictor for LUTS development (99). Other reports also support such conclusions (100, 108).
Notably, dyslipidemia is also associated with BPH. Patients with lower HDL cholesterol show increased prostate volume and a higher annual BPH growth rate compared with higher HDL cholesterol values (109). Also, patients with BPH have higher total and low-density lipoprotein (LDL) cholesterol than those without BPH (110) and an association between LDL cholesterol and BPH has been already shown (111). Also, among patients with diabetes, those with the highest LDL cholesterol levels have a 4-fold higher risk of developing BPH (111). These findings suggest that dyslipidemia strongly associates with BPH in the presence of other signs of MetS.
Results from 2,372 patients enrolled in the Third National Health and Nutrition Examination Survey (NHANES III) provide a role for the association between hypertension and LUTS-BPH. Particularly, those with a positive anamnesis for hypertension have increased odds for LUTS compared to those with no hypertension (112). These findings have been largely confirmed also elsewhere (100). Moreover, patients with hypertension showed larger prostate volume and a higher annual BPH growth rate compared with controls (109).
In addition to mechanisms closely associated to hyperinsulinemia, the impairment of nitric oxide (NO) and NO synthase (NOS) activity, the Rho kinase system, the pro-inflammatory status and the abnormality of sexual hormones seems also to be involved in BPH/LUTS pathogenesis in patients with MetS (102, 113). However, the hypothesis of the existence of a male polycystic ovary syndrome (PCOS) equivalent may represent an additional pathogenic mechanism deserving of consideration (please see Male PCOS Equivalent Exists: A New Syndrome)?.
Several lines of experimental evidence have pointed to the role of insulin in the enhancement of prostate carcinogenesis, particularly in advanced PCa. More in detail, androgens are known to induce epithelial differentiation in prostate cells (114). Androgen deprivation therapy (ADT), usually prescribed in patients with advanced PCa, leads to cellular de-differentiation and trans-differentiation (114–116). Under such conditions, hyperinsulinemia, a common side effect of ADT, enhances PCa cell plasticity, thus increasing tumor migration and invasiveness, by upregulating the Forkhead Box Protein C2 (FOXC2) transcription factor (117). Furthermore, PCa cell lines have an increased ratio of pyruvate dehydrogenase flux to citrate synthase flux following exposure to insulin, which correlates with an insulin dose-dependent increase in cell division (118). This experimental evidence suggests that ADT-induced hyperinsulinemia may likely impact on cancer progression and metastasis.
In line with these in-vitro findings, hyperinsulinemia has been associated with a greater PCa mortality (119–122) and treatment failure (122). A retrospective study evaluating the effects of MetS on the time to develop tumor progression in patients with PCa and ADT reported a significantly shorter time to PSA progression (16 vs. 36 months) and a trend toward a shorter overall survival (36.5 vs. 46.7 months) when MetS is present compared with those who do not have it (122). Some Authors have even proposed to define PCa as a component of MetS, since hyperisulinemia prospectively is a risk factor for PCa deaths (119). A long-term survival analysis performed on 2,546 patients who developed PCa, found that patients with weight excess and high C-peptide levels have a four-fold higher risk of mortality compared with those with a BMI <25 kg/m2 and low C-peptide levels, independently of other clinical predictors (120).
Supporting these data, the IR is expressed in PCa cells and, interestingly, its expression increases with the Gleason score (74, 123) and ADT (124).
Despite such findings, whether treatment with metformin or other insulin-sensitizing drugs may play a role in PCa therapy is still a matter of debate. An in-vitro study reported that GSK1838705A, a potent insulin-like growth factor-1 receptor (IGF1R)/IR inhibitor, is capable of decreasing docetaxel-resistant PCa cell viability and migration, as well as in-vivo tumor growth. Therefore, a role for this drug for the treatment of advanced resistant PCa has been postulated (125). Also, by blocking IGF1R, metformin has been reported to significantly inhibit PCa cell proliferation, migration and invasiveness, suggesting a role in PCa treatment (126). Furthermore, tyrphostin NT157, which inhibits the insulin receptor substrates 1 and 2 (IRS1 and IRS2), was shown to decrease proliferation and increase apoptosis of PCa cell lines (127).
On these premises, a population-based study carried out in a cohort of 1,001 patients with PCa and 942 controls showed a lower PCa prevalence in patients using metformin compared with non-users. Also, it showed an inverse relationship between PCa risk and metformin length of treatment, intensity of use and cumulative dosage (128). Similarly, a population-based study on 24,723 case-control pairs found a lower PCa risk in men using antidiabetic medication (129). Other evidence supports a beneficial effect of metformin in lowering PCa incidence and overall survival (130–132).
In conclusion, despite the evidence on the possible role of antidiabetic drugs as adjuvant therapy in patients with PCa, particularly in those on ADT, is growing, there is still insufficient data from randomized trials to suggest their use in the clinical practice (133).
PCOS is a very common endocrine disorder in women of reproductive age, with a prevalence of 6-15% (134). A genetic background and environmental factors are involved in its etiology. Despite PCOS diagnostic criteria mainly include hyperandrogenism, oligo-ovulation, or anovulation and polycystic ovaries (135), the role of metabolic dysfunction in the pathogenesis of this syndrome is widely accepted. Indeed, up to 75% of patients with PCOS are insulin-resistant and some are hyperinsulinemic (135). Accordingly, the presence of polycystic ovaries, which gave the name to the syndrome, is only one of the many downstream clinical manifestations of PCOS and it is not the pivotal pathogenic factor leading to the development of this syndrome (136, 137).
Since a genetic background has been observed in PCOS, this hereditary predisposition can be potentially inherited by the male sibling of the affected patients. Interestingly, the brothers and the relatives of women with PCOS have a high prevalence of hormonal and metabolic abnormalities (138–140). They also show a greater prevalence of early-onset androgenetic alopecia (AGA) (141), which has been suggested as a clinical sign of the male PCOS equivalent (142–144). The occurrence of hormonal and metabolic abnormalities in men with early-onset AGA (younger than 35 years) has been reported (145, 146) (Table 1). A meta-analytic study performed in 1009 unrelated men found increased luteotropic hormone (LH) and dehydroepiandrosterone (DHEAS), decreased sex hormone-binding globulin (SHBG), a downward trend for FSH and an upward trend for the LH/FSH ratio in patients with early-onset AGA compared with controls. This hormonal pattern somewhat resembles that found in female PCOS. The same meta-analysis showed a significant increase in insulin levels and HOMA index, total and LDL cholesterol and triglycerides in patients vs. controls, already before the age of 35 (147).
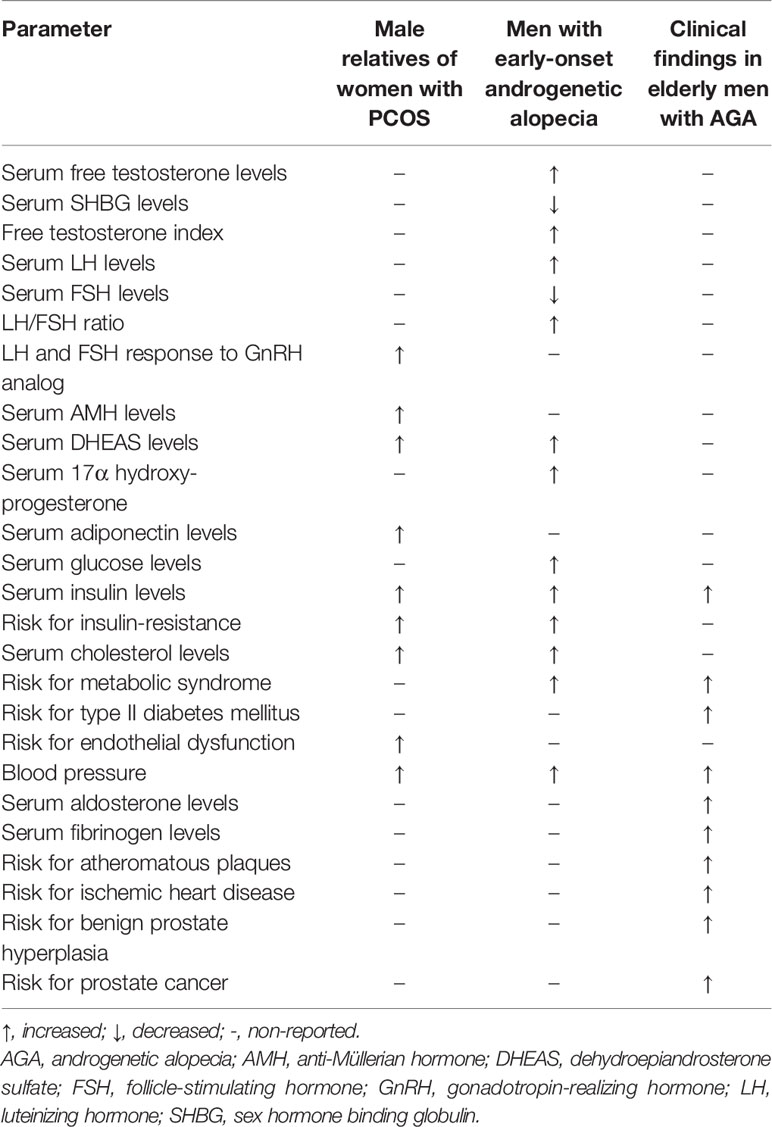
Table 1 Biochemical and clinical parameters in male relatives of women with polycystic ovarian syndrome, in young and elderly men with early androgenetic alopecia (146).
Large cohort studies in older men with early-onset AGA have clearly shown a higher prevalence of type II diabetes mellitus (148) and a greater risk for cardiovascular disease (CVD) (149). Thus, AGA has been suggested as an independent predictor of mortality for diabetes mellitus and CVD (150). Curiously, these men have also a higher prevalence of prostate diseases, such as BPH (151–155), PCa (156), and prostatitis (157), which are known to be influenced by the metabolic status. In this view, prostate diseases may be seen as long-term complications of the male PCOS equivalent syndrome (Table 2) (146). This concept supports the association between metabolic abnormalities and prostate diseases in the elderly. The acknowledgment and timely diagnosis of this syndrome may be of great utility to early identify and treat metabolic disorders in men, preventing the long-term complications, including diabetes, CVD and prostate diseases.

Table 2 Age-related features of the male polycystic ovarian syndrome (PCOS) equivalent.
In conclusion, the receptors for sex hormones, IGF1, THs and insulin expressed in the prostate cells indicate the close relationship between the prostate and hormones (Figure 1). Also, serum levels of these hormones vary with aging and are influenced by several comorbidities such as gonadal dysfunction, thyroid disease, obesity, MetS, insulin-resistance and diabetes mellitus. In particular, the evidence attributes a role in the pathogenesis of prostate inflammation, BPH and PCa to the age-related change in T levels as well as the increase in obesity-related estrogen levels. Furthermore, the increase in IGF1, which occurs in diabetic or acromegalic patients, leads to the proliferation of prostate cells. In contrast, the age-related decline in serum TSH levels is unlikely to be involved in the pathogenesis of prostate disease. Finally, metabolic abnormalities can lead to insulin (but also to IGF1 and E2)-mediated prostate inflammation and hyperplasia. This justifies the importance of endocrine counseling in patients with prostate diseases, which primarily allows the identification of endocrine or metabolic comorbidities responsible for an increased cardiovascular risk. Further research is needed to confirm the existence of a male-PCOS equivalent.
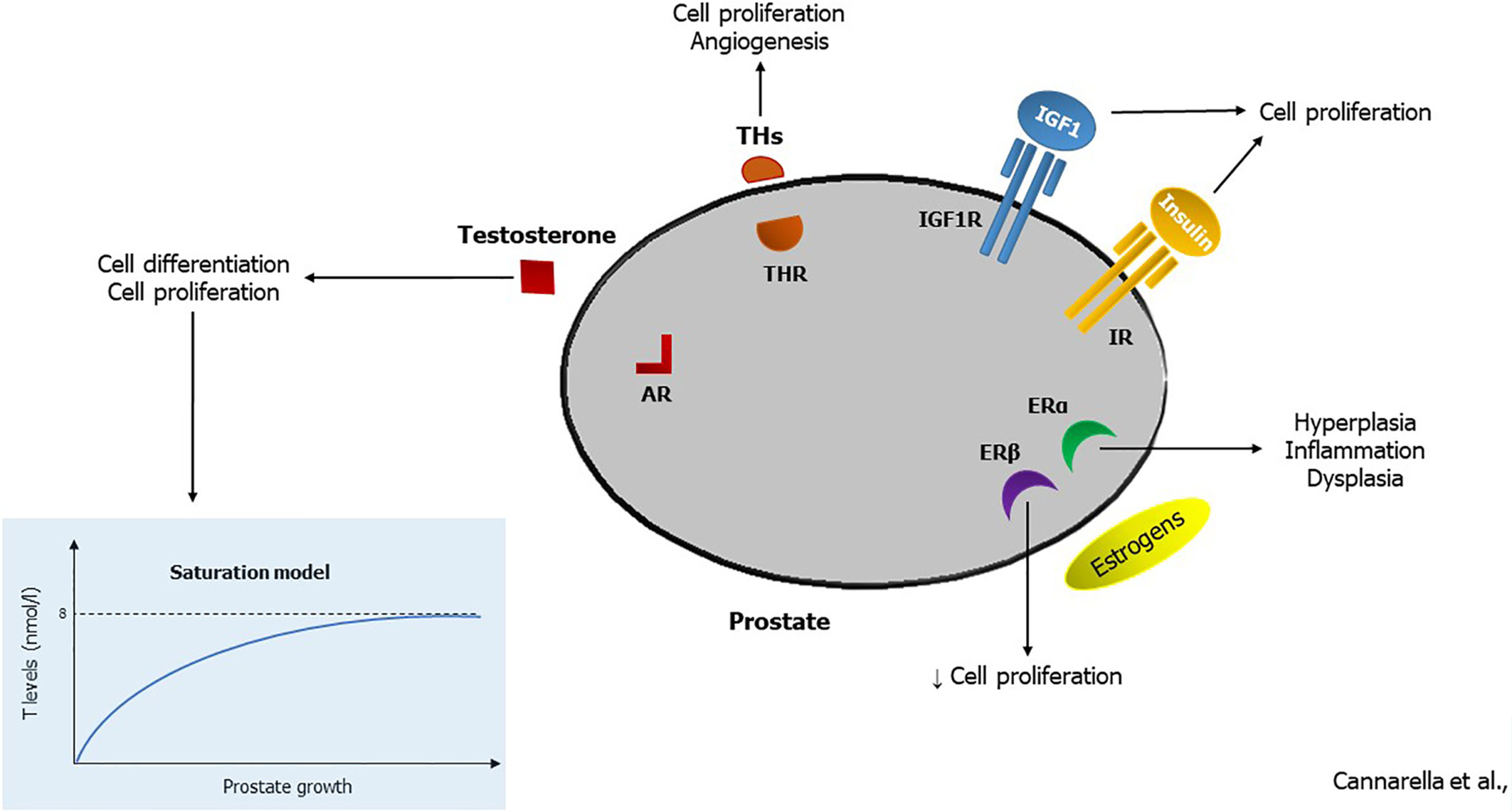
Figure 1 Hormonal effects in the prostatic tissue. By binding to the androgen receptor (AR), testosterone (T) stimulates cell differentiation in prenatal life. According to the saturation model, it induces cell proliferation when its serum levels are in the hypogonadal range (<8 nmol/l). Thyroid hormones (THs) enhance prostate cell proliferation and angiogenesis, triggering the TH receptor (THR). Insulin-like growth factor 1 (IGF1) and insulin induce cell proliferation by interacting with their receptors (IGF1R and IR, respectively). Finally, estrogens can trigger the estrogen receptor α (ERα) that stimulates cell hyperplasia, inflammation, and dysplasia, or the estrogen receptor β (ERβ) that hinders cell proliferation.
RC conceived the study and contributed to writing the draft of the paper. RAC contributed to write the draft of the paper and critically revised the final version of the paper. FB searched the studies and contributed to write the draft of the paper. SL contributed to writing the final version of the paper and critically revised it. AC was the project administrator. He contributed to writing the final version of the paper and critically revised it. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Vuichoud C, Loughlin KR. Benign prostatic hyperplasia: epidemiology, economics and evaluation. Can J Urol (2015) 22(1):1–6.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin (2016) 66:7–30. doi: 10.3322/caac.21332
3. De Santis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI, et al. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin (2016) 66(4):290–308. doi: 10.3322/caac.21340
4. Daniyal M, Siddiqui ZA, Akram M, Asif HM, Sultana S, Khan A. Epidemiology, etiology, diagnosis and treatment of prostate cancer. Asian Pac J Cancer Prev (2014) 15(22):9575–8. doi: 10.7314/APJCP.2014.15.22.9575
5. Feldman HA, Longcope C, Derby CA, Johannes CB, Araujo AB, Coviello, et al. Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts male aging study. J Clin Endocrinol Metab (2002) 87(2):589–98. doi: 10.1210/jcem.87.2.8201
6. O’Donnell AB, Araujo AB, McKinlay JB. The health of normally aging men: The Massachusetts Male Aging Study (1987–2004). Exp Gerontol (2004) 39:975–84. doi: 10.1016/j.exger.2004.03.023
7. Tajar A, Huhtaniemi IT, O’Neill TW, Finn JD, Pye SR, Lee DM, et al. Characteristics of androgen deficiency in late-onset hypogonadism: results from the European Male Aging Study (EMAS). J Clin Endocrinol Metab (2012) 97(5):1508–16. doi: 10.1210/jc.2011-2513
8. Buvat J, Maggi M, Guay A, Torres LO. Testosterone deficiency in men: systematic review and standard operating procedures for diagnosis and treatment. J Sex Med (2013) 10(1):245–84. doi: 10.1111/j.1743-6109.2012.02783.x
9. Huhtaniemi I. Late-onset hypogonadism: Current concepts and controversies of pathogenesis, diagnosis and treatment. Asian J Androl (2014) 16:192–202. doi: 10.4103/1008-682X.122336
10. Corona G, Vignozzi L, Rastrelli G, Lotti F, Cipriani S, Maggi M. Benign prostatic hyperplasia: a new metabolic disease of the aging male and its correlation with sexual dysfunctions. Int J Endocrinol (2014) 2014:329456. doi: 10.1155/2014/329456
11. Bartsch G, Rittmaster RS, Klocker H. Dihydrotestosterone and the concept of 5alpha-reductase inhibition in human benign prostatic hyperplasia. Eur Urol (2000) 37(4):367–80. doi: 10.1159/000020181
12. Rastrelli G, Vignozzi L, Corona G, Maggi M. Testosterone and Benign Prostatic Hyperplasia. Sex Med Rev (2019) 7(2):259–71. doi: 10.1016/j.sxmr.2018.10.006
13. Kellokumpu-Lehtinen P, Santti R, Pelliniemi LJ. Correlation of early cytodifferentiation of the human fetal prostate and Leydig cells. Anat Rec (1980) 196:263–73. doi: 10.1002/ar.1091960302
14. Cunha GR, Alarid ET, Turner T, Donjacour AA, Boutin EL, Foster BA. Normal and abnormal development of the male urogenital tract. Role of androgens, mesenchymal-epithelial interactions, and growth factors. J Androl (1992) 13(6):465–75.
15. Schauer IG, Rowley DR. The functional role of reactive stroma in benign prostatic hyperplasia. Differentiation (2011) 82:200–10. doi: 10.1016/j.diff.2011.05.007
16. Liu CC, Huang SP, Li WM, Wang CJ, Chou YH, Li CC, et al. Relationship between serum testosterone and measures of benign prostatic hyperplasia in aging men. Urology (2007) 70(4):677–80. doi: 10.1016/j.urology.2007.05.025
17. Miranda EP, Torres LO. Late-onset hypogonadism: Prostate safety. Andrology (2020) 8(6):1606–13. doi: 10.1111/andr.12772 (in press).
18. Morgentaler A, Traish AM. Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth. Eur Urol (2009) 55(2):310–20. doi: 10.1016/j.eururo.2008.09.024
19. Rastrelli G, Corona G, Vignozzi L, Maseroli E, Silverii A, Monami M, et al. Serum PSA as a predictor of testosterone deficiency. J Sex Med (2013) 10(10):2518–28. doi: 10.1111/jsm.12266
20. Karazindiyanoğlu S, Cayan S. The effect of testosterone therapy on lower urinary tract symptoms/bladder and sexual functions in men with symptomatic late-onset hypogonadism. Aging Male (2008) 11(3):146–9. doi: 10.1080/13685530802290438
21. Haider A, Gooren LJ, Padungtod P, Saad F. Concurrent improvement of the metabolic syndrome and lower urinary tract symptoms upon normalisation of plasma testosterone levels in hypogonadal elderly men. Andrologia (2009) 41(1):7–13. doi: 10.1111/j.1439-0272.2008.00880.x
22. Shigehara K, Sugimoto K, Konaka H, Iijima M, Fukushima M, Maeda Y, et al. Androgen replacement therapy contributes to improving lower urinary tract symptoms in patients with hypogonadism and benign prostate hypertrophy: a randomised controlled study. Aging Male (2011) 14(1):53–8. doi: 10.3109/13685538.2010.518178
23. Kohn TP, Mata DA, Ramasamy R, Lipshultz LI. Effects of Testosterone Replacement Therapy on Lower Urinary Tract Symptoms: A Systematic Review and Meta-analysis. Eur Urol (2016) 69(6):1083–90. doi: 10.1016/j.eururo.2016.01.043
24. Vignozzi L, Cellai I, Santi R, Lombardelli L, Morelli A, Comeglio P, et al. Antiinflammatory effect of androgen receptor activation in human benign prostatic hyperplasia cells. J Endocrinol (2012) 214(1):31–43. doi: 10.1530/JOE-12-0142
25. Vignozzi L, Morelli A, Sarchielli E, Comeglio P, Filippi S, Cellai I, et al. Testosterone protects from metabolic syndrome-associated prostate inflammation: an experimental study in rabbit. J Endocrinol (2012) 212(1):71–84. doi: 10.1530/JOE-11-0289
26. Morelli A, Comeglio P, Filippi S, Sarchielli E, Vignozzi L, Maneschi E, et al. Mechanism of action of phosphodiesterase type 5 inhibition in metabolic syndrome-associated prostate alterations: an experimental study in the rabbit. Prostate (2013) 73(4):428–41. doi: 10.1002/pros.22584
27. Ho CH, Fan CK, Yu HJ, Wu CC, Chen KC, Liu SP, et al. Testosterone suppresses uropathogenic Escherichia coli invasion and colonization within prostate cells and inhibits inflammatory responses through JAK/STAT-1 signaling pathway. PloS One (2017) 12(6):e0180244. [published correction appears in PLoS One. 12 (12), e0190317]. doi: 10.1371/journal.pone.0180244
28. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin (1972) 22(4):232–40. doi: 10.3322/canjclin.22.4.232
29. Rodriguez KM, Pastuszak AW, Khera M. The Role of Testosterone Therapy in the Setting of Prostate Cancer. Curr Urol Rep (2018) 19(8):67. doi: 10.1007/s11934-018-0812-1
30. Morgentaler A, Bruning CO, DeWolf WC. Occult prostate cancer in men with low serum testosterone levels. JAMA (1996) 276(23):1904–6. doi: 10.1001/jama.276.23.1904
31. Shin BS, Hwang EC, Im CM, Kim SO, Jung SI, Kang TW, et al. Is a decreased serum testosterone level a risk factor for prostate cancer? A cohort study of Korean men. Korean J Urol (2010) 51(12):819–23. doi: 10.4111/kju.2010.51.12.819
32. García-Cruz E, Piqueras M, Huguet J, Peri L, Izquierdo L, Musquera M, et al. Low testosterone levels are related to poor prognosis factors in men with prostate cancer prior to treatment. BJU Int (2012) 110(11):541–6. doi: 10.1111/j.1464-410X.2012.11232.x
33. Mearini L, Zucchi A, Nunzi E, Villirillo T, Bini V, Porena M. Low serum testosterone levels are predictive of prostate cancer. World J Urol (2013) 31(2):247–52. doi: 10.1007/s00345-011-0793-x
34. Bayar G, Şirin H, Aydın M, Özağarı A, Tanrıverdi O, Kadıhasanoğlu M, et al. Low free and bioavailable testosterone levels may predict pathologically-proven high-risk prostate cancer: a prospective, clinical trial. Turk J Urol (2017) 43(3):289–96. doi: 10.5152/tud.2017.35467
35. Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, et al. Testosterone Therapy in Men With Hypogonadism: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2018) 103(5):1715–44. doi: 10.1210/jc.2018-00229
36. Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T, et al. EAU guidelines on prostate cancer. part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur Urol (2014) 65(1):124–37. doi: 10.1016/j.eururo.2013.09.046
37. Ridlon JM, Ikegawa S, Alves JM, Zhou B, Kobayashi A, Iida T, et al. Clostridium scindens: a human gut microbe with a high potential to convert glucocorticoids into androgens. J Lipid Res (2013) 54(9):2437–49. doi: 10.1194/jlr.M038869
38. Ly LK, Rowles JL, Paul HM, Alves JMP, Yemm C, Wolf PM, et al. Bacterial steroid-17,20-desmolase is a taxonomically rare enzymatic pathway that converts prednisone to 1,4-androstanediene-3,11,17-trione, a metabolite that causes proliferation of prostate cancer cells. J Steroid Biochem Mol Biol (2019) 199:105567. doi: 10.1016/j.jsbmb.2019.105567
39. Bacher M, Rausch U, Goebel HW, Polzar B, Mannherz HG, Aumüller G. Stromal and epithelial cells from rat ventral prostate during androgen deprivation and estrogen treatment–regulation of transcription. Exp Clin Endocrinol (1993) 101(2):78–86. doi: 10.1055/s-0029-1211211
40. Gupta C. The role of estrogen receptor, androgen receptor and growth factors in diethylstilbestrol-induced programming of prostate differentiation. Urol Res (2000) 28(4):223–9. doi: 10.1007/s002400000107
41. Lau KM, To KF. Importance of Estrogenic Signaling and Its Mediated Receptors in Prostate Cancer. Int J Mol Sci (2016) 17(9):E1434. doi: 10.3390/ijms17091434
42. Vermeulen A, Kaufman JM, Goemaere S, van Pottelberg I. Estradiol in elderly men. Aging Male (2002) 5:98–102. doi: 10.1080/713604678
43. Belanger A, Candas B, Dupont A, Cusan L, Diamond P, Gomez JL, et al. Changes in serum concentrations of conjugated and unconjugated steroids in 40- to 80-year-old men. J Clin Endocrinol Metab (1994) 79:1086–90. doi: 10.1210/jcem.79.4.7962278
44. Nicholson TM, Ricke WA. Androgens and estrogens in benign prostatic hyperplasia: past, present and future. Differentiation (2011) 82(4-5):184–99. doi: 10.1016/j.diff.2011.04.006
45. Ho SM, Tang WY, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res (2006) 66(11):5624–32. doi: 10.1158/0008-5472.CAN-06-0516
46. Ellem SJ, Risbridger GP. The dual, opposing roles of estrogen in the prostate. Ann N Y Acad Sci (2009) 1155:174–86. doi: 10.1111/j.1749-6632.2009.04360.x
47. Comeglio P, Morelli A, Cellai I, Vignozzi L, Sarchielli E, Filippi S, et al. Opposite effects of tamoxifen on metabolic syndrome-induced bladder and prostate alterations: a role for GPR30/GPER? Prostate (2014) 74(1):10–28. doi: 10.1002/pros.22723
48. Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A (1998) 95(26):15677–82. doi: 10.1073/pnas.95.26.15677
49. Marmorston J, Lombardo LJ, Myers SM, Gierson H, Stern E, Hopkins CE. Urinary excretion of estrone, estradiol and estriol by patients with prostatic cancer and benign prostate hypertrophy. J Urol (1965) 93(2):287–95. doi: 10.1016/S0022-5347(17)63760-1
50. Roberts RO, Jacobson DJ, Rhodes T, Klee GG, Leiber MM, Jacobsen SJ. Serum sex hormones and measures of benign prostatic hyperplasia. Prostate (2004) 61(2):124–31. doi: 10.1002/pros.20080
51. Hammarsten J, Damber JE, Karlsson M, Knutson T, Ljunggren O, Ohlsson C, et al. Insulin and free oestradiol are independent risk factors for benign prostatic hyperplasia. Prostate Cancer Prostatic Dis (2009) 12(2):160–5. doi: 10.1038/pcan.2008.50
52. Habib CN, Al-Abd AM, Tolba MF, Khalifa AE, Khedr A, Mosli HA, et al. Leptin influences estrogen metabolism and accelerates prostate cell proliferation. Life Sci (2015) 121:10–5. doi: 10.1016/j.lfs.2014.11.007
53. Nomura H, Kawashima H, Masaki S, Hosono TY, Matsumura K, Tamada S, et al. Effect of selective estrogen receptor modulators on cell proliferation and estrogen receptor activities in normal human prostate stromal and epithelial cells. Prostate Cancer Prostatic Dis (2009) 12:375–81. doi: 10.1038/pcan.2009.20
54. Noble RL. Production of NB rat carcinoma of the dorsal prostate and response of estrogen-dependent transplants to sex hormones and tamoxifen. Cancer Res (1980) 40:3547–50.
55. Ellem SJ, Risbridger GP. Aromatase and regulating the estrogen: androgen ratio in the prostate gland. J Steroid Biochem Mol Biol (2010) 118:246–51. doi: 10.1016/j.jsbmb.2009.10.015
56. Raghow S, Hooshdaran MZ, Katiyar S, Steiner MS. Toremifene prevents prostate cancer in the transgenic adenocarcinoma of mouse prostate model. Cancer Res (2002) 62:1370–6.
57. Price D, Stein B, Sieber P, Tutrone R, Bailen J, Goluboff E, et al. Toremifene for the prevention of prostate cancer in men with high grade prostatic intraepithelial neoplasia: results of a double-blind, placebo-controlled, phase IIB clinical trial. J Urol (2006) 176:965–70. doi: 10.1016/j.juro.2006.04.011
58. Ricke WA, McPherson SJ, Bianco JJ, Cunha GR, Wang Y, Risbridger GP. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J (2008) 22:1512–20. doi: 10.1096/fj.07-9526com
59. Di Zazzo E, Galasso G, Giovannelli P, Di Donato M, Castoria G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front Oncol (2018) 8:2:2. doi: 10.3389/fonc.2018.00002
60. Ross RK, Bernstein L, Lobo RA, Shimizu H, Stanczyk FZ, Pike MC, et al. 5-alpha-reductase activity and risk of prostate cancer among Japanese and US white and black males. Lancet (1992) 339:887–9. doi: 10.1016/0140-6736(92)90927-U
61. Takizawa I, Lawrence MG, Balanathan P, Rebello R, Pearson HB, Garg E, et al. Estrogen receptor alpha drives proliferation in PTEN-deficient prostate carcinoma by stimulating survival signaling, MYC expression and altering glucose sensitivity. Oncotarget (2015) 6:604–16. doi: 10.18632/oncotarget.2820
62. Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Loss of ERbeta expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer (2004) 11:537–51. doi: 10.1677/erc.1.00800
63. Sonntag WE, Lynch CD, Cefalu WT, Ingram RL, Bennett SA, Thornton PL, et al. Pleiotropic effects of growth hormone and insulin-like growth factor (IGF)-1 on biological aging: inferences from moderate caloric-restricted animals. J Gerontol A Biol Sci Med Sci (1999) 54(12):521–38. doi: 10.1093/gerona/54.12.B521
64. Aguirre GA, De Ita JR, de la Garza RG, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med (2016) 14:3. doi: 10.1186/s12967-015-0762-z
65. Neam Ţu MC, Avramescu ET, Marcu IR, Turcu-Ştiolică A, Boldeanu MV, NeamŢu OM, et al. The correlation between insulin-like growth factor with glycemic control, glomerular filtration rate, blood pressure, hematological changes or body mass index in patients with type 2 diabetes mellitus. Rom J Morphol Embryol (2017) 58(3):857–61.
66. Mantzoros CS, Tzonou A, Signorello LB, Stampfer M, Trichopoulos D, Adami HO. Insulin-like growth factor 1 in relation to prostate cancer and benign prostatic hyperplasia. Br J Cancer (1997) 76(9):1115–8. doi: 10.1038/bjc.1997.520
67. Colao A, Marzullo P, Spiezia S, Ferone D, Giaccio A, Cerbone G, et al. Effect of growth hormone (GH) and insulin-like growth factor I on prostate diseases: an ultrasonographic and endocrine study in acromegaly, GH deficiency, and healthy subjects. J Clin Endocrinol Metab (1999) 84(6):1986–91. doi: 10.1210/jcem.84.6.5776
68. Corrêa LL, Lima GA, Paiva HB, Silva CM, Cavallieri SA, Miranda LC, et al. Prostate cancer and acromegaly. Arq Bras Endocrinol Metabol (2009) 53(8):963–8. doi: 10.1590/S0004-27302009000800009
69. Corrêa LL, Balarini Lima GA, Cavallieri SA, Miranda LC, Gadelha MR. Prostatic disorders in acromegalic patients experience of a Brazilian center. Int Braz J Urol (2013) 39(3):393–401. doi: 10.1590/S1677-5538.IBJU.2013.03.13
70. Ahearn TU, Peisch S, Pettersson A, Ebot EM, Zhou CK, Graff RE, et al. Expression of IGF/insulin receptor in prostate cancer tissue and progression to lethal disease. Carcinogenesis (2018) 39(12):1431–7. doi: 10.1093/carcin/bgy112
71. Kumar S, Yadav RN, Gupta P, Gaspar BL, Kakkar N, Verma A, et al. Prostatic hyperplasia in acromegaly, a myth or reality: a case-control study. Eur J Endocrinol (2015) 172(2):97–106. doi: 10.1530/EJE-14-0698
72. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8(12):915–28. doi: 10.1038/nrc2536
73. Price AJ, Allen NE, Appleby PN, Crowe FL, Travis RC, Tipper SJ, et al. Insulin-like growth factor-I concentration and risk of prostate cancer: results from the European Prospective Investigation into Cancer and Nutrition. Cancer Epidemiol Biomarkers Prev (2012) 21(9):1531–41. doi: 10.1158/1055-9965.EPI-12-0481-T
74. Cox ME, Gleave ME, Zakikhani M, Bell RH, Piura E, Vickers E, et al. Insulin receptor expression by human prostate cancers. Prostate (2009) 69:33–40. doi: 10.1002/pros.20852
75. L-López F, Sarmento-Cabral A, Herrero-Aguayo V, Gahete MD, Castaño JP, Luque RM. Obesity and metabolic dysfunction severely influence prostate cell function: role of insulin and IGF1. J Cell Mol Med (2017) 21(9):1893–904. doi: 10.1111/jcmm.13109
76. Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, et al. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol (2009) 56(2):275–86. doi: 10.1016/j.eururo.2009.04.036
77. Yanase T, Fan W. Modification of androgen receptor function by IGF-1 signaling implications in the mechanism of refractory prostate carcinoma. Vitam Horm (2009) 80:649–66. doi: 10.1016/S0083-6729(08)00623-7
78. Levy DE, Gilliland DG. Divergent roles of STAT1 and STAT5 in malignancy as revealed by gene disruptions in mice. Oncogene (2000) 19(21):2505–10. doi: 10.1038/sj.onc.1203480
79. Rick FG, Schally AV, Block NL, Nadji M, Szepeshazi K, Zarandi M, et al. Antagonists of growth hormone-releasing hormone (GHRH) reduce prostate size in experimental benign prostatic hyperplasia. Proc Natl Acad Sci U S A (2011) 108(9):3755–60. doi: 10.1073/pnas.1018086108
80. Moeller LC, Broecker-Preuss M. Transcriptional regulation by nonclassical action of thyroid hormone. Thyroid Res (2011) 4(1):6. doi: 10.1186/1756-6614-4-S1-S6
81. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab (2002) 87(2):489–99. doi: 10.1210/jcem.87.2.8182
82. Laurberg P, Pedersen KM, Vestergaard H, Sigurdsson G. High incidence of multinodular toxic goitre in the elderly population in a low iodine intake area vs. high incidence of Graves’ disease in the young in a high iodine intake area: Comparative surveys of thyrotoxicosis epidemiology in East-Jutland Denmark and Iceland. J Intern Med (1991) 229:415–20. doi: 10.1111/j.1365-2796.1991.tb00368.x
83. Sakurai A, Nakai A, DeGroot LJ. Expression of three forms of thyroid hormone receptor in human tissues. Mol Endocrinol (1989) 3(2):392–9. doi: 10.1210/mend-3-2-392
84. Lehrer S, Diamond EJ, Bajwa AM, Kornreich R, Stagger S, Stone NN, et al. Association between serum triiodothyronine (t3) level and risk of disease recurrence in men with localized prostate cancer. Prostate Cancer Prostatic Dis (2001) 4(4):232–4. doi: 10.1038/sj.pcan.4500542
85. Lehrer S, Diamond EJ, Stone NN, Droller MJ, Stock RG. Serum triiodothyronine is increased in men with prostate cancer and benign prostatic hyperplasia. J Urol (2002) 168(6):2431–3. doi: 10.1016/S0022-5347(05)64161-4
86. Eldhose A, Nandeesha H, Dorairajan LN, Sreenivasulu K, Arul Vijaya Vani S. Thyroid and parathyroid hormones in benign prostatic hyperplasia. Br J Biomed Sci (2016) 73(2):94–6. doi: 10.1080/09674845.2016.1173333
87. Mondul AM, Weinstein SJ, Bosworth T, Remaley AT, Virtamo J, Albanes D. Circulating thyroxine, thyroid-stimulating hormone, and hypothyroid status and the risk of prostate cancer. PloS One (2012) 7(10):e47730. doi: 10.1371/journal.pone.0047730
88. Tran TV, Kitahara CM, de Vathaire F, Boutron-Ruault MC, Journy N. Thyroid dysfunction and cancer incidence: a systematic review and meta-analysis. Endocr Relat Cancer (2020) 27(4):245–59. doi: 10.1530/ERC-19-0417 pii: ERC-19-0417.R2.
89. Hsieh ML, Juang HH. Cell growth effects of triiodothyronine and expression of thyroid hormone receptor in prostate carcinoma cells. J Androl (2005) 26(3):422–8. doi: 10.2164/jandrol.04162
90. Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP model of human prostatic carcinoma. Cancer Res (1983) 43(4):1809–18.
91. Hedlund TE, Miller GJ. A serum-free defined medium capable of supporting growth of four established human prostatic carcinoma cell lines. Prostate (1994) 24(5):221–8. doi: 10.1002/pros.2990240502
92. Zhang S, Hsieh ML, Zhu W, Klee GG, Tindall DJ, Young CY. Interactive effects of triiodothyronine and androgens on prostate cell growth and gene expression. Endocrinology (1999) 140(4):1665–71. doi: 10.1210/endo.140.4.6666
93. Zhu W, Young CY. Androgen-Dependent transcriptional regulation of the prostate-specific antigen gene by thyroid hormone 3,5,3′-L-triiodothyronine. J Androl (2001) 22(1):136–41.
94. Tsui KH, Hsieh WC, Lin MH, Chang PL, Juang HH. Triiodothyronine modulates cell proliferation of human prostatic carcinoma cells by downregulation of the B-cell translocation gene 2. Prostate (2008) 68(6):610–9. doi: 10.1002/pros.20725
95. Pinto M, Soares P, Ribatti D. Thyroid hormone as a regulator of tumor-induced angiogenesis. Cancer Lett (2011) 301(2):119–26. doi: 10.1016/j.canlet.2010.11.011
96. Vikram A, Jena GB, Ramarao P. Increased cell proliferation and contractility of prostate in insulin-resistant rats: linking hyperinsulinemia with benign prostate hyperplasia. Prostate (2010) 70:79–89. doi: 10.1002/pros.21041
97. Vikram A, Tripathi DN, Ramarao P, Jena GB. Intervention of D-glucose ameliorates the toxicity of streptozotocin in accessory sex organs of rat. Toxicol Appl Pharmacol (2008) 226:84–93. doi: 10.1016/j.taap.2007.09.006
98. Hammarsten J, Högstedt B. Hyperinsulinaemia as a risk factor for developing benign prostatic hyperplasia. Eur Urol (2001) 39:151–8. doi: 10.1159/000052430
99. Seim A, Hoyo C, Ostbye T, Vatten L. The prevalence and correlates of urinary tract symptoms in Norwegian men: the HUNT study. BJU Int (2005) 96:88–92. doi: 10.1111/j.1464-410X.2005.05573.x
100. Joseph MA, Harlow SD, Wei JT, Sarma AV, Dunn RL, Taylor JM, et al. Risk factors for lower urinary tract symptoms in a population-based sample of African-American men. Am J Epidemiol (2003) 157:906–14. doi: 10.1093/aje/kwg051
101. Russo GI, Cimino S, Fragalà E, Privitera S, La Vignera S, Condorelli R, et al. Insulin resistance is an independent predictor of severe lower urinary tract symptoms and erectile dysfunction: results from a cross-sectional study. J Sex Med (2014) 11(8):2074–82. doi: 10.1111/jsm.12587
102. Abdollah F, Briganti A, Suardi N, Castiglione F, Gallina A, Capitanio U, et al. Metabolic syndrome and benign prostatic hyperplasia: evidence of a potential relationship, hypothesized etiology, and prevention. Korean J Urol (2011) 52(8):507–16. doi: 10.4111/kju.2011.52.8.507
103. Xia BW, Zhao SC, Chen ZP, Chen C, Liu TS, Yang F, et al. The underlying mechanism of metabolic syndrome on benign prostatic hyperplasia and prostate volume. Prostate (2020) 80(6):481–90. doi: 10.1002/pros.23962
104. Russo GI, Castelli T, Urzì D, Privitera S, Fragalà E, La Vignera S, et al. Connections between lower urinary tract symptoms related to benign prostatic enlargement and metabolic syndrome with its components: a systematic review and meta-analysis. Aging Male (2015) 18(4):207–16. doi: 10.3109/13685538.2015.1062980
105. Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech (2009) 2:231–7. doi: 10.1242/dmm.001180
106. Parsons JK, Carter HB, Partin AW, Windham BG, Metter EJ, Ferrucci L, et al. Metabolic factors associated with benign prostatic hyperplasia. J Clin Endocrinol Metab (2006) 91:2562–8. doi: 10.1210/jc.2005-2799
107. Kristal AR, Arnold KB, Schenk JM, Neuhouser ML, Weiss N, Goodman P, et al. Race/ethnicity, obesity, health-related behaviors and the risk of symptomatic benign prostatic hyperplasia: results from the prostate cancer prevention trial. J Urol (2007) 177:1395–400. doi: 10.1016/j.juro.2006.11.065
108. Laven BA, Orsini N, Andersson SO, Johansson JE, Gerber GS, Wolk A. Birth weight, abdominal obesity and the risk of lower urinary tract symptoms in a population-based study of Swedish men. J Urol (2008) 179:1891–5. doi: 10.1016/j.juro.2008.01.029
109. Hammarsten J, Högstedt B, Holthuis N, Mellström D. Components of the metabolic syndrome-risk factors for the development of benign prostatic hyperplasia. Prostate Cancer Prostatic Dis (1998) 1:157–62. doi: 10.1038/sj.pcan.4500221
110. Nandeesha H, Koner BC, Dorairajan LN, Sen SK. Hyperinsulinemia and dyslipidemia in non-diabetic benign prostatic hyperplasia. Clin Chim Acta (2006) 370:89–93. doi: 10.1016/j.cca.2006.01.019
111. Parsons JK, Bergstrom J, Barrett-Connor E. Lipids, lipoproteins and the risk of benign prostatic hyperplasia in community-dwelling men. BJU Int (2008) 101:313–8. doi: 10.1111/j.1464-410X.2007.07332.x
112. Rohrmann S, Smit E, Giovannucci E, Platz EA. Association between markers of the metabolic syndrome and lower urinary tract symptoms in the Third National Health and Nutrition Examination Survey (NHANES III). Int J Obes (Lond) (2005) 29:310–6. doi: 10.1038/sj.ijo.0802881
113. Russo GI, Cimino S, Castelli T, Favilla V, Gacci M, Carini M, et al. Benign Prostatic Hyperplasia, Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease: Is Metaflammation the Link? Prostate (2016) 76(16):1528–35. doi: 10.1002/pros.23237
114. Nouri M, Ratther E, Stylianou N, Nelson CC, Hollier BG, Williams ED. Androgen-targeted therapy-induced epithelial-mesenchymal plasticity and neuroendocrine transdifferentiation in prostate cancer: an opportunity for intervention. Front Oncol (2014) 4:370:370. doi: 10.3389/fonc.2014.00370
115. Li P, Yang R, Gao WQ. Contributions of epithelial-mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol Cancer (2014) 13:55. doi: 10.1186/1476-4598-13-55
116. Bishop JL, Davies A, Ketola K, Zoubeidi A. Regulation of tumor cell plasticity by the androgen receptor in prostate cancer. Endocr Relat Cancer (2015) 22:165–82. doi: 10.1530/ERC-15-0137
117. Chaki SP, Barhoumi R, Rivera GM. Nck adapter proteins promote podosome biogenesis facilitating extracellular matrix degradation and cancer invasion. Cancer Med (2019) 8(17):7385–98. doi: 10.1002/cam4.2640
118. Rabin-Court A, Rodrigues MR, Zhang XM, Perry RJ. Obesity-associated, but not obesity-independent, tumors respond to insulin by increasing mitochondrial glucose oxidation. PloS One (2019) 14(6):e0218126. doi: 10.1371/journal.pone.0218126
119. Hammarsten J, Hogstedt B. Hyperinsulinaemia: a prospective risk factor for lethal clinical prostate cancer. Eur J Cancer (2005) 41:2887–95. doi: 10.1016/j.ejca.2005.09.003
120. Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol (2008) 9:1039–47. doi: 10.1016/S1470-2045(08)70235-3
121. Faris JE, Smith MR. Metabolic sequelae associated with androgen deprivation therapy for prostate cancer. Curr Opin Endocrinol Diabetes Obes (2010) 17:240–6. doi: 10.1097/MED.0b013e3283391fd1
122. Flanagan J, Gray PK, Hahn N, Hayes J, Myers LJ, Carney-Doebbeling C, et al. Presence of the metabolic syndrome is associated with shorter time to castration-resistant prostate cancer. Ann Oncol (2011) 22:801–7. doi: 10.1093/annonc/mdq443
123. Heni M, Hennenlotter J, Scharpf M, Lutz SZ, Schwentner C, Todenhofer T, et al. Insulin receptor isoforms A and B as well as insulin receptor substrates1 and–2 are differentially expressed in prostate cancer. PloS One (2012) 7:e50953. doi: 10.1371/journal.pone.0050953
124. Lubik AA, Gunter JH, Hollier BG, Fazli L, Ettinger S, Stylianou N, et al. Insulin-like growth factor-2 increases de novo steroidogenesis in prostate cancer cells. Endocr Relat Cancer (2013) 20:173–86. doi: 10.1530/ERC-12-0250
125. Zhou F, Chen X, Fan S, Tai S, Jiang C, Zhang Y, et al. GSK1838705A, an insulin-like growth factor-1 receptor/insulin receptor inhibitor, induces apoptosis and reduces viability of docetaxel-resistant prostate cancer cells both in vitro and in vivo. Onco Targets Ther (2015) 8:753–60. doi: 10.2147/OTT.S79105
126. Kato H, Sekine Y, Furuya Y, Miyazawa Y, Koike H, Suzuki K. Metformin inhibits the proliferation of human prostate cancer PC-3 cells via the downregulation of insulin-like growth factor 1 receptor. Biochem Biophys Res Commun (2015) 461(1):115–21. doi: 10.1016/j.bbrc.2015.03.178
127. Ibuki N, Ghaffari M, Reuveni H, Pandey M, Fazli L, Azuma H, et al. The tyrphostin NT157 suppresses insulin receptor substrates and augments therapeutic response of prostate cancer. Mol Cancer Ther (2014) 13(12):2827–39. doi: 10.1158/1535-7163.MCT-13-0842
128. Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control (2009) 20(9):1617–22. doi: 10.1007/s10552-009-9407-y
129. Murtola TJ, Tammela TL, Lahtela J, Auvinen A. Antidiabetic medication and prostate cancer risk: a population-based case-control study. Am J Epidemiol (2008) 168(8):925–31. doi: 10.1093/aje/kwn190
130. He XX, Tu SM, Lee MH, Yeung SJ. Thiazolidinediones and metformin associated with improved survival of diabetic prostate cancer patients. Ann Oncol (2011) 22(12):2640–5. doi: 10.1093/annonc/mdr020
131. Ruiter R, Visser LE, van Herk-Sukel MP, Coebergh JW, Haak HR, Geelhoed-Duijvestijn PH, et al. Lower risk of cancer in patients on metformin in comparison with those on sulfonylurea derivatives: results from a large population-based follow-up study. Diabetes Care (2012) 35(1):119–24. doi: 10.2337/dc11-0857
132. Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol (2013) 31(25):3069–75. doi: 10.1200/JCO.2012.46.7043
133. Whitburn J, Edwards CM, Sooriakumaran P. Metformin and Prostate Cancer: a New Role for an Old Drug. Curr Urol Rep (2017) 18(6):46. doi: 10.1007/s11934-017-0693-8
134. Yu HF, Chen HS, Rao DP, Gong J. Association between polycystic ovary syndrome and the risk of pregnancy complications: A PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) (2016) 95(51):e4863. doi: 10.1097/MD.0000000000004863
135. Goodman NF, Cobin RH, Futterweit W, Glueck JS, Legro RS, Carmina E, et al. American association of clinical endocrinologists, American college of endocrinology, and androgen excess and PCOS society disease state clinical review: guide to the best practices in the evaluation and treatment of polycystic ovary syndrome. Endocr Pract (2015) 21(12):1415–26. doi: 10.4158/EP15748.DSCPT2
136. Benítez R, Sir-Petermann T, Palomino A, Angel B, Maliqueo M, Pérez F, et al. Prevalence of metabolic disorders among family members of patients with polycystic ovary syndrome. Rev Med Chil (2001) 129(7):707–12. doi: 10.4067/S0034-98872001000700001
137. Yilmaz M, Bukan N, Ersoy R, Karakoç A, Yetkin I, Ayvaz G, et al. Glucose intolerance, insulin resistance and cardiovascular risk factors in first degree relatives of women with polycystic ovary syndrome. Hum Reprod (2005) 20(9):2414–20. doi: 10.1093/humrep/dei070
138. Legro RS, Driscoll D, Strauss JF, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci USA (1998) 95(25):14956–60. doi: 10.1073/pnas.95.25.14956
139. Legro RS, Kunselman AR, Demers L, Wang SC, Bentley-Lewis R, Dunaif A. Elevated dehydroepiandrosterone sulfate levels as the reproductive phenotype in the brothers of women with polycystic ovary syndrome. J Clin Endocrinol Metab (2002) 87:2134–8. doi: 10.1210/jcem.87.5.8387
140. Lenarcik A, Bidzińska-Speichert B, Tworowska-Bardzińska U, Krępuła K. Hormonal abnormalities in first-degree relatives of women with polycystic ovary syndrome (PCOS). Endokrynol Pol (2011) 62(2):129–33.
141. Lunde O, Magnus P, Sandvik L, Høglo S. Familial clustering in the Polycystic Ovarian syndrome. Gynecol Obstet Invest (1989) 28:23–30. doi: 10.1159/000293493
142. Carey AH, Chan KL, Short F, White D, Williamson R, Franks S. Evidence for a single gene effect causing polycystic ovaries and male pattern baldness. Clin Endocrinol (1993) 38:653–8. doi: 10.1111/j.1365-2265.1993.tb02150.x
143. Govind A, Obhrai MS, Clayton RN. Polycystic ovaries are inherited as an autosomal dominant trait: analysis of 29 polycystic ovary syndrome and 10 control families. J Clin Endocrinol Metab (1999) 84:38–53. doi: 10.1210/jcem.84.1.5382
144. Legro RS. Is there a male phenotype in polycystic ovary syndrome families? J Pediatr Endocrinol Metab (2000) 13:1307–9.
145. Cannarella R, Condorelli RA, Dall’Oglio F, La Vignera S, Mongioì LM, Micali G, et al. Increased DHEAS and Decreased Total Testosterone Serum Levels in a Subset of Men with Early-Onset Androgenetic Alopecia: Does a Male PCOS-Equivalent Exist? Int J Endocrinol (2020) 2020:1942126. doi: 10.1155/2020/1942126
146. Cannarella R, Condorelli RA, Mongioì LM, La Vignera S, Calogero AE. Does a male polycystic ovarian syndrome equivalent exist? J Endocrinol Invest (2018) 41(1):49–57. doi: 10.1007/s40618-017-0728-5
147. Cannarella R, La Vignera S, Condorelli RA, Calogero AE. Glycolipid and hormonal profiles in young men with early-onset androgenetic alopecia: A meta-analysis. Sci Rep (2017) 7(1):7801. doi: 10.1038/s41598-017-08528-3
148. Lotufo PA, Chae CU, Ajani UA, Hennekens CH, Manson JE. Male pattern baldness and coronary heart disease: the Physicians’ Health Study. Arch Intern Med (2000) 160(2):165–71. doi: 10.1001/archinte.160.2.165
149. Lesko SM, Rosenberg L, Shapiro S. A case-control study of baldness in relation to myocardial infarction in men. JAMA (1993) 269(8):998–1003. doi: 10.1001/jama.269.8.998
150. Su LH, Chen LS, Lin SC, Chen HH. Association of androgenetic alopecia with mortality from diabetes mellitus and heart disease. JAMA Dermatol (2013) 149:601–6. doi: 10.1001/jamadermatol.2013.130
151. Oh BR, Kim SJ, Moon JD, Kim HN, Kwon DD, Won YH, et al. Association of benign prostatic hyperplasia with male pattern baldness. Urology (1998) 51(5):744–8. doi: 10.1016/S0090-4295(98)00108-3
152. Chen W, Yang CC, Chen GY, Wu MC, Sheu HM, Tzai TS. Patients with a large prostate show a higher prevalence of androgenetic alopecia. Arch Dermatol Res (2004) 296(6):245–9. doi: 10.1007/s00403-004-0514-z
153. Kaplan SA. Patients with a large prostate show a higher prevalence of androgenetic alopecia. J Urol (2005) 174(5):1905. doi: 10.1016/S0022-5347(01)68832-3
154. Kaplan SA. Re: Androgenetic alopecia as an early marker of benign prostatic hyperplasia. J Urol (2012) 188(5):1846–7. doi: 10.1016/j.juro.2012.07.079
155. Arias-Santiago S, Arrabal-Polo MA, Buendía-Eisman A, Arrabal-Martín M, Gutiérrez-Salmerón MT, Girón-Prieto MS, et al. Androgenetic alopecia as an early marker of benign prostatic hyperplasia. J Am Acad Dermatol (2012) 66:401–8. doi: 10.1016/j.jaad.2010.12.023
156. Amoretti A, Laydner H, Bergfeld W. Androgenetic alopecia and risk of prostate cancer: a systematic review and meta-analysis. J Am Acad Dermatol (2013) 68(6):937–43. doi: 10.1016/j.jaad.2012.11.034
Keywords: low urinary tract symptoms, metabolic syndrome, aging, insulin, male PCOS-equivalent, benign prostate hyperplasia
Citation: Cannarella R, Condorelli RA, Barbagallo F, La Vignera S and Calogero AE (2021) Endocrinology of the Aging Prostate: Current Concepts. Front. Endocrinol. 12:554078. doi: 10.3389/fendo.2021.554078
Received: 21 April 2020; Accepted: 05 January 2021;
Published: 22 February 2021.
Edited by:
Marc R. Blackman, Washington DC VA Medical Center, United StatesReviewed by:
Carla Pelusi, University of Bologna, ItalyCopyright © 2021 Cannarella, Condorelli, Barbagallo, La Vignera and Calogero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandro La Vignera, c2FuZHJvbGF2aWduZXJhQHVuaWN0Lml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.