Erratum: Case Report: Sellar Ependymomas: A Clinic-Pathological Study and Literature Review
 Liyan Zhao1
Liyan Zhao1 Yining Jiang
Yining Jiang Yubo Wang
Yubo Wang- 1Department of Clinical Laboratory, Second Hospital of Jilin University, Changchun, China
- 2Department of Neurosurgery, First Hospital of Jilin University, Changchun, China
Ependymomas are primary glial tumors arising from cells related to the ependymal lining of the ventricular system. They are classified into at least nine different molecular subtypes according to molecular phenotype, histological morphology, and tumor location. Primary sellar ependymoma is an extremely rare malignancy of the central nervous system, with only 12 known cases reported in humans. We herein report a case of ependymoma located at the pituitary region in a 44-year-old female patient and discuss the molecular subtype, natural history, clinical presentation, radiological findings, histological features, immunohistochemical characteristics, ultrastructural examinations, treatment, and prognosis of sellar ependymoma. This case report may serve as a helpful reference for clinicians and radiologists in clinical practice.
Introduction
Ependymomas are relatively uncommon primary glial tumors, accounting for 2% to 6% of the central nervous system (CNS) tumors (1), that usually arise from the ependymal lining of the cerebral ventricles, the central cord of the spinal canal, and/or terminal ventricle cells in the terminal filum (2–4). Ependymomas that develop in brain parenchyma are rare, and exceptionally rare in saddle areas (2, 3, 5). Upon reviewing all the primary ependymoma reports available on PubMed, only 12 cases were reported in humans (3, 5–15). Notably, all reported cases were diagnosed by pathological confirmation after surgical resection. Here, we report a case of sellar ependymoma (SE) involving a 44-year-old female with a history of 7 months of secondary amenorrhea and bitemporal hemianopsia. Magnetic resonance imaging (MRI) showed a mass located in the sellar and suprasellar region of the brain. The patient underwent total microsurgical removal and was found to have an ependymoma of Grade II classification. In addition, we reviewed the available medical and scientific literature and summarized all the reported SE cases, as well as the etiology, radiological features, histopathological characteristics, differential diagnoses, current treatment landscape, and prognosis of this rare tumor entity.
Case Report
History and Examination
A 44-year-old female was admitted to the Neurosurgery Department with a 7-month history of intermittent headaches, secondary amenorrhea, and visual blurring. The patient reported not having galactorrhea, hyponatremia, hyposexuality, weakness, or any other sensory or motor dysfunctions. A review of the family history indicated no history of tumor or cancer in close family members. Moreover, neuro-ophthalmologic examination disclosed bitemporal hemianopia, which was worse in the right eye, and no papilledema was found. A Snellen chart indicated a 0.4 value in the unaided right eye and 0.7 in the left eye. A preoperative laboratory examination showed a notable increase in the prolactin (PRL) level (1,370.51 mIU/L, range: 70.81-566) and no evidence of pituitary dysfunction or hypoadrenocorticism.
Neuroimaging Findings
MRI scanning revealed a 1.94×1.91×2.16-cm (anterior-posterior×transverse×cranial-caudal) well-defined mass located in the sellar and suprasellar regions of the brain (Knosp II and Hardy-Wilson Grade III), with a noticeable expansion of the sella turcica. Furthermore, the tumor was isointense on the T1-weighted imaging (T1WI; Figure 1A), slightly hyperintense on the T2-weighted imaging (T2WI; Figure 1B), with strong and irregular contrast enhancement after gadolinium administration (Figure 1C). Notably, the tumor compressed the overlying optic chiasm, and there was no evidence of cavernous sinus invasion. In addition, the pituitary stalk was difficult to locate as the lesion protruded to the front of the brain third ventricle.
FIGURE 1
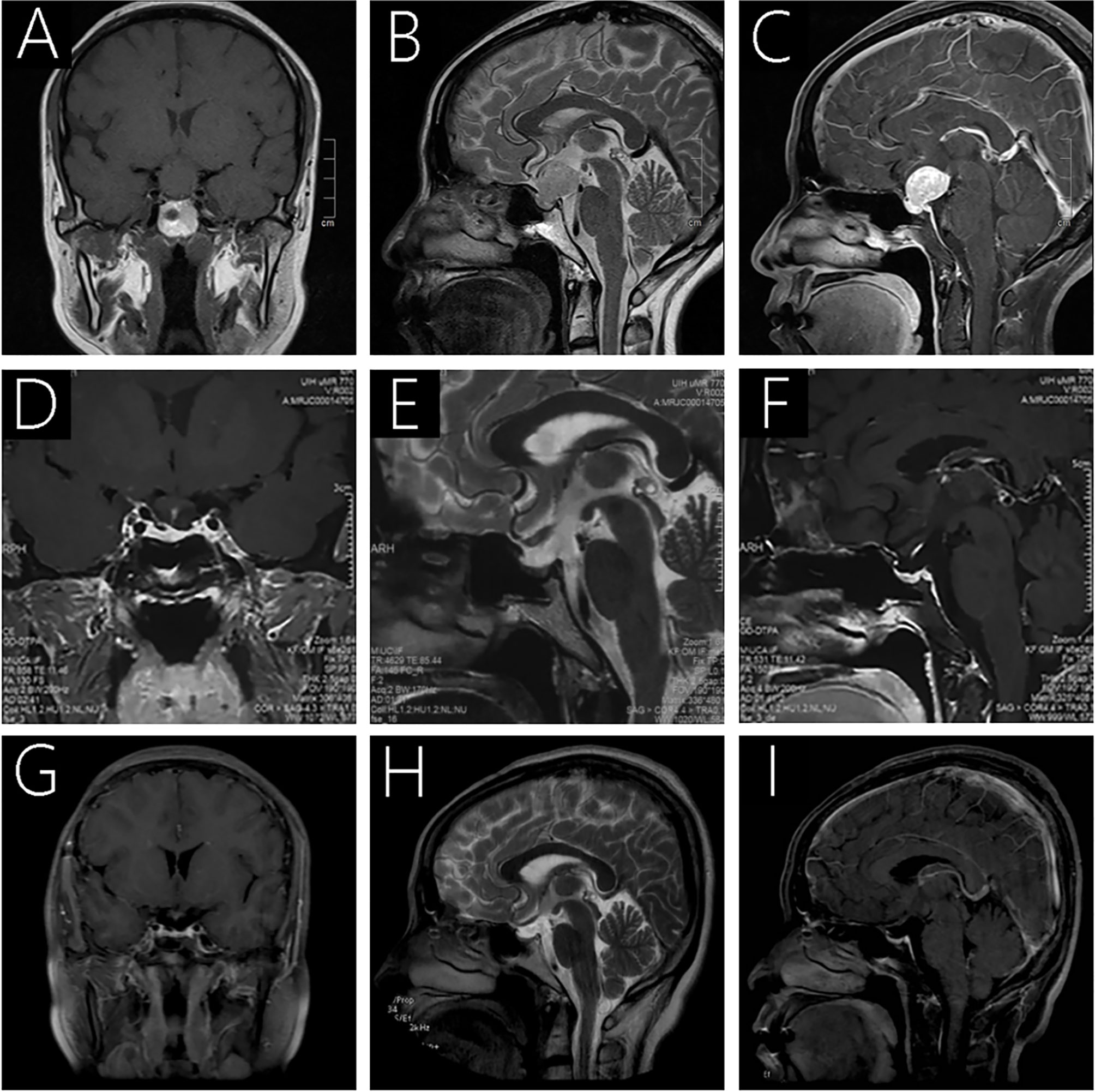
Figure 1 Preoperative MRI (A-C) revealed a 1.94×1.91×2.16-cm well-defined mass located in the sellar and suprasellar regions. It was isointense on T1WI (A) and slightly hyperintense on T2WI (B), and the tumor was strongly and irregularly contrast-enhanced after gadolinium administration (C). Follow-up MRI 3 months (D–F) and 27 months after surgery (G–I) showed complete removal of the lesion, with no sign of recurrence.
Surgery
A preoperative clinical diagnosis of pituitary adenoma was made and a total surgical resection was performed by the corresponding author using a transfrontal-temporal approach. The tumor appeared pinkish in color, soft, friable, rich in vascular supply and was found located near the junction with the optic chiasm. The surgery was uneventful, with no noticeable leakage of cerebrospinal fluid or substantial venous bleeding. Follow-up MRI analysis demonstrated a total excision of the tumor (Figures 1D–I).
Histopathology Findings
The postoperative pathology manifested SE of World Health Organization (WHO) Grade II classification. Moreover, gross specimen sections showed a gray-brown, soft, and friable mass. When examining hematoxylin and eosin (H&E)-stained paraffin sections, we observed characteristic and well-formed perivascular anucleate zones (pseudorosettes) and (true) ependymal rosettes composed of bland cuboidal or columnar tumor cells (Figure 2A). Moreover, immunohistochemical analysis showed that the Ki-67 labeling index for cell division was approximately 3%. Positive staining was found for thyroid transcription factor-1 (TTF-1), neuron-specific enolase, vimentin, and epithelial membrane antigen (EMA; Figure 2B), as well as S-100 (Figure 2C). In addition, our staining results were negative for glial fibrillary acidic protein (GFAP), synaptophysin, and pituitary hormones. Unfortunately, no further genetic tests were performed.
FIGURE 2

Figure 2 Well-formed perivascular anucleate zones (pseudorosettes) and (true) ependymal rosettes composed of bland cuboidal or columnar tumor cells seen throughout the tumor (A). Immunolabeling was positive for EMA (B) and S-100 (C).
Postoperative Course
Visual blurring significantly improved after surgery, and the patient was discharged without any neurological deficits on day 14. A postoperative laboratory examination revealed adrenocortical axis and thyroid axis hypofunction, and the patient underwent disciplinary hormone replacement therapy for six months. Follow-up MRI scans were taken three months postoperatively (Figures 1D–F), demonstrating that the lesion had been completely removed, with no sign of recurrence. During the last follow-up in April 2021 (27 months postoperatively), the patient reported leading a normal daily life without noticeable symptoms, and the follow-up MRI exhibited no recurrence (Figure 1G-I). We believe that her condition is stable and will continue to monitor her progress with routine follow-up MRIs.
Discussion
Ependymomas are primary glial tumors that arise from cells related to the ependymal lining of the cerebrospinal fluid circulatory system (9, 16). The saddle area is an exceedingly rare location for ependymomas, first reported by Sarkisian and Schultz in 1956 (6). Since then, only 12 cases of primary SEs have been reported in humans (Tables 1, 2). Interestingly, Heath et al. also reported a case of a pituitary ependymoma occurrence in a horse (17). In 2017 WHO classification of pituitary tumors, SE, pituicytoma, granular cell tumor, and spindle cell oncocytoma were classified as nosological entities, called primary posterior pituitary tumors, a distinct group of low-grade and non-neuroendocrine neoplasms with TTF-1 immunopositivity (18, 19). All these tumors are believed to arise from pituicytes (20). Among them, SE is the least described, and scant effective information is available (18–22). According to the latest histopathological classification of WHO in 2016, ependymoma was classified into three grades (23). Grade I: subependymoma and myxopapillary ependymoma; Grade II: papillary ependymoma, clear cell ependymoma, and tanycytic ependymoma; Grade II-III: ependymoma, RELA fusion-positive; Grade III: anaplastic ependymoma. For ependymomas of Grade I, the prognosis is usually good, and it can be cured by surgery alone (24). For ependymomas in Grades II and III, the differential diagnosis is difficult. Increased cell density, nuclear atypia, a high mitosis rate, and varying degrees of angiogenesis are the main features of ependymomas in Grade III for differentiation (25). However, in some clinical settings, some literature presented a poor association between tumor grading and outcome (25).
TABLE 1
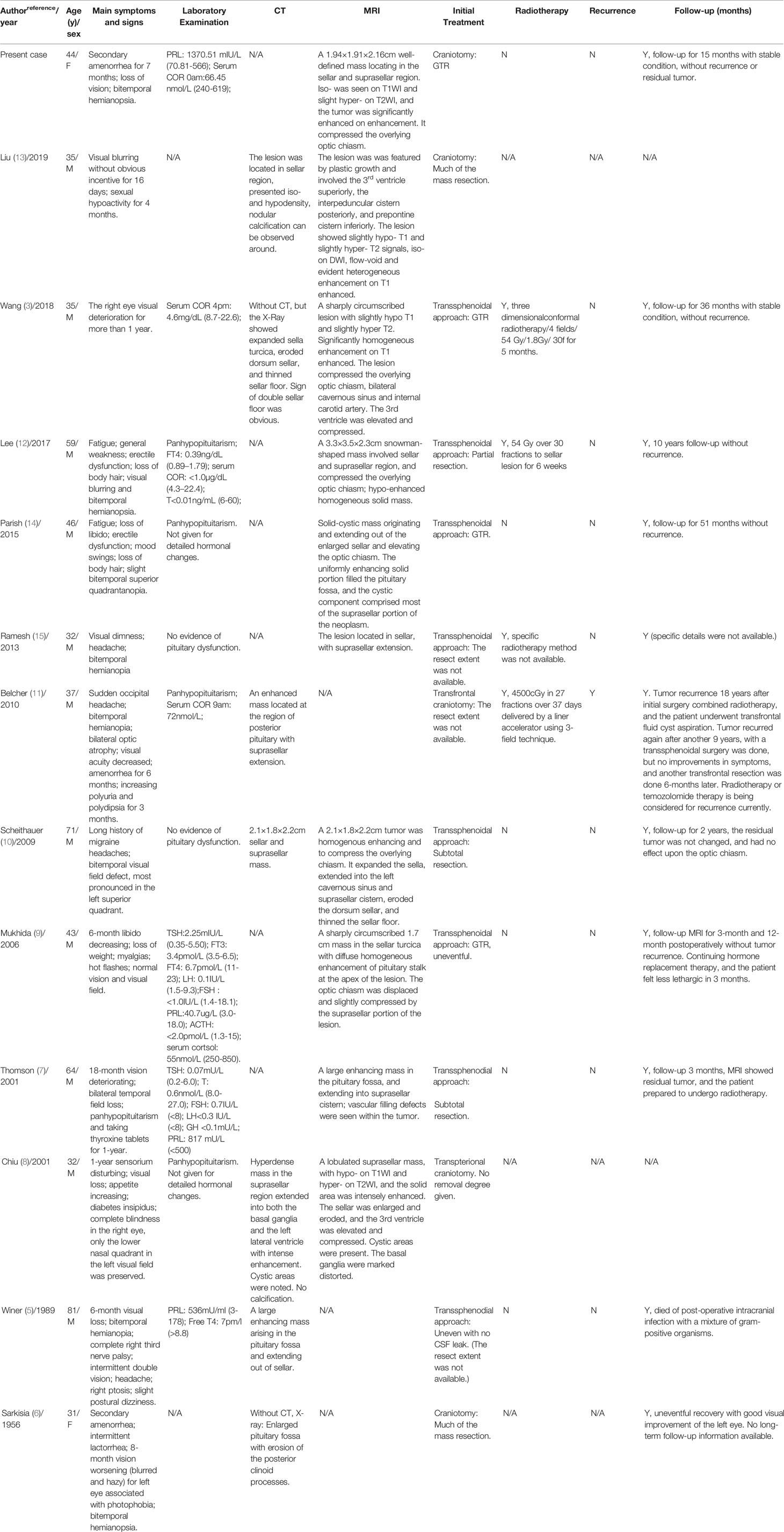
Table 1 Patients’ characteristics of reported case in the literature.
TABLE 2
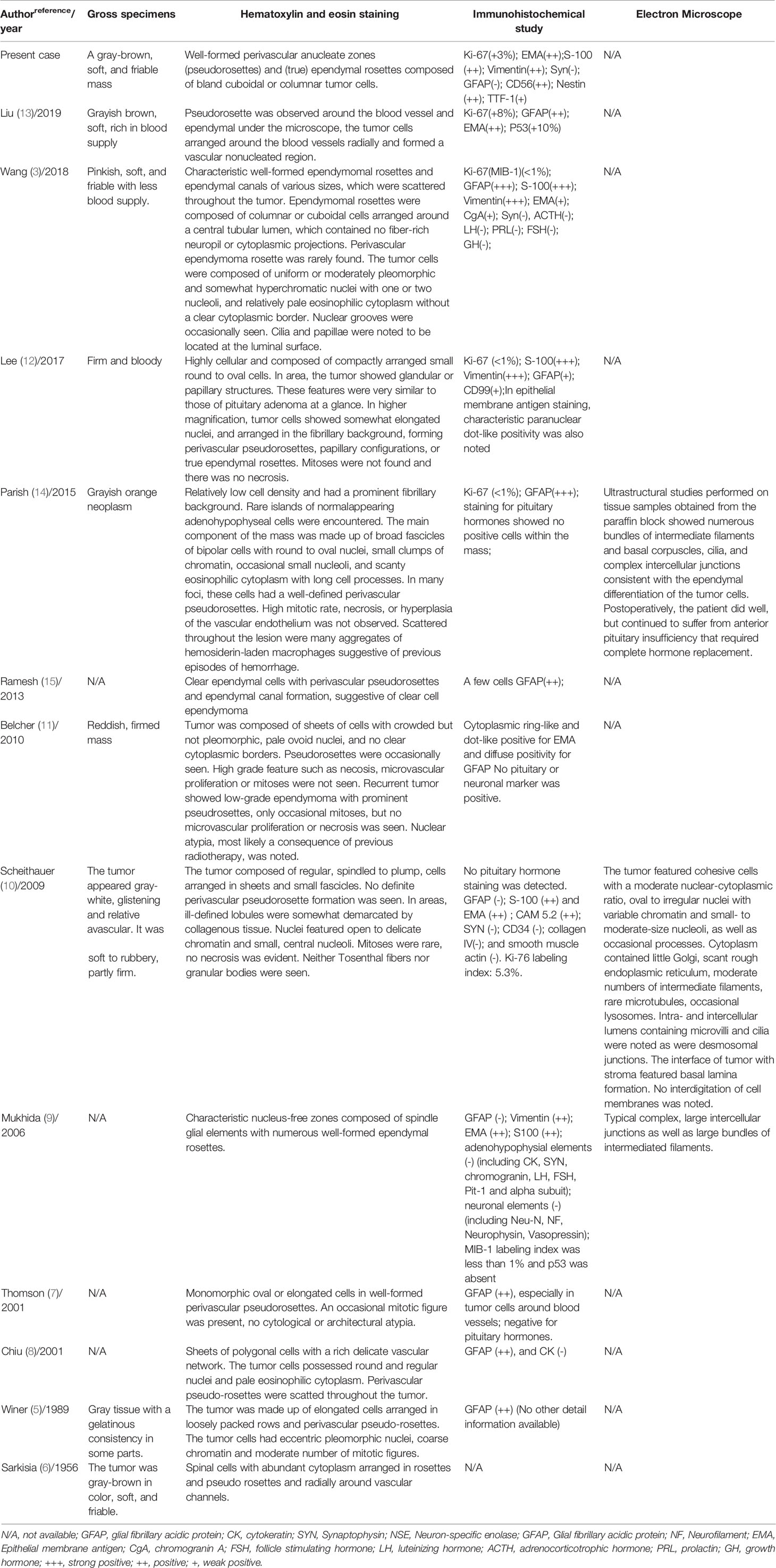
Table 2 Pathological features of reported cases in the literature.
Molecular Subtypes of Ependymomas
Along with developments in molecular biology techniques, many studies have been carried out on the occurrence and development mechanisms of ependymoma. Pajtler et al. published a DNA methylation profile in 2015, which analyzed the molecular types of ependymomas in different anatomical locations (26), and the classification was adopted by the WHO classification of CNS tumors in 2016. In 2018, Pajtler et al. refined the classification further (27). At present, ependymomas are usually regarded as a molecularly heterogeneous disease entity and are classified into nine distinct molecular subtypes (Table 3), according to DNA methylation and gene expression profiling, which have reached a broad consensus (26–28). Supratentorial ependymoma (ST-EPN) includes two molecular subtypes, ST-EPN-RELA (RELA-C110RF95 fusion type) and ST-EPN-YAP1 (YAP1 fusing with other genes, with a high frequency of YAP1-MAMLD1 fusion) (29, 30). ST-EPN-RELA accounts for 72% of ST-EPN and occurs in infants, children, and adults (28). RELA-C110RF95 fusion protein leads to the abnormal activation of NF-κB pathway, leading to a poor prognosis of this subtype (30). Gessi et al. suggested that p65/RELA combining with L1CAM antibody is also a valuable index to diagnose ST-EPN-RELA (31). Moreover, ependymomas with RELA fusion-positive were added to the newly revised WHO classification in 2016 (WHO Grade II-III). ST-EPN-YAP1 occurs primarily in infants and children and usually has an excellent outcome after surgery combined with radiotherapy (26, 27, 30). A European retrospective study showed a 5-year progression-free survival (PFS) rate of <30% and a 5-year overall survival (OS) rate of 75% for ST-EPN-RELA, compared to a 5-year PFS rate of 66% and a 5-year OS rate of 100% for ST-EPN-YAP1 (26, 27). However, these findings need further verification in prospective studies.
TABLE 3
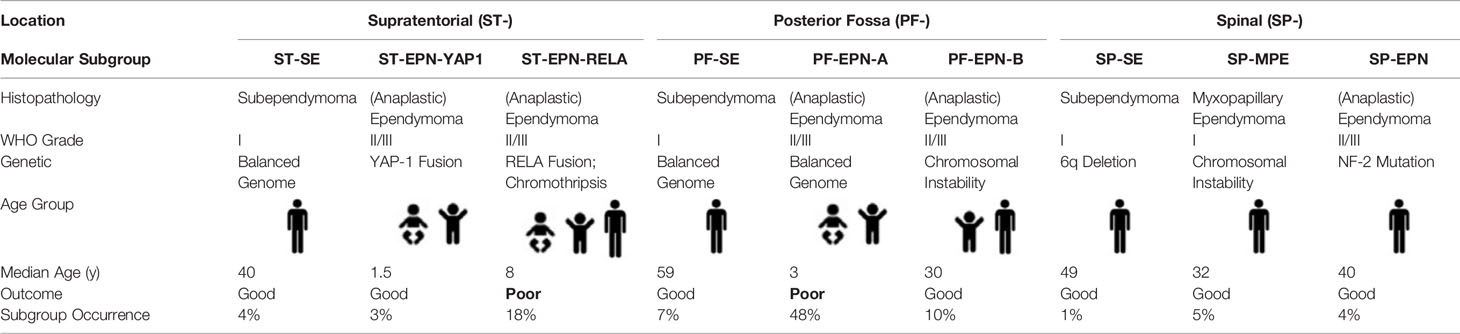
Table 3 Summary of 9 molecular subtype and clinical characteristics.
Posterior fossa ependymoma (PF-EPN) was classified into two molecular subtypes of PF-EPN-A and PF-EPN-B, according to the methylator phenotype of the promoter CpG island (27). The former exhibits hypermethylation of the CpG island and the latter exhibits hypomethylation. The grouping could also be divided by the global level of lysine 27 on histone 3 (H3K27), with the low epigenetic mark in PF-EPN-A and high in PF-EPN-B (32). PF-EPN-A accounts for 74% of all PF-EPNs and occurs primarily in infants and young children. Moreover, tumors are mainly located in the midline, with high recurrence and metastasis rates, despite gross tumor resection (GTR) (27, 33–35). Chromosome 1q amplification (15-20% PF-EPN) and the absence of H3K27me3 trimethylation are considered to be associated with a poor prognosis (27, 32, 36, 37). PF-EPN-B occurs primarily in adolescents and adults, with a more favorable prognosis (27, 33). Some PF-EPN-Bs could be cured by GTR alone, and tumor relapse could be controlled by radiotherapy effectively (35). The prognosis is usually favorable for spinal ependymomas, and 43% of cases exhibit NF-2 mutation (38). However, some recent studies on aggressive spinal ependymomas have characterized them with MYCN amplification, anaplastic morphology, and early dissemination along conus medullaris, with usually a poor prognosis (39, 40), necessitating further studies. Vera-Bolanos et al. also reported that spinal myxopapillary ependymomas should be assigned to WHO grade II because the clinical studies do not support a WHO grade I clinical behavior (41).
Besides, some researchers have also reported some other differences in the molecular phenotype. Johnson et al. reported that ST-EPN also exhibits EPHB2 amplification and INK4A absence (42). Some literature suggested that the tumor suppressor gene RASSF1A could be expressed in most ependymomas, with high methylation and site-specification (42, 43). The molecular alterations could be discovered with a range of diagnostic tests below. Fluorescence in situ hybridization could demonstrate RELA-C110RF95 fusion and YAP1 rearrangement of ST-EPN in association with MYCN amplification in aggressive spinal ependymomas (30, 44). Immunohistochemistry assessment of the expression of H3K27-trimethylation could be used to distinguish two types of PF-EPN (32).
Pathogenesis
Due to their rarity, little is known about the underlying causes mediating the development of SEs. One theoretical explanation is that, their occurrence may be a consequence of a neoplastic transformation during the development of either heterotopic ependymal lining cells or embryological remnants of the ependymal lining within the sellar, especially in the infundibular region (6, 7, 10, 13, 14). From an anatomical perspective, in a 10-week (45-mm) human fetus, neurohypophysis develops as an elongated out-pouching of neuroepithelial cells, which encloses a cavity that is continuous with the cavity of the neural tube. At this stage, the infundibulum consists of undifferentiated ependymal cell precursors (3). Particularly, the ependymal cleft, which is not present at birth, is readily identified at 13-weeks during development (60-mm), although it recedes by 16-weeks (112-mm). It is plausible that isolated ependymal cells or embryonic ependymoma remnants are left behind in the infundibulum and neurohypophysis. These cells might undergo subsequent neoplastic transformation (7, 8, 13), giving rise to an ependymoma (3, 5–7, 10). Other hypothesis speculates that SE might be a variant of pituicytoma (10, 22). The hypothesis has been supported by the ultrastructural evidence of pituicyte, a specialized intrinsic cell type of the posterior pituitary gland (3, 45). Pituicytes are classified as major, dark, ependymal, oncocytic, and granular cell types (45). All these are considered different functional forms of a unique cell line with an ependymal phylogenetic origin (21). The ependymal pituicyte remnants might undergo neoplastic transformation (22).
Tumor Growth Characteristics
Subsequently, we analyzed the tumor growth characteristics of all the reported cases of SEs. First, the tumor mass can be solid [9/13, (69.23%)] or cystic-solid (3/13, [23.08%]) (8, 13, 14), with the cystic component always involving the suprasellar region of the brain (2/3, [66.67%]) (8, 14). Lesions mostly involve both the sellar and suprasellar regions of the brain (12/13, [92.31%], presenting a prominent enlarged sella turcica (11/12, [84.62%]). The pituitary stem appeared consistently raised or displaced (12/13, [92.31%]), and the tumor mass was found to often end up compressing the optic chiasm and optic tract (12/13, [92.31%]). Furthermore, the tumor mass can, in some cases, protrude upward into the third ventricle (2/13, [15.38%]) (8, 13) and affect the basal ganglia regions (1/13, [7.69%]) (8). Ependymoma refers to a ‘plastic’ growth pattern or desmoplastic development that typically protrudes through the outlet foramina into adjacent cisterns (7, 8, 13).
Clinical Presentation
The mean outset age of SE has been reported to be approximately 46.9 years of age (range: 31-81). In our analysis, we found two female patients and 11 male patients, suggesting a male inclination, consistent with previous reports, suggesting that males have a higher incidence of ependymoma occurrences than females (1, 4). As for the clinical presentation, SEs are often symptomatically similar to nonfunctional pituitary adenomas (18), with some studies reporting that the duration of symptoms also depends on the histological grade of tumors (7). We divided the condition into two aspects here. The first aspect is associated with the mass effect, leading to debilitating symptoms, including headaches, visual deteriorating, bitemporal hemianopsia, and oculomotor paralysis, among others (3). Moreover, cases in which the tumor might grow superiorly can lead to the elevation and compression of the third ventricle of the brain, which might result in hydrocephalus (8). The second aspect is associated with hypopituitarism, leading to weakness, mood swings, hot flashes, weight loss, fatigue, increased appetite, decreased libido, erectile dysfunction, and loss of the body hair. Moreover, patients could have polydipsia and diabetes insipidus if ependymomas involves the posterior pituitary gland (3). However, diabetes insipidus has only been reported in two patients (11, 46). Loh et al. suggested that it can be attributed to the slow growth of SEs (47). In addition, PRL levels could be slightly higher than normal, which is possible due to the so-called “stalk effect” (3), but galactorrhea has been reported only once (6), necessitating further studies. In our patient, PRL levels were found to be significantly raised (1,370.51 mIU/L) compared to normal levels (range: 70.81-566), with no galactorrhea. Furthermore, some reports revealed that the duration of symptoms depends on the histological grade of the tumor (7).
Radiologic Characteristics
The rarity of SE has led to limited radiographic characteristics, and we have summarized imaging analysis of all 13 clinical cases discussed in this study in Table 1. CT scanning frequently shows an enhanced mass arising from the pituitary fossa, extending to the suprasellar region of the brain, and presenting an enlarged sella turcica (3). These ependymomas appear isodense or hyperdense, which can make them difficult to distinguish from pituitary adenoma (3, 7). MRI is regarded as the primary imaging technique for the identification of SEs (3). Although not sufficiently sensitive to diagnose SE on its own, MRI is significant in assessing the location of SEs, demonstrating tumor vascularity, as well as displacement of normal vessels, which are important for surgical planning, especially on sagittal and coronal planes. The lesion is often solid or cystic-solid, with the cystic component usually located in the suprasellar region. Moreover, the tumor solid components appear slightly hypointense on T1WI and slightly hyperintense on T2WI, and the cystic components appear hypointense on T1WI and hyperintense on T2WI (8, 9, 14). Upon enhanced MRI analysis, the tumor margins appear well-defined. In addition, the solid part and cyst wall observed in these tumors are frequently enhanced. Intermixing of poorly enhanced areas is frequently shown in solid parts, suggesting a vascular filling defect (7, 48), not usually seen in pituitary adenomas (1, 7). In some studies, it was suggested that the vascular filling defects observed on MRI scans might help distinguish pituitary adenomas from ependymomas (7). Furthermore, the cystic area of a lesion may follow fluid signal intensity on both T2WI and on the fluid-attenuated inversion recovery (FLAIR) sequence or might remain hyperintense on FLAIR due to the presence of proteinaceous fluid contents (7). Lukashova-v Zangen et al. (49) demonstrated that the isointense signal is shown on the diffusion-weighted imaging. Another characteristic of SEs is the flow-void phenomenon, which can be seen on the plain view of the MRI (13).
Pathology Features
We summarized the pathological examination of all 13 cases, including our case in Table 2. SEs are characterized by various-sized and well-formed true ependymoma rosettes and/or perivascular pseudorosettes scattered throughout the tumor as seen under H&E staining examination (3, 9, 10, 12, 13). Using light microscopy, these rosettes provide strong evidence of ependymal differentiation. The histological features of these rosettes were observed in all 13 cases studied (13/13 [100.0%]), with 5 cases (5/13 [38.46%]) presenting both pseudorosettes and true ependymoma rosettes, 6 cases (6/13, [46.15%]) presenting pseudorosettes, 1 case (9)(1/13 [7.69%]) presenting true ependymoma rosettes only, and 1 other case (10) (1/13 [7.69%]) presenting neither ependymoma rosettes nor pseudorosettes. Perivascular pseudorosettes seem to be more sensitive for the diagnosis of ependymomas than true ependymal rosettes (2, 50). On the other hand, perivascular pseudorosettes are less specific in that they can also be found in medulloblastomas, glioblastomas, and central neurocytoma malignancies (50). Importantly, with the exception of rosette-like structures, the nuclear groove is also a specific cytologic finding for the diagnosis of SEs, which has not been detected in other CNS tumors (3, 51). Kumar et al. (51) compared the cytologic findings of 21 cases of classic ependymomas with other CNS tumors and found ependymal rosettes (100%), nuclear grooves (71.4%), perivascular pseudorosettes (52.4%), and acinar structures (33.3%) throughout these cases.
Immunostaining analysis provides additional significant information for the correct diagnosis of ependymomas, with positive expression levels of TTF-1, GFAP, S-100, vimentin, EMA, and negative expression levels for secreted pituitary hormones and neural markers, such as synaptophysin (10–12, 18, 21). In some cases, GFAP did not appear to be immunoreactive (52), consistent with 3 out of 13 reported cases of SE, including our patient. Moreover, expression levels of the low molecular weight protein and keratin could be observed in approximately 20% of ependymomas (9). Another notable consideration is that the diagnosis of SEs cannot always be made solely based on histological examinations and immunohistochemical analyses. As in the case reported by Scheithauer et al. (10), histological examination and immunohistochemical analysis of the patient were not straightforward. In difficult-to-diagnose cases, ultrastructural analysis by electron microscopy is imperative.
We found that electron microscopy examination was carried out in 3 cases (3/13, [23.08%]) of SE (9, 10, 14). Here, we have reviewed previous reports of SEs and summarized important ultrastructural features. Cilia and microvilli structures located at the luminal surface of these neoplastic ependymal cells, as well as intercellular junctional complexes located on their lateral surfaces, can be usually observed using an electron microscope (9, 10, 14, 45). In addition, the cytoplasm of these tumor cells contains a moderate to a large number of intermediated filaments and few Golgi, rough endoplasmic reticulum, or microtubules (10, 14, 45). A basal lamina formation can also be present at the interface between tumor cells and vascularized stroma (10, 14), whereas neurosecretory granules have been noticeably absent (3).
The different diagnoses obtained are quite extensive, including pituitary adenoma, craniopharyngioma, chordomas, Rathke’s cleft cyst, meningioma, glioma, and aneurysms, among others (8, 10, 14, 53). Here, we analyzed the preoperative diagnosis of the 13 cases evaluated, of which, 9 cases (9/13 [69.23%]) of pituitary adenomas, 4 cases (4/9 [44.44%]) of pituitary macroadenoma among them, 1 case (1/13 [7.69%]) of glioma, and 1 case (1/13 [7.69%]) of craniopharyngioma, were identified.
Treatment
Surgery
Despite advances in many other tumor types in past decades, maximal safe tumor resection followed by postoperative adjuvant radiotherapy remains the most viable treatment modality for ependymomas (21, 24, 54, 55). Currently, there is no established treatment protocol for SE patients. Surgical resection is still the primary treatment choice with the aim of GTR, as the extent of resection is one of the most important predictors of recurrence and survival (3, 11, 12). Surgical paradigms include the transsphenoidal and craniotomy approach, and the choice depends on the tumor location and its suprasellar extension. All of the 13 clinical cases studied here (including our patient) underwent surgical resection, 8 cases (8/13 [61.54%]) using the transsphenoidal approach and 5 cases (5/13 [38.46%]) using the craniotomy approach, respectively (Table 1). When using the transsphenoidal approach, three cases (3/8 [37.5%]) underwent GTR, two cases had a subtotal resection (2/8 [25.0%]), and one case had a partial resection (1/8 [12.5%]). Notably, for two of these cases (2/8 [25.0%]), the extent of resection was not reported. Using the craniotomy approach, one case underwent GTR (1/5 [20.0%]), two cases (2/8 [40.0%]) underwent subtotal resection, and for two cases (2/8 [40.0%]), the extent of the resection was not reported. Our patient underwent a transfrontal-temporal approach of total tumor resection. Compared with incomplete resection, local recurrence and metastatic seeding significantly reduced, and the 5- and 10-year survival rates were prolonged after GTR (16, 56). Relapse of ependymomas is usually treated with surgery or re-radiotherapy, and no chemotherapeutics or targeted agents have shown significant survival benefits to date (57, 58).
Radiotherapy
The use of radiotherapy modalities to treat ependymoma remains controversial, especially in children (59). Whipple et al. reported that radiotherapy is imperative for residual tumors (18); however, no objective data currently supports this theory (3). As for SE, 4 cases (4/13 [30.77%]) underwent postoperative radiotherapy, and only one case (1/13 [7.69%]) showed tumor recurrence (11). Due to the limited number of patients reported, we do not know whether postoperative radiotherapy is a feasible and effective therapy for patients with SE. In some reports, it was indicated that postoperative local radiotherapy could be considered if the residual tumor was distant from the optic apparatus based on postoperative MRI scan analysis (11, 12), and this might be feasible. The patient reported here did not accept radiotherapy as the postoperative CT and follow-up MRI showed complete tumor resection and no tumor recurrence sign.
Chemotherapy
Chemotherapy has not been a routine treatment paradigm for ependymomas, and no patient with SE has been reported to undergo chemotherapy (2). However, chemotherapy is advised for some ependymomas following relapse, particularly when the patient has already undergone radiotherapy or as an attempt to defer radiotherapy and associated adverse effects, including stroke, neurocognitive deficits, and secondary malignancy, among others (59). Generally, chemotherapy for SE remains to be studied, although it might have some benefits as salvage treatment when there are no other options (2, 11). We do not advocate immediate postoperative chemotherapy. However, when dealing with recurrent disease and/or complex SE, according to a report by Belcher et al., the chemotherapeutic agent temozolomide could be considered a treatment option (11).
Molecular Targeted Therapy
Drugs targeting mutant fusion genes are not available and are under investigation (21, 24, 54, 55). The roles of NF-κB inhibitor and YAP-1 inhibitor have been verified in the pre-clinical studies in animal models (60). The hypermethylation of CpG island and aberrant histone modification were confirmed as a reversible chemical modification. Deepening of basic research will verify whether they are expected to be an effective, potential drug target. Moreover, DNA methyltransferase inhibitors (5-Azacytidine and decitabine) and histone deacetylase inhibitors (HDACIs, suberoylanilide hydroxamic acid [SAHA], and romidepsin) are currently used in clinical practice, mainly to treat hematological malignancies (61, 62). Mack et al. also verified that decitabine, SAHA, and GSK343 (the competitive inhibitor of HDACI) could inhibit the growth of ependymoma cells during in vitro and in vivo tests (34).
Prognosis
Patients with ST-EPN-RELA and PF-EPN-A comprise the largest molecular subtypes and usually have a poor prognosis (63). The prognosis of SEs is difficult to assess given the small number of reported cases in the medical literature, and no study is available on the molecular subtypes of SE. Nevertheless, as shown in Table 1, the overall reported outcome for these patients is promising. The prognosis of ependymoma is associated with the age at onset, tumor location, the extent of initial surgical resection, and histological tumor grade (2, 4, 11). The only factor that a care provider can truly influence is the extent of tumor resection; therefore, maximum tumor resection should be the final surgical aim. Another consideration worth noting is that the recurrence of ependymomas is usually relatively late (23). Belcher et al. reported a patient of SE, who exhibited recurrence twice, and the first and second recurrence cases occurred 18 and 27 years after the initial surgery, respectively (11). Moreover, distant and cerebrospinal metastases appear to be associated with an incomplete initial excision and high-grade tumor histology (11). In summary, maximum tumor resection combined with radiotherapy and close follow-up using MRI analysis is highly encouraged in these patients.
Limitations
The main limitation of this study is a lack of genetic analysis and molecular subtyping.
Conclusion
In this study, we reported a patient with SE, which is exceedingly rare, with only 12 cases having been reported to date. GTR usually provides the best chance for long-term survival. However, the pathogenesis and recurrence rate remain unknown. Besides, the therapeutic modality has not been standardized, requiring further validation. Thus, more reported cases and long-term follow-up studies are necessary to increase knowledge about this disease. Moreover, the classification of ependymoma that combines molecular phenotype and clinical characteristics will help determine the prognosis and risk classification of patients more accurately, providing an important basis for individualized treatment in the future. The establishment of different molecular subtypes of ependymoma cells and animal models is the key to screening new treatment modalities.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of the First Hospital of Jilin University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LZ and YJ made study design, data collection, data analysis and interpretation, and composed the manuscript and literature review. YL and YW were the surgeon that performed the surgery and did data collection, data analysis, and interpretation. LL and YB made English and grammar corrections, critical revisions, and approved final version. LZ made great effort in the modification of the article. LZ and YL had the acquisition, analysis or interpretation of data for the work, revising it critically for important intellectual content, final approval of the version to be published, and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CNS, central nervous system; CT, computerized tomography; EMA, epithelial membrane antigen; FLAIR, fluid attenuated inversion recovery; GFAP, glial fibrillary acidic protein; GTR, gross tumor resection; H&E, hematoxylin and eosin; H3K27, lysine 27 on histone 3; HDACI, histone deacetylase inhibitor; OS, overall survival; PF-EPN, posterior fossa ependymoma; PFS, progression-free survival; PRL, prolactin; MRI, magnetic resonance imaging; SAHA, suberoylanilide hydroxamic acid; SE, sellar ependymoma; ST-EPN, Supratentorial ependymoma; T1WI, T1-weighted imaging; T2WI, T2-weighted imaging; TTF-1, thyroid transcription factor-1; WHO, World Health Organization.
References
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol (2007) 114(2):97–109. doi: 10.1007/s00401-007-0243-4
2. Van Gompel JJ, Koeller KK, Meyer FB, Marsh WR, Burger PC, Roncaroli F, et al. Cortical Ependymoma: An Unusual Epileptogenic Lesion. J Neurosurg (2011) 114(4):1187–94. doi: 10.3171/2010.12.Jns10846
3. Wang S, Zong W, Li Y, Wang B, Ke C, Guo D. Pituitary Ependymoma: A Case Report and Review of the Literature. World Neurosurg (2018) 110:43–54. doi: 10.1016/j.wneu.2017.10.134
4. McGuire CS, Sainani KL, Fisher PG. Incidence Patterns for Ependymoma: A Surveillance, Epidemiology, and End Results Study. J Neurosurg (2009) 110(4):725–9. doi: 10.3171/2008.9.Jns08117
5. Winer JB, Lidov H, Scaravilli F. An Ependymoma Involving the Pituitary Fossa. J Neurol Neurosurg Psychiatry (1989) 52(12):1443–4. doi: 10.1136/jnnp.52.12.1443
6. Sarkisian SS, Schultz AL. Pituitary Ependymoma. United States Armed Forces Med J (1956) 7(12):1813–6.
7. Thomson S, Chakrabarty A, Marks P. Ependymoma of the Neurohypophysis. Br J Neurosurg (2001) 15(3):277–8. doi: 10.1080/026886901750353764
8. Chiu V, Chan C, Chan P, Chiu H, Cheung Y. Chan Cjjohkcor. Suprasellar Ependymoma (2001) 4(2):169–72.
9. Mukhida K, Asa S, Gentili F, Shannon P. Ependymoma of the Pituitary Fossa. Case Report and Review of the Literature. J Neurosurg (2006) 105(4):616–20. doi: 10.3171/jns.2006.105.4.616
10. Scheithauer BW, Swearingen B, Whyte ET, Auluck PK, Stemmer-Rachamimov AO. Ependymoma of the Sella Turcica: A Variant of Pituicytoma. Hum Pathol (2009) 40(3):435–40. doi: 10.1016/j.humpath.2008.08.013
11. Belcher R, Chahal HS, Evanson J, Afshar F, Marino S, Grossman AB. Recurrent Pituitary Ependymoma: A Complex Clinical Problem. Pituitary (2010) 13(2):176–82. doi: 10.1007/s11102-008-0139-x
12. Lee JS, Cho KH, Hong EK, Shin SH. Pituitary Ependymoma, 10-Year Follow-Up After Partial Resection and Radiation Therapy. Brain Tumor Res Treat (2017) 5(2):94–8. doi: 10.14791/btrt.2017.5.2.94
13. Liu MQ, Liu Y, Chen ZY. [Ependymoma in Sellar Region:Report of One Case]. Zhongguo yi xue ke xue yuan xue bao Acta Academiae Med Sinicae (2019) 41(1):139–42. doi: 10.3881/j.issn.1000-503X.10397
14. Parish JM, Bonnin JM, Goodman JM, Cohen-Gadol AA. Intrasellar Ependymoma: Clinical, Imaging, Pathological, and Surgical Findings. J Clin Neurosci (2015) 22(4):638–41. doi: 10.1016/j.jocn.2014.10.026
15. Ramesh VG, Karthikeyan KV, Rajaraman S, Rao R. Pituitary Ependymoma: Report of a Rare Case With an Insight Into the Histogenesis. Neurol India (2013) 61(5):545–6. doi: 10.4103/0028-3886.121948
16. Schwartz TH, Kim S, Glick RS, Bagiella E, Balmaceda C, Fetell MR, et al. Supratentorial Ependymomas in Adult Patients. Neurosurgery (1999) 44(4):721–31. doi: 10.1097/00006123-199904000-00018
17. Heath SE, Peter AT, Janovitz EB, Selvakumar R, Sandusky GE. Ependymoma of the Neurohypophysis and Hypernatremia in a Horse. J Am Vet Med Assoc (1995) 207(6):738–41.
18. Whipple SG, Savardekar AR, Rao S, Mahadevan A, Guthikonda B, Kosty JA. Primary Tumors of the Posterior Pituitary Gland: A Systematic Review of the Literature in Light of the New 2017 World Health Organization Classification of Pituitary Tumors. World Neurosurg (2021) 145:148–58. doi: 10.1016/j.wneu.2020.09.023
19. Lopes MBS. The 2017 World Health Organization Classification of Tumors of the Pituitary Gland: A Summary. Acta Neuropathol (2017) 134(4):521–35. doi: 10.1007/s00401-017-1769-8
20. Mete O, Lopes MB. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocrine Pathol (2017) 28(3):228–43. doi: 10.1007/s12022-017-9498-z
21. Guerrero-Pérez F, Marengo AP, Vidal N, Iglesias P, Villabona C. Primary Tumors of the Posterior Pituitary: A Systematic Review. Rev Endocrine Metab Disord (2019) 20(2):219–38. doi: 10.1007/s11154-019-09484-1
22. Shibuya M. Welcoming the New WHO Classification of Pituitary Tumors 2017: Revolution in TTF-1-positive Posterior Pituitary Tumors. Brain Tumor Pathol (2018) 35(2):62–70. doi: 10.1007/s10014-018-0311-6
23. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
24. Nabors LB, Portnow J, Ammirati M, Baehring J, Brem H, Butowski N, et al. Nccn Guidelines Insights: Central Nervous System Cancers, Version 1.2017. J Natl Compr Cancer Net JNCCN (2017) 15(11):1331–45. doi: 10.6004/jnccn.2017.0166
25. Ellison DW, Kocak M, Figarella-Branger D, Felice G, Catherine G, Pietsch T, et al. Histopathological Grading of Pediatric Ependymoma: Reproducibility and Clinical Relevance in European Trial Cohorts. J Neg Results Biomed (2011) 10:7. doi: 10.1186/1477-5751-10-7
26. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular Classification of Ependymal Tumors Across All Cns Compartments, Histopathological Grades, and Age Groups. Cancer Cell (2015) 27(5):728–43. doi: 10.1016/j.ccell.2015.04.002
27. Pajtler KW, Mack SC, Ramaswamy V, Smith CA, Witt H, Smith A, et al. The Current Consensus on the Clinical Management of Intracranial Ependymoma and its Distinct Molecular Variants. Acta Neuropathol (2017) 133(1):5–12. doi: 10.1007/s00401-016-1643-0
28. Delgado-López PD, Corrales-García EM, Alonso-García E, García-Leal R, González-Rodrigálvarez R, Araus-Galdós E, et al. Central Nervous System Ependymoma: Clinical Implications of the New Molecular Classification, Treatment Guidelines and Controversial Issues. Clin Trans Oncol Off Publ Fed Spanish Oncol Soc Natl Cancer Inst Mexico (2019) 21(11):1450–63. doi: 10.1007/s12094-019-02082-2
29. Andreiuolo F, Varlet P, Tauziède-Espariat A, Jünger ST, Dörner E, Dreschmann V, et al. Childhood Supratentorial Ependymomas With YAP1-MAMLD1 Fusion: An Entity With Characteristic Clinical, Radiological, Cytogenetic and Histopathological Features. Brain Pathol (2019) 29(2):205–16. doi: 10.1111/bpa.12659
30. Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, et al. C11orf95-RELA Fusions Drive Oncogenic NF-κb Signalling in Ependymoma. Nature (2014) 506(7489):451–5. doi: 10.1038/nature13109
31. Gessi M, Giagnacovo M, Modena P, Elefante G, Gianno F, Buttarelli FR, et al. Role of Immunohistochemistry in the Identification of Supratentorial C11orf95-Rela Fused Ependymoma in Routine Neuropathology. Am J Surg Pathol (2019) 43(1):56–63. doi: 10.1097/pas.0000000000000979
32. Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, et al. Immunohistochemical Analysis of H3K27me3 Demonstrates Global Reduction in Group-a Childhood Posterior Fossa Ependymoma and is a Powerful Predictor of Outcome. Acta Neuropathol (2017) 134(5):705–14. doi: 10.1007/s00401-017-1752-4
33. Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, et al. Delineation of Two Clinically and Molecularly Distinct Subgroups of Posterior Fossa Ependymoma. Cancer Cell (2011) 20(2):143–57. doi: 10.1016/j.ccr.2011.07.007
34. Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, et al. Epigenomic Alterations Define Lethal CIMP-positive Ependymomas of Infancy. Nature (2014) 506(7489):445–50. doi: 10.1038/nature13108
35. Ramaswamy V, Hielscher T, Mack SC, Lassaletta A, Lin T, Pajtler KW, et al. Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(21):2468–77. doi: 10.1200/jco.2015.65.7825
36. Merchant TE, Bendel AE, Sabin ND, Burger PC, Shaw DW, Chang E, et al. Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol Off J Am Soc Clin Oncol (2019) 37(12):974–83. doi: 10.1200/jco.18.01765
37. Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, et al. Lowered H3K27me3 and DNA Hypomethylation Define Poorly Prognostic Pediatric Posterior Fossa Ependymomas. Sci Trans Med (2016) 8(366):366ra161. doi: 10.1126/scitranslmed.aah6904
38. Pollack IF, Agnihotri S, Broniscer A. Childhood Brain Tumors: Current Management, Biological Insights, and Future Directions. J Neurosurg Pediatr (2019) 23(3):261–73. doi: 10.3171/2018.10.Peds18377
39. Swanson AA, Raghunathan A, Jenkins RB, Messing-Jünger M, Pietsch T, Clarke MJ, et al. Spinal Cord Ependymomas With Mycn Amplification Show Aggressive Clinical Behavior. J Neuropathol Exp Neurol (2019) 78(9):791–7. doi: 10.1093/jnen/nlz064
40. Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, et al. MYCN Amplification Drives an Aggressive Form of Spinal Ependymoma. Acta Neuropathol (2019) 138(6):1075–89. doi: 10.1007/s00401-019-02056-2
41. Vera-Bolanos E, Aldape K, Yuan Y, Wu J, Wani K, Necesito-Reyes MJ, et al. Clinical Course and Progression-Free Survival of Adult Intracranial and Spinal Ependymoma Patients. Neuro-oncology (2015) 17(3):440–7. doi: 10.1093/neuonc/nou162
42. Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, et al. Cross-Species Genomics Matches Driver Mutations and Cell Compartments to Model Ependymoma. Nature (2010) 466(7306):632–6. doi: 10.1038/nature09173
43. Hamilton DW, Lusher ME, Lindsey JC, Ellison DW, Clifford SC. Epigenetic Inactivation of the RASSF1A Tumour Suppressor Gene in Ependymoma. Cancer Lett (2005) 227(1):75–81. doi: 10.1016/j.canlet.2004.11.044
44. Ellison DW, Aldape KD, Capper D, Fouladi M, Gilbert MR, Gilbertson RJ, et al. cIMPACT-NOW Update 7: Advancing the Molecular Classification of Ependymal Tumors. Brain Pathol (2020) 30(5):863–6. doi: 10.1111/bpa.12866
45. Takei Y, Seyama S, Pearl GS, Tindall GT. Ultrastructural Study of the Human Neurohypophysis. II. Cellular Elements of Neural Parenchyma, the Pituicytes. Cell Tissue Res (1980) 205(2):273–87. doi: 10.1007/bf00234685
46. Choi JY, Chang KH, Yu IK, Kim KH, Kwon BJ, Han MH, et al. Intracranial and Spinal Ependymomas: Review of MR Images in 61 Patients. Korean J Radiol (2002) 3(4):219–28. doi: 10.3348/kjr.2002.3.4.219
47. Loh JA, Verbalis JG. Diabetes Insipidus as a Complication After Pituitary Surgery. Nat Clin Pract Endocrinol Metab (2007) 3(6):489–94. doi: 10.1038/ncpendmet0513
48. Brat DJ, Scheithauer BW, Staugaitis SM, Holtzman RN, Morgello S, Burger PC. Pituicytoma: A Distinctive Low-Grade Glioma of the Neurohypophysis. Am J Surg Pathol (2000) 24(3):362–8. doi: 10.1097/00000478-200003000-00004
49. Lukashova-v Zangen I, Kneitz S, Monoranu CM, Rutkowski S, Hinkes B, Vince GH, et al. Ependymoma Gene Expression Profiles Associated With Histological Subtype, Proliferation, and Patient Survival. Acta Neuropathol (2007) 113(3):325–37. doi: 10.1007/s00401-006-0190-5
50. Wippold FJ,2, Perry A. Neuropathology for the Neuroradiologist: Rosettes and Pseudorosettes. AJNR Am J Neuroradiol (2006) 27(3):488–92.
51. Kumar PV. Nuclear Grooves in Ependymoma. Cytologic Study of 21 Cases. Acta Cytol (1997) 41(6):1726–31. doi: 10.1159/000333176
52. Ng HK, Tse CC, Lo ST. Ependymomas–an Immunohistochemical Study of 14 Cases. Histopathology (1988) 13(4):462–5. doi: 10.1111/j.1365-2559.1988.tb02063.x
53. Valassi E, Biller BM, Klibanski A, Swearingen B. Clinical Features of Nonpituitary Sellar Lesions in a Large Surgical Series. Clin Endocrinol (2010) 73: (6):798–807. doi: 10.1111/j.1365-2265.2010.03881.x
54. Wang M, Zhang R, Liu X, Li D, Qiu C, Zhao P, et al. Supratentorial Extraventricular Ependymomas: A Retrospective Study Focused on Long-Term Outcomes and Prognostic Factors. Clin Neurol Neurosurg (2018) 165:1–6. doi: 10.1016/j.clineuro.2017.12.013
55. Takeda N, Nishihara M, Harada T, Kidoguchi K, Hashimoto K. Supratentorial Extraventricular WHO Grade III (Anaplastic) Ependymoma 17 Years After Total Removal of WHO Grade II Ependymoma of the Fourth Ventricle. Br J Neurosurg (2017) 31(2):270–2. doi: 10.1080/02688697.2016.1187251
56. Paulino AC, Wen BC, Buatti JM, Hussey DH, Zhen WK, Mayr NA, et al. Intracranial Ependymomas: An Analysis of Prognostic Factors and Patterns of Failure. Am J Clin Oncol (2002) 25(2):117–22. doi: 10.1097/00000421-200204000-00003
57. Wu J, Armstrong TS, Gilbert MR. Biology and Management of Ependymomas. Neuro-oncology (2016) 18(7):902–13. doi: 10.1093/neuonc/now016
58. Lee SH, Chung CK, Kim CH, Yoon SH, Hyun SJ, Kim KJ, et al. Long-Term Outcomes of Surgical Resection With or Without Adjuvant Radiation Therapy for Treatment of Spinal Ependymoma: A Retrospective Multicenter Study by the Korea Spinal Oncology Research Group. Neuro-oncology (2013) 15(7):921–9. doi: 10.1093/neuonc/not038
59. Udaka YT, Packer RJ. Pediatric Brain Tumors. Neurol Clinics (2018) 36(3):533–56. doi: 10.1016/j.ncl.2018.04.009
60. Johnson R, Halder G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat Rev Drug Discovery (2014) 13(1):63–79. doi: 10.1038/nrd4161
61. Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, et al. DNA Methylation Predicts Survival and Response to Therapy in Patients With Myelodysplastic Syndromes. J Clin Oncol Off J Am Soc Clin Oncol (2010) 28(4):605–13. doi: 10.1200/jco.2009.23.4781
62. Armstrong TS, Vera-Bolanos E, Bekele BN, Aldape K, Gilbert MR. Adult Ependymal Tumors: Prognosis and the M. D. Anderson Cancer Center Experience. Neuro-oncology (2010) 12(8):862–70. doi: 10.1093/neuonc/noq009
Keywords: diagnosis, ependymoma, molecular subtype, pituitary tumor, sellar ependymoma, treatment
Citation: Zhao L, Jiang Y, Wang Y, Bai Y, Liu L and Li Y (2021) Case Report: Sellar Ependymomas: A Clinic-Pathological Study and Literature Review. Front. Endocrinol. 12:551493. doi: 10.3389/fendo.2021.551493
Received: 15 April 2020; Accepted: 19 April 2021;
Published: 08 June 2021.
Edited by:
Veronica Vella, University of Catania, ItalyReviewed by:
Fady Hannah-Shmouni, National Institutes of Health (NIH), United StatesCarl Friedrich Classen, University Hospital Rostock, Germany
Copyright © 2021 Zhao, Jiang, Wang, Bai, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunqian Li, eXVucWlhbkBqbHUuZWR1LmNu