 Leonie Rieger
Leonie Rieger Rosemary O’Connor
Rosemary O’Connor
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 29 January 2021
Sec. Molecular and Structural Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.620013
This article is part of the Research Topic The Role of the IGF/Insulin-IGFBP Axis in Normal Physiology and Disease View all 10 articles
Ligand-induced activation of the IGF-1 receptor triggers plasma-membrane-derived signal transduction but also triggers receptor endocytosis, which was previously thought to limit signaling. However, it is becoming ever more clear that IGF-1R endocytosis and trafficking to specific subcellular locations can define specific signaling responses that are important for key biological processes in normal cells and cancer cells. In different cell types, specific cell adhesion receptors and associated proteins can regulate IGF-1R endocytosis and trafficking. Once internalized, the IGF-1R may be recycled, degraded or translocated to the intracellular membrane compartments of the Golgi apparatus or the nucleus. The IGF-1R is present in the Golgi apparatus of migratory cancer cells where its signaling contributes to aggressive cancer behaviors including cell migration. The IGF-1R is also found in the nucleus of certain cancer cells where it can regulate gene expression. Nuclear IGF-1R is associated with poor clinical outcomes. IGF-1R signaling has also been shown to support mitochondrial biogenesis and function, and IGF-1R inhibition causes mitochondrial dysfunction. How IGF-1R intracellular trafficking and compartmentalized signaling is controlled is still unknown. This is an important area for further study, particularly in cancer.
Insulin-like growth factor-1 (IGF-1) stimulates essential cellular processes including proliferation, differentiation, survival and metabolism and thereby is essential for normal growth and development. Upon IGF-1 binding to the IGF-1 receptor (IGF-1R), the kinase domain becomes activated, leading to autophosphorylation of specific tyrosine residues (1–4). The subsequent recruitment and phosphorylation of Insulin-receptor-substrate (IRS-1 and IRS-2) proteins (5, 6) facilitates recruitment of PI3-Kinase and activation of the AKT-mTOR pathway (Figure 1A). This conserved signaling pathway regulates metabolism and transcription to promote cell survival growth or proliferation (7, 8). Activated IGF-1R may also recruit Src homology and Collagen (SHC) adaptor proteins (6, 9), and IGF-1-induced SHC phosphorylation leads to activation of RAS and the MAPK pathways that mediate mitogenic, differentiation, and migratory signals (10, 11).
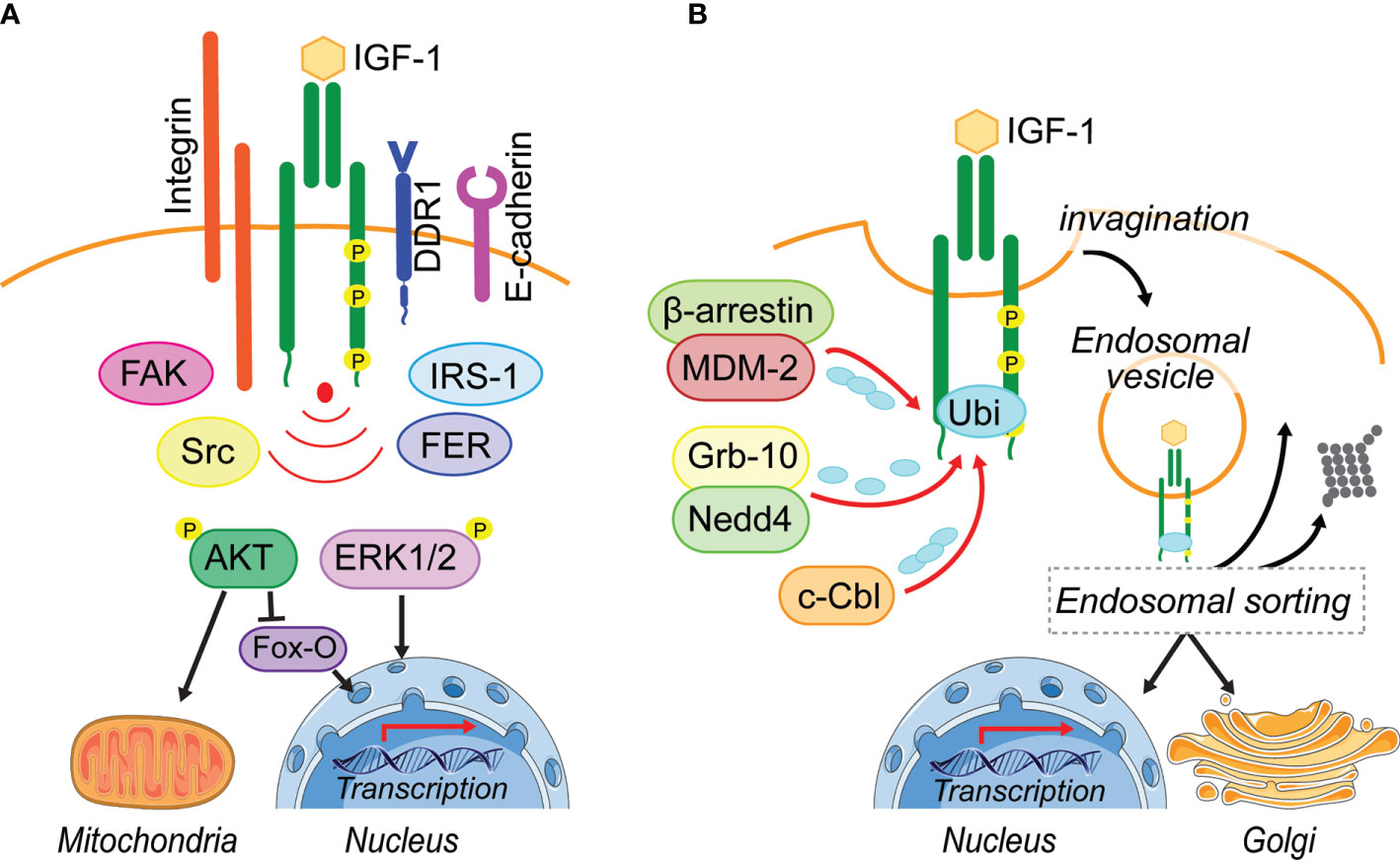
Figure 1 Leaving the plasma membrane. (A) Located on the plasma membrane, activated IGF-1R induces two major pathways, PI3-K/AKT and MAPK/ERK1/2, to regulate cellular processes including metabolism and transcription. Different adhesion related kinases (FAK, Src, FER) and interacting proteins (IRS-1, DDR1) regulate IGF-1R endocytosis and thereby prolong or reduce IGF-1R signaling from the cell surface. In addition, these IGF-1R interacting proteins can enhance bias IGF-1R signaling or their cooperation is needed for the activation of IGF-1-induced pathways (Integrin). (B) Ligand-induced IGF-1R activation leads to the recruitment of E3-liages (MDM-2, Nedd4, c-Cbl) that can initiate IGF-1R poly- and mono-ubiquitination. Via membrane invagination and formation of clathrin- and caveolin-coated pits, the IGF-1R enters the cell in endosomal vesicles. It is assumed that the endosomal sorting system decides, whether IGF-1R gets degraded, travels back to the plasma membrane or translocates to intracellular membrane compartments. To this day it is unknown how the post-endocytotic IGF-1R translocation to intracellular membrane compartments, such as the Golgi and the nucleus is regulated and whether IGF-1R regulation of mitochondrial function is exclusively due to signaling transduction. Figure elements adapted from Servier Medical Art (https://smart.servier.com/), under license CC-BY3.0.
IGF-1R activity can facilitate tumorigenesis, maintenance of the transformed phenotype and cancer progression (12, 13). Furthermore, IGF-1 may stimulate cancer cell migration, acquisition of epithelial-mesenchymal transformation (EMT) and chemotherapy resistance. Unsurprisingly, targeting the IGF-1R has been extensively investigated as a strategy in cancer therapy. Several kinase inhibitors and blocking monoclonal antibodies that inhibit ligand binding and signal transduction, while also triggering downregulation of the receptor have been tested (14, 15). However, the fact that these inhibitors have been largely unsuccessful in clinical trials renewed attention on how regulation of IGF-1R internalization, subcellular location and signaling are controlled in normal and cancer cells.
Although once thought that when cell surface RTKs are internalized, their signal transduction is terminated, it is now generally accepted that internalized receptors, including the IGF-1R may signal from endosomal and intracellular membrane compartments, or may also regulate gene transcription by translocating to the nucleus (16–22). However, the mechanisms of intracellular trafficking and which signals determine the subcellular localization of the IGF-1R or its compartmentalization with other signaling proteins are not known. Recent studies suggest that these events are regulated in a cell type-specific way and that cell-specific signals may influence the recruitment and activation of effector proteins (20, 22). Therefore, the cell-specific IGF-1R trafficking, compartmentalization and its subcellular location may define how cells respond to different extracellular stimuli.
Here, we review recent work on IGF-1R endocytosis, post-endocytotic trafficking and IGF-1R signaling to and from intracellular membrane compartments. We review how a non-canonical trafficking pathway via translocation of the receptor to internal membrane compartments and its signaling from the Golgi apparatus may contribute to its activity in cancer cells. Finally, we review the functions of IGF-1R presence in the nucleus and its effects of IGF1 signaling on mitochondrial activity.
Whether the IGF-1R undergoes ligand-induced endocytosis or remains on the plasma membrane is determined by the recruitment of interacting proteins (Figure 1A). It has been suggested that under pathological conditions like cancer, the IGF-1R associates with a range of other receptor and signaling complexes at the plasma membrane (23, 24). In particular, adhesion receptors and kinases, known to associate with the IGF-1R include E-cadherin (25), β1-Integrin (26), the discoidin domain receptor 1 (DDR1) (27), focal adhesion kinase (FAK) (28, 29), Src (30), the feline-sacroma-related kinase (FER) (31). All of these have been implicated in modulating IGF-1R stability or endocytosis to promote specific cellular responses (Figure 1A). However, it is unknown whether or how they might influence IGF-1R endosomal trafficking.
As with other RTKs, IGF-1R endocytosis is initiated by vesicle formation on the membrane (Figure 1B), and endocytosis via clathrin-coated-pits (CCP) is considered to be the fastest and predominant mode of internalization (23, 24, 32). The formation of CCPs requires recruitment of proteins that contain a ubiquitin-interacting motif, such as epsin, Eps15, or AP-2, to the activated receptor (23, 24, 32). Once clathrin-dependent endocytosis is saturated due to a large number of surface receptors being activated, it has been proposed that alternative endocytosis mechanisms subsequently facilitate IGF-1R internalization (33–35).
A clathrin-independent mechanism of endocytosis has been described for ligand-activated EGFR via micro- and macropinocytic vesicles. This involves the reorganization of the cytoskeleton and dynamic membrane ruffling (36–38). Although a similar process could be possible for IGF 1R endocytosis, it has not been demonstrated. However, clathrin independent IGF-1R endocytosis also involves the formation of lipid rafts/caveolae, which are generally described as plasma membrane invaginations. Indeed, IGF-1R has been shown to co-localize with the phosphorylated version of caveolin-1, the main component of these lipid rafts (35, 39).
Ubiquitination of the β-subunit of the IGF-1R is associated with initiation of IGF-1R endocytosis (24, 35, 40). This is dependent on IGF-1R kinase activity and requires the presence of the receptor C-terminal tail (35, 41).
Four E3 ligases have been described to either directly or indirectly interact with IGF-1R to facilitate its ubiquitination. The least studied in the context of IGF-1R is HRD1, which functions in the endoplasmic reticulum (42, 43), whereas the others, Nedd4 (40, 44), MDM2 (35, 45–47) and c-Cbl (39), are well studied (Figure 1B). IGF-1R ubiquitination can be observed within the first 5 min of ligand-binding. Two IGF-1R ubiquitination sites at Lys1138 and Lys1141 located within the kinase domain are believed to be the key lysine residues for ubiquitination (48). It is proposed that MDM2 recruitment to the IGF-1R occurs when low amounts of IGF-1are available, leading to IGF-1R endocytosis via clathrin, while high IGF-1 concentrations may initiate c-Cbl-mediated ubiquitination of the receptor followed by endocytosis using the caveolin/lipid raft route (39). This supports the idea that alternative endocytosis mechanisms are activated to internalize the IGF-1R, once clathrin-dependent endocytosis is saturated (33–35). A protein complex consisting of MDM2 and the β-arrestin protein links K63-conjugated ubiquitin polypeptide chains to the IGF-1R. This mode of ubiquitination is generally associated with cell signaling responses, DNA repair and protein trafficking (49–51) (Figure 1B). c-Cbl attaches K48-conjugated ubiquitin polypeptide chains to the IGF-1R, which may initiate degradation of the receptor (51) (Figure 1B). Thus, it is possible that depending on available IGF-1 levels, different E3 ligases are recruited to the receptor to initiate ubiquitination.
Although IGF-1R kinase activity is clearly essential for recruiting the proteins that facilitate receptor internalization and ubiquitination, it is not understood how the C-terminal tail contributes to ubiquitin-mediated IGF-1R trafficking and degradation. Our recent study showed that IGF-1-promoted phosphorylation of the Tyr1250/1251 site in the IGF-1R C-terminal results in enhanced IGF-1R internalization and proteosomal degradation (22). However, whether the Tyr1250/1251 phospho-site is involved in or modulates IGF-1R ubiquitination is still unknown. The C-terminal tail contains three lysines that are putative sites for ubiquitination, but this has not been demonstrated in cells. It remains possible that phosphorylated Tyr1250/1251 could provide a binding site for adaptor proteins or an E3 ligase that targets these sites. This would implicate the activity of domains of the receptor other than the kinase in regulating IGF-1R internalization and trafficking.
CCP/caveolin-vesicles that contain internalized IGF-1R become fused with early endosomes (27, 40, 44, 52). Here the IGF-1R proteins are sorted, either targeted for degradation (24, 35), transported toward the Golgi network (22), transported to the nucleus (20, 53–56), or recycled back to the plasma membrane (57) (Figure 1B). Internalized ubiquitinated proteins can be detected by distinct multiprotein complexes that comprise the endosomal sorting complex required for transport (ESCRT) (58–61) and serve as signal for cargo sorting (58). The fate of internalized proteins to either undergo degradation or recycling is determined within the endosomal sorting network (61). Before membrane cargo within the early endosomes, is submitted to several rounds of cargo sorting, as the early endosome matures into a late endosome (62), cargo destined for the fast recycling route is sorted and delivered back to the cell surface (63). There is also a slow recycling route where proteins first traffic through the recycling compartments before moving back to the cell surface.
Emerging evidence indicates that cargo may also enter a retrograde trafficking route where it is transported back to the Golgi apparatus, a process that serves to maintain a robust membrane protein delivery along the Golgi-associated microtubules (18, 64–66). This particular transport route is important for β1-Integrin-promoted cell migration and adhesion (65). Although precise details of IGF-1R sorting mechanisms and which proteins are involved is still unknown, it is clear that the endosomal network is essential for selecting internalized IGF-1R and its trafficking to distinct cellular compartments. The IGF-1R also travels on a path to the Golgi apparatus as a response to IGF-1-induced phosphorylation at Tyr1250/1251 (22). This enhances the potential for distinct intracellular signaling responses from the IGF-1R in different cells and different physiological or pathological settings.
The Golgi apparatus has a long-understood function in distribution, modification and secretion of newly synthesized proteins. However, it is also intimately involved in cellular processes such as cell polarization (67), directional migration (68), stress (69) and DNA repair (70). Cell migration requires coordinated communication between the plasma membrane and the Golgi apparatus (68). This may be facilitated by the retrograde trafficking of internalized plasma membrane proteins back to the Golgi apparatus (65, 71). This retrograde trafficking enables persistent cell migration because Golgi-derived microtubules act as a fast-track lane to deliver essential proteins to cell migration hot-spots, such as the sites of focal cell adhesion (Figure 2A) (64–66). Several key signaling proteins including Ras/MAPK (72–74) and RTKs, including MET, KIT, VGFR2, EGFR, FGFR (21, 75–77) and IGF-1R (22) have been demonstrated to locate to the Golgi apparatus, which acts as a signaling hub in normal and cancer cells (Figure 2A).
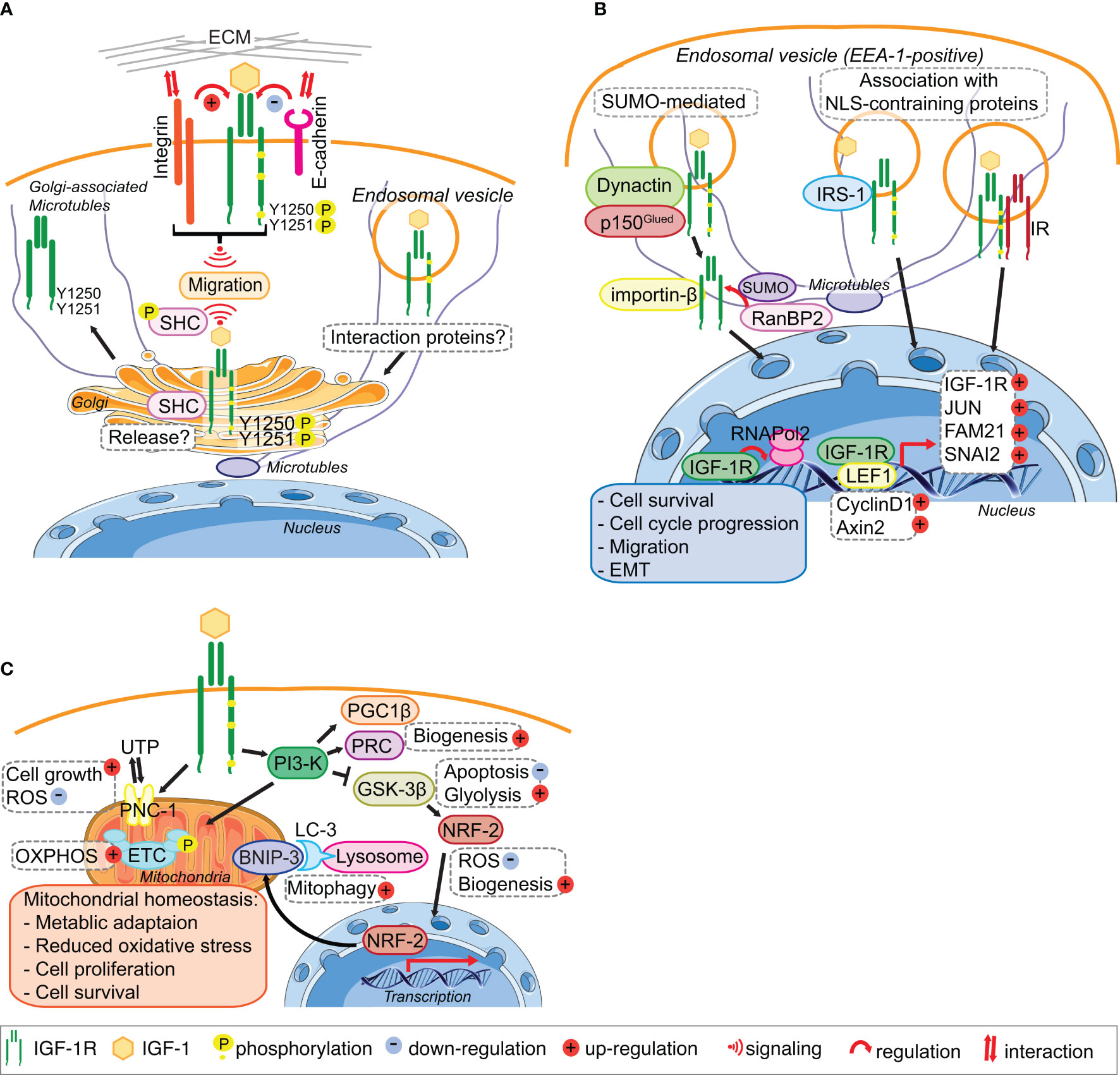
Figure 2 IGF-1R trafficking routes and signaling to the mitochondria and from the Golgi and the Nucleus. (A) The IGF-1R translocates to the Golgi apparatus. In migratory cell lines, IGF-1R autophosphorylates Tyr1250/1251 in an adhesion dependent manner. Phosphorylation of Tyr1250/1251 IGF-1R leads to rapid IGF-1R endocytosis leads to activation of the MAPK pathway and results in translocation of the IGF-1R to the Golgi which promotes sustained SHC activation to facilitate migration. points. The release and retention of IGF-1R in the Golgi may be regulated by β1-Integrin and its interaction with the ECM. In cells with low or no migratory capacity, IGF-1R remains on the surface inducing signaling from the membrane. The interaction with other proteins, including E-cadherin, stabilizes the adhesion points and internalization rate of the IGF-1R is low. (B) IGF-1R translocates to the nucleus. IGF-1 binding to the IGF-1R induces the translocation of the membrane receptor to the nucleus. Various mechanisms have been proposed for the import of the IGF-1R to the nucleus. Nuclear IGF-1R can bind to DNA and enhance or initiate the transcription of various genes, leading to cell survival, migration, EMT and cell cycle progression. (C) IGF-1 signaling regulates mitochondrial function. The activation of the PI3-K pathway in response to IGF-1 induces the expression of the mitophagy regulators PGC1β and PRC. Inhibition of GSK-3 β by PI3-K activation leads to the release of NFE2L2/Nrf2, which translocates to the nucleus to enhance the expression of the mitophagy receptor BNIP-3. Activation of IGF1-R also enhances the expression of the UTP importer PNC-1, which was linked to cell growth and the reduction of ROS. Through these pathways IGF-1 signaling contributes to the maintenance of mitochondrial homeostasis. Figure elements adapted from Servier Medical Art (https://smart.servier.com/), under license CC-BY3.0.
The rapid endocytosis and subsequent translocation of the IGF-1R to the Golgi in fibroblasts and cancer cell lines requires an adhesion-dependent autophosphorylation on Tyr1250/1251 in the C-terminal tail (Figure 2A). Although evident in all cells tested, Golgi-localized IGF-1R is however a particular feature of migratory cancer cells, because cancer cell lines with low or no migratory capacity exhibit little less Golgi-localized IGF-1R. Golgi-derived IGF-1R signaling might therefore contribute to aggressive cancer cell behavior (22). In migratory cancer cell lines, IGF-1-induced SHC phosphorylation, which is required for cell migration, is dependent on an intact Golgi apparatus and also requires cell contact with the extra-cellular matrix (ECM), suggesting that the IGF-1R mediates communication between the plasma membrane and Golgi. IGF-1-induced cell migration also requires an intact Golgi apparatus (22), as well as cooperative signaling between the IGF-1R and β1-Integrin (26, 78–81) (Figure 2A). β1-Integrin connects the ECM with the actin cytoskeleton of cells and thereby has both a structural and signaling function in cell adhesion and migration (82, 83). This suggests, that in migrating normal and cancer cell lines β1-Integrin signaling from the plasma membrane can influence IGF-1R distribution within cells and determine its presence at the Golgi apparatus (Figure 2A).
While β1-Integrin is a strong candidate for determining IGF-1R translocation to and its release from the Golgi in migratory cells (Figure 2A), E-cadherin is a strong candidate for enhancing IGF-1R stability and plasma membrane location in low- or non-migratory cell lines. E-cadherin, which is often repressed in migratory cancer cell lines and upon EMT, especially in triple negative breast cancer cells, is readily detectable in a complex with the IGF-1R at sites of cell–cell contact in cancer cells with no or low migratory capacity (25, 84). However, in confluent migratory cancer cells (with evident high levels of cell-cell contact), and under conditions where cells are unable to migrate, the IGF-1R remains in the Golgi apparatus. Therefore, E-cadherin expression in cancer cells with no or low migratory capacity may limit IGF-1R translocation to the Golgi apparatus. Regulated and exclusive expression of cadherins and Integrins has been linked to the migratory capacity of cells during embryonic development, tumor invasion and metastasis (85–87).
Thus, it is likely that IGF-1R function in facilitating cell migration through its translocation to and signaling from the Golgi is influenced by adhesion related proteins that are expressed differently depending on cell type, which may be influenced by their hormone receptor expression, and fate, as it has already been proposed (23, 24). However, the mechanisms of this interplay between adhesion receptors and IGF-1R trafficking to and from the Golgi are still unknown. It is not known how phosphorylation and or dephosphorylation of key residues on the receptor control this and how the array of signaling proteins present at the Golgi interact.
Several RTKs have been observed in the nucleus of cancer cells. These include EGFR family members (88–90), FGFR1 and 3 (91, 92), the IR (93, 94), VEGFR (95, 96), and IGF-1R (19, 20, 52–54, 97).
Translocation of the IGF 1R to the nucleus in cancer cells is induced by IGF-1 (20, 53, 98). Nuclear IGF-1R is more pronounced in cancer cell lines, including breast cancer, prostate cancer and sarcoma cells, compared to non-transformed cells (97). Furthermore, nuclear IGF-1R has been linked to a poor outcome for cancer patients and suggested to promote a more advanced disease stage (20, 53, 98, 99). Nuclear IGF-1R traffics from the plasma membrane (97) and the levels of IGF-1R nuclear translocation are proportional to ligand-induced kinase activation, because its translocation in cancer cells can be inhibited by xentuzumab, an IGF-1/2 neutralizing antibody, or by inhibition of IGF-1R endocytosis (20, 53, 54).
The precise mechanisms of IGF-1R import into the nucleus of normal and cancer cells are still unclear because the IGF-1R does not have a nuclear localization sequence (NLS) (53, 54) (Figure 2B). SUMOylation of the IGF-1R induced by IGF-1R internalization was proposed to be important (54), and IGF-1R translocation in cancer cells is facilitated by a specific subunit of dynactin p150Glued (52) (Figure 2B). The latter study showed that IGF-1-bound and internalized IGF-1R is transported within early endosome antigen 1 (EEA1)-positive vesicles (Figure 2B), it becomes positioned in the nuclear pore complex by β-importin, and is subsequently SUMOylated by RanBP2 for translocation into the nucleus (52). Suppression of any of the proteins involved in this import, leads to a significant decrease in nuclear IGF-1R. However, mutation of the SUMOylation lysine sites on IGF-1R did not abolish accumulation of IGF-1R in the nucleus (54), suggesting that additional import mechanisms exist. IGF-1R association with other proteins containing an NLS, such as IRS-1, which was previously shown to translocate to the nucleus in response to IGF-1, could also promote the import (100). It has also been suggested that heterodimerization with the IR, which occurs rapidly in response to Insulin stimulation (93) could promote nuclear import (55).
Nuclear IGF-1R may associate with DNA to enhance transcription (19, 54–56, 101), for example, by mediating the recruitment of RNAPol2 (20). Nuclear IGF-1R autoregulates its own expression in breast cancer cells depending on their estrogen receptor (ER) status (102) and binds the LEF1 transcription factor, which subsequently leads to upregulated cyclinD1 and axin 2 and cell proliferation (56). In HeLa cells, nuclear IGF-1R can increase the expression of SNAI2 (55), which is involved in EMT by suppressing E-cadherin expression (103). In prostate cancer cells nuclear IGF-1R facilitates expression of JUN and FAM 21, which are linked to cell survival, anchorage independent growth and cell migration, all of which are associated with advanced cancer stage (20). Nuclear IGF-1R is associated with proliferation of alveolar rhabdomyosarcoma cells (104) and contributes to chemoresistance in sarcomas and hepatocellular carcinoma (105, 106).
Overall, the results from recent studies suggest that nuclear IGF-1R facilitates an aggressive cancer phenotype. However, Aleksic et al. suggest that the sites of IGF-1R binding in DNA, and therefore the genes influenced by nuclear IGF-1R, might be cell type specific and that this could be defined by nuclear structure and chromatin organization (20). This is supported by the result of Sarfstein et al., which suggests that in presence of ER, nuclear IGF-1R cannot enhance its own expression (102).
While IGF-1 signaling in metabolism has been well studied (107), its contributions to mitochondrial function, maintenance and turnover is an emerging topic. Mitochondrial metabolism and oxidative phosphorylation (OXPHOS) provide building blocks and energy for all cellular functions (108). At the same time, reactive oxygen species (ROS), which are a normal by-product of OXPHOS are neutralized to avoid accumulation and cell damage (109). Mitochondria synthesis (mitochondrial biogenesis) and the regulation of numbers and quality (mitophagy linked to mitochondrial fission and fusion) are well-orchestrated processes. The importance of mitochondrial quality control and mitochondrial homeostasis in the maintenance of healthy tissues is well documented (108, 110). Impaired mitophagy can lead to the accumulation of dysfunctional mitochondria and oxidative stress, which is associated with various diseases including neurodegeneration, diabetes, heart disease and cancer (108, 111–115).
IGF-1 signaling and a functional IGF-1R is essential for mitochondrial biogenesis through inducing the transcriptional mediators Peroxisome proliferator-activated receptor gamma coactivator 1 β (PGC1β) and PGC-1-related coactivator (PRC) (116, 117) (Figure 2C). Suppression of the IGF-1Ror the PI3-Kpathway using the IGF-1R kinase inhibitor BMS-754807 or LY294002, respectively, leads to a reduction in mitochondrial mass and biogenesis (116). IGF-1 also induces the mitophagy receptor BNIP-3 (116) through GSK-3β mediated activation of NFE2L2/Nrf2 (118) (Figure 2C). This highly conserved signaling pathway is conserved from C. elegans where it coordinates mitochondrial biogenesis with mitophagy and thereby controls cellular metabolism that is ultimately linked with lifespan (119, 120). In mammalian cells (normal or transformed), IGF-1-mediated regulation of mitochondrial biogenesis and mitophagy is more complex that in C. elegans. In metazoans, it needs to be integrated with metabolic status and IGF-1-stimulated mTORC1 actions in suppressing cellular macro-autophagy (121, 122). Although IGF-1 signaling may be critical for both mitochondrial biogenesis and basal mitophagy, it is not however easy to distinguish specific signals for mitophagy from general autophagy. Moreover, IGF-1 signals may control basal mitochondria health and the triggering of mitophagy in very specific cellular contexts such as cell division or differentiation.
Further evidence for an essential IGF-1 signal in maintaining healthy mitochondria comes from the IGF-1-inducible mitochondrial UTP importer, pyrimidine nucleotide carrier 1 (SLC25A33/PNC1) that is required for maintaining mitochondrial RNA and DNA (123, 124) (Figure 2A). Suppression of PNC-1 results in cellular accumulation of ROS under normal oxygen conditions, an increase in glycolysis and a profound induction of EMT in cancer cells (124).
Overall, it will be important to establish how IGF-1 signals and IGF-1R activity support mitochondrial function in normal cells and in phenotypically distinct cancer cells, and whether an essential component of these signals is to maintain a healthy pool of mitochondria that would prevent cancer aggressiveness that is associated with hypoxia, mitochondria dysfunction and an accumulation of cellular ROS.
This review summarizes current knowledge on IGF-1R trafficking and signaling to and from intracellular compartments. Overall, the potential for intracellular IGF-1R signaling adds complexity to understanding and modulating IGF-1 actions in physiological and patho-physiological conditions. For example, efforts to inhibit IGF-1R signaling at the plasma membrane are not very effective, as is evident from the poor success of mAb in targeting the IGF-1R in cancer. One explanation for this is that continued signaling from intracellular pools of IGF-1R in association with specific organelles or protein signaling complexes may circumvent plasma membrane targeting. Correlating IGF-1R location and activity at the Golgi or in the nucleus with a specific subset of cancer may be a valuable biomarker for targeting IGF-1R in cancer (125). Therefore, if IGF-1R trafficking to and signaling from intracellular compartments determines its activity in cancer and contributes to an aggressive cancer behavior (20, 22), it is now important to identify the molecular regulators of IGF-1R trafficking. The functions of these proteins in selecting incoming receptors and regulating their cellular distribution and localization may the key to cellular signaling responses. Illuminating the mechanisms of IGF-1R trafficking and endosomal sorting would provide new insights on IGF signaling in normal cells and cancer cells, and may also identify potential co-targets for pharmacological intervention in cancer. Targeted therapy against proteins facilitating IGF-1R location and activity in the Golgi or the nucleus, or enhancing IGF-1R sorting toward proteosomal degradation may be beneficial in certain subtypes of cancer. Moreover, the presence of the IGF-1R at the Golgi may have potential to identify cancer subtypes where membrane targeting would not be effective. Our data on IGF-1R derived Golgi signaling also suggest that removing the receptor is important to suppress IGF-1 signaling. However, it is not yet clear whether specific antibodies that promote IGF-1R internalization could be used to direct it to the degradation machinery. It may be necessary to identify the key regulators of receptor trafficking to achieve selectivity here. 125.
LR and RO’C contributed to the writing of the article. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J (1997) 16(18):5572–81. doi: 10.1093/emboj/16.18.5572
2. Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol (2001) 8(12):1058–63. doi: 10.1038/nsb721
3. Kavran JM, McCabe JM, Byrne PO, Connacher MK, Wang Z, Ramek A, et al. How IGF-1 activates its receptor. eLife (2014) 3:e03772. doi: 10.7554/eLife.03772
4. Xu Y, Kong GKW, Menting JG, Margetts MB, Delaine CA, Jenkin LM, et al. How ligand binds to the type 1 insulin-like growth factor receptor. Nat Commun (2018) 9(1):821. doi: 10.1038/s41467-018-03219-7
5. Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature (1991) 352(6330):73–7. doi: 10.1038/352073a0
6. Cai W, Sakaguchi M, Kleinridders A, Gonzalez-Del Pino G, Dreyfuss JM, O’Neill BT, et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat Commun (2017) 8(1):14892. doi: 10.1038/ncomms14892
7. Fukushima T, Nakamura Y, Yamanaka D, Shibano T, Chida K, Minami S, et al. Phosphatidylinositol 3-Kinase (PI3K) Activity Bound to Insulin-like Growth Factor-I (IGF-I) Receptor, which Is Continuously Sustained by IGF-I Stimulation, Is Required for IGF-I-induced Cell Proliferation. J Biol Chem (2012) 287(35):29713–21. doi: 10.1074/jbc.M112.393074
8. Hakuno F, Takahashi SI. IGF1 receptor signaling pathways. J Mol Endocrinol (2018) 61(1):T69–t86. doi: 10.1530/JME-17-0311
9. Gustafson TA, He W, Craparo A, Schaub CD, O’Neill TJ. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Mol Cell Biol (1995) 15(5):2500–8. doi: 10.1128/MCB.15.5.2500
10. Leahy M, Lyons A, Krause D, O’Connor R. Impaired Shc, Ras, and MAPK Activation but Normal Akt Activation in FL5.12 Cells Expressing an Insulin-like Growth Factor I Receptor Mutated at Tyrosines 1250 and 1251. J Biol Chem (2004) 279(18):18306–13. doi: 10.1074/jbc.M309234200
11. Cox OT, O’Shea S, Tresse E, Bustamante-Garrido M, Kiran-Deevi R, O’Connor R. IGF-1 Receptor and Adhesion Signaling: An Important Axis in Determining Cancer Cell Phenotype and Therapy Resistance. Front Endocrinol (2015) 6:106. doi: 10.3389/fendo.2015.00106
12. Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A (1993) 90(23):11217–21. doi: 10.1073/pnas.90.23.11217
13. Sell C, Dumenil G, Deveaud C, Miura M, Coppola D, DeAngelis T, et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol (1994) 14(6):3604–12. doi: 10.1128/MCB.14.6.3604
14. Arteaga CL, Kitten LJ, Coronado EB, Jacobs S, Kull FC Jr., Allred DC, et al. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice. J Clin Invest (1989) 84(5):1418–23. doi: 10.1172/JCI114315
15. Osher E, Macaulay VM. Therapeutic Targeting of the IGF Axis. Cells (2019) 8(8):895. doi: 10.3390/cells8080895
16. Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol (2006) 174(4):593–604. doi: 10.1083/jcb.200602080
17. Brankatschk B, Wichert SP, Johnson SD, Schaad O, Rossner MJ, Gruenberg J. Regulation of the EGF Transcriptional Response by Endocytic Sorting. Sci Signaling (2012) 5(215):ra21–ra. doi: 10.1126/scisignal.2002351
18. Ye Q-H, Zhu W-W, Zhang J-B, Qin Y, Lu M, Lin G-L, et al. GOLM1 Modulates EGFR/RTK Cell-Surface Recycling to Drive Hepatocellular Carcinoma Metastasis. Cancer Cell (2016) 30(3):444–58. doi: 10.1016/j.ccell.2016.07.017
19. Waraky A, Lin Y, Warsito D, Haglund F, Aleem E, Larsson O. Nuclear insulin-like growth factor 1 receptor phosphorylates proliferating cell nuclear antigen and rescues stalled replication forks after DNA damage. J Biol Chem (2017) 292(44):18227–39. doi: 10.1074/jbc.M117.781492
20. Aleksic T, Gray N, Wu X, Rieunier G, Osher E, Mills J, et al. Nuclear IGF1R Interacts with Regulatory Regions of Chromatin to Promote RNA Polymerase II Recruitment and Gene Expression Associated with Advanced Tumor Stage. Cancer Res (2018) 78(13):3497–509. doi: 10.1158/0008-5472.CAN-17-3498
21. Frazier NM, Brand T, Gordan JD, Grandis J, Jura N. Overexpression-mediated activation of MET in the Golgi promotes HER3/ERBB3 phosphorylation. Oncogene (2019) 38(11):1936–50. doi: 10.1038/s41388-018-0537-0
22. Rieger L, O’Shea S, Godsmark G, Stanicka J, Kelly G, O’Connor R. IGF-1 receptor activity in the Golgi of migratory cancer cells depends on adhesion-dependent phosphorylation of Tyr 1250 and Tyr 1251. Sci Signaling (2020) 13:eaba3176. doi: 10.1126/scisignal.aba3176
23. Goh LK, Sorkin A. Endocytosis of Receptor Tyrosine Kinases. Cold Spring Harbor Perspect Biol (2013) 5(5):a017459. doi: 10.1101/cshperspect.a017459
24. Crudden C, Song D, Cismas S, Trocmé E, Pasca S, Calin GA, et al. Below the Surface: IGF-1R Therapeutic Targeting and Its Endocytic Journey. Cells (2019) 8(10):1223. doi: 10.3390/cells8101223
25. Nagle AM, Levine KM, Tasdemir N, Scott JA, Burlbaugh K, Kehm J, et al. Loss of E-cadherin Enhances IGF1–IGF1R Pathway Activation and Sensitizes Breast Cancers to Anti-IGF1R/InsR Inhibitors. Clin Cancer Res (2018) 024(20):5165–77. doi: 10.1158/1078-0432.CCR-18-0279
26. Kiely PA, Leahy M, O’Gorman D, O’Connor R. RACK1-mediated Integration of Adhesion and Insulin-like Growth Factor I (IGF-I) Signaling and Cell Migration Are Defective in Cells Expressing an IGF-I Receptor Mutated at Tyrosines 1250 and 1251. J Biol Chem (2005) 280(9):7624–33. doi: 10.1074/jbc.M412889200
27. Belfiore A, Malaguarnera R, Nicolosi ML, Lappano R, Ragusa M, Morrione A, et al. A novel functional crosstalk between DDR1 and the IGF axis and its relevance for breast cancer. Cell Adhesion Migration (2018) 12(4):305–14. doi: 10.1080/19336918.2018.1445953
28. Taliaferro-Smith L, Oberlick E, Liu T, McGlothen T, Alcaide T, Tobin R, et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget (2015) 6(7):4757–72. doi: 10.18632/oncotarget.3023
29. Kiely PA, Baillie GS, Barrett R, Buckley DA, Adams DR, Houslay MD, et al. Phosphorylation of RACK1 on Tyrosine 52 by c-Abl Is Required for Insulin-like Growth Factor I-mediated Regulation of Focal Adhesion Kinase. J Biol Chem (2009) 284(30):20263–74. doi: 10.1074/jbc.M109.017640
30. Peterson JE, Kulik G, Jelinek T, Reuter CWM, Shannon JA, Weber MJ. Src Phosphorylates the Insulin-like Growth Factor Type I Receptor on the Autophosphorylation Sites: REQUIREMENT FOR TRANSFORMATION BY src. J Biol Chem (1996) 271(49):31562–71. doi: 10.1074/jbc.271.49.31562
31. Stanicka J, Rieger L, O’Shea S, Cox O, Coleman M, O’Flanagan C, et al. FES-related tyrosine kinase activates the insulin-like growth factor-1 receptor at sites of cell adhesion. Oncogene (2018) 37(23):3131–50. doi: 10.1038/s41388-017-0113-z
32. Yoneyama Y, Lanzerstorfer P, Niwa H, Umehara T, Shibano T, Yokoyama S, et al. IRS-1 acts as an endocytic regulator of IGF-I receptor to facilitate sustained IGF signaling. eLife (2018) 7:e32893. doi: 10.7554/eLife.32893
33. Backer JM, Shoelson SE, Haring E, White MF. Insulin receptors internalize by a rapid, saturable pathway requiring receptor autophosphorylation and an intact juxtamembrane region. J Cell Biol (1991) 115(6):1535–45. doi: 10.1083/jcb.115.6.1535
34. Prager D, Li HL, Yamasaki H, Melmed S. Human insulin-like growth factor I receptor internalization. Role of the juxtamembrane domain. J Biol Chem (1994) 269(16):11934–7.
35. Sehat B, Andersson S, Vasilcanu R, Girnita L, Larsson O. Role of Ubiquitination in IGF-1 Receptor Signaling and Degradation. PloS One (2007) 2(4):e340. doi: 10.1371/journal.pone.0000340
36. Haigler HT, McKanna JA, Cohen S. Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J Cell Biol (1979) 83(1):82–90. doi: 10.1083/jcb.83.1.82
37. Yamazaki T, Zaal K, Hailey D, Presley J, Lippincott-Schwartz J, Samelson LE. Role of Grb2 in EGF-stimulated EGFR internalization. J Cell Sci (2002) 115(Pt 9):1791–802.
38. Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res (2008) 314(17):3093–106. doi: 10.1016/j.yexcr.2008.08.013
39. Sehat B, Andersson S, Girnita L, Larsson O. Identification of c-Cbl as a New Ligase for Insulin-like Growth Factor-I Receptor with Distinct Roles from Mdm2 in Receptor Ubiquitination and Endocytosis. Cancer Res (2008) 68(14):5669–77. doi: 10.1158/0008-5472.CAN-07-6364
40. Monami G, Emiliozzi V, Morrione A. Grb10/Nedd4-mediated multiubiquitination of the insulin-like growth factor receptor regulates receptor internalization. J Cell Physiol (2008) 216(2):426–37. doi: 10.1002/jcp.21405
41. Marshall S. Kinetics of insulin receptor internalization and recycling in adipocytes. Shunting of receptors to a degradative pathway by inhibitors of recycling. J Biol Chem (1985) 260(7):4136–44.
42. Xu Y-M, Wang H-J, Chen F, Guo W-H, Wang Y-Y, Li H-Y, et al. HRD1 suppresses the growth and metastasis of breast cancer cells by promoting IGF-1R degradation. Oncotarget (2015) 6(40):42854–67. doi: 10.18632/oncotarget.5733
43. Yan C, Xu W, Huang Y, Li M, Shen Y, You H, et al. HRD1-Mediated IGF-1R Ubiquitination Contributes to Renal Protection of Resveratrol in db/db Mice. Mol Endocrinol (2016) 30(6):600–13. doi: 10.1210/me.2015-1277
44. Vecchione A, Marchese A, Henry P, Rotin D, Morrione A. The Grb10/Nedd4 Complex Regulates Ligand-Induced Ubiquitination and Stability of the Insulin-Like Growth Factor I Receptor. Mol Cell Biol (2003) 23(9):3363–72. doi: 10.1128/MCB.23.9.3363-3372.2003
45. Girnita L, Girnita A, Larsson O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci (2003) 100(14):8247–52. doi: 10.1073/pnas.1431613100
46. Girnita L, Shenoy SK, Sehat B, Vasilcanu R, Vasilcanu D, Girnita A, et al. β-Arrestin and Mdm2 Mediate IGF-1 Receptor-stimulated ERK Activation and Cell Cycle Progression. J Biol Chem (2007) 282(15):11329–38. doi: 10.1074/jbc.M611526200
47. Worrall C, Suleymanova N, Crudden C, Trocoli Drakensjö I, Candrea E, Nedelcu D, et al. Unbalancing p53/Mdm2/IGF-1R axis by Mdm2 activation restrains the IGF-1-dependent invasive phenotype of skin melanoma. Oncogene (2017) 36(23):3274–86. doi: 10.1038/onc.2016.472
48. Mao Y, Shang Y, Pham VC, Ernst JA, Lill JR, Scales SJ, et al. Polyubiquitination of Insulin-like Growth Factor I Receptor (IGF-IR) Activation Loop Promotes Antibody-induced Receptor Internalization and Down-regulation. J Biol Chem (2011) 286(48):41852–61. doi: 10.1074/jbc.M111.288514
49. Spence J, Sadis S, Haas AL, Finley D. A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol (1995) 15(3):1265–73. doi: 10.1128/MCB.15.3.1265
50. Hoege C, Pfander B, Moldovan G-L, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature (2002) 419(6903):135–41. doi: 10.1038/nature00991
51. Acconcia F, Sigismund S, Polo S. Ubiquitin in trafficking: The network at work. Exp Cell Res (2009) 315(9):1610–8. doi: 10.1016/j.yexcr.2008.10.014
52. Packham S, Warsito D, Lin Y, Sadi S, Karlsson R, Sehat B, et al. Nuclear translocation of IGF-1R via p150Glued and an importin-β/RanBP2-dependent pathway in cancer cells. Oncogene (2015) 34(17):2227–38. doi: 10.1038/onc.2014.165
53. Aleksic T, Chitnis MM, Perestenko OV, Gao S, Thomas PH, Turner GD, et al. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res (2010) 70(16):6412–9. doi: 10.1158/0008-5472.CAN-10-0052
54. Sehat B, Tofigh A, Lin Y, Trocmé E, Liljedahl U, Lagergren J, et al. SUMOylation Mediates the Nuclear Translocation and Signaling of the IGF-1 Receptor. Sci Signaling (2010) 3(108):ra10–ra. doi: 10.1126/scisignal.2000628
55. Warsito D, Lin Y, Gnirck A-C, Sehat B, Larsson O. Nuclearly translocated insulin-like growth factor 1 receptor phosphorylates histone H3 at tyrosine 41 and induces SNAI2 expression via Brg1 chromatin remodeling protein. Oncotarget (2016) 7(27):42288–302. doi: 10.18632/oncotarget.9785
56. Warsito D, Sjöström S, Andersson S, Larsson O, Sehat B. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Reports (2012) 13(3):244–50. doi: 10.1038/embor.2011.251
57. Romanelli RJ, LeBeau AP, Fulmer CG, Lazzarino DA, Hochberg A, Wood TL. Insulin-like Growth Factor Type-I Receptor Internalization and Recycling Mediate the Sustained Phosphorylation of Akt. J Biol Chem (2007) 282(31):22513–24. doi: 10.1074/jbc.M704309200
58. Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell (2001) 106(2):145–55. doi: 10.1016/S0092-8674(01)00434-2
59. Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem Sci (2017) 42(1):42–56. doi: 10.1016/j.tibs.2016.08.016
60. Stuffers S, Brech A, Stenmark H. ESCRT proteins in physiology and disease. Exp Cell Res (2009) 315(9):1619–26. doi: 10.1016/j.yexcr.2008.10.013
61. Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol (2018) 19(11):679–96. doi: 10.1038/s41580-018-0053-7
62. Klumperman J, Raposo G. The complex ultrastructure of the endolysosomal system. Cold Spring Harbor Perspect Biol (2014) 6(10):a016857–a. doi: 10.1101/cshperspect.a016857
63. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol (2009) 10(9):597–608. doi: 10.1038/nrm2755
64. Hao H, Niu J, Xue B, Su QP, Liu M, Yang J, et al. Golgi-associated microtubules are fast cargo tracks and required for persistent cell migration. EMBO Rep (2020) 21(3):e48385. doi: 10.15252/embr.201948385
65. Shafaq-Zadah M, Gomes-Santos CS, Bardin S, Maiuri P, Maurin M, Iranzo J, et al. Persistent cell migration and adhesion rely on retrograde transport of β1 integrin. Nat Cell Biol (2016) 18(1):54–64. doi: 10.1038/ncb3287
66. Fourriere L, Kasri A, Gareil N, Bardin S, Bousquet H, Pereira D, et al. RAB6 and microtubules restrict protein secretion to focal adhesions. J Cell Biol (2019) 218(7):2215–31. doi: 10.1083/jcb.201805002
67. Kupfer A, Dennert G, Singer SJ. Polarization of the Golgi apparatus and the microtubule-organizing center within cloned natural killer cells bound to their targets. Proc Natl Acad Sci U S A (1983) 80(23):7224–8. doi: 10.1073/pnas.80.23.7224
68. Millarte V, Farhan H. The Golgi in Cell Migration: Regulation by Signal Transduction and Its Implications for Cancer Cell Metastasis. Sci World J (2012) 2012:11. doi: 10.1100/2012/498278
69. Sasaki K, Yoshida H. Organelle autoregulation—stress responses in the ER, Golgi, mitochondria and lysosome. J Biochem (2015) 157(4):185–95. doi: 10.1093/jb/mvv010
70. Farber-Katz S, Dippold H, Buschman M, Peterman M, Xing M, Noakes C, et al. DNA Damage Triggers Golgi Dispersal via DNA-PK and GOLPH3. Cell (2014) 156(3):413–27. doi: 10.1016/j.cell.2013.12.023
71. Johannes L, Popoff V. Tracing the Retrograde Route in Protein Trafficking. Cell (2008) 135(7):1175–87. doi: 10.1016/j.cell.2008.12.009
72. Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol (2002) 4(5):343–50. doi: 10.1038/ncb783
73. Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol (2006) 24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723
74. Farhan H, Wendeler MW, Mitrovic S, Fava E, Silberberg Y, Sharan R, et al. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J Cell Biol (2010) 189(6):997–1011. doi: 10.1083/jcb.200912082
75. Citores L, Bai L, Sørensen V, Olsnes S. Fibroblast growth factor receptor-induced phosphorylation of STAT1 at the golgi apparatus without translocation to the nucleus. J Cell Physiol (2007) 212(1):148–56. doi: 10.1002/jcp.21014
76. Warren CM, Ziyad S, Briot A, Der A, Iruela-Arispe ML. A Ligand-Independent VEGFR2 Signaling Pathway Limits Angiogenic Responses in Diabetes. Sci Signaling (2014) 7(307):ra1–ra. doi: 10.1126/scisignal.2004235
77. Obata Y, Horikawa K, Shiina I, Takahashi T, Murata T, Tasaki Y, et al. Oncogenic Kit signalling on the Golgi is suppressed by blocking secretory trafficking with M-COPA in gastrointestinal stromal tumours. Cancer Lett (2018) 415:1–10. doi: 10.1016/j.canlet.2017.11.032
78. Brodt P, Fallavollita L, Khatib A-M, Samani AA, Zhang D. Cooperative Regulation of the Invasive and Metastatic Phenotypes by Different Domains of the Type I Insulin-like Growth Factor Receptor β Subunit. J Biol Chem (2001) 276(36):33608–15. doi: 10.1074/jbc.M102754200
79. Hermanto U, Zong CS, Li W, Wang L-H. RACK1, an Insulin-Like Growth Factor I (IGF-I) Receptor-Interacting Protein, Modulates IGF-I-Dependent Integrin Signaling and Promotes Cell Spreading and Contact with Extracellular Matrix. Mol Cell Biol (2002) 22(7):2345–65. doi: 10.1128/MCB.22.7.2345-2365.2002
80. Kiely PA, O’Gorman D, Luong K, Ron D, O’Connor R. Insulin-Like Growth Factor I Controls a Mutually Exclusive Association of RACK1 with Protein Phosphatase 2A and β1 Integrin To Promote Cell Migration. Mol Cell Biol (2006) 26(11):4041–51. doi: 10.1128/MCB.01868-05
81. Kiely PA, Baillie GS, Lynch MJ, Houslay MD, O’Connor R. Tyrosine 302 in RACK1 Is Essential for Insulin-like Growth Factor-I-mediated Competitive Binding of PP2A and β1 Integrin and for Tumor Cell Proliferation and Migration. J Biol Chem (2008) 283(34):22952–61. doi: 10.1074/jbc.M800802200
82. Huttenlocher A, Lakonishok M, Kinder M, Wu S, Truong T, Knudsen KA, et al. Integrin and Cadherin Synergy Regulates Contact Inhibition of Migration and Motile Activity. J Cell Biol (1998) 141(2):515–26. doi: 10.1083/jcb.141.2.515
83. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell (2002) 110(6):673–87. doi: 10.1016/S0092-8674(02)00971-6
84. Bowe RA, Cox OT, Ayllón V, Tresse E, Healy NC, Edmunds SJ, et al. PDLIM2 regulates transcription factor activity in epithelial-to-mesenchymal transition via the COP9 signalosome. Mol Biol Cell (2014) 25(1):184–95. doi: 10.1091/mbc.e13-06-0306
85. Hynes RO, Lander AD. Contact and adhesive specificities in the associations, migrations, and targeting of cells and axons. Cell (1992) 68(2):303–22. doi: 10.1016/0092-8674(92)90472-O
86. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer (2018) 18(9):533–48. doi: 10.1038/s41568-018-0038-z
87. Yu W, Yang L, Li T, Zhang Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front Oncol (2019) 9:989. doi: 10.3389/fonc.2019.00989
88. Lin S-Y, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol (2001) 3(9):802–8. doi: 10.1038/ncb0901-802
89. Offterdinger M, Weipoltshammer K, Grunt TW. c-erbB-3 : a nuclear protein in mammary epithelial cells. J Cell Biol (2002) 157(6):929–40. doi: 10.1083/jcb.200109033
90. Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell (2004) 6(3):251–61. doi: 10.1016/j.ccr.2004.07.012
91. Johnston CL, Cox HC, Gomm JJ, Coombes RC. Fibroblast Growth Factor Receptors (FGFRs) Localize in Different Cellular Compartments: A SPLICE VARIANT OF FGFR-3 LOCALIZES TO THE NUCLEUS. J Biol Chem (1995) 270(51):30643–50. doi: 10.1074/jbc.270.51.30643
92. Stachowiak EK, Fang X, Myers J, Dunham S, Stachowiak MK. cAMP-induced differentiation of human neuronal progenitor cells is mediated by nuclear fibroblast growth factor receptor-1 (FGFR1). J Neurochem (2003) 84(6):1296–312. doi: 10.1046/j.1471-4159.2003.01624.x
93. Kim S-J, Kahn CR. Insulin induces rapid accumulation of insulin receptors and increases tyrosine kinase activity in the nucleus of cultured adipocytes. J Cell Physiol (1993) 157(2):217–28. doi: 10.1002/jcp.1041570203
94. Seol KC, Kim SJ. Nuclear matrix association of insulin receptor and IRS-1 by insulin in osteoblast-like UMR-106 cells. Biochem Biophys Res Commun (2003) 306(4):898–904. doi: 10.1016/S0006-291X(03)01046-5
95. Feng Y, Venema VJ, Venema RC, Tsai N, Caldwell RB. VEGF Induces Nuclear Translocation of Flk-1/KDR, Endothelial Nitric Oxide Synthase, and Caveolin-1 in Vascular Endothelial Cells. Biochem Biophys Res Commun (1999) 256(1):192–7. doi: 10.1006/bbrc.1998.9790
96. Mayo LD, Kessler KM, Pincheira R, Warren RS, Donner DB. Vascular Endothelial Cell Growth Factor Activates CRE-binding Protein by Signaling through the KDR Receptor Tyrosine Kinase. J Biol Chem (2001) 276(27):25184–9. doi: 10.1074/jbc.M102932200
97. Deng H, Lin Y, Badin M, Vasilcanu D, Stromberg T, Jernberg-Wiklund H, et al. Over-accumulation of nuclear IGF-1 receptor in tumor cells requires elevated expression of the receptor and the SUMO-conjugating enzyme Ubc9. Biochem Biophys Res Commun (2011) 404(2):667–71. doi: 10.1016/j.bbrc.2010.12.038
98. Codony-Servat J, Cuatrecasas M, Asensio E, Montironi C, Martínez-Cardús A, Marín-Aguilera M, et al. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br J Cancer (2017) 117(12):1777–86. doi: 10.1038/bjc.2017.279
99. van Gaal JC, Roeffen MH, Flucke UE, van der Laak JA, van der Heijden G, de Bont ES, et al. Simultaneous targeting of insulin-like growth factor-1 receptor and anaplastic lymphoma kinase in embryonal and alveolar rhabdomyosarcoma: a rational choice. Eur J Cancer (2013) 49(16):3462–70. doi: 10.1016/j.ejca.2013.06.022
100. Prisco M, Santini F, Baffa R, Liu M, Drakas R, Wu A, et al. Nuclear translocation of insulin receptor substrate-1 by the simian virus 40 T antigen and the activated type 1 insulin-like growth factor receptor. J Biol Chem (2002) 277(35):32078–85. doi: 10.1074/jbc.M204658200
101. Lin Y, Liu H, Waraky A, Haglund F, Agarwal P, Jernberg-Wiklund H, et al. SUMO-modified insulin-like growth factor 1 receptor (IGF-1R) increases cell cycle progression and cell proliferation. J Cell Physiol (2017) 232(10):2722–30. doi: 10.1002/jcp.25818
102. Sarfstein R, Pasmanik-Chor M, Yeheskel A, Edry L, Shomron N, Warman N, et al. Insulin-like growth factor-I receptor (IGF-IR) translocates to nucleus and autoregulates IGF-IR gene expression in breast cancer cells. J Biol Chem (2012) 287(4):2766–76. doi: 10.1074/jbc.M111.281782
103. Rukstalis JM, Habener JF. Snail2, a mediator of epithelial-mesenchymal transitions, expressed in progenitor cells of the developing endocrine pancreas. Gene Expression Patterns (2007) 7(4):471–9. doi: 10.1016/j.modgep.2006.11.001
104. Aslam MI, Hettmer S, Abraham J, Latocha D, Soundararajan A, Huang ET, et al. Dynamic and nuclear expression of PDGFRα and IGF-1R in alveolar Rhabdomyosarcoma. Mol Cancer Res (2013) 11(11):1303–13. doi: 10.1158/1541-7786.MCR-12-0598
105. Asmane I, Watkin E, Alberti L, Duc A, Marec-Berard P, Ray-Coquard I, et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: a predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur J Cancer (2012) 48(16):3027–35. doi: 10.1016/j.ejca.2012.05.009
106. Bodzin AS, Wei Z, Hurtt R, Gu T, Doria C. Gefitinib resistance in HCC mahlavu cells: upregulation of CD133 expression, activation of IGF-1R signaling pathway, and enhancement of IGF-1R nuclear translocation. J Cell Physiol (2012) 227(7):2947–52. doi: 10.1002/jcp.23041
107. Clemmons DR. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol Metab Clin North Am (2012) 41(2):425–43, vii-viii. doi: 10.1016/j.ecl.2012.04.017
108. Sádaba MC, Martín-Estal I, Puche JE, Castilla-Cortázar I. Insulin-like growth factor 1 (IGF-1) therapy: Mitochondrial dysfunction and diseases. Biochim Biophys Acta (BBA) - Mol Basis Dis (2016) 1862(7):1267–78. doi: 10.1016/j.bbadis.2016.03.010
109. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
110. Rambold AS, Lippincott-Schwartz J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle (2011) 10(23):4032–8. doi: 10.4161/cc.10.23.18384
111. Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of Subsarcolemmal Mitochondria in Obesity and Type 2 Diabetes. Diabetes (2005) 54(1):8–14. doi: 10.2337/diabetes.54.1.8
112. Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol (2007) 83(1):84–92. doi: 10.1016/j.yexmp.2006.09.008
113. Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (2010) 88(10):993–1001. doi: 10.1007/s00109-010-0663-9
114. Sheng B, Wang X, Su B, H-g L, Casadesus G, Perry G, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem (2012) 120(3):419–29. doi: 10.1111/j.1471-4159.2011.07581.x
115. Macleod KF. Mitophagy and Mitochondrial Dysfunction in Cancer. Annu Rev Cancer Biol (2020) 4(1):41–60. doi: 10.1146/annurev-cancerbio-030419-033405
116. Lyons A, Coleman M, Riis S, Favre C, O’Flanagan CH, Zhdanov AV, et al. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem (2017) 292(41):16983–98. doi: 10.1074/jbc.M117.792838
117. Chang CY, Kazmin D, Jasper JS, Kunder R, Zuercher WJ, McDonnell DP. The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell (2011) 20(4):500–10. doi: 10.1016/j.ccr.2011.08.023
118. Riis S, Murray JB, O’Connor R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β–Nrf2–BNIP3 Pathway. Cells (2020) 9(1):147. doi: 10.3390/cells9010147
119. Palikaras K, Lionaki E, Tavernarakis N. Coupling mitogenesis and mitophagy for longevity. Autophagy (2015) 11(8):1428–30. doi: 10.1080/15548627.2015.1061172
120. Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature (2015) 521(7553):525–8. doi: 10.1038/nature14300
121. Dossou AS, Basu A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel) (2019) 11(10):1422. doi: 10.3390/cancers11101422
122. Bareja A, Lee DE, White JP. Maximizing Longevity and Healthspan: Multiple Approaches All Converging on Autophagy. Front Cell Dev Biol (2019) 7:183. doi: 10.3389/fcell.2019.00183
123. Floyd S, Favre C, Lasorsa FM, Leahy M, Trigiante G, Stroebel P, et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol Biol Cell (2007) 18(9):3545–55. doi: 10.1091/mbc.e06-12-1109
124. Favre C, Zhdanov A, Leahy M, Papkovsky D, O’Connor R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene (2010) 29(27):3964–76. doi: 10.1038/onc.2010.146
Keywords: insulin-like growth factor 1 receptor (IGF-1R), signaling, endosomes, nucleus, Golgi
Citation: Rieger L and O’Connor R (2021) Controlled Signaling—Insulin-Like Growth Factor Receptor Endocytosis and Presence at Intracellular Compartments. Front. Endocrinol. 11:620013. doi: 10.3389/fendo.2020.620013
Received: 21 October 2020; Accepted: 02 December 2020;
Published: 29 January 2021.
Edited by:
Shin-Ichiro Takahashi, The University of Tokyo, JapanReviewed by:
Haim Werner, Tel Aviv University, IsraelCopyright © 2021 Rieger and O’Connor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosemary O’Connor, ci5vY29ubm9yQHVjYy5pZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.