 Erin M. MacFarlane
Erin M. MacFarlane Jennifer E. Bruin
Jennifer E. Bruin- Department of Biology & Institute of Biochemistry, Carleton University, Ottawa, ON, Canada
Diabetes prevalence is increasing worldwide, and epidemiological studies report an association between diabetes incidence and environmental pollutant exposure. There are >84,000 chemicals in commerce, many of which are released into the environment without a clear understanding of potential adverse health consequences. While in vivo rodent studies remain an important tool for testing chemical toxicity systemically, we urgently need high-throughput screening platforms in biologically relevant models to efficiently prioritize chemicals for in depth toxicity analysis. Given the increasing global burden of obesity and diabetes, identifying chemicals that disrupt metabolism should be a high priority. Pancreatic endocrine cells are key regulators of systemic metabolism, yet often overlooked as a target tissue in toxicology studies. Immortalized β-cell lines and primary human, porcine, and rodent islets are widely used for studying the endocrine pancreas in vitro, but each have important limitations in terms of scalability, lifespan, and/or biological relevance. Human pluripotent stem cell (hPSC) culture is a powerful tool for in vitro toxicity testing that addresses many of the limitations with other β-cell models. Current in vitro differentiation protocols can efficiently generate glucose-responsive insulin-secreting β-like cells that are not fully mature, but still valuable for high-throughput toxicity screening in vitro. Furthermore, hPSCs can be applied as a model of developing pancreatic endocrine cells to screen for chemicals that influence endocrine cell formation during critical windows of differentiation. Given their versatility, we recommend using hPSCs to identify potential β-cell toxins, which can then be prioritized as chemicals of concern for metabolic disruption.
Introduction
Diabetes Pathogenesis
Diabetes is a chronic disease characterized by high blood sugar levels and devastating secondary health complications (1). In 2019, there were >460 million people with diabetes worldwide, which translates to roughly 1 in 11 adults ages 20-79 years. For those over the age of 65, diabetes rates further increase to 1 in 5 (1). Moreover, the International Diabetes Federation projects that diabetes incidence will increase by 51% over the next 25 years to exceed 700 million adults worldwide.
Glucose homeostasis is maintained by the exquisite balance of hormones secreted from pancreatic islets. The predominant islet cell type is the β-cell, which secretes insulin in a tightly regulated manner in response to glucose and other stimuli (2, 3). Type 1 diabetes (T1D), accounting for ~10% of patients with diabetes, is caused by autoimmune destruction of β-cells leading to insufficient insulin production. Type 2 diabetes (T2D) accounts for ~90% of cases and was classically thought of as a disease of insulin resistance. However, we now appreciate that β-cell dysfunction and loss of β-cell mass are also central to T2D pathogenesis (4–6). The critical role of β-cells in driving diabetes risk is further confirmed by genome-wide association studies, which find that most loci influencing T2D risk are involved in regulating insulin secretion (7–9). These studies also emphasize that rising diabetes rates cannot simply be explained by genetics, but rather must be influenced by environmental factors (9, 10). For example, there is strong epidemiological evidence linking exposure to persistent organic pollutants (POPs) with increased T2D incidence (11–29) and β-cell dysfunction (28–30) in humans. However, basic research in clinically relevant models is needed to understand the potential causal role for environmental contaminants in diabetes pathogenesis and to explore underlying tissue- and cell-specific mechanisms of toxicity.
Environmental Contaminants
Environmental pollutants are a major global concern due to their wide-ranging acute and chronic adverse effects on human health (31). With over 84,000 chemicals in commerce, there is an urgent need to develop tools for extensive chemical screening and toxicity testing (32). Environmental contaminants fall within a wide range of classes, including but not limited to POPs (e.g., pesticides, polychlorinated biphenyls (PCBs), dioxin-like compounds), estrogen analogues (e.g., bisphenol A (BPA), used in polycarbonate plastics), phthalates (used in cosmetics, paints, textiles), heavy metals, perfluorinated chemicals (e.g., perfluorooctane sulfonate (PFOS) used in food packaging and fire-fighting foams), and flame retardants (e.g., polybrominated diphenyl ethers, organohalogen compounds, organophosphates esters) (33–35). Contaminants can be further classified by their mechanism of action. For example, chemicals that impair proper hormone function are referred to as endocrine-disrupting chemicals (EDCs) (33, 36–40) and those that disrupt metabolism are classified as metabolism-disrupting chemicals (MDCs) (41–43). Despite restrictions on many environmental pollutants, these chemicals continue to persist in the environment, contaminating food and water sources, and remain detectable in human tissues (44, 45).
Biomonitoring is essential for tracking human contaminant exposure and predicting adverse health outcomes (46, 47). However, this is a reactive approach to evaluating the impact of toxins on human health. Ideally, we need to efficiently screen chemicals for toxicity in relevant model systems prior to their release into the environment. Since pollutants often accumulate in tissues, effectively creating a chemical mixture cocktail (48), we also need to consider the combined effects of complex chemical mixtures. Dose and duration of exposure add additional layers of complexity. For example, POPs have long half-lives of years to decades (49), but the shorter lifespan of other chemicals such as BPA and phthalates is also not trivial. Much like hormones, EDCs can exert their effects on the human body at low concentrations over an extended period of time (50). Despite being excreted within days, frequent consumption of these pollutants results in chronic, low dose exposure over time (51, 52). Furthermore, nonlinear dose-responses are frequently seen with EDCs, so acute high dose studies may not accurately predict adverse health outcomes of chronic or subacute low dose exposures (42, 53, 54). The need to consider chemical exposures ranging from acute high doses to chronic low doses, as well as individual chemicals and complex mixtures, further emphasizes the importance of scaling up toxicity testing capacity.
Developmental Origins of Disease
Another important consideration for toxicology studies is the timing of exposure to environmental contaminants. Gestational or early life stressors, such as undernutrition or overnutrition, are linked to a variety of adult-onset diseases - termed developmental origins of health and disease (DOHaD) (55, 56). For instance, low birth weight and early life “catch up” growth are well-established risk factors for developing metabolic disease later in life (57, 58). Maternal-fetal exposure to POPs has been linked to adverse outcomes such as reduced birth weight, disruption of hormone levels in cord blood, and changes in epigenetic markers of development (59–61). There is also mounting epidemiological evidence suggesting a possible link between early-life environmental contaminant exposure and long-term metabolic dysfunction (62–66). More epidemiology is needed and important cohort studies like the Maternal-Infant Research on Environmental Chemicals (MIREC) continue to track long-term metabolic outcomes in offspring (67–70). However, it takes decades to truly establish a link between early-life exposure and long-term adverse health outcomes. In vitro model systems that allow for toxicity screening in developing human cells will be a powerful starting point for studying DOHaD.
Perspective Overview
There is an urgent need to identify environmental contaminants, specifically EDCs or MDCs, that contribute to diabetes pathogenesis. To do so, we must consider non-classical toxicological endpoints in a wide variety of tissues involved in regulating metabolic homeostasis. This means thinking beyond typical hepatoxicity endpoints and considering diverse metabolic targets such as neuroendocrine cells, enteroendocrine cells, white or brown adipocytes, skeletal muscle, thyroid gland, and pancreatic endocrine cells (38, 40, 71). While injury to any of these tissues would potentially disrupt energy homeostasis, we propose that pancreatic endocrine cells should be a high priority for toxicity testing to identify MDCs of concern for diabetes pathogenesis. In this Perspective Article, we discuss a range of endpoints that could be considered in the context of β-cell toxicity. We also discuss various model systems available for toxicity testing, including the numerous advantages of human pluripotent stem cells (hPSCs). In particular, we propose hPSCs as a unique model system for evaluating toxicity both during critical windows of β-cell development and in glucose-responsive adult β-like cells (Figure 1).
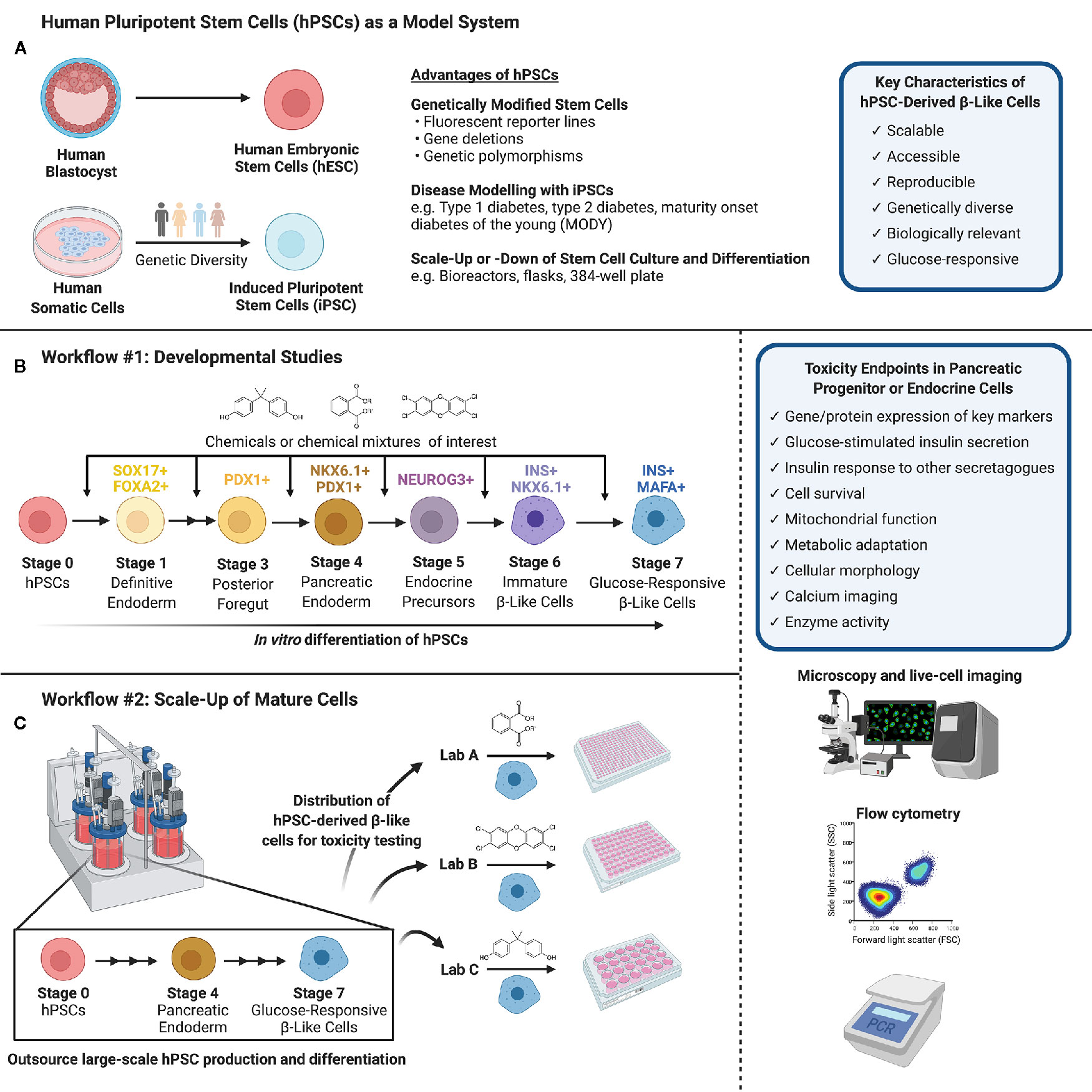
Figure 1 (A) Human pluripotent stem cells (hPSCs) can be isolated from the inner cell mass of a human blastocyst (human embryonic stem cells, hESCs) or obtained via reprogramming of human somatic cells obtained from genetically diverse donors (induced pluripotent stem cells, iPSCs). hPSCs are versatile in their capacity for genetic modifications and disease modeling and may be scaled up or down to suit a variety of experimental conditions. (B) Workflow #1 illustrates how hPSCs may be used to screen chemicals or chemical mixtures of interest throughout pancreas development. hPSCs can be differentiated into pancreatic endoderm using published protocols or commercially available differentiation kits, and further into maturing, glucose-responsive β-like cells. Chemicals can be introduced at different days or stages of differentiation to mimic environmental exposures at different windows of pancreas development. (C) Workflow #2 demonstrates the capacity to outsource hPSC expansion and large-scale differentiation, allowing individual labs to conduct toxicity screening of specific chemicals/chemical mixtures using glucose-responsive β-like cells generated in a central location. (B, C) We suggest a number of potential toxicity endpoints, such as cell survival, insulin secretion, and mitochondrial function. Common analytical methods include but are not limited to microscopy and live-cell imaging, flow cytometry to quantify cell populations throughout differentiation, and PCR to assess gene expression (Created with BioRender.com).
Toxicity Testing in Pancreatic β-Cells
Despite mounting evidence implicating pollutants as metabolic disruptors, the pancreas has not been extensively studied in the toxicology field (40, 42). Interestingly, the occasional biodistribution studies that include pancreas tissue report a slower elimination of lipophilic pollutants in the pancreas compared to liver or adipose (72, 73). Xenobiotic metabolism enzymes, such as cytochrome P450 (Cyp) enzymes, are useful biomarkers for direct cellular exposure to pollutants. We have reported induction of Cyp1a1 in mouse and human islets following direct exposure to TCDD/dioxin or dioxin-like pollutants in vitro and in mouse islets following systemic administration of TCDD in vivo (73). Moreover, in pregnant TCDD-exposed mice, Cyp1a1 was induced 17-fold in pancreas compared to only 3-fold and 7-fold in liver and adipose, respectively (74). Therefore, pancreatic cells are not only directly exposed to pollutants in vivo, but may even act as a “sink” for long-term storage of lipophilic chemicals, similar to adipose depots.
There is mounting evidence that a wide range of environmental contaminants can directly impact β-cell function. For example, BPA, a non-persistent additive commonly used in plastic products, acutely increases insulin secretion in mouse and human islets via inhibition of KATP channels and increased Ca2+ signaling (75), whereas longer-term BPA exposure inhibits Ca2+ entry and reduces insulin secretion (76). Newer BPA-replacement chemicals, BPS and BPF, also disrupt mouse β-cell function (77). Exposure to POPs, including organochlorine pesticides and a PCB mixture, directly inhibited insulin secretion in a rat β-cell line (INS-1E cells) (30). A “northern contaminant mixture”, containing 20 different POPs at environmentally relevant concentrations, also suppressed insulin secretion in rats in vivo and in a rodent β-cell line (MIN6 cells) in vitro (78). Additionally, we and others have shown that dioxin suppresses insulin secretion in rodent islets (73, 79, 80) and human islets (73). Interestingly, acute high-dose dioxin exposure caused β-cell apoptosis in male mice but not female mice (81), whereas prolonged low-dose dioxin exposure led to impaired β-cell adaptation to high fat diet feeding in female but not male mice (74, 82).
Given the critical role for β-cells in diabetes pathogenesis, environmental toxins that adversely impact β-cells are likely to disrupt overall glucose homeostasis or at minimum, increase diabetes risk. Importantly, there are numerous plausible avenues for toxin-induced β-cell injury that could lead to adverse metabolic health outcomes. Below, we propose key toxicity endpoints for β-cells. This is not intended to be a comprehensive list of potential adverse outcomes, but rather examples that should be considered as a starting point for identifying MDCs that act as β-cell toxins.
Insulin Secretory Defects
Pancreatic β-cells are highly specialized to synthesize, process, store, and secrete insulin rapidly and sustainably in response to numerous physiological stimuli, including glucose (2). There is a rapid first phase of insulin secretion within minutes of a glucose stimulus, followed by a sustained second phase that can last for several hours. Glucose-stimulated insulin secretion is amplified by other nutrients, such as fatty acids and amino acids, and binding of gut-derived hormones (GLP-1, GIP) to incretin receptors. Dysregulated glucose-induced insulin secretion is a well-established feature of T2D (83–87) and also reported in T1D patients prior to disease onset (88–92), suggesting a possible link between insulin secretory defects and activation of inappropriate autoimmune responses. Therefore, environmental contaminants that interfere with any aspect of the complex insulin secretory pathway (e.g., glucose sensing, mitochondrial metabolism, ion flux, exocytotic machinery, sensitivity to amplification signals) could adversely affect the fine-tuned ability of β-cells to couple insulin secretion with a nutrient secretagogue. Furthermore, defects in the timing of insulin release, either the rapid first-phase or the sustained second-phase response, could impact overall glycemic control. Importantly, β-cell dysfunction is not just insufficient or decreased insulin secretion, but also refers to overproduction of insulin. Hyperinsulinemia is not only an adaptation to insulin resistance but can also be the primary defect that drives obesity and insulin resistance (93–96). Therefore, toxins that increase insulin secretion – either inappropriate insulin release under basal glucose conditions or excessive insulin secretion following a nutrient stimulus – should also be considered potential MDCs.
Loss of β-Cell Mass
Patients with T1D display near complete loss of β-cell mass (> 80% reduction) at the time of disease onset (6). Although less extreme, individuals with T2D also have reduced β-cell mass (5, 6, 97, 98), which may be caused by a combination of increased β-cell death, insufficient β-cell proliferation, or impaired β-cell neogenesis. β-cell mass is generally increased in overweight or obese non-diabetic subjects compared to lean controls but reduced by 24%–65% in patients with T2D (5, 6, 99). Loss of β-cell mass can be detected by measuring the α-cell to β-cell ratio, which is consistently higher in patients with T2D (97). Therefore, environmental toxins that disrupt β-cell mass, for example by inducing β-cell apoptosis or preventing β-cell expansion, should be flagged as potential MDCs.
Impaired β-Cell Adaptation
Healthy β-cells have fine-tuned mechanisms for adapting to fluctuations in energy supply and insulin demand to maintain appropriate glucose homeostasis (100). Examples of complex compensatory mechanisms include a) regulation of key β-cell transcription factors that control the β-cell transcriptome, b) altered activity or expression of metabolic enzymes to allow for flexibility in nutrient metabolism, and c) increased β-cell proliferation to expand functional mass of β-cells. Failed compensatory insulin secretion and expansion of β-cell mass during insulin resistance are important predictors of diabetes susceptibility (99, 101, 102). Therefore, toxicology studies should consider the ability of β-cells to appropriately compensate under conditions of fasting or insulin resistance. If only direct cellular toxicity is tested without considering interactions with other metabolic challenges, potential MDCs will be overlooked.
Impaired β-Cell Development
The number of pancreatic progenitors present throughout embryonic development is a critical determinant of β-cell mass and pancreas size in adulthood, unlike the closely related endoderm-derived liver which can fully compensate following partial progenitor cell ablation (103). Therefore, an infant born with reduced β-cell mass may have a compromised ability to adapt to metabolic stressors later in life. Additionally, overproduction of insulin at birth caused by inappropriate β-cell expansion or accelerated maturation could lead to the development of insulin resistance and obesity (as described above) (94–96, 104). Therefore, a starting point for predicting long-term adverse metabolic health outcomes following intrauterine chemical exposure would be screening for chemicals that alter β-cell development. We propose that “developmental MDCs” could be prioritized, in part, based on whether they influence the formation of β-cells – either decreasing or increasing numbers – during critical windows of development.
Traditional Models for Toxicity Testing in Pancreatic Endocrine Cells
In Vivo Rodent Models
In vivo rodent models are an important tool for toxicity testing, but pose a significant technical barrier to high throughput screening (105) and are limited in their ability to predict human outcome. In a largescale study of pharmaceutical toxicity testing, rodents were predictive of human toxicity for only 43% of tested compounds, and demonstrated poor concordance for liver and endocrine toxicity (106). Further, human populations are genetically diverse and exist amongst variable exogenous factors, whereas laboratory animals are genetically uniform and housed within controlled environments to support reproducibility. While in vivo testing is necessary for assessing the impact of chemicals on a whole organism rather than just a single cell or tissue type in isolation, there has been a shift towards first prioritizing chemicals using in vitro model systems with higher throughput capacity (107, 108).
Immortalized β-Cells
Rodent Cell Lines
Immortalized rodent β-cell lines are robust in culture and highly proliferative, making them a useful tool for large-scale studies. Unfortunately, their replication capacity limits their applicability as a model for human β-cells, which have minimal ability to proliferate (99, 109). In addition, commonly used insulin-secreting rodent β-cell lines, including β-TC-6 (mouse), MIN6 (mouse), and INS-1 (rat) cells, have varying degrees of glucose-responsiveness (110, 111). Immortalized cell lines also tend to be genetically unstable in culture for extended periods of time. Clonal INS-1E cells have higher stability than the INS-1 parental line and maintain their insulin content in passages >40 (112), but both INS-1/INS-1E cells are cultured with the toxic reducing agent, 2-mercaptoethanol, which further limits their biological relevance for toxicity testing.
Despite these limitations, INS-1 823/13 cells were comprehensively evaluated as a potential pollutant-screening system. This cell line was found to be adequately glucose-responsive, but the insulin secretory responses to key control compounds and pollutants deviated substantially from previous reports (113). The authors concluded that INS-1 823/13 cells were lacking key β-cell characteristics and deemed inadequate as a diabetogenic pollutant screening system (113). We reached a similar conclusion after testing immortalized pancreatic endocrine cells for their response to TCDD/dioxin, a POP that acts via the aryl hydrocarbon receptor (AhR). In primary mouse and human islets, TCDD significantly upregulated CYP1A1 gene expression and enzyme activity, whereas immortalized β-cell (INS-1, MIN6, β-TC6) and α-cell (α-TC1 and α-TC3) lines showed no evidence of AhR activation by TCDD (73). Our findings confirm that important discrepancies exist in the cellular machinery between primary and immortalized cell lines.
Human Cell Lines
Novel engineered human β-cell lines, EndoC-βH1 and EndoC-βH2, are a useful tool for studying β-cell physiology and drug responses (114, 115). EndoC-βH1 cells were engineered from human fetal pancreatic buds transduced with an SV40LT-expressing lentiviral vector under the control of an insulin promoter. These cell lines are glucose-responsive in vitro and have minimal expression of other pancreatic genes (114–116). Their main drawback is the limited capacity for expansion in vitro, which is more biologically appropriate than rodent β-cell lines, but less practical. With a doubling time of approximately 7 days, their potential for high-throughput toxicity studies is limited. While proliferation can be stimulated with SerpinA6, STC1, and APOH (114), using compounds that alter cellular physiology is not ideal for toxicity testing. Additionally, as EndoC β-cells are a product of oncogenesis, there is undoubtedly alterations to normal pathways (117).
Primary Isolated Islets
Non-Human Islets
Primary rodent islets are more biologically relevant than immortalized rodent β-cell lines, but there are important distinctions between human and rodent islets that must be considered. For example, the distribution and composition of endocrine cells, vasculature, innervation, proliferation, and insulin secretion all differ between human and rodent islets (118, 119). Pig islets are more similar to human islets (120). Interestingly, islets isolated from juvenile porcine pancreata have greater expression of β-cell-related genes compared to those isolated from adult pigs (121), but are functionally immature and require in vitro maturation following isolation (122–124). Further, it is possible to isolate up to 5,000 islets/g juvenile porcine pancreas (122), compared to a typical yield of ~200–400 islets total per mouse pancreas and 600–800 islets per rat pancreas depending on the strain (125–127). Thus, pig islets may be a useful tool for toxicity screening, although species differences will always remain a concern for translation.
Human Islets
Human islets are currently the gold standard for a physiologically relevant model to study the endocrine pancreas in vitro due to their cellular composition, human origin, and genetic diversity. Human islets are harvested from deceased organ donors and great strides have been made to ensure that high quality donor islets are broadly available for research (128, 129). However, even with the highest quality isolation procedures, the pancreas begins to autodigest after death, resulting in decreased cell viability and sample quality (119). Human islets have a limited functional in vitro lifespan with current tissue culture protocols, although advances in the field are ongoing. For example, islets cultured on specific matrices maintain glucose-stimulated insulin secretion for at least 7 days in culture (130). The number of purified islets per donor also varies; while an average healthy adult pancreas houses over 3 million islets (118), between 200,000 and 500,000 islets typically remain post-purification (131). This, coupled with the limited proliferation of human β-cells, presents a critical barrier to scalability and longevity for toxicology testing. Therefore, human islets are an excellent resource for in vitro chemical testing at a smaller scale, wherein endpoints such as glucose-stimulated insulin secretion, islet morphology, mitochondrial function, and gene expression can be assessed in biologically diverse organ donors. Numerous factors, such as donor sex, age, and body mass index, will influence islet function ex vivo and thus impact biological reproducibility. However, the genetic and environmental diversity of human organ donors (132, 133) also offer a unique opportunity for toxicity testing.
Stem Cells are a Unique Tool for Pancreas Toxicology Studies
HPSC culture offers a unique in vitro solution to address the need for high-throughput screening of environmental toxins in a variety of biologically relevant mature cell types, as well as in differentiating or “developing” immature progenitor cells. HPSCs can be obtained from either the inner cell mass of a human blastocyst, termed human embryonic stem cells (hESCs), or from human somatic cells that have been reprogrammed to a pluripotent state, termed induced pluripotent stem cells (iPSCs) (Figure 1A).
Remarkable progress has been made over the past decade unraveling the developmental cues involved in islet cell formation. We now have robust step-wise differentiation protocols that mimic the key fate decisions for directing hPSCs into pancreatic endocrine cells using small molecules and growth factors in vitro (134–139). These differentiation protocols efficiently guide hPSCs towards pancreatic endoderm cells (PDX1+/NKX6.1+) in four “stages”, followed by commitment to the pancreatic endocrine lineage (NEUROG3+), then insulin-secreting endocrine cells (INS+/NKX6.1+), and finally to β-like cells capable of glucose-induced insulin secretion (INS+/MAFA+/UCN3+) (3) (Figure 1B). The challenge in recent years has been understanding the final stages of human β-cell maturation so we can generate fully mature β-cells with a rapid and robust insulin secretory response to various secretagogues. Despite these limitations, we believe that hPSCs are an excellent tool for studying adverse effects of environmental contaminants both during pancreas development and in adult pancreatic endocrine cells. Indeed, a recent study by Zhou et al. in Nature Communications beautifully demonstrated the diverse and powerful applicability of hPSCs for high-content screening of potential β-cell toxins, exploring gene-environment interactions, and comparing toxicity in diverse cell types (140). The authors differentiated hESCs into INS+ cells in a 384-well plate format and screened a U.S. Environmental Protection Agency (EPA) ToxCast library of ~ 2,000 compounds for “hits” that impaired survival of INS+ cells (140). Using this study as an example, we highlight the numerous benefits of using hPSCs, whether hESCs or iPSCs, for exploring MDC toxicity.
Flexibility to Model Developing or Adult Cells
HPSCs offer a flexible model to test for MDCs that impact either the early formation of β-cells during fetal development or the function and survival of adult insulin-secreting β-like cells. Depending on the research question, we propose two different workflow approaches. For developmental studies (Figure 1B, Workflow #1), environmental toxin(s) can be introduced to differentiating hPSCs at critical days or “stages” of differentiation. Thus, a critical aspect of this workflow is establishing hPSC differentiation protocols within the toxicology lab conducting chemical testing. The impact of toxins can be assessed by measuring key pancreatic cell markers by flow cytometry, image-based analysis, qPCR, or other techniques that are amenable to high throughput analysis. For example, the proportion of cells expressing markers of pancreatic commitment (% PDX1+), pancreatic endoderm (% PDX1+/NKX6.1+), and induction of the endocrine program (% NEUROG3+) are excellent benchmarks for early stages of differentiation. At later stages, the proportion of cells that acquire insulin (% INS+/NKX6.1+) is as an indicator of commitment to the β-cell lineage, and subsequently the proportion of INS+ cells co-expressing critical β-cell markers such as MAFA is an important indicator of β-cell maturity. Fluorescent reporter hPSC lines generated by genome editing – for example, NEUROG3-EGFP or INS-GFP hESCs – will be particularly useful for efficient image-based screening or high-content flow cytometry applications to identify MDCs that disrupt the formation of key pancreatic cell populations (141–144).
The workflow and endpoints for toxicity studies in adult cells could differ considerably from developmental studies (Figure 1C, Workflow #2). First, it is feasible for hPSC-derived β-like cells to be mass-produced in large quantities at a central location to generate a reproducible starting point for toxicology screening studies. Once hPSC-derived β-like cells are validated, they can be distributed to toxicology laboratories for testing of individual chemicals or complex chemical mixtures. This is important because it separates the need for toxicity testing capacity and stem cell differentiation expertise to be housed within the same lab. As with the progenitor cell model, there are numerous potential outcomes that could be assessed in a high-throughput screening platform, such as the expression of key β-cell markers using live-cell imaging or flow cytometry, β-cell survival as in Zhou et al. (140), and basal or glucose-induced insulin secretion. Any of the outcome measures described in the section on “Toxicity Testing in Pancreatic β-cells” could be assessed in hPSC-derived β-like cells, although not necessarily in a high-content format.
It is important to recognize that current differentiation protocols generate human β-like cells with a blunted insulin secretory response to glucose compared to primary human islets (134–139). For the purpose of identifying MDCs that cause β-cell dysfunction or apoptosis, we propose that generating fully mature human β-cells in vitro may not be a necessary milestone. Instead, the benefits of a large-scale source of expandable stem cells that can generate large quantities of moderately glucose-responsive insulin-secreting cells outweighs the downside of working with a slightly immature β-like cell. This has certainly proven true for toxicity studies in other cell types, such as cardiomyocytes, where differentiation protocols currently generate immature cardiomyocytes, but recapitulate sufficient features of adult cells to study adverse drug reactions in specific aspects of cardiotoxicity (145–148).
Scalability and Reproducibility
The scalability of hPSCs is a significant advantage for high-content screening. Importantly, hPSCs share the proliferative advantage of immortalized β-cells, but subsequently lose this capacity as they differentiate into pancreatic lineage cells (139). The highly proliferative nature of hPSCs allows them to be substantially expanded before differentiation, 50–100 fold per week, particularly when grown in suspension format (149). Large batches of hPSCs can then be differentiated into a mass-produced cell product, which can be carefully validated with well-defined QA/QC protocols before being frozen down and distributed for toxicity testing (Figure 1C, Workflow #2). This is similar to the model proposed by the diabetes cell therapy field for mass-production of a GMP-grade cell product for transplantation (3, 149). Alternatively, more modest scale-up approaches can be established within the same lab that will perform toxicity endpoint assessments (Figure 1B, Workflow #1). For example, Zhou and colleagues expanded hESCs in a more traditional adherent format with Matrigel-coated plates before seeding dissociated cells into 384-well plates for pancreatic differentiation and chemical screening (140). The incredible flexibility to both scale-up hPSC production and differentiation or to miniaturize pancreatic differentiation is an important benefit of using hPSCs for toxicity studies.
Unique Capacity for Disease Modeling
Stem cells offer remarkable capacity for disease modeling through both the natural genetic diversity of iPSCs (150) and the ability to create isogenic hPSC lines using genome editing (151). The use of hPSCs for disease modeling in diabetes has been reviewed elsewhere (152), but here, we briefly discuss the benefits of toxicity testing in human β-cells with diverse genetic backgrounds. There is much to be learned from comparing the impact of environmental contaminants on β-like cells generated using iPSCs from a spectrum of patients with different types of diabetes (T1D, T2D, maturity onset diabetes of the young (MODY), or neonatal diabetes) or known genetic risk factors for diabetes (153–160), relative to iPSCs derived from control subjects. One particularly exciting avenue to explore in the context of T1D is how environmental toxins influence immune interactions between iPSC-derived β-cells and autologous immune cells from the same donor (159). Zhou and colleagues also demonstrated the potential for using iPSCs to explore mechanisms of toxicity (140). They used 10 different iPSC lines with heterogeneous expression of a phase 2 xenobiotic metabolism enzyme, GSTT1, and found that pesticide-induced INS+ cell death was significantly higher in lines lacking GSTT1 compared to those with at least one copy of GSTT1 (140). Their results were also validated in isogenic hESC lines with GSTT1 deletion by CRISPR-based genome editing; INS+ cells generated from GSTT1-/- hESCs were more susceptible to pesticide-induced cell death than INS+ cells from wildtype hESCs. Importantly, with the advent of CRISPR-Cas9 technology, modifying the genome of hPSCs has become broadly accessible and the number of gene-edited hPSC lines that effectively recapitulate different aspects of diabetes-related phenotypes is increasing rapidly (140, 151, 155, 161–164).
One final consideration for disease modeling is that despite being reprogrammed back to their embryonic/pluripotent state, iPSCs retain DNA methylation marks, lineage bias, and other memory of previous environmental exposures (165). For this reason, there is a strong argument for developmental models of pancreatic toxicity being limited to hESCs rather than iPSCs. On the other hand, the genetic variability of iPSCs, combined with the ability to create targeted genome-edited hPSC lines with isogenic wildtype controls, should be harnessed to explore the biological diversity of gene-environment interactions in adult β-like cells.
Diversity of Human Cell Types
Another unique advantage of hPSCs is their ability to be directed into diverse cell types. For example, the toxicology field is already using hPSCs to test for adverse drug reactions in iPSC-derived hepatocyte-like cells to model hepatoxicity (166–168) and iPSC-derived cardiomyocytes to model cardiotoxicity (145–147, 168–172). While our Perspective focused on the application of hPSCs for toxicity testing in pancreatic lineage cells specifically, there is immense value in a more integrated approach to screen for MDCs that adversely impact different metabolic target tissues, all derived from the same hPSC source. For example, Zhou and colleagues differentiated hESCs into CD29+/CD73+ mesenchymal stem cells, CTNT+ cardiomyocytes, A1AT+ hepatocytes, and HuC/D+ neurons (140). They found that much like hESC-derived INS+ cells, HuC/D+ neurons were also highly susceptible to pesticide-induced cell death, suggesting that the pesticide flagged in their high-content screening could be involved in the pathogenesis of both diabetes and Parkinson’s disease. An even more complex, but intriguing application of hPSCs is the potential to develop multi-organ systems in a microfluidic device (173) or other platform containing numerous hPSC-derived metabolic tissues such as liver, adipose, and β-cells to determine how environmental contaminants influence metabolic tissue cross-talk.
Conclusion
Despite the critical importance of pancreatic endocrine cells for maintaining metabolic homeostasis, the pancreas has not traditionally been studied as a key target tissue of chemical toxicity. Given the metabolic-disrupting nature of many environmental pollutants, we propose that islet toxicity should be considered a key toxicological endpoint. With the staggering number of poorly studied chemicals in commerce, physiologically relevant models that can be scaled up for efficient chemical screening are urgently needed. Human stem cells offer a unique solution to many of the limitations posed by other in vitro model systems of pancreatic endocrine cells. Most importantly, hPSCs are scalable and amenable to high-throughput screening for assessing the impact of environmental contaminants on either adult β-like cells or critical windows of pancreas development.
Author Contributions
EM and JB conceived the review topic and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by a Canadian Institutes of Health Research (CIHR) Project Grant (#PJT-2018-159590).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Myriam Hoyeck for her valuable insight during the review of this manuscript.
References
2. Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol (2013) 75:155–79. doi: 10.1146/annurev-physiol-030212-183754
3. Bruin JE, Rezania A, Kieffer TJ. Replacing and safeguarding pancreatic beta cells for diabetes. Sci Transl Med (2015) 7:316ps23. doi: 10.1126/scitranslmed.aaa9359
4. Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia (2009) 52:1003–12. doi: 10.1007/s00125-009-1321-z
5. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
6. Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab (2017) 6:943–57. doi: 10.1016/j.molmet.2017.06.019
7. Gaulton KJ, Ferreira T, Lee Y, Raimondo A, Magi R, Reschen ME, et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet (2015) 47:1415–25. doi: 10.1038/ng.3437
8. DIAbetes Genetics Replication Meta-analysis (DIAGRAM) Consortium. Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) Consortium. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet (2014) 46:234–44. doi: 10.1038/ng.2897
9. Franks PW, McCarthy MI. Exposing the exposures responsible for type 2 diabetes and obesity. Science (2016) 354:69–73. doi: 10.1126/science.aaf5094
10. Maher B. Personal genomes: The case of the missing heritability. Nature (2008) 456:18–21. doi: 10.1038/456018a
11. Bertazzi PA, Consonni D, Bachetti S, Rubagotti M, Baccarelli A, Zocchetti C, et al. Health effects of dioxin exposure: a 20-year mortality study. Am J Epidemiol (2001) 153:1031–44. doi: 10.1093/aje/153.11.1031
12. Wang SL, Tsai PC, Yang CY, Guo YL. Increased risk of diabetes and polychlorinated biphenyls and dioxins: a 24-year follow-up study of the Yucheng cohort. Diabetes Care (2008) 31:1574–9. doi: 10.2337/dc07-2449
13. Vena J, Boffetta P, Becher H, Benn T, Bueno-de-Mesquita HB, Coggon D, et al. Exposure to dioxin and nonneoplastic mortality in the expanded IARC international cohort study of phenoxy herbicide and chlorophenol production workers and sprayers. Environ Health Perspect (1998) 106(Suppl 2):645–53. doi: 10.1289/ehp.98106645
14. Calvert GM, Sweeney MH, Deddens J, Wall DK. Evaluation of diabetes mellitus, serum glucose, and thyroid function among United States workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Occup Environ Med (1999) 56:270–6. doi: 10.1136/oem.56.4.270
15. Yi SW, Hong JS, Ohrr H, Yi JJ. Agent Orange exposure and disease prevalence in Korean Vietnam veterans: the Korean veterans health study. Environ Res (2014) 133:56–65. doi: 10.1016/j.envres.2014.04.027
16. Kim JS, Lim HS, Cho SI, Cheong HK, Lim MK. Impact of Agent Orange exposure among Korean Vietnam veterans. Ind Health (2003) 41:149–57. doi: 10.2486/indhealth.41.149
17. Henriksen GL, Ketchum NS, Michalek JE, Swaby JA. Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology (1997) 8:252–8. doi: 10.1097/00001648-199705000-00005
18. Longnecker MP, Michalek JE. Serum dioxin level in relation to diabetes mellitus among Air Force veterans with background levels of exposure. Epidemiology (2000) 11:44–8. doi: 10.1097/00001648-200001000-00010
19. Kuo CC, Moon K, Thayer KA, Navas-Acien A. Environmental chemicals and type 2 diabetes: an updated systematic review of the epidemiologic evidence. Curr Diabetes Rep (2013) 13:831–49. doi: 10.1007/s11892-013-0432-6
20. Porta M. Persistent organic pollutants and the burden of diabetes. Lancet (2006) 368:558–9. doi: 10.1016/S0140-6736(06)69174-5
21. Thayer KA, Heindel JJ, Bucher JR, Gallo MA. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect (2012) 120:779–89. doi: 10.1289/ehp.1104597
22. Lee DH, Lee IK, Song K, Steffes M, Toscano W, Baker BA, et al. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999-2002. Diabetes Care (2006) 29:1638–44. doi: 10.2337/dc06-0543
23. Pal S, Blais JM, Robidoux MA, Haman F, Krummel E, Seabert TA, et al. The association of type 2 diabetes and insulin resistance/secretion with persistent organic pollutants in two First Nations communities in northern Ontario. Diabetes Metab (2013) 39:497–504. doi: 10.1016/j.diabet.2013.01.006
24. Everett CJ, Frithsen IL, Diaz VA, Koopman RJ, Simpson WM Jr., Mainous AG. 3rd. Association of a polychlorinated dibenzo-p-dioxin, a polychlorinated biphenyl, and DDT with diabetes in the 1999-2002 National Health and Nutrition Examination Survey. Environ Res (2007) 103:413–8. doi: 10.1016/j.envres.2006.11.002
25. Lee DH, Porta M, Jacobs DR Jr., Vandenberg LN. Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocr Rev (2014) 35:557–601. doi: 10.1210/er.2013-1084
26. Taylor KW, Novak RF, Anderson HA, Birnbaum LS, Blystone C, Devito M, et al. Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ Health Perspect (2013) 121:774–83. doi: 10.1289/ehp.1205502
27. Roh E, Kwak SH, Jung HS, Cho YM, Pak YK, Park KS, et al. Serum aryl hydrocarbon receptor ligand activity is associated with insulin resistance and resulting type 2 diabetes. Acta Diabetol (2015) 52:489–95. doi: 10.1007/s00592-014-0674-z
28. Dirinck EL, Dirtu AC, Govindan M, Covaci A, Van Gaal LF, Jorens PG. Exposure to persistent organic pollutants: relationship with abnormal glucose metabolism and visceral adiposity. Diabetes Care (2014) 37:1951–8. doi: 10.2337/dc13-2329
29. Jorgensen ME, Borch-Johnsen K, Bjerregaard P. A cross-sectional study of the association between persistent organic pollutants and glucose intolerance among Greenland Inuit. Diabetologia (2008) 51:1416–22. doi: 10.1007/s00125-008-1066-0
30. Lee YM, Ha CM, Kim SA, Thoudam T, Yoon YR, Kim DJ, et al. Low-Dose Persistent Organic Pollutants Impair Insulin Secretory Function of Pancreatic beta-Cells: Human and In Vitro Evidence. Diabetes (2017) 66:2669–80. doi: 10.2337/db17-0188
31. Government of Canada. The Canadian Environmental Protection Act, 1999. (1999). Available at: https://www.canada.ca/en/environment-climate-change/services/canadian-environmental-protection-act-registry/publications/canadian-environmental-protection-act-1999.html.
32. United States Government Accountability Office. Toxic substances: EPA has increased efforts to assess and control chemicals but could strengthen its approach. GAO-13-249. (2013). Available at: https://www.gao.gov/products/GAO-13-249GAO-13-249.
33. Schug TT, Janesick A, Blumberg B, Heindel JJ. Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol (2011) 127:204–15. doi: 10.1016/j.jsbmb.2011.08.007
34. Manzetti S, van der Spoel ER, van der Spoel D. Chemical properties, environmental fate, and degradation of seven classes of pollutants. Chem Res Toxicol (2014) 27:713–37. doi: 10.1021/tx500014w
35. Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature (1999) 401:763–4. doi: 10.1038/44517
36. Lecomte S, Habauzit D, Charlier TD, Pakdel F. Emerging Estrogenic Pollutants in the Aquatic Environment and Breast Cancer. Genes (Basel) (2017) 8:1–21. doi: 10.3390/genes8090229
37. Gao H, Yang BJ, Li N, Feng LM, Shi XY, Zhao WH, et al. and hormone-associated cancers: current progress and perspectives. Med (Baltimore) (2015) 94:e211. doi: 10.1097/MD.0000000000000211
38. Nadal A, Quesada I, Tuduri E, Nogueiras R, Alonso-Magdalena P. Endocrine-disrupting chemicals and the regulation of energy balance. Nat Rev Endocrinol (2017) 13:536–46. doi: 10.1038/nrendo.2017.51
39. Meeker JD. Exposure to environmental endocrine disruptors and child development. Arch Pediatr Adolesc Med (2012) 166:E1–7. doi: 10.1001/archpediatrics.2012.241
40. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev (2009) 30:293–342. doi: 10.1210/er.2009-0002
41. Mimoto MS, Nadal A, Sargis RM. Polluted Pathways: Mechanisms of Metabolic Disruption by Endocrine Disrupting Chemicals. Curr Environ Health Rep (2017) 4:208–22. doi: 10.1007/s40572-017-0137-0
42. Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol (2017) 68:3–33. doi: 10.1016/j.reprotox.2016.10.001
43. Sargis RM, Heindel JJ, Padmanabhan V. Interventions to Address Environmental Metabolism-Disrupting Chemicals: Changing the Narrative to Empower Action to Restore Metabolic Health. Front Endocrinol (Lausanne) (2019) 10:33. doi: 10.3389/fendo.2019.00033
44. Jing R, Fusi S, Kjellerup BV. Remediation of Polychlorinated Biphenyls (PCBs) in Contaminated Soils and Sediment: State of Knowledge and Perspectives. Front Environ Sci (2018) 6:79. doi: 10.3389/fenvs.2018.00079
45. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. Fourth National Report on Human Exposure to Environmental Chemicals, Updated Tables Volume 1 and 2. (2019). Available at: https://www.cdc.gov/exposurereport/index.html.
46. Government of Canada. Uses of human biomonitoring in data risk assessment. (2018). Available at: https://www.canada.ca/en/health-canada/services/chemical-substances/fact-sheets/human-biomonitoring-data-risk-assessment.html.
47. Centers for Disease Control and Prevention. National biomonitoring program. (2017). Available at: https://www.cdc.gov/biomonitoring/about.html.
48. Dewailly E, Ayotte P, Pereg D, Dery S, Dallaire R, et al. Exposure to heavy metals and persistant organic pollutants in Nunavik: the Nunavik Health Study (Human Health). Smith S., Stow J. (Eds) Synopsis of Research conducted under the 2005–2006 Northern Contaminants Program Indian and Northern Affairs Canada (2006) 44–64. Available at: http://pubs.aina.ucalgary.ca/ncp/60582.pdf.
49. Ashraf MA. Persistent organic pollutants (POPs): a global issue, a global challenge. Environ Sci Pollut Res Int (2017) 24:4223–7. doi: 10.1007/s11356-015-5225-9
50. Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ Health Perspect (2010) 118:1055–70. doi: 10.1289/ehp.0901716
51. Meeker JD, Sathyanarayana S, Swan SH. Phthalates and other additives in plastics: human exposure and associated health outcomes. Philos Trans R Soc Lond B Biol Sci (2009) 364:2097–113. doi: 10.1098/rstb.2008.0268
52. Main KM, Mortensen GK, Kaleva MM, Boisen KA, Damgaard IN, Chellakooty M, et al. Human breast milk contamination with phthalates and alterations of endogenous reproductive hormones in infants three months of age. Environ Health Perspect (2006) 114:270–6. doi: 10.1289/ehp.8075
53. Zoeller RT, Vandenberg LN. Assessing dose-response relationships for endocrine disrupting chemicals (EDCs): a focus on non-monotonicity. Environ Health (2015) 14:42. doi: 10.1186/s12940-015-0029-4
54. Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr., Lee DH, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev (2012) 33:378–455. doi: 10.1210/er.2011-1050
55. Barker DJ. The origins of the developmental origins theory. J Intern Med (2007) 261:412–7. doi: 10.1111/j.1365-2796.2007.01809.x
56. Vickers MH. Developmental programming and transgenerational transmission of obesity. Ann Nutr Metab (2014) 64(Suppl 1):26–34. doi: 10.1159/000360506
57. Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia (1993) 36:62–7. doi: 10.1007/BF00399095
58. Eriksson JG, Osmond C, Kajantie E, Forsen TJ, Barker DJ. Patterns of growth among children who later develop type 2 diabetes or its risk factors. Diabetologia (2006) 49:2853–8. doi: 10.1007/s00125-006-0459-1
59. Ouidir M, Buck Louis GM, Kanner J, Grantz KL, Zhang C, Sundaram R, et al. Association of Maternal Exposure to Persistent Organic Pollutants in Early Pregnancy With Fetal Growth. JAMA Pediatr (2019) 174(2):149–61. doi: 10.1001/jamapediatrics.2019.5104
60. Papalou O, Kandaraki EA, Papadakis G, Diamanti-Kandarakis E. Endocrine Disrupting Chemicals: An Occult Mediator of Metabolic Disease. Front Endocrinol (Lausanne) (2019) 10:112. doi: 10.3389/fendo.2019.00112
61. Kappil MA, Li Q, Li A, Dassanayake PS, Xia Y, Nanes JA, et al. In utero exposures to environmental organic pollutants disrupt epigenetic marks linked to fetoplacental development. Environ Epigenet (2016) 2:1–7. doi: 10.1093/eep/dvv013
62. Inadera H. Developmental origins of obesity and type 2 diabetes: molecular aspects and role of chemicals. Environ Health Prev Med (2013) 18:185–97. doi: 10.1007/s12199-013-0328-8
63. Iszatt N, Stigum H, Govarts E, Murinova LP, Schoeters G, Trnovec T, et al. Perinatal exposure to dioxins and dioxin-like compounds and infant growth and body mass index at seven years: A pooled analysis of three European birth cohorts. Environ Int (2016) 94:399–407. doi: 10.1016/j.envint.2016.04.040
64. Smink A, Ribas-Fito N, Garcia R, Torrent M, Mendez MA, Grimalt JO, et al. Exposure to hexachlorobenzene during pregnancy increases the risk of overweight in children aged 6 years. Acta Paediatr (2008) 97:1465–9. doi: 10.1111/j.1651-2227.2008.00937.x
65. Karmaus W, Osuch JR, Eneli I, Mudd LM, Zhang J, Mikucki D, et al. Maternal levels of dichlorodiphenyl-dichloroethylene (DDE) may increase weight and body mass index in adult female offspring. Occup Environ Med (2009) 66:143–9. doi: 10.1136/oem.2008.041921
66. Verhulst SL, Nelen V, Hond ED, Koppen G, Beunckens C, Vael C, et al. Intrauterine exposure to environmental pollutants and body mass index during the first 3 years of life. Environ Health Perspect (2009) 117:122–6. doi: 10.1289/ehp.0800003
67. Lee WC, Fisher M, Davis K, Arbuckle TE, Sinha SK. Identification of chemical mixtures to which Canadian pregnant women are exposed: The MIREC Study. Environ Int (2017) 99:321–30. doi: 10.1016/j.envint.2016.12.015
68. Rawn DFK, Sadler AR, Casey VA, Breton F, Sun WF, Arbuckle TE, et al. Dioxins/furans and PCBs in Canadian human milk: 2008-2011. Sci Total Environ (2017) 595:269–78. doi: 10.1016/j.scitotenv.2017.03.157
69. Fisher M, Arbuckle TE, Liang CL, LeBlanc A, Gaudreau E, Foster WG, et al. Concentrations of persistent organic pollutants in maternal and cord blood from the maternal-infant research on environmental chemicals (MIREC) cohort study. Environ Health (2016) 15:59. doi: 10.1186/s12940-016-0143-y
70. Arbuckle TE, Fraser WD, Fisher M, Davis K, Liang CL, Lupien N, et al. Cohort profile: the maternal-infant research on environmental chemicals research platform. Paediatr Perinat Epidemiol (2013) 27:415–25. doi: 10.1111/ppe.12061
71. Marraudino M, Bonaldo B, Farinetti A, Panzica G, Ponti G, Gotti S. Metabolism Disrupting Chemicals and Alteration of Neuroendocrine Circuits Controlling Food Intake and Energy Metabolism. Front Endocrinol (Lausanne) (2018) 9:766. doi: 10.3389/fendo.2018.00766
72. Diliberto JJ, Akubue PI, Luebke RW, Birnbaum LS. Dose-response relationships of tissue distribution and induction of CYP1A1 and CYP1A2 enzymatic activities following acute exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Toxicol Appl Pharmacol (1995) 130:197–208. doi: 10.1006/taap.1995.1025
73. Ibrahim M, MacFarlane EM, Matteo G, Hoyeck MP, Rick KRC, Farokhi SB, et al. Functional cytochrome P450 1a enzymes are induced in mouse and human islets following pollutant exposure. Diabetologia (2020) 63:162–78. doi: 10.1007/s00125-019-05035-0
74. Hoyeck MP, Merhi RC, Blair HL, Spencer CD, Payant MA, Martin Alfonso DI, et al. Female mice exposed to low doses of dioxin during pregnancy and lactation have increased susceptibility to diet-induced obesity and diabetes. Mol Metab (2020) 101104:1–4. doi: 10.1016/j.molmet.2020.101104
75. Soriano S, Alonso-Magdalena P, Garcia-Arevalo M, Novials A, Muhammed SJ, Salehi A, et al. Rapid insulinotropic action of low doses of bisphenol-A on mouse and human islets of Langerhans: role of estrogen receptor beta. PLoS One (2012) 7:e31109. doi: 10.1371/journal.pone.0031109
76. Villar-Pazos S, Martinez-Pinna J, Castellano-Munoz M, Alonso-Magdalena P, Marroqui L, Quesada I, et al. Molecular mechanisms involved in the non-monotonic effect of bisphenol-a on ca2+ entry in mouse pancreatic beta-cells. Sci Rep (2017) 7:11770. doi: 10.1038/s41598-017-11995-3
77. Martinez-Pinna J, Marroqui L, Hmadcha A, Lopez-Beas J, Soriano S, Villar-Pazos S, et al. Bisphenol-S and Bisphenol-F alter mouse pancreatic β-cell ion channel expression and activity and insulin release through an estrogen receptor ERβ mediated pathway. Chemosphere (2020) 129051. doi: 10.1016/j.chemosphere.2020.129051
78. Mailloux R, Fu A, Florian M, Petrov I, Chen Q, Coughlan MC, et al. A Northern contaminant mixture impairs pancreas function in obese and lean JCR rats and inhibits insulin secretion in MIN6 cells. Toxicology (2015) 334:81–93. doi: 10.1016/j.tox.2015.06.001
79. Novelli M, Piaggi S, De Tata V. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced impairment of glucose-stimulated insulin secretion in isolated rat pancreatic islets. Toxicol Lett (2005) 156:307–14. doi: 10.1016/j.toxlet.2004.12.004
80. Kurita H, Yoshioka W, Nishimura N, Kubota N, Kadowaki T, Tohyama C. Aryl hydrocarbon receptor-mediated effects of TCDD on glucose-stimulated insulin secretion in mice. J Appl Toxicol (2009) 29:689–94. doi: 10.1002/jat.1459
81. Hoyeck M, Blair H, Ibrahim M, Solanky S, Elsawy M, Matteo G, et al. Long-term metabolic consequences of acute dioxin exposure differ between male and female mice. Sci Reports (2020) 10:1448. doi: 10.1038/s41598-020-57973-0
82. Matteo G, Hoyeck M, Blair H, Rick KRC, Williams A, Buick JK, et al. Chronic low-dose dioxin exposure accelerates high fat diet-induced hyperglycemia in female mice. BioRxiv (2020) 2020.09.12.294587. doi: 10.1101/2020.09.12.294587
83. Fu J, Githaka JM, Dai X, Plummer G, Suzuki K, Spigelman AF, et al. A glucose-dependent spatial patterning of exocytosis in human beta-cells is disrupted in type 2 diabetes. JCI Insight (2019) 5:e127896. doi: 10.1101/534669
84. Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Meneur C, Bernal-Mizrachi E. Natural history of beta-cell adaptation and failure in type 2 diabetes. Mol Aspects Med (2015) 42:19–41. doi: 10.1016/j.mam.2014.12.002
85. Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, et al. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab (2020) 31:1017–31.e4. doi: 10.1016/j.cmet.2020.04.005
86. Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes (2005) 54:727–35. doi: 10.2337/diabetes.54.3.727
87. Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab (2004) 89:5535–41. doi: 10.1210/jc.2004-0150
88. Scott FW, Pound LD, Patrick C, Eberhard CE, Crookshank JA. Where genes meet environment-integrating the role of gut luminal contents, immunity and pancreas in type 1 diabetes. Transl Res (2017) 179:183–98. doi: 10.1016/j.trsl.2016.09.001
89. Krogvold L, Skog O, Sundstrom G, Edwin B, Buanes T, Hanssen KF, et al. Function of Isolated Pancreatic Islets From Patients at Onset of Type 1 Diabetes: Insulin Secretion Can Be Restored After Some Days in a Nondiabetogenic Environment In Vitro: Results From the DiViD Study. Diabetes (2015) 64:2506–12. doi: 10.2337/db14-1911
90. Koskinen MK, Helminen O, Matomaki J, Aspholm S, Mykkanen J, Makinen M, et al. Reduced beta-cell function in early preclinical type 1 diabetes. Eur J Endocrinol (2016) 174:251–9. doi: 10.1530/EJE-15-0674
91. Keskinen P, Korhonen S, Kupila A, Veijola R, Erkkila S, Savolainen H, et al. First-phase insulin response in young healthy children at genetic and immunological risk for Type I diabetes. Diabetologia (2002) 45:1639–48. doi: 10.1007/s00125-002-0981-8
92. Chase HP, Cuthbertson DD, Dolan LM, Kaufman F, Krischer JP, Schatz DA, et al. First-phase insulin release during the intravenous glucose tolerance test as a risk factor for type 1 diabetes. J Pediatr (2001) 138:244–9. doi: 10.1067/mpd.2001.111274
93. Templeman NM, Clee SM, Johnson JD. Suppression of hyperinsulinaemia in growing female mice provides long-term protection against obesity. Diabetologia (2015) 58:2392–402. doi: 10.1007/s00125-015-3676-7
94. Templeman NM, Skovso S, Page MM, Lim GE, Johnson JD. A causal role for hyperinsulinemia in obesity. J Endocrinol (2017) 232:R173–R83. doi: 10.1530/JOE-16-0449
95. Thomas DD, Corkey BE, Istfan NW, Apovian CM. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J Endocr Soc (2019) 3:1727–47. doi: 10.1210/js.2019-00065
96. Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab (2012) 16:723–37. doi: 10.1016/j.cmet.2012.10.019
97. Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab (2003) 88:2300–8. doi: 10.1210/jc.2002-020735
98. Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, et al. {beta}-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology (2010) 151:1462–72. doi: 10.1210/en.2009-1277
99. Linnemann AK, Baan M, Davis DB. Pancreatic beta-cell proliferation in obesity. Adv Nutr (2014) 5:278–88. doi: 10.3945/an.113.005488
100. Wortham M, Sander M. Mechanisms of beta-cell functional adaptation to changes in workload. Diabetes Obes Metab (2016) 18(Suppl 1):78–86. doi: 10.1111/dom.12729
101. Cnop M, Vidal J, Hull RL, Utzschneider KM, Carr DB, Schraw T, et al. Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care (2007) 30:677–82. doi: 10.2337/dc06-1834
102. Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest (1999) 104:787–94. doi: 10.1172/JCI7231
103. Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature (2007) 445:886–91. doi: 10.1038/nature05537
104. Elsakr JM, Gannon M. Developmental programming of the pancreatic islet by in utero overnutrition. Trends Dev Biol (2017) 10:79–95.
105. Soldatow VY, Lecluyse EL, Griffith LG, Rusyn I. In vitro models for liver toxicity testing. Toxicol Res (Camb) (2013) 2:23–39. doi: 10.1039/C2TX20051A
106. Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol (2000) 32:56–67. doi: 10.1006/rtph.2000.1399
107. Sass N. Humane endpoints and acute toxicity testing. ILAR J (2000) 41:114–23. doi: 10.1093/ilar.41.2.114
108. Krewski D, Acosta D Jr., Andersen M, Anderson H, Bailar JC,3, Boekelheide K, et al. Toxicity testing in the 21st century: a vision and a strategy. J Toxicol Environ Health B Crit Rev (2010) 13:51–138. doi: 10.1080/10937404.2010.483176
109. Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, et al. Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab (2012) 97:3197–206. doi: 10.1210/jc.2012-1206
110. Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, et al. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia (1993) 36:1139–45. doi: 10.1007/BF00401058
111. Poitout V, Olson LK, Robertson RP. Insulin-secreting cell lines: classification, characteristics and potential applications. Diabetes Metab (1996) 22:7–14.
112. Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology (2004) 145:667–78. doi: 10.1210/en.2003-1099
113. Hectors TL, Vanparys C, Pereira-Fernandes A, Martens GA, Blust R. Evaluation of the INS-1 832/13 cell line as a beta-cell based screening system to assess pollutant effects on beta-cell function. PLoS One (2013) 8:e60030. doi: 10.1371/journal.pone.0060030
114. Tsonkova VG, Sand FW, Wolf XA, Grunnet LG, Kirstine Ringgaard A, Ingvorsen C, et al. The EndoC-betaH1 cell line is a valid model of human beta cells and applicable for screenings to identify novel drug target candidates. Mol Metab (2018) 8:144–57. doi: 10.1016/j.molmet.2017.12.007
115. Andersson LE, Valtat B, Bagge A, Sharoyko VV, Nicholls DG, Ravassard P, et al. Characterization of stimulus-secretion coupling in the human pancreatic EndoC-betaH1 beta cell line. PLoS One (2015) 10:e0120879. doi: 10.1371/journal.pone.0120879
116. Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest (2011) 121:3589–97. doi: 10.1172/JCI58447
117. Oleson BJ, McGraw JA, Broniowska KA, Annamalai M, Chen J, Bushkofsky JR, et al. Distinct differences in the responses of the human pancreatic beta-cell line EndoC-betaH1 and human islets to proinflammatory cytokines. Am J Physiol Regul Integr Comp Physiol (2015) 309:R525–34. doi: 10.1152/ajpregu.00544.2014
118. Da Silva Xavier G. The Cells of the Islets of Langerhans. J Clin Med (2018) 7:1–17. doi: 10.3390/jcm7030054
119. Hart NJ, Powers AC. Use of human islets to understand islet biology and diabetes: progress, challenges and suggestions. Diabetologia (2019) 62:212–22. doi: 10.1007/s00125-018-4772-2
120. Kim S, Whitener RL, Peiris H, Gu X, Chang CA, Lam JY, et al. Molecular and genetic regulation of pig pancreatic islet cell development. Development (2020) 147:1–14. doi: 10.1242/dev.186213
121. Nagaya M, Hayashi A, Nakano K, Honda M, Hasegawa K, Okamoto K, et al. Distributions of endocrine cell clusters during porcine pancreatic development. PLoS One (2019) 14:e0216254. doi: 10.1371/journal.pone.0216254
122. Zhu HT, Wang WL, Yu L, Wang B. Pig-islet xenotransplantation: recent progress and current perspectives. Front Surg (2014) 1:7. doi: 10.3389/fsurg.2014.00007
123. Lamb M, Laugenour K, Liang O, Alexander M, Foster CE, Lakey JR. In vitro maturation of viable islets from partially digested young pig pancreas. Cell Transpl (2014) 23:263–72. doi: 10.3727/096368912X662372
124. Hardikar AA, Wang XY, Williams LJ, Kwok J, Wong R, Yao M, et al. Functional maturation of fetal porcine beta-cells by glucagon-like peptide 1 and cholecystokinin. Endocrinology (2002) 143:3505–14. doi: 10.1210/en.2001-211344
125. Saliba Y, Bakhos JJ, Itani T, Fares N. An optimized protocol for purification of functional islets of Langerhans. Lab Invest (2017) 97:70–83. doi: 10.1038/labinvest.2016.123
126. Yin N, Chen T, Yu Y, Han Y, Yan F, Zheng Z, et al. Facile mechanical shaking method is an improved isolation approach for islet preparation and transplantation. Exp Ther Med (2016) 12:3658–64. doi: 10.3892/etm.2016.3844
127. Carter JD, Dula SB, Corbin KL, Wu R, Nunemaker CS. A practical guide to rodent islet isolation and assessment. Biol Proced Online (2009) 11:3–31. doi: 10.1007/s12575-009-9021-0
128. Lyon J, Manning Fox JE, Spigelman AF, Kim R, Smith N, O’Gorman D, et al. Research-Focused Isolation of Human Islets From Donors With and Without Diabetes at the Alberta Diabetes Institute IsletCore. Endocrinology (2016) 157:560–9. doi: 10.1210/en.2015-1562
129. Brissova M, Niland JC, Cravens J, Olack B, Sowinski J, Evans-Molina C. The Integrated Islet Distribution Program answers the call for improved human islet phenotyping and reporting of human islet characteristics in research articles. Diabetologia (2019) 62:1312–4. doi: 10.1007/s00125-019-4876-3
130. Sigmundsson K, Ojala JRM, Ohman MK, Osterholm AM, Moreno-Moral A, Domogatskaya A, et al. Culturing functional pancreatic islets on alpha5-laminins and curative transplantation to diabetic mice. Matrix Biol (2018) 70:5–19. doi: 10.1016/j.matbio.2018.03.018
131. Yeh CC, Wang LJ, McGarrigle JJ, Wang Y, Liao CC, Omami M, et al. Effect of Manufacturing Procedures on Human Islet Isolation From Donor Pancreata Standardized by the North American Islet Donor Score. Cell Transpl (2017) 26:33–44. doi: 10.3727/096368916X692834
132. Ihm SH, Matsumoto I, Sawada T, Nakano M, Zhang HJ, Ansite JD, et al. Effect of donor age on function of isolated human islets. Diabetes (2006) 55:1361–8. doi: 10.2337/db05-1333
133. Wang Y, Danielson KK, Ropski A, Harvat T, Barbaro B, Paushter D, et al. Systematic analysis of donor and isolation factor’s impact on human islet yield and size distribution. Cell Transpl (2013) 22:2323–33. doi: 10.3727/096368912X662417
134. Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, et al. Acquisition of Dynamic Function in Human Stem Cell-Derived beta Cells. Stem Cell Rep (2019) 12:351–65. doi: 10.1016/j.stemcr.2018.12.012
135. Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, Millman JR. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat Biotechnol (2020) 38:460–70. doi: 10.1038/s41587-020-0430-6
136. Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic beta cells in vitro. Cell (2014) 159:428–39. doi: 10.1016/j.cell.2014.09.040
137. Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J (2015) 34:1759–72. doi: 10.15252/embj.201591058
138. Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JH, Harb G, et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature (2019) 569:368–73. doi: 10.1038/s41586-019-1168-5
139. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol (2014) 32:1121–33. doi: 10.1038/nbt.3033
140. Zhou T, Kim TW, Chong CN, Tan L, Amin S, Sadat Badieyan Z, et al. Chen S. A hPSC-based platform to discover gene-environment interactions that impact human beta-cell and dopamine neuron survival. Nat Commun (2018) 9:4815. doi: 10.1038/s41467-018-07201-1
141. Krentz NA, Nian C, Lynn FC. TALEN/CRISPR-mediated eGFP knock-in add-on at the OCT4 locus does not impact differentiation of human embryonic stem cells towards endoderm. PLoS One (2014) 9:e114275. doi: 10.1371/journal.pone.0114275
142. Krentz NAJ, van Hoof D, Li Z, Watanabe A, Tang M, Nian C, et al. Phosphorylation of NEUROG3 Links Endocrine Differentiation to the Cell Cycle in Pancreatic Progenitors. Dev Cell (2017) 41:129–42.e6. doi: 10.1016/j.devcel.2017.02.006
143. Micallef SJ, Li X, Schiesser JV, Hirst CE, Yu QC, Lim SM, et al. INS(GFP/w) human embryonic stem cells facilitate isolation of in vitro derived insulin-producing cells. Diabetologia (2012) 55:694–706. doi: 10.1007/s00125-011-2379-y
144. Krentz NAJ, Lee MYY, Xu EE, Sproul SLJ, Maslova A, Sasaki S, et al. Single-Cell Transcriptome Profiling of Mouse and hESC-Derived Pancreatic Progenitors. Stem Cell Rep (2018) 11:1551–64. doi: 10.1016/j.stemcr.2018.11.008
145. Breckwoldt K, Letuffe-Breniere D, Mannhardt I, Schulze T, Ulmer B, Werner T, et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat Protoc (2017) 12:1177–97. doi: 10.1038/nprot.2017.033
146. Ye L, Zhang S, Greder L, Dutton J, Keirstead SA, Lepley M, et al. Effective cardiac myocyte differentiation of human induced pluripotent stem cells requires VEGF. PLoS One (2013) 8:e53764. doi: 10.1371/journal.pone.0053764
147. Zhang J, Klos M, Wilson GF, Herman AM, Lian X, Raval KK, et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res (2012) 111:1125–36. doi: 10.1161/CIRCRESAHA.112.273144
148. Denning C, Borgdorff V, Crutchley J, Firth KS, George V, Kalra S, et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim Biophys Acta (2016) 1863:1728–48. doi: 10.1016/j.bbamcr.2015.10.014
149. Schulz TC, Young HY, Agulnick AD, Babin MJ, Baetge EE, Bang AG, et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One (2012) 7:e37004. doi: 10.1371/journal.pone.0037004
150. Warren CR, Cowan CA. Humanity in a Dish: Population Genetics with iPSCs. Trends Cell Biol (2018) 28:46–57. doi: 10.1016/j.tcb.2017.09.006
151. Balboa D, Prasad RB, Groop L, Otonkoski T. Genome editing of human pancreatic beta cell models: problems, possibilities and outlook. Diabetologia (2019) 62:1329–36. doi: 10.1007/s00125-019-4908-z
152. Balboa D, Saarimaki-Vire J, Otonkoski T. Concise Review: Human Pluripotent Stem Cells for the Modeling of Pancreatic beta-Cell Pathology. Stem Cells (2019) 37:33–41. doi: 10.1002/stem.2913
153. Teo AK, Windmueller R, Johansson BB, Dirice E, Njolstad PR, Tjora E, et al. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J Biol Chem (2013) 288:5353–6. doi: 10.1074/jbc.C112.428979
154. Braverman-Gross C, Nudel N, Ronen D, Beer NL, McCarthy MI, Benvenisty N. Derivation and molecular characterization of pancreatic differentiated MODY1-iPSCs. Stem Cell Res (2018) 31:16–26. doi: 10.1016/j.scr.2018.06.013
155. Balboa D, Saarimaki-Vire J, Borshagovski D, Survila M, Lindholm P, Galli E, et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. Elife (2018) 7:e38519. doi: 10.7554/eLife.38519
156. Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes (2014) 63:923–33. doi: 10.2337/db13-0717
157. Teo AK, Lau HH, Valdez IA, Dirice E, Tjora E, Raeder H, et al. Early Developmental Perturbations in a Human Stem Cell Model of MODY5/HNF1B Pancreatic Hypoplasia. Stem Cell Rep (2016) 6:357–67. doi: 10.1016/j.stemcr.2016.01.007
158. Hosokawa Y, Toyoda T, Fukui K, Baden MY, Funato M, Kondo Y, et al. Insulin-producing cells derived from ‘induced pluripotent stem cells’ of patients with fulminant type 1 diabetes: Vulnerability to cytokine insults and increased expression of apoptosis-related genes. J Diabetes Investig (2017) 9(3):481–93. doi: 10.1111/jdi.12727
159. Leite NC, Sintov E, Meissner TB, Brehm MA, Greiner DL, Harlan DM, et al. Modeling Type 1 Diabetes In Vitro Using Human Pluripotent Stem Cells. Cell Rep (2020) 32:107894. doi: 10.1016/j.celrep.2020.107894
160. Millman JR, Xie C, Van Dervort A, Gurtler M, Pagliuca FW, Melton DA. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat Commun (2016) 7:11463. doi: 10.1038/ncomms11463
161. Beer NL, Gloyn AL. Genome-edited human stem cell-derived beta cells: a powerful tool for drilling down on type 2 diabetes GWAS biology. F1000Res (2016) 5:1–12. doi: 10.12688/f1000research.8682.1
162. Zhu Z, Li QV, Lee K, Rosen BP, Gonzalez F, Soh CL, et al. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell (2016) 18:755–68. doi: 10.1016/j.stem.2016.03.015
163. McGrath PS, Watson CL, Ingram C, Helmrath MA, Wells JM. The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes (2015) 64:2497–505. doi: 10.2337/db14-1412
164. Guo D, Liu H, Ruzi A, Gao G, Nasir A, Liu Y, et al. Modeling Congenital Hyperinsulinism with ABCC8-Deficient Human Embryonic Stem Cells Generated by CRISPR/Cas9. Sci Rep (2017) 7:3156. doi: 10.1038/s41598-017-03349-w
165. Liang G, Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell (2013) 13:149–59. doi: 10.1016/j.stem.2013.07.001
166. Goldring C, Antoine DJ, Bonner F, Crozier J, Denning C, Fontana RJ, et al. Stem cell-derived models to improve mechanistic understanding and prediction of human drug-induced liver injury. Hepatology (2017) 65:710–21. doi: 10.1016/j.toxlet.2017.07.124
167. Lynch S, Pridgeon CS, Duckworth CA, Sharma P, Park BK, Goldring CEP. Stem cell models as an in vitro model for predictive toxicology. Biochem J (2019) 476:1149–58. doi: 10.1042/BCJ20170780
168. Grimm FA, Iwata Y, Sirenko O, Bittner M, Rusyn I. High-Content Assay Multiplexing for Toxicity Screening in Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Hepatocytes. Assay Drug Dev Technol (2015) 13:529–46. doi: 10.1089/adt.2015.659
169. Lahti AL, Kujala VJ, Chapman H, Koivisto AP, Pekkanen-Mattila M, Kerkela E, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech (2012) 5:220–30. doi: 10.1242/dmm.008409
170. Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J (2011) 32:952–62. doi: 10.1093/eurheartj/ehr073
171. Itzhaki I, Maizels L, Huber I, Gepstein A, Arbel G, Caspi O, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol (2012) 60:990–1000. doi: 10.1016/j.jacc.2012.02.066
172. Sirenko O, Cromwell EF, Crittenden C, Wignall JA, Wright FA, Rusyn I. Assessment of beating parameters in human induced pluripotent stem cells enables quantitative in vitro screening for cardiotoxicity. Toxicol Appl Pharmacol (2013) 273:500–7. doi: 10.1016/j.taap.2013.09.017
Keywords: stem cells, diabetes, pollution, beta cells, pancreas development, toxicology
Citation: MacFarlane EM and Bruin JE (2021) Human Pluripotent Stem Cells: A Unique Tool for Toxicity Testing in Pancreatic Progenitor and Endocrine Cells. Front. Endocrinol. 11:604998. doi: 10.3389/fendo.2020.604998
Received: 11 September 2020; Accepted: 27 November 2020;
Published: 19 January 2021.
Edited by:
Holger Andreas Russ, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Barak Blum, University of Wisconsin-Madison, United StatesAdrian Kee Keong Teo, Institute of Molecular and Cell Biology (A*STAR), Singapore
Copyright © 2021 MacFarlane and Bruin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer E. Bruin, amVubnkuYnJ1aW5AY2FybGV0b24uY2E=