 Rebekah J. Nicholson
Rebekah J. Nicholson Marcus G. Pezzolesi
Marcus G. Pezzolesi Scott A. Summers
Scott A. Summers
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 28 January 2021
Sec. Diabetes: Molecular Mechanisms
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.622692
This article is part of the Research Topic The Role of Sphingolipid Metabolism in the Development of Type 2 Diabetes and Obesity View all 7 articles
Diabetic kidney disease (DKD) is a prevalent and progressive comorbidity of diabetes mellitus that increases one’s risk of developing renal failure. Progress toward development of better DKD therapeutics is limited by an incomplete understanding of forces driving and connecting the various features of DKD, which include renal steatosis, fibrosis, and microvascular dysfunction. Herein we review the literature supporting roles for bioactive ceramides as inducers of local and systemic DKD pathology. In rodent models of DKD, renal ceramides are elevated, and genetic and pharmacological ceramide-lowering interventions improve kidney function and ameliorate DKD histopathology. In humans, circulating sphingolipid profiles distinguish human DKD patients from diabetic controls. These studies highlight the potential for ceramide to serve as a central and therapeutically tractable lipid mediator of DKD.
Diabetes is the global leading cause of chronic kidney disease (CKD) and end-stage renal disease (ESRD) (1, 2). An estimated 30–40% of patients with type 1 and type 2 diabetes develop CKD throughout the course of their disease (3, 4). Improvements over the last decades in the treatment of diabetes to improve blood glucose control (i.e. continuous blood glucose monitoring) have led to a general decrease in diabetes-related complications. However, kidney disease and failure have persisted as primary drivers of excess morbidity and mortality in diabetic patients (5, 6). Importantly, increases in diabetes and obesity prevalence are responsible for the projected increase in all-cause ESRD incidence in the United States (7).
Diabetic kidney disease (DKD) is thought to result from a progression of systemic and local insults, clinically represented by impaired renal function with or without elevated urinary albumin excretion (8, 9). Current DKD prevention and treatment strategies primarily target glycemic and hypertensive control; however, these therapeutic options are only capable of modest delays in disease progression toward renal failure (4). A deeper understanding of kidney-specific DKD disease mechanisms and development of more targeted therapeutics to meaningfully slow or halt disease progression are urgently needed. Investigations in human and animal studies have characterized glomerular and tubulointerstitial hypertrophy, inflammation, and sclerosis as key disease processes of DKD. Exciting evidence has qualified sphingolipids as potential drivers of renal pathology, with implicated roles in multiple features of DKD including altered lipid metabolism, insulin resistance, mitochondrial dysfunction, fibrosis, apoptosis, and vascular damage. The purpose of this review is to present relevant findings supporting the role of sphingolipids, with a focus on ceramide, in kidney-specific pathologies.
Sphingolipids make up a non-abundant, yet highly diverse, lipid class. The central metabolites of sphingolipid metabolism, ceramides, are produced via a de novo synthesis pathway in the endoplasmic reticulum (ER). Ceramides are subjected to further processing in the Golgi apparatus to produce complex sphingolipid molecules such as glycosphingolipids and sphingomyelins (SM). Importantly, dynamic breakdown and salvaging of select sphingolipids can also contribute to the re-synthesis of ceramides. As a whole, the sphingolipid pool influences cell membrane dynamics as well as cell growth, function, and programmed death (10). Studies manipulating enzymes involved in sphingolipid synthesis and metabolism continue to provide valuable information regarding the roles of sphingolipid flux and signaling in metabolism and disease.
De novo synthesis of ceramide begins in the endoplasmic reticulum with the condensation of an amino acid (i.e. L-serine) with an acyl-CoA (i.e. palmitoyl-CoA) to form a sphingoid backbone, a reaction that is catalyzed by serine palmitoyl transferase (SPT) (11). A family of six enzymes termed (dihydro)ceramide synthase (CERS1-6) adds a second, variable acyl group to sphinganine to form the dihydroceramides. The CERS enzymes are distinguishable by their substrate specificity and tissue distribution. CERS5 and 6 are upregulated by stress stimuli and promote the incorporation of long-chain (e.g. C16) acyl groups, which produce the deleterious C16-ceramides often implicated in metabolic dysfunction (12–19). Alternatively, CERS2 is more ubiquitously expressed, with particularly high expression in the kidney and liver. This enzyme produces very-long-chain (i.e. C20–26) ceramide species, which are considered benign or protective (20, 21). The fact that mammals and other species contain numerous, highly conserved CERS enzymes is noteworthy, and highlights the importance of the acyl chain length in ceramide signaling. As such, the CERS enzymes contribute significantly to the structural and functional diversity of sphingolipids (22). Regulatory activity of ceramides is also mediated by the addition of a double bond to the delta-4 carbon of the dihydroceramides by dihydroceramide desaturase (DES1 and 2). Animal studies reveal that genetic or pharmacological inhibition of DES1 reduces ceramides, induces accumulation of dihydroceramides, and attenuates metabolic dysfunction (23–25). This de novo ceramide synthesis pathway is upregulated in conditions of nutrient excess (i.e. oversupply of palmitate and serine) and in response to inflammatory agonists and glucocorticoids (26).
Ceramides can be further metabolized in the Golgi apparatus by a series of additional enzymes, which produce the complex species that make up the majority of the cellular sphingolipidome. For example, sphingomyelin synthases add a phosphocholine headgroup to ceramides, producing the most abundant sphingolipid class (i.e. the sphingomyelins) (27). Alternatively, glucosylceramide synthase adds a glucose moiety, producing the glucosylceramides (27). Glucosylceramides can receive additional carbohydrates through a series of enzymes that produce the complex ganglioside family. Ceramides can also be deacylated by a family of ceramidases, which produce sphingosine. Curiously, the beneficial adipokine adiponectin activates a ceramidase activity within adiponectin receptors (AdipoRs), improving metabolic homeostasis and blocking apoptosis by degrading ceramides (28). Both sphingosine and ceramide can be phosphorylated by specific kinases, which produce sphingosine-1-phosphate and ceramide-1-phosphate (29–31). These phosphorylated species have additional signaling functions, often opposing the actions of ceramides.
DKD shares similar risk factors and disease mechanisms to diabetes, cardiovascular disease (CVD), obesity, and steatohepatitis, conditions associated with deregulation and accumulation of bioactive sphingolipids (14, 27). As such, considerable attention has turned to the role of sphingolipids in DKD.
Numerous studies have demonstrated that circulating sphingolipid profiles differ in humans with DKD versus diabetic controls. In subjects with type 1 diabetes recruited as part of the Diabetes Control and Complications Trial (DCCT) and long-term follow-up (EDIC) cohort, lower baseline levels of very-long-chain ceramides (C20–26:1) were associated with worsening albuminuria over 14–20 years (32). A second report by the same group noted distinct associations between specific glycosphingolipid species and development of macroalbuminuria and chronic kidney disease (33). A metabolomic screen in type 2 diabetes patients with early and overt DKD reported positive associations between C16 ceramide, C16 SM, C18 glucosylceramide, and sphingosine with urinary albumin-creatinine ratio (34). Notably, several reports have documented associations of SM and kidney disease in type 1 diabetes, wherein SM correlates positively with albuminuria (35), rapid estimated glomerular filtration rate (eGFR) decline, and progression toward ESRD (36). Conversely, Tofte and colleagues reported inverse associations of several SM species and longitudinal kidney disease endpoints including renal impairment, ESRD, and all-cause mortality (37).
Whether differences in serum sphingolipids are reflective of altered kidney sphingolipid metabolism or are attributable to changes in non-renal tissues remains unanswered by human studies. The one piece of supportive data from patients is by Eckes et al., who found that CERS5 and CERS6 were upregulated in kidney cortices of humans (and mice) with non-diabetic kidney fibrosis (38). Nonetheless, numerous studies have reported that kidney-specific changes in sphingolipids and/or sphingolipid-metabolizing enzymes occur in animal and cell models of DKD. For example, investigators have shown that enzymes controlling ceramide production are upregulated in kidneys from mouse models of DKD (e.g. the SPTLC2 subunit of the SPT complex in db/db mice) (39). Exposing cultured glomerular cells to elevated glucose, free fatty acids, and angiotensin II, which recapitulate the environment in diabetes, elevates ceramides (40) and hexosylceramides (41). Additionally, several studies have demonstrated sphingolipid accumulation in kidney cortices of C57BLKS db/db diabetic mice (41, 42) or rats with streptozotocin (STZ)-induced diabetes (43, 44). In db/db eNOS deficient mice, MALDI imaging mass spectrometry was utilized to visualize GM3 and sulfoglycosphingolipid accumulation in kidney glomeruli and tubules, respectively (45). Notably, Sas et al. reported ceramide depletion in kidney cortices of mice with more advanced DKD (39). This corresponds to recent work tracking increases in urine ceramides in human patients with early DKD (i.e. CKD stages 1–3), but not CKD stage 4, compared to diabetic and healthy controls (46). Lastly, the Huang group has built a body of work to demonstrate modulation of sphingosine kinase activity in regulating mesangial cell proliferation and extracellular matrix (ECM) deposition under conditions of elevated glucose or advanced glycation end products (AGEs) (47–49).
DKD is clinically diagnosed by a persistent reduction in eGFR less than 60 ml/min per 1.73 m2 and/or the presence of a chronically high urinary albumin-to-creatinine ratio above 30 mg/g (4). However, analysis of DKD presentation between the time periods of 1988 to 1994 and 2009 to 2014 suggests that fewer DKD patients present with albuminuria; more manifest declining eGFR, and some develop histologically distinguishable DKD before presentation of clinical markers (50, 51). Both type 1 and type 2 diabetes are characterized by chronic hyperglycemia, which is a risk factor of DKD and often accompanied by other systemic perturbations, such as hypertension and obesity. Additionally, DKD is associated with marked hyperlipidemia with elevated levels of circulating LDL, oxidized-LDL, VLDL, triglyceride, and free-fatty acids (FFA) and low HDL in both type 1 and type 2 diabetes (8, 52).
The kidney is a highly metabolic tissue and houses a multitude of terminally differentiated cell types. As such, developing a comprehensive understanding of DKD onset and progression in different cell types is a monumental challenge. Nevertheless, distinguishable metabolic and histopathological alterations of DKD have been documented, and some are well-described (Figure 1). Structurally, DKD is distinguishable by the early thickening of the glomerular basement membrane (GBM) (4, 8, 52, 53). Additionally, mesangial cells within the glomerulus undergo marked hypertrophy and fibrosis. Podocytes, cells essential for maintaining the size-restriction barrier of the glomerulus, experience foot process effacement, hypertrophy, and apoptosis. Diabetic kidneys are also defined by atrophy of the brush border within renal tubules, tubular epithelial cell (TEC) loss, and TEC dedifferentiation. Tubular and glomerular cells are susceptible to lipid accumulation, and lipid-rich lesions were noted in initial reports of DKD (54). The renal interstitium, defined as the ECM, fluid, and cells surrounding nephrons and capillaries, is also subject to pathological changes. Activation and migration of peripheral or local immune cells, as well as mesenchymal-TECs accumulate in DKD kidneys and induce inflammation and ECM deposition. Progression of DKD drives interstitial and tubular fibrosis, as well as glomerulosclerosis (4, 8, 52, 53).
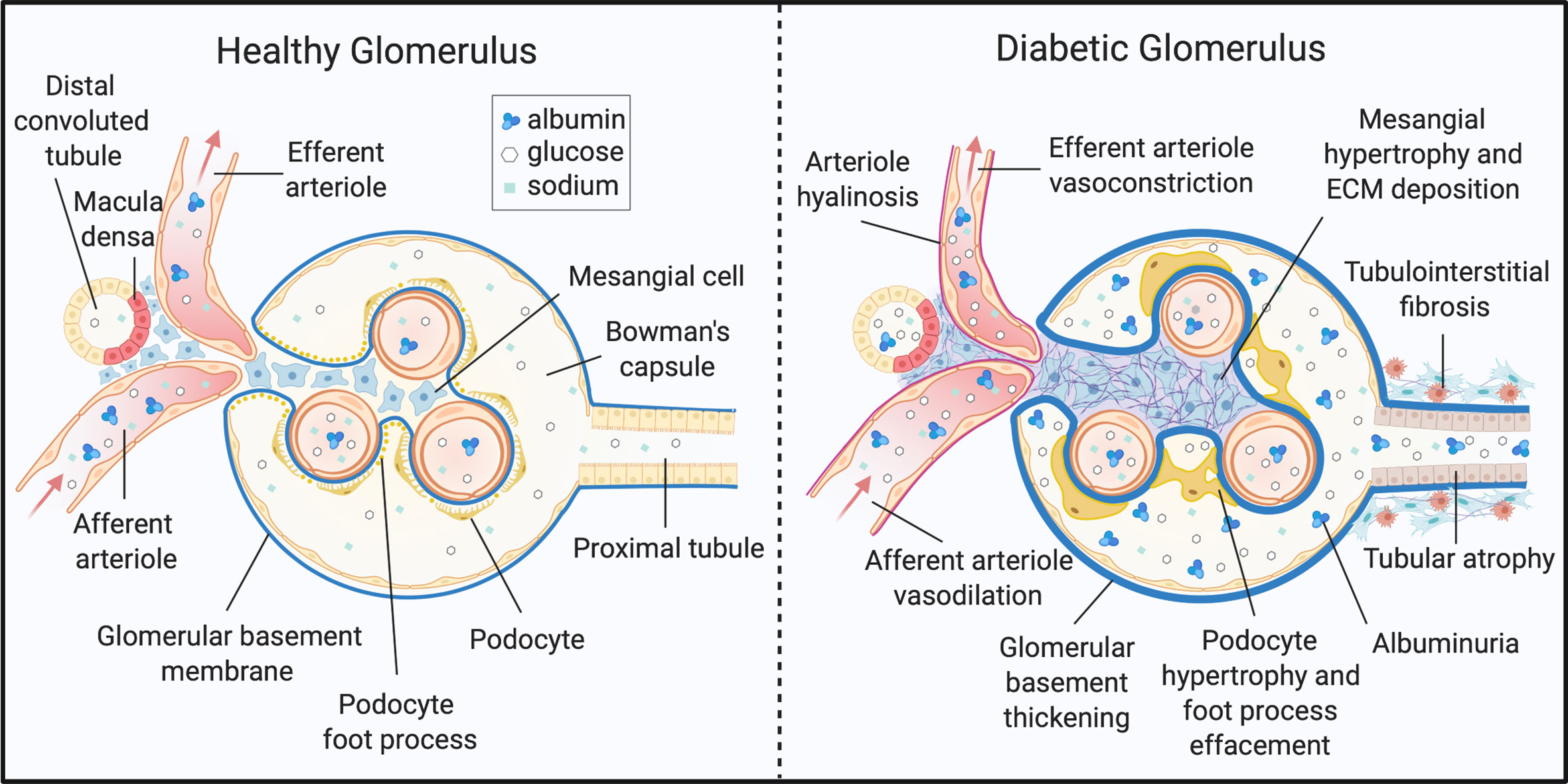
Figure 1 Histopathology of diabetic kidney disease. DKD elicits structural and functional changes to the glomerulus, tubules, and microvasculature of the kidney. ECM, extracellular matrix. Created with BioRender.com.
Recently, attention has been paid to renal steatosis as a marker of lipotoxicity in the diabetic kidney. Healthy renal cells—particularly proximal TECs, which are responsible for the majority of active reabsorption of sodium, glucose, and other metabolites from blood filtrate—rely primarily on fatty acid oxidation for ATP production (55, 56). Furthermore, terminally differentiated podocytes and TECs have limited metabolic flexibility and are highly susceptible to ATP depletion (55). Lipid accumulation in renal cells stems from both increased lipid uptake and impaired fatty acid oxidation (57). Exposure of kidney cells to free fatty acids could increase with the onset of proteinuria, in which albumin-bound FFAs are present in kidney filtrate (58). Additionally, podocytes and TECs contain swollen mitochondria with poorly defined cristae which undergo rapid fission and fusion, yet are not recycled via mitophagy (56, 59). The kidney also exhibits exceptional rates of protein turnover and fractional synthesis (53, 60). As such, renal cells are vulnerable to greater endoplasmic reticulum (ER) stress in addition to onslaughts from mitochondrial reactive oxygen species (ROS) production, systemic pro-inflammatory cytokines, and aggregation of advanced glycation end products (53, 57).
Lastly, the diabetic kidney undergoes multiple hemodynamic shifts. Hypertension is both a potential precursor and product of kidney injury. In the diabetic kidney, increased delivery of glucose and reabsorption in the proximal tubule by the SGLT2 sodium-glucose co-transporter decreases sodium concentrations reaching the macula densa (8). The resulting chronic activation of the systemic and local renin-angiotensin-aldosterone system (RAAS) stimulates blood vessel vasoconstriction and sodium resorption to increase blood pressure and induce glomerular hyperfiltration (61, 62). Additionally, decreased production of nitric oxide production by eNOS, elevated VEGF-B signaling, and increased endothelin-1 (ET-1) production results in hemodynamic imbalance, exacerbating pro-inflammatory and pro-fibrotic signaling (61, 63, 64).
The question of whether ceramide plays a causative role in DKD pathology has been partially addressed in preclinical investigations. Notably, whole-body ceramide-lowering interventions improve kidney outcomes (40, 42). Diabetic rats and high fat diet (HFD)-fed mice subjected to 4 weeks of treatment with myriocin, an SPT inhibitor, had lower body weight, fasting blood glucose, plasma insulin, and circulating free fatty acid levels compared to diabetic and obese controls (40). Myriocin lowered kidney cortex ceramide and attenuated diabetes- or HFD-induced proteinuria, mesangial matrix and GBM thickening, and podocytopathy. Oral administration of AdipoRon, an adiponectin mimetic, effectively lowered ceramide in kidney cortices of db/db mice and improved glomerular mesangial thickening, steatosis, sclerosis, and immune cell infiltration (42). Importantly, AdipoRon treatment reversed kidney histopathology without changing body weight or whole-body glucose metabolism. Similarly, prevention of ceramide accumulation by intraperitoneal administration of an acid sphingomyelinase (ASM) inhibitor in db/db mice prevented proteinuria and glomerular injury (65). Use of rapamycin, an mTOR inhibitor, to treat rats with STZ-induced diabetes ameliorated mesangial matrix thickening and glomerular cell death (43). Interestingly, rapamycin treatment decreased SPT expression and cortical ceramide and SM levels in diabetic animals. This observation supports the idea that mTOR influences sphingolipid synthesis by regulating SPT and ceramide synthases (66).
Few studies have reported kidney-specific sphingolipid-lowering interventions in the setting of DKD. In vitro models have demonstrated that myriocin and AdipoRon have protective actions, lowering ceramide and preventing mitochondrial ROS production and cell apoptosis (40, 42). Additionally, promoting the production of anti-apoptotic sphingosine-1-phosphate (S1P) in HEK-293 cells significantly decreased rates of ceramide- or TNFα-induced apoptosis (67). At the time of this review, Guangbi and colleagues are the first and only group to produce a kidney-targeted in vivo model to study manipulation of renal ceramide metabolism (68). Specifically, knockout of acid ceramidase in podocytes (Asah1fl/fl/PodoCre) of healthy mice led to glomerular ceramide accumulation and a concomitant increase in albuminuria, podocyte foot process effacement, and glomerular permeability. Furthermore, depleting glomerular ceramides with the additional knockout of ASM (Smpd1−/− Asahfl/fl/PodoCre) normalized proteinuria and podocytopenia.
The role of ceramide as a lipotoxic mediator of metabolic dysfunction in CVD, diabetes, and fatty liver disease has been well-characterized (69). These conditions share common risk factors and disease processes with DKD. As such, we propose a comparable mechanism for ceramide-mediated pathology in kidney disease processes (Figure 2). Below, we detail various elements of a mechanism for ceramides in DKD:
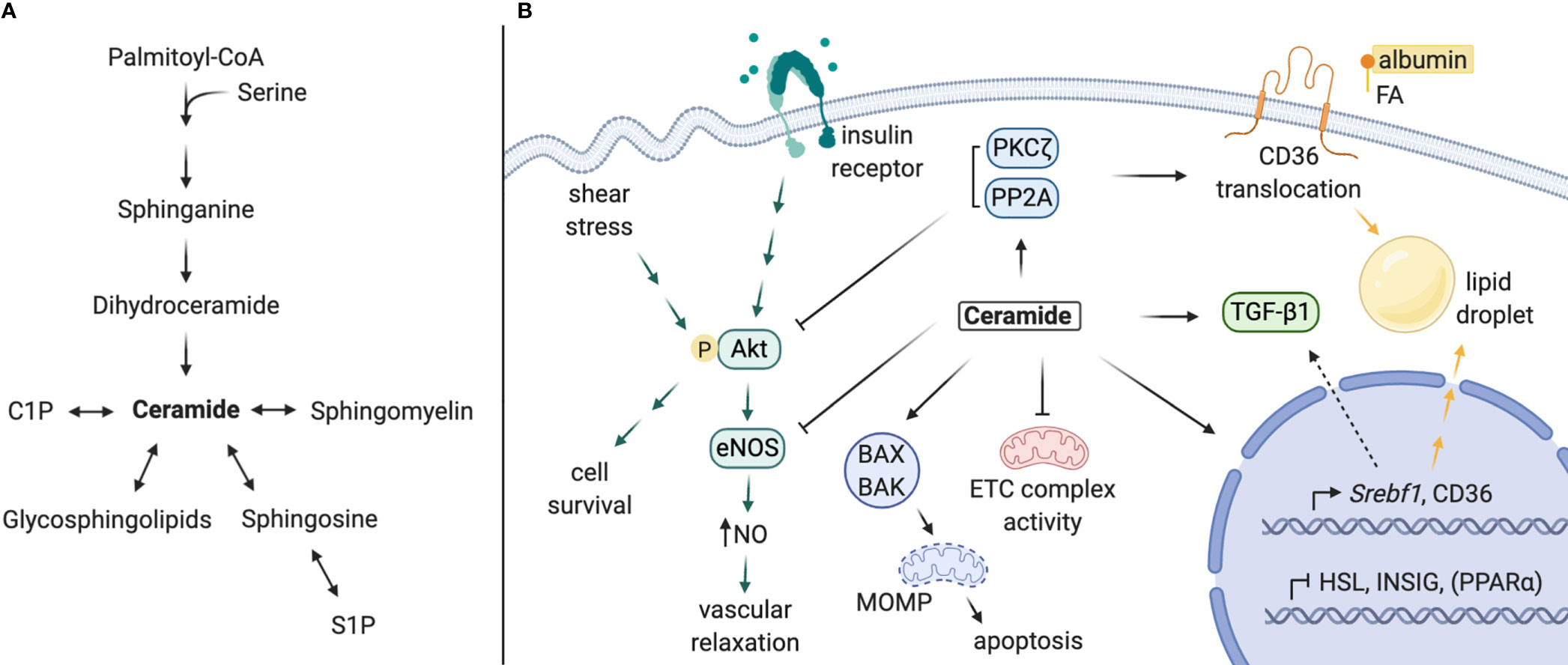
Figure 2 Proposed mechanisms of local ceramides in DKD pathology. (A) Schematic of sphingolipid metabolism. (B) Ceramides promote lipid accumulation via i) upregulation of CD36 and Srebf1 expression, ii) stimulation of CD36 translocation and SREBP-1 cleavage, and iii) inhibition of intracellular lipase expression. Ceramide and its targets PP2A and PKCζ inhibit Akt activation, impeding downstream cell survival (most cell types) or eNOS-dependent NO production (in endothelial cells). Ceramide antagonizes mitochondrial function by inhibiting ETC complex activity and permeabilizing mitochondrial membranes, leading to programmed cell death. Lastly, ceramide contributes to TGF-β signaling and tissue fibrosis, which may be partially mediated by SREBP-1. C1P, ceramide-1-phosphate; S1P, sphingosine-1-phosphate; FA, fatty acid; NO, nitric oxide; ETC, electron transport chain; MOMP, mitochondrial outer membrane permeabilization. Created with BioRender.com.
Transcriptomic analyses of kidney biopsies from human patients have revealed elevated expression of SREBF and CD36 in the setting of DKD, which facilitate transcriptional upregulation of genes involved in de novo lipogenesis, triglyceride synthesis, and lipid uptake (13, 23, 70). Studies in other tissues (e.g. the liver) show that ceramide accumulation or depletion is sufficient to regulate Srebf1 gene expression, as well as sterol response element (SRE)-mediated transcriptional targets (13, 21, 23, 71, 72). The SREBP-1 protein is also post-translationally activated by long-chain ceramides, which decrease INSIG-1 expression in an ER stress-dependent manner to promote SREBP-1 cleavage and maturation (21). Secondly, expression of fatty acid transporters (i.e. CD36, FABP, FATP2) and translocation of CD36 are regulated by ceramide (23, 73). CD36 translocation is thought to be mediated through ceramide activation of PKCζ (72). Lastly, direct regulation of genes involved in lipid metabolism by ceramide synthase enzymes has been reported. Notably, schlank, the drosophila CERS isoform, and mammalian CERS2 are capable of responding to intracellular fatty acid levels with direct binding and upregulation of Srebf expression and, conversely, downregulated expression of intracellular lipases (74, 75). Most of these actions have not been confirmed in the kidney; nonetheless, each could contribute to the lipotoxic effects of lipid accumulation observed with DKD in podocytes, TECs, and other kidney cell types (76–79).
Dysregulation of podocyte insulin signaling contributes to podocytopathy in DKD (80). Mice with podocyte-specific knockout of the insulin receptor or Akt2 develop characteristic DKD features including albuminuria, glomerular sclerosis, fibrosis, apoptosis, and mesangial matrix thickening (81, 82). These can occur in the absence of systemic hyperglycemia (81). Indeed, podocytes from db/db diabetic mice have decreased insulin-stimulated phosphorylation of Akt and are prone to apoptosis (83). The action of ceramide to inhibit Akt activation downstream of insulin signaling is well characterized (84, 85). Ceramides trigger an inhibitory phosphorylation of Akt by PKCζ, which prevents Akt/PKB translocation to the cell membrane (86–88). In addition, ceramide promotes dephosphorylation of Akt/PKB by PP2A to inhibit its activation (89–91). Aside from podocytes, insulin is a potent mediator of cellular function and survival in various kidney cell types and modulates important processes such as sodium reabsorption and gluconeogenesis in the proximal tubule (92). Systemically, ceramide-mediated insulin resistance within peripheral tissues (i.e. pancreas, liver, adipose, and muscle) can exacerbate chronic elevations in circulating glucose, inflammatory cytokines, and free fatty acids, all of which could add additional insult to the diabetic kidney (8, 93, 94). As such, ceramide’s antagonism of insulin signaling could induce a multitude of potential renal consequences.
DKD is characterized by impaired fatty acid oxidation and mitochondrial dynamics (56, 59). Patients with DKD have lower renal expression of fatty acid oxidation (FAO) genes (i.e. CPT1a, CPT2, ACOX1, ACOX2), as well as their transcriptional regulators PPARa and PGC1a (95). This phenomenon is also apparent in mouse models of DKD. Rescue of FAO in TECs via overexpression of PGC1α or treatment with the PPARα agonist fenofibrate prevents histopathological progression of nephropathy in mouse models of kidney fibrosis (95).
Several studies indicate that ceramides impair mitochondrial function in a wide range of experimental systems, including the kidney. Genetic and pharmacological ceramide-lowering interventions are sufficient to recover kidney PPARα expression (13, 42), although the mechanism is poorly understood. Several other studies have demonstrated that ceramides impair mitochondrial electron transport chain (ETC) activity. Hepatic mitochondrial complex inhibition was driven in Cers2 haploinsufficient (96) and null (97) mice via accumulation of C16 ceramide. Accordingly, CERS6 overexpression in primary hepatocytes elicited the same effect (96), whereas CERS6 knockout prevented HFD-induced disruption of mitochondrial dynamics and function (98). Mechanistically, the CERS6-derived ceramides induce mitochondrial fragmentation by interacting with mitochondrial fission factor, which contributes to the dysfunction of the organelle in mouse models of metabolic disease (98).
Kidney cell death and tissue fibrosis are common endpoints of the varying forms of CKD. Although DKD-specific studies are limited, the influence of ceramide in apoptosis is well defined (10, 99, 100). Indeed, ceramides elicit mitochondrial Bax-dependent apoptosis in cultured kidney cells in response to C(2)-ceramide or TNFα (67, 101, 102). Inhibition of oxidative stress in carbon tetrachloride-induced kidney fibrosis depleted kidney ceramide and prevented cell injury and death (103). Ceramides are also suspected to interact with and regulate TGF-β, a key regulator in fibrosis development (104–106). Recently, Dorotea et al. detailed a proposed mechanism for a positive feedback loop between SREBP1 and TGF-β, in which SREBP1 increases TGF-β expression and prevents exosomal degradation of the TGF-β receptor (70). This was demonstrated in transgenic mice overexpressing transcriptionally active SREBP-1a, which was sufficient to upregulate renal TGF-β expression and induce glomerulosclerosis and collagen deposition (79). As such, ceramides may indirectly activate TGF-β signaling and kidney fibrosis via their regulation of SREBP.
DKD is strongly associated with CVD, in part due to the prevalence of macro- and microvascular insults (107). Bioactivity of ceramide is thought to contribute to vascular dysfunction, both by inducing blood vessel vasoconstriction and increasing vascular permeability (108). Early studies demonstrated impairment of vasodilation in small blood vessels treated with C(2)-ceramide (109, 110), ceramide (109, 110), or palmitate (111), which corresponded with increased ROS production, decreased eNOS phosphorylation, and decreased NO. The Symons group confirmed that endothelial ceramide accumulation induces PP2A-eNOS co-localization, which prevents eNOS activation by Akt (111, 112). Additionally, accumulation of ceramide in human arterioles favors pathogenic H2O2-mediated flow-induced dilation (FID), rather than NO-mediated FID observed in healthy vessels (113). Inhibition of eNOS by ceramide, along with stimulation of mitochondrial ROS production (114), effectively decreases NO bioavailability and increases vascular susceptibility to sheer stress and damage (108). Ceramide also mediates disruptions to vascular integrity induced by inflammatory stimuli by disturbing endothelial tight junctions and inducing apoptosis (108, 115). Both systemic and local vascular impairments in vasodilation could exacerbate interglomerular pressure and injury (8, 108). Furthermore, in the DKD milieu of circulating glucose and inflammatory cytokines, increased permeability of the kidney endothelial barrier potentiates further damage.
Though our review has focused on ceramides, which have established roles in cellular stress, alternative bioactive sphingolipids could contribute to DKD. Within glomerular mesangial cells, elevated glucose or advanced glycation end products (AGEs) upregulate glycosphingolipid production (41) and/or sphingosine kinase activity (48) to promote mesangial proliferation and glomerular fibrosis. Beyond the general perceptions that S1P antagonizes ceramide as an anti-apoptotic, pro-survival signal (116, 117), its role in kidney disease is controversial. Several studies have supported a relationship between S1P, myofibroblast transdifferentiation, and ECM synthesis in kidney disease (reviewed in 118). Alternatively, loss of sphingosine kinase 1 activity led to poorer kidney outcomes in mouse models of DKD and kidney fibrosis (119, 120). Notably, S1P signaling outcomes are dependent upon downstream effectors of select G-protein-coupled S1P receptors, which are differentially expressed in diabetic kidneys (121). C1P has also been implicated as a regulator of mesangial cell proliferation (122) and podocytopathy (123). Lastly, gangliosides, namely GM3 and Gb3, accumulate in diabetic kidneys and may contribute to diabetic renal pathogenesis (124). Though we cannot cover these alternative sphingolipids in depth, we encourage the reader to further explore these alternative and compelling mechanisms, which have been reviewed elsewhere (118, 125–127).
Thus far, the frontier of research delineating ceramides as bioactive drivers of DKD remains minimally explored. We present a unifying scaffold identifying ceramide as a potential central lipid mediator of DKD pathology. The scientific premise of the proposed action of ceramide in DKD is compelling. Additional work is needed, however, to demarcate the function of ceramide and sphingolipids in the onset of DKD and progression toward renal failure. Better use of kidney cell-specific animal models with genetic manipulation of ceramide accumulation and degradation will be useful to identify which cell types are most affected by the action of ceramide. Additionally, manipulation of ceramide synthases in the kidney will determine which ceramide species are most implicated in disease. The current natural history of DKD onset and advancement cannot explain the vast heterogeneity in DKD presentation and rate of kidney functional decline. We look forward to targeted mechanistic investigations of ceramide in DKD processes to improve the state of knowledge regarding DKD pathophysiology and to inform therapeutic development and treatment strategies.
RN, MP, and SS drafted the manuscript in collaboration with one another. RN generated the figures. All authors contributed to the article and approved the submitted version.
The authors received research support from the National Institutes of Health (DK115824, DK116888, and DK116450 to SS), the Juvenile Diabetes Research Foundation (JDRF 3-SRA-2019-768-A-B to SS), the American Diabetes Association (to SS), the American Heart Association (to SS), and the Margolis Foundation (to SS).
SS is a consultant and shareholder with Centaurus Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Toth-Manikowski S, Atta MG. Diabetic Kidney Disease: Pathophysiology and Therapeutic Targets. J Diabetes Res (2015) 2015:697010. doi: 10.1155/2015/697010
2. Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, et al. US Renal Data System 2016 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis (2017) 69(3 Suppl 1):A7–a8. doi: 10.1053/j.ajkd.2016.12.004
3. Gheith O, Farouk N, Nampoory N, Halim MA, Al-Otaibi T. Diabetic kidney disease: world wide difference of prevalence and risk factors. J Nephropharmacol (2016) 5(1):49–56. doi: 10.4103/1110-9165.197379
4. Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin J Am Soc Nephrol (2017) 12(12):2032–45. doi: 10.2215/cjn.11491116
5. Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, et al. Changes in diabetes-related complications in the United States, 1990-2010. N Engl J Med (2014) 370(16):1514–23. doi: 10.1056/NEJMoa1310799
6. Afkarian M, Sachs MC, Kestenbaum B, Hirsch IB, Tuttle KR, Himmelfarb J, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol (2013) 24(2):302–8. doi: 10.1681/asn.2012070718
7. McCullough KP, Morgenstern H, Saran R, Herman WH, Robinson BM. Projecting ESRD Incidence and Prevalence in the United States through 2030. J Am Soc Nephrol (2019) 30(1):127–35. doi: 10.1681/asn.2018050531
8. Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, et al. Diabetic kidney disease. Nat Rev Dis Primers (2015) 1:15018. doi: 10.1038/nrdp.2015.18
9. Krolewski AS, Skupien J, Rossing P, Warram JH. Fast renal decline to end-stage renal disease: an unrecognized feature of nephropathy in diabetes. Kidney Int (2017) 91(6):1300–11. doi: 10.1016/j.kint.2016.10.046
10. Nikolova-Karakashian MN, Rozenova KA. Ceramide in stress response. Adv Exp Med Biol (2010) 688:86–108. doi: 10.1007/978-1-4419-6741-1_6
11. Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta (2003) 1632(1–3):16–30. doi: 10.1016/s1388-1981(03)00059-3
12. Raichur S, Brunner B, Bielohuby M, Hansen G, Pfenninger A, Wang B, et al. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol Metab (2019) 21:36–50. doi: 10.1016/j.molmet.2018.12.008
13. Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab (2014) 20(4):678–86. doi: 10.1016/j.cmet.2014.08.002
14. Li Y, Tippetts TS, Chaurasia B. Ceramide dependent lipotoxicity in metabolic diseases. Nutr Healthy Aging (2019) 5(1):1–12. doi: 10.3233/NHA-170032
15. Fekry B, Jeffries KA, Esmaeilniakooshkghazi A, Ogretmen B, Krupenko SA, Krupenko NI. CerS6 Is a Novel Transcriptional Target of p53 Protein Activated by Non-genotoxic Stress. J Biol Chem (2016) 291(32):16586–96. doi: 10.1074/jbc.M116.716902
16. Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res (2010) 70(3):1120–9. doi: 10.1158/0008-5472.Can-09-4043
17. Eberle M, Ebel P, Wegner MS, Männich J, Tafferner N, Ferreiros N, et al. Regulation of ceramide synthase 6 in a spontaneous experimental autoimmune encephalomyelitis model is sex dependent. Biochem Pharmacol (2014) 92(2):326–35. doi: 10.1016/j.bcp.2014.08.016
18. Bai X, He T, Liu M, Li L, Chen J, Cao M, et al. Integrative Analysis of MicroRNAs and mRNAs in LPS-Induced Macrophage Inflammation Based on Adipose Tissue Stem Cell Therapy. Inflammation (2020) 44)(1):407–20. doi: 10.1007/s10753-020-01345-3
19. Hernández-Corbacho MJ, Canals D, Adada MM, Liu M, Senkal CE, Yi JK, et al. Tumor Necrosis Factor-α (TNFα)-induced Ceramide Generation via Ceramide Synthases Regulates Loss of Focal Adhesion Kinase (FAK) and Programmed Cell Death. J Biol Chem (2015) 290(42):25356–73. doi: 10.1074/jbc.M115.658658
20. Stiban J, Perera M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim Biophys Acta (2015) 1848(2):561–7. doi: 10.1016/j.bbamem.2014.11.018
21. Kim YR, Lee EJ, Shin KO, Kim MH, Pewzner-Jung Y, Lee YM, et al. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp Mol Med (2019) 51(11):1–16. doi: 10.1038/s12276-019-0340-1
22. Zelnik ID, Rozman B, Rosenfeld-Gur E, Ben-Dor S, Futerman AH. A Stroll Down the CerS Lane. Adv Exp Med Biol (2019) 1159:49–63. doi: 10.1007/978-3-030-21162-2_4
23. Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science (2019) 365(6451):386–92. doi: 10.1126/science.aav3722
24. Siddique MM, Li Y, Chaurasia B, Kaddai VA, Summers SA. Dihydroceramides: From Bit Players to Lead Actors. J Biol Chem (2015) 290(25):15371–9. doi: 10.1074/jbc.R115.653204
25. Casasampere M, Ordoñez YF, Pou A, Casas J. Inhibitors of dihydroceramide desaturase 1: Therapeutic agents and pharmacological tools to decipher the role of dihydroceramides in cell biology. Chem Phys Lipids (2016) 197:33–44. doi: 10.1016/j.chemphyslip.2015.07.025
26. Chaurasia B, Talbot CL, Summers SA. Adipocyte Ceramides—The Nexus of Inflammation and Metabolic Disease. Front Immunol (2020) 11:576347(2282). doi: 10.3389/fimmu.2020.576347
27. Chaurasia B, Summers SA. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol Metab (2015) 26(10):538–50. doi: 10.1016/j.tem.2015.07.006
28. Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab (2017) 6(3):267–75. doi: 10.1016/j.molmet.2017.01.002
29. Cartier A, Hla T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science (2019) 366(6463):eaar5551-63. doi: 10.1126/science.aar5551
30. Gomez-Muñoz A, Presa N, Gomez-Larrauri A, Rivera IG, Trueba M, Ordoñez M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res (2016) 61:51–62. doi: 10.1016/j.plipres.2015.09.002
31. Mitrofanova A, Mallela SK, Ducasa GM, Yoo TH, Rosenfeld-Gur E, Zelnik ID, et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat Commun (2019) 10(1):2692. doi: 10.1038/s41467-019-10584-4
32. Klein RL, Hammad SM, Baker NL, Hunt KJ, Al Gadban MM, Cleary PA, et al. Decreased plasma levels of select very long chain ceramide species are associated with the development of nephropathy in type 1 diabetes. Metabolism (2014) 63(10):1287–95. doi: 10.1016/j.metabol.2014.07.001
33. Lopes-Virella MF, Baker NL, Hunt KJ, Hammad SM, Arthur J, Virella G, et al. Glycosylated sphingolipids and progression to kidney dysfunction in type 1 diabetes. J Clin Lipidol (2019) 13(3):481–91.e1. doi: 10.1016/j.jacl.2019.03.005
34. Liu JJ, Ghosh S, Kovalik JP, Ching J, Choi HW, Tavintharan S, et al. Profiling of Plasma Metabolites Suggests Altered Mitochondrial Fuel Usage and Remodeling of Sphingolipid Metabolism in Individuals With Type 2 Diabetes and Kidney Disease. Kidney Int Rep (2017) 2(3):470–80. doi: 10.1016/j.ekir.2016.12.003
35. Mäkinen VP, Tynkkynen T, Soininen P, Forsblom C, Peltola T, Kangas AJ, et al. Sphingomyelin is associated with kidney disease in type 1 diabetes (The FinnDiane Study). Metabolomics (2012) 8(3):369–75. doi: 10.1007/s11306-011-0343-y
36. Pongrac Barlovic D, Harjutsalo V, Sandholm N, Forsblom C, Groop PH. Sphingomyelin and progression of renal and coronary heart disease in individuals with type 1 diabetes. Diabetologia (2020) 63(9):1847–56. doi: 10.1007/s00125-020-05201-9
37. Tofte N, Suvitaival T, Ahonen L, Winther SA, Theilade S, Frimodt-Møller M, et al. Lipidomic analysis reveals sphingomyelin and phosphatidylcholine species associated with renal impairment and all-cause mortality in type 1 diabetes. Sci Rep (2019) 9(1):16398. doi: 10.1038/s41598-019-52916-w
38. Eckes T, Trautmann S, Djudjaj S, Beyer S, Patyna S, Schwalm S, et al. Consistent alteration of chain length-specific ceramides in human and mouse fibrotic kidneys. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866(1):158821. doi: 10.1016/j.bbalip.2020.158821
39. Sas KM, Nair V, Byun J, Kayampilly P, Zhang H, Saha J, et al. Targeted Lipidomic and Transcriptomic Analysis Identifies Dysregulated Renal Ceramide Metabolism in a Mouse Model of Diabetic Kidney Disease. J Proteomics Bioinform (2015) Suppl 14:1–8. doi: 10.4172/jpb.S14-002
40. Woo CY, Baek JY, Kim AR, Hong CH, Yoon JE, Kim HS, et al. Inhibition of Ceramide Accumulation in Podocytes by Myriocin Prevents Diabetic Nephropathy. Diabetes Metab J (2020) 44(4):581–91. doi: 10.4093/dmj.2019.0063
41. Subathra M, Korrapati M, Howell LA, Arthur JM, Shayman JA, Schnellmann RG, et al. Kidney glycosphingolipids are elevated early in diabetic nephropathy and mediate hypertrophy of mesangial cells. Am J Physiol Renal Physiol (2015) 309(3):F204–15. doi: 10.1152/ajprenal.00150.2015
42. Choi SR, Lim JH, Kim MY, Kim EN, Kim Y, Choi BS, et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism (2018) 85:348–60. doi: 10.1016/j.metabol.2018.02.004
43. Liu G, Han F, Yang Y, Xie Y, Jiang H, Mao Y, et al. Evaluation of sphingolipid metabolism in renal cortex of rats with streptozotocin-induced diabetes and the effects of rapamycin. Nephrol Dial Transplant (2011) 26(5):1493–502. doi: 10.1093/ndt/gfq633
44. Hou B, He P, Ma P, Yang X, Xu C, Lam SM, et al. Comprehensive Lipidome Profiling of the Kidney in Early-Stage Diabetic Nephropathy. Front Endocrinol (Lausanne) (2020) 11:359. doi: 10.3389/fendo.2020.00359
45. Grove KJ, Voziyan PA, Spraggins JM, Wang S, Paueksakon P, Harris RC, et al. Diabetic nephropathy induces alterations in the glomerular and tubule lipid profiles. J Lipid Res (2014) 55(7):1375–85. doi: 10.1194/jlr.M049189
46. Morita Y, Kurano M, Sakai E, Nishikawa T, Nishikawa M, Sawabe M, et al. Analysis of urinary sphingolipids using liquid chromatography-tandem mass spectrometry in diabetic nephropathy. J Diabetes Invest (2020) 11(2):441–9. doi: 10.1111/jdi.13154
47. Geoffroy K, Wiernsperger N, Lagarde M, El Bawab S. Bimodal effect of advanced glycation end products on mesangial cell proliferation is mediated by neutral ceramidase regulation and endogenous sphingolipids. J Biol Chem (2004) 279(33):34343–52. doi: 10.1074/jbc.M403273200
48. Lan T, Liu W, Xie X, Xu S, Huang K, Peng J, et al. Sphingosine kinase-1 pathway mediates high glucose-induced fibronectin expression in glomerular mesangial cells. Mol Endocrinol (2011) 25(12):2094–105. doi: 10.1210/me.2011-0095
49. Chen C, Huang K, Hao J, Huang J, Yang Z, Xiong F, et al. Polydatin attenuates AGEs-induced upregulation of fibronectin and ICAM-1 in rat glomerular mesangial cells and db/db diabetic mice kidneys by inhibiting the activation of the SphK1-S1P signaling pathway. Mol Cell Endocrinol (2016) 427:45–56. doi: 10.1016/j.mce.2016.03.003
50. Afkarian M, Zelnick LR, Hall YN, Heagerty PJ, Tuttle K, Weiss NS, et al. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988-2014. Jama (2016) 316(6):602–10. doi: 10.1001/jama.2016.10924
51. Klessens CQ, Woutman TD, Veraar KA, Zandbergen M, Valk EJ, Rotmans JI, et al. An autopsy study suggests that diabetic nephropathy is underdiagnosed. Kidney Int (2016) 90(1):149–56. doi: 10.1016/j.kint.2016.01.023
52. Opazo-Ríos L, Mas S, Marín-Royo G, Mezzano S, Gómez-Guerrero C, Moreno JA, et al. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int J Mol Sci (2020) 21(7):2632–62. doi: 10.3390/ijms21072632
53. Nishi H, Higashihara T, Inagi R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients (2019) 11(7):1664–81. doi: 10.3390/nu11071664
54. Kimmelstiel P, Wilson C. Intercapillary Lesions in the Glomeruli of the Kidney. Am J Pathol (1936) 12(1):83–98.7.
55. Jang HS, Noh MR, Kim J, Padanilam BJ. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front Med (Lausanne) (2020) 7:65. doi: 10.3389/fmed.2020.00065
56. Wei PZ, Szeto CC. Mitochondrial dysfunction in diabetic kidney disease. Clin Chim Acta (2019) 496:108–16. doi: 10.1016/j.cca.2019.07.005
57. Thongnak L, Pongchaidecha A, Lungkaphin A. Renal Lipid Metabolism and Lipotoxicity in Diabetes. Am J Med Sci (2020) 359(2):84–99. doi: 10.1016/j.amjms.2019.11.004
58. Ruggiero C, Elks CM, Kruger C, Cleland E, Addison K, Noland RC, et al. Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am J Physiol Renal Physiol (2014) 306(8):F896–906. doi: 10.1152/ajprenal.00484.2013
59. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int (2013) 83(4):568–81. doi: 10.1038/ki.2012.441
60. Garibotto G, Tessari P, Robaudo C, Zanetti M, Saffioti S, Vettore M, et al. Protein turnover in the kidney and the whole body in humans. Miner Electrolyte Metab (1997) 23(3–6):185–8.
61. Rüster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol (2006) 17(11):2985–91. doi: 10.1681/asn.2006040356
62. Nishiyama A, Kobori H. Independent regulation of renin-angiotensin-aldosterone system in the kidney. Clin Exp Nephrol (2018) 22(6):1231–9. doi: 10.1007/s10157-018-1567-1
63. Cheng H, Harris RC. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc Hematol Disord Drug Targets (2014) 14(1):22–33. doi: 10.2174/1871529x14666140401110841
64. Tessari P. Nitric oxide in the normal kidney and in patients with diabetic nephropathy. J Nephrol (2015) 28(3):257–68. doi: 10.1007/s40620-014-0136-2
65. Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villarreal R, et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J Am Soc Nephrol (2015) 26(1):133–47. doi: 10.1681/asn.2013111213
66. Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell (2017) 32(6):807–23.e12. doi: 10.1016/j.ccell.2017.11.011
67. Pastore D, Della-Morte D, Coppola A, Capuani B, Lombardo MF, Pacifici F, et al. SGK-1 protects kidney cells against apoptosis induced by ceramide and TNF-α. Cell Death Dis (2015) 6(9):e1890. doi: 10.1038/cddis.2015.232
68. Li G, Kidd J, Kaspar C, Dempsey S, Bhat OM, Camus S, et al. Podocytopathy and Nephrotic Syndrome in Mice with Podocyte-Specific Deletion of the Asah1 Gene: Role of Ceramide Accumulation in Glomeruli. Am J Pathol (2020) 190(6):1211–23. doi: 10.1016/j.ajpath.2020.02.008
69. Summers SA, Chaurasia B, Holland WL. Metabolic Messengers: ceramides. Nat Metab (2019) 1(11):1051–8. doi: 10.1038/s42255-019-0134-8
70. Dorotea D, Koya D, Ha H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front Pharmacol (2020) 11:265. doi: 10.3389/fphar.2020.00265
71. Worgall TS, Juliano RA, Seo T, Deckelbaum RJ. Ceramide synthesis correlates with the posttranscriptional regulation of the sterol-regulatory element-binding protein. Arterioscler Thromb Vasc Biol (2004) 24(5):943–8. doi: 10.1161/01.atv.0000125703.20434.4d
72. Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab (2015) 22(2):266–78. doi: 10.1016/j.cmet.2015.06.007
73. Konstantynowicz-Nowicka K, Harasim E, Baranowski M, Chabowski A. New evidence for the role of ceramide in the development of hepatic insulin resistance. PLoS One (2015) 10(1):e0116858. doi: 10.1371/journal.pone.0116858
74. Bauer R, Voelzmann A, Breiden B, Schepers U, Farwanah H, Hahn I, et al. Schlank, a member of the ceramide synthase family controls growth and body fat in Drosophila. EMBO J (2009) 28(23):3706–16. doi: 10.1038/emboj.2009.305
75. Sociale M, Wulf AL, Breiden B, Klee K, Thielisch M, Eckardt F, et al. Ceramide Synthase Schlank Is a Transcriptional Regulator Adapting Gene Expression to Energy Requirements. Cell Rep (2018) 22(4):967–78. doi: 10.1016/j.celrep.2017.12.090
76. Zhao J, Rui HL, Yang M, Sun LJ, Dong HR, Cheng H. CD36-Mediated Lipid Accumulation and Activation of NLRP3 Inflammasome Lead to Podocyte Injury in Obesity-Related Glomerulopathy. Mediators Inflamm (2019) 2019:3172647. doi: 10.1155/2019/3172647
77. Li LC, Yang JL, Lee WC, Chen JB, Lee CT, Wang PW, et al. Palmitate aggravates proteinuria-induced cell death and inflammation via CD36-inflammasome axis in the proximal tubular cells of obese mice. Am J Physiol Renal Physiol (2018) 315(6):F1720–f31. doi: 10.1152/ajprenal.00536.2017
78. de Vries AP, Ruggenenti P, Ruan XZ, Praga M, Cruzado JM, Bajema IM, et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol (2014) 2(5):417–26. doi: 10.1016/s2213-8587(14)70065-8
79. Sun L, Halaihel N, Zhang W, Rogers T, Levi M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem (2002) 277(21):18919–27. doi: 10.1074/jbc.M110650200
80. Lay AC, Coward RJM. The Evolving Importance of Insulin Signaling in Podocyte Health and Disease. Front Endocrinol (Lausanne) (2018) 9:693. doi: 10.3389/fendo.2018.00693
81. Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab (2010) 12(4):329–40. doi: 10.1016/j.cmet.2010.08.015
82. Canaud G, Bienaimé F, Viau A, Treins C, Baron W, Nguyen C, et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med (2013) 19(10):1288–96. doi: 10.1038/nm.3313
83. Tejada T, Catanuto P, Ijaz A, Santos JV, Xia X, Sanchez P, et al. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int (2008) 73(12):1385–93. doi: 10.1038/ki.2008.109
84. Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem (1998) 273(26):16568–75. doi: 10.1074/jbc.273.26.16568
85. Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol (1998) 18(9):5457–64. doi: 10.1128/MCB.18.9.5457
86. Stratford S, DeWald DB, Summers SA. Ceramide dissociates 3’-phosphoinositide production from pleckstrin homology domain translocation. Biochem J (2001) 354(Pt 2):359–68. doi: 10.1042/0264-6021:3540359
87. Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J (2004) 382(Pt 2):619–29. doi: 10.1042/bj20040139
88. Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol (2003) 23(21):7794–808. doi: 10.1128/mcb.23.21.7794-7808.2003
89. Kasumov T, Li L, Li M, Gulshan K, Kirwan JP, Liu X, et al. Ceramide as a mediator of non-alcoholic Fatty liver disease and associated atherosclerosis. PLoS One (2015) 10(5):e0126910. doi: 10.1371/journal.pone.0126910
90. Sharma AX, Holland WL. Adiponectin and its Hydrolase-Activated Receptors. J Nat Sci (2017) 3(6):e396–402. doi: 10.1016/j.molmet.2017.01.002
91. Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem (2004) 279(35):36608–15. doi: 10.1074/jbc.M406499200
92. Artunc F, Schleicher E, Weigert C, Fritsche A, Stefan N, Häring HU. The impact of insulin resistance on the kidney and vasculature. Nat Rev Nephrol (2016) 12(12):721–37. doi: 10.1038/nrneph.2016.145
93. Sokolowska E, Blachnio-Zabielska A. The Role of Ceramides in Insulin Resistance. Front Endocrinol (Lausanne) (2019) 10:577. doi: 10.3389/fendo.2019.00577
94. Magee C, Grieve DJ, Watson CJ, Brazil DP. Diabetic Nephropathy: a Tangled Web to Unweave. Cardiovasc Drugs Ther (2017) 31(5-6):579–92. doi: 10.1007/s10557-017-6755-9
95. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med (2015) 21(1):37–46. doi: 10.1038/nm.3762
96. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab (2014) 20(4):687–95. doi: 10.1016/j.cmet.2014.09.015
97. Zigdon H, Kogot-Levin A, Park JW, Goldschmidt R, Kelly S, Merrill AH Jr., et al. Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J Biol Chem (2013) 288(7):4947–56. doi: 10.1074/jbc.M112.402719
98. Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, et al. CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell (2019) 177(6):1536–52.e23. doi: 10.1016/j.cell.2019.05.008
99. Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science (1993) 259(5102):1769–71. doi: 10.1126/science.8456305
100. Dupre TV, Siskind LJ. The role of sphingolipids in acute kidney injury. Adv Biol Regul (2018) 70:31–9. doi: 10.1016/j.jbior.2018.11.003
101. Iwayama H, Ueda N. Role of mitochondrial Bax, caspases, and MAPKs for ceramide-induced apoptosis in renal proximal tubular cells. Mol Cell Biochem (2013) 379(1–2):37–42. doi: 10.1007/s11010-013-1624-8
102. Ueda N. Ceramide-induced apoptosis in renal tubular cells: a role of mitochondria and sphingosine-1-phoshate. Int J Mol Sci (2015) 16(3):5076–124. doi: 10.3390/ijms16035076
103. Ma JQ, Liu CM, Yang W. Protective effect of rutin against carbon tetrachloride-induced oxidative stress, inflammation and apoptosis in mouse kidney associated with the ceramide, MAPKs, p53 and calpain activities. Chem Biol Interact (2018) 286:26–33. doi: 10.1016/j.cbi.2018.03.003
104. Poss AM, Summers SA. Too Much of a Good Thing? An Evolutionary Theory to Explain the Role of Ceramides in NAFLD. Front Endocrinol (Lausanne) (2020) 11:505. doi: 10.3389/fendo.2020.00505
105. Shea BS, Tager AM. Sphingolipid regulation of tissue fibrosis. Open Rheumatol J (2012) 6:123–9. doi: 10.2174/1874312901206010123
106. Vestri A, Pierucci F, Frati A, Monaco L, Meacci E. Sphingosine 1-Phosphate Receptors: Do They Have a Therapeutic Potential in Cardiac Fibrosis? Front Pharmacol (2017) 8:296. doi: 10.3389/fphar.2017.00296
107. de Boer IH, Bakris GL. Diabetic Kidney Disease: A Determinant of Cardiovascular Risk in Type 1 Diabetes. Diabetes Care (2018) 41(4):662–3. doi: 10.2337/dci17-0053
108. Cogolludo A, Villamor E, Perez-Vizcaino F, Moreno L. Ceramide and Regulation of Vascular Tone. Int J Mol Sci (2019) 20(2):411–33. doi: 10.3390/ijms20020411
109. Zhang DX, Zou AP, Li PL. Ceramide reduces endothelium-dependent vasodilation by increasing superoxide production in small bovine coronary arteries. Circ Res (2001) 88(8):824–31. doi: 10.1161/hh0801.089604
110. Didion SP, Faraci FM. Ceramide-induced impairment of endothelial function is prevented by CuZn superoxide dismutase overexpression. Arterioscler Thromb Vasc Biol (2005) 25(1):90–5. doi: 10.1161/01.ATV.0000149868.74075.5d
111. Zhang QJ, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes (2012) 61(7):1848–59. doi: 10.2337/db11-1399
112. Bharath LP, Ruan T, Li Y, Ravindran A, Wan X, Nhan JK, et al. Ceramide-Initiated Protein Phosphatase 2A Activation Contributes to Arterial Dysfunction In Vivo. Diabetes (2015) 64(11):3914–26. doi: 10.2337/db15-0244
113. Schulz ME, Katunaric B, Hockenberry JC, Gutterman DD, Freed JK. Manipulation of the Sphingolipid Rheostat Influences the Mediator of Flow-Induced Dilation in the Human Microvasculature. J Am Heart Assoc (2019) 8(17):e013153. doi: 10.1161/jaha.119.013153
114. Corda S, Laplace C, Vicaut E, Duranteau J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am J Respir Cell Mol Biol (2001) 24(6):762–8. doi: 10.1165/ajrcmb.24.6.4228
115. Pandolfi R, Barreira B, Moreno E, Lara-Acedo V, Morales-Cano D, Martínez-Ramas A, et al. Role of acid sphingomyelinase and IL-6 as mediators of endotoxin-induced pulmonary vascular dysfunction. Thorax (2017) 72(5):460–71. doi: 10.1136/thoraxjnl-2015-208067
116. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med (2011) 17(1):55–63. doi: 10.1038/nm.2277
117. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol (2012) 22(1):50–60. doi: 10.1016/j.tcb.2011.09.003
118. Zhang X, Ritter JK, Li N. Sphingosine-1-phosphate pathway in renal fibrosis. Am J Physiol Renal Physiol (2018) 315(4):F752–f6. doi: 10.1152/ajprenal.00596.2017
119. Ren S, Babelova A, Moreth K, Xin C, Eberhardt W, Doller A, et al. Transforming growth factor-beta2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int (2009) 76(8):857–67. doi: 10.1038/ki.2009.297
120. Du C, Ren Y, Yao F, Duan J, Zhao H, Du Y, et al. Sphingosine kinase 1 protects renal tubular epithelial cells from renal fibrosis via induction of autophagy. Int J Biochem Cell Biol (2017) 90:17–28. doi: 10.1016/j.biocel.2017.07.011
121. Imasawa T, Kitamura H, Ohkawa R, Satoh Y, Miyashita A, Yatomi Y. Unbalanced expression of sphingosine 1-phosphate receptors in diabetic nephropathy. Exp Toxicol Pathol (2010) 62(1):53–60. doi: 10.1016/j.etp.2009.02.068
122. Pastukhov O, Schwalm S, Römer I, Zangemeister-Wittke U, Pfeilschifter J, Huwiler A. Ceramide kinase contributes to proliferation but not to prostaglandin E2 formation in renal mesangial cells and fibroblasts. Cell Physiol Biochem (2014) 34(1):119–33. doi: 10.1159/000362989
123. Mallela SK, Mitrofanova A, Merscher S, Fornoni A. Regulation of the amount of ceramide-1-phosphate synthesized in differentiated human podocytes. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(12):158517. doi: 10.1016/j.bbalip.2019.158517
124. Mather AR, Siskind LJ. Glycosphingolipids and kidney disease. Adv Exp Med Biol (2011) 721:121–38. doi: 10.1007/978-1-4614-0650-1_8
125. Mitrofanova A, Drexler Y, Merscher S, Fornoni A. Role of Sphingolipid Signaling in Glomerular Diseases: Focus on DKD and FSGS. J Cell Signal (2020) 1(3):56–69. doi: 10.33696/Signaling.1.013
126. Abou Daher A, El Jalkh T, Eid AA, Fornoni A, Marples B, Zeidan YH. Translational Aspects of Sphingolipid Metabolism in Renal Disorders. Int J Mol Sci (2017) 18(12):2528–51. doi: 10.3390/ijms18122528
Keywords: ceramide, sphingolipids, diabetic kidney disease, diabetic nephropathy, lipid metabolism, lipotoxicity
Citation: Nicholson RJ, Pezzolesi MG and Summers SA (2021) Rotten to the Cortex: Ceramide-Mediated Lipotoxicity in Diabetic Kidney Disease. Front. Endocrinol. 11:622692. doi: 10.3389/fendo.2020.622692
Received: 29 October 2020; Accepted: 11 December 2020;
Published: 28 January 2021.
Edited by:
Eric Hajduch, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Herve Le Stunff, Université Paris-Saclay, FranceCopyright © 2021 Nicholson, Pezzolesi and Summers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott A. Summers, c2NvdHQuYS5zdW1tZXJzQGhlYWx0aC51dGFoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.