 Jon D. Piganelli
Jon D. Piganelli Mark J. Mamula
Mark J. Mamula Eddie A. James
Eddie A. James- 1Division of Pediatric Surgery, Department of Surgery, Children’s Hospital of Pittsburgh, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 2Section of Rheumatology, Department of Medicine, Yale School of Medicine, New Haven, CT, United States
- 3Translational Research Program, Benaroya Research Institute at Virginia Mason, Seattle, WA, United States
Due to their secretory function, β cells are predisposed to higher levels of endoplasmic reticulum (ER) stress and greater sensitivity to inflammation than other cell types. These stresses elicit changes in β cells that alter their function and immunogenicity, including defective ribosomal initiation, post-translational modifications (PTMs) of endogenous β cell proteins, and alternative splicing. Multiple published reports confirm the presence of not only CD8+ T cells, but also autoreactive CD4+ T cells within pancreatic islets. Although the specificities of T cells that infiltrate human islets are incompletely characterized, they have been confirmed to include neo-epitopes that are formed through stress-related enzymatic modifications of β cell proteins. This article summarizes emerging knowledge about stress-induced changes in β cells and data supporting a role for neo-antigen formation and cross-talk between immune cells and β cells that provokes autoimmune attack - leading to a breakdown in tissue-specific tolerance in subjects who develop type 1 diabetes.
Introduction
Type 1 diabetes (T1D) is a chronic immune mediated disease in which insulin-producing β cells are destroyed leading to lifelong insulin deficiency (1, 2). The autoimmune etiology of T1D is clear and both CD4+ and CD8+ T cells have been shown to recognize a wide variety of beta cell derived epitopes (3, 4). However, there is an increasing appreciation that β-cell dysfunction also plays a crucial role in disease (5–7). Emerging published work demonstrates that inflammatory cytokines and/or reactive oxygen species (ROS) can trigger ER stress, HLA Class I upregulation, and other deleterious changes in β cells (8, 9). ER stress, in turn, has been shown to promote post-translational modifications and alternative mRNA splicing, thereby generating neo-sequences that have been shown to be recognized by autoreactive T cells and autoantibodies in patients with type 1 diabetes and animal models of disease (10, 11). Importantly, such neo-epitopes are not genetically encoded and thought to be underrepresented in healthy tissue. Therefore, neo-epitope responses may be less subject to the central or peripheral tolerance mechanisms that limit autoimmunity. Clearly, native self-antigens are represented in the thymus (12), though post-translational modifications (PTMs) of self-antigens can generate a novel autoantigenic proteome to which tolerance has not been developed by the immune system. This theme has been previously described as “autoantigenesis”, a process to indicate how proteins acquire PTMs over the progression of disease and stimulate B and T cell autoimmunity (13). This phenomenon is observed with a number of autoimmune diseases, including multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus (SLE), and type 1 diabetes (T1D) (13, 14). This brief review will emphasize the relevance of PTMs that are generated within the insulin producing β cells of pancreatic islets. In addition, we will address how inflammatory stresses, such as cytokines and ROS, have the ability to reduce β-cell function through impaired insulin production, processing, handling, and export. Such stresses would perpetuate a continuum of immunologic epitope spreading leading to β-cell dysfunction and waves of immune attack over time (Figure 1).
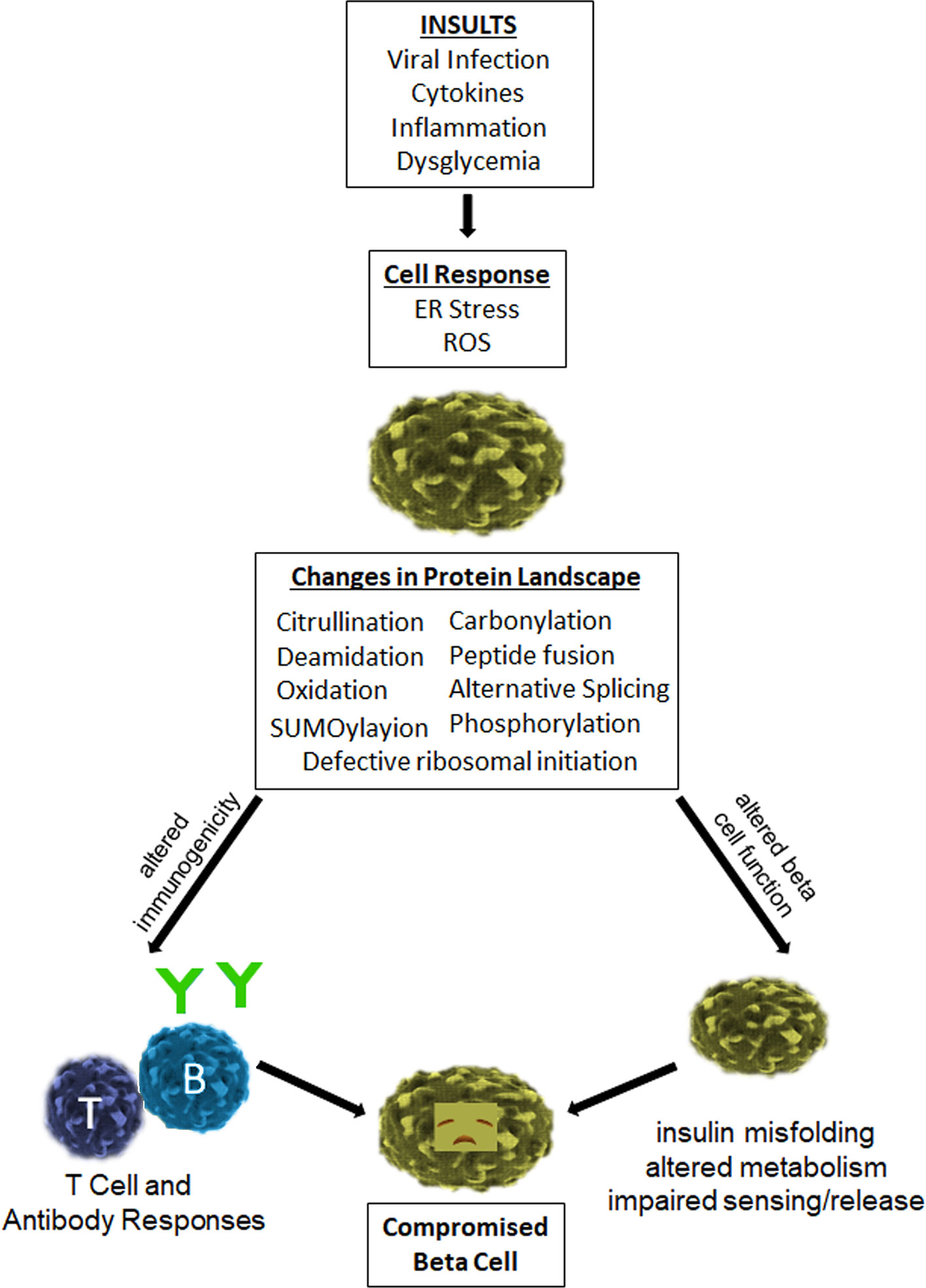
Figure 1 Impact of inflammatory stresses on β-cell immunogenicity and function. Human β-cells are vulnerable to various insults that generate a cellular stress response and deleterious effects. In particular, viral infection and exposure to inflammatory cytokines elicit ER stress and ROS, which have been shown to promote enzymatic and non-enzymatic processes that lead to the generation and release of neo-epitopes. These epitopes increase the antigenicity of β-cells and provoke the activity of autoreactive T cells and B cells. These same processes elicit alternative splicing, defective ribosomal initiation, SUMOylayion, phosphorylation, and other enzymatic modifications of key β-cell proteins, all of which can have a deleterious impact on beta cell health and function. The cumulative result of these changes is a compromised beta cell.
Pancreatic β Cells Are Vulnerable to Stress
ER stress and activation of the UPR can occur in any human cell under increased demand for protein translation. Because of their function as professional secretory cells, β cells must carry out extremely high levels of protein translation. What this means for the β cell is nearly constant synthesis and processing of proinsulin into active insulin in the ER. The mature insulin is stored in secretory granules awaiting release upon increases in blood glucose levels (15). The insulin granules are released dynamically to maintain normal blood glucose levels (16). This dynamic glucose flux places the β cell in a constant state of secretion readiness to ensure there is a perpetual flux of insulin at the ready to maintain glucose homeostasis. To achieve this, the β cell contains a large pool of cytoplasmic proinsulin mRNA (~20% of the total mRNA), one of the most abundant mRNA species (16). One profound example of physiological fluctuation in the protein-folding load in the ER is the unique translational response of pancreatic β cells to variations in blood glucose (17). In response to increased blood glucose, β cells increase translation of preproinsulin by 50-fold (18), reaching a production rate of 1 million molecules per minute (18). The preproinsulin molecules flood the ER lumen for proper folding and disulfide bond formation, causing tremendous ER stress. The active production and packaging of insulin maintaining glucose homeostasis places the β cell in a constant state of ER stress. Although the ER of the β cell is encumbered with the major task of insulin production in the face of dynamic glucose sensing (described above), the ER has adapted a fail-safe program of intracellular signaling pathways (also active in other cell types), termed the unfolded protein response (UPR). The activation of the UPR initiates a cascade of signaling events to quell the activity of protein processing and folding in order to resolve the ER burden and restore homeostasis. The adaptive unfolded protein response (aUPR) promotes adaptation in cells experiencing increased levels of ER stress to ensure that the cellular production is regulated and manageable (19). To regulate ER stress, the chaperone GRP78 releases the protein sensors of ER stress (20). As part of this response, activated PERK phosphorylates eIF2α to generally suppress mRNA translation and reduces the protein burden on the ER. Also, active ATF6 initiates new chaperone synthesis to aid with proper protein folding in the ER, and coincident phosphorylation of IRE1 leads to splicing of XBP1 mRNA and further chaperone synthesis [recently reviewed in detail elsewhere (21, 22)]. This aUPR serves to alleviate ER stress during times of high protein load (22, 23). However, it is known that the UPR has two modes of the unfolded protein response (UPR) (19, 24) which begins with protein sensors of stress in the ER membrane (25). The aUPR (described above) occurs early to alleviate ER stress and restore normal cellular function. However, if ER stress is too great, prolonged or further induced by environmental or physiological triggers, then the aUPR-mediated recovery fails and induction of the terminal (tUPR) initiates a specialized programmed cell death pathway (19, 24, 26–28). This switch from aUPR to tUPR has been further described (24, 26, 28, 29), and may ultimately result from the unmet need in bioenergetics and reducing equivalents needed for β cell day-to-day operation leading the programmed cell death (30, 31).
Emerging work suggests that in addition to being subject to high levels of ER stress due to their function, β cells may be predisposed to increased stress and damage in subjects at high risk of developing T1D due to disease associated genetic variants. Indeed, a substantial proportion of candidate genes within T1D susceptibility loci are expressed in β cells (32). Several of these, including PTPN2, MDA5, and BACH2 have an implicated role in modulating islet inflammation, β-cell apoptosis, and responses to cytokines and viruses (33, 34). These observations support a paradigm in which genetically susceptible individuals experience higher levels of β cell stress under physiologic conditions, leading to a greater probability of β-cell dysfunction and immune attack. Indeed, a model was recently proposed in which beta-cell defects may significantly contribute to T1D (35). A key element of that paradigm is the concept that there is an intrinsic (and to some degree genetically driven) vulnerability of beta-cells to death and dysfunction, which continues to drive the loss of insulin secretion after the establishment of persistent anti-islet autoimmunity. Indeed, it there is some evidence to suggest that beta cell dysfunction is present even in the absence of overt insults. For example, recent work shows that β-cell dysfunction (evidenced by an abnormal proinsulin/insulin ratio) precedes disease and is a common feature in subjects with T1D (36, 37) and yet pronounced insulitis in human islets is rare (38). One potential consequence of such intrinsic β cell fragility and vulnerability is a continued propagation of dysfunction in even after the resolution or diminution of active immune attack.
ER Stress in β Cells Is Influenced by Immunologic and Environmental Factors
It understood that cytokines play a crucial role in β cell damage. Although the precise mechanisms of responsiveness to cytokines are species specific, the induction of ER stress and/or apoptosis by cytokines in β cells is indeed important and key general aspects of cytokine-induced apoptosis are conserved in mice, rats, and humans (39). In particular, proinflammatory factors such as IL-1β, TNF-α and IFN-γ have been shown to play important roles in eliciting ER stress. Many other physiological and environmental triggers that are associated with T1D have also been shown to enhance ER stress in β cells, including viral infection (40–42), exposure to chemicals (43–46), dysglycemia (47), and the intrinsic demands of insulin secretion (as delineated above).
ER stress may be a common downstream pathway that contributes to the development of T1D. However there is some disconnect, in that the progression of immune cell infiltration leads to the deposition of cytokines at the β cell. In all likelihood, an intricate interplay between genetic predisposition, the immune system, and environmental factors precipitates T1D in humans. As described above, one feasible bridge between genetics, the immune system, and environmental factors are type 1 interferons (T1-IFNs) (48). Type 1 interferons (T1-IFNs) are well known for inducing antiviral factors that limit infection by regulating innate and adaptive immune responses. Furthermore, as described above, several T1D genetic risk loci coincide with genes that are associated with innate and adaptive responses to T1-IFN (33, 34). Additional support that T1-IFN play a role in T1D is that these cytokines are a known constituent of the autoinflammatory milieu within the pancreas of patients with T1D. The presence of IFNα/β is correlated with characteristic MHC class I (MHC-I) hyperexpression found in the islets of patients with T1D, suggesting that T1-IFNs enhance β cell recognition by autoreactive cytotoxic CD8+ T lymphocytes and insulin-producing pancreatic β cells through increasing MHC I expression (48). Of course, cytokine induced β cell apoptosis is only one of the possible outcomes. An important scenario in which cytokine related effects could occur is through viral infection—specifically enteroviral infection with Coxsackie virus (CVB). It is well known that viral infection leads to a type I interferon response at the target tissue site (49). This serves to mobilize the immune system to the site of infection to initiate the clearance of the pathogen. Viral infection facilitates the recruitment of accessory cells and T cells to the islets (50) leading to site directed production of inflammatory cytokines, particularly INF-α, INF-β, IFN-γ, tumor necrosis factor (TNF) and IL-1β (51). The importance of these cytokines in β-cell destruction has been exhaustively demonstrated in NOD mice and rat models of diabetes mellitus (52–54). CVB infection accelerates disease onset in young non-obese diabetic (NOD) mice with established insulitis—likely acting as an accelerant to the break in tolerance as a result of type 1 and type 2 cytokines (55–57)—and elicits bot ER stress (58–60) and the release of intracellular Ca2+ (41, 42, 61) upon entry into β cells (62, 63). Interestingly, Ca2+ flux also facilitates the induction of ER stress during CVB infection (40, 62–66). While ironclad proof of direct causality has remained elusive, CVB infection is highly associated with T1D onset in humans (67–75). Furthermore, a number of studies have defined footprints of CVB infection in the islets, demonstrated through the presence of RNA and VP1 antibody staining (70, 74, 76–80). More specifically, evidence is mounting that enteroviruses such as CVB could be involved in perpetuating the break in self-tolerance by increasing islet β cell specific inflammation (28, 81–84), thereby providing a more inflammatory milieu (85, 86) that promotes the optimal activation of virus reactive and self-reactive T cells (87–94). This idea has been supported by a number of elegant studies demonstrating that CVB infection selectively activates certain pathways that allow a tunable ER-stress and unfolded protein response (UPR) that favors viral amplification (60, 75) and persistent infection (67, 73, 74, 95–98) without the induction of premature apoptosis and death. Finally, recent work demonstrates that enterovirus family members show a strong association with islet autoimmunity in human T1D patients (99), are capable of infecting islets, and show that a sizeable percentage of type 1 diabetic patients have prolonged/persistent enterovirus infection associated with gut mucosa inflammation (98, 100).
Stressed β Cells Exhibit Increased Immunogenicity
A key observation in murine studies that has been subsequently supported by parallel studies of human beta cell lines is that there is increased immune recognition of stressed β cells. For example, multiple studies show that endoplasmic reticulum (ER) stress in β cells increases cytosolic calcium Ca2+ and the activity of tissue transglutaminase 2 (tTG2), leading to the generation of deamidated neo-epitopes (10, 101–103). Our work and other published studies demonstrated increased immunogenicity of beta cell peptides following enzymatic modifications at specific residues which are crucial for HLA binding and presentation, T cell receptor recognition, or both (10, 104, 105). Indeed, the progression of T1D and beta cell dysfunction are characterized by an accumulation of autoantibodies against beta cell antigens (106) and the activation of auto-reactive T cells, which have been shown to infiltrate pancreatic islets (107, 108). Furthermore, we have shown that subjects with T1D have elevated frequencies of T cells that recognize citrullinated and deamidated epitopes from β cell antigens, and that T cells with some of these specificities can be found in the pancreatic lymph nodes of organ donors with T1D (10, 105, 108). Hence, it is plausible that T cells that recognize citrullinated and deamidated epitopes, as a result of ER stress induced Ca2+ flux and activation of tTG2 enzymes, become activated and expanded in subjects who progress to develop T1D, likely playing a role in the pathogenesis of the disease. Notably, many of the enzymes which are responsible for the introduction of protein modifications, including tTG2 (responsible for deamidation), peptidyl arginine deaminase (PAD) enzymes (responsible for the introduction of citrullinate into proteins and peptides), and various cysteine proteases (e.g. calpains), which may participate in peptide transpeptidation-reactions (leading to the formation of hybrid insulin peptides or “HIPs”) are Ca2+-dependent enzymes (109–111). HIPs belong to a new family of autoantigens in T1D, which are targeted by diabetes triggering T cells in mice, and that have been shown to be recognized by T cells in the peripheral blood of T1D patients, and by T cells identified in the residual pancreatic islets organ donors with T1D (112, 113).
A central pathway that contributes to the enhanced immunogenicity of stressed β cells is the development of neo-antigens and epitopes, which has been shown to occur by a variety of enzymatic and non-enzymatic processes that have been reviewed elsewhere (114). Several published studies illustrate the genetic risk factors associated with autoimmune diabetes, particularly the associations with susceptible HLA class II haplotypes (115). The most likely contribution of HLA class II proteins to disease is through selection of a potentially autoreactive CD4+ T cell repertoire (116). These same HLA class II molecules have been shown to have an increased capacity to bind and present peptides with post-translational modifications (10, 117). Furthermore, it has been clearly shown that autoantibodies and autoreactive T cells recognize multiple beta cell antigens, including novel stress-related specificities formed through alternative splicing and defective ribosomal initiation that have been only recently appreciated (11, 118, 119). Therefore, mounting evidence implicates the formation of neo-epitopes as one important means of circumventing immune tolerance.
PTMs also change many other features of protein chemistry, including primary and tertiary structure, biological (and/or enzymatic) functions, and proteolytic degradation (antigen processing) that are important in creating both toleragenic and immunogenic self-peptides. Clearly, the way in which a self-protein is processed by antigen presenting cells may break immune tolerance (120, 121). The modification of amino acid(s) critical for the recognition and cleavage by certain proteases can affect the peptides generated or the rate in which they are generated. For example, the lack of N-glycosylation of the neuronal glutamate receptor subunit in Rasmussen’s encephalitis (a severe form of pediatric epilepsy) exposes a granzyme B cleavage site that is otherwise inaccessible to the enzyme (121). Additionally, the presence of citrulline residues in peptides of myelin basic protein (MBP) increases its rate of digestion by cathepsin D (122). Tissue stress, both cytokines and ROS, amplifies the accumulation of PTMs that induce disease in the host. Several factors control the ability and rate of PTMs that occur in a given protein. Flanking residues near an epitope sequence of amino acids significantly influence how the site may be modified. Spontaneous isoaspartyl modification occurs most frequently at Asp/Asn-Ser or Asp/Asn-Gly amino acid motifs where serine or glycine adjacent sites are critical for modification (123–125). The environment of a modifying enzyme (such as in the pancreatic islet) is also important since they are compartmentalized in intracellular organelles, the endoplasmic reticulum, or in extracellular spaces. For example, protein and/or DNA methylation require both the presence and cellular proximity of methylases (DNA methyltransferases or protein methyltransferases) along with the cellular source of methyl donor groups, S-adenosylmethionine (SAM) (126). Finally, features within the beta cell protein itself, such as previous modifications, will affect how a particular residue is modified (127).
It could be said that T1D is an autoimmune disease for which evidence of how modified autoantigens contribute to pathogenesis is currently emerging. One key example of this is the evolving understanding of chromogranin A as a disease relevant antigen. Studies by Stadinski and colleagues demonstrated that chromogranin A is recognized by disease relevant T cells; specifically, the WE14 peptide (a natural cleavage product derived from chromogranin A) stimulated diabetogenic CD4 T cell clones and reactivity of those clones with islet preparations was abrogated by knocking out chromogranin A (128). However, further studies showed that the antigenicity of WE14 (chromogranin A fragment) is greatly increased by treatment with transglutaminase; this enzyme that is known to modify peptides through deamidation and also through cross-linking so either could contribute to the observed change in immunogenicity (129). Studies that are more recent strongly suggested that the most potent ligand for that T cell clone is a hybrid peptide formed between WE14 and a fragment of insulin (111, 112), implicating cross linking as the most likely mechanism.
Certain self-antigens appear to elude central and peripheral tolerance mechanisms. Previous work from our laboratory and others has identified the presence of autoreactive T and B cells even in the peripheral repertoire of normal mice and healthy human subjects (11, 130). Autoreactive cells can escape deletion because both cryptic peptides and posttranslationally modified proteins are underrepresented in the thymus, leading to impaired negative selection of these potentially self-reactive T cells (131, 132). We have shown that protein modifications alter both the antigenicity of self-proteins and the intracellular signaling properties of lymphocytes, leading to aberrant autoimmune responses (131, 133). As one example, the spontaneous conversion of an aspartic acid to an isoaspartic acid induces both T and B cell immunity to model self-antigens (14, 134). The presence of isoaspartyl modifications alter the immune processing and presentation of self peptides as indicated earlier since proteases and peptidases are not able to cleave on the carboxyl side of the isoaspartic acid modifications (135). Isoaspartyl modifications alter the structural integrity of histone H2B as well as trigger autoantibodies to H2B, characteristic of systemic lupus erythematosus (SLE) (136). Similarly, isoaspartyl modification of the SLE autoantigen Sm snRNP amplifies lupus autoimmunity and is bound by SLE patient autoantibodies (137). Finally, T lymphocytes that acquire isoaspartyl protein modifications have a hyperproliferative phenotype due to increased phosphorylation of ERK and Akt, characteristic of human SLE and murine models of disease (138–142).
PTMs often arise spontaneously, but are amplified as a consequence of cellular activation, inflammation, and cellular stress. These modifications include deamination, acetylation, glycosylation, citrullination, phosphorylation, and isoaspartylation. One unique form of modification, carbonylation, is the non-enzymatic addition of aldehydes or ketones to amino acid residues via a metal-catalyzed reaction. This PTM has not fully been studied in the initiation of T1D, although extensive oxidative carbonylation is a component of diabetic complications (143). For example, it has been demonstrated that islet lysates treated with copper and ascorbate generate new glutamic acid decarboxylase (GAD65) aggregates that react with T1DM patient sera (144). Importantly, neo-epitopes formed in peripheral tissues by these diverse mechanisms but underrepresented in the thymus could be expected to be recognized with high affinity by self-reactive T cells. As an example, certain HIP epitopes activate T cell clones at extremely low peptide doses (111, 112).
Posttranslational Protein Modifications May Alter The Biologic Functions of Cells
Beyond the autoimmune responses clearly defining ‘biomarkers’ of the onset and perpetuation of T1D, PTMs also have the potential to alter the metabolic pathways and function of the beta cell. As one example, glucose metabolism in humans is carefully regulated by the activity of glucokinase (GCK), a glucose sensor and a protein highly expressed by pancreatic beta cells. GCK catalyzes a principle rate controlling step of glucose metabolism needed to trigger insulin release, and in the liver, where it has a role in glycogen synthesis; reviewed in (145). While not the topic of the present review, a number of genetic mutations of GCK lead to a variety of clinical manifestations including MODY (maturity onset diabetes of the young), hyperinsulinism, and loss of function (145).
Several metabolites, including glucose itself and insulin, influence the transcription of GCK. GCK regulation is a network of cellular processes that coordinate with the metabolic state of the beta cell. PTMs clearly alter the tertiary structure of proteins, particularly relevant to the metabolic properties of GCK. In particular, GCK assumes an “open” and “closed” conformational state that regulates binding to glucose and to other allosteric macromolecules. A number of PTMs of GCK that both increase and decrease metabolic activity have recently been described (145). For example, attachment of Small Ubiquitin-like Modifier proteins (SUMOylation) or S-nitrosylation can shift the conformational state of GCK to increase its catalytic activity for glucose (though only a small fraction, about 5%, of the GCK pool is SUMOylated in beta cells). Interestingly, SUMOylation has been shown to have broader effects in controlling beta cell survival and oxidative stress, such that either its overexpression or conditional ablation leads to imparted β cell function (146). Glucokinase regulatory protein (GKRP) inhibition of GCK is modulated by PTMs. Nuclear translocation of GCK is impaired by SUMOlyation, though this PTM also stabilizes GCK catalytic activity. Identification of the specific PTMs that alter the association of GCK with Ubiquitin-like domain (ULD) are a target of therapeutic intervention by several groups (145). All of these conformational states of GCK alter the downstream interactions with GKRP, phosphofructokinase biphosphatase-2, ULD, and propionyl-CoA Carboxylase β subunit. Ubiquitin-like domain (ULD) proteins also interact with and reduce GCK activity over the course of glucose metabolic pathways (145). ULD protein interactions with GCK are highly dependent on native structure. Recent studies (Yang, James, and Mamula, in preparation) have identified the presence of citrulline modifications that alter the Km and Vmax of GCK, in addition to the presence of autoantibodies and T cells specific for citrulline GCK epitopes. The repair of citrulline modifications may indeed be yet another therapeutic strategy to maintain the normal metabolic state of beta cells under inflammatory stress.
Conclusions
In this brief review, we have recounted how protein and peptide modifications, prompted by β cell stress and responsible for the formation of neo-epitopes appear to play a role in in the immunopathology of type 1 diabetes. Changes in β cell immunogenicity are easily attributable to recognition of neo-epitopes and antigens, but further research could reveal additional means through which stress-induced changes can encourage immune attack. Emerging research increasingly supports that stress and protein modification can compromise β cell function. Therefore, stress related pathways appear to elicit relevant changes in β cells altering both their immunogenicity and biological function. These effects appear to combine with genetic variants that promote β cell fragility and susceptible HLA haplotypes that are more prone to select a potentially autoreactive repertoire. This paves the way for a self-reinforcing dialogue between immune cells and β cells that provokes either sustained or recurrent autoimmune attack, eventually leading to the clinical onset of diabetes. The most elusive factor that remains to be elucidated are environmental factors such as viral infection, the footprints of which can be seen in the islet, which probably play a crucial role in initiating progression toward disease.
Author Contributions
Each of the authors (JP, MM, and EJ) made contributions to this work by conceiving, writing, and editing the manuscript. All authors agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version
Funding
This work was supported by finding from the JDRF (Grant Key 2-SRA-2020-910-S-B to JP, Grant Key 1-SRA-2020-978-S-B to EJ, and 3-SRA-2017-345-S-B to MM) and the NIH (AR41032 to MM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge the sustained efforts of their research groups and administrative support at their respective institutions, which have been crucial to gain the insights communicated in this review.
References
1. Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2:1–19. doi: 10.1101/cshperspect.a007641
2. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
3. Tsai S, Shameli A, Santamaria P. CD8+ T cells in type 1 diabetes. Adv Immunol (2008) 100:79–124. doi: 10.1016/S0065-2776(08)00804-3
4. James EA MR, Kent SC, DiLorenzo TP. T cell epitopes and neo-epitopes in type 1 diabetes: a comprehensive update and reappraisal. Diabetes (2020) 69(7):1311–35. doi: 10.2337/dbi19-0022
5. Mathis D, Vence L, Benoist C. beta-Cell death during progression to diabetes. Nature (2001) 414:792–8. doi: 10.1038/414792a
6. Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, et al. How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited. Diabetes (2011) 60:1370–9. doi: 10.2337/db10-1797
7. Mallone R ED. Presumption of innocence for beta cells: Why are they vulnerable autoimmune targets in type 1 diabetes? Diabetologia (2020) 63(10):1999–2006. doi: 10.1007/s00125-020-05176-7
8. Marroqui L, Dos Santos RS, Op de Beeck A, Coomans de Brachene A, Marselli L, Marchetti P, et al. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia (2017) 60:656–67. doi: 10.1007/s00125-016-4201-3
9. Ramos-Rodriguez M, Raurell-Vila H, Colli ML, Alvelos MI, Subirana-Granes M, Juan-Mateu J, et al. The impact of proinflammatory cytokines on the beta-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat Genet (2019) 51:1588–95. doi: 10.1101/560193
10. Marre ML, McGinty JW, Chow IT, DeNicola ME, Beck NW, Kent SC, et al. Modifying Enzymes Are Elicited by ER Stress, Generating Epitopes That Are Selectively Recognized by CD4(+) T Cells in Patients With Type 1 Diabetes. Diabetes (2018) 67:1356–68. doi: 10.2337/db17-1166
11. Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze JV, Nigi L, et al. Conventional and Neo-antigenic Peptides Presented by beta Cells Are Targeted by Circulating Naive CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab (2018) 28:946–60.e6. doi: 10.1016/j.cmet.2018.07.007
12. Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol (2001) 2:1032–9. doi: 10.1038/ni723
13. Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol (2012) 24:112–8. doi: 10.1016/j.coi.2011.12.003
14. Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Ann N Y Acad Sci (2005) 1050:1–9. doi: 10.1196/annals.1313.001
15. Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol (1998) 8:189–94. doi: 10.1016/S0959-440X(98)80037-7
16. Van Lommel L, Janssens K, Quintens R, Tsukamoto K, Vander Mierde D, Lemaire K, et al. Probe-independent and direct quantification of insulin mRNA and growth hormone mRNA in enriched cell preparations. Diabetes (2006) 55:3214–20. doi: 10.2337/db06-0774
17. Goodge KA, Hutton JC. Translational regulation of proinsulin biosynthesis and proinsulin conversion in the pancreatic beta-cell. Semin Cell Dev Biol (2000) 11:235–42. doi: 10.1006/scdb.2000.0172
18. Schuit FC, In’t Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci USA (1988) 85:3865–9. doi: 10.1073/pnas.85.11.3865
19. Papa FR. Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes. Cold Spring Harb Perspect Med (2012) 2:a007666. doi: 10.1101/cshperspect.a007666
20. Tsai YC, Weissman AM. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer (2010) 1:764–78. doi: 10.1177/1947601910383011
21. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
22. Liu M, Weiss MA, Arunagiri A, Yong J, Rege N, Sun J, et al. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes Metab (2018) 20 Suppl 2:28–50. doi: 10.1111/dom.13378
23. Arunagiri A, Haataja L, Pottekat A, Pamenan F, Kim S, Zeltser LM, et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. Elife (2019) 8. doi: 10.7554/eLife.44532
24. Eizirik DL, Cnop M. ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci Signal (2010) 3:pe7. doi: 10.1126/scisignal.3110pe7
25. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13:89–102. doi: 10.1038/nrm3270
26. Brozzi F, Eizirik DL. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J Med Sci (2016) 121:133–9. doi: 10.3109/03009734.2015.1135217
27. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev (2008) 29:42–61. doi: 10.1210/er.2007-0015
28. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia (2013) 56:234–41. doi: 10.1007/s00125-012-2762-3
29. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev (2008) 29:42–61. doi: 10.1210/er.2007-0015
30. Okamoto H. Molecular basis of experimental diabetes: Degeneration, oncogenesis and regeneration of pancreatic B-cells of islets of Langerhans. BioEssays (1985) 2:15–21. doi: 10.1002/bies.950020106
31. Okamoto H, Takasawa S. Recent advances in the Okamoto model: the CD38-cyclic ADP-ribose signal system and the regenerating gene protein (Reg)-Reg receptor system in beta-cells. Diabetes (2002) 51 Suppl 3:S462–73. doi: 10.2337/diabetes.51.2007.S462
32. Fløyel T, Kaur S, Pociot F. Genes affecting β-cell function in type 1 diabetes. Curr Diabetes Rep (2015) 15:97. doi: 10.1007/s11892-015-0655-9
33. Santin I, Eizirik DL. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and beta-cell apoptosis. Diabetes Obes Metab (2013) 15 Suppl 3:71–81. doi: 10.1111/dom.12162
34. Colli ML, Moore F, Gurzov EN, Ortis F, Eizirik DL. MDA5 and PTPN2, two candidate genes for type 1 diabetes, modify pancreatic beta-cell responses to the viral by-product double-stranded RNA. Hum Mol Genet (2010) 19:135–46. doi: 10.1093/hmg/ddp474
35. Liston A, Todd JA, Lagou V. Beta-Cell Fragility As a Common Underlying Risk Factor in Type 1 and Type 2 Diabetes. Trends Mol Med (2017) 23:181–94. doi: 10.1016/j.molmed.2016.12.005
36. Sims EK, Chaudhry Z, Watkins R, Syed F, Blum J, Ouyang F, et al. Elevations in the Fasting Serum Proinsulin-to-C-Peptide Ratio Precede the Onset of Type 1 Diabetes. Diabetes Care (2016) 39:1519–26. doi: 10.2337/dc15-2849
37. Sims EK, Bahnson HT, Nyalwidhe J, Haataja L, Davis AK, Speake C, et al. Proinsulin Secretion Is a Persistent Feature of Type 1 Diabetes. Diabetes Care (2019) 42:258–64. doi: 10.2337/dc17-2625
38. In’t Veld P. Insulitis in human type 1 diabetes: a comparison between patients and animal models. Semin Immunopathol (2014) 36:569–79. doi: 10.1007/s00281-014-0438-4
39. Brozzi F, Nardelli TR, Lopes M, Millard I, Barthson J, Igoillo-Esteve M, et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia (2015) 58:2307–16. doi: 10.1007/s00125-015-3669-6
40. Campanella M, de Jong AS, Lanke KW, Melchers WJ, Willems PH, Pinton P, et al. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J Biol Chem (2004) 279:18440–50. doi: 10.1074/jbc.M309494200
41. van Kuppeveld FJ, de Jong AS, Melchers WJ, Willems PH. Enterovirus protein 2B po(u)res out the calcium: a viral strategy to survive? Trends Microbiol (2005) 13:41–4. doi: 10.1016/j.tim.2004.12.005
42. van Kuppeveld FJ, Hoenderop JG, Smeets RL, Willems PH, Dijkman HB, Galama JM, et al. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J (1997) 16:3519–32. doi: 10.1093/emboj/16.12.3519
43. Heikkila RE, Winston B, Cohen G. Alloxan-induced diabetes-evidence for hydroxyl radical as a cytotoxic intermediate. Biochem Pharmacol (1976) 25:1085–92. doi: 10.1016/0006-2952(76)90502-5
44. Takasu N, Komiya I, Asawa T, Nagasawa Y, Yamada T. Streptozocin- and alloxan-induced H2O2 generation and DNA fragmentation in pancreatic islets. H2O2 as mediator for DNA fragmentation. Diabetes (1991) 40:1141–5. doi: 10.2337/diab.40.9.1141
45. Bedoya FJ, Solano F, Lucas M. N-monomethyl-arginine and nicotinamide prevent streptozotocin-induced double strand DNA break formation in pancreatic rat islets. Experientia (1996) 52:344–7. doi: 10.1007/BF01919538
46. Sandler S, Swenne I. Streptozotocin, but not alloxan, induces DNA repair synthesis in mouse pancreatic islets in vitro. Diabetologia (1983) 25:444–7. doi: 10.1007/BF00282526
47. Sosenko JM, Palmer JP, Rafkin-Mervis L, Krischer JP, Cuthbertson D, Mahon J, et al. Incident dysglycemia and progression to type 1 diabetes among participants in the Diabetes Prevention Trial-Type 1. Diabetes Care (2009) 32:1603–7. doi: 10.2337/dc08-2140
48. Newby BN, Mathews CE. Type I Interferon Is a Catastrophic Feature of the Diabetic Islet Microenvironment. Front Endocrinol (Lausanne) (2017) 8:232. doi: 10.3389/fendo.2017.00232
49. Davidson S, Maini MK, Wack A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J Interferon Cytokine Res (2015) 35:252–64. doi: 10.1089/jir.2014.0227
50. Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA (2007) 104:5115–20. doi: 10.1073/pnas.0700442104
51. Slifka MK, Rodriguez F, Whitton JL. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature (1999) 401:76–9. doi: 10.1038/43454
52. Sandberg JO, Eizirik DL, Sandler S. IL-1 receptor antagonist inhibits recurrence of disease after syngeneic pancreatic islet transplantation to spontaneously diabetic non-obese diabetic (NOD) mice. Clin Exp Immunol (1997) 108:314–7. doi: 10.1046/j.1365-2249.1997.3771275.x
53. von Herrath MG, Oldstone MB. Interferon-gamma is essential for destruction of beta cells and development of insulin-dependent diabetes mellitus. J Exp Med (1997) 185:531–9. doi: 10.1084/jem.185.3.531
54. Campbell IL, Kay TW, Oxbrow L, Harrison LC. Essential role for interferon-gamma and interleukin-6 in autoimmune insulin-dependent diabetes in NOD/Wehi mice. J Clin Invest (1991) 87:739–42. doi: 10.1172/JCI115055
55. Horwitz MS, Ilic A, Fine C, Balasa B, Sarvetnick N. Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin Immunol (Orlando Fla) (2004) 110:134–44. doi: 10.1016/j.clim.2003.09.014
56. Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes (2000) 49:708–11. doi: 10.2337/diabetes.49.5.708
57. Serreze DV, Wasserfall C, Ottendorfer EW, Stalvey M, Pierce MA, Gauntt C, et al. Diabetes acceleration or prevention by a coxsackievirus B4 infection: critical requirements for both interleukin-4 and gamma interferon. J Virol (2005) 79:1045–52. doi: 10.1128/JVI.79.2.1045-1052.2005
58. Zhang HM, Ye X, Su Y, Yuan J, Liu Z, Stein DA, et al. Coxsackievirus B3 infection activates the unfolded protein response and induces apoptosis through downregulation of p58IPK and activation of CHOP and SREBP1. J Virol (2010) 84:8446–59. doi: 10.1128/JVI.01416-09
59. Luo XN, Yao HL, Song J, Song QQ, Shi BT, Xia D, et al. Coxsackievirus B3 Infection Triggers Autophagy through 3 Pathways of Endoplasmic Reticulum Stress. BioMed Environ Sci (2018) 31:867–75. doi: 10.3967/bes2018.115
60. Colli ML, Paula FM, Marselli L, Marchetti P, Roivainen M, Eizirik DL, et al. Coxsackievirus B Tailors the Unfolded Protein Response to Favour Viral Amplification in Pancreatic beta Cells. J Innate Immun (2019) 11:375–90. doi: 10.1159/000496034
61. van Kuppeveld FJ, Melchers WJ, Willems PH, Gadella TW Jr. Homomultimerization of the coxsackievirus 2B protein in living cells visualized by fluorescence resonance energy transfer microscopy. J Virol (2002) 76:9446–56. doi: 10.1128/JVI.76.18.9446-9456.2002
62. Bozym RA, Patel K, White C, Cheung KH, Bergelson JM, Morosky SA, et al. Calcium signals and calpain-dependent necrosis are essential for release of coxsackievirus B from polarized intestinal epithelial cells. Mol Biol Cell (2011) 22:3010–21. doi: 10.1091/mbc.e11-02-0094
63. Bozym RA, Morosky SA, Kim KS, Cherry S, Coyne CB. Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PloS Pathog (2010) 6:e1001135. doi: 10.1371/journal.ppat.1001135
64. Stutzmann GE, Mattson MP. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol Rev (2011) 63:700–27. doi: 10.1124/pr.110.003814
65. van Kuppeveld FJ, de Jong AS, Melchers WJ, Willems PH. Enterovirus protein 2B po(u)res out the calcium: a viral strategy to survive? Trends Microbiol (2005) 13:41–4. doi: 10.1016/j.tim.2004.12.005
66. van Kuppeveld FJ, Hoenderop JG, Smeets RL, Willems PH, Dijkman HB, Galama JM, et al. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J (1997) 16:3519–32. doi: 10.1093/emboj/16.12.3519
67. Chehadeh W, Kerr-Conte J, Pattou F, Alm G, Lefebvre J, Wattre P, et al. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J Virol (2000) 74:10153–64. doi: 10.1128/JVI.74.21.10153-10164.2000
68. Clements GB, Galbraith DN, Taylor KW. Coxsackie B virus infection and onset of childhood diabetes. Lancet (1995) 346:221–3. doi: 10.1016/S0140-6736(95)91270-3
69. Gallagher GR, Brehm MA, Finberg RW, Barton BA, Shultz LD, Greiner DL, et al. Viral infection of engrafted human islets leads to diabetes. Diabetes (2015) 64:1358–69. doi: 10.2337/db14-1020
71. Hyoty H, Leon F, Knip M. Developing a vaccine for type 1 diabetes by targeting coxsackievirus B. Expert Rev Vaccin (2018) 17:1071–83. doi: 10.1080/14760584.2018.1548281
72. Op de Beeck A, Eizirik DL. Viral infections in type 1 diabetes mellitus — why the β cells? Nat Rev Endocrinol (2016) 12:263–73. doi: 10.1038/nrendo.2016.30
73. Roep BO. A viral link for type 1 diabetes. Nat Med (2019) 25:1816–8. doi: 10.1038/s41591-019-0689-7
74. van der Werf N, Kroese FG, Rozing J, Hillebrands JL. Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev (2007) 23:169–83. doi: 10.1002/dmrr.695
75. Wang R, Moniruzzaman M, Shuffle E, Lourie R, Hasnain SZ. Immune regulation of the unfolded protein response at the mucosal barrier in viral infection. Clin Trans Immunol (2018) 7:e1014. doi: 10.1002/cti2.1014
76. Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes (2014) 63:446–55. doi: 10.2337/db13-0619
77. Nyalwidhe JO, Gallagher GR, Glenn LM, Morris MA, Vangala P, Jurczyk A, et al. Coxsackievirus-Induced Proteomic Alterations in Primary Human Islets Provide Insights for the Etiology of Diabetes. J Endocr Soc (2017) 1:1272–86. doi: 10.1210/js.2017-00278
78. Stene LC, Oikarinen S, Hyoty H, Barriga KJ, Norris JM, Klingensmith G, et al. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes (2010) 59:3174–80. doi: 10.2337/db10-0866
79. Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ (2011) 342:d35. doi: 10.1136/bmj.d35
80. Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, et al. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia (2004) 47:225–39. doi: 10.1007/s00125-003-1297-z
81. Colli ML, Ramos-Rodriguez M, Nakayasu ES, Alvelos MI, Lopes M, Hill JLE, et al. An integrated multi-omics approach identifies the landscape of interferon-alpha-mediated responses of human pancreatic beta cells. Nat Commun (2020) 11:2584. doi: 10.1038/s41467-020-16327-0
82. Eizirik DL, Op de Beeck A. Coxsackievirus and Type 1 Diabetes Mellitus: The Wolf’s Footprints. Trends Endocrinol Metab (2018) 29:137–9. doi: 10.1016/j.tem.2017.12.002
83. Miani M, Barthson J, Colli ML, Brozzi F, Cnop M, Eizirik DL. Endoplasmic reticulum stress sensitizes pancreatic beta cells to interleukin-1beta-induced apoptosis via Bim/A1 imbalance. Cell Death Dis (2013) 4:e701. doi: 10.1038/cddis.2013.236
84. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The Human Pancreatic Islet Transcriptome: Expression of Candidate Genes for Type 1 Diabetes and the Impact of Pro-Inflammatory Cytokines. PloS Genet (2012) 8:e1002552. doi: 10.1371/journal.pgen.1002552
85. Burg AR, Das S, Padgett LE, Koenig ZE, Tse HM. Superoxide Production by NADPH Oxidase Intensifies Macrophage Antiviral Responses during Diabetogenic Coxsackievirus Infection. J Immunol (2018) 200:61–70. doi: 10.4049/jimmunol.1700478
86. Burg AR, Tse HM. Redox-Sensitive Innate Immune Pathways During Macrophage Activation in Type 1 Diabetes. Antioxid Redox Signal (2018) 29:1373–98. doi: 10.1089/ars.2017.7243
87. Delmastro-Greenwood MM, Tse HM, Piganelli JD. Effects of metalloporphyrins on reducing inflammation and autoimmunity. Antioxid Redox Signal (2014) 20:2465–77. doi: 10.1089/ars.2013.5257
88. Padgett LE, Anderson B, Liu C, Ganini D, Mason RP, Piganelli JD, et al. Loss of NOX-Derived Superoxide Exacerbates Diabetogenic CD4 T-Cell Effector Responses in Type 1 Diabetes. Diabetes (2015) 64:4171–83. doi: 10.2337/db15-0546
89. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci (2013) 1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x
90. Piganelli JD, Delmastro-Greenwood MM, Tse HM. Effects of Metalloporphyrins on Reducing Inflammation and Autoimmunity. Antioxid Redox Signal (2014) 20(15):2465–77. doi: 10.1089/ars.2013.5257
91. Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, et al. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes (2011) 60:2144–51. doi: 10.2337/db10-1222
92. Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic Biol Med (2004) 36:233–47. doi: 10.1016/j.freeradbiomed.2003.10.029
93. Tse HM, Milton MJ, Schreiner S, Profozich JL, Trucco M, Piganelli JD. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J Immunol (2007) 178:908–17. doi: 10.4049/jimmunol.178.2.908
94. Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol (2010) 185:5247–58. doi: 10.4049/jimmunol.1001472
95. Christen U, Edelmann KH, McGavern DB, Wolfe T, Coon B, Teague MK, et al. A viral epitope that mimics a self antigen can accelerate but not initiate autoimmune diabetes. J Clin Invest (2004) 114:1290–8. doi: 10.1172/JCI200422557
96. Rodriguez-Calvo T. Enterovirus infection and type 1 diabetes: unraveling the crime scene. Clin Exp Immunol (2019) 195:15–24. doi: 10.1111/cei.13223
97. Tracy S, Smithee S, Alhazmi A, Chapman N. Coxsackievirus can persist in murine pancreas by deletion of 5’ terminal genomic sequences. J Med Virol (2015) 87:240–7. doi: 10.1002/jmv.24039
98. Vehik K, Lynch KF, Wong MC, Tian X, Ross MC, Gibbs RA, et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat Med (2019) 25:1865–72. doi: 10.1038/s41591-019-0667-0
99. Kim KW, Horton JL, Pang CNI, Jain K, Leung P, Isaacs SR, et al. Higher abundance of enterovirus A species in the gut of children with islet autoimmunity. Sci Rep (2019) 9:1749. doi: 10.1038/s41598-018-38368-8
100. Oikarinen M, Tauriainen S, Oikarinen S, Honkanen T, Collin P, Rantala I, et al. Type 1 Diabetes Is Associated With Enterovirus Infection in Gut Mucosa. Diabetes (2012) 61:687–91. doi: 10.2337/db11-1157
101. Marre ML, Piganelli JD. Environmental Factors Contribute to beta Cell Endoplasmic Reticulum Stress and Neo-Antigen Formation in Type 1 Diabetes. Front Endocrinol (Lausanne) (2017) 8:262. doi: 10.3389/fendo.2017.00262
102. Marre ML, Profozich JL, Coneybeer JT, Geng X, Bertera S, Ford MJ, et al. Inherent ER stress in pancreatic islet beta cells causes self-recognition by autoreactive T cells in type 1 diabetes. J Autoimmun (2016) 72:33–46. doi: 10.1016/j.jaut.2016.04.009
103. Marre ML, James EA, Piganelli JD. beta cell ER stress and the implications for immunogenicity in type 1 diabetes. Front Cell Dev Biol (2015) 3:67. doi: 10.3389/fcell.2015.00067
104. Buitinga M, Callebaut A, Marques Camara Sodre F, Crevecoeur I, Blahnik-Fagan G, Yang ML, et al. Inflammation-Induced Citrullinated Glucose-Regulated Protein 78 Elicits Immune Responses in Human Type 1 Diabetes. Diabetes (2018) 67:2337–48. doi: 10.2337/db18-0295
105. McGinty JW, Chow IT, Greenbaum C, Odegard J, Kwok WW, James EA. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes (2014) 63:3033–40. doi: 10.2337/db13-1952
106. Yu L, Rewers M, Gianani R, Kawasaki E, Zhang Y, Verge C, et al. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J Clin Endocrinol Metab (1996) 81:4264–7. doi: 10.1210/jc.81.12.4264
107. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209:51–60. doi: 10.1084/jem.20111187
108. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22:1482–7. doi: 10.1038/nm.4203
109. Berkers CR, de Jong A, Ovaa H, Rodenko B. Transpeptidation and reverse proteolysis and their consequences for immunity. Int J Biochem Cell Biol (2009) 41:66–71. doi: 10.1016/j.biocel.2008.08.036
110. Wiles TA, Powell R, Michel R, Beard KS, Hohenstein A, Bradley B, et al. Identification of Hybrid Insulin Peptides (HIPs) in Mouse and Human Islets by Mass Spectrometry. J Proteome Res (2019) 18:814–25. doi: 10.1021/acs.jproteome.8b00875
111. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351:711–4. doi: 10.1126/science.aad2791
112. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes (2019) 68:1830–40. doi: 10.2337/db19-0128
113. Babon JAB, DeNicola ME, Blodgett DM, Crèvecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22:1482–7. doi: 10.1038/nm.4203
114. James EA, Pietropaolo M, Mamula MJ. Immune Recognition of beta-Cells: Neoepitopes as Key Players in the Loss of Tolerance. Diabetes (2018) 67:1035–42. doi: 10.2337/dbi17-0030
115. Noble JA, Valdes AM, Varney MD, Carlson JA, Moonsamy P, Fear AL, et al. HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes (2010) 59:2972–9. doi: 10.2337/db10-0699
116. Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol (2004) 172:5967–72. doi: 10.4049/jimmunol.172.10.5967
117. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol (2003) 171:538–41. doi: 10.4049/jimmunol.171.2.538
118. Knip M, Siljander H, Ilonen J, Simell O, Veijola R. Role of humoral beta-cell autoimmunity in type 1 diabetes. Pediatr Diabetes (2016) 17 Suppl 22:17–24. doi: 10.1111/pedi.12386
119. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med (2017) 23:501–7. doi: 10.1038/nm.4289
120. Manoury B, Mazzeo D, Fugger L, Viner N, Ponsford M, Streeter H, et al. Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol (2002) 3:169–74. doi: 10.1038/ni754
121. Gahring L, Carlson NG, Meyer EL, Rogers SW. Granzyme B proteolysis of a neuronal glutamate receptor generates an autoantigen and is modulated by glycosylation. J Immunol (2001) 166:1433–8. doi: 10.4049/jimmunol.166.3.1433
122. Pritzker LB, Joshi S, Gowan JJ, Harauz G, Moscarello MA. Deimination of myelin basic protein. 1. Effect of deimination of arginyl residues of myelin basic protein on its structure and susceptibility to digestion by cathepsin D. Biochemistry (2000) 39:5374–81. doi: 10.1021/bi9925569
123. Clarke S, Stephenson RC, Lowenson JD. Liability of asparagine and aspartic acid residues in proteins and peptides. In: Aher TJ, Manning MC, editors. Stability of Protein Pharmaceuticals, Part A: Chemical and Physical Pathways of Protein Degradation. New York: Plenum Press (1992).
124. Lowenson J, Clarke S. Does the chemical instability of aspartyl and asparaginyl residues in proteins contribute to erythrocyte aging? The role of protein carboxyl methylation reactions. Blood Cells (1988) 14:103–18.
125. Aswad DW, Paranandi MV, Schurter BT. Isoaspartate in peptides and proteins: formation, significance, and analysis. J Pharm BioMed Anal (2000) 21:1129–36. doi: 10.1016/S0731-7085(99)00230-7
126. Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell (2009) 33:1–13. doi: 10.1016/j.molcel.2008.12.013
127. Wold F. In vivo chemical modification of proteins (post-translational modification). Annu Rev Biochem (1981) 50:783–814. doi: 10.1146/annurev.bi.50.070181.004031
128. Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol (2010) 11:225–31. doi: 10.1038/ni.1844
129. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes (2012) 61:3239–46. doi: 10.2337/db12-0112
130. Yan J, Mamula MJ. Autoreactive T cells revealed in the normal repertoire: escape from negative selection and peripheral tolerance. J Immunol (2002) 168:3188–94. doi: 10.4049/jimmunol.168.7.3188
131. Doyle HA, Gee RJ, Mamula MJ. Altered immunogenicity of isoaspartate containing proteins. Autoimmunity (2007) 40:131–7. doi: 10.1080/08916930601165180
132. Mamula MJ, Gee RJ, Elliott JI, Sette A, Southwood S, Jones PJ, et al. Isoaspartyl post-translational modification triggers autoimmune responses to self-proteins. J Biol Chem (1999) 274:22321–7. doi: 10.1074/jbc.274.32.22321
133. Yang ML, Gee AJP, Gee RJ, Zurita-Lopez CI, Khare S, Clarke S, et al. Lupus autoimmunity altered by cellular methylation metabolism. Autoimmunity (2013) 46(1):21–31. doi: 10.3109/08916934.2012.732133
134. Doyle HA, Mamula MJ. Posttranslational protein modifications: new flavors in the menu of autoantigens. Curr Opin Rheumatol (2002) 14:244–9. doi: 10.1097/00002281-200205000-00009
135. Johnson BA, Aswad DW. Fragmentation of isoaspartyl peptides and proteins by carboxypeptidase Y: release of isoaspartyl dipeptides as a result of internal and external cleavage. Biochemistry (1990) 29:4373–80. doi: 10.1021/bi00470a017
136. Young AL, Carter WG, Doyle HA, Mamula MJ, Aswad DW. Structural integrity of histone H2B in vivo requires the activity of protein L-isoaspartate O-methyltransferase, a putative protein repair enzyme. J Biol Chem (2001) 276:37161–5. doi: 10.1074/jbc.M106682200
137. Bockenstedt LK, Gee RJ, Mamula MJ. Self-peptides in the initiation of lupus autoimmunity. J Immunol (1995) 154:3516–24.
138. Doyle HA, Gee RJ, Mamula MJ. A failure to repair self-proteins leads to T cell hyperproliferation and autoantibody production. J Immunol (2003) 171:2840–7. doi: 10.4049/jimmunol.171.6.2840
139. Farrar C, Houser CR, Clarke S. Activation of the PI3K/Akt signal transduction pathway and increased levels of insulin receptor in protein repair-deficient mice. Aging Cell (2005) 4:1–12. doi: 10.1111/j.1474-9728.2004.00136.x
140. Salvador JM, Hollander MC, Nguyen AT, Kopp JB, Barisoni L, Moore JK, et al. Mice lacking the p53-effector gene Gadd45a develop a lupus-like syndrome. Immunity (2002) 16:499–508. doi: 10.1016/S1074-7613(02)00302-3
141. Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol (2000) 165:4773–7. doi: 10.4049/jimmunol.165.9.4773
142. Vratsanos GS, Jung S, Park YM, Craft J. CD4(+) T cells from lupus-prone mice are hyperresponsive to T cell receptor engagement with low and high affinity peptide antigens: a model to explain spontaneous T cell activation in lupus. J Exp Med (2001) 193:329–37. doi: 10.1084/jem.193.3.329
143. Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes (1999) 48:1–9. doi: 10.2337/diabetes.48.1.1
144. Trigwell SM, Radford PM, Page SR, Loweth AC, James RF, Morgan NG, et al. Islet glutamic acid decarboxylase modified by reactive oxygen species is recognized by antibodies from patients with type 1 diabetes mellitus. Clin Exp Immunol (2001) 126:242–9. doi: 10.1046/j.1365-2249.2001.01653.x
145. Sternisha SM, Miller BG. Molecular and cellular regulation of human glucokinase. Arch Biochem Biophys (2019) 663:199–213. doi: 10.1016/j.abb.2019.01.011
Keywords: ER stress, neo-antigen, post-translational modification, type 1 diabetes (T1D), Beta Cell (β cell), immune cells
Citation: Piganelli JD, Mamula MJ and James EA (2021) The Role of β Cell Stress and Neo-Epitopes in the Immunopathology of Type 1 Diabetes. Front. Endocrinol. 11:624590. doi: 10.3389/fendo.2020.624590
Received: 31 October 2020; Accepted: 23 December 2020;
Published: 18 February 2021.
Edited by:
Roberto Mallone, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Arnaud Zaldumbide, Leiden University Medical Center, NetherlandsSarah J. Richardson, University of Exeter, United Kingdom
Copyright © 2021 Piganelli, Mamula and James. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eddie A. James, ZWphbWVzQGJlbmFyb3lhcmVzZWFyY2gub3Jn