 Christopher R. Shepard
Christopher R. Shepard
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 29 January 2021
Sec. Obesity
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.613639
This article is part of the Research TopicNon-Alcoholic Steatohepatitis (NASH) – Metabolic Contributors and Therapeutic TargetsView all 25 articles
Toll-Like Receptor 9 (TLR9) is an ancient receptor integral to the primordial functions of inflammation and metabolism. TLR9 functions to regulate homeostasis in a healthy system under acute stress. The literature supports that overactivation of TLR9 under the chronic stress of obesity is a critical driver of the pathogenesis of NASH and NASH-associated fibrosis. Research has focused on the core contributions of the parenchymal and non-parenchymal cells in the liver, adipose, and gut compartments. TLR9 is activated by endogenous circulating mitochondrial DNA (mtDNA). Chronically elevated circulating levels of mtDNA, caused by the stress of overnutrition, are observed in obesity, metabolic dysfunction-associated fatty liver disease (MAFLD), and NASH. Clinical evidence is supportive of TLR9 overactivation as a driver of disease. The role of TLR9 in metabolism and energy regulation may have an underappreciated contribution in the pathogenesis of NASH. Antagonism of TLR9 in NASH and NASH-associated fibrosis could be an effective therapeutic strategy to target both the inflammatory and metabolic components of such a complex disease.
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of liver disease worldwide (1). NAFLD is a progressive condition most closely associated with overnutrition. Obesity is an overwhelming condition for normal biological processes. The simplest distillation is that when lipid input exceeds the body’s ability for lipid disposal, a number of physiological stresses are put on the organs under metabolic overload: including the liver, the adipose compartment, and the gut. How the physiological stresses manifest disease is acknowledged as complex, and recently the term metabolic dysfunction-associated fatty liver disease (MAFLD) was recommended instead of NAFLD as a more appropriate overarching term for a disease that is a multifaceted confluence of metabolic and inflammatory processes (2, 3).
Non-alcoholic steatohepatitis (NASH) is the more progressive form of the disease that includes hepatocellular injury in addition to hepatic steatosis and mild inflammation. NASH may progress to hepatic fibrosis, which is, by far, the most critical determinant outcome for a course of disease at risk of progression to cirrhosis and hepatocellular cancer. NASH is an unmet medical need still lacking any approved therapeutics (4–6).
Toll-Like Receptor 9 (TLR9) is a pattern recognition receptor, so-called because this family of receptors is activated by recognizing common structures or motifs across biological molecules (7). There are at least ten TLRs present in mammals responsible for cell-extrinsic and -intrinsic activities (8, 9). Characterization of the common motifs and their physiological association have spawned several names and their associated acronyms, such as pathogen-associated molecular patterns (PAMPs), associated with microorganisms; damage-associated molecular patterns (DAMPs), associated with endogenous molecules released from damaged cells; and metabolism-associated molecular patters (MAMPS), associated with danger molecules resulting from metabolic overload (10, 11).
TLR9 detects the “CpG” motif, unmethylated cytosine-phosphate-guanine (CpG) dinucleotides. CpG is a common motif in bacterial and viral DNA, but uncommon in the vertebrate genome. However, mitochondria, the energy center of every vertebrate cell, is an organelle of bacterial origin and has its own mitochondrial DNA (mtDNA). Mitochondria are the source for endogenous molecules that activate TLR9 (12). Upon cell stress or damage, mtDNA is ejected from the mitochondria and into the cell’s surrounding environment to activate TLR9 as paracrine or endocrine signals for danger or stress.
The breadcrumbs that led to the discovery of TLR function, and eventually TLR9, are relevant because they are important in the evaluation of TLR9 in NASH. O’Neill et al. have conducted comprehensive work in summarizing the research history of TLRs (13), of which a small slice will be focused on here to uncover the core biological functions of TLR9.
The discovery of the interleukin-1 (IL-1) family of molecules, called necrosin by Menkin in his original characterization in 1943 because of the observed inflammatory tissue injury (14), opened the door to the signaling of the innate immune system. By 1975, the connection between innate immunity and metabolism had been made with the molecular players still needing to be identified (15). Infection of rabbits with various bacterial pathogens caused marked changes in lipid levels (16), and carbohydrate-regulating hormones triggered by the host response were associated with biochemical and ultrastructural changes in the liver (17).
The soluble signaling molecules of the IL-1 family were characterized in seminal work at Harvard Medical School by Dayer, Krane and Robinson et al. and Mizel and Mergenhagen at the National Institutes of Health in the last years of the 1970s (18, 19). The receptor for those signaling molecules, IL-1R, was cloned in 1988 by Sims et al. while at Immunex, a private biotechnology company eventually acquired by Amgen. In that 1988 report, the authors write, “How the cytoplasmic domain functions in signal transduction is unknown. Computer searches of the 1987 edition of Genbank [and other databases] have no revealed significant similarity to any currently available sequences” (20). Research on the role of IL-1 signaling in inflammation continued concurrently with investigations into the effects on energy balance. Particularly prescient was a report by Kitade et al. in 1996 that IL-1b regulates metabolism in hepatocytes (21).
The discovery that launched the dissection of TLR signaling in mammals was catalyzed by a short communication in Nature in 1991 from the Department of Biochemistry at the University of Cambridge that the cytoplasmic domain of IL-1R was homologous to the cytoplasmic domain of the D. melanogaster Toll protein, which was only studied at the time in the development of dorsoventral polarity in fruit flies (22, 23). The shared domain came to be known as the Toll/interleukin-1 receptor (TIR) domain.
In 1996, when it was discovered that Drosophila does use Toll for immunity, a kind of parallax was created that moved Toll’s involvement in immunity to the forefront of research (24). Discovery of the human TLR homologs starting in 1997 occurred on this backdrop, and although no function was yet ascribed to the mammalian TLR, immunity seemed the most likely function (25). Recentered efforts by researchers on innate immunity minimized research efforts devoted to TLR’s potential involvement in energy balance. Relative to functioning in immune processes, TLR’s role as a mediator of metabolism became largely overlooked. TLR4 was identified as the signaling receptor for LPS in 1998 (26). TLR9, including its sub-family members TLR7 and TLR8, were cloned in 2000 with CpG-DNA identified as a ligand for TLR9 in the same year (27–30). The next decade of research swiftly confirmed the TLR family’s undoubted importance in immunity, inflammation, and involvement in autoimmune diseases.
It was not until 2009 that the connection was made in Drosophila that Toll pathway activation in the insect compartment analogous to the human liver leads to inhibition of insulin signaling that results in decreased triglyceride storage (31). In the human system, only a handful of publications in the intervening years have focused on the relationship between TLR9’s signaling, metabolism, and energy expenditure.
In the mid-2000s, IL-1b and TLR9 adaptor protein MyD88 were implicated in weight loss-associated activation of the innate immune receptors independent from the inflammatory cascade and associated inflammatory pathology, however the mechanism was unknown (32). A seminal paper on TLR9 signaling was published in 2010 that reported the bifurcation of TLR9’s signaling pathway into pro-inflammatory and non-inflammatory pathways (33). Three years later, TLR9 was directly implicated in modulating energy metabolism independent of its proinflammatory function (34). It was not until 2020 that TLR9 was found upstream of a master regulator of energy homeostasis, AMPK activation (35), a topic to which this review will later return. The observations of TLR9’s importance in regulating metabolism are consistent with the discoveries that the signature signaling domain of TLR9, TIR, is a primordial metabolic regulatory enzyme (36, 37).
The role of TLRs as the gateway to the innate immune system is certain, and many articles summarize the history of this research in detail (13, 24, 38, 39). The slice of history presented here illustrates that TLR9’s role in metabolism is a relatively recent discovery, and perhaps underappreciated. The majority of research on TLR9 has investigated its function as an immune receptor (Figure 1). In the next section, we will find that TLR9 is an ancient protein. There is only one other primordial function as important as immunity, and that is metabolism.

Figure 1 Historical developments in the TLR9 field. IL-1R’s role in immunity was known during the same period as Toll’s role in Drosophila development. The homology of their common signaling domain, TIR, was discovered in 1991. In 1996, a role for hepatocyte metabolic regulation for IL-1R and Toll’s role for immunity was discovered in Drosophila. TLR9 was cloned and identified as the immunological pattern recognition sensor for CpG oligonucleotides in 2000. It was not until 2009 that Toll and TLR9 were found to have direct roles in metabolism and energy homeostasis. Adapted from a figure by Beutler (38). Immun, Immunity; Dev, Development; Metab, Metabolism.
TLR9 is one component of a tiered system of DNA pattern sensing by the cell (40). Emming and Schroder do an excellent job of offering a unifying hypothesis of the tiered structure that includes the endosomal TLR9, cytosolic or nuclear cGAS-STING, and cytosolic AIM2, all of which transduce inflammatory signals according to the level of threat. TLR9 functions as the canary—the leading signaling component for what is happening in the environment outside the cell. This system, or at least components of the system, appears throughout the tree of life (41). It is suspected that non-TLR9 elements evolved the ability to sense DNA relatively recently (42). The metabolic effects of DNA sensing are also a common feature even by extracellular sensing of DNA in protozoa and insects (43).
TLR9 has the lowest genetic drift of any of the components of the nucleic acid-sensing system and any TLR in mammals (41, 44–47). It is speculated that more intense selective pressures on other pattern recognition receptors may have left TLR9 intact (48).
All of this is to say that the extracellular sensing of DNA by TLR9 is a primordial program in response to danger. The group that initially characterized TLR9 as a sensor of bacterial DNA hypothesized that its specific action was to distinguish bacterial DNA from self-DNA (27). Polly Matzinger at the NIH urged the research community to change their point of view: she hypothesized that the primary driving force of the immune system is not to discriminate between self and non-self, but rather to protect and detect against danger (49). That TLR9 expression and upregulation are reported in nonimmune cells, including cardiomyocytes, neurons, hepatocytes, adipocytes, endothelial and epithelial, is suggestive of a critical role beyond immunity (50–55).
One of the foremost dangers of the 21st century to population health is overnutrition that disturbs metabolic homeostasis. The clinical manifestation is the epidemic of obesity and the metabolic syndrome. On the physiological level, it is the insult of chronic positive energy balance that stresses those metabolic organs most involved in nutrient handling: the liver, adipose tissue, and gut. Connor et al. describe the blurred line in obesity between pathological and protective mechanisms in the cell (56). It has been known that diets enriched with lard prime the immune system (including TLRs) for response (57).
In TLR9’s role as a metabolic regulator, and more generally as a sensor of danger, surely overnutrition was not a selection pressure in its evolution in man. The remaining content of this article will review the investigation into TLR9 in MAFLD and NASH.
TLR9 is expressed in a broad range of immune cells with varying ability across tissues in the normal physiological state to respond to CpG nucleotides (58, 59). There is some evidence of differential expression of TLR9 isoforms across healthy immune-cell rich tissues and peripheral blood cell types with limited data that the isoforms may differ in cellular localization (60). The classical pro-inflammatory role of TLR9 is through the activation of NF-κB. Results from clinical survey studies and more focused basic research investigations suggest that TLR9 drives expansion of resident and patrolling monocytes and monocyte-derived dendritic cells in inflammatory diseases.
TLR9 expression in non-immune cell types is curious. Non-immune cells, such as hepatocytes and adipocytes, can produce inflammatory molecules, but not at similar levels to immune cells (61, 62). Similarly, the tissue cell-type-specific responses to TLR9 agonists is also conserved in non-immune cells. For example, in a rodent model of renal-ischemia reperfusion that produces mtDNA release from stressed cells, TLR9 on hepatocytes respond to the CpG mtDNA, while proximal tubular epithelial cells do not—despite both having increased TLR9 expression (52). These cell and tissue regulatory systems make sense, as it could result in a catastrophic inflammatory response if tubular epithelial cells responded to stress and danger signals the same as immune cells. TLR expression in non-immune cells could be serving another purpose—TLR activation in adipose tissue and liver in inflammatory states leads to the downregulation of metabolism-related genes (63).
TLR9 is normally expressed in organs of the alimentary tract, including the liver and intestines. Expression is higher in the small and large intestine than in the liver, a pattern that exists across normal human, conventional CD-1 mice, and C57BL/6 germ-free mice (64). There is a degree of tolerance in TLR9 signaling in the intestinal epithelium, which one would anticipate given the organ’s constant exposure to bacterial products. The tolerance is maintained through differential apical and basolateral signaling of TLR9 (65, 66). When intestinal epithelial cells are exposed to pathogenic DNA in vitro, TLR9 mRNA is upregulated and increases in surface localization (67). The resulting transient increase in TLR9 signaling until the danger has passed likely maintains cellular and physiological homeostasis. For example, that intestinal FXR is modulated by TLR9 should not be a surprise, but more studies are needed to resolve regulation in normal physiological conditions versus TLR9-mediated FXR downregulation in chronic inflammation (68, 69).
TLR9 is also expressed in two less obvious compartments important to the innate immune system: adipose tissue and the liver. Several observations were made in the early 2000s about the involvement of macrophages in adipose tissue [a good summary of the studies from this period is contained in the introduction in Cinti et al. (70)]. In lean populations of both rodents and humans, adipose tissue macrophages are M2-polarized (71, 72). It was a surprise in 2007 when TLR9 was also detected on cultured adipocytes (55), though at the time it was already known to be a functional immunological organ, secreting a variety of proinflammatory cytokines and modulating monocyte and macrophage function through adipokines (73).
The liver is a metabolic organ, and also the first line of defense against infection (74–76). Not only does one need to consider the functional cells of the liver, hepatocytes, but also the supporting architecture (sinusoidal endothelial cells) and the mix of resident immune cells (Kupffer, the resident macrophage of the liver; dendritic, and stellate cells). Hepatocytes have low TLR9 protein expression levels compared with the other resident immune cells of the liver (52). Functional TLR9 is undoubtedly expressed in the nonparenchymal cells of the liver, which comprise around 40% of the organ’s cells (77–80). TLR9 expression is similar in the livers of conventional (CD-1) mice and humans, with germ-free (C57BL/6 strain) mice having lower levels of expression (64). Interesting is the significantly greater expression of TLR7, a TLR9 family member that is both an RNA and CpG DNA pattern recognition sensor, in the mouse liver compared with humans. The minimal sequence requirement for activation of mouse-TLR9 and human-TLR9 is also slightly different, but the physiological consequence, if any, is unclear (81). Such species-specific differences are important in translating TLR-focused biology to humans.
TLR9 protein expression is dynamic. In resting cells, TLR9 is localized to the endoplasmic reticulum, and the shuttling to initiate signal transduction is complex (82). In cells stimulated with bacterial DNA, TLR9 mRNA expression increases and endosomal TLR9 protein is shuttled toward the surface (67). The upregulation in TLR9 gene expression also occurs with cell exposure to LPS (83).
TLR9 upregulation in organs with relatively low basal levels is also observed in systems stressed with chronic obesity. TLR9 is significantly increased in visceral compared to subcutaneous adipose tissue in non-diabetic obese patients (84). It should be noted that most of the expression comes from the stroma-vascular cell fractions of the compartment (e.g., adipose tissue macrophages), not the adipocytes. In liver, Geoffrey Farrell’s lab at the Australian National University found that TLR9 expression is significantly upregulated in patients with biopsy-verified NASH, but not in liver with bland steatosis (85). This result was similar to the upregulation of TLR9 in two other mouse models of NASH: atherogenic-diet fed and foz/foz mice. TLR9 expression was observed primarily in aggregates of inflammatory cells, but expression was also observed in binucleate cells that were most likely hepatocytes.
In the severely obese, the chaperone protein UNC93B, required for successful TLR9 trafficking, is also upregulated (86, 87). The upregulation of TLR9 and its chaperones in concert in obesity suggests an organized effort by the cell for increased TLR9 signaling.
Charles Janeway coined the term “Signal 0” in 1989 to describe non-antigen stimuli outside the sequence of steps (“Signals 1, 2, and 3”) that lead to adaptive immunity (88). Sterile inflammation, which occurs when DAMPs are released into the microenvironment, is a Signal 0 and prototypically underlies NASH.
Circulating mitochondrial DNA (mtDNA) is the principal suspect for sterile inflammation in NASH, allowing these danger signals to reach remote organs through the circulation. Various biological features contribute to normal basal fluctuations of circulating cell-free DNA in humans (89). While acute trauma, liver injury, or even strenuous exercise can significantly raise levels of circulating mtDNA temporarily (89–94), significant chronically elevated levels of circulating mtDNA exist in patients with obesity and T2DM (95, 96).
mtDNA copy numbers in subjects with metabolic dysfunction are also significantly increased in adipose tissue compared with patients with normal BMIs (97). Bariatric surgery significantly reduced elevated urinary mtDNA copy number in patients with obesity (96). The elevated levels of circulating mtDNA in patients with metabolic dysfunction is biologically meaningful. Elevated mtDNA levels were associated with IL-1b levels in patients with T2DM in two different studies (98, 99). Plasma concentration of cell-free DNA was significantly higher in patients with visceral adiposity and positively correlated with visceral fat area and insulin resistance (100).
Obese patients with liver dysfunction or patients with biopsy-confirmed NASH have elevated levels of circulating mtDNA. The observation of elevated circulating levels of mtDNA in obese patients who also had elevated ALT as a marker of chronic liver injury was first made by Wajahat Mehal’s group at Yale in 2016 (101) with preliminary data reported at the meeting of the American Association for the Study of Liver Diseases two years earlier (102). They reported that obese patients with ALT elevations had a greater percentage of mitochondria inside microparticles than lean subjects, and the fraction of microparticles containing mitochondria was also larger in obese subjects with ALT elevations. They determined that the microparticles were of hepatocyte origin.
Mehal’s group linked their observations with a previous finding that oxidized DNA increases the CpG motif-dependent response of TLR9 (103). They found a significantly larger fraction of oxidized DNA in the microparticles from plasma of obese subjects with ALT elevations. In circulating mitochondria not contained in microparticles, a fraction with an origin that could not be traced, they did not see similarly elevated levels of oxidized DNA. The group noted that the hepatocyte origin of the fraction of plasma microparticles containing mitochondria with increased oxidation is consistent with the observation of increased oxidation of hepatocyte DNA in NASH (104). Indeed, the microparticles could activate TLR9 in a reporter cell line.
In a different study, it was also demonstrated that in vitro treatment of hepatocytes with palmitic acid, to model lipotoxic overload, causes the release of mtDNA into the cytosol (105). Incubating the cultured hepatocytes with a superoxide scavenger prevented the release of mtDNA. Lipotoxic overload in hepatocytes is also known to impair autophagy, which may function in the progression from bland steatosis to NASH (106). shRNA-mediated knockdown of a master regulator of autophagy, BECN1, enhanced release of mtDNA into hepatocyte cytosol. Similarly, rapamycin, an inducer of autophagy, attenuated mtDNA release. Bafilomycin A1, an inhibitor of autophagy, enhanced mtDNA release. Therefore, mtDNA is intimately connected to hepatocyte fate.
The presence of elevated circulating mtDNA levels in NASH was validated in a later study by Yury Popov’s group at Harvard in 2020, who reported preliminary results at the meeting of the American Association for the Study of Liver Diseases three years earlier (107, 108). Rather than the relative fold-induction qPCR technique Mehal previously used to determine elevated levels of circulating mtDNA in cases of presumptive NASH, Popov determined absolute circulating mtDNA copy number in patients with biopsy-proven NASH and NASH-fibrosis. In separate pilot and validation cohorts, the group demonstrated that patients with biopsy-confirmed NASH had significantly elevated circulating mtDNA levels compared with healthy control patients. In the larger validation cohort of 114 samples, the group reported statistically significant elevations in mtDNA copy number in patients with NAS of 4 or greater compared with patients with NAS of less than 4 (p = 0.0334). With respect to fibrosis stage, patients with F2-4 had significantly elevated mtDNA compared with F0-1 patients (p = 0.0003). Based on the means and standard deviations from Popov’s source data, only about 20% of NASH patients (patients with NAS of 4 or higher) have levels of circulating mtDNA that are less than the 99th percentile of circulating mtDNA in healthy patients.
Mehal and Popov’s results were further validated in a report from China, also in 2020 (105). This report is a valuable addition to the literature, as it has been shown that Chinese individuals have higher body fat percentages than Caucasians controls for any given BMI, with attendant increased visceral adiposity. Compared with other ethnic backgrounds, Chinese patients are affected by NAFLD at a lower BMI (109, 110). If chronic mtDNA elevation is a product of adipose tissue stress from overnutrition, one should see elevated circulating mtDNA levels in this population at lower BMIs. Indeed, Gao et al. observed elevated plasma levels of mtDNA in patients with BMIs in the lower 20s, consistent with healthcare professionals adopting a lower BMI threshold for clinical assessment and referral of NAFLD/NASH in these patients (111). In this relatively small report of 61 patients, divided into healthy control, NAFL, borderline-NASH (not defined in the paper, but generally defined as patients with an NAS of 3 (112)), and NASH, Chinese patients with borderline-NASH and NASH had significantly higher levels of mtDNA compared to healthy controls and non-NASH subjects.
Other diseases associated with elevated circulating mtDNA and NASH, independent from obesity, are intriguing. Sarcopenia, a muscle-wasting disorder associated with myocyte-specific mitochondrial dysfunction and circulating mtDNA, is associated with NAFLD independently of obesity and insulin resistance (113–116). The skeletal muscle compartment’s putative involvement in NASH disease deserves further exploration, as the inflammatory paracrine loop involving TLR9 in the sarcopenic muscle compartment may have analogs in other metabolic compartments (117–119). A similar pattern of NASH without metabolic risk factors exists in patients with HIV infection independent of combination antiretroviral therapy that causes liver damage (120, 121). Elevated adipose mtDNA levels are observed in HIV patients not on antiretroviral therapy, and elevated circulating mtDNA in HIV infected patients is a common observation (122, 123). One could hypothesize the presence of elevated circulating mtDNA levels links sarcopenia and HIV to attendant cases of NASH without the observation of obesity or metabolic dysfunction.
The gut supplies the liver with the majority of its blood, which is likely a source for TLR9-activating molecules into the portal circulation. Richard Flavell’s laboratory tested the hypothesis that gut microbiota has a central role in the pathogenesis of NASH (124). They demonstrated that TLR9 is necessary for mice to be susceptible to NASH when they are co-housed with mice that harbor transmissible colitogenic gut microbiota. In contrast, TLR5, a receptor for bacterial flagellin, did not have an effect in mediating disease severity. In the same rodent model, they found that TLR9 agonist influx into the portal circulation increased the severity of NASH. Finally, they demonstrated that microbiota-dependent, subclinical inflammation of the colon caused by the induction and secretion of CCL5 by the colonic epithelium was significantly associated with the influx of TLR9 agonist into the portal circulation.
In contrast, there was no difference in the amount of TLR2 agonists, which encompass a wide range of microbial cell wall components from Gram-positive and Gram-negative bacteria, in the portal circulation of mice with colonic inflammation. That TLR9 agonists are elevated, but not TLR2 agonists, is consistent with the immune system killing and disintegrating microbes with the release of microbial nucleic acids (125–129). The data indicate that bacterial products derived from the intestine, most likely microbial cell-free DNA, enter the hepatic portal circulation and trigger TLR9 activation.
Translocation of bacterial contents from the gut may only be relevant in a subset of NASH patients. A meta-analysis of 128 NAFLD patients across five single-center studies employing the same intestinal permeability assay found that 39.1% of NAFLD patients had evidence of increased intestinal permeability compared with 6.8% of healthy controls (130). In the same meta-analysis, it was found that 49.2% of patients with NASH had increased intestinal permeability. The data in total suggest that alterations in intestinal permeability in at least a significant subset of NAFLD and NASH patients may contribute to liver injury via TLR9 activation.
In a collaboration between Akita University and UCSD with Bernd Schnabl, Jerrold Olefsky, David Brenner, and Ekihiro Seki, Miura et al. first published on NASH in TLR9 knockouts (KOs) motivated by their interest in the TLR-MyD88 pathway in 2010 (131). They reported that TLR9-/- mice showed less steatohepatitis and liver fibrosis than wild-type (WT) mice when both were fed a choline-deficient, L-amino acid-defined (CDAA) diet. Later studies revealed that mice on a CDAA diet display a significantly different metabolic profile from human NASH, and therefore this diet model is not suitable as the surrogate for human disease (132). Nonetheless, the severity of the inflammatory profile produced by this diet yields intriguing insights into the inflammation-related biology associated with NASH pathogenesis
The group demonstrated that Kupffer cells produced the inflammatory cytokine IL-1β in response to CpG oligonucleotides. IL-1β increased apoptosis and necrosis in lipid-accumulated hepatocytes isolated from mice fed the CDAA diet, while CpG-mediated TLR9 signaling had little effect on cell death in either normal or lipid-accumulated hepatocytes. They next examined IL-1β’s effect on the fibrogenic response of hepatic stellate cells (HSC). While numerical increases in the levels of molecular markers of fibrogenic activation when HSC were treated with CpG DNA were observed, IL-1β increased the same markers by a significantly larger fraction. Employing experiments with conditioned media from Kupffer cells, their conclusion was TLR9-mediated IL-1β release from Kupffer cells was essential for HSC activation. They also found that a knockout of MyD88, the adaptor molecule shared by TLR9 and the IL-1β receptor IL-1R, is crucial for signaling that promotes NASH and fibrosis. TLR9 knockout mice on the CDAA diet, unlike the IL-1R and My88 knockout mice on the same diet, had a significant reduction on HOMA-IR.
Therefore, Miura et al.’s model involves the TLR9-dependent activation of Kupffer cells through endogenous TLR9 ligands, which in turn stimulates IL-1β release that acts on both hepatocytes and stellate cells. The model is consistent with observation of increased IL-1β in patients with NASH and fibrosis (133, 134); increased number of IL-1β positive liver cells in mice fed a high fat and cholesterol diet (135); that the lack of IL-1β inhibits the transformation of steatosis to steatohepatitis (136); and that deficiency of hepatic- rather than bone marrow-derived IL-1β protected mice against development of steatohepatitis and liver fibrosis on an atherogenic diet (136). Also consistent with Miura et al.’s model is interventional data in humans with the IL-1β neutralizing antibody canakinumab in a population with high cardiovascular risk who were borderline obese. Canakinumab tempered metabolic-related inflammation, but it did not affect plasma lipoprotein levels or new-onset diabetes (137, 138). Therefore, while IL-1β therapy in NASH subjects has yet to be tested, the data suggests it may not be a node that significantly impacts both the inflammatory and dysfunctional metabolic components of the disease.
Wajahat Mehal’s group at Yale continued the investigation into the identities of the TLR9 ligands and NASH pathogenesis with a report in 2016 (101). Mehal’s group used a high-fat diet (HFD) model (45% fat), which results in a metabolic and histological profile similar to human NASH (139). Mehal’s work with a HFD was key to bridging previous work to produce results considered more translationally relevant in humans. The group’s experiments that led them to focus on hepatocyte mtDNA as the critical TLR9 ligand in this system was described in the Section titled Signal 0 in NASH. The determination of whether TLR9 is signaling to NF-κB or IRF-7 dependent type 1 IFN is key to determining TLR9’s role in the pathogenesis of NASH. While the paths are not mutually exclusive, the NF-κB pathway results in the production of proinflammatory cytokines, while the IRF-7 pathway can upregulate the anti-inflammatory IL-1 receptor antagonist (IL-1RA). mtDNA from hepatocytes fed a HFD, when cultured with primary murine macrophages, resulted in selective upregulation of pro-inflammatory cytokines, but not type 1 IFN. The group confirmed hepatic macrophage (Kupffer cell) NF-κB activation in vivo using an NF-κB reporter mouse. HFD for twelve weeks induced the upregulation of NF-κB on cells excised from liver tissue with macrophage markers.
The group generated mice in which TLR9 was selectively removed from lysozyme producing cells (Lysm-Cre Tlr9fl/fl), including neutrophils, monocytes, and tissue macrophages. WT, Lysm-Cre Tlr9fl/fl mice, and total TLR9 knockout mice were placed on a HFD. The three groups had no difference in food intake as determined by paired-feeding experiment. The wild type mice on a 12-week HFD developed hepatosteatosis, balloon cells, and inflammation with elevated ALT, while both the Lysm-Cre Tlr9fl/fl mice and TLR9 KO mice exhibited NASH component histology that was significantly less severe.
Pharmacological inhibition of TLR9 with a TLR7/9 oligonucleotide antagonist was tested next by Mehal’s group (101). Treatment with the TLR9 antagonist at 5mg/kg once weekly prevented NASH component histology, ALT elevations, and cytokine transcript levels when the treatment was administered concurrently with the HFD. The TLR9 antagonist also reversed NASH component histology, ALT elevations, and cytokine transcript levels (pro-Il1b, Il6, Tnfa) when administered after eight weeks of HFD once weekly for four weeks at the same dose while the mice remained on the HFD. The treatment had no effect on food intake.
Geoffrey Farrell’s lab in 2017 determined the effects of TLR9 deletion in NASH pathogenesis in mice fed an atherosclerotic diet (23% fat, 0.2% cholesterol w/w) (85). This diet contained a much lower level of cholesterol than the typical 1–1.25% cholesterol often used in atherosclerotic diets and no cholate. Therefore, this diet is also translationally relevant to human NASH. TLR9 KO mice fed an atherosclerotic diet gained weight proportionally, including visceral fat mass, to WT mice. There were no differences in serum insulin between WT and TLR9 KO, and fasting blood glucose in response to atherosclerotic diet feeding was similarly elevated. Serum total cholesterol and hepatic free cholesterol were lower than WT mice after atherosclerotic diet feeding. Intriguingly, ALT values failed to increase when the TLR9 knockouts were fed the atherosclerotic diet. WT mice fed the atherosclerotic diet had a significant decrease in serum adiponectin that was not observed in TLR9 KO mice.
The decrease in adiponectin in the diet-fed WT, but not the TLR9 KO mice, is fascinating because it immediately draws attention to dysfunctional TLR9 signaling in the primary compartment from which adipokine signaling originates, the adipose tissue. Adiponectin is an incredibly unique pleiotropic signaling molecule that modulates insulin target tissues (140, 141). In the liver, adiponectin decreases fat accumulation, glucose output, and inflammation; in adipose tissue, it functions to decrease inflammation and increase insulin-stimulated glucose uptake; and, in skeletal muscle, decreases fat accumulation, increases glucose uptake and energy expenditure. Adiponectin is not secreted by fat cells alone—hepatic stellate cells secrete adiponectin in the resting state, and activated stellate cells produce apoptosis when treated with adiponectin (142). Therefore, normally elevated adiponectin levels are a systemic regulator of inflammatory and metabolic intra- and inter-organ homeostasis (143). More on the relationship between TLR9 and adiponectin is discussed in the Sections titled Adipose Tissue and Cutting the Endocrine Brakes on TLR9 Signaling.
The group next looked at liver histological features of inflammation and fibrosis. Despite exhibiting equivalent levels of steatosis, hardly any inflammation was observed in the TLR9 KOs fed the atherosclerotic diet. Abundant inflammation was present in the diet-fed wild-type mice, a difference that was also supported by markers for matrix deposition and activated hepatic stellate cells.
On the molecular level, NF-κB and JNK activation was evident in hepatic nuclei, including hepatocyte nuclei, in WT atherosclerotic-diet fed mice. Similar activation in the TLR9 KO mice was absent. The observation of significantly less inflammatory recruitment (macrophages and neutrophils) in the diet-fed TLR9 KOs was consistent. The fatty livers of TLR9 KO mice expressed less MCP-1 and Th1 cytokines (Mcp1, Tnf, iNos, Il-6). Markers for Th2 cytokines (Il-4, Ym1, Arg, circulating IL-10) increased in both groups similarly, but the authors noted that the imbalance in Th1 cytokines shifted the cytokine balance to one of protection in the TLR9 KO mice.
Isolation of bone marrow macrophages and neutrophils from WT and TLR9 KO mice confirmed their findings. Macrophages from TLR9 KO mice lost the ability to generate TNF when cultured with necrotic hepatocyte media, and neutrophils exhibited less chemotaxis. Isolated macrophages from TLR9 KO mice did not lose the ability to respond to the M2 stimulatory IL-4.
To further investigate if TLR9 expressed on bone marrow-derived cells was essential for NASH pathogenesis in this model, they created chimeric WT mice with TLR9-/- myeloid cells. When fed an atherosclerotic diet, the chimeric mice with TLR9-/- myeloid cells had impaired levels of circulating pro-inflammatory cytokines compared with chimeric WT mice with WT-TLR9+/+ myeloid cells.
The group also found decreased cytochrome c in the liver lysates of atherosclerotic diet-fed TLR9 KO mice compared with WT mice. The explanation for the observation was unclear at the time, but now makes sense with the discovery three years later that an AMPK-caspase-6 regulated mechanism activates a feed-forward loop fueled by cytochrome c release resulting in hepatocyte death (139).
Nishimoto first reported that genetic ablation of TLR9 improves insulin resistance through decreased macrophage accumulation in adipose tissue at the European Society of Cardiology in 2013 (144). In the publication that followed, the group demonstrated in C57BL/6 mice on a HFD that obesity-related adipocyte degeneration causes the release of cell-free DNA (cfDNA) in the visceral adipocyte compartment (100). The release of cfDNA was associated with increased visceral adipose tissue (VAT) weight, but not liver weight. Compared with lean mice, the HFD enhanced adipocyte degeneration. Concurrently, TLR9 transcript by RT-PCR increased in the VAT and was dominant in the macrophage population of the VAT. Using transwell co-culture experiments, the group established that TLR9 activation by cfDNA released from degenerated adipocytes increased monocyte chemoattractant protein-1 (MCP-1) expression.
To determine if TLR9 promoted adipose tissue inflammation by accelerating macrophage infiltration into the tissue, they used WT and TLR9 KO mice. After 12 weeks of feeding, body weight, VAT weight, and food intake were similar between the two groups. Compared with the WT mice, the TLR9 knockouts showed reduced macrophage infiltration and reduced expression of MCP-1 and TNF-α. In epididymal fat tissue of TLR9 KO mice, data suggested the macrophages were M2 polarized, while in the WT mice, the macrophages were M1 polarized. Additionally, the VAT of the TLR9 KOs showed less NF-κB activation and better insulin sensitivity. Both adiponectin and PPARγ were significantly higher in the VAT of the TLR9 KO mice fed a HFD than the WT counterparts. The observations on adiponectin in this HFD model were consistent with the TLR9-KO mice fed the atherosclerotic diet mentioned in the previous section (85).
The creation of chimeric TLR9 knockout mice with the bone marrow of wild-type (Tlr+/+) mice demonstrated that chimeric mice have more macrophage infiltration into adipose tissue, higher levels of inflammatory molecules, higher NF-κB activation, and more insulin resistance compared with control animals on the same diet without TLR9 expressing bone marrow.
Administration of a TLR9 inhibitory oligonucleotide at approximately 5 mg/kg, three times a week, resulted in reduced accumulation of macrophages in adipose tissue and improved insulin resistance. The treatment also decreased the level of plasma triglycerides with no difference in food intake.
A group from the Diabetes Research Group at the Toronto General Research Institute observed that the adipocytes in the VAT compartment may not be the only source of TLR9-activating molecules (145). VAT macrophages were also observed to expel extracellular traps (ETs) composed of nucleic acids. HFD-fed mice had increased formation of ETs in VAT, and TLR9 KOs had fewer M1 macrophages, fewer crown-like structures, and improved glucose homeostasis and insulin signaling during HFD feeding. Despite no difference in body weight to WT controls, the TLR9 KO mice fed a HFD showed decreased liver weights and decreased hepatic steatosis. Detailed metabolic profiling demonstrated that TLR9 KO mice also had similar food intake, oxygen consumption, CO2 output, respiratory exchange ratio, and energy expenditure to WT control mice.
Plasmacytoid dendritic cells (pDCs) are nearly absent in VAT but are present in the liver, where they can lead to prolonged inflammation and hepatocyte damage. TLR9 KO mice fed a HFD had decreased numbers of pDCs in the liver. They identified IFNα as a possible agent of hepatic insulin resistance and noted the number of IFNα-positive pDCs was consistently decreased in TLR9 KO mice fed a HFD compared with wild type mice fed a HFD. Exogenous introduction of TLR9 agonist in 20-week-old NCD mice worsened glucose tolerance, increased the number of hepatic pDCs, and decreased the number of tolerogenic pDCs.
Finally, obese mice treated for three weeks with ~3.3 mg/kg/week TLR9 oligonucleotide antagonist had improved glucose tolerance and a tendency for lower fasting insulin compared with PBS-injected controls.
Unlike other reports of TLR9 KOs, Hong et al. reported that TLR9 deficiency accelerates HFD-induced weight gain, insulin resistance, and adipocyte dysfunction (146). The observation of similar food intake between the control and knockout group was the same as previous studies. It is unknown why this study is an outlier. However, one possible reason is that it is the one report employing TLR9 knockouts initially developed on a 129P2 background before being backcrossed to B6 [as the difference in weight between WT and TLR9 KO employing the same transgenic model was seemingly replicated in the recovery phase of a study performed by a different lab (147)]. In normal physiological conditions, TLR9 certainly has a protective role. The results of this TLR9 KO study may be more illustrative of the importance of TLR9’s protective function in normal physiology, but it is difficult to know without an investigation into how the methods differed between studies.
The stressed parenchyma of the liver and adipose compartments along with possible gut leakage are not the only source for TLR9-activating molecules. It was observed that lymphocytes (B cells, T cells, NK cells), as well as monocytes and neutrophils, can secrete mtDNA webs in response to CpG oligonucleotides (148). mtDNA webs are distinct from neutrophil extracellular traps, which are expelled genomic DNA complexed with antibacterial proteins (149). The secretion of mtDNA webs upon stimulus with CpG oligonucleotides was not dependent on TLR9, as targeted TLR9 inhibition or other techniques to prevent TLR9 endosomal signaling did not impact mtDNA secretion. This result seems to indicate the existence of at least one feed-forward loop to increase the amount of mtDNA present in the hepatic milieu, independent of TLR9.
A different group documented a TLR9-dependent negative feedback loop that limited neutrophil overactivation upon stimulation with mtDNA, which could putatively function in the same system as the feed-forward loop (150). TLR9 activation upregulated the main actor of the negative feedback loop, miR-223. miR-233 knockout mice were more susceptible to activation of inflammatory mediators and NF-κB by TLR9 agonists. The same group later observed that miR-223 is initially elevated in the hepatocytes of rodents on a HFD and in human NASH samples indicative of a protective function, but that expression levels likely deteriorate as NASH disease worsens and progresses to cirrhosis and hepatocellular cancer (151). Therefore, NASH disease progression could be modulated by the concurrent downregulation of miR-233 that allows TLR9-ligand-activated signals to increase (152–154).
Last, hepatic free cholesterol accumulation in the liver alters normal transport of cellular cargo, including endosomal TLR9. The association between the accumulation of hepatic free cholesterol and NAFLD and fibrosis is well characterized (155, 156). In hepatic sinusoidal endothelial cells, free cholesterol accumulation exacerbated TLR9 signaling in a model of acetaminophen (APAP)-induced liver injury in obese animals (157). APAP injury is directly tied to the release of mtDNA in mice and humans (158). The authors observed that elevated free cholesterol levels in endolysosomes impaired the trafficking of TLR9 from late endosomes to lysosomes via Rab7. TLR9 escaped degradation and accumulated, thus enhancing TLR9 signaling. In Tlr9-/- mice, the effects of increased intake of cholesterol on APAP injury disappeared. Treatment of mice with ~3.3 mg/kg of oligonucleotide TLR9 antagonist 4 h after APAP treatment significantly ameliorated cholesterol-loading induced TLR9 signaling.
Free cholesterol accumulation in hepatocytes is also a likely source for TLR9 activation. In free cholesterol loaded hepatocytes, HMGB1 was released into the culture medium (158). It had already been reported that HMGB1 is an important modulator of TLR9 activation by CpG containing DNA (159–161). Extracellular HMGB1 accelerates the formation of the CpG-DNA–TLR9 complex to lower the effective concentration of CpG DNA necessary for activating cellular responses.
In addition to the core function of sensing danger signals in the cell’s environment, TLR9 also senses mtDNA that originates from its own cell. In an in vivo model of mitochondrial dysfunction caused by deficiency of Opa1, a regulator of mitochondrial fusion and functional compartment formation (162), Opa-1 deficiency driven inflammation required mtDNA and was independent of cGAS. mtDNA ejected from the mitochondria was not present in the cytosol and was detected by TLR9 in the endosome. Incubation of the cells with a TLR9 oligonucleotide antagonist attenuated the expression of NF-κB genes. The exact mechanism of how TLR9 interacts with mtDNA from its own cell needs to be clarified. The authors speculate that the interaction could occur via mitochondrial-derived vesicles under conditions of stress.
The strongest data that ties TLR9 activation to NASH disease is data from the Sanyal lab that directly associates TLR9 activation with NASH and fibrosis disease severity in human patients. The authors analyzed hepatic gene expression and coordinately regulated pathways in disease and control cohorts characterized by biopsy across the full histological spectrum of NASH disease (163). Incredibly valuable was the availability of the supplementary data from this study on the Gene Set Variation Analysis (GSVA). The GSVA analysis allowed identifying specific pathways differentially regulated with increasing histological severity in NASH and NASH-associated fibrosis. In the GSVA analysis, most of the identified pathways that appear within the first several hundred pathways that meet the statistical threshold for discoveries by the false discovery rate approach are too broadly described to be specifically druggable (e.g., “Intrinsic Pathway for Apoptosis”, “Cellular Responses to External Stimuli”). Of the handful of pathways listed that are directly targetable, differential activation of TLR9 and TLR9 signaling and adaptor proteins are all significantly associated with NASH disease severity, all with adjusted p-values of <0.0001. The TLR9 cascade is also significantly associated with NASH-fibrosis severity with an adjusted p-value of 0.01. The GSVA analysis cannot distinguish between pathways that are drivers of progressive disease and pathways activated as secondary to disease severity. Therefore, the results must be placed in the appropriate context with interventional studies. In combination with the interventional studies described in the previous section titled Liver the GSVA offers compelling evidence from human biopsies that TLR9 signaling is directly associated with the severity of NASH disease.
There are naturally occurring loss-of-function variants of TLR9 (164). However, the variants may be too rare (< 1%) to test the hypothesis that they are protective in NASH. As the only known subjects of this rare variant have been detected in North-Western Europe, it may be worthwhile to investigate further the specific hypothesis in NASH and control databases that intersect with that geography (165).
Instead of investigating a loss-of-function population, Alegre et al. took the approach of identifying patients with matched parameters of metabolic dysfunction (including BMI, HOMA-IR, lipids), but who were diagnosed by biopsy with either simple steatosis or NASH (166). They focused on TLR9 expression in T cells, as intrahepatic T cells’ role in NASH progression was confirmed in several studies. They also investigated the T cell production of IFN-γ via activation of TLR9 in cells from the matched patient cohorts. T cell production of IFN-γ is critical for the differentiation of proinflammatory macrophages. They found that reduced expression of TLR9 in T cells, both hepatic and peripheral, was associated with lower liver necroinflammatory activity and fibrosis. When they co-stimulated T-cells via TLR9, the cells from the patients with simple steatosis produced a limited number of IFN-γ producing CD8+ T cells compared with the T cells from patients with NASH. They concluded that limited expression, or active downregulation, of TLR9 on T cells is protective. In turn, this would also favor the differentiation of anti-inflammatory (M2-polarized) Kupffer cells. Patients with NASH may have limited endogenous expression of TLR9 or failure of the downregulation mechanism. The observation is strikingly similar to TLR9 expression in surgical lung biopsies differentiating rapidly from slowly progressing forms of idiopathic pulmonary fibrosis (167). This single study does not parse correlation versus causation with a high enough level of evidence for generalization, but the results are intriguing. The study certainly supports the role of TLR9 activation on yet another type of immune cell that could be driving NASH disease.
Evidence suggests that two factors simultaneously contribute to NASH progression: elevations in circulating TLR9 agonists in response to organ stress because of overnutrition; and upregulation of the TLR9 receptor in both immune and non-immune cells. Most likely, the time-integrated exposure of chronic TLR9 activation across the liver, adipose, and gut drives progression of the disease over a period of years (Figure 2).
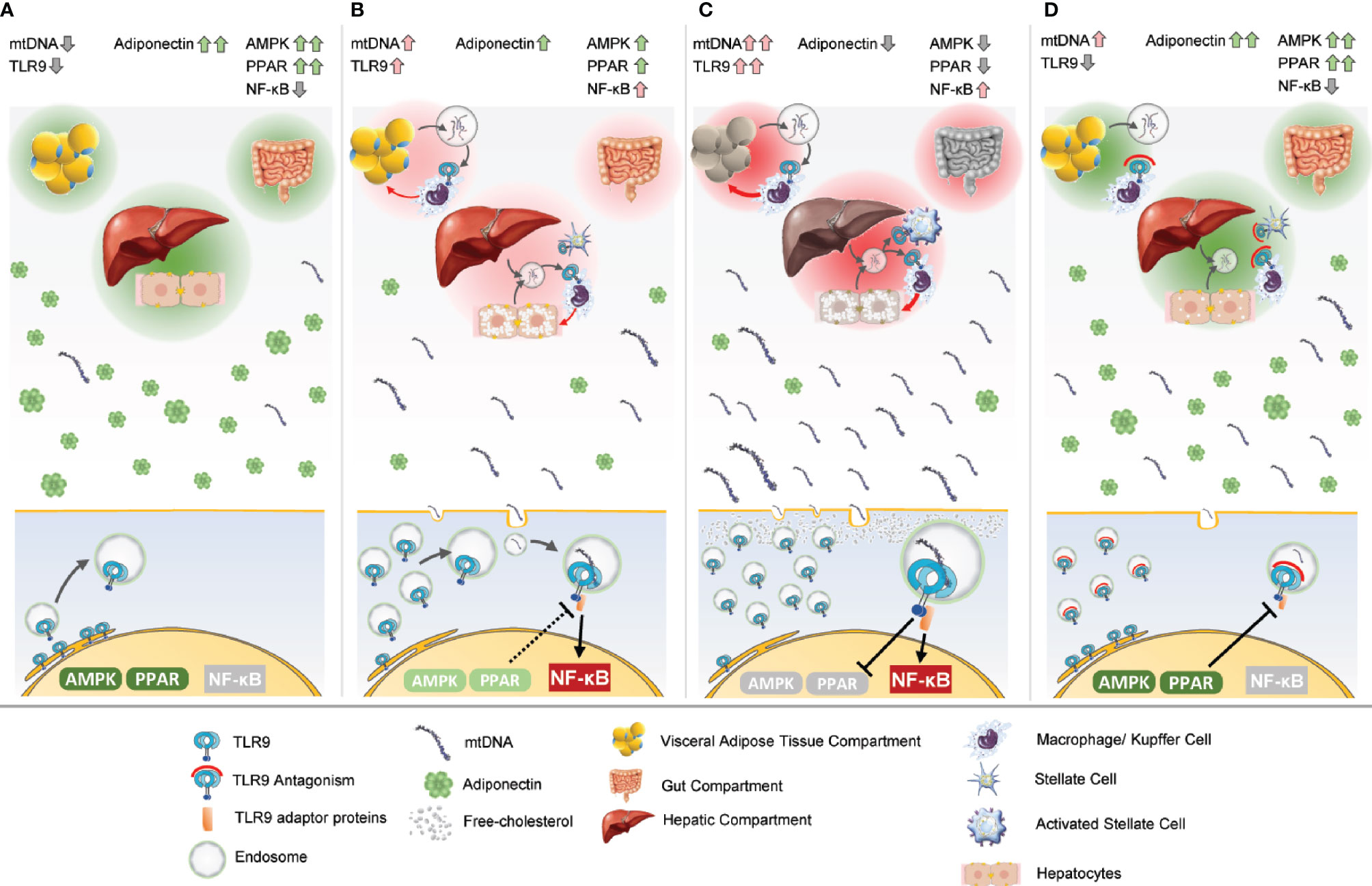
Figure 2 Unification of the theory of TLR9’s integral role in NASH pathogenesis. Three critical tissue compartments to NASH pathogenesis are pictured at the top of each frame, the adipose, gut, and liver. Endocrine and paracrine signals work at the compartment level and the level of the individual cell, pictured at the bottom. (A) The adipose, gut, and liver tissues are healthy and unstressed in a lean individual. Adiponectin levels are normally elevated. Transient elevations in mtDNA may occur in the course of normal physiology. PPAR is functioning normally, which dampens any transient TLR9 activation. TLR9 and AMPK are coupled so the cell can appropriately regulate energy expenditure if TLR9 is transiently activated, but in the resting state, most of the TLR9 is localized to the ER. NF-κB is not activated. (B) The adipose, gut, and liver tissues become increasingly stressed with overnutrition. Hepatosteatosis is evident. The secretion of mtDNA as response to the stress causes TLR9 upregulation. TLR9-dependent adipose infiltration and activation of macrophages and Kupffer cells occurs in the hepatic compartment. Gut leakage may allow bacterial product translocation into the portal circulation, either priming or activating TLR9 in the liver. A pro-inflammatory positive feedback paracrine loop forms between the infiltrating immune cells and the parenchyma. mtDNA levels increase in the systemic circulation. As adipocytes become more stressed, the levels of adiponectin decrease, causing AMPK activation to also decrease. Concurrently, increased TLR9 activation dampens PPAR activity. TLR9 activates pro-inflammatory NF-κB. (C) The various tissue compartments are even more stressed, and the TLR9-dependent pro-inflammatory positive feedback loop is robust. There are high levels of circulating mtDNA, and adiponectin levels are low. TLR9 activation may actively be suppressing PPAR activation, and adiponectin levels are too low to dampen TLR9 signaling. Free cholesterol present in the cell may further amplify TLR9 signaling. Stellate cells are activated in the liver, and hepatic fibrosis begins. (D) TLR9 antagonism prevents the inflammatory paracrine loop in the tissue compartments. Fibrosis is attenuated by TLR9 antagonism of stellate cell activation. PPAR signaling rises to dampen TLR9 activation as the system regains homeostatic function. Adiponectin works systemically to increase energy expenditure, decrease fat accumulation, and decrease inflammation and fibrosis.
In normal physiology, TLR9 is expressed in both immune and non-immune cells to varying degrees and is localized to the endoplasmic reticulum on resting cells. In the liver, Kupffer cells are M2 polarized, which TLR9 may even facilitate in normal physiological states in conjunction with normal PPAR functioning (168, 169). Hepatic dendritic cells are tolerogenic and immature. Other surveilling immune cells, such as neutrophils and lymphocytes (liver natural killer cells) are ready to respond if attracted from the periphery or in the early defense against pathogens (170).
Outside the liver, the gut is healthy and under TLR9 homeostatic control. The visceral adipose tissue surrounding the liver is healthy and circulating adiponectin levels act on the liver and systemically to control the metabolism of glucose and lipids by stimulating AMPK and PPARα (171). In hepatocytes, AMPK activity is acting to sustain a healthy hepatic parenchyma (139).
In states of obesity and overnutrition, the liver is under a considerable amount of immune stress even if hepatosteatosis is not harmful per se. Liver fat flux is relatively fast, hepatic steatosis is often self-limited, and hepatic energy metabolism in patients newly diagnosed with fatty liver is not different than controls (172, 173). However, there is evidence that the complement system is activated in obese patients with steatosis without NASH (174, 175). NASH may play out on the stage of the liver by virtue of being a first-pass organ. The liver is exposed to the highest concentration of TLR9 agonists from the portal circulation. NAFLD, in its subclinical phase in states of obesity, is thought to condition hepatic cells for the transition from a normal physiological state to a disease state.
It could be that TLR9 activation in Kupffer cells and other hepatic cells are primed by LPS leakage from the gut. In vitro, it was demonstrated that LPS-pretreated mouse bone marrow-derived macrophages produced significantly more TNF and IL-6 when stimulated with CpG DNA (176). The effects were still evident 12 h post-LPS treatment, meaning it was not a requirement that the LPS and CpG signals be administered at the same time for the signal amplification to occur. The levels of LPS used in the in vitro study were consistent with levels observed in NASH subjects (177, 178). Recent evidence even supports intrahepatic residence of bacteria that could also be priming TLR9 in the liver (179).
These studies are consistent with LPS’ downregulating effects on IL-1R8, an IL-1 receptor family member that can put the brakes on TLR9 signaling (180). It is unclear in what subset of patients LPS may incrementally contribute to TLR9 priming. In a study of pediatric patients who were either obese or had biopsy-proven NASH, endotoxin levels were increased in only 42.1% (8 of 19) in the NASH group (181). The reports on the association of endotoxemia with NASH histological severity is mixed (182, 183).
Gao et al. reported that mtDNA from mice fed a HFD, when combined with LPS stimulation, caused the release of significantly higher amounts of pro-inflammatory IL-33 from cultured bone-marrow-derived macrophages than LPS alone (105). The amount of IL-33 released into the media when mtDNA from mice on a chow diet was combined with LPS stimulus was no greater than LPS stimulus alone. Production of IL-33 was reduced by more than 50% by treatment with either a TLR9 oligonucleotide antagonist or TLR9 knockdown with siRNA.
By the time hepatosteatosis is evident, the visceral adipose tissue surrounding the liver is surely stressed and in a state of metainflammation (184). Circulating adiponectin levels are reduced (185). As the adipose inflammation worsens, adipocytes exhibit necrotic-like abnormalities that trigger the recruitment of inflammatory cells such as macrophages. mtDNA is released by the stressed adipocytes as a damage-associated molecule pattern (DAMP), which not only exacerbates inflammation at the local tissue level but is released into the circulation. TLR9 expression increases in the visceral adipose tissue (84).
At the same time TLR9 activation priming is occurring, subclinical conditioning during NAFLD may also be tipping the balance towards favoring greater levels of TLR9 activation. One of those regulatory mechanisms involves the peroxisome proliferators-activated receptors (PPARs). The PPARs act as fatty acid sensors and act as master regulators of metabolism, energy homeostasis, and inflammation (186, 187). Regulation of the various PPAR subfamilies is complex, but PPAR silencing is a common observation in obesity (188–193). In a morbidly obese population, a high fat meal resulted in a significant decrease of PPARγ mRNA expression (194). The ratio of a naturally occurring dominant-negative splice isoform PPARγΔ5 to PPARγ in humans correlates with BMI in overweight or obese and diabetic patients (195). PPARα transcription and immunofluorescence staining in liver tissue were significantly reduced in a cohort of Chinese NAFLD patients that was height- and weight-matched to a healthy cohort (196).
The significance of normal PPAR functioning is that it inhibits TLR9 signaling. The PPARγ-activating thiazolidinedione (TZD) troglitazone, when added to cultured peripheral blood adhering monocytes stimulated with a TLR9 family member ligand, prevented IL-6 release and decreased stimulatory capacity (197). TLR9 family member-associated MAPK signaling (p38 and p42) is significantly blunted when treated with PPARγ’s natural ligand, 15-deoxy- PGJ2. In fact, the connection between PPARs and TLRs is well documented (198). This is also consistent with the transrepression of TLR9 signaling by a different member of the nuclear-receptor family, the glucocorticoid receptor (199). NAFLD has been associated with signaling changes that reduce glucocorticoid receptor signaling (200, 201). Indeed, impairment of glucocorticoid receptor signaling causes steatosis, and if restored, reverses NAFLD in mice (202, 203). That the glucocorticoid receptor transrepresses TLR9 signaling is consistent with a model wherein nuclear receptor families, like PPARs, are silenced, which removes the brakes from and augments TLR9 signaling.
TLR4, which functions through a common adaptor protein with TLR9 and similarly activates NF-κB, inhibits PPARγ mRNA synthesis when activated via a negative feedback loop involving NF-κB (204). It seems likely that TLR9-mediated NF-κB activation also inhibits PPAR mRNA synthesis.
Augmented TLR9 signaling has consequences in both immune and non-immune cells. In both the liver and adipose compartments, TLR9 activation on immune cells causes the release of cytokines and chemokines (205). TLR9 dependent overactivation of the immune component, particularly the activation of hepatic stellate cells, potentiates fibrosis (78). On non-immune cells, such as parenchymal hepatocytes and adipocytes, dysfunctional TLR9 signaling may be directly tied to disturbed energy homeostasis.
TLR9 signaling is directly coupled to the master regulator of energy homeostasis, AMPK (206). In the context of exercise and glucose starvation under normal physiological conditions, TLR9 is required to activate AMPK via an association with beclin1 and simultaneous TLR9 binding to endogenous mtDNA (35). They found that TLR9 expression is also required for AMPK-regulated effects on glucose metabolism during the stress of acute exercise. Therefore, the data suggest that under normal physiological conditions, transient TLR9 activation by mtDNA to activate AMPK is part of a normal physiological process. A biological mechanism that controls the transient activation of AMPK in normal physiology may be important, as it was demonstrated that a constitutively activated AMPK in mice induced obesity and reduced beta cell function (207).
The connection between beclin1, a regulator of autophagy, and TLR9 was the result of a screen to identify proteins that interacted with a region of beclin1 that is sufficient to promote autophagy when introduced exogenously (208). Beclin1 is a key part of phosphatidylinositol 3-kinase complex (PI3KC3) signaling in the endosome (209). The association with beclin1 led the group to find that TLR9 is also required for the association of beclin1 and UVRAG. UVRAG is another key component of the Class II phosphatidylinositol 3-kinase complex (PI3KC3-C2) crucial for endosomal signaling. The conclusion was that TLR9 regulates the assembly of PI3KC3-C2, which in turn regulates AMPK activity.
In a separate stream of work, Nemazanyy et al. found that a PI3KC3 complex containing UVRAG was a key node in the negative feedback inhibition of metabolic signaling (210). The PI3KC3 complex they investigated contained UVRAG, beclin1, and Vps15, common core components to Class II and Class III PI3KC3 (211). Vps15 acts as the regulatory subunit of PI3KC3. Through experiments that interfered with the expression of Vps15, they made the novel finding that PI3KC3 containing UVRAG and beclin1 had a previously unappreciated role in whole body nutrient homeostasis and control of metabolic adaptation. In vivo¸ hepatic downregulation of Vps15 significantly improved glucose tolerance in ob/ob mice, decreased liver steatosis, and decreased hepatic triglyceride levels without the observation of a change in plasma metabolite levels. The findings were consistent with reports that established Vps15 as part of a complex thought to integrate environmental cues through an AMPK-dependent mechanism (212). Changing AMPK cellular localization, ordinarily present in both the cytoplasm and nucleus but in periods of stress (including cellular oxidants) shifts to the nucleus, is surely a part of this story (213). That TLR9 is a likely component, even rate-limiting component, of the PI3KC3 complex puts the receptor front and center in metabolic regulation through environmental cues.
One possibility is that chronic stimulation of TLR9 by endogenous mtDNA in states of overnutrition leads to the constitutive negative feedback of metabolic or autophagy signals through TLR9’s regulation of PI3KC3 assembly. This is consistent with AMPK downregulation and its role in liver damage in NASH (139, 214, 215). The regulation of expression of the TLR9-interacting protein beclin1 is significantly different in lean and obese states. When HFD-fed obese and lean mice are maintained on a 15 day 40% caloric restriction, then returned to ad libitum feeding on their original diets, refeeding led to a greater than 2-fold increase in beclin1 in the visceral adipose tissue in the obese mice whereas an 80% reduction in beclin1 was observed in the lean mice (216). Similarly, in humans, beclin expression was significantly higher in the adipose tissue of both non-diabetic and diabetic obese subjects than lean controls. Following gastric bypass, a significant drop in the expression of beclin was observed in both the obese groups. It is unknown how Vps15 responds in states of overnutrition. However, the discoveries that deficiency in other adaptor protein components of the Beclin1-Vps15 complexes in the AMPK pathway leads to lipid accumulation in the liver echoes the importance of this regulatory system to which TLR9 belongs (217). The two-way regulation of PI3KC3 complexes by AMPK is complex (212), and more investigation into the role TLR9 and TLR9 stimulation in both lean and obese states play in this pathway is needed.
Other mechanisms that modulate TLR9’s involvement between inflammation and energy modulation in non-immune cells have also been reported. In normal physiological systems, it was reported that TLR9 reduces energy substrates (intracellular ATP) in stressed cardiomyocytes by activating AMPK (34). AMPK activation may be turned off in disease-state TLR9 activation by the pivotal switch, Unc93b1. The shRNA-mediated knockdown of Unc93b1 in macrophages could replicate the AMPK activation observed in the cardiomyocytes instead of observing the more prototypical inflammatory response. Conversely, overexpression of Unc93b1 in cardiomyocytes reduced TLR9-induced AMPK activation and activated inflammatory signaling. Unc93b1 overexpression also transformed the trafficking of both TLR9 and endocytosed CpG DNA so the agonist and cognate receptor could successfully meet in the endosome. They validated the results in a completely different kind of non-immune neuroblastoma cell line. Therefore, Unc93b1 may be an additional regulatory component in the switch from TLR9 activation of AMPK out of self-protection, to AMPK silencing in a disease state. Indeed, in the severely obese, UNC93B is upregulated (Lawless and Greene 2012, Clayton 2016). While there is no reported direct interaction between UNC93B1 and regulation of the PI3KC3 complex, they have shared involvement in TLR9 stabilization, endosomal transport, and modulation of AMPK (218). The possible decoupling of TLR9 and AMPK through TLR9 overaction needs to be investigated.
Impaired AMPK activation is intimately tied to another impaired global signaling system in NASH, the adipokine adiponectin. Adiponectin receptor activation increases AMPK and PPARs, resulting in increased fatty acid oxidation and glucose utilization (219, 220). Adiponectin also has anti-inflammatory properties targeted toward both hepatic and immune cells (221). The hormone targets the key organs involved in metabolic regulation, including the liver, heart, pancreatic β cells, kidney, and skeletal muscle. Scherer’s landmark discovery in 1995 of adiponectin marked the beginning of understanding the hormone’s intimate ties to the metabolic syndrome (222–224). Metabolic syndrome is strongly associated with decreased levels of circulating adiponectin, “hypoadiponectinemia”. A number of studies, too many for inclusive citation here, have demonstrated the strong association between NASH and decreased levels of circulating adiponectin in both adults and pediatrics (225–228). NASH patients have lower circulating adiponectin levels than patients with NAFLD (229).
Adiponectin is an inhibitor of TLR9 signaling. Yamaguchi et al. found that pretreatment of macrophages with globular adiponectin significantly inhibited NF-κB activation after CpG DNA stimulation (230). The adiponectin levels that suppressed TLR9 activation in vitro were consistent with those observed in metabolically healthy, non-obese Caucasian subjects and also Asian subjects without NAFLD (231, 232). Therefore, adiponectin is yet another braking mechanism for TLR9 activation that is removed in subjects with NASH. Studies to determine a direct association between circulating adiponectin levels, TLR9 activation and the severity of NASH and fibrosis should be pursued.
By this moment in time, the totality of the literature suggests a robust positive feedback loop exists between the liver, gut, and adipose compartments involving circulating mtDNA triggering TLR9-dependent inflammatory activation in immune cells and upregulation of TLR9 in non-immune cells. The putative decoupling of TLR9 from AMPK may further dysregulate macrophage polarization in adipose tissue, further amplifying the paracrine loop between adipocytes and infiltrating macrophages (233, 234). Outside the liver-VAT-gut axis, TLR9 overactivation may induce podocyte apoptosis, accelerating insulin resistance and leading to the metabolic syndrome (235, 236).
Concurrent with the upregulation of TLR9 across various tissues, the molecular brakes tempering TLR9 activation have been removed, such as PPAR signaling, which forms a feed-forward loop with aberrant TLR9 trafficking to result in TLR9 signal amplification. The inflammatory milieu attracts neutrophils into the liver, and subsequent neutrophil overaction leads to the secretion of mtDNA webs.
The positive feedback loop continues to exacerbate hepatocyte cell degeneration with aberrant AMPK signaling leading to hepatocyte cell death (237, 238). Apoptotic hepatocyte DNA provides both a stop signal and stationary phenotype‐associated up‐regulation of collagen, both dependent on TLR9, in stellate cells (239). Gabele demonstrated that CpG stimulation of both human and murine hepatic stellate cells increases levels of the profibrogenic chemokine monocyte chemotactic protein 1 (MCP-1) and that TLR9-/- rodents had significantly less MCP-1 and α1(I) collagen mRNA expression, and less fibrosis by histology when challenged with bile duct ligation (78). Hypoadiponectinemia makes stellate cells more susceptible to activation (240).
Evidence supports that mtDNA from degenerated, injured, or apoptotic hepatocytes leads to hepatic fibrosis. Ballooned hepatocyte cells are independently associated with both sinusoidal fibrosis and perivenular fibrosis in NASH patients (241). Popov demonstrated that the failure to clear dead hepatocytes by persistent macrophage infiltrates (impaired efferocytosis) led to fibrosis in a thioacetamide (TAA)-induced model of fibrosis in mice (107). In the TAA model, they observed a 3-fold elevation of circulating mtDNA in serum levels post-TAA treatment in a mouse strain that was particularly susceptible to fibrosis because of impaired efferocytosis. The same elevation in circulating mtDNA post-treatment was not observed in a mouse strain resistant to TAA-induced fibrosis that also had functioning efferocytosis. The group could recapitulate the severity of fibrosis of the susceptible mice in the resistant mice by injecting the resistant mice with mtDNA post-TAA treatment. The injection of mtDNA in the resistant mice mimicked the prolonged exposure by susceptible mice to circulating mtDNA. The “resistant” mice developed significant liver fibrosis.
When Popov isolated hepatic stellate cells in vitro and treated them with increasing doses of purified mtDNA from hepatocytes, dose-dependent changes were observed in morphology characteristic of activation, increased proliferation, and profibrogenic gene expression. Therefore, stellate cell fibrogenesis seems directly linked to circulating mtDNA. TLR9 on stellate cells may be the primary receptor for detecting the prolonged elevations in circulating mtDNA in progressive NASH, and hepatic fibrogenesis advances.
The cumulative data supports that TLR9 antagonism is a promising therapeutic approach to treating NASH. TLR9’s place at the intersection of metabolism and inflammation is an important node for promising therapeutic intervention. Any therapy developed for this indication needs to be safe and well-tolerated across a broad population of people, and the safety database for TLR9 antagonism is supportive (242).
The most promising cornerstone strategies for NASH are likely those with comprehensive biological activity that match the multifactorial pathogenesis of NASH disease. Advances in FGF21 and GLP-1 analogs with clinical action on body weight, lipids, and adipokines are particularly interesting (243, 244). Both strategies fall under the category of metabolic agonists. Other late-stage pipeline candidates address narrower biology and disease nodes less proximal to positive energy balance, such as inflammation and cellular stress (ASK1 and CCR2/5 antagonists), lipid metabolism (thyroid hormone analogs), and de novo lipogenesis (FXR agonists) (245). The landscape covers the various therapeutic hypotheses of the main pathogenic mechanisms of NASH and NASH-associated fibrosis. The question remains what level of information exists to indicate that a patient will have a superior benefit-risk to a prescribed therapy.
The multi-factorial nature of NASH pathogenesis has made the afflicted population difficult to subset. The clinical interest in finding subsets of a disease population is in selecting patients more likely to respond to a therapeutic strategy, one component of precision medicine (246). That patients more likely to respond to TLR9 antagonism may be identified by measurement of circulating levels of TLR9 agonist or by observation of TLR9 (over)-expression in hepatic or adipose tissue, or by a combination of both, could allow for a degree of precision medicine in such a complex disease.
However, the data supports that circulating mtDNA levels are significantly elevated in the vast majority of NASH and NASH-fibrosis patients who are obese, and TLR9 activation is significantly associated with disease severity in unenriched cohorts of NASH and NASH-fibrosis patients. There may not be a bright line separating minimal and maximal responders—it may be more of a “ragged edge,” as Fleck calls it (247). The existing data suggest that TLR9 antagonism would benefit the broad majority of patients with NASH and NASH-fibrosis. The hypothesis merits clinical testing.
One of the primary advantages of TLR9 antagonism in NASH may be in the therapeutic index. Upon reviewing the research, we see that many of the molecular players coupled to TLR9 signaling are therapeutic targets in the current pipeline for NASH and NASH-fibrosis (248, 249). PPAR and AMPK signaling are compelling targets. While targeting those pathways with molecular agonists have shown effectiveness, they come with questionable safety profiles for use in broad populations (250, 251). Further, these molecular agonists are small molecules with systemic bioavailability, and therefore have pharmacologic (and potentially toxicologic) effects on critical organs not fundamental to NASH and NASH-fibrosis pathogenesis. The TLR9 antagonists used most often in the studies described here are oligonucleotide antagonists. The liver-gut-adipose-centric bioavailability of this mode of delivering TLR9 antagonism would seem to have an advantage in keeping the highest free drug concentrations limited to those organs most involved in the pathogenesis of NASH disease (252).
While adiponectin is a tempting target, there is no current strategy that allows for the direct drugging for adiponectin elevation (253). Adiponectin elevations secondary to other drug targets have proven clinically impactful. Observations of adiponectin elevations upon pharmacological PPARγ activation go back to 2002 (254). Merck Research Laboratories was quick to use adiponectin elevations as an example of a “putative biomarker” for PPARγ activity even in the nascent stage of biomarker application to drug development (255). Later, Cusi and colleagues demonstrated in clinical trials of the PPARγ activator pioglitazone that adiponectin elevation is the best predictor of histologic response in NASH and fibrosis (256, 257). Clinically relevant adiponectin elevations secondary to TLR9 antagonism would be a significant therapeutic advance.
Finally, TLR9 antagonism is compelling as a therapeutic strategy in NASH because it targets a positive feedback loop dependent on a chronic disease-specific signal, circulating mtDNA. Allowing the system to reset by pharmacologically returning to a more homeostatic state of inflammation and metabolism, one without chronic activation of TLR9 by mtDNA, is a plausible strategy for efficacious therapy. Antagonizing a disease-specific signal limits the safety liabilities associated with metabolic agonists.
The critical mass of research supporting TLR9’s importance in the pathogenesis of NASH and NASH-associated fibrosis includes an integral role in the inflammatory process that fuels NASH, as well as a metabolic one. Elevated levels of circulating mtDNA in patients with NASH and NASH-associated fibrosis, along with the association between TLR9 pathway activation and NASH disease severity, is strongly suggestive when combined with the mechanistic animal models of disease. TLR9’s role in hypoadiponectinemia has implications for insulin sensitive tissues throughout the body. The evidence suggests that TLR9 functions as a critical node that modulates at least three master regulators of NASH pathogenesis: AMPK, PPAR, and NF-κB.
Much more is known about TLR9’s role in the inflammatory process than in dysregulated metabolism. There are undoubtedly unexplored research areas in how TLR9 coordinates the PI3KC complex that could prove valuable in elucidating new therapeutic targets or strategies to target TLR9 signaling. The evidence supports that TLR9 is an important node in the inflammatory and dysfunctional metabolic components of NASH and NASH-associated fibrosis. Targeting TLR9 in NASH may prove an efficacious clinical strategy for a disease that is still an unmet medical need for a large fraction of the population.
CS conceived, summarized the main conceptual ideas, made intellectual contributions, prepared the figures, and wrote the manuscript.
No payment has been offered or will be offered to the author for authoring this publication and no compensation will be provided for the time he spent on the publication development. Structured BioEquity (SBE) funded CS’s participation as an Organizer of the 2019 Keystone Symposia Integrated Pathways of Disease in NASH and NAFLD.
CS is the founder of the company Avogadro Development Corp that has research and development interests in TLR9 antagonism in NASH.
The author would like to acknowledge David Shapiro and Amanda M. Burns, who reviewed the manuscript and offered helpful suggestions.
1. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA (2015) 313:2263–73. doi: 10.1001/jama.2015.5370
2. Eslam M, Sanyal AJ, George J, International Consensus P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology (2020) 158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
3. Tilg H, Effenberger M. From NAFLD to MAFLD: when pathophysiology succeeds. Nat Rev Gastroenterol Hepatol (2020) 17:387–8. doi: 10.1038/s41575-020-0316-6
4. Geier A, Rinella M, Balp MM, McKenna SJ, Brass C, Przybysz R, et al. Real-World Burden of Nonalcoholic Steatohepatitis. Clin Gastroenterol Hepatol (2020). doi: 10.1016/j.cgh.2020.06.064
5. Povsic M, Wong OY, Perry R, Bottomley J. A Structured Literature Review of the Epidemiology and Disease Burden of Non-Alcoholic Steatohepatitis (NASH). Adv Ther (2019) 36:1574–94. doi: 10.1007/s12325-019-00960-3
6. Balp MM, Krieger N, Przybysz R, Way N, Cai J, Zappe D, et al. The burden of non-alcoholic steatohepatitis (NASH) among patients from Europe: A real-world patient-reported outcomes study. JHEP Rep (2019) 1:154–61. doi: 10.1016/j.jhepr.2019.05.009
7. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
8. Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
9. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol (2018) 37:20–36. doi: 10.1080/08830185.2017.1380200
10. Wang X, Wang Y, Antony V, Sun H, Liang G. Metabolism-Associated Molecular Patterns (MAMPs). Trends Endocrinol Metab (2020) 31:712–24. doi: 10.1016/j.tem.2020.07.001
11. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
12. Meyer A, Laverny G, Bernardi L, Charles AL, Alsaleh G, Pottecher J, et al. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front Immunol (2018) 9:536. doi: 10.3389/fimmu.2018.00536
13. O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol (2013) 13:453–60. doi: 10.1038/nri3446
14. Menkin V. Studies on the Isolation of the Factor Responsible for Tissue Injury in Inflammation. Science (1943) 97:165–7. doi: 10.1126/science.97.2511.165
15. Beisel WR. Metabolic response to infection. Annu Rev Med (1975) 26:9–20. doi: 10.1146/annurev.me.26.020175.000301
16. Farshtchi D, Lewis VJ. Effects of three bacterial infections on serum lipids of rabbits. J Bacteriol (1968) 95:1615–21. doi: 10.1128/JB.95.5.1615-1621.1968
17. Tsung PK, Paretsky D. Biochemical and ultrastructural changes in liver endoplasmic reticular fractions during Q-fever. Acta Virol (1968) 12:49–53.
18. Mizel SB, Dayer JM, Krane SM, Mergenhagen SE. Stimulation of rheumatoid synovial cell collagenase and prostaglandin production by partially purified lymphocyte-activating factor (interleukin 1). Proc Natl Acad Sci U S A (1981) 78:2474–7. doi: 10.1073/pnas.78.4.2474
19. Dayer JM, Stephenson ML, Schmidt E, Karge W, Krane SM. Purification of a factor from human blood monocyte-macrophages which stimulates the production of collagenase and prostaglandin E2 by cells cultured from rheumatoid synovial tissues. FEBS Lett (1981) 124:253. doi: 10.1016/0014-5793(81)80149-4
20. Sims JE, March CJ, Cosman D, Widmer MB, MacDonald HR, McMahan CJ, et al. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science (1988) 241:585–9. doi: 10.1126/science.2969618
21. Kitade H, Kanemaki T, Sakitani K, Inoue K, Matsui Y, Kamiya T, et al. Regulation of energy metabolism by interleukin-1beta, but not by interleukin-6, is mediated by nitric oxide in primary cultured rat hepatocytes. Biochim Biophys Acta (1996) 1311:20–6. doi: 10.1016/0167-4889(95)00188-3
22. Gay NJ, Keith FJ. Drosophila Toll and IL-1 receptor. Nature (1991) 351:355–6. doi: 10.1038/351355b0
23. Valanne S, Wang JH, Ramet M. The Drosophila Toll signaling pathway. J Immunol (2011) 186:649–56. doi: 10.4049/jimmunol.1002302
25. Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A (1998) 95:588–93. doi: 10.1073/pnas.95.2.588
26. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
27. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature (2000) 408:740–5. doi: 10.1038/35047123
28. Chuang TH, Ulevitch RJ. Cloning and characterization of a sub-family of human toll-like receptors: hTLR7, hTLR8 and hTLR9. Eur Cytokine Netw (2000) 11:372–8.
29. Jurk M, Kritzler A, Debelak H, Vollmer J, Krieg AM, Uhlmann E. Structure-activity relationship studies on the immune stimulatory effects of base-modified CpG toll-like receptor 9 agonists. ChemMedChem (2006) 1:1007–14. doi: 10.1002/cmdc.200600064
30. Yasuda K, Richez C, Uccellini MB, Richards RJ, Bonegio RG, Akira S, et al. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J Immunol (2009) 183:3109–17. doi: 10.4049/jimmunol.0900399
31. DiAngelo JR, Bland ML, Bambina S, Cherry S, Birnbaum MJ. The immune response attenuates growth and nutrient storage in Drosophila by reducing insulin signaling. Proc Natl Acad Sci U S A (2009) 106:20853–8. doi: 10.1073/pnas.0906749106
32. Ogimoto K, Harris MK Jr, Wisse BE. MyD88 is a key mediator of anorexia, but not weight loss, induced by lipopolysaccharide and interleukin-1 beta. Endocrinology (2006) 147:4445–53. doi: 10.1210/en.2006-0465
33. Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science (2010) 329:1530–4. doi: 10.1126/science.1187029
34. Shintani Y, Kapoor A, Kaneko M, Smolenski RT, D’Acquisto F, Coppen SR, et al. TLR9 mediates cellular protection by modulating energy metabolism in cardiomyocytes and neurons. Proc Natl Acad Sci U S A (2013) 110:5109–14. doi: 10.1073/pnas.1219243110
35. Liu Y, Nguyen PT, Wang X, Zhao Y, Meacham CE, Zou Z, et al. TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature (2020) 578:605–9. doi: 10.1038/s41586-020-1992-7
36. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature (2009) 458:1056–60. doi: 10.1038/nature07813
37. Essuman K, Summers DW, Sasaki Y, Mao X, Yim AKY, DiAntonio A, et al. TIR Domain Proteins Are an Ancient Family of NAD(+)-Consuming Enzymes. Curr Biol (2018) 28:421–30.e4. doi: 10.1016/j.cub.2017.12.024
38. Beutler BA. TLRs and innate immunity. Blood (2009) 113:1399–407. doi: 10.1182/blood-2008-07-019307
39. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol (2004) 5:987–95. doi: 10.1038/ni1112
40. Emming S, Schroder K. Tiered DNA sensors for escalating responses. Science (2019) 365:1375–6. doi: 10.1126/science.aay2701
41. Gallucci S, Maffei ME. DNA Sensing across the Tree of Life. Trends Immunol (2017) 38:719–32. doi: 10.1016/j.it.2017.07.012
42. Margolis SR, Wilson SC, Vance RE. Evolutionary Origins of cGAS-STING Signaling. Trends Immunol (2017) 38:733–43. doi: 10.1016/j.it.2017.03.004
43. Mazzoleni S, Carteni F, Bonanomi G, Senatore M, Termolino P, Giannino F, et al. Inhibitory effects of extracellular self-DNA: a general biological process? New Phytol (2015) 206:127–32. doi: 10.1111/nph.13306
44. Hancks DC, Hartley MK, Hagan C, Clark NL, Elde NC. Overlapping Patterns of Rapid Evolution in the Nucleic Acid Sensors cGAS and OAS1 Suggest a Common Mechanism of Pathogen Antagonism and Escape. PLoS Genet (2015) 11:e1005203. doi: 10.1371/journal.pgen.1005203
45. Mozzi A, Pontremoli C, Forni D, Clerici M, Pozzoli U, Bresolin N, et al. OASes and STING: adaptive evolution in concert. Genome Biol Evol (2015) 7:1016–32. doi: 10.1093/gbe/evv046
46. Cagliani R, Forni D, Biasin M, Comabella M, Guerini FR, Riva S, et al. Ancient and recent selective pressures shaped genetic diversity at AIM2-like nucleic acid sensors. Genome Biol Evol (2014) 6:830–45. doi: 10.1093/gbe/evu066
47. Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One (2013) 8:e77846. doi: 10.1371/journal.pone.0077846
48. Kloch A, Wenzel MA, Laetsch DR, Michalski O, Bajer A, Behnke JM, et al. Signatures of balancing selection in toll-like receptor (TLRs) genes - novel insights from a free-living rodent. Sci Rep (2018) 8:8361. doi: 10.1038/s41598-018-26672-2
49. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
50. Crack PJ, Bray PJ. Toll-like receptors in the brain and their potential roles in neuropathology. Immunol Cell Biol (2007) 85:476–80. doi: 10.1038/sj.icb.7100103
51. Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res (2006) 72:384–93. doi: 10.1016/j.cardiores.2006.09.011
52. Bakker PJ, Scantlebery AM, Butter LM, Claessen N, Teske GJ, van der Poll T, et al. TLR9 Mediates Remote Liver Injury following Severe Renal Ischemia Reperfusion. PLoS One (2015) 10:e0137511. doi: 10.1371/journal.pone.0137511
53. El Kebir D, Damlaj A, Makhezer N, Filep JG. Toll-like receptor 9 signaling regulates tissue factor and tissue factor pathway inhibitor expression in human endothelial cells and coagulation in mice. Crit Care Med (2015) 43:e179–89. doi: 10.1097/CCM.0000000000001005
54. McClure R, Massari P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front Immunol (2014) 5:386. doi: 10.3389/fimmu.2014.00386
55. Khazen W, M’Bika JP, Collinet M, Tramoni M, Chany C, Achour A, et al. Differentiation-dependent expression of interferon gamma and toll-like receptor 9 in 3T3-F442A adipocytes. Biochimie (2007) 89:669–75. doi: 10.1016/j.biochi.2007.01.003
56. Connor T, Martin SD, Howlett KF, McGee SL. Metabolic remodelling in obesity and type 2 diabetes: pathological or protective mechanisms in response to nutrient excess? Clin Exp Pharmacol Physiol (2015) 42:109–15. doi: 10.1111/1440-1681.12315
57. Qiu H, Schlegel V. Impact of nutrient overload on metabolic homeostasis. Nutr Rev (2018) 76:693–707. doi: 10.1093/nutrit/nuy023
58. Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, et al. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol (2002) 168:4531–7. doi: 10.4049/jimmunol.168.9.4531
59. Sato R, Reuter T, Hiranuma R, Shibata T, Fukui R, Motoi Y, et al. The impact of cell maturation and tissue microenvironments on the expression of endosomal Toll-like receptors in monocytes and macrophages. Int Immunol (2020). doi: 10.1093/intimm/dxaa055
60. McKelvey KJ, Highton J, Hessian PA. Cell-specific expression of TLR9 isoforms in inflammation. J Autoimmun (2011) 36:76–86. doi: 10.1016/j.jaut.2010.11.001
61. Sell H, Dietze-Schroeder D, Eckardt K, Eckel J. Cytokine secretion by human adipocytes is differentially regulated by adiponectin, AICAR, and troglitazone. Biochem Biophys Res Commun (2006) 343:700–6. doi: 10.1016/j.bbrc.2006.03.010
62. Dong W, Simeonova PP, Gallucci R, Matheson J, Fannin R, Montuschi P, et al. Cytokine expression in hepatocytes: role of oxidant stress. J Interferon Cytokine Res (1998) 18:629–38. doi: 10.1089/jir.1998.18.629
63. Feingold KR, Moser A, Shigenaga JK, Grunfeld C. Inflammation inhibits the expression of phosphoenolpyruvate carboxykinase in liver and adipose tissue. Innate Immun (2012) 18:231–40. doi: 10.1177/1753425911398678
64. Huhta H, Helminen O, Kauppila JH, Salo T, Porvari K, Saarnio J, et al. The Expression of Toll-like Receptors in Normal Human and Murine Gastrointestinal Organs and the Effect of Microbiome and Cancer. J Histochem Cytochem (2016) 64:470–82. doi: 10.1369/0022155416656154
65. Pedersen G, Andresen L, Matthiessen MW, Rask-Madsen J, Brynskov J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin Exp Immunol (2005) 141:298–306. doi: 10.1111/j.1365-2249.2005.02848.x
66. Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol (2006) 8:1327–36. doi: 10.1038/ncb1500
67. Ewaschuk JB, Backer JL, Churchill TA, Obermeier F, Krause DO, Madsen KL. Surface expression of Toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenic bacterial DNA. Infect Immun (2007) 75:2572–9. doi: 10.1128/IAI.01662-06
68. Renga B, Mencarelli A, Cipriani S, D’Amore C, Carino A, Bruno A, et al. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS One (2013) 8:e54472. doi: 10.1371/journal.pone.0054472
69. Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim Biophys Acta (2011) 1812:851–8. doi: 10.1016/j.bbadis.2011.04.005
70. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res (2005) 46:2347–55. doi: 10.1194/jlr.M500294-JLR200
71. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi: 10.1172/JCI29881
72. Hill AA, Reid Bolus W, Hasty AH. A decade of progress in adipose tissue macrophage biology. Immunol Rev (2014) 262:134–52. doi: 10.1111/imr.12216
73. Schaffler A, Scholmerich J, Salzberger B. Adipose tissue as an immunological organ: Toll-like receptors, C1q/TNFs and CTRPs. Trends Immunol (2007) 28:393–9. doi: 10.1016/j.it.2007.07.003
74. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol (2013) 14:996–1006. doi: 10.1038/ni.2691
75. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
76. Kubes P, Jenne C. Immune Responses in the Liver. Annu Rev Immunol (2018) 36:247–77. doi: 10.1146/annurev-immunol-051116-052415
77. Schuchmann M, Hermann F, Herkel J, van der Zee R, Galle PR, Lohse AW. HSP60 and CpG-DNA-oligonucleotides differentially regulate LPS-tolerance of hepatic Kupffer cells. Immunol Lett (2004) 93:199–204. doi: 10.1016/j.imlet.2004.03.016
78. Gabele E, Muhlbauer M, Dorn C, Weiss TS, Froh M, Schnabl B, et al. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem Biophys Res Commun (2008) 376:271–6. doi: 10.1016/j.bbrc.2008.08.096
79. Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, et al. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect Immun (2002) 70:3433–42. doi: 10.1128/IAI.70.7.3433-3442.2002
80. Martin-Armas M, Simon-Santamaria J, Pettersen I, Moens U, Smedsrod B, Sveinbjornsson B. Toll-like receptor 9 (TLR9) is present in murine liver sinusoidal endothelial cells (LSECs) and mediates the effect of CpG-oligonucleotides. J Hepatol (2006) 44:939–46. doi: 10.1016/j.jhep.2005.09.020
81. Pohar J, Lainscek D, Fukui R, Yamamoto C, Miyake K, Jerala R, et al. Species-Specific Minimal Sequence Motif for Oligodeoxyribonucleotides Activating Mouse TLR9. J Immunol (2015) 195:4396–405. doi: 10.4049/jimmunol.1500600
82. Marongiu L, Gornati L, Artuso I, Zanoni I, Granucci F. Below the surface: The inner lives of TLR4 and TLR9. J Leukoc Biol (2019) 106:147–60. doi: 10.1002/JLB.3MIR1218-483RR
83. An H, Xu H, Yu Y, Zhang M, Qi R, Yan X, et al. Up-regulation of TLR9 gene expression by LPS in mouse macrophages via activation of NF-kappaB, ERK and p38 MAPK signal pathways. Immunol Lett (2002) 81:165–9. doi: 10.1016/S0165-2478(02)00010-X
84. Thomalla M, Schmid A, Neumann E, Pfefferle PI, Muller-Ladner U, Schaffler A, et al. Evidence of an anti-inflammatory toll-like receptor 9 (TLR 9) pathway in adipocytes. J Endocrinol (2019) 240:325–43. doi: 10.1530/JOE-18-0326
85. Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, et al. TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci (Lond) (2017) 131:2145–59. doi: 10.1042/CS20160838
86. Clayton BC. The Implications of Different Types of Diet and Exercise on Human Health”. Masters Theses Specialist Projects Paper (2016) 1617.
87. Lawless MW, Greene CM. Toll-like receptor signalling in liver disease: ER stress the missing link? Cytokine (2012) 59:195–202. doi: 10.1016/j.cyto.2012.04.003
88. Janeway C. Immunogenicity signals 1,2,3 ... and 0. Immunol Today (1989) 10:283–6. doi: 10.1016/0167-5699(89)90081-9
89. Aucamp J, Bronkhorst AJ, Badenhorst CPS, Pretorius PJ. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol Rev Camb Philos Soc (2018) 93:1649–83. doi: 10.1111/brv.12413
90. Ohlsson L, Hall A, Lindahl H, Danielsson R, Gustafsson A, Lavant E, et al. Increased level of circulating cell-free mitochondrial DNA due to a single bout of strenuous physical exercise. Eur J Appl Physiol (2020) 120:897–905. doi: 10.1007/s00421-020-04330-8
91. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
92. Thurairajah K, Briggs GD, Balogh ZJ. The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur J Trauma Emerg Surg (2018) 44:325–34. doi: 10.1007/s00068-018-0954-3
93. Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology (2012) 56:1971–82. doi: 10.1002/hep.25801
94. Marques PE, Oliveira AG, Pereira RV, David BA, Gomides LF, Saraiva AM, et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology (2015) 61:348–60. doi: 10.1002/hep.27216
95. Yuzefovych LV, Pastukh VM, Ruchko MV, Simmons JD, Richards WO, Rachek LI. Plasma mitochondrial DNA is elevated in obese type 2 diabetes mellitus patients and correlates positively with insulin resistance. PLoS One (2019) 14:e0222278. doi: 10.1371/journal.pone.0222278
96. Lee H, Oh S, Yang W, Park R, Kim H, Jeon JS, et al. Bariatric Surgery Reduces Elevated Urinary Mitochondrial DNA Copy Number in Patients With Obesity. J Clin Endocrinol Metab (2019) 104:2257–66. doi: 10.1210/jc.2018-01935
97. Litvinova L, Zatolokin P, Vulf M, Mazunin I, Skuratovskaia D. The relationship between the mtDNA copy number in insulin-dependent tissues and markers of endothelial dysfunction and inflammation in obese patients. BMC Med Genomics (2019) 12:41. doi: 10.1186/s12920-019-0486-7
98. Bae JH, Jo SI, Kim SJ, Lee JM, Jeong JH, Kang JS, et al. Circulating Cell-Free mtDNA Contributes to AIM2 Inflammasome-Mediated Chronic Inflammation in Patients with Type 2 Diabetes. Cells (2019) 8:328 doi: 10.3390/cells8040328
99. Catano Canizales YG, Uresti Rivera EE, Garcia Jacobo RE, Portales Perez DP, Yadira B, Rodriguez Rivera JG, et al. Increased Levels of AIM2 and Circulating Mitochondrial DNA in Type 2 Diabetes. Iran J Immunol (2018) 15:142–55. doi: 10.22034/IJI.2018.39378
100. Nishimoto S, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Murata C, et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci Adv (2016) 2:e1501332. doi: 10.1126/sciadv.1501332
101. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest (2016) 126:859–64. doi: 10.1172/JCI83885
102. Garcia-Martinez I, Ouyang X, Santoro N, Shlomchik MJ, Mehal WZ. Elevated plasma DNA in patients with NASH and reduced liver injury in mice with absence of TLR9 on Kupffer cells. 65th Annu Meeting Am Assoc Study Liver Dis: Liver Meeting (2014) 2014:654 doi: 10.1002/hep.27508
103. Yoshida H, Nishikawa M, Kiyota T, Toyota H, Takakura Y. Increase in CpG DNA-induced inflammatory responses by DNA oxidation in macrophages and mice. Free Radic Biol Med (2011) 51:424–31. doi: 10.1016/j.freeradbiomed.2011.04.035
104. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab (2015) 21:739–46. doi: 10.1016/j.cmet.2015.04.004
105. Gao Y, Wang Y, Liu H, Liu Z, Zhao J. Mitochondrial DNA from hepatocytes induces upregulation of interleukin-33 expression of macrophages in nonalcoholic steatohepatitis. Dig Liver Dis (2020) 52:637–43. doi: 10.1016/j.dld.2020.03.021
106. Lavallard VJ, Gual P. Autophagy and non-alcoholic fatty liver disease. BioMed Res Int (2014), 2014:120179. doi: 10.1155/2014/120179
107. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun (2020) 11:2362. doi: 10.1038/s41467-020-16092-0
108. Zhao S, An P, Ikenaaa N, Vaid KA, Lai M, Popov YV. Hepatocyte-derived mitochondrial DNA induces fibrogenic activation of stellate cells and promotes fibrosis in non-alcoholic steatohepatitis. In: 68th Annual Meeting of the American Association for the Study of Liver Diseases: The Liver Meeting, vol. (2017) HEPATOLOGY 66. 1
109. Wong RJ, Ahmed A. Obesity and non-alcoholic fatty liver disease: Disparate associations among Asian populations. World J Hepatol (2014) 6:263–73. doi: 10.4254/wjh.v6.i5.263
110. Szanto KB, Li J, Cordero P, Oben JA. Ethnic differences and heterogeneity in genetic and metabolic makeup contributing to nonalcoholic fatty liver disease. Diabetes Metab Syndr Obes (2019) 12:357–67. doi: 10.2147/DMSO.S182331
111. Fan JG, Saibara T, Chitturi S, Kim BI, Sung JJ, Chutaputti A, et al. What are the risk factors and settings for non-alcoholic fatty liver disease in Asia-Pacific? J Gastroenterol Hepatol (2007) 22:794–800. doi: 10.1111/j.1440-1746.2007.04952.x
112. Juluri R, Vuppalanchi R, Olson J, Unalp A, Van Natta ML, Cummings OW, et al. Generalizability of the nonalcoholic steatohepatitis Clinical Research Network histologic scoring system for nonalcoholic fatty liver disease. J Clin Gastroenterol (2011) 45:55–8. doi: 10.1097/MCG.0b013e3181dd1348
113. Lee YH, Jung KS, Kim SU, Yoon HJ, Yun YJ, Lee BW, et al. Sarcopaenia is associated with NAFLD independently of obesity and insulin resistance: Nationwide surveys (KNHANES 2008-2011). J Hepatol (2015) 63:486–93. doi: 10.1016/j.jhep.2015.02.051
114. Lee YH, Kim SU, Song K, Park JY, Kim DY, Ahn SH, et al. Sarcopenia is associated with significant liver fibrosis independently of obesity and insulin resistance in nonalcoholic fatty liver disease: Nationwide surveys (KNHANES 2008-2011). Hepatology (2016) 63:776–86. doi: 10.1002/hep.28376
115. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Bossola M, et al. Circulating Mitochondrial DNA at the Crossroads of Mitochondrial Dysfunction and Inflammation During Aging and Muscle Wasting Disorders. Rejuvenation Res (2018) 21:350–9. doi: 10.1089/rej.2017.1989
116. Li AA, Kim D, Ahmed A. Association of Sarcopenia and NAFLD: An Overview. Clin Liver Dis (Hoboken) (2020) 16:73–6. doi: 10.1002/cld.900
117. Lyu AK, Zhu SY, Chen JL, Zhao YX, Pu D, Luo C, et al. Inhibition of TLR9 attenuates skeletal muscle fibrosis in aged sarcopenic mice via the p53/SIRT1 pathway. Exp Gerontol (2019) 122:25–33. doi: 10.1016/j.exger.2019.04.008
118. Koo BK, Kim D, Joo SK, Kim JH, Chang MS, Kim BG, et al. Sarcopenia is an independent risk factor for non-alcoholic steatohepatitis and significant fibrosis. J Hepatol (2017) 66:123–31. doi: 10.1016/j.jhep.2016.08.019
119. Nachit M, Leclercq IA. Emerging awareness on the importance of skeletal muscle in liver diseases: time to dig deeper into mechanisms! Clin Sci (Lond) (2019) 133:465–81. doi: 10.1042/CS20180421
120. van Welzen BJ, Mudrikova T, El Idrissi A, Hoepelman AIM, Arends JE. A Review of Non-Alcoholic Fatty Liver Disease in HIV-Infected Patients: The Next Big Thing? Infect Dis Ther (2019) 8:33–50. doi: 10.1007/s40121-018-0229-7
121. Hulgan T, Gerschenson M. HIV and mitochondria: more than just drug toxicity. J Infect Dis (2012) 205:1769–71. doi: 10.1093/infdis/jis105
122. Morse CG, Voss JG, Rakocevic G, McLaughlin M, Vinton CL, Huber C, et al. HIV infection and antiretroviral therapy have divergent effects on mitochondria in adipose tissue. J Infect Dis (2012) 205:1778–87. doi: 10.1093/infdis/jis101
123. Pinti M, Mussini C, Cossarizza A. Mitochondrial DNA: a proinflammatory ‘enemy from within’ during HIV infection? Cell Death Dis (2012) 3:307. doi: 10.1038/cddis.2012.47
124. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature (2012) 482:179–85. doi: 10.1038/nature10809
125. Nielsen KM, Johnsen PJ, Bensasson D, Daffonchio D. Release and persistence of extracellular DNA in the environment. Environ Biosafety Res (2007) 6:37–53. doi: 10.1051/ebr:2007031
126. van der Vaart M, Pretorius PJ, Circulating DNA. Its origin and fluctuation. Ann N Y Acad Sci (2008) 1137:18–26. doi: 10.1196/annals.1448.022
127. Wichmann D, Panning M, Quack T, Kramme S, Burchard GD, Grevelding C, et al. Diagnosing schistosomiasis by detection of cell-free parasite DNA in human plasma. PLoS Negl Trop Dis (2009) 3:e422. doi: 10.1371/journal.pntd.0000422
128. Schaack S, Gilbert C, Feschotte C. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol (2010) 25:537–46. doi: 10.1016/j.tree.2010.06.001
129. Burnham P, Kim MS, Agbor-Enoh S, Luikart H, Valantine HA, Khush KK, et al. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci Rep (2016) 6:27859s. doi: 10.1038/srep27859
130. Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol Gastroenterol Hepatol (2015) 1:222–32. doi: 10.1016/j.jcmgh.2015.01.001
131. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology (2010) 139:323–34.e7. doi: 10.1053/j.gastro.2010.03.052
132. Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol (2018) 68:230–7. doi: 10.1016/j.jhep.2017.10.031
133. Negrin KA, Roth Flach RJ, DiStefano MT, Matevossian A, Friedline RH, Jung D, et al. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS One (2014) 9:e107265. doi: 10.1371/journal.pone.0107265
134. Moschen AR, Molnar C, Enrich B, Geiger S, Ebenbichler CF, Tilg H. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol Med (2011) 17:840–5. doi: 10.2119/molmed.2010.00108
135. Stanton MC, Chen SC, Jackson JV, Rojas-Triana A, Kinsley D, Cui L, et al. Inflammatory Signals shift from adipose to liver during high fat feeding and influence the development of steatohepatitis in mice. J Inflamm (2011) 8:8. doi: 10.1186/1476-9255-8-8
136. Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol (2011) 55:1086–94. doi: 10.1016/j.jhep.2011.01.048
137. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
138. Tilg H, Effenberger M, Adolph TE. A role for IL-1 inhibitors in the treatment of non-alcoholic fatty liver disease (NAFLD)? Expert Opin Invest Drugs (2020) 29:103–6. doi: 10.1080/13543784.2020.1681397
139. Zhao P, Sun X, Chaggan C, Liao Z, In Wong K, He F, et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science (2020) 367:652–60. doi: 10.1126/science.aay0542
140. Scherer PE. The many secret lives of adipocytes: implications for diabetes. Diabetologia (2019) 62:223–32. doi: 10.1007/s00125-018-4777-x
141. Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. J Mol Cell Biol (2016) 8:101–9. doi: 10.1093/jmcb/mjw014
142. Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol (2005) 166:1655–69. doi: 10.1016/S0002-9440(10)62476-5
143. Dadson K, Liu Y, Sweeney G. Adiponectin action: a combination of endocrine and autocrine/paracrine effects. Front Endocrinol (Lausanne) (2011) 2:62. doi: 10.3389/fendo.2011.00062
144. Nishimoto S, Fukada D, Shimabukuro M, Matsumoto S, Ishida H, Yagi S, et al. Genetic ablation of TLR9 improves insulin resistance through macrophage accumulation in adipose tissue. Amsterdam: European Society of Cardiology (2013). doi: 10.1093/eurheartj/eht309.P3267
145. Revelo XS, Ghazarian M, Chng MH, Luck H, Kim JH, Zeng K, et al. Nucleic Acid-Targeting Pathways Promote Inflammation in Obesity-Related Insulin Resistance. Cell Rep (2016). 16(3):717–30 doi: 10.1016/j.celrep.2016.06.024
146. Hong CP, Yun CH, Lee GW, Park A, Kim YM, Jang MH. TLR9 regulates adipose tissue inflammation and obesity-related metabolic disorders. Obesity (Silver Spring) (2015) 23:2199–206. doi: 10.1002/oby.21215
147. Rose WA2, Sakamoto K, Leifer CA. TLR9 is important for protection against intestinal damage and for intestinal repair. Sci Rep (2012) 2:574. doi: 10.1038/srep00574
148. Ingelsson B, Soderberg D, Strid T, Soderberg A, Bergh AC, Loitto V, et al. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc Natl Acad Sci U S A (2018) 115:E478–87. doi: 10.1073/pnas.1711950115
149. Li T, Zhang Z, Li X, Dong G, Zhang M, Xu Z, et al. Neutrophil Extracellular Traps: Signaling Properties and Disease Relevance. Mediators Inflamm (2020) 2020:9254087. doi: 10.1155/2020/9254087
150. He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology (2017) 66:220–34. doi: 10.1002/hep.29153
151. He Y, Hwang S, Cai Y, Kim SJ, Xu M, Yang D, et al. MicroRNA-223 Ameliorates Nonalcoholic Steatohepatitis and Cancer by Targeting Multiple Inflammatory and Oncogenic Genes in Hepatocytes. Hepatology (2019) 70:1150–67. doi: 10.1002/hep.30645
152. Ye D, Zhang T, Lou G, Liu Y. Role of miR-223 in the pathophysiology of liver diseases. Exp Mol Med (2018) 50:128. doi: 10.1038/s12276-018-0153-7
153. Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, et al. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1beta production in macrophages by targeting STAT3. PLoS One (2012) 7:e42971. doi: 10.1371/journal.pone.0042971
154. Zhang N, Fu L, Bu Y, Yao Y, Wang Y. Downregulated expression of miR-223 promotes Toll-like receptor-activated inflammatory responses in macrophages by targeting RhoB. Mol Immunol (2017) 91:42–8. doi: 10.1016/j.molimm.2017.08.026
155. Ho CM, Ho SL, Jeng YM, Lai YS, Chen YH, Lu SC, et al. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J Inflammation (2019) 16:7. doi: 10.1186/s12950-019-0211-5
156. Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta (2015) 1852:1765–78. doi: 10.1016/j.bbadis.2015.05.015
157. Teratani T, Tomita K, Suzuki T, Furuhashi H, Irie R, Hida S, et al. Free cholesterol accumulation in liver sinusoidal endothelial cells exacerbates acetaminophen hepatotoxicity via TLR9 signaling. J Hepatol (2017) 67:780–90. doi: 10.1016/j.jhep.2017.05.020
158. McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest (2012) 122:1574–83. doi: 10.1172/JCI59755
160. Ivanov S, Dragoi AM, Wang X, Dallacosta C, Louten J, Musco G, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood (2007) 110:1970–81. doi: 10.1182/blood-2006-09-044776
161. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol (2007) 8:487–96. doi: 10.1038/ni1457
162. Rodriguez-Nuevo A, Diaz-Ramos A, Noguera E, Diaz-Saez F, Duran X, Munoz JP, et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J (2018) 37(10):e96553. doi: 10.15252/embj.201796553
163. Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, et al. Gene Expression Predicts Histological Severity and Reveals Distinct Molecular Profiles of Nonalcoholic Fatty Liver Disease. Sci Rep (2019) 9:12541. doi: 10.1038/s41598-019-48746-5
164. Kubarenko AV, Ranjan S, Rautanen A, Mills TC, Wong S, Vannberg F, et al. A naturally occurring variant in human TLR9, P99L, is associated with loss of CpG oligonucleotide responsiveness. J Biol Chem (2010) 285:36486–94. doi: 10.1074/jbc.M110.117200
165. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort(). J Hepatol (2020) 73:505–15. doi: 10.1016/j.jhep.2020.04.003
166. Alegre NS, Garcia CC, Billordo LA, Ameigeiras B, Poncino D, Benavides J, et al. Limited expression of TLR9 on T cells and its functional consequences in patients with nonalcoholic fatty liver disease. Clin Mol Hepatol (2020) 26:216–26. doi: 10.3350/cmh.2019.0074
167. Trujillo G, Meneghin A, Flaherty KR, Sholl LM, Myers JL, Kazerooni EA, et al. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med (2010) 2:57ra82. doi: 10.1126/scitranslmed.3001510
168. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep (2017) 7:44612. doi: 10.1038/srep44612
169. Celhar T, Pereira-Lopes S, Thornhill SI, Lee HY, Dhillon MK, Poidinger M, et al. TLR7 and TLR9 ligands regulate antigen presentation by macrophages. Int Immunol (2016) 28:223–32. doi: 10.1093/intimm/dxv066
170. Freitas-Lopes MA, Mafra K, David BA, Carvalho-Gontijo R, Menezes GB. Differential Location and Distribution of Hepatic Immune Cells. Cells (2017) 6(4):48. doi: 10.3390/cells6040048
171. Gamberi T, Magherini F, Modesti A, Fiaschi T. Adiponectin Signaling Pathways in Liver Diseases. Biomedicines (2018) 6(2):52. doi: 10.3390/biomedicines6020052
172. Lattuada G, Radaelli MG, De Cobelli F, Esposito A, Manzoni G, Perra S, et al. Fasting Whole-Body Energy Homeostasis and Hepatic Energy Metabolism in Nondiabetic Humans with Fatty Liver. Oxid Med Cell Longev (2019) 2019:9796175. doi: 10.1155/2019/9796175
173. Drinda S, Grundler F, Neumann T, Lehmann T, Steckhan N, Michalsen A, et al. Effects of Periodic Fasting on Fatty Liver Index-A Prospective Observational Study. Nutrients (2019) 11(11):2601. doi: 10.3390/nu11112601
174. Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, et al. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology (2009) 50:1809–17. doi: 10.1002/hep.23228
175. Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol (2010) 31:154–63. doi: 10.1016/j.it.2010.01.002
176. De Nardo D, De Nardo CM, Nguyen T, Hamilton JA, Scholz GM. Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J Immunol (2009) 183:8110–8. doi: 10.4049/jimmunol.0901031
177. Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (2010) 7:15. doi: 10.1186/1476-9255-7-15
178. Ozkul C, Yalinay M, Karakan T, Yilmaz G. Determination of certain bacterial groups in gut microbiota and endotoxin levels in patients with nonalcoholic steatohepatitis. Turk J Gastroenterol (2017) 28:361–9. doi: 10.5152/tjg.2017.17033
179. Sookoian S, Salatino A, Castano GO, Landa MS, Fijalkowky C, Garaycoechea M, et al. Intrahepatic bacterial metataxonomic signature in non-alcoholic fatty liver disease. Gut (2020) 69:1483–91. doi: 10.1136/gutjnl-2019-318811
180. Molgora M, Barajon I, Mantovani A, Garlanda C. Regulatory Role of IL-1R8 in Immunity and Disease. Front Immunol (2016) 7:149. doi: 10.3389/fimmu.2016.00149
181. Yuan J, Baker SS, Liu W, Alkhouri R, Baker RD, Xie J, et al. Endotoxemia unrequired in the pathogenesis of pediatric nonalcoholic steatohepatitis. J Gastroenterol Hepatol (2014) 29:1292–8. doi: 10.1111/jgh.12510
182. Pang J, Xu W, Zhang X, Wong GL, Chan AW, Chan HY, et al. Significant positive association of endotoxemia with histological severity in 237 patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther (2017) 46:175–82. doi: 10.1111/apt.14119
183. du Plessis J, Korf H, van Pelt J, Windmolders P, Vander Elst I, Verrijken A, et al. Pro-Inflammatory Cytokines but Not Endotoxin-Related Parameters Associate with Disease Severity in Patients with NAFLD. PLoS One (2016) 11:e0166048. doi: 10.1371/journal.pone.0166048
184. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature (2017) 542:177–85. doi: 10.1038/nature21363
185. Kim AY, Park YJ, Pan X, Shin KC, Kwak SH, Bassas AF, et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat Commun (2015) 6:7585. doi: 10.1038/ncomms8585
186. Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol (2017) 13:36–49. doi: 10.1038/nrendo.2016.135
187. Christofides A, Konstantinidou E, Jani C, Boussiotis VA. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism (2020) 114:154338. doi: 10.1016/j.metabol.2020.154338
188. Motawi TK, Shaker OG, Ismail MF, Sayed NH. Peroxisome Proliferator-Activated Receptor Gamma in Obesity and Colorectal Cancer: the Role of Epigenetics. Sci Rep (2017) 7:10714. doi: 10.1038/s41598-017-11180-6
189. Fujiki K, Kano F, Shiota K, Murata M. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol (2009) 7:38. doi: 10.1186/1741-7007-7-38
190. Macias-Gonzalez M, Cardona F, Queipo-Ortuno M, Bernal R, Martin M, Tinahones FJ. PPARgamma mRNA expression is reduced in peripheral blood mononuclear cells after fat overload in patients with metabolic syndrome. J Nutr (2008) 138:903–7. doi: 10.1093/jn/138.5.903
191. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol (2015) 63:164–73. doi: 10.1016/j.jhep.2015.02.019
192. Diradourian C, Girard J, Pegorier JP. Phosphorylation of PPARs: from molecular characterization to physiological relevance. Biochimie (2005) 87:33–8. doi: 10.1016/j.biochi.2004.11.010
193. Sugii S, Evans RM. Epigenetic codes of PPARgamma in metabolic disease. FEBS Lett (2011) 585:2121–8. doi: 10.1016/j.febslet.2011.05.007
194. Garcia-Fuentes E, Murri M, Garrido-Sanchez L, Garcia-Serrano S, Garcia-Almeida JM, Moreno-Santos I, et al. PPARgamma expression after a high-fat meal is associated with plasma superoxide dismutase activity in morbidly obese persons. Obesity (Silver Spring) (2010) 18:952–8. doi: 10.1038/oby.2009.314
195. Aprile M, Cataldi S, Ambrosio MR, D’Esposito V, Lim K, Dietrich A, et al. PPARgammaDelta5, a Naturally Occurring Dominant-Negative Splice Isoform, Impairs PPARgamma Function and Adipocyte Differentiation. Cell Rep (2018) 25:1577–92.e6. doi: 10.1016/j.celrep.2018.10.035
196. Ju J, Huang Q, Sun J, Zhao X, Guo X, Jin Y, et al. Correlation between PPAR-alpha methylation level in peripheral blood and atherosclerosis of NAFLD patients with DM. Exp Ther Med (2018) 15:2727–30. doi: 10.3892/etm.2018.5730
197. Appel S, Mirakaj V, Bringmann A, Weck MM, Grunebach F, Brossart P. PPAR-gamma agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-kappaB pathways. Blood (2005) 106:3888–94. doi: 10.1182/blood-2004-12-4709
198. Dana N, Vaseghi G, Haghjooy Javanmard S. Crosstalk between Peroxisome Proliferator-Activated Receptors and Toll-Like Receptors: A Systematic Review. Adv Pharm Bull (2019) 9:12–21. doi: 10.15171/apb.2019.003
199. Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell (2005) 122:707–21. doi: 10.1016/j.cell.2005.06.029
200. Targher G, Bertolini L, Rodella S, Zoppini G, Zenari L, Falezza G. Associations between liver histology and cortisol secretion in subjects with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf) (2006) 64:337–41. doi: 10.1111/j.1365-2265.2006.02466.x
201. Lonardo A, Carani C, Carulli N, Loria P. ‘Endocrine NAFLD’ a hormonocentric perspective of nonalcoholic fatty liver disease pathogenesis. J Hepatol (2006) 44:1196–207. doi: 10.1016/j.jhep.2006.03.005
202. Mueller KM, Kornfeld JW, Friedbichler K, Blaas L, Egger G, Esterbauer H, et al. Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology (2011) 54:1398–409. doi: 10.1002/hep.24509
203. Koorneef LL, van den Heuvel JK, Kroon J, Boon MR, t Hoen PAC, Hettne KM, et al. Selective Glucocorticoid Receptor Modulation Prevents and Reverses Nonalcoholic Fatty Liver Disease in Male Mice. Endocrinology (2018) 159:3925–36. doi: 10.1210/en.2018-00671
204. Necela BM, Su W, Thompson EA. Toll-like receptor 4 mediates cross-talk between peroxisome proliferator-activated receptor gamma and nuclear factor-kappaB in macrophages. Immunology (2008) 125:344–58. doi: 10.1111/j.1365-2567.2008.02849.x
205. Jarrar MH, Baranova A, Collantes R, Ranard B, Stepanova M, Bennett C, et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol Ther (2008) 27:412–21. doi: 10.1111/j.1365-2036.2007.03586.x
206. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19:121–35. doi: 10.1038/nrm.2017.95
207. Yavari A, Stocker CJ, Ghaffari S, Wargent ET, Steeples V, Czibik G, et al. Chronic Activation of gamma2 AMPK Induces Obesity and Reduces beta Cell Function. Cell Metab (2016) 23:821–36. doi: 10.1016/j.cmet.2016.04.003
208. Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
209. Wirth M, Joachim J, Tooze SA. Autophagosome formation–the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol (2013) 23:301–9. doi: 10.1016/j.semcancer.2013.05.007
210. Nemazanyy I, Montagnac G, Russell RC, Morzyglod L, Burnol AF, Guan KL, et al. Class III PI3K regulates organismal glucose homeostasis by providing negative feedback on hepatic insulin signalling. Nat Commun (2015) 6:8283. doi: 10.1038/ncomms9283
211. Hurley JH, Young LN. Mechanisms of Autophagy Initiation. Annu Rev Biochem (2017) 86:225–44. doi: 10.1146/annurev-biochem-061516-044820
212. Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell (2013) 152:290–303. doi: 10.1016/j.cell.2012.12.016
213. Kodiha M, Rassi JG, Brown CM, Stochaj U. Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK–>ERK1/2 pathway. Am J Physiol Cell Physiol (2007) 293:C1427–36. doi: 10.1152/ajpcell.00176.2007
214. Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem (2006) 281:18933–41. doi: 10.1074/jbc.M512831200
215. Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, et al. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol (2010) 45:276–95. doi: 10.3109/10409238.2010.488215
216. Nunez CE, Rodrigues VS, Gomes FS, Moura RF, Victorio SC, Bombassaro B, et al. Defective regulation of adipose tissue autophagy in obesity. Int J Obes (Lond) (2013) 37:1473–80. doi: 10.1038/ijo.2013.27
217. Zhao Y, Wang Q, Qiu G, Zhou S, Jing Z, Wang J, et al. RACK1 Promotes Autophagy by Enhancing the Atg14L-Beclin 1-Vps34-Vps15 Complex Formation upon Phosphorylation by AMPK. Cell Rep (2015) 13:1407–17. doi: 10.1016/j.celrep.2015.10.011
218. Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, et al. The Chaperone UNC93B1 Regulates Toll-like Receptor Stability Independently of Endosomal TLR Transport. Immunity (2018) 48:911–22.e7. doi: 10.1016/j.immuni.2018.04.011
219. Fang H, Judd RL. Adiponectin Regulation and Function. Compr Physiol (2018) 8:1031–63. doi: 10.1002/cphy.c170046
220. Astapova O, Leff T. Adiponectin and PPARgamma: cooperative and interdependent actions of two key regulators of metabolism. Vitam Horm (2012) 90:143–62. doi: 10.1016/B978-0-12-398313-8.00006-3
221. Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun (2004) 323:630–5. doi: 10.1016/j.bbrc.2004.08.145
222. Trujillo ME, Scherer PE. Adiponectin–journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med (2005) 257:167–75. doi: 10.1111/j.1365-2796.2004.01426.x
223. Wang ZV, Scherer PE. Adiponectin, the past two decades. J Mol Cell Biol (2016) 8:93–100. doi: 10.1093/jmcb/mjw011
224. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem (1995) 270:26746–9. doi: 10.1074/jbc.270.45.26746
225. Aygun C, Senturk O, Hulagu S, Uraz S, Celebi A, Konduk T, et al. Serum levels of hepatoprotective peptide adiponectin in non-alcoholic fatty liver disease. Eur J Gastroenterol Hepatol (2006) 18:175–80. doi: 10.1097/00042737-200602000-00010
226. Arvaniti VA, Thomopoulos KC, Tsamandas A, Makri M, Psyrogiannis A, Vafiadis G, et al. Serum adiponectin levels in different types of non alcoholic liver disease. Correlation Steatosis Necroinflammation Fibrosis Acta Gastroenterol Belg (2008) 71:355–60.
227. Jiang LL, Li L, Hong XF, Li YM, Zhang BL. Patients with nonalcoholic fatty liver disease display increased serum resistin levels and decreased adiponectin levels. Eur J Gastroenterol Hepatol (2009) 21:662–6. doi: 10.1097/MEG.0b013e328317f4b5
228. Lebensztejn DM, Wojtkowska M, Skiba E, Werpachowska I, Tobolczyk J, Kaczmarski M. Serum concentration of adiponectin, leptin and resistin in obese children with non-alcoholic fatty liver disease. Adv Med Sci (2009) 54:177–82. doi: 10.2478/v10039-009-0047-y
229. Polyzos SA, Toulis KA, Goulis DG, Zavos C, Kountouras J. Serum total adiponectin in nonalcoholic fatty liver disease: a systematic review and meta-analysis. Metabolism (2011) 60:313–26. doi: 10.1016/j.metabol.2010.09.003
230. Yamaguchi N, Argueta JG, Masuhiro Y, Kagishita M, Nonaka K, Saito T, et al. Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett (2005) 579:6821–6. doi: 10.1016/j.febslet.2005.11.019
231. Ahl S, Guenther M, Zhao S, James R, Marks J, Szabo A, et al. Adiponectin Levels Differentiate Metabolically Healthy vs Unhealthy Among Obese and Nonobese White Individuals. J Clin Endocrinol Metab (2015) 100:4172–80. doi: 10.1210/jc.2015-2765
232. Kim YS, Lee SH, Park SG, Won BY, Chun H, Cho DY, et al. Low levels of total and high-molecular-weight adiponectin may predict non-alcoholic fatty liver in Korean adults. Metabolism (2020) 103:154026. doi: 10.1016/j.metabol.2019.154026
233. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol (2005) 25:2062–8. doi: 10.1161/01.ATV.0000183883.72263.13
234. Weng SY, Schuppan D. AMPK regulates macrophage polarization in adipose tissue inflammation and NASH. J Hepatol (2013) 58:619–21. doi: 10.1016/j.jhep.2012.09.031
235. Bao W, Xia H, Liang Y, Ye Y, Lu Y, Xu X, et al. Toll-like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci Rep (2016) 6:22579. doi: 10.1038/srep22579
236. Lonardo A, Ballestri S, Marchesini G, Angulo P, Loria P. Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig Liver Dis (2015) 47:181–90. doi: 10.1016/j.dld.2014.09.020
237. Hirsova P, Gores GJ. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol (2015) 1:17–27. doi: 10.1016/j.jcmgh.2014.11.005
238. Guicciardi ME, Nakao Y, Gores GJ. The Metabolic Sensor AMKP Regulates Apoptosis in NASH. Hepatology (2020). 72(3):1139–41. doi: 10.1002/hep.31294
239. Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology (2007) 46:1509–18. doi: 10.1002/hep.21867
240. Shafiei MS, Shetty S, Scherer PE, Rockey DC. Adiponectin regulation of stellate cell activation via PPARgamma-dependent and -independent mechanisms. Am J Pathol (2011) 178:2690–9. doi: 10.1016/j.ajpath.2011.02.035
241. Gramlich T, Kleiner DE, McCullough AJ, Matteoni CA, Boparai N, Younossi ZM. Pathologic features associated with fibrosis in nonalcoholic fatty liver disease. Hum Pathol (2004) 35:196–9. doi: 10.1016/j.humpath.2003.09.018
242. Gao W, Xiong Y, Li Q, Yang H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front Physiol (2017) 8:508. doi: 10.3389/fphys.2017.00508
243. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N Engl J Med (2020). doi: 10.1056/NEJMoa2028395
244. Zarei M, Pizarro-Delgado J, Barroso E, Palomer X, Vazquez-Carrera M. Targeting FGF21 for the Treatment of Nonalcoholic Steatohepatitis. Trends Pharmacol Sci (2020) 41:199–208. doi: 10.1016/j.tips.2019.12.005
245. Qureshi K, Neuschwander-Tetri BA. The molecular basis for current targets of NASH therapies. Expert Opin Invest Drugs (2020) 29:151–61. doi: 10.1080/13543784.2020.1703949
246. Nielsen J. Systems Biology of Metabolism: A Driver for Developing Personalized and Precision Medicine. Cell Metab (2017) 25:572–9. doi: 10.1016/j.cmet.2017.02.002
247. Fleck LM. Personalized medicine’s ragged edge. Hast Cent Rep (2010) 40:16–8. doi: 10.1353/hcr.2010.0005
248. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
249. Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab (2016) 311:E730–40. doi: 10.1152/ajpendo.00225.2016
250. Issa D, Wattacheril J, Sanyal AJ. Treatment options for nonalcoholic steatohepatitis - a safety evaluation. Expert Opin Drug Saf (2017) 16:903–13. doi: 10.1080/14740338.2017.1343299
251. Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science (2017) 357:507–11. doi: 10.1126/science.aah5582
252. Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev (2015) 87:46–51. doi: 10.1016/j.addr.2015.01.008
253. Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in nonalcoholic fatty liver disease. Metabolism (2016) 65:1062–79. doi: 10.1016/j.metabol.2015.11.006
254. Combs TP, Wagner JA, Berger J, Doebber T, Wang WJ, Zhang BB, et al. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology (2002) 143:998–1007. doi: 10.1210/endo.143.3.8662
255. Wagner JA. Overview of biomarkers and surrogate endpoints in drug development. Dis Markers (2002) 18:41–6. doi: 10.1155/2002/929274
256. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med (2016) 165:305–15. doi: 10.7326/M15-1774
Keywords: non-alcoholic fatty liver disease, fibrosis, CpG DNA, mitochondrial DNA, PPAR, AMPK, adiponectin, NF-kappa B
Citation: Shepard CR (2021) TLR9 in MAFLD and NASH: At the Intersection of Inflammation and Metabolism. Front. Endocrinol. 11:613639. doi: 10.3389/fendo.2020.613639
Received: 02 October 2020; Accepted: 10 December 2020;
Published: 29 January 2021.
Edited by:
Claire Joanne Stocker, Aston University, United KingdomReviewed by:
Sergei Gryaznov, Maia Biotechnology Inc, United StatesCopyright © 2021 Shepard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher R. Shepard, Y3NoZXBhcmRAYXZvYmlvLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.