 Erik J. Tillman
Erik J. Tillman Tim Rolph
Tim Rolph- Akero Therapeutics, South San Francisco, CA, United States
The rising global prevalence of obesity, metabolic syndrome, and type 2 diabetes has driven a sharp increase in non-alcoholic fatty liver disease (NAFLD), characterized by excessive fat accumulation in the liver. Approximately one-sixth of the NAFLD population progresses to non-alcoholic steatohepatitis (NASH) with liver inflammation, hepatocyte injury and cell death, liver fibrosis and cirrhosis. NASH is one of the leading causes of liver transplant, and an increasingly common cause of hepatocellular carcinoma (HCC), underscoring the need for intervention. The complex pathophysiology of NASH, and a predicted prevalence of 3–5% of the adult population worldwide, has prompted drug development programs aimed at multiple targets across all stages of the disease. Currently, there are no approved therapeutics. Liver-related morbidity and mortality are highest in more advanced fibrotic NASH, which has led to an early focus on anti-fibrotic approaches to prevent progression to cirrhosis and HCC. Due to limited clinical efficacy, anti-fibrotic approaches have been superseded by mechanisms that target the underlying driver of NASH pathogenesis, namely steatosis, which drives hepatocyte injury and downstream inflammation and fibrosis. Among this wave of therapeutic mechanisms targeting the underlying pathogenesis of NASH, the hormone fibroblast growth factor 21 (FGF21) holds considerable promise; it decreases liver fat and hepatocyte injury while suppressing inflammation and fibrosis across multiple preclinical studies. In this review, we summarize preclinical and clinical data from studies with FGF21 and FGF21 analogs, in the context of the pathophysiology of NASH and underlying metabolic diseases.
Introduction
A marked rise in the global prevalence of obesity and associated metabolic pathologies has been observed over the past 30 years (1). The burden of managing a chronic excess of energy over demand falls on the metabolic organs, including adipose tissue, pancreas, and liver. Ultimately the capacity of these organs to manage the surfeit of energy is exceeded, manifesting as hyperlipidemia/dyslipidemia, type 2 diabetes, and nonalcoholic fatty liver disease (NAFLD), defined as >5% liver fat content not attributable to other chronic liver conditions.
Globally, more than 1 billion people are estimated to have NAFLD, including 83 million patients in the United States, making NAFLD the most common chronic liver disease in the US (2, 3). Fifteen to twenty percent of patients with NAFLD progress to the more severe nonalcoholic steatohepatitis (NASH) (4), characterized by hepatocyte stress, injury and apoptosis, inflammation and fibrosis (5). Scarring of the liver (i.e., fibrosis) occurs in response to chronic injury and inflammation. Left unaddressed, NASH fibrosis may advance to cirrhosis, which in turn may cause end-stage liver disease or hepatocellular carcinoma (HCC) (6). The prevalence of NASH, and in particular severe F3 and F4 fibrosis, is rapidly increasing in the US and globally (7). By 2030, there are predicted to be 27 million NASH patients in the US, including 3 million with NASH cirrhosis (8). As fibrosis progresses, the risk of liver-related morbidity and mortality increases (9). NASH-related liver failure and HCC have become leading causes of liver transplantation (10, 11), particularly with the emergence of effective treatment for hepatitis C. Besides the widely recognized morbidities, pre-cirrhotic NAFLD and NASH also negatively impact health-related quality of life, further underscoring the need for effective treatments (12).
NASH is diagnosed histologically, based on the extent of steatosis, hepatocyte stress and damage (hepatocyte ballooning), immune infiltration (lobular inflammation), and degree of fibrosis (13). Within the liver, lipotoxicity-induced endoplasmic reticulum (ER) stress- and oxidative stress-induced tissue injury (14) release pro-inflammatory damage-associated molecular patterns (DAMPs) that recruit and activate Kupffer cells, liver-resident macrophages (15). Kupffer cells amplify the cycle of necroinflammation (16) while directly and indirectly activating hepatic stellate cells (HSCs) to differentiate and proliferate to a myofibroblast-like, collagen-producing phenotype that promotes fibrogenesis in NASH (17). Liver damage and ensuing inflammation may also contribute, at a whole-body level, to metabolic dysfunction in other organs and tissues (18). An ideal disease-modifying therapeutic agent needs to target the primary driver of NASH: elevated intrahepatocyte fat deposition. By removing the underlying pathogenic insult, hepatocyte oxidative and ER stress and lipotoxicity will diminish, resulting in less activation of pathways driving hepatocyte dedifferentiation and death, in turn allowing the downstream sequelae of liver inflammation and fibrosis to resolve.
While late-stage NASH greatly increases the risk of liver-related morbidity, NAFLD and NASH with less advanced fibrosis (i.e., F0–F2) are associated with greatly increased risk of cardiovascular disease and related mortality (19). Indeed, the leading cause of death among patients with NASH is cardiovascular disease, even after correcting for the contribution of other known CV risk factors and metabolic comorbidities (20–22). These highly prevalent metabolic comorbidities, including hypertriglyceridemia (83% of NASH patients), obesity (82%), dyslipidemia (72%), metabolic syndrome (71%), and type 2 diabetes (44%), underscore the critical need to holistically address the imbalanced metabolic state underlying NASH (23).
Despite the unmet medical need, there is currently no approved therapy for NASH. Lifestyle modifications, including diet and exercise, that lead to significant weight loss have demonstrated clinical benefit, but long-term compliance is poor (24, 25). The first wave of therapeutic approaches in NASH targeted downstream pathologies, specifically necroinflammation and fibrosis, either inadequately addressing or in some cases exacerbating the underlying drivers of NASH pathology: steatosis, insulin resistance, and lipotoxicity. Among numerous mid-to-late-stage failures across a variety of mechanisms of action (26–29), the only positive interim phase 3 readout in the last decade has been a trial of the bile acid analog, obeticholic acid (30), implying therapeutic target selection has been suboptimal. Moreover, none of the therapeutics in development has demonstrated both fibrosis improvement and resolution of NASH in a phase 3 clinical study, the two histological endpoints accepted by the FDA and EMA as sufficient to support accelerated or expedited regulatory approval. While the shift to address the underlying drivers of NASH holds promise, many of the mechanisms under investigation are limited to the liver as their site of action, because of safety constraints arising from systemic exposure (Table 1). As a consequence, these mechanisms have not improved whole-body metabolism, and in some cases have been associated with adverse effects such as increases in serum low-density lipoprotein (LDL)-cholesterol (30–32), serum triglycerides (33, 34), and body weight (35, 36).
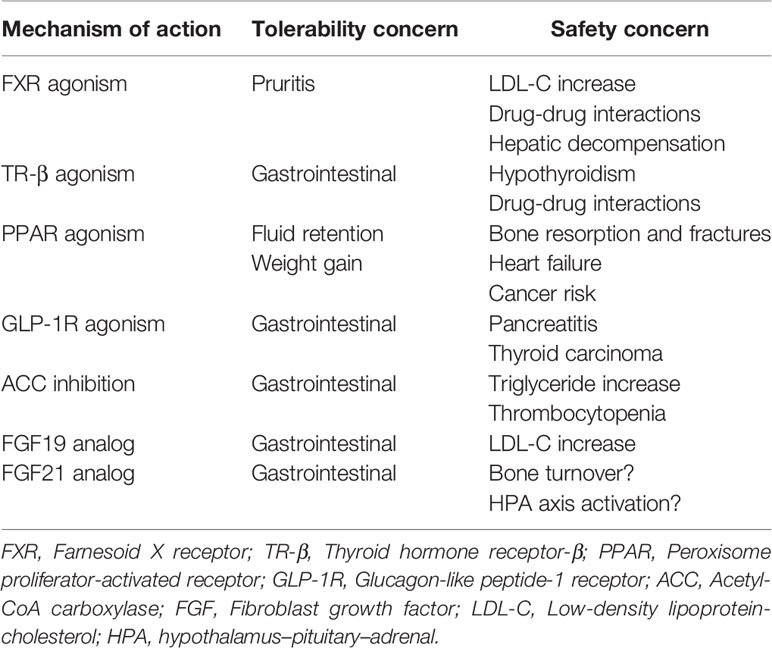
Table 1 Safety and tolerability across various mechanisms of action under investigation in NASH.
On the other hand, endocrine fibroblast growth factor (FGF) analogs have emerged as a promising class because of their ability to not only act directly on liver, but also to shift whole-body metabolism to a healthier state. The FGF19 subfamily of FGFs, consisting of FGF19, FGF21, and FGF23, is characterized by reduced affinity for heparan sulfate (HS) (37). In the absence of HS binding, these FGFs may diffuse from their site of secretion, enabling endocrine actions via the systemic circulation, as well as canonical FGF paracrine signaling on their cell of origin or nearby cells (38). Whereas HS in extracellular matrix serves as a coreceptor for autocrine and paracrine FGFs, the endocrine FGFs rely on high-affinity interactions with either α-Klotho (FGF23) or β-Klotho (FGF19 and FGF21) for recruitment and localization to the cell surface, where they engage canonical FGF receptors (FGFRs) (39–42). Dysregulation of endocrine FGF signaling, particularly FGF19 and FGF21, has been implicated in metabolic disease, suggesting their potential as therapeutic mechanisms (43–46). While gut-secreted FGF19 and liver-secreted FGF21 (47) share some overlapping physiological roles, including regulation of glucose and lipid metabolism (48), significant differences exists between them, primarily in the ability of FGF19 to stimulate hepatocyte proliferation and to suppress bile acid synthesis (49). These divergent actions result from different agonist profiles across the FGFRs: whereas the FGF19/β-Klotho receptor complex is able to bind and activate FGFR1c, FGFR2c, FGFR3c, and FGFR4, the FGF21/β-Klotho complex signals only through FGFR1c, FGFR2c, and FGFR3c (41).
This review examines the unique physiology of FGF21 signaling at the cellular, tissue, and whole-body level, presenting preclinical and clinical evidence that FGF21-based therapeutics exert a broad set of metabolic actions and also suppress inflammation and fibrosis. In particular, the discussion focuses on the promise of FGF21 analogs as foundational treatments for NASH.
Metabolic Dysregulation Underlying Early Steatohepatitis Pathology
Insulin resistance, obesity, and dyslipidemia are the clinical manifestations of a chronic excess of energy. To reduce energy uptake, peripheral organs including adipose tissue and skeletal muscle respond by reducing sensitivity to insulin. As a result, dietary energy is diverted to the liver, leading to excessive accumulation of lipid droplets (LD) in hepatocytes (hepatic fat fraction of up to 20–30%, vs <5% in healthy liver) (5), and hyperlipidemia (50). In NASH patients, the sources of hepatic fatty acid flux are presented in Figure 1A: 45–55% is accounted for by adipose tissue lipolysis; 25–35% by de novo lipogenesis (DNL) in the liver; and 10–20% by dietary fat (51, 52). Impaired peripheral insulin sensitivity (52) contributes substantially to the increased flux of fatty acid in liver. Post prandial suppression of adipose tissue lipolysis by insulin is impaired (48), while uptake by adipose tissue of dietary fat as chylomicrons is also attenuated. Insulin-dependent glucose uptake by adipose tissue and skeletal muscle is likewise reduced, redirecting glucose to the liver where it fuels DNL by activating the carbohydrate response element-binding protein (ChREBP), a master transcriptional regulator of lipogenic pathway proteins (53). ChREBP is also induced in liver by dietary fructose, a major source of calories in “Western” obesogenic diets (54, 55). Increased esterification of fatty acid by liver is associated with higher secretion of triglyceride into the circulation as VLDL. Accumulation of fat in the circulation manifests as elevated serum triglyceride and LDL-cholesterol, in combination with low levels of high-density lipoprotein (HDL), characteristic of the dyslipidemia prevalent in NASH patients (56).
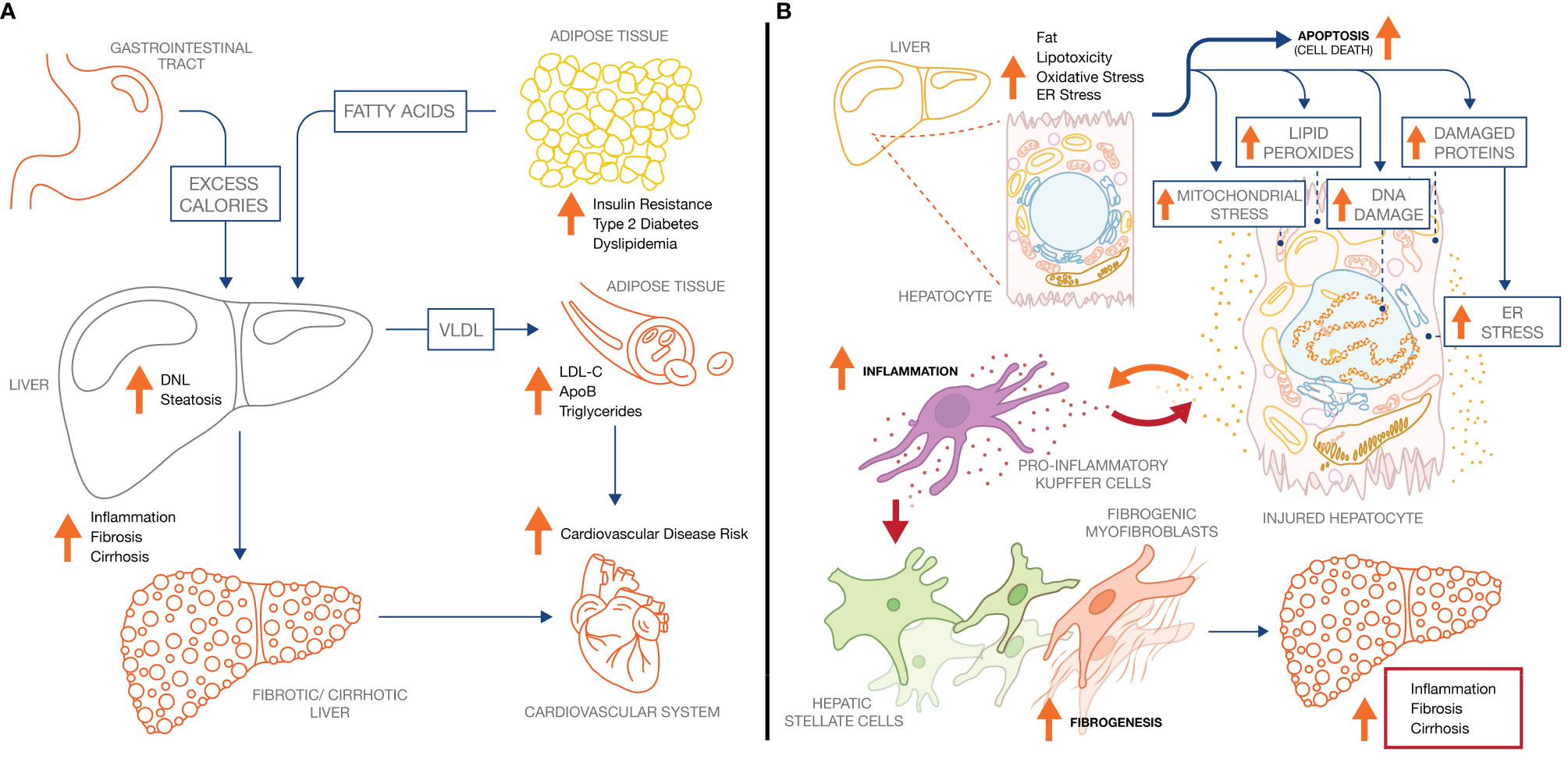
Figure 1 NASH pathology and pathophysiology. (A) Increased adipose tissue lipolysis-derived fatty acid flux and increased hepatic DNL drive hepatic steatosis in NASH, which increases both liver-related mortality and cardiovascular mortality. (B) Increased intrahepatic fat drives pathophysiological processes across the hepatocyte, inflammatory Kupffer cell, and collagen-producing hepatic stellate cell. Ultimately, continuous exposure to the underlying stressors (metabolic dysregulation, lipotoxicity) sustain the inflammatory and fibrotic phenotype leading to progressive fibrosis and cirrhosis.
Chronic accumulation of triglyceride in liver elevates the basal rate of fatty acid β-oxidation in hepatocytes. Conditions in which energy supply from fat oxidation exceeds energy demand lead to formation of reactive oxygen species, in turn causing oxidative stress (57, 58). This stress, which damages hepatocytes, also induces transcription and secretion of FGF21, which appears able to act directly on hepatocytes as a paracrine or autocrine hormone (59, 60).
Preclinical characterization of FGF21 in rodents and primates has established its role in restoring metabolic homeostasis in obese or metabolically challenged animals, reducing body weight, liver and circulating triglycerides, fasting plasma insulin and glucose, and increasing energy expenditure (43, 61). Consistent with a protective role, endogenous FGF21 serum concentration may be elevated by up to 10–20-fold in patients with NAFLD (62, 63), NASH (64), obesity (64–66), type 2 diabetes (67), chronic kidney disease (68), diabetic nephropathy (69), atherosclerosis (70, 71), or coronary heart disease (72). However, this elevation of FGF21 observed in chronic, pathological metabolic states in humans does not appear sufficient to ameliorate disease.
While decreased FGF21 receptor expression has been proposed to underlie this “FGF21-resistant state,” (73), the dramatic upregulation of fibroblast activation protein (FAP), an endopeptidase that cleaves and inactivates endogenous FGF21 (74), has been reported in liver and serum of patients with metabolic liver disease (75–77). Together, these observations have guided the design and development of FGF21 analogs that may restore metabolic homeostasis. The physiology and dysregulation of FGF21 signaling in NASH is discussed in detail below, particularly in the context of FGF21 resolving discrete aspects of NASH pathology.
Fibroblast Growth Factor 21 Physiology
FGF21 relies on its obligate co-receptor, β-Klotho, for recruitment to the extracellular surface of plasma membrane (78). The β-Klotho/FGF21 receptor complex interacts specifically with the cognate receptors: FGFR1c, FGFR2c, or FGFR3c (40, 41), enabling downstream FGFR signaling via pathways including the mitogen-activated protein kinase (MAPK) and AKT signaling networks (79).
Endocrine Role of Fibroblast Growth Factor 21 Physiology
Because FGF21 enters the systemic circulation, it is able to integrate metabolism across liver, adipose tissue, skeletal muscle, pancreas, and other metabolic organs (Figure 2), by controlling expression of transcriptional programs that shape cellular phenotype and tissue metabolic function, ultimately exerting anti-obesity, anti-diabetic, and anti-hyperlipidemic effects in rodents and primates (43, 44).
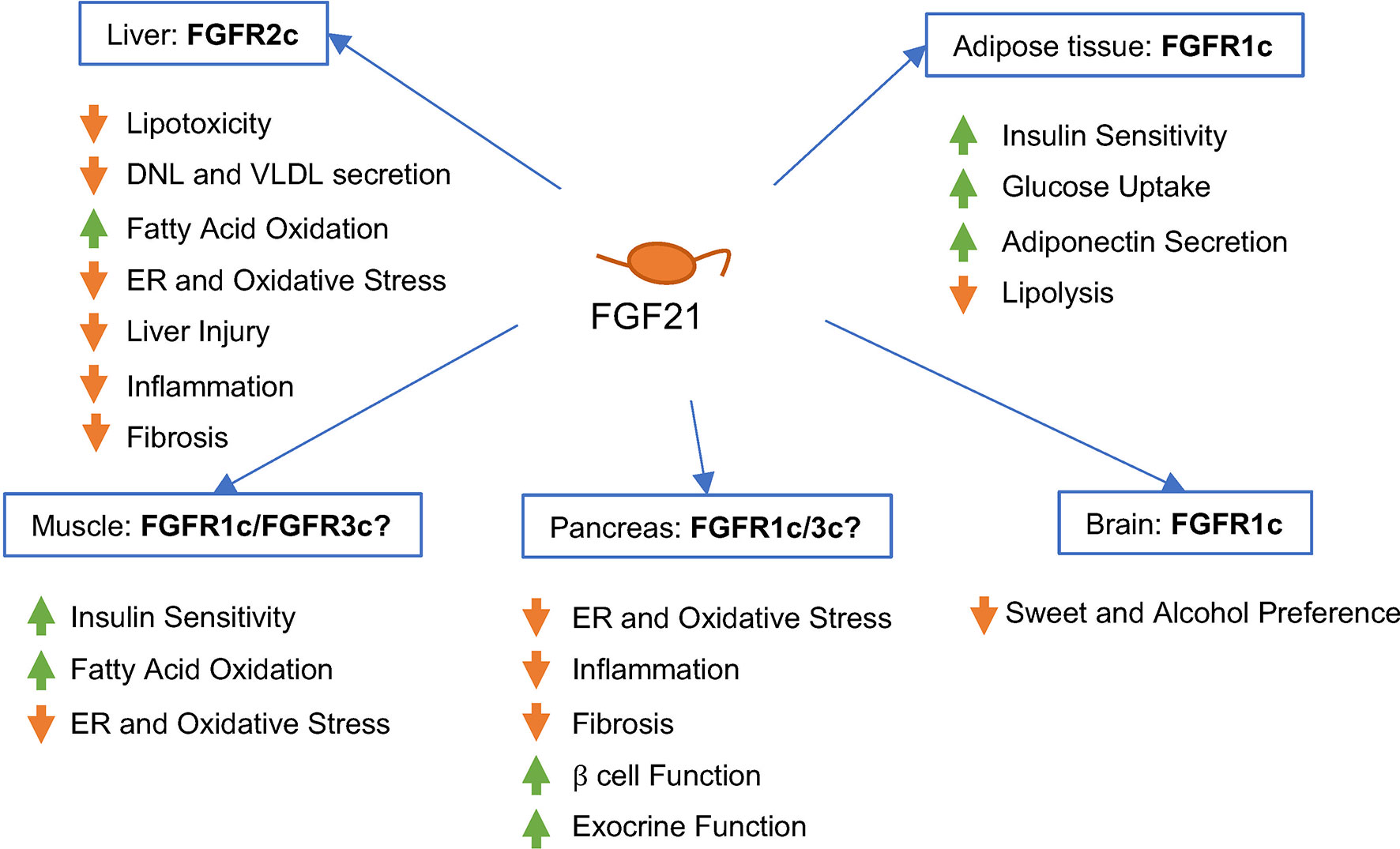
Figure 2 Predicted effects of FGF21 signaling in different human tissues and cell types in vivo. FGF21 binds -Klotho on the cell surface, enabling interaction of the ligand/receptor complex with the FGFR isoform present on that cell type. FGF21 signaling appears to be primarily mediated by FGFR1c in adipose tissue, FGFR2c in liver, and FGFR1c or FGFR3c in pancreas. The contribution of FGFR1c signaling in the brain to the metabolic effects of FGF21 in humans remains to be elucidated.
Expression of FGF21 is regulated by metabolic cues. Changes in organismal nutritional state, such as those induced by fasting or starvation (80), high-carbohydrate diets (81, 82) or low-protein diets (83–86) activate FGF21 expression. These diverse states activate transcription from the FGF21 locus via well-characterized nutritional and metabolite-responsive transcription factors, including fatty acid-responsive nuclear hormone receptors peroxisome proliferator-actived receptor- (PPAR)α and PPARγ (87–92), and the glucose and lipid homeostasis-controlling transcription factor CREBH (93, 94).
FGF21 appears to be mainly synthesized in the liver (95), with dynamic contributions from the pancreas, adipose tissue, and skeletal muscle (96). The target tissues of FGF21’s endocrine actions are determined by overlapping tissue expression patterns for β-Klotho and cognate FGFRs.
In mice, β-Klotho is primarily expressed across metabolic tissues including liver, pancreas, and adipose tissue (96, 97). FGFR1c is much more highly expressed than other FGFR isoforms in adipose tissue and pancreas (41, 73, 96, 98, 99). FGFR2c is more highly expressed than FGFR1c or FGFR3c in liver (96, 100). FGFR2c and KLB (the gene encoding β-Klotho) are induced upon exposure to FGF21 or an FGF21 analog, consistent with enhanced sensitivity to FGF21’s metabolic effects (101).
In humans, FGFR1c is also the predominantly expressed receptor in adipose tissue, with mRNA levels up to two orders of magnitude higher than for FGFR2c or FGFR3c (99, 102). FGF21, FGFR1c, FGFR2c, and FGFR3c are also expressed in skeletal muscle (103, 104). FGFR2c and, to a lesser extent, FGFR3c are expressed basally in liver (102), and FGFR2c is more highly expressed in NAFLD relative to healthy liver (99). Obesity increases expression of FGF21, KLB, FGFR1c, and FGFR3c in the liver, although FGFR2c remains the most highly expressed of FGF21’s receptors in the liver. In contrast, obesity reduces the expression of KLB in subcutaneous and visceral adipose tissue (102).
As an endocrine factor, FGF21 has been shown in preclinical models to exert effects in diverse tissues beyond liver, including adipose tissue, pancreas, skeletal muscle, and the central nervous system. These diverse extrahepatic actions provide broad metabolic benefits and help to resolve hepatic steatosis in patients with NASH. In particular, agonism of FGFR1c appears essential to mediating FGF21’s suppression of fatty acid flux from adipose tissue to liver. On the other hand, direct modulation of metabolism and mediation of FGF21’s cytoprotective effects in liver, described below, likely requires agonism of FGFR2c and possibly FGFR3c. Consequently, analogs of FGF21 designed for treating NASH should posess balanced agonism across FGFR1c, FGFR2c, and FGFR3c.
Integration of Endocrine Actions With Other Hormones
The interaction of FGF21 with other hormones that regulate metabolism is complex. On one hand, FGF21 appears to increase peripheral anabolic signaling, for example through muscle and adipose tissue insulin sensitization, under conditions of adequate or excessive energy intake, as described in detail below. On the other hand, FGF21 appears to augment catabolic hormone signalling under conditions of inadequate energy intake, for example by increasing adrenal gland sensitivity to adrenocorticotropic hormone by upregulating levels of steroidogenic proteins (105). FGF21 also appears to mediate some of the effects of hepatic glucagon and thyroid hormone signaling, since both activation of glucagon receptor (GCGR) (106) and thyroid hormone receptor-β (TR-β) (107) induce secretion of FGF21 by hepatocytes. The cooperative interaction between FGF21, glucagon and thyroid hormone would be expected to increase glucose output and oxidation of fat by liver (108, 109). In the setting of NASH, increasing liver fat oxidation should help reduce steatosis, while exporting energy from the liver as glucose aids in unburdening the liver of excess energy. Simultaneously, the released glucose is taken up more effectively by peripheral organs due to FGF21-enhanced insulin sensitivity, described in detail below.
Autocrine/Paracrine Role of Fibroblast Growth Factor 21 Physiology
Expression and secretion of FGF21 is activated in many cell types experiencing ER stress and oxidative stress. FGF21 induces a number of pathways that serve to protect cells against these stressors, while simultaneously inhibiting pro-cell death pathways. Mitochondrial dysfunction leading to oxidative stress also induces FGF21 expression in preclinical disease models (110), while levels of FGF21 are elevated in the circulation and skeletal muscle of patients with myopathies caused by mutations in components of the mitochondrial oxidative phosphorylation machinery (111).
Enhanced secretory load, imbalances in redox homeostasis, or perturbations in protein homeostasis triggered by lysosomal or autophagosomal dysfunction may all cause ER stress and trigger the unfolded protein response (UPR). Across multiple tissues, FGF21 appears to counteract ER stress (101, 112, 113).
Potential of Fibroblast Growth Factor 21 Physiology as A Therapy For Non-Alcoholic Steatohepatitis
Steatosis and Lipotoxicity in Non-Alcoholic Steatohepatitis
Steatosis in hepatocytes is characterized microscopically by accumulation of triglyceride in numerous cytosolic LDs. Recent studies of human genetic associations with NAFLD and NASH have confirmed the central role of aberrant LD biology in disease etiology. Three risk alleles uncovered in genome-wide association studies on NASH patients include polymorphisms in the LD-associated genes HSD17B13 (114), PNPLA3 (115), and TM6SF2 (116). These risk alleles correlate with increased rates of liver injury, fibrosis, HCC, and liver-related mortality. How these genotypes influence LD homeostasis is the subject of considerable ongoing research. The pathogenic variant of TM6SF2 increases the risk and severity of NASH but reduces cardiovascular disease, seemingly by reducing triglyceride secretion from the liver as VLDL (117). While accumulation of esterified triglyceride in hepatocyte LDs are not directly lipotoxic, their constant turnover releases lipotoxic species including fatty acids, diacylglycerol, free cholesterol and ceramide (118–120). Saturated fatty acids liberated from LDs, or taken up from circulation into the liver, may directly promote tissue injury, leading to additional macrophage recruitment and activation (121). Unesterified fatty acids also destabilize lysosomes leading to activation of NF-κB-dependent tumor necrosis factor α (TNF-α) expression which potentiates lipotoxicity-driven inflammation (122). Palmitate, a major saturated fatty acid, has also been reported to contribute directly and indirectly to proteotoxic stress and ER stress (123). Unsaturated fatty acids under conditions of oxidative stress form highly reactive epoxides resulting in formation of protein adducts (124), which also increase ER stress (125).
Fibroblast Growth Factor 21 Reduces Hepatic Steatosis and Lipotoxicity
Administration of FGF21, FGF21 analogs or adenoviral delivery of FGF21 reduces hepatic steatosis in diverse rodent models of NAFLD and NASH (101, 126–129). The reduction in liver fat results from pleiotropic actions of FGF21, particularly suppressing caloric burden in the liver, reducing de novo lipogenesis, and increasing fat oxidation in the liver. In diet-induced obese (101, 129) or non-obese (130) mice, FGF21 or an FGF21 analog reduces expression of lipogenic genes, including SCD1, FASN, and/or SREBF1, a master regulator of the lipogenic transcriptional network that is overexpressed on a high-fat diet. The reduction in lipogenic gene expression appears attributable to both weight loss and mechanisms independent of weight loss, since a similar magnitude of weight loss induced by a low-calorie diet elicited a smaller reduction in lipogenic gene expression than FGF21 (101). Conversely, an FGF21 analog increases expression of genes involved in mitochondrial β-oxidation of fatty acids (131). The reduction in hepatic steatosis by FGF21 is independent of AMPK-dependent inhibition of acetyl CoA carboxylase (ACC) (132).
Oxidative and Endoplasmic Reticulum Stress in Non-Alcoholic Steatohepatitis
Another of the genetic variants associated strongly with NASH, the I148M variant of patatin-like phospholipase domain-containing protein 3 (PNPLA3) appears to disrupt release of mono-unsaturated fatty acids (MUFAs), which activate peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α) and nuclear factor erythroid 2-related factor 2 (NRF2). These transcription factors mediate increases in fatty acid oxidation capacity and pathways protective against oxidative stress (86–88). NADPH oxidase 4 (NOX4), a ROS-producing enzyme, is highly expressed in the livers of NASH patients and contributes to pathological inflammation and fibrosis in a mouse model of diet-induced NASH (94). Chronic oxidative stress may exacerbate LD accumulation as a means to regenerate NAD from NADH under conditions of excess energy supply (Figure 1B), as observed in cultured mouse hepatocytes in which expression of fatty acid synthase (FASN) and sterol response element-binding protein 1c (SREBF1) are induced (133). Moreover in vivo knockout of genes encoding antioxidant enzymes SOD1 or GPX1 induces hepatic expression of SREBP1c and SREBP2, and increases liver triglyceride content by over 50% (134).
An increase in oxidative species and altered redox leads to an accumulation of misfolded or damaged proteins and in turn ER stress (135). In healthy hepatocytes, autophagy degrades damaged proteins, but this pathway appears disrupted in liver of patients with NAFLD (98). Stabilization and activation of mammalian target of rapamycin complex 1 (mTORC1) by acetyl CoA, the product of fatty acid oxidation, may contribute to disruption of autophagy (136).
The UPR has three canonical branches (PERK/ATF4, IRE1a/XBP1, and ATF6), which play a central role in mitigating cellular stress by enhancing protein folding capacity and secretion, activating the oxidative stress response, and inducing autophagy, thereby restoring protein homeostasis (137). In preclinical studies, UPR dysregulation contributes to oxidative damage and hepatocyte death in a mouse model of fructose-induced NAFLD (138). UPR function also appears compromised in maintaining redox balance within hepatocytes in NAFLD and NASH patients (139). Two branches of the UPR appear, at least in part, to suppress steatosis in animal models (140). Loss of either the ATF6 or the IRE1α pathways sensitizes animals to ER stress-induced liver steatosis, metabolic dysregulation and mortality (140). Ablation of either of these branches increases hepatic lipogenic gene expression and steatosis while leading to unresolved ER stress, characterized by sustained UPR activation and, in particular, elevated ATF4/CHOP signaling. One potential mechanism underlying this steatosis may be upregulation of hepatic VLDL receptor expression by PERK/eIF2α signaling, thereby enhancing hepatocyte triglyceride uptake (141). In addition to contributing to the steatotic drive associated with sustained UPR signaling (140), CHOP mediates the pro-apoptotic effects of the UPR in the post-adaptive phase (137).
Unresolved lipotoxicity, oxidative stress, and ER stress ultimately trigger apoptosis of hepatocytes (Figure 1B). Apoptotic cell death is observed both in liver and in peripheral tissues in NAFLD patients (142, 143). CHOP drives hepatocyte apoptosis in part via transcriptional regulation of autophagy (144). Toxic lipids may themselves stimulate apoptosis via IRE1α-dependent activation of the pro-apoptotic c-Jun N-terminal kinase, JNK1, a MAPK (145). Intracellular free cholesterol levels, which trigger apoptosis of hepatocytes in a JNK1-dependent manner, are elevated in NAFLD patients, correlating with degree of histological disease activity (146). Pro-apoptotic signaling of JNK1 is mediated by the upstream MAP3K apoptosis signal-regulating kinase (ASK1) cascade (147). Apoptotic and lipo-apoptotic cell death is dependent on mitochondrial disruption by pro-apoptotic mediators (e.g., BAX, BAD, etc) that in turn release molecules that activate caspase-mediated degradation and cell death. Upon cell death, hepatocytes release DAMPs, the degradation products of endogenous subcellular structures and proteins that recruit and activate Kupffer cells, thereby initiating and progressing inflammatory liver disease (148).
Fibroblast Growth Factor 21 Reduces Oxidative Stress and Endoplasmic Reticulum Stress
Expression and secretion of FGF21 is induced by oxidative stress. For example, blocking hepatic oxidation of long-chain fatty acids induces oxidative stress and expression of FGF21 (149). Furthermore, acute oxidative stress induced by acetaminophen markedly upregulates expression of FGF21, β-Klotho and antioxidant enzymes in hepatocytes (150). Acting as an autocrine agent, FGF21 activates pathways that protect against oxidative stress. Consistent with potent antioxidant effects in liver, FGF21 protects mouse liver from acetaminophen-induced oxidative damage (150). In wild-type mice, FGF21 treatment increases hepatic transcription of the canonical oxidative stress response and antioxidant genes Sod2 (superoxide dismutase 2), Cat (catalase), Gpx1 (glutathione peroxidase 1), Sirt1 (sirtuin), and Foxo3 (forkhead box transcription factor 3) (151). In liver of obese diabetic mice, FGF21 reduces oxidative damage and lipid peroxidation (152).
FGF21 is also induced by the UPR. In diet-induced obese mice and in NAFLD patients, activation of all three branches of the UPR is observed, with increased nuclear ATF4, ATF6, and XBP1 protein levels along with increased whole-lysate phosphorylated eIF2α, phosphorylated IRE1α, and FGF21 (153). In mice, this induction of FGF21 expression requires IRE1-XBP1 signaling. Acting as an autocrine or paracrine hormone, FGF21 appears both to induce pathways that restore protein homeostasis, for example by enhancing TFEB-dependent lysosomal biogenesis to enable turnover of damaged proteins (154, 155); and also to act as a negative feedback loop to suppress chronic activation of the ATF4/CHOP-mediated, pro-steatotic and pro-apoptotic pathway. The eIF2α/PERK/ATF4 branch of the UPR also activates FGF21 under different conditions of ER stress, including change in ER redox state upon dithiothreitol treatment, or perturbation of ER calcium homeostasis with thapsigargin (156). Because lipid accumulation, lipotoxicity, oxidative stress, and ER stress all appear to be fundamental drivers of NASH pathogenesis, FGF21-based therapies may be broadly useful in a potentially heterogeneous NASH patient population.
Fibroblast Growth Factor 21 Reprograms Hepatocyte Gene Expression
FGF21 signaling in hepatocytes may promote epigenetic reprogramming to a healthier metabolic phenotype. In mice, fasting-induced FGF21 signaling leads to PKA-dependent recruitment of a histone demethylase, JMJD3, to the promoters of autophagy genes (157). JMJD3 demethylation of these promoters de-represses target autophagy genes, which in turn restores functional autophagy and lipid catabolism in obese animals. In fibrotic and cirrhotic liver tissue from patients with NASH (158, 159), reduced expression of HNF4α, a master regulator of mature hepatocyte cell fate, has been observed. In high-fat diet-induced obese mice, FGF21 treatment increases hepatocyte HNF4α by a weight loss-independent mechanism (101). Moreover, transgenic overexpression of FGF21 rescues rats from liver failure by restoring a mature hepatocyte transcriptional profile to dedifferentiated hepatocytes, in turn improving their function (160).
Inflammation and Inflammatory Signaling in Non-Alcoholic Steatohepatitis
Liver resident macrophages, known as Kupffer cells, are activated by DAMPs and pro-inflammatory cytokines released by stressed and apoptotic hepatocytes (15). DAMPs bind and activate pattern recognition receptors (PRRs) on Kupffer cells, including Toll-like receptor 4 (TLR4), which is upregulated in NASH livers, leading to nuclear translocation of a master transcriptional regulator of inflammation, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (161). Mitochondrial DNA released from damaged or dying hepatocytes also activates Kupffer cells via TLR9 receptors (117). Transcription of NF-κB target genes activates Kupffer cells, leading to secretion of pro-inflammatory cytokines and chemokines including TNFα (162), IL-1β (163), IL-6 (164), monocyte chemoattractant protein-1 (MCP-1)/CCL2 (165), and C-X-C motif chemokine ligand-10 CXCL10 (166). These signals serve to recruit monocyte-derived macrophages, neutrophils, and T-helper cells (Th1 and Th17) to the inflammatory niche (167, 168). The resulting, locally elevated concentration of pro-inflammatory molecules directly causes hepatocyte injury and apoptosis, for example via TNFα-mediated death receptor activation (169), initiating and perpetuating a “vicious cycle” of necroinflammation leading to loss of hepatocytes and of the liver’s functional capacity (170).
In addition to release of PRR ligands, hepatocyte stress and death are associated with release of pro-inflammatory cytokines, including inflammasome-mediated interleukin-1β (IL-1β), which further contributes to NASH-associated hepatitis (171). Components of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome are more highly expressed in liver samples from NASH patients than those without NASH (172). The NLRP3 inflammasome cleaves and activates caspase 1, which not only activates IL-1β but also IL-18 (171). In mice, inactivation of NLRP3 or inflammasome signaling suppresses inflammation and fibrosis in models of NASH (173, 174) and alcoholic steatohepatitis (175) by reducing recruitment and activation of Kupffer cells (174). NLRP3 activation in hepatocytes can also lead to caspase 1-mediated pyroptosis, or inflammasome-mediated lytic cell death, amplifying the release of DAMPs beyond just apoptotis-associated DAMP release (176).
Fibroblast Growth Factor-21 Exerts Direct Anti-Inflammatory Effects
FGF21 suppresses inflammation in NASH models, as well as in models of other inflammatory diseases. Consistent with a direct anti-inflammatory effect in multiple cell types, FGF21 inhibits macrophage expression of the pro-inflammatory cytokines TNFα, IL-6, IL-1β, and IFN-γ by inducing NRF2 nuclear translocation, thereby blocking NF-κB activation (177). FGF21 also reduces fatty acid-induced hepatocyte expression of TNFα and the NF-κB subunit p65, and LPS-induced hepatocyte expression of IL-6 in vitro (178). The broad anti-inflammatory effects of FGF21 have also been demonstrated in cell-based models of inflammatory cytokine secretion by pulmonary endothelial cells (179) and human lung cells (180).
These same inflammatory pathways are suppressed in vivo upon FGF21 administration in animal models of NASH and other metabolic diseases. In obese diabetic mice with elevated inflammatory signaling in liver, administration of an FGF21 analog reduces hepatic activation of NF-κB and JNK1/2, inhibiting TNFα expression and consequent macrophage recruitment (152). Conversely, loss of FGF21 increases both liver and plasma cytokine levels in mice, including TNFα, IL-6, and MCP-1, upon chronic alcohol exposure (181). Loss of FGF21 also potentiates fructose-induced hepatic expression of the inflammatory genes MCP-1/CCL2, MIP1α/CCL3, and CD68, suggesting that FGF21 maintains liver health during chronic fructose exposure (182). In liver tissue of high-fat diet-induced obese mice, FGF21 treatment augments expression of the immunosuppressive gene IKBKE and reduces expression of the pro-inflammatory gene IL-18, independent of FGF21-induced weight loss (101). Additionally, an FGF21 analog reduces expression of pro-inflammatory and increases expression of anti-inflammatory genes in non-human primate adipose tissue (183). Finally, FGF21 reduces expression and activation of the NLRP3 inflammasome in vascular endothelial cells in diabetic mice (184), which both protects the cells themselves and reduces further inflammatory damage by suppressing pyroptosis and immune cell recruitment (185).
Consistent with its suppression of pro-inflammatory gene expression, FGF21 inhibits immune cell recruitment and activation in vivo. An FGF21 analog reduces neutrophil and macrophage infiltration in the liver of obese non-human primates with NAFLD (186). Adenovirus-mediated long-term overexpression of FGF21 in high-fat diet-induced obese mice suppresses hepatic and adipose inflammatory cell infiltration and activation (187). FGF21 also indirectly suppresses recruitment of CD4+ Th17 cells and secretion of IL-17 in a choline-deficient, high-fat diet model of NASH, by enhancing adiponectin release from adipose tissue (128). Consistent with suppression of the Th17/IL-17 axis in NASH models, FGF21 reduces the expression of IL-17 and the expansion of Th17 cells in a mouse model of rheumatoid arthritis (188). FGF21 significantly suppresses immune infiltration and inflammatory gene expression in cerulein-induced pancreatitis in mice (189), protects against high-fat diet-induced pancreatic lymphocytic inflammation and islet dysfunction (190), and reduces macrophage infiltration in murine pancreatic ductal adenocarcinoma (191).
In summary, FGF21’s anti-inflammatory actions likely arise from both direct suppression of proinflammatory signalling, by innate and possibly adaptive immune cell types as well as attenuation of immune-effector cell infiltration into the liver, and its indirect inhibition of pro-inflammatory signals released by injured hepatocytes. Because inflammation is a core component and driver of NASH pathology, the anti-inflammatory actions of FGF21-based therapies are likely to benefit the general NASH patient population.
Fibrosis in Non-Alcoholic Steatohepatitis
In addition to exacerbating hepatocyte injury and apoptosis, activation of Kupffer cells and expansion of the inflammatory niche stimulates differentiation of liver-resident HSCs into pro-fibrogenic myofibroblasts (192) (Figure 1B). In particular, hepatocyte- or Kupffer cell-derived TGF-β, platelet-derived growth factor (PDGF) and TNFα stimulate and sustain proliferation and differentiation of HSCs. HSC activation initially promotes wound healing and liver regeneration (193), including elevated production of type I and type III collagen, as well as proteases and cytokines that drive remodeling of extracellular matrix (ECM). However, chronic inflammation sustains this fibrogenic signaling in the HSC niche, leading to progressive liver fibrosis and ultimately cirrhosis (193–196). HSCs are also activated by increased attachment to the ECM, mediated by integrin/focal adhesion kinase signaling, suggesting a self-sustaining program of collagen deposition and HSC activation (197). By secreting inflammatory cytokines and cell adhesion molecules (198), activated HSCs in turn further amplify and perpetuate the inflammatory cycle by promoting macrophage activation and infiltration.
Inflammation is not the only pathogenic mechanism underlying fibrosis in livers of NASH patients, as dysregulation of a number of developmental and metabolic pathways also contributes to fibrosis (199). Notch ligands NOTCH1 and NOTCH2 and their corresponding receptors are upregulated in liver of patients with type 2 diabetes or NASH (200) compared to those without (201). Hepatocytes in particular demonstrate marked upregulation of the canonical Notch target gene, HES1, compared to nonparenchymal cells, correlating with disease severity (132). In a mouse model of diet-induced NASH, Notch signaling in hepatocytes induces osteopontin secretion, paracrine HSC activation and hepatic fibrosis, which are abrogated by Notch inhibition (132). Like Notch signaling components, levels of osteopontin protein and gene expression are positively correlated with NAFLD disease severity and extent of fibrosis (202).
Hepatocyte triglyceride accumulation and cholesterol dysregulation also directly drive HSC activation via induction of the Hippo pathway (146, 199). Marked upregulation of the Hippo pathway transcriptional activator, TAZ, is evident in liver samples from NASH patients, but not from patients with simple steatosis (149). Elevated levels of hepatocyte free cholesterol in NASH (203) stabilize TAZ, which is constitutively degraded (204). Stabilized TAZ directly activates transcription of the Hedgehog pathway ligand, Indian Hedgehog (IHH), which is secreted from hepatocytes and activates HSCs, thereby promoting collagen secretion and fibrogenesis (149). In mice, overexpression of hepatocyte TAZ is sufficient to drive NASH progression, whereas TAZ silencing attenuates inflammation and fibrosis (149). Hepatocyte Hippo pathway activity also upregulates expression of the pro-inflammatory and pro-fibrotic gene, Cyr61/CCN1, which is highly expressed in livers of NASH patients (205).
Stressed and injured NASH hepatocytes express and secrete a second Hedgehog ligand, Sonic Hedgehog (SHH), which is positively correlated with hepatocyte ballooning, steatosis, pericellular fibrosis, and fibrosis stage in liver samples from NASH patients (206, 207). Hepatocytes that highly express SHH appear colocalized within fibrotic areas in livers of NASH patients (208), and are proximal to areas of hepatic progenitor cells (209), consistent with the observation that SHH signaling promotes and maintains a de-differentiated liver cell state (210). SHH-positive livers from NASH patients express higher levels of fibrogenic and immunomodulatory genes compared to SHH-negative NASH liver samples, whose transcriptomes resemble steatotic, non-NASH livers (209). Decreases in SHH expression are significantly associated with improvement in histological features of NASH and reduced prevalence of hepatic progenitor cells in patients treated with vitamin E in the PIVENS trial (211). These data are consistent with a clear pro-fibrotic paracrine signal originating from the injured, ballooned hepatocyte.
In summary, the reprogramming of hepatocytes associated with a profibrotic milieu results in loss of mature parenchymal cells, characterized by loss of the master hepatocyte transcription factor HNF4α (158, 212) and upregulation of the developmental Notch, Hippo (213), and Hedgehog (214) signaling pathways. In addition to accelerating the loss of metabolically competent hepatocytes and liver function, advanced fibrosis and cirrhosis restrict hepatic blood flow, increasing the risk of acute liver failure, HCC, and liver-related mortality (215). The extensive cross-talk between liver cell types during NASH pathogenesis and progression underscores the importance of targeting multiple aspects of liver physiology and pathophysiology in treating this complex disease.
Fibroblast Growth Factor 21 Exerts Direct Anti-Fibrotic Effects
FGF21 and its analogs have demonstrated consistent anti-fibrotic effects in cell culture systems and animal models of NASH, liver injury, or metabolic disease. An FGF21 analog reduces expression of fibrogenic type I collagen and α-SMA in the LX-2 human HSC line induced by either succinate, palmitate, or culture medium devoid of methionine and choline (216). Consistent with these gene expression changes, FGF21 significantly reduces HSC activation and proliferation in vitro (216). FGF21 also inhibits ethanol- or PDGF-induced expression of type I collagen and α-SMA in the T6 rat HSC line (217).
Examining hepatic gene expression and histology in vivo corroborates FGF21’s direct inhibition of fibrosis arising from liver injury induced by diverse insults. For example, in obese diabetic mice fed a methionine and choline-deficient diet (MCD), an FGF21 analog significantly reduces fibrogenic expression and inhibits hepatic fibrosis (152). In the same model, FGF21 also reduces the cholesterol level in the liver, thereby suppressing activation of the Hippo pathway, which in turn attenuates Indian Hedgehog (IHH)-dependent activation of HSCs (204). In contrast, loss of FGF21 potentiates MCD-induced liver injury and fibrosis, which is prevented by administering exogenous FGF21 (218). Similarly, in an alcohol-induced model of hepatic fibrosis, loss of FGF21 exacerbates liver injury and fibrosis (219). FGF21 also inhibits fibrogenesis in two mouse models of chemically-induced liver injury, dimethylnitrosamine- (217) or thioacetamide-induced fibrogenesis (220), by blocking TGF-β expression and signaling, and NF-κB activation.
Additional evidence for FGF21’s direct anti-fibrotic action is provided by multiple models of tissue injury. FGF21 reduces cerulein-induced exocrine pancreatitis in mice by activating FGFR signaling in pancreatic acini and suppressing fibrogenic gene expression (221). Administration or overexpression of FGF21 reduces murine renal fibrosis in a diabetic model (222) and a ureteral obstruction model (223). Likewise, FGF21 administration inhibits formation of bleomycin-induced pulmonary fibrosis in vivo by suppressing oxidative stress via NRF2 activation, and inhibiting TGF-β-induced activation of pulmonary myofibroblasts (224).
FGF21’s breadth of antifibrotic effects are likely mediated both by direct suppression of HSC activation, and by reductions in expression and secretion of pro-fibrotic factors derived from Kupffer cells and injured hepatocytes. With these diverse anti-fibrotic effects, FGF21-based therapies may deliver clinically meaningful histologic improvements independent of the extent of fibrosis at baseline.
Fibroblast Growth Factor 21 Improves Whole-Body Metabolic Health
In contrast to many candidates being clinically tested as potential NASH therapeutics, FGF21 not only exerts direct effects on the liver to improve its metabolic state, but also acts on whole body metabolism to lighten the metabolic load on the liver.
Fibroblast Growth Factor 21 Improves Peripheral Insulin Sensitivity and Promotes Uptake of Energy by Adipose Tissue and Skeletal Muscle
FGF21 stimulates insulin-mediated glucose uptake into adipose tissue (43, 95, 225) in part by increasing expression of the glucose transporter GLUT1 (43, 226). In cultured skeletal muscle myotubes, elevated insulin stimulated FGF21 expression, and FGF21 facilitated insulin-dependent energy uptake by upregulating GLUT1 (227). FGF21 also stimulates adipose tissue uptake of fatty acids derived from lipoproteins (i.e., VLDL and chylomicron) (228), which together with increased glucose uptake functions to direct energy to adipose tissue and effectively redirect calories away from the liver. FGF21 signaling suppresses adipose tissue lipolysis (94, 229), particularly in the fed state (230), and promotes adipocyte lipid homeostasis by ameliorating lipogenesis-associated ER stress (231). FGF21 also increases energy expenditure in mice via UCP1-dependent (232) and UCP1-independent (233) mechanisms, which include browning of white adipose tissue by upregulating PGC1α (234). Together, these effects on adipose tissue and skeletal muscle contribute to the role of FGF21 in mediating protection against metabolic disease in numerous models (230, 235). In diet-induced obese mice, exogenous FGF21 decreases body weight and adiposity while improving whole-body insulin sensitivity and lowering serum glucose, insulin, triglycerides, and cholesterol (129, 235, 236). Similar improvements of insulin sensitivity and glycemic control, have been observed in obese non-diabetic, or diabetic non-human primates (44, 183, 237–239). Consistent with these beneficial effects being mediated through adipose tissue, lipodystrophic mice with severely depleted adiposity, or mice with adipose-specific deletion of FGFR1 have blunted glycemic responses to FGF21 (240, 241).
In addition to directly enhancing energy uptake in adipose tissue, FGF21 potently stimulates secretion of adiponectin from adipocytes (44, 100, 225, 236, 242, 243). Increased levels of adiponectin in non-human primates and humans with metabolic disease (type 2 diabetes, obesity and NASH) have been consistently observed following treatment with various FGF21 analogs (183, 244–246). As an adipokine, adiponectin signals from adipose tissue to distal tissues including liver (247). Adiponectin has demonstrated antidiabetic and insulin sensitizing actions (248), in part by suppressing endogenous glucose production (249) and enhancing peripheral fat uptake (250) by increasing lipoprotein lipase activity and enhancing VLDL catabolism (251).
Consistent with improving uptake of energy by peripheral tissues, adiponectin suppresses hepatic steatosis (252) and inflammation (253) in animal models. Based on in vitro models of NASH, adiponectin may also exert beneficial effects by acting directly on the liver to reduce hepatic stellate cell activation and migration (254–256). Adiponectin’s biological effects in the liver of NASH patients may be reduced, since expression of its receptors is lower than in liver of patients with simple steatosis (257). In summary, FGF21 enhances peripheral uptake of energy both directly through FGFR-mediated pathways, and indirectly via increased adiponectin.
Fibroblast Growth Factor 21 Ameliorates Dyslipidemia
The earliest and most consistent preclinical observation with FG21 was its amelioration of dyslipidemia. Elevations in plasma LDL-cholesterol, non-HDL-cholesterol, ApoB, and triglycerides are well-established, independent, causal risk factors for cardiovascular disease and major adverse cardiovascular events (258). In diabetic, obese or MCD-induced NAFLD non-human primates, FGF21 (44) or FGF21 analogs (237) robustly reduce serum triglycerides, LDL-cholesterol, and VLDL-cholesterol while increasing HDL-cholesterol. Conversely, knockdown of FGF21 in mice fed a ketogenic diet significantly increases serum lipemia and cholesterolemia (87). The consistent, robust effects of FGF21 analogs are described in greater detail below.
Fibroblast Growth Factor 21 Protects Other Organs Stressed by Chronic Energy Excess and Metabolic Dysfunction
Pancreas
By sensitizing adipose tissue and skeletal muscle to insulin, FGF21 reduces demand for insulin secretion, in turn lessening demand for energy generation by β cells. FGF21 also improves β cell homeostasis by promoting autophagy and resistance to ER stress (259, 260). In obese or diabetic mice, FGF21 restores the insulin secretion capacity of β cells in vivo (261) via several pathways: first, by activating AMPK signaling and autophagy (262); second, by reducing the accumulation of lipotoxic lipids in the pancreas by upregulating expression of CPT1 (which facilitates oxidation of long-chain fatty acids) and downregulating the expression of lipogenic genes SREBF1 and FASN (263); third, by restoring the expression of core pancreatic genes including PDX1, INS (insulin), and MafA (264); and finally, by reducing immune cell infiltration and activation within islets (190).
FGF21 also protects the exocrine pancreas from pancreatitis induced by cerulein, associated with accumulation of reactive oxygen species, immune infiltration, and exocrine dysfunction (265). FGF21 mRNA and protein expression is induced within hours of cerulein-induced supraphysiological exocrine secretion, consistent with an adaptive stress response program (221). Transgenic expression of FGF21 in pancreatic acini reduces tissue damage, lipid accumulation, and fibrosis induced by cerulein, all of which were exacerbated in animals lacking FGF21 (221, 266). Further, because the functional cells of the pancreas (both exocrine acini and endocrine islets) are highly secretory, FGF21 plays a protective role by enhancing the response to ER stress via activation of the UPR (189, 259). By restoring protein folding homeostasis and secretory enzyme trafficking, FGF21 enhances digestive enzyme secretion without increasing protein synthesis (259). These protective responses are not limited to cerulein-induced pancreatitis, but are also observed in mouse models of alcohol-induced pancreatitis and obstructive pancreatitis (189). Together, these stress-responsive, anti-inflammatory, and anti-fibrotic actions of FGF21 preserve endocrine and exocrine function.
Cardiac and Skeletal Muscle
As with other cell types, FGF21 protects myocytes under cell stress, particularly ER stress and oxidative stress. Because protein chaperone function requires maintenance of intracellular calcium homeostasis, overloaded myocytes are susceptible to ER stress (267). Increased cytoplasmic calcium levels in cardiomyocytes, arising from chronic overloading or mitochondrial dysfunction, are associated with altered protein folding, ER stress, and induction of FGF21 (113).
By mediating expression of the oxidative stress response genes, FGF21 also protects cardiomyocytes from ischemic cell death (268) and oxidative injury caused by LPS-induced inflammation (269). In addition, FGF21 has been reported to protect the heart from inflammation and fibrosis caused by high-fat diet-induced cardiac steatosis (154). Consistent with this, FGF21 is induced by lipid accumulation in cardiomyocytes (112).
Nervous System
The cytoprotective effects of FGF21, whether direct or indirect, are also evident in the nervous system, where FGF21 treatment attenuates neuroinflammation, neuronal oxidative stress, and neurodegeneration and preserves cognitive function in diabetic mice and high fat fed rats (270, 271). As in other tissues, FGF21 exerts anti-inflammatory effects by suppressing microglial NF-κB signaling, thereby attenuating LPS-induced inflammatory cytokine expression (272). FGF21 also reduces mitochondrial stress in neurons in vitro (273) and in vivo (271), and improves pathology in mouse models of Parkinson’s (274) and Alzheimer’s disease (275).
Fibroblast Growth Factor 21 Modulates Food Preference and Fuel Utilization
FGF21 acts via three distinct mechanisms to modulate whole-body metabolism. It modifies dietary preferences, regulates metabolism at the level of individual organs, and integrates whole-body metabolism of different substrates.
Firstly, modification of dietary preferences by FGF21 are mediated by behavioral changes consistent with altered taste preference in preclinical models and in humans. Consumption of sucrose by both mice and humans induces FGF21, which serves as a negative feedback mechanism to inhibit consumption (82, 276). In mice, FGF21 deletion increases sucrose intake, whereas FGF21 administration or overexpression suppresses consumption of sweetened food and beverages (276, 277). These actions appear to be mediated by β-Klotho-expressing glutamatergic neurons in the ventromedial hypothalamus (276) and paraventricular nucleus (278). FGF21 administration to non-human primates also reduces sweet preference, demonstrating a conserved effect on macronutrient preference (277). FGF21 reduces murine preference for ethanol while suppressing dopamine signaling (277). Conversely, a putative loss-of-function FGF21 allele in humans is associated with greater intake of alcohol (82), preference for sweet foods (279), and altered macronutrient intake (280), but has no effect on total caloric consumption. Additionally, a genome-wide meta-analysis also linked a KLB genetic variant with altered alcohol consumption (281).
Secondly, FGF21 appears to modulate substrate utilization by individual organs. For example, it appears to switch cardiomyocyte energy production from glucose utilization to fatty acid oxidation in mice, dependent on UCP2 induction (282). In myocytes isolated from myopathy patients with an iron-sulfur cluster deficiency, elevated FGF21 expression and secretion correlate with increased expression of ketogenic enzymes (111).
Thirdly, protein restriction in humans (283) and rodents (84), particularly with concomitant high-carbohydrate dietary content (284), increases circulating FGF21. Protein restriction may contribute to perturbations in cellular protein homeostasis, resulting in GCN2-mediated, ATF4-dependent activation of FGF21 expression (84). Higher levels of FGF21 may help to rebalance amino acid availability by enhancing TFEB-dependent lysosomal biogenesis, thereby enabling turnover of proteins within tissues and organs. On a high-fat diet, expression of FGF21 increases whole-body energy expenditure and increases carbohydrate, relative to fatty acid, catabolism (223). Similarly, in diabetic mice, a PEGylated FGF21 analog increases the contribution of glucose to whole-body energy consumption, potentiating glycemic control without affecting body weight (285). On the other hand, mice on a control diet with transgenic overexpression of FGF21 significantly increase ketogenesis in the fed state, consistent with enhanced fat oxidation (89, 286).
In mice, FGF21 appears to improve glycemic control and lipid homeostasis via centrally-mediated pathways (287, 288). For example, selective knock out of β-Klotho in regions of the central nervous system abolishes induction of adipose tissue browning, while central infusion of FGF21 increased energy expenditure (289). While endogenous FGF21 is detectable in human cerebrospinal fluid at much lower concentrations than plasma FGF21, it is positively correlated with adiposity (290). However, the extent of FGF21’s centrally mediated effects remains to be determined in humans. For example, while FGF21 stimulates sympathetic activation in mice, elevations in cortisol that would suggest hypothalamic-pituitary-adrenal activation have not been observed in monkeys (239). In mice, the bulk of the metabolic improvement seen upon FGF21 treatment derives from centrally mediated weight loss, whereas FGF21 analogs elicit limited weight loss in humans. Overall, these differences suggest the contributions of FGF21’s diverse metabolic effects vary across species.
In summary, suppression of dietary intake of fructose and sucrose by FGF21 could both reduce liver fat and maintain it at a lower level. In addition, rebalancing of substrate utilization at both organ and whole body level by FGF21 should contribute to improving and maintaining whole body metabolic homeostasis.
Fibroblast Growth Factor 21 Analogs: Clinical Experience in Patients with Metabolic Disease
In humans, FGF21 analogs have recapitulated many of the lipid effects and some of the glycemic effects observed in preclinical species, though notably not with all compounds tested (Figure 3B). A glycosylated FGF21 variant, LY2405319, maintains the molecular size of endogenous FGF21 but improves its thermal stability and reduces aggregation by engineering an additional disulfide bond and a small number of point mutations into the molecule (291). While the half-life of LY2405319 was not reported, the fact that it was administered daily, coupled with the lack of half-life-extending modifications, suggest a short half-life. Notably, the C-terminal FAP cleavage site remains unmodified, suggesting that LY2405319 is likely susceptible to cleavage and inactivation in human serum via loss affinity for KLB (Figure 3A). Daily administration of LY2405319 to type 2 diabetes patients for 4 weeks significantly reduced triglycerides by about 45% and LDL-cholesterol by 20–30% from baseline, while increasing HDL-cholesterol by 15–20% and adiponectin by up to 80% (244). LY2405319 administration also reduced levels of ApoB from baseline by up to 20% and ApoC-III by up to 35%, consistent with a shift to a less atherogenic lipoprotein profile. There were slight but not statistically significant decreases in body weight, fasting insulin, and fasting glucose from baseline compared to placebo.
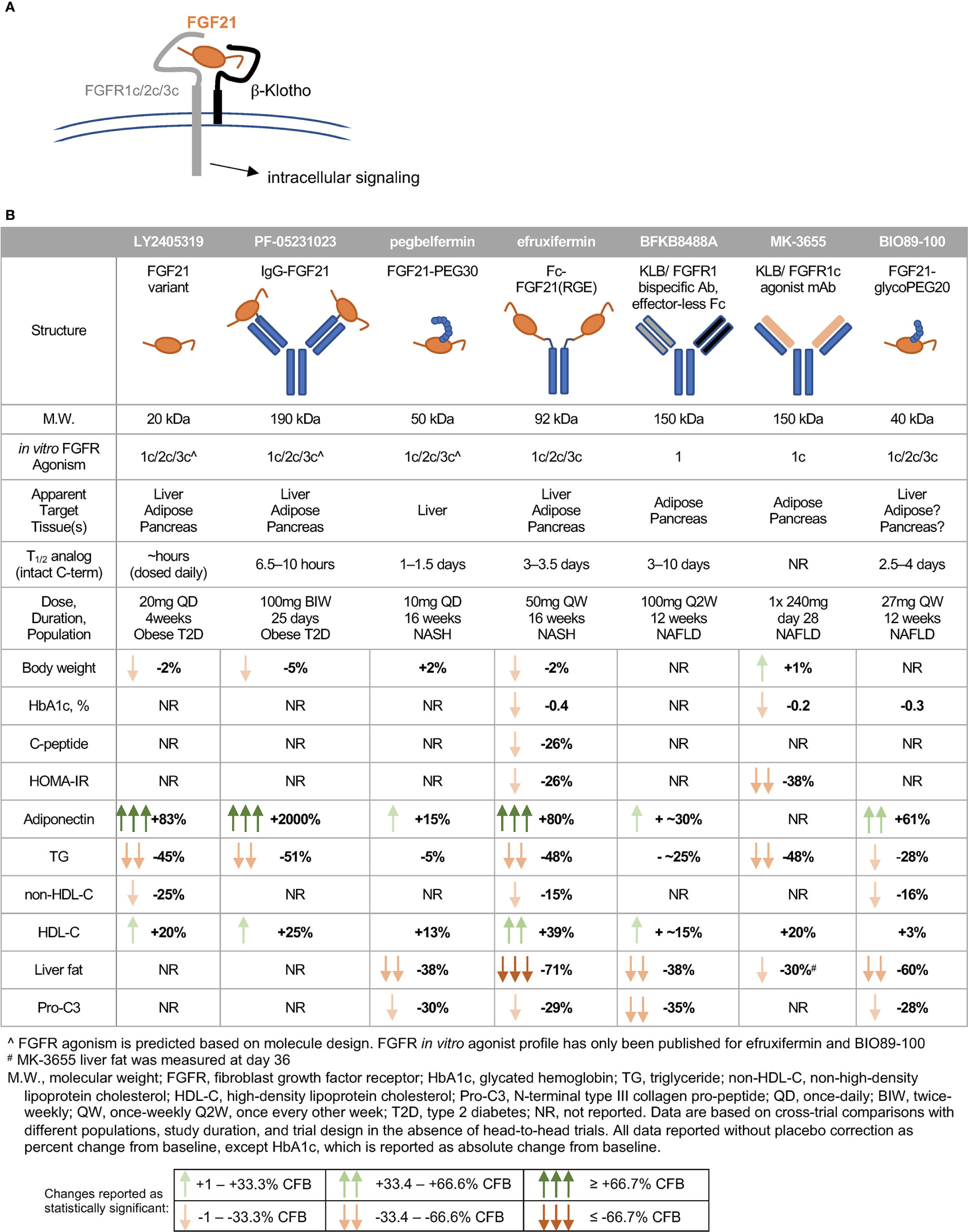
Figure 3 FGF21 analogs demonstrate non-overlapping pharmacological effects in humans. (A) FGF21’s C-terminal domain binds -Klotho, enabling N-terminal residues to interact with the c-isoform of the cognate FGFR. (B) The clinical pharmacodynamic profile of an FGF21 analog is determined by intrinsic potency as an agonist of FGFR1c/2c/3c, pattern of expression of FGFRs in target tissues, and concentration of intact FGF21 analog at each target tissue/FGFR.
Another analog, PF-05231023, is a 190 kDa molecule consisting of human FGF21 covalently linked to each Fab region of a humanized IgG1κ mAb scaffold, resulting in a 2:1 stoichiometry of FGF21 molecules per scaffold (292). In type 2 diabetes patients, twice-weekly administration of PF-05231023 for 4 weeks reduced body weight by about 5%, triglycerides by about 50%, and LDL-cholesterol by about 20% from baseline, while increasing HDL-cholesterol by about 15% and adiponectin 15- to 20-fold (183). However, modest decreases in glucose, insulin, and HOMA-IR (homeostatic model assessment for insulin resistance) did not reach statistical significance (183). Weekly administration of the same molecule for 4 weeks in a second study with obese hypertriglyceridemic subjects demonstrated similar effects on triglycerides, HDL- and non-HDL cholesterol, and adiponectin, but without affecting body weight (293). Although PF-05231023 demonstrated pharmacodynamic effects when dosed weekly or twice-weekly in humans, the unmodified N- and C-termini of the FGF21 moities resulted in a short (approximately 8 h) half-life for the intact C-terminal domain of FGF21 (183). As with LY2405319, it appears unlikely that a high level of agonism of FGF21’s receptors was sustained between doses.
A PEGylated FGF21, pegbelfermin (formerly BMS-986036), consists of a 30 kDa PEG moiety attached to residue 108 of FGF21, yielding a 50 kDa analog (285). One of only two FGF21 analogs tested in a study of biopsy-confirmed NASH patients, pegbelfermin administered daily or weekly for 16 weeks reduced triglycerides by 5% and LDL-cholesterol by about 10% from baseline, while increasing HDL-cholesterol by about 12% and adiponectin by 15% (245). No effects on body weight or glycemic control were reported in this study. While the changes in lipid parameters are smaller compared to those elicited by LY2405319 or PF-05231023, pegbelfermin is the first FGF21 analog to demonstrate beneficial hepatic effects in NASH patients, with daily administration yielding approximately a 30% relative reduction in liver fat content. Pegbelfermin additionally reduced levels of serum alanine aminotransferase (ALT) and Pro-C3 (a marker of fibrogenesis) by up to 30%, consistent with improved hepatic health. Notably, daily dosing of 10 mg pegbelfermin delivered stronger improvements in clinical parameters than weekly dosing of 20 mg pegbelfermin, consistent with a reported intact C-terminal half-life of 1–1.5 days in humans (294). Daily or weekly administration of pegbelfermin for 12 weeks in type 2 diabetes patients recapitulated these modest effects on lipid parameters without altering body weight, and did not elicit clinically or statistically significant changes in markers of glycemic control (295). Because of the location of the PEG moiety and absence of additional modifications at the C-terminus of the FGF21 moiety, pegbelfermin remains susceptible to FAP cleavage and inactivation.
Efruxifermin (formerly AKR-001, AMG 876), the only other FGF21 analog studied in a clinical trial of biopsy-confirmed NASH patients, is a 92 kDa Fc-FGF21fusion protein with a 3–3.5 day half-life (based on intact C-terminal domain of the FGF21 moiety), which is substantially extended relative to the FGF21 analogs described above. This longer half-life results both from endosomal recycling mediated by the neonatal Fc receptor, and from stabilization of the N- and C-termini of the FGF21 moiety. Because the Fc domain is translated directly upstream of a peptide linker and N-terminus of the FGF21 variant, the N-terminus is protected from degradation by dipeptidyl peptidase (DPP) enzymes (296). Protection of the C-terminus is achieved via substitution of a glycine for the native proline at residue 171 (P171G) relative to mature (i.e., signal peptide removed) human FGF21, preventing degradation by FAP. In the context of a potential therapy for NASH, stabilization against FAP degradation is noteworthy because expression of FAP is greatly elevated in liver from NASH patients compared to appropriate control liver (297). Two additional substitutions further enhance the in vivo activity of efruxifermin: A180E, which increases affinity for KLB, and L98R, which decreases aggregation (238). Efruxifermin retains in vitro balanced potency as an agonist of FGFR1c, FGFR2c, and FGFR3c, a characteristic that may be required to elicit maximal effects on liver and extra-hepatic tissues. In a 4-week study in type 2 diabetes patients, weekly administration of up to 70 mg efruxifermin reduced triglycerides by 60–70%, non-HDL-cholesterol by about 35%, fasting insulin about 50%, and fasting glucose about 20% compared to baseline, while increasing HDL-cholesterol by 60% and adiponectin by about 95% (246). Efruxifermin did not significantly alter body weight over this 4-week trial but did significantly reduce the atherogenic lipoproteins ApoB and ApoC-III to a greater extent than did LY2405319 over the same period. Intriguingly, weekly but not every-other-weekly administration of efruxifermin demonstrated for the first time that an FGF21 analog could deliver improved glycemic control in humans, suggesting that the limitations of previous molecules may have been rooted in inadequate exposure. In a subsequent, 16-week study in biopsy-confirmed F1–F3 NASH patients, approximately half of whom were type 2 diabetes patients, efruxifermin improved glycemic control with significant reductions in HbA1c at doses up to 70 mg weekly. Improvements in lipid and lipoprotein profile were also observed, consistent with those seen after 4 weeks’ treatment. Efruxifermin reduced liver fat content by over 70% after 12 weeks dosing. For the first time, an FGF21 analog induced a strong reduction in liver fat that appeared to be associated with resolution of histological features of NASH, including fibrosis (298). An observed rapid and sustained 30% reduction in serum Pro-C3 levels is consistent with preclinical literature demonstrating FGF21’s antifibrotic activity, and with the reduction in Pro-C3 observed upon pegbelfermin treatment. Finally, the longer 16-week dosing revealed efruxifermin-associated weight loss.
A second PEGylated FGF21, BIO89-100, prevents C-terminal degradation by attachment of a PEG moiety to the amino acid residue adjacent to the FAP cleavage site, resulting in a 2.5–4 day half-life in healthy volunteers. This analog retains balanced in vitro potency at FGFR1c, FGFR2c, and FGFR3c (299). In a multiple-ascending dose study in NAFLD subjects, about 20% of whom had biopsy-confirmed NASH, weekly or every-other-weekly administration of BIO89-100 for 12 weeks reduced serum triglycerides by 18–28%, reduced liver fat by 36–60%, and increased adiponectin by 23–61% relative to baseline (299). Notably, only the highest dose of BIO89-100 administered weekly appeared to elicit strong pharmacodynamic effects mediated via FGFR1c activity in adipose tissue, i.e., significantly increasing systemic levels of adiponectin. Every-other-weekly administration of BIO89-100 did not improve markers of glycemic control, with only the highest once-weekly dose showing a trend towards improvements (299). The smaller magnitude of effect on liver fat, HbA1c and lipoprotein profile for BIO89-100 with every-other-weekly dosing appears to limit the value of extending the dosing regimen (299).
Two antibody-based approaches have been developed to mimic FGF21 signaling through FGFR1c only. BFKB8488A is a humanized bispecific antibody that specifically activates the FGFR1/β-Klotho complex (300). In a 12-week study in NAFLD patients, every-other-weekly administration of up to 100 mg BFKB8488A, the highest adequately tolerated dose, reduced liver fat by about 40%, serum triglycerides and Pro-C3 by up to 25 to 30% from baseline, while increasing adiponectin by up to 40% and HDL-cholesterol by up to 20% (301). A parallel study of BFKB8488A in type 2 diabetes patients recapitulated improvements in HDL-cholesterol, triglycerides, and adiponectin, but did not demonstrate improvements in HbA1c, fasting glucose, or fasting insulin (302). Changes in body weight were not reported in either study.
Another monoclonal antibody targeted to β-Klotho and FGFR1c, MK-3655 (formerly NGM313), has been tested in a single-dose study by comparison with pioglitazone (303). It has an extended half-life, and appeared to improve glycemic control in obese, non-diabetic subjects. Liver fat content was reduced by 30–35% relative to baseline at day 36 after administration, while serum triglycerides and LDL-cholesterol were reduced and HDL-cholesterol increased (303). As observed with pioglitazone, MK-3655 was associated with statistically significant weight gain from baseline to day 36. Based on these data, monthly dosing may be achievable with MK3655.
Factors Underlying Differences Between Fibroblast Growth Factor 21 Analogs
While LY2405319 (244), PF-05231023 (183), efruxifermin (246), BFKB8488A (301), and MK-3655 (303) were observed to ameliorate dyslipidemia, with strong reductions in triglycerides and increases in HDL-cholesterol, only efruxifermin and MK-3655 have so far demonstrated the potential to increase insulin sensitivity and to improve glycemic control. Further, while pegbelfermin (245), BFKB8488A (301), BIO89-100 (299), MK-3655 (303), and efruxifermin (298) have all been associated with reductions in ALT, Pro-C3, and hepatic fat fraction, the observed magnitude of liver fat reduction was greatest following efruxifermin treatment. Several structural parameters differentiate these molecules that may explain the different clinical profiles (Figure 3B).
Maximal reduction of liver fat appears to require both inhibition of hepatic de novo lipogenesis and suppression of adipose tissue lipolysis (51, 52). For FGF21 analogs, this is likely to require balanced agonism of FGFR1c, FGFR2c and possibly FGFR3c. The almost 2-fold greater reduction in liver fat with efruxifermin than with FGFR1-selective BFKB8488A is consistent with this argument, as FGFR1c agonism would mainly reduce steatosis by suppressing adipose tissue lipolysis. While the in vitro potency of pegbelfermin as an agonist of FGFR1c, FGFR2c and FGFR3c has not been reported, the modest induction of adiponectin in humans suggests that it exerts minimal action through adipose tissue FGFR1c in humans. As a result, suppression of adipose tissue lipolysis by pegbelfermin is likely limited. Consistent with this, pegbelfermin reduced liver fat by about half the extent of efruxifermin, and serum triglyceride by about 10%, compared to about 30–50% for other FGF21 analogs, including FGFR1c selective antibodies. BIO89-100, on the other hand, appears to deliver relative hepatic fat loss in between efruxifermin and pegbelfermin, with the highest dose reducing fat content by 60% in NAFLD patients, and all other doses reducing fat content by 36–50% (299). While BIO89-100 has been reported to be a balanced agonist of FGF21’s receptors in vitro, the smaller magnitude of effect on adiponectin compared to efruxifermin suggest that BIO89-100 may not have maximally stimulated FGFR1c signaling in adipose tissue at the doses tested in humans (Figure 3B). Notably, comparisons of these analogs are limited because efruxifermin and pegbelfermin have been evaluated in patients with confirmed NASH, while BIO89-100 has been studied in patients with less severe NAFLD.
FGF21’s effects on glycemic control and insulin sensitivity are likely mediated predominantly by FGFR1c signaling in adipose tissue, which should normally serve as one of the body’s main depots for excess calories in response to post-meal insulin. To enhance insulin sensitivity and improve glycemic control, it appears that FGF21 analogs must maintain FGFR1c activation in adipose tissue above a threshold level throughout the inter-dose interval. For example, 70 mg efruxifermin dosed weekly, corresponding to two pharmacokinetic half-lives, appeared to improve HOMA-IR, fasting glucose, and fasting insulin in type 2 diabetes patients (246). By comparison, every-other-weekly dosing of 140 mg achieved equivalent total exposure over 4 weeks, but with four pharmacokinetic half-lives between doses, did not appear to improve glycemic control, despite comparable effects on serum triglycerides and lipoproteins. Consistent with this, the FGR1c-selective MK-3655, with the long half-life of a monoclonal antibody, is the only other FGF21 analog that appears to improve glycemic control, though repeat-dose data has not been reported. The β-Klotho/FGFR1 bispecific agonist antibody, BFKB8488A, failed to improve glycemic control, suggesting that there may have been insufficient agonism of FGFR1c throughout the every-other-weekly dosing interval.
Differentiation Between Analogs of Fibroblast Growth Factor 21 and Fibroblast Growth Factor 19
An FGF19 analog, aldafermin (formerly NGM282, M70) has a five-amino acid deletion and three amino acid substitutions in the N-terminal region of human FGF19 (304). This engineering was necessary to reduce the proliferative effect of native FGF19 on hepatocytes, mediated via signaling through FGFR4—the only FGF receptor at which FGF19 and FGF21 differ in agonist activity (305). When administered to patients with NASH for 12 weeks, aldafermin (up to 6 mg daily) reduced liver fat content by up to 60% relative to baseline, reduced ALT by about 50%, and decreased Pro-C3 by about 20–25% (31). However, consistent with FGFR4-mediated suppression of CYP7A1, which catalyzes the rate limiting step of cholesterol metabolism to bile acid, aldafermin significantly increased LDL-cholesterol and non-HDL cholesterol without exerting a consistent effect on insulin, glucose, or HbA1c. In a longer-term 24-week study in NASH patients, daily administration of 1 mg aldafermin led to an approximately 30% placebo-adjusted relative reduction in liver fat while reducing ALT by about 45% from baseline. However, statin co-administration was required in more than 95% of treated subjects to mitigate the increase in LDL-cholesterol, which was also observed previously at higher doses (306). This study confirmed that aldafermin treatment did not improve glycemic control. The lack of effect of FGF19 on glycemic control is consistent with preclinical studies. For example, selective FGFR4 agonism, either peripherally or in liver, does not affect fasting glucose in obese diabetic mice, but increases triglycerides and total cholesterol (307, 308). Conversely, either germline knockout or antisense oligonucleotide knockdown of FGFR4 expression in mice enhances hepatic fat oxidation, glucose metabolism, and insulin sensitivity (308–310) while reducing hepatic steatosis (308, 309). While these metabolic effects were likely to have been mediated by compensatory upregulation of FGF19 and/or FGF21 (308), they were clearly independent of FGFR4 agonism, instead being mediated by agonism of FGFR1c, 2c or 3c.
As elevated bile acids are associated with fibrotic liver disease, and FGF19–FGFR4 signaling suppresses bile acid synthesis, it has been posited that reducing bile acids may deliver anti-fibrotic actions (311). However, anti-fibrotic actions of FGF19 signaling do not appear to depend on FGFR4 signaling, based on three lines of evidence. First, both FGF21 (216, 217) and FGF19 (312) directly inhibit HSC activation and proliferation in vitro. Second, FGF21 suppresses TGF-β release from pro-inflammatory macrophages in vitro (177), which would be expected to reduce Kupffer cell activation of HSCs in vivo. While a similar effect does not appear to have been reported for FGF19, this indirect anti-fibrotic effect of FGF21 suggests that FGFR4 is unnecessary for inhibition of Kupffer cell-HSC signaling. Third, analogs of FGF21 and FGF19 appear to reduce markers of liver injury (ALT) and fibrosis (Pro-C3) to a similar extent in humans (245, 306), consistent with FGFR4 agonism not being required in vivo, while being associated with an undesirable increase in LDL-C.
Safety and Tolerability of Fibroblast Growth Factor 21 Analogs in Clinical Trials
Preclinical studies have suggested that FGF21 increases tone of the hypothalamic-pituitary-adrenal (HPA) axis (287, 288). However, whether FGF21 analogs modulate the HPA axis in humans remains incompletely characterized. Increased levels of corticotropin-releasing hormone or corticosterone/cortisol with FGF21 or FGF21 analog treatment have been reported in rodents, but not in non-human primates nor in any human studies (183, 293, 313). Likewise, reports of FGF21 increasing water intake and blood pressure in rodents, potentially by stimulating sympathetic pathways (313, 314) do not appear to translate consistently to humans, with one study (293) but no others (183, 244–246, 315) suggesting an effect. The lack of effect is consistent with blood pressure phenotypes of humans with FGF21 genetic variants. The minor A allele of rs838133 corresponds to a putative FGF21 loss-of-function allele, which is associated with 0.29 mmHg higher systolic blood pressure per allele (279). This study also identified a rare variant encoding a truncated, putatitive loss-of-function FGF21 protein, associated with 7.5 mmHg higher blood pressure. Additionally, administration or overexpression of FGF21 reduces blood pressure in various preclinical models of hypertension (316–318). Overall, the preclinical and clinical data do not support a likely hypertensive effect of FGF21 analogs downstream of increased sympathetic tone in humans.
The apparent differences between rodents and primates may reflect differences in blood-brain barrier permeability across species (319). Mice in particular (320) may be more sensitive to systemically administered FGF21 and FGF21 analogs than humans. Larger molecules based on antibody scaffolds and fragments, including PF-05231023 (190 kDa), BFKB8488A and MK-3655 (150 kDa), and efruxifermin (92 kDa) may be less likely to cross the blood-brain barrier at therapeutic doses, but their tissue distribution has not been described. Emerging clinical observations with smaller FGF21 analogs including pegbelfermin (50 kDa) and BIO89-100 (40 kDa), may provide insights into effects possibly mediated via the central nervous system.
While some preclinical studies with FGF21 and FGF21 analogs have suggested effects on bone turnover, clinical data have not demonstrated a consistent effect. One group demonstrated that FGF21 increased bone resorption and decreased bone mass in mice (321), in part via induction of hepatic insulin-like growth factor binding protein-1 (IGFBP1) (322). However, a more recent preclinical study did not find evidence of FGF21-induced bone loss in a diet-induced model of obesity (323). While the first clinical trial of PF-05231023 demonstrated a decrease in markers of bone formation and an increase in markers of bone resorption, this was accompanied by a significant reduction in body weight. A subsequent study of PF-05231023 did not demonstrate weight loss, and there were no changes observed in markers of bone formation or resorption, IGFBP1–3, or free IGF1 (293). No subsequent clinical studies of FGF21 analogs in adults have demonstrated a significant effect on bone density or markers of bone resorption and formation in adults (245, 246, 295). Notably, while markers of type I collagen synthesis and degradation are frequently used to indicate changes in bone turnover, they may also reflect changes in liver fibrogenesis or fibrinolysis (324, 325). Overall, the weight of evidence to date does not support a role for FGF21 in mediating bone remodeling in adult humans.
The most prevalent and consistent dose-limiting side effects observed across studies of FGF21 analogs in patients with metabolic disease are gastrointestinal (GI), including mild-to-moderate nausea and diarrhea (183, 245, 246, 298, 301). These side effects were also seen in trials of the FGF19 analog, aldafermin (31, 306). However, preclinical studies of these various endocrine FGF analogs in rodents and monkeys did not reveal GI symptoms, therefore the potential mechanisms underlying adverse GI effects have not been investigated (Table 2). It is possible that FGF21 exerts some direct or indirect effect on the GI tract, but experiments directly testing this hypothesis have not been reported to date.

Table 2 Readthrough from preclinical to clinical safety and tolerability for FGF21 analogs.
Whether GI tolerability affects patient adherence to or uptake of FGF21-based therapies in NASH remains to be seen. However, precedent suggests that mild to moderate adverse GI events are not a major barrier to uptake of a promising metabolic therapy. While GLP-1 analogs for treatment of type 2 diabetes cause mild-to-moderate nausea and vomiting in a significant proportion of patients, their benefits in terms of weight loss and improved glycemic control have allowed the class to overcome these tolerability concerns and become one of the most-prescribed classes of type 2 diabetes medications in the US and Europe (326). Second, up-titration regimens for GLP-1 analogs have mitigated adverse GI side effects, reducing their prevalence and severity in both clinical trials and post-marketing surveillance (327). FGF21 analogs have not so far employed up-titration, but given the reportedly mild-to-moderate as well as the transient nature of the nausea and diarrhea, such an approach could reasonably be expected to mitigate the GI side effects.
Conclusions and Outlook
Despite uninspiring outcomes in clinical trials of the first wave of potential NASH therapies, a spate of promising mid-to-late stage clinical trials has provided a needed morale boost to NASH patients, endocrinologists and hepatologists, and caregivers. The complex pathophysiology of NASH mandates a holistic approach to treating patients, focusing not only on liver histology but also cardiovascular risk and glycemic control, which has proven to be a high bar (Table 1).
An FXR agonist, obeticholic acid, is the first molecule to deliver a statistically significant improvement in one of the FDA’s surrogate endpoints for accelerated approval of a treatment for NASH. However, it is associated with a risk of liver injury in patients with primary biliary cholangitis (328), and significantly elevates LDL-cholesterol (30, 32). FGF19, like FXR agonists, has demonstrated potential to improve liver histology, but also exacerbates dyslipidemia already common in NASH patients (306). Inhibitors of acetyl-CoA carboxylase demonstrate a reduction in hepatic fat fraction, but significantly increase serum triglyceride levels (34). PPAR agonists have demonstrated insulin sensitizing effects and some liver-specific improvements, but are associated with increased body weight (329). The GLP-1 analog semaglutide demonstrated a strong ability to resolve NASH, and is associated with weight loss and improved cardiovascular health, but did not statistically significantly improve fibrosis (326). TR-β agonists have demonstrated improvements in liver fat and biomarkers of cardiovascular disease along with significant resolution of NASH, but have not demonstrated a strong anti-fibrotic effect (330). Across the field of potential NASH drugs, each mechanistic class appears to deliver improvements in a subset of liver histology, whole-body metabolism, or cardiovascular risk profile, but not all three. Moreover, some mechanisms worsen cardiovascular risk profile despite improving liver health, which appears incompatible with long-term use in patients with increased cardiovascular risk.
FGF21 analogs therefore represent a promising emerging class of NASH therapeutics, as a number of them have demonstrated pleiotropic effects consistent with improved liver health and whole-body metabolism. Because of an elevated risk of cardiovascular morbidity and mortality in NASH patients prior to advanced fibrosis, as well as hepatic decompensation, HCC, and/or liver failure, therapeutic options must address the complex pathophysiology of this disease. Analogs of FGF21 that are able to maintain balanced and sustained agonism of FGFR1c, FGFR2c, and FGFR3c throughout the inter-dose interval appear to show the most promising pharmacological profiles, with among the largest reported decreases in liver fat of any therapeutic mechanism tested in NASH patients, and with encouraging signs of resolving NASH histology and fibrosis after relatively short-term treatment. FGF21 analogs with balanced receptor agonism and sustained exposure have also been shown to improve glycemic control, ameliorate dyslipidemia and reduce body weight. Should this pharmacological profile be confirmed in late-stage clinical studies, optimized FGF21 analogs have the potential to not only resolve liver pathology, but also to reduce risk of cardiovascular morbidity and mortality, and incidence of organ damage associated with inadequate glycemic control.
Author Contributions
ET and TR wrote the first draft, edited, and wrote the final draft of this article. All authors contributed to the article and approved the submitted version.
Conflict of Interest
ET is an employee and shareholder of Akero Therapeutics. TR is a co-founder, employee, and shareholder of Akero Therapeutics, and a shareholder of Pfizer.
Acknowledgments
We thank Kerry af Forselles of afPharmative Insights for critical reading of this review.
References
1. The GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med (2017) 377:13–27. doi: 10.1056/NEJMoa1614362
2. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
3. Albhaisi S, Sanyal A. Recent advances in understanding and managing non-alcoholic fatty liver disease. F1000Res (2018) 7:720. doi: 10.12688/f1000research.14421.1
4. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology (2010) 52:1836–46. doi: 10.1002/hep.24001
5. Bril F, Barb D, Portillo-Sanchez P, Biernacki D, Lomonaco R, Suman A, et al. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology (2017) 65:1132–44. doi: 10.1002/hep.28985
6. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
7. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
8. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology (2018) 67:123–33. doi: 10.1002/hep.29466
9. Hagström H, Nasr P, Ekstedt M, Hammar U, Stål P, Hultcrantz R, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol (2017) 67:1265–73. doi: 10.1016/j.jhep.2017.07.027
10. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology (2015) 148:547–55. doi: 10.1053/j.gastro.2014.11.039
11. Noureddin M, Vipani A, Bresee C, Todo T, Kim IK, Alkhouri N, et al. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. Am J Gastroenterol (2018) 113:1649–59. doi: 10.1038/s41395-018-0088-6
12. Golabi P, Otgonsuren M, Cable R, Felix S, Koenig A, Sayiner M, et al. Non-alcoholic Fatty Liver Disease (NAFLD) is associated with impairment of Health Related Quality of Life (HRQOL). Health Qual Life Outcomes (2016) 14:1–7. doi: 10.1186/s12955-016-0420-z
13. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (2005) 41:1313–21. doi: 10.1002/hep.20701
14. Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol (2018) 69:927–47. doi: 10.1016/j.jhep.2018.06.008
15. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol (2018) 34:274. doi: 10.1038/s41575-018-0082-x
16. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol (2018) 15:738–52. doi: 10.1038/s41575-018-0065-y
17. Bian Z, Ma X. Liver fibrogenesis in non-alcoholic steatohepatitis. Front Physiol (2012) 3:248. doi: 10.3389/fphys.2012.00248
18. Gehrke N, Schattenberg JM. Metabolic Inflammation-A Role for Hepatic Inflammatory Pathways as Drivers of Comorbidities in Nonalcoholic Fatty Liver Disease? Gastroenterology (2020) 158:1929–47.e6. doi: 10.1053/j.gastro.2020.02.020
19. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology (2017) 65:1557–65. doi: 10.1002/hep.29085
20. Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology (2005) 129:113–21. doi: 10.1053/j.gastro.2005.04.014
21. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med (2010) 363:1341–50. doi: 10.1056/NEJMra0912063
22. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology (2018) 67:328–57. doi: 10.1002/hep.29367
23. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (2016) 64:73–84. doi: 10.1002/hep.28431
24. Vilar-Gomez E, Martinez-Perez Y, Calzadilla Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology (2015) 149:367–78.e5– quiz e14–5. doi: 10.1053/j.gastro.2015.04.005
25. Glass LM, Dickson RC, Anderson JC, Suriawinata AA, Putra J, Berk BS, et al. Total body weight loss of ≥ 10 % is associated with improved hepatic fibrosis in patients with nonalcoholic steatohepatitis. Dig Dis Sci (2015) 60:1024–30. doi: 10.1007/s10620-014-3380-3
26. Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology (2018) 155:1140–53. doi: 10.1053/j.gastro.2018.07.006
27. Harrison SA, Wong VWS, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatol (2020) 73:26–39. doi: 10.1016/j.jhep.2020.02.027
28. Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol (2020) 72:816–27. doi: 10.1016/j.jhep.2019.11.024
29. Patel K, Harrison SA, Elkashab M, Trotter JF, Herring R, Rojter S, et al. Cilofexor, a Nonsteroidal FXR Agonist, in Non-Cirrhotic Patients with Nonalcoholic Steatohepatitis: A Phase 2 Randomized Controlled Trial. Hepatology (2020) 72:58–71. doi: 10.1002/hep.31205
30. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (2019) 394:2184–96. doi: 10.1016/S0140-6736(19)33041-7
31. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet (2018) 391:1174–85. doi: 10.1016/S0140-6736(18)30474-4
32. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet (2015) 385:956–65. doi: 10.1016/S0140-6736(14)61933-4
33. Kim C-W, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab (2017) 26:394–406.e6. doi: 10.1016/j.cmet.2017.07.009
34. Alkhouri N, Lawitz E, Noureddin M, DeFronzo R, Shulman GI. GS-0976 (Firsocostat): an investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin Invest Drugs (2020) 29:135–41. doi: 10.1080/13543784.2020.1668374
35. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J Hepatol (2019) 72:613–26. doi: 10.1016/j.jhep.2019.10.023
36. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med (2010) 362:1675–85. doi: 10.1056/NEJMoa0907929
37. Wu X, Ge H, Lemon B, Vonderfecht S, Baribault H, Weiszmann J, et al. Separating mitogenic and metabolic activities of fibroblast growth factor 19 (FGF19). Proc Natl Acad Sci (2010) 107:14158–63. doi: 10.1073/pnas.1009427107
38. Itoh N, Nakayama Y, Konishi M. Roles of FGFs As Paracrine or Endocrine Signals in Liver Development, Health, and Disease. Front Cell Dev Biol (2016) 4:30. doi: 10.3389/fcell.2016.00030
39. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol (2007) 27:3417–28. doi: 10.1128/MCB.02249-06
40. Ogawa Y, Kurosu H, Yamamoto M, Nandi A, Rosenblatt KP, Goetz R, Kuro-O M, et al. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci (2007) 104:7432–7. doi: 10.1073/pnas.0701600104
41. Kurosu H, Choi M, Ogawa Y, Dickson AS, Goetz R, Eliseenkova AV, Kuro-O M, et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem (2007) 282:26687–95. doi: 10.1074/jbc.M704165200
42. Makoto K-O. The Klotho proteins in health and disease. Nat Rev Nephrol (2018) 15:27–44. doi: 10.1038/s41581-018-0078-3.
43. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest (2005) 115:1627–35. doi: 10.1172/JCI23606
44. Kharitonenkov A, Wroblewski VJ, Koester A, Chen Y-F, Clutinger CK, Tigno XT, et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology (2007) 148:774–81. doi: 10.1210/en.2006-1168
45. Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology (2002) 143:1741–7. doi: 10.1210/endo.143.5.8850
46. Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab (2011) 13:729–38. doi: 10.1016/j.cmet.2011.03.019
47. Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta (2000) 1492:203–6. doi: 10.1016/s0167-4781(00)00067-1
48. Degirolamo C, Sabbà C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov (2016) 15:51–69. doi: 10.1038/nrd.2015.9
49. Wu A-L, Coulter S, Liddle C, Wong A, Eastham-Anderson J, French DM, et al. FGF19 regulates cell proliferation, glucose and bile acid metabolism via FGFR4-dependent and independent pathways. PLoS One (2011) 6:e17868. doi: 10.1371/journal.pone.0017868
50. Finck BN. Targeting Metabolism, Insulin Resistance, and Diabetes to Treat Nonalcoholic Steatohepatitis. Diabetes (2018) 67:2485–93. doi: 10.2337/dbi18-0024
51. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest (2005) 115:1343–51. doi: 10.1172/JCI23621
52. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology (2014) 146:726–35. doi: 10.1053/j.gastro.2013.11.049
53. Eissing L, Scherer T, Tödter K, Knippschild U, Greve JW, Buurman WA, et al. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat Commun (2013) 4:1528–11. doi: 10.1038/ncomms2537
54. Kim M-S, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest (2016) 126:4372–86. doi: 10.1172/JCI81993
55. Softic S, Cohen DE, Kahn CR. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig Dis Sci (2016) 61:1282–93. doi: 10.1007/s10620-016-4054-0
56. Chatrath H, Vuppalanchi R, Chalasani N. Dyslipidemia in patients with nonalcoholic fatty liver disease. Semin Liver Dis (2012) 32:22–9. doi: 10.1055/s-0032-1306423
57. Videla LA, Rodrigo R, Orellana M, Fernández V, Tapia G, Quiñones L, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci (2004) 106:261–8. doi: 10.1042/CS20030285
58. Bellanti F, Villani R, Facciorusso A, Vendemiale G, Serviddio G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic Biol Med (2017) 111:173–85. doi: 10.1016/j.freeradbiomed.2017.01.023
59. Lin X, Li G, He X, Ma X, Zhang K, Zhang H, et al. FGF21 inhibits apolipoprotein(a) expression in HepG2 cells via the FGFR1-ERK1/2-Elk-1 pathway. Mol Cell Biochem (2014) 393:33–42. doi: 10.1007/s11010-014-2044-0
60. Do HT, Tselykh TV, Mäkelä J, Ho TH, Olkkonen VM, Bornhauser BC, et al. Fibroblast growth factor-21 (FGF21) regulates low-density lipoprotein receptor (LDLR) levels in cells via the E3-ubiquitin ligase Mylip/Idol and the Canopy2 (Cnpy2)/Mylip-interacting saposin-like protein (Msap). J Biol Chem (2012) 287:12602–11. doi: 10.1074/jbc.M112.341248
61. Adams AC, Kharitonenkov A. FGF21: The center of a transcriptional nexus in metabolic regulation. Curr Diabetes Rev (2012) 8:285–93. doi: 10.2174/157339912800840505
62. Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol (2010) 53:934–40. doi: 10.1016/j.jhep.2010.05.018
63. Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, et al. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Invest (2010) 40:887–92. doi: 10.1111/j.1365-2362.2010.02338.x
64. Barb D, Bril F, Kalavalapalli S, Cusi K. Plasma Fibroblast Growth Factor 21 Is Associated With Severity of Nonalcoholic Steatohepatitis in Patients With Obesity and Type 2 Diabetes. J Clin Endocrinol Metab (2019) 104:3327–36. doi: 10.1210/jc.2018-02414
65. Zhang X, Yeung DCY, Karpisek M, Stejskal D, Zhou Z-G, Liu F, et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes (2008) 57:1246–53. doi: 10.2337/db07-1476
66. Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology (2010) 139:456–63. doi: 10.1053/j.gastro.2010.04.054
67. Woo YC, Lee CH, Fong CHY, Xu A, Tso AWK, Cheung BMY, et al. Serum fibroblast growth factor 21 is a superior biomarker to other adipokines in predicting incident diabetes. Clin Endocrinol (Oxf) (2017) 86:37–43. doi: 10.1111/cen.13229
68. Marchelek-Myśliwiec M, Dziedziejko V, Nowosiad-Magda M, Dołęgowska K, Dołęgowska B, Pawlik A, et al. Chronic Kidney Disease Is Associated with Increased Plasma Levels of Fibroblast Growth Factors 19 and 21. Kidney Blood Press Res (2019) 44:1207–18. doi: 10.1159/000502647
69. Lee CH, Hui EYL, Woo YC, Yeung CY, Chow WS, Yuen MMA, et al. Circulating fibroblast growth factor 21 levels predict progressive kidney disease in subjects with type 2 diabetes and normoalbuminuria. J Clin Endocrinol Metab (2015) 100:1368–75. doi: 10.1210/jc.2014-3465
70. Chow W-S, Xu A, Woo YC, Tso AWK, Cheung SCW, Fong CHY, et al. Serum fibroblast growth factor-21 levels are associated with carotid atherosclerosis independent of established cardiovascular risk factors. Arterioscler Thromb Vasc Biol (2013) 33:2454–9. doi: 10.1161/ATVBAHA.113.301599
71. Wu L, Qian L, Zhang L, Zhang J, Zhou J, Li Y, et al. Fibroblast Growth Factor 21 is Related to Atherosclerosis Independent of Nonalcoholic Fatty Liver Disease and Predicts Atherosclerotic Cardiovascular Events. J Am Heart Assoc (2020), 9:e015226. doi: 10.1161/JAHA.119.015226
72. Lin Z, Wu Z, Yin X, Liu Y, Yan X, Lin S, et al. Serum levels of FGF-21 are increased in coronary heart disease patients and are independently associated with adverse lipid profile. PLoS One (2010) 5:e15534. doi: 10.1371/journal.pone.0015534
73. Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, et al. Obesity Is a Fibroblast Growth Factor 21 (FGF21)-Resistant State. Diabetes (2010) 59:2781–9. doi: 10.2337/db10-0193
74. Dunshee DR, Bainbridge TW, Kljavin NM, Zavala-Solorio J, Schroeder AC, Chan R, et al. Fibroblast Activation Protein Cleaves and Inactivates Fibroblast Growth Factor 21. J Biol Chem (2016) 291:5986–96. doi: 10.1074/jbc.M115.710582
75. Levy MT, McCaughan GW, Abbott CA, Park JE, Cunningham AM, Müller E, et al. Fibroblast activation protein: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology (1999) 29:1768–78. doi: 10.1002/hep.510290631
76. Williams KH, Viera de Ribeiro AJ, Prakoso E, Veillard AS, Shackel NA, Bu Y, et al. Lower serum fibroblast activation protein shows promise in the exclusion of clinically significant liver fibrosis due to non-alcoholic fatty liver disease in diabetes and obesity. Diabetes Res Clin Pract (2015) 108:466–72. doi: 10.1016/j.diabres.2015.02.024
77. Uitte de Willige S, Keane FM, Bowen DG, Malfliet JJMC, Zhang HE, Maneck B, et al. Circulating fibroblast activation protein activity and antigen levels correlate strongly when measured in liver disease and coronary heart disease. PLoS One (2017) 12:e0178987. doi: 10.1371/journal.pone.0178987
78. Lee S, Choi J, Mohanty J, Sousa LP, Tome F, Pardon E, et al. Structures of β-klotho reveal a “zip code-” like mechanism for endocrine FGF signalling. Nature (2018) 553:501–5. doi: 10.1038/nature25010
79. Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol (2015) 4:215–66. doi: 10.1002/wdev.176
80. Fazeli PK, Lun M, Kim SM, Bredella MA, Wright S, Zhang Y, et al. FGF21 and the late adaptive response to starvation in humans. J Clin Invest (2015) 125:4601–11. doi: 10.1172/JCI83349
81. Lundsgaard A-M, Fritzen AM, Sjøberg KA, Myrmel LS, Madsen L, Wojtaszewski JFP, et al. Circulating FGF21 in humans is potently induced by short term overfeeding of carbohydrates. Mol Metab (2017) 6:22–9. doi: 10.1016/j.molmet.2016.11.001
82. Søberg S, Sandholt CH, Jespersen NZ, Toft U, Madsen AL, Holstein-Rathlou von S, et al. FGF21 Is a Sugar-Induced Hormone Associated with Sweet Intake and Preference in Humans. Cell Metab (2017) 25:1045–53.e6. doi: 10.1016/j.cmet.2017.04.009
83. De Sousa-Coelho AL, Relat J, Hondares E, Pérez-Martí A, Ribas F, Villarroya F, et al. FGF21 mediates the lipid metabolism response to amino acid starvation. J Lipid Res (2013) 54:1786–97. doi: 10.1194/jlr.M033415
84. Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest (2014) 124:3913–22. doi: 10.1172/JCI74915
85. Maida A, Zota A, Sjøberg KA, Schumacher J, Sijmonsma TP, Pfenninger A, et al. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. J Clin Invest (2016) 126:3263–78. doi: 10.1172/JCI85946
86. Vinales KL, Begaye B, Bogardus C, Walter M, Krakoff J, Piaggi P. FGF21 is a Hormonal Mediator of the Human “Thrifty” Metabolic Phenotype. Diabetes (2018) 68:318–23. doi: 10.2337/db18-0696
87. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab (2007) 5:426–37. doi: 10.1016/j.cmet.2007.05.002
88. Lundasen T, Hunt MC, Nilsson L-M, Sanyal S, Angelin B, Alexson SEH, et al. PPARalpha is a key regulator of hepatic FGF21. Biochem Biophys Res Commun (2007) 360:437–40. doi: 10.1016/j.bbrc.2007.06.068
89. Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab (2007) 5:415–25. doi: 10.1016/j.cmet.2007.05.003
90. Muise ES, Azzolina B, Kuo DW, El-Sherbeini M, Tan Y, Yuan X, et al. Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor gamma and altered metabolic states. Mol Pharmacol (2008) 74:403–12. doi: 10.1124/mol.108.044826
91. Gälman C, Lundåsen T, Kharitonenkov A, Bina HA, Eriksson M, Hafström I, et al. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab (2008) 8:169–74. doi: 10.1016/j.cmet.2008.06.014
92. Oishi K, Tomita T. Thiazolidinediones are potent inducers of fibroblast growth factor 21 expression in the liver. Biol Pharm Bull (2011) 34:1120–1. doi: 10.1248/bpb.34.1120
93. Nakagawa Y, Shimano H. CREBH Regulates Systemic Glucose and Lipid Metabolism. Int J Mol Sci (2018) 19:1396. doi: 10.3390/ijms19051396
94. Park J-G, Xu X, Cho S, Hur KY, Lee M-S, Kersten S, et al. CREBH-FGF21 axis improves hepatic steatosis by suppressing adipose tissue lipolysis. Sci Rep (2016) 6:1–13. doi: 10.1038/srep27938
95. Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes (2014) 63:4057–63. doi: 10.2337/db14-0595
96. Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, Kuro-O M, et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol (2010) 24:2050–64. doi: 10.1210/me.2010-0142
97. Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, et al. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech Dev (2000) 98:115–9. doi: 10.1016/s0925-4773(00)00439-1
98. Fisher FM, Estall JL, Adams AC, Antonellis PJ, Bina HA, Flier JS, et al. Integrated Regulation of Hepatic Metabolism by Fibroblast Growth Factor 21 (FGF21) in Vivo. Endocrinology (2011) 152:2996–3004. doi: 10.1210/en.2011-0281
99. Woolsey SJ, Beaton MD, Mansell SE, Leon-Ponte M, Yu J, Pin CL, Tirona RG, et al. A Fibroblast Growth Factor 21-Pregnane X Receptor Pathway Downregulates Hepatic CYP3A4 in Nonalcoholic Fatty Liver Disease. Mol Pharmacol (2016) 90:437–46. doi: 10.1124/mol.116.104687
100. Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y, et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation (2015) 131:1861–71. doi: 10.1161/CIRCULATIONAHA.115.015308
101. Keinicke H, Sun G, Mentzel CMJ, Fredholm M, John LM, Andersen B, et al. FGF21 regulates hepatic metabolic pathways to improve steatosis and inflammation. Endocr Connect (2020) 9:755–68. doi: 10.1530/EC-20-0152
102. Gallego-Escuredo JM, Gómez-Ambrosi J, Catalan V, Domingo P, Giralt M, Frühbeck G, et al. Opposite alterations in FGF21 and FGF19 levels and disturbed expression of the receptor machinery for endocrine FGFs in obese patients. Int J Obes (Lond) (2015) 39:121–9. doi: 10.1038/ijo.2014.76
103. Kruse R, Vienberg SG, Vind BF, Andersen B, Højlund K. Effects of insulin and exercise training on FGF21, its receptors and target genes in obesity and type 2 diabetes. Diabetologia (2017) 60:2042–51. doi: 10.1007/s00125-017-4373-5
104. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science (2015) 347:1260419–1260419. doi: 10.1126/science.1260419
105. Patel R, Bookout AL, Magomedova L, Owen BM, Consiglio GP, Shimizu M, et al. Glucocorticoids regulate the metabolic hormone FGF21 in a feed-forward loop. Mol Endocrinol (2015) 29:213–23. doi: 10.1210/me.2014-1259
106. Habegger KM, Stemmer K, Cheng C, Müller TD, Heppner KM, Ottaway N, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes (2013) 62:1453–63. doi: 10.2337/db12-1116
107. Adams AC, Astapova I, Fisher FM, Badman MK, Kurgansky KE, Flier JS, et al. Thyroid hormone regulates hepatic expression of fibroblast growth factor 21 in a PPARalpha-dependent manner. J Biol Chem (2010) 285:14078–82. doi: 10.1074/jbc.C110.107375
108. Senese R, Lasala P, Leanza C, de Lange P. New avenues for regulation of lipid metabolism by thyroid hormones and analogs. Front Physiol (2014) 5:475. doi: 10.3389/fphys.2014.00475
109. Adeva-Andany MM, Funcasta-Calderón R, Fernández-Fernández C, Castro-Quintela E, Carneiro-Freire N. Metabolic effects of glucagon in humans. J Clin Transl Endocrinol (2019) 15:45–53. doi: 10.1016/j.jcte.2018.12.005
110. Fu T, Xu Z, Liu L, Guo Q, Wu H, Liang X, et al. Mitophagy Directs Muscle-Adipose Crosstalk to Alleviate Dietary Obesity. Cell Rep (2018) 23:1357–72. doi: 10.1016/j.celrep.2018.03.127
111. Crooks DR, Natarajan TG, Jeong SY, Chen C, Park SY, Huang H, et al. Elevated FGF21 secretion, PGC-1α and ketogenic enzyme expression are hallmarks of iron-sulfur cluster depletion in human skeletal muscle. Hum Mol Genet (2014) 23:24–39. doi: 10.1093/hmg/ddt393
112. Brahma MK, Adam RC, Pollak NM, Jaeger D, Zierler KA, Pöcher N, et al. Fibroblast growth factor 21 is induced upon cardiac stress and alters cardiac lipid homeostasis. J Lipid Res (2014) 55:2229–41. doi: 10.1194/jlr.M044784
113. Dogan SA, Pujol C, Maiti P, Kukat A, Wang S, Hermans S, et al. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab (2014) 19:458–69. doi: 10.1016/j.cmet.2014.02.004
114. Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med (2018) 378:1096–106. doi: 10.1056/NEJMoa1712191
115. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet (2008) 40:1461–5. doi: 10.1038/ng.257
116. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet (2014) 46:352–6. doi: 10.1038/ng.2901
117. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology (2015) 61:506–14. doi: 10.1002/hep.27490
118. Watt MJ, Barnett AC, Bruce CR, Schenk S, Horowitz JF, Hoy AJ. Regulation of plasma ceramide levels with fatty acid oversupply: evidence that the liver detects and secretes de novo synthesised ceramide. Diabetologia (2012) 55:2741–6. doi: 10.1007/s00125-012-2649-3
119. Chaurasia B, Tippetts TS, Monibas RM, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science (2019) 365:386–92. doi: 10.1126/science.aav3722
120. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology (2010) 52:774–88. doi: 10.1002/hep.23719
121. Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr Opin Lipidol (2009) 20:379–85. doi: 10.1097/MOL.0b013e32832fa5c4
122. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology (2004) 40:185–94. doi: 10.1002/hep.20283
123. Piccolis M, Bond LM, Kampmann M, Pulimeno P, Chitraju C, Jayson CBK, et al. Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol Cell (2019) 74:32–44. doi: 10.1016/j.molcel.2019.01.036
124. Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol (2009) 44:625–33. doi: 10.1016/j.exger.2009.07.003
125. Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res (2016) 57:1329–38. doi: 10.1194/jlr.R067595
126. Tanaka N, Takahashi S, Zhang Y, Krausz KW, Smith PB, Patterson AD, et al. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim Biophys Acta (2015) 1852:1242–52. doi: 10.1016/j.bbadis.2015.02.012
127. Zarei M, Barroso E, Palomer X, Dai J, Rada P, Quesada-López T, et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol Metab (2018) 8:117–31. doi: 10.1016/j.molmet.2017.12.008
128. Bao L, Yin J, Gao W, Wang Q, Yao W, Gao X. A long-acting FGF21 alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis partly through an FGF21-adiponectin-IL17A pathway. Br J Pharmacol (2018) 175:3379–93. doi: 10.1111/bph.14383
129. Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes (2009) 58:250–9. doi: 10.2337/db08-0392
130. Chen MM, Hale C, Stanislaus S, Xu J, Véniant MM. FGF21 acts as a negative regulator of bile acid synthesis. J Endocrinol (2018) 237:139–52. doi: 10.1530/JOE-17-0727
131. Lee J, Choi J, Selen Alpergin ES, Zhao L, Hartung T, Scafidi S, et al. Loss of Hepatic Mitochondrial Long-Chain Fatty Acid Oxidation Confers Resistance to Diet-Induced Obesity and Glucose Intolerance. Cell Rep (2017) 20:655–67. doi: 10.1016/j.celrep.2017.06.080
132. Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med (2018) 10:eaat0344. doi: 10.1126/scitranslmed.aat0344
133. Lee J, Homma T, Kurahashi T, Kang ES, Fujii J. Oxidative stress triggers lipid droplet accumulation in primary cultured hepatocytes by activating fatty acid synthesis. Biochem Biophys Res Commun (2015) 464:229–35. doi: 10.1016/j.bbrc.2015.06.121
134. Wang L, Jiang Z, Lei XG. Knockout of SOD1 alters murine hepatic glycolysis, gluconeogenesis, and lipogenesis. Free Radic Biol Med (2012) 53:1689–96. doi: 10.1016/j.freeradbiomed.2012.08.570
135. González-Rodríguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis (2014) 5:e1179–9. doi: 10.1038/cddis.2014.162
136. He A, Chen X, Tan M, Chen Y, Lu D, Zhang X, et al. Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Mol Cell (2020) 79:30–42. doi: 10.1016/j.molcel.2020.05.007
137. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science (2011) 334:1081–6. doi: 10.1126/science.1209038
138. Choi W-G, Han J, Kim J-H, Kim M-J, Park J-W, Song B, et al. eIF2α phosphorylation is required to prevent hepatocyte death and liver fibrosis in mice challenged with a high fructose diet. Nutr Metab (Lond) (2017) 14:48. doi: 10.1186/s12986-017-0202-6
139. Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology (2008) 134:568–76. doi: 10.1053/j.gastro.2007.10.039
140. Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell (2008) 15:829–40. doi: 10.1016/j.devcel.2008.10.015
141. Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, et al. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology (2013) 57:1366–77. doi: 10.1002/hep.26126
142. Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology (2006) 44:27–33. doi: 10.1002/hep.21223
143. Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol (2011) 5:201–12. doi: 10.1586/egh.11.6
144. Lei Y, Wang S, Ren B, Wang J, Chen J, Lu J, et al. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS One (2017) 12:e0183680. doi: 10.1371/journal.pone.0183680
145. Akazawa Y, Cazanave S, Mott JL, Elmi N, Bronk SF, Kohno S, et al. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J Hepatol (2010) 52:586–93. doi: 10.1016/j.jhep.2010.01.003
146. Min H-K, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab (2012) 15:665–74. doi: 10.1016/j.cmet.2012.04.004
147. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep (2001) 2:222–8. doi: 10.1093/embo-reports/kve046
148. Mihm S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int J Mol Sci (2018) 19:3104. doi: 10.3390/ijms19103104
149. Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab (2016) 24:848–62. doi: 10.1016/j.cmet.2016.09.016
150. Ye D, Wang Y, Li H, Jia W, Man K, Lo CM, et al. Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1α-mediated antioxidant capacity in mice. Hepatology (2014) 60:977–89. doi: 10.1002/hep.27060
151. Boparai RK, Arum O, Miquet JG, Masternak MM, Bartke A, Khardori RK. Resistance to the Beneficial Metabolic Effects and Hepatic Antioxidant Defense Actions of Fibroblast Growth Factor 21 Treatment in Growth Hormone-Overexpressing Transgenic Mice. Int J Endocrinol (2015) 2015:282375–11. doi: 10.1155/2015/282375
152. Lee JH, Kang YE, Chang JY, Park KC, Kim H-W, Kim JT, et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am J Transl Res (2016) 8:4750–63.
153. Jiang S, Yan C, Fang Q-C, Shao M-L, Zhang Y-L, Liu Y, et al. Fibroblast growth factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J Biol Chem (2014) 289:29751–65. doi: 10.1074/jbc.M114.565960
154. Rupérez C, Lerin C, Ferrer-Curriu G, Cairo M, Mas-Stachurska A, Sitges M, et al. Autophagic control of cardiac steatosis through FGF21 in obesity-associated cardiomyopathy. Int J Cardiol (2018) 260:163–70. doi: 10.1016/j.ijcard.2018.02.109
155. Chen L, Wang K, Long A, Jia L, Zhang Y, Deng H, et al. Fasting-induced hormonal regulation of lysosomal function. Cell Res (2017) 27:748–63. doi: 10.1038/cr.2017.45
156. Schaap FG, Kremer AE, Lamers WH, Jansen PLM, Gaemers IC. Fibroblast growth factor 21 is induced by endoplasmic reticulum stress. Biochimie (2013) 95:692–9. doi: 10.1016/j.biochi.2012.10.019
157. Byun S, Seok S, Kim Y-C, Zhang Y, Yau P, Iwamori N, et al. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat Commun (2020) 11:807. doi: 10.1038/s41467-020-14384-z
158. Guzman-Lepe J, Cervantes-Alvarez E, Collin de l’Hortet A, Wang Y, Mars WM, Oda Y, et al. Liver-enriched transcription factor expression relates to chronic hepatic failure in humans. Hepatol Commun (2018) 2:582–94. doi: 10.1002/hep4.1172
159. Florentino RM, Fraunhoffer NA, Morita K, Takeishi K, Ostrowska A, Achreja A, et al. Cellular Location of HNF4αis Linked With Terminal Liver Failure in Humans. Hepatol Commun (2020) 66:1–17. doi: 10.1002/hep4.1505
160. Nishikawa T, Bell A, Brooks JM, Setoyama K, Melis M, Han B, et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest (2015) 125:1533–44. doi: 10.1172/JCI73137
161. Sharifnia T, Antoun J, Verriere TGC, Suarez G, Wattacheril J, Wilson KT, et al. Hepatic TLR4 signaling in obese NAFLD. Am J Physiol Gastrointest Liver Physiol (2015) 309:G270–8. doi: 10.1152/ajpgi.00304.2014
162. Tosello-Trampont A-C, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem (2012) 287:40161–72. doi: 10.1074/jbc.M112.417014
163. Mirea A-M, Tack CJ, Chavakis T, Joosten LAB, Toonen EJM. IL-1 Family Cytokine Pathways Underlying NAFLD: Towards New Treatment Strategies. Trends Mol Med (2018) 24:458–71. doi: 10.1016/j.molmed.2018.03.005
164. Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol (2008) 103:1372–9. doi: 10.1111/j.1572-0241.2007.01774.x
165. Haukeland JW, Damås JK, Konopski Z, Løberg EM, Haaland T, Goverud I, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol (2006) 44:1167–74. doi: 10.1016/j.jhep.2006.02.011
166. Zhang X, Shen J, Man K, Chu ESH, Yau TO, Sung JCY, et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J Hepatol (2014) 61:1365–75. doi: 10.1016/j.jhep.2014.07.006
167. Gao B, Tsukamoto H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology (2016) 150:1704–9. doi: 10.1053/j.gastro.2016.01.025
168. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The Differential Roles of T Cells in Non-alcoholic Fatty Liver Disease and Obesity. Front Immunol (2019) 10:82. doi: 10.3389/fimmu.2019.00082
169. Yoon J-H, Gores GJ. Death receptor-mediated apoptosis and the liver. J Hepatol (2002) 37:400–10. doi: 10.1016/s0168-8278(02)00209-x
170. Diehl AM, Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med (2017) 377:2063–72. doi: 10.1056/NEJMra1503519
171. Wu X, Dong L, Lin X, Li J. Relevance of the NLRP3 Inflammasome in the Pathogenesis of Chronic Liver Disease. Front Immunol (2017) 8:1728. doi: 10.3389/fimmu.2017.01728
172. Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (2014) 92:1069–82. doi: 10.1007/s00109-014-1170-1
173. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (2011) 54:133–44. doi: 10.1002/hep.24341
174. Wree A, Eguchi A, McGeough MD, Peña CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (2014) 59:898–910. doi: 10.1002/hep.26592
175. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest (2012) 122:3476–89. doi: 10.1172/JCI60777
176. Gautheron J, Gores GJ, Rodrigues CMP. Lytic cell death in metabolic liver disease. J Hepatol (2020) 73:394–408. doi: 10.1016/j.jhep.2020.04.001
177. Yu Y, He J, Li S, Song L, Guo X, Yao W, et al. Fibroblast growth factor 21 (FGF21) inhibits macrophage-mediated inflammation by activating Nrf2 and suppressing the NF-κB signaling pathway. Int Immunopharmacol (2016) 38:144–52. doi: 10.1016/j.intimp.2016.05.026
178. Zhou L, Li Q, Chen A, Liu N, Chen N, Chen X, et al. KLF15-activating Twist2 ameliorated hepatic steatosis by inhibiting inflammation and improving mitochondrial dysfunction via NF-κB–FGF21 or SREBP1c-FGF21 pathway. FASEB J (2019) 33:14254–69. doi: 10.1096/fj.201901347RR
179. Liu J, Cai G, Li M, Fan S, Yao B, Ping W, et al. Fibroblast growth factor 21 attenuates hypoxia-induced pulmonary hypertension by upregulating PPARγ expression and suppressing inflammatory cytokine levels. Biochem Biophys Res Commun (2018) 504:478–84. doi: 10.1016/j.bbrc.2018.09.004
180. Gao J, Liu Q, Li J, Hu C, Zhao W, Ma W, et al. Fibroblast Growth Factor 21 dependent TLR4/MYD88/NF-κB signaling activation is involved in lipopolysaccharide-induced acute lung injury. Int Immunopharmacol (2020) 80:106219. doi: 10.1016/j.intimp.2020.106219
181. Liu Y, Zhao C, Xiao J, Liu L, Zhang M, Wang C, et al. Fibroblast growth factor 21 deficiency exacerbates chronic alcohol-induced hepatic steatosis and injury. Sci Rep (2016) 6:31026–13. doi: 10.1038/srep31026
182. Fisher FM, Kim M, Doridot L, Cunniff JC, Parker TS, Levine DM, et al. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol Metab (2017) 6:14–21. doi: 10.1016/j.molmet.2016.11.008
183. Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab (2016) 23:427–40. doi: 10.1016/j.cmet.2016.02.001
184. Wei W, Li X-X, Xu M. Inhibition of vascular neointima hyperplasia by FGF21 associated with FGFR1/Syk/NLRP3 inflammasome pathway in diabetic mice. Atherosclerosis (2019) 289:132–42. doi: 10.1016/j.atherosclerosis.2019.08.017
185. Zeng Z, Zheng Q, Chen J, Tan X, Li Q, Ding L, et al. FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp Cell Res (2020) 393:112108. doi: 10.1016/j.yexcr.2020.112108
186. Cui A, Li J, Ji S, Ma F, Wang G, Xue Y, et al. The Effects of B1344, a Novel Fibroblast Growth Factor 21 Analog, on Nonalcoholic Steatohepatitis in Nonhuman Primates. Diabetes (2020) 69:1611–23. doi: 10.2337/db20-0209
187. Jimenez V, Jambrina C, Casana E, Sacristan V, Muñoz S, Darriba S, et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol Med (2018) 10:e8791. doi: 10.15252/emmm.201708791
188. Li S-M, Yu Y-H, Li L, Wang W-F, Li D-S. Treatment of CIA Mice with FGF21 Down-regulates TH17-IL-17 Axis. Inflammation (2016) 39:309–19. doi: 10.1007/s10753-015-0251-9
189. Hernandez G, Luo T, Javed TA, Wen L, Kalwat MA, Vale K, et al. Pancreatitis is an FGF21-deficient state that is corrected by replacement therapy. Sci Transl Med (2020) 12:eaay5186. doi: 10.1126/scitranslmed.aay5186
190. Singhal G, Fisher FM, Chee MJ, Tan TG, Ouaamari El A, Adams AC, et al. Fibroblast Growth Factor 21 (FGF21) Protects against High Fat Diet Induced Inflammation and Islet Hyperplasia in Pancreas. PLoS One (2016) 11:e0148252. doi: 10.1371/journal.pone.0148252
191. Luo Y, Yang Y, Liu M, Wang D, Wang F, Bi Y, et al. Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet. Gastroenterology (2019) 157:1413–28.e11. doi: 10.1053/j.gastro.2019.07.030
192. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
193. Yin C, Evason KJ, Asahina K, Stainier DYR. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest (2013) 123:1902–10. doi: 10.1172/JCI66369
194. Cai X, Wang J, Wang J, Zhou Q, Yang B, He Q, et al. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol Res (2020) 155:104720. doi: 10.1016/j.phrs.2020.104720
195. Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab (2020) 31:406–21.e7. doi: 10.1016/j.cmet.2019.11.013
196. Pradere J-P, Kluwe J, De Minicis S, Jiao J-J, Gwak G-Y, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (2013) 58:1461–73. doi: 10.1002/hep.26429
197. Zhao X-K, Yu L, Cheng M-L, Che P, Lu Y-Y, Zhang Q, et al. Focal Adhesion Kinase Regulates Hepatic Stellate Cell Activation and Liver Fibrosis. Sci Rep (2017) 7:4032–12. doi: 10.1038/s41598-017-04317-0
198. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest (2017) 127:55–64. doi: 10.1172/JCI88881
199. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology (2020) 1–16. doi: 10.1053/j.gastro.2019.11.311
200. Valenti L, Mendoza RM, Rametta R, Maggioni M, Kitajewski C, Shawber CJ, et al. Hepatic notch signaling correlates with insulin resistance and nonalcoholic fatty liver disease. Diabetes (2013) 62:4052–62. doi: 10.2337/db13-0769
201. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284:770–6. doi: 10.1126/science.284.5415.770
202. Syn W-K, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology (2011) 53:106–15. doi: 10.1002/hep.23998
203. Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta (2015) 1852:1765–78. doi: 10.1016/j.bbadis.2015.05.015
204. Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab (2020) 31:969–86.e7. doi: 10.1016/j.cmet.2020.03.010
205. Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, et al. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator With PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology (2020) 66:1–18. doi: 10.1002/hep.30928
206. Estep M, Mehta R, Bratthauer G, Alaparthi L, Monge F, Ali S, et al. Hepatic sonic hedgehog protein expression measured by computer assisted morphometry significantly correlates with features of non-alcoholic steatohepatitis. BMC Gastroenterol (2019) 19:1–7. doi: 10.1186/s12876-019-0951-y
207. Zhou X, Wang P, Ma Z, Li M, Teng X, Sun L, et al. Novel Interplay Between Sonic Hedgehog and Transforming Growth Factor-β1 in Human Nonalcoholic Steatohepatitis. Appl Immunohistochem Mol Morphol (2020) 28:154–60. doi: 10.1097/PAI.0000000000000724
208. Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, et al. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol (2011) 224:401–10. doi: 10.1002/path.2888
209. Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Ünalp A, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology (2012) 55:1711–21. doi: 10.1002/hep.25559
210. Sicklick JK, Li Y-X, Melhem A, Schmelzer E, Zdanowicz M, Huang J, et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am J Physiol Gastrointest Liver Physiol (2006) 290:G859–70. doi: 10.1152/ajpgi.00456.2005
211. Guy CD, Suzuki A, Abdelmalek MF, Burchette JL, Diehl AM, for the NASH CRN. Treatment response in the PIVENS trial is associated with decreased hedgehog pathway activity. Hepatology (2014) 61:98–107. doi: 10.1002/hep.27235
212. Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, et al. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology (2016) 64:261–75. doi: 10.1002/hep.28450
213. Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell (2014) 157:1324–38. doi: 10.1016/j.cell.2014.03.060
214. Omenetti A, Choi S, Michelotti G, Diehl AM. Hedgehog signaling in the liver. J Hepatol (2011) 54:366–73. doi: 10.1016/j.jhep.2010.10.003
215. Taylor RS, Taylor RJ, Bayliss S, Hagström H, Nasr P, Schattenberg JM, et al. Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology (2020) 158:1611–25.e12. doi: 10.1053/j.gastro.2020.01.043
216. Le CT, Nguyen G, Park SY, Choi DH, Cho E-H. LY2405319, an analog of fibroblast growth factor 21 ameliorates α-smooth muscle actin production through inhibition of the succinate-G-protein couple receptor 91 (GPR91) pathway in mice. PLoS One (2018) 13:e0192146. doi: 10.1371/journal.pone.0192146
217. Xu P, Zhang Y, Liu Y, Yuan Q, Song L, Liu M, et al. Fibroblast growth factor 21 attenuates hepatic fibrogenesis through TGF-β/smad2/3 and NF-κB signaling pathways. Toxicol Appl Pharmacol (2016) 290:43–53. doi: 10.1016/j.taap.2015.11.012
218. Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, et al. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology (2014) 147:1073–83.e6. doi: 10.1053/j.gastro.2014.07.044
219. Desai BN, Singhal G, Watanabe M, Stevanovic D, Lundasen T, Fisher FM, et al. Fibroblast growth factor 21 (FGF21) is robustly induced by ethanol and has a protective role in ethanol associated liver injury. Mol Metab (2017) 6:1395–406. doi: 10.1016/j.molmet.2017.08.004
220. Opoku YK, Liu Z, Afrifa J, Kumi AK, Liu H, Ghartey-Kwansah G, et al. Fibroblast Growth Factor-21 ameliorates hepatic encephalopathy by activating the STAT3-SOCS3 pathway to inhibit activated hepatic stellate cells. EXCLI J (2020) 19:567–81. doi: 10.17179/excli2020-1287
221. Johnson CL, Weston JY, Chadi SA, Fazio EN, Huff MW, Kharitonenkov A, et al. Fibroblast growth factor 21 reduces the severity of cerulein-induced pancreatitis in mice. Gastroenterology (2009) 137:1795–804. doi: 10.1053/j.gastro.2009.07.064
222. Weng W, Ge T, Wang Y, He L, Liu T, Wang W, et al. Therapeutic Effects of Fibroblast Growth Factor-21 on Diabetic Nephropathy and the Possible Mechanism in Type 1 Diabetes Mellitus Mice. Diabetes Metab J (2020) 44:566–80. doi: 10.4093/dmj.2019.0089
223. Davidsohn N, Pezone M, Vernet A, Graveline A, Oliver D, Slomovic S, et al. A single combination gene therapy treats multiple age-related diseases. Proc Natl Acad Sci (2019) 116:23505–11. doi: 10.1073/pnas.1910073116
224. Zhang S, Yu D, Wang M, Huang T, Wu H, Zhang Y, et al. FGF21 attenuates pulmonary fibrogenesis through ameliorating oxidative stress in vivo and in vitro. BioMed Pharmacother (2018) 103:1516–25. doi: 10.1016/j.biopha.2018.03.100
225. Minard AY, Tan S-X, Yang P, Fazakerley DJ, Domanova W, Parker BL, et al. mTORC1 Is a Major Regulatory Node in the FGF21 Signaling Network in Adipocytes. Cell Rep (2016) 17:29–36. doi: 10.1016/j.celrep.2016.08.086
226. Ge X, Chen C, Hui X, Wang Y, Lam KSL, Xu A. Fibroblast growth factor 21 induces glucose transporter-1 expression through activation of the serum response factor/Ets-like protein-1 in adipocytes. J Biol Chem (2011) 286:34533–41. doi: 10.1074/jbc.M111.248591
227. Mashili FL, Austin RL, Deshmukh AS, Fritz T, Caidahl K, Bergdahl K, et al. Direct effects of FGF21 on glucose uptake in human skeletal muscle: implications for type 2 diabetes and obesity. Diabetes Metab Res Rev (2011) 27:286–97. doi: 10.1002/dmrr.1177
228. Schlein C, Talukdar S, Heine M, Fischer AW, Krott LM, Nilsson SK, et al. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab (2016) 23:441–53. doi: 10.1016/j.cmet.2016.01.006
229. Arner P, Pettersson A, Mitchell PJ, Dunbar JD, Kharitonenkov A, Rydén M. FGF21 attenuates lipolysis in human adipocytes - a possible link to improved insulin sensitivity. FEBS Lett (2008) 582:1725–30. doi: 10.1016/j.febslet.2008.04.038
230. Mottillo EP, Desjardins EM, Fritzen AM, Zou VZ, Crane JD, Yabut JM, et al. FGF21 does not require adipocyte AMP-activated protein kinase (AMPK) or the phosphorylation of acetyl-CoA carboxylase (ACC) to mediate improvements in whole-body glucose homeostasis. Mol Metab (2017) 6:471–81. doi: 10.1016/j.molmet.2017.04.001
231. Ye M, Lu W, Wang X, Wang C, Abbruzzese JL, Liang G, et al. FGF21-FGFR1 Coordinates Phospholipid Homeostasis, Lipid Droplet Function, and ER Stress in Obesity. Endocrinology (2016) 157:4754–69. doi: 10.1210/en.2016-1710
232. Kwon MM, O’Dwyer SM, Baker RK, Covey SD, Kieffer TJ. FGF21-Mediated Improvements in Glucose Clearance Require Uncoupling Protein 1. Cell Rep (2015) 13:1521–7. doi: 10.1016/j.celrep.2015.10.021
233. Samms RJ, Smith DP, Cheng CC, Antonellis PP, Perfield JW, Kharitonenkov A, et al. Discrete Aspects of FGF21 In Vivo Pharmacology Do Not Require UCP1. Cell Rep (2015) 11:991–9. doi: 10.1016/j.celrep.2015.04.046
234. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev (2012) 26:271–81. doi: 10.1101/gad.177857.111
235. Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology (2008) 149:6018–27. doi: 10.1210/en.2008-0816
236. Camporez JPG, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, Serr J, et al. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology (2013) 154:3099–109. doi: 10.1210/en.2013-1191
237. Adams AC, Halstead CA, Hansen BC, Irizarry AR, Martin JA, Myers SR, et al. LY2405319, an Engineered FGF21 Variant, Improves the Metabolic Status of Diabetic Monkeys. PLoS One (2013) 8:e65763. doi: 10.1371/journal.pone.0065763
238. Stanislaus S, Hecht R, Yie J, Hager T, Hall M, Spahr C, et al. A Novel Fc-FGF21 With Improved Resistance to Proteolysis, Increased Affinity Toward β-Klotho, and Enhanced Efficacy in Mice and Cynomolgus Monkeys. Endocrinology (2017) 158:1314–27. doi: 10.1210/en.2016-1917
239. Andersen B, Straarup EM, Heppner KM, Takahashi DL, Raffaele V, Dissen GA, et al. FGF21 decreases body weight without reducing food intake or bone mineral density in high-fat fed obese rhesus macaque monkeys. Int J Obes (Lond) (2018) 42:1151–60. doi: 10.1038/s41366-018-0080-7
240. Véniant MM, Hale C, Helmering J, Chen MM, Stanislaus S, Busby J, et al. FGF21 promotes metabolic homeostasis via white adipose and leptin in mice. PLoS One (2012) 7:e40164. doi: 10.1371/journal.pone.0040164
241. Adams AC, Yang C, Coskun T, Cheng CC, Gimeno RE, Luo Y, et al. The breadth of FGF21’s metabolic actions are governed by FGFR1 in adipose tissue. Mol Metab (2012) 2:31–7. doi: 10.1016/j.molmet.2012.08.007
242. Zhang Y, Xie Y, Berglund ED, Coate KC, He TT, Katafuchi T, et al. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. eLife Sci (2012) 1:e00065. doi: 10.7554/eLife.00065
243. Lin Z, Tian H, Lam KSL, Lin S, Hoo RCL, Konishi M, et al. Adiponectin Mediates the Metabolic Effects of FGF21 on Glucose Homeostasis and Insulin Sensitivity in Mice. Cell Metab (2013) 17:779–89. doi: 10.1016/j.cmet.2013.04.005
244. Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, et al. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab (2013) 18:333–40. doi: 10.1016/j.cmet.2013.08.005
245. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet (2019) 392:2705–17. doi: 10.1016/S0140-6736(18)31785-9
246. Kaufman A, Abuqayyas L, Denney WS, Tillman EJ, Rolph T. AKR-001, an Fc-FGF21 Analog, Showed Sustained Pharmacodynamic Effects on Insulin Sensitivity and Lipid Metabolism in Type 2 Diabetes Patients. Cell Rep Med (2020) 1:100057. doi: 10.1016/j.xcrm.2020.100057
247. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab (2013) 17:790–7. doi: 10.1016/j.cmet.2013.03.019
248. Achari AE, Jain SK. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int J Mol Sci (2017) 18:1321. doi: 10.3390/ijms18061321
249. Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest (2001) 108:1875–81. doi: 10.1172/JCI14120
250. Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab (2002) 13:84–9. doi: 10.1016/s1043-2760(01)00524-0
251. Qiao L, Zou C, van der Westhuyzen DR, Shao J. Adiponectin reduces plasma triglyceride by increasing VLDL triglyceride catabolism. Diabetes (2008) 57:1824–33. doi: 10.2337/db07-0435
252. Awazawa M, Ueki K, Inabe K, Yamauchi T, Kaneko K, Okazaki Y, et al. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem Biophys Res Commun (2009) 382:51–6. doi: 10.1016/j.bbrc.2009.02.131
253. Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta (2007) 380:24–30. doi: 10.1016/j.cca.2007.01.026
254. Shafiei MS, Shetty S, Scherer PE, Rockey DC. Adiponectin regulation of stellate cell activation via PPARγ-dependent and -independent mechanisms. Am J Pathol (2011) 178:2690–9. doi: 10.1016/j.ajpath.2011.02.035
255. Ramezani-Moghadam M, Wang J, Ho V, Iseli TJ, Alzahrani B, Xu A, et al. Adiponectin reduces hepatic stellate cell migration by promoting tissue inhibitor of metalloproteinase-1 (TIMP-1) secretion. J Biol Chem (2015) 290:5533–42. doi: 10.1074/jbc.M114.598011
256. Kumar P, Raeman R, Chopyk DM, Smith T, Verma K, Liu Y, et al. Adiponectin inhibits hepatic stellate cell activation by targeting the PTEN/AKT pathway. Biochim Biophys Acta Mol Basis Dis (2018) 1864:3537–45. doi: 10.1016/j.bbadis.2018.08.012
257. Kaser S, Moschen A, Cayon A, Kaser A, Crespo J, Pons-Romero F, et al. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut (2005) 54:117–21. doi: 10.1136/gut.2003.037010
258. Musunuru K, Kathiresan S. Surprises From Genetic Analyses of Lipid Risk Factors for Atherosclerosis. Circ Res (2016) 118:579–85. doi: 10.1161/CIRCRESAHA.115.306398
259. Coate KC, Hernandez G, Thorne CA, Sun S, Le TDV, Vale K, et al. FGF21 Is an Exocrine Pancreas Secretagogue. Cell Metab (2017) 25:472–80. doi: 10.1016/j.cmet.2016.12.004
260. Chen Q, Li J, Ma J, Yang X, Ni M, Zhang Y, et al. Fibroblast growth factor 21 alleviates acute pancreatitis via activation of the Sirt1-autophagy signalling pathway. J Cell Mol Med (2020) 24:5341–51. doi: 10.1111/jcmm.15190
261. Wente W, Efanov AM, Brenner M, Kharitonenkov A, Köster A, Sandusky GE, et al. Fibroblast Growth Factor-21 Improves Pancreatic β-Cell Function and Survival by Activation of Extracellular Signal–Regulated Kinase 1/2 and Akt Signaling Pathways. Diabetes (2006) 55:2470–8. doi: 10.2337/db05-1435
262. Cheng STW, Li SYT, Leung PS. Fibroblast Growth Factor 21 Stimulates Pancreatic Islet Autophagy via Inhibition of AMPK-mTOR Signaling. Int J Mol Sci (2019) 20:2517. doi: 10.3390/ijms20102517
263. Xie T, So WY, Li XY, Leung PS. Fibroblast growth factor 21 protects against lipotoxicity-induced pancreatic β-cell dysfunction via regulation of AMPK signaling and lipid metabolism. Clin Sci (2019) 133:2029–44. doi: 10.1042/CS20190093
264. Pan Y, Wang B, Zheng J, Xiong R, Fan Z, Ye Y, et al. Pancreatic fibroblast growth factor 21 protects against type 2 diabetes in mice by promoting insulin expression and secretion in a PI3K/Akt signaling-dependent manner. J Cell Mol Med (2019) 23:1059–71. doi: 10.1111/jcmm.14007
265. Kim H. Cerulein pancreatitis: oxidative stress, inflammation, and apoptosis. Gut Liver (2008) 2:74–80. doi: 10.5009/gnl.2008.2.2.74
266. Johnson CL, Mehmood R, Laing SW, Stepniak CV, Kharitonenkov A, Pin CL. Silencing of the Fibroblast growth factor 21 gene is an underlying cause of acinar cell injury in mice lacking MIST1. Am J Physiol Endocrinol Metab (2014) 306:E916–28. doi: 10.1152/ajpendo.00559.2013
267. Wang S, Binder P, Fang Q, Wang Z, Xiao W, Liu W, et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br J Pharmacol (2018) 175:1293–304. doi: 10.1111/bph.13888
268. Liu SQ, Roberts D, Kharitonenkov A, Zhang B, Hanson SM, Li YC, et al. Endocrine protection of ischemic myocardium by FGF21 from the liver and adipose tissue. Sci Rep (2013) 3:2767–11. doi: 10.1038/srep02767
269. Planavila A, Redondo-Angulo I, Ribas F, Garrabou G, Casademont J, Giralt M, et al. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc Res (2015) 106:19–31. doi: 10.1093/cvr/cvu263
270. Kang K, Xu P, Wang M, Chunyu J, Sun X, Ren G, et al. FGF21 attenuates neurodegeneration through modulating neuroinflammation and oxidant-stress. Biomed Pharmacother (2020) 129:110439. doi: 10.1016/j.biopha.2020.110439
271. Sa-nguanmoo P, Tanajak P, Kerdphoo S, Jaiwongkam T, Wang X, Liang G, et al. FGF21 and DPP-4 inhibitor equally prevents cognitive decline in obese rats. BioMed Pharmacother (2018) 97:1663–72. doi: 10.1016/j.biopha.2017.12.021
272. Wang X, Zhu L, Hu J, Guo R, Ye S, Liu F, et al. FGF21 Attenuated LPS-Induced Depressive-Like Behavior via Inhibiting the Inflammatory Pathway. Front Pharmacol (2020) 11:154. doi: 10.3389/fphar.2020.00154
273. Mäkelä J, Tselykh TV, Maiorana F, Eriksson O, Do HT, Mudò G, et al. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. Springerplus (2014) 3:2–12. doi: 10.1186/2193-1801-3-2
274. Fang X, Ma J, Mu D, Li B, Lian B, Sun C. FGF21 Protects Dopaminergic Neurons in Parkinson’s Disease Models Via Repression of Neuroinflammation. Neurotox Res (2020) 34:1–12. doi: 10.1007/s12640-019-00151-6
275. Chen S, Chen S-T, Sun Y, Xu Z, Wang Y, Yao S-Y, et al. Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer’s disease. Redox Biol (2019) 22:101133. doi: 10.1016/j.redox.2019.101133
276. Holstein-Rathlou von S, BonDurant LD, Peltekian L, Naber MC, Yin TC, Claflin KE, et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab (2016) 23:335–43. doi: 10.1016/j.cmet.2015.12.003
277. Talukdar S, Owen BM, Song P, Hernandez G, Zhang Y, Zhou Y, et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab (2016) 23:344–9. doi: 10.1016/j.cmet.2015.12.008
278. Jensen-Cody SO, Flippo KH, Claflin KE, Yavuz Y, Sapouckey SA, Walters GC, et al. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab (2020) 32:273–86. doi: 10.1016/j.cmet.2020.06.008
279. Frayling TM, Beaumont RN, Jones SE, Yaghootkar H, Tuke MA, Ruth KS, et al. A Common Allele in FGF21 Associated with Sugar Intake Is Associated with Body Shape, Lower Total Body-Fat Percentage, and Higher Blood Pressure. Cell Rep (2018) 23:327–36. doi: 10.1016/j.celrep.2018.03.070
280. Chu AY, Workalemahu T, Paynter NP, Rose LM, Giulianini F, Tanaka T, et al. Novel locus including FGF21 is associated with dietary macronutrient intake. Hum Mol Genet (2013) 22:1895–902. doi: 10.1093/hmg/ddt032
281. Schumann G, Liu C, O’Reilly P, Gao H, Song P, Xu B, et al. KLB is associated with alcohol drinking, and its gene product β-Klotho is necessary for FGF21 regulation of alcohol preference. Proc Natl Acad Sci U S A (2016) 113:14372–7. doi: 10.1073/pnas.1611243113
282. Kukat A, Dogan SA, Edgar D, Mourier A, Jacoby C, Maiti P, et al. Loss of UCP2 Attenuates Mitochondrial Dysfunction without Altering ROS Production and Uncoupling Activity. PLoS Genet (2014) 10:e1004385–14. doi: 10.1371/journal.pgen.1004385
283. Harris L-ALS, Smith GI, Patterson BW, Ramaswamy RS, Okunade AL, Kelly SC, et al. Alterations in 3-Hydroxyisobutyrate and FGF21 Metabolism Are Associated With Protein Ingestion-Induced Insulin Resistance. Diabetes (2017) 66:1871–8. doi: 10.2337/db16-1475
284. Solon-Biet SM, Cogger VC, Pulpitel T, Heblinski M, Wahl D, McMahon AC, et al. Defining the Nutritional and Metabolic Context of FGF21 Using the Geometric Framework. Cell Metab (2016) 24:555–65. doi: 10.1016/j.cmet.2016.09.001
285. Mu J, Pinkstaff J, Li Z, Skidmore L, Li N, Myler H, et al. FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes (2012) 61:505–12. doi: 10.2337/db11-0838
286. Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci (2009) 106:10853–8. doi: 10.1073/pnas.0904187106
287. Bookout AL, de Groot MHM, Owen BM, Lee S, Gautron L, Lawrence HL, et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med (2013) 19:1147–52. doi: 10.1038/nm.3249
288. Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, et al. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab (2014) 20:670–7. doi: 10.1016/j.cmet.2014.07.012
289. Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, et al. Central Fibroblast Growth Factor 21 Browns White Fat via Sympathetic Action in Male Mice. Endocrinology (2015) 156:2470–81. doi: 10.1210/en.2014-2001
290. Tan BK, Hallschmid M, Adya R, Kern W, Lehnert H, Randeva HS. Fibroblast growth factor 21 (FGF21) in human cerebrospinal fluid: relationship with plasma FGF21 and body adiposity. Diabetes (2011) 60:2758–62. doi: 10.2337/db11-0672
291. Kharitonenkov A, Beals JM, Micanovic R, Strifler BA, Rathnachalam R, Wroblewski VJ, et al. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLoS One (2013) 8:e58575. doi: 10.1371/journal.pone.0058575
292. Huang J, Ishino T, Chen G, Rolzin P, Osothprarop TF, Retting K, et al. Development of a Novel Long-Acting Antidiabetic FGF21 Mimetic by Targeted Conjugation to a Scaffold Antibody. J Pharmacol Exp Ther (2013) 346:270–80. doi: 10.1124/jpet.113.204420
293. Kim AM, Somayaji VR, Dong JQ, Rolph TP, Weng Y, Chabot JR, et al. Once-weekly administration of a long-acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non-human primates. Diabetes Obes Metab (2017) 19:1762–72. doi: 10.1111/dom.13023
294. Tirucherai G, Mora J, Revankar R, Charles E. SAT-359-Pharmacokinetics and safety of pegbelfermin (BMS-986036) administered in the abdomen and upper arm to normal, overweight, and obese healthy participants. J Hepatol (2019) 70:e798. doi: 10.1016/S0618-8278(19)31589-0
295. Charles ED, Neuschwander-Tetri BA, Pablo Frias J, Kundu S, Luo Y, Tirucherai GS, et al. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity (Silver Spring) (2019) 27:41–9. doi: 10.1002/oby.22344
296. Zhen EY, Jin Z, Ackermann BL, Thomas MK, Gutierrez JA. Circulating FGF21 proteolytic processing mediated by fibroblast activation protein. Biochem J (2016) 473:605–14. doi: 10.1042/BJ20151085
297. Lay AJ, Zhang HE, McCaughan GW, Gorrell MD. Fibroblast activation protein in liver fibrosis. Front Biosci (Landmark Ed.) (2019) 24:1–17.
298. Akero Therapeutics. Biopsy Data and Safety/Tolerability for 16-Week Phase 2a BALANCED Study in NASH Patients. (2020). https://ir.akerotx.com/static-files/e46c72bf-eb43-45df-9ef8-462345e17bdb. [Accessed 15 August 2020].
299. 89bio, Inc. BIO89-100 Phase 1b/2a Topline Results (2020). https://ir.89bio.com/static-files/0da30221-2ed4-4ada-b666-1c34e400b6c2 [Accessed October 20, 2020].
300. Kolumam G, Chen MZ, Tong R, Zavala-Solorio J, Kates L, van Bruggen N, et al. Sustained Brown Fat Stimulation and Insulin Sensitization by a Humanized Bispecific Antibody Agonist for Fibroblast Growth Factor Receptor 1/βKlotho Complex. EBioMedicine (2015) 2:730–43. doi: 10.1016/j.ebiom.2015.05.028
301. Kunder R, Frederickson J, Yeh FL, Chinn L, Dash A, Lewin-Koh N, et al. Multiple Doses of an Anti-FGFR1/KLB Bispecific Antibody (BFKB8488A) are Associated with a Decrease in Hepatic Fat in Patients in NAFLD. AASLD: The Liver Meeting (2019). Poster LP8
302. Wong C, Yeh FL, Chinn L, Dash A, Lewin-Koh N, Yoshida K, et al. A Bispecific Antibody to FGFR1/KLB, BFKB8488A, Improves Lipid Profile, Markers of Fibrogenesis and Liver Health in Patients with Type 2 Diabetes–Preliminary Results from a Phase 1b Study. AASLD: The Liver Meeting (2019) Poster 2310.
303. DePaoli A, Phung V, Bashir MR, Morrow L, Beysen C, Yan A, et al. NGM313, a Novel Activator of b-Klotho/FGFR1c, Improves Insulin Resistance and Reduces Hepatic Fat in Obese, Nondiabetic Subjects. ADA Annual Meeting (2019) Poster, 140–LB. doi: 10.2337/db19-140-LB
304. Luo J, Ko B, Elliott M, Zhou M, Lindhout DA, Phung V, et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci Transl Med (2014) 6:247ra100. doi: 10.1126/scitranslmed.3009098
305. Wu X, Ge H, Lemon B, Vonderfecht S, Weiszmann J, Hecht R, et al. FGF19-induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem (2010) 285:5165–70. doi: 10.1074/jbc.M109.068783
306. Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology (2020) In Press. doi: 10.1053/j.gastro.2020.08.004
307. Wu X, Ge H, Lemon B, Weiszmann J, Gupte J, Hawkins N, et al. Selective activation of FGFR4 by an FGF19 variant does not improve glucose metabolism in ob/ob mice. Proc Natl Acad Sci (2009) 106:14379–84. doi: 10.1073/pnas.0907812106
308. Wu X, Ge H, Baribault H, Gupte J, Weiszmann J, Lemon B, et al. Dual actions of fibroblast growth factor 19 on lipid metabolism. J Lipid Res (2013) 54:325–32. doi: 10.1194/jlr.M027094
309. Huang X, Yang C, Luo Y, Jin C, Wang F, McKeehan WL. FGFR4 prevents hyperlipidemia and insulin resistance but underlies high-fat diet induced fatty liver. Diabetes (2007) 56:2501–10. doi: 10.2337/db07-0648
310. Yu XX, Watts LM, Manchem VP, Chakravarty K, Monia BP, McCaleb ML, et al. Peripheral reduction of FGFR4 with antisense oligonucleotides increases metabolic rate and lowers adiposity in diet-induced obese mice. PLoS One (2013) 8:e66923. doi: 10.1371/journal.pone.0066923
311. Harrison SA, Rossi SJ, Paredes AH, Trotter JF, Bashir MR, Guy CD, et al. NGM282 Improves Liver Fibrosis and Histology in 12 Weeks in Patients With Nonalcoholic Steatohepatitis. Hepatology (2019) 5:303. doi: 10.1002/hep.30590
312. Schumacher JD, Kong B, Wu J, Rizzolo D, Armstrong LE, Chow MD, et al. Direct and Indirect Effects of Fibroblast Growth Factor (FGF) 15 and FGF19 on Liver Fibrosis Development. Hepatology (2019) 121:27. doi: 10.1002/hep.30810
313. Turner T, Chen X, Zahner M, Opsahl A, DeMarco G, Boucher M, et al. FGF21 increases water intake, urine output and blood pressure in rats. PLoS One (2018) 13:e0202182. doi: 10.1371/journal.pone.0202182
314. Song P, Zechner C, Hernandez G, Cánovas J, Xie Y, Sondhi V, et al. The Hormone FGF21 Stimulates Water Drinking in Response to Ketogenic Diet and Alcohol. Cell Metab (2018) 27:1338–47.e4. doi: 10.1016/j.cmet.2018.04.001
315. Carroll HA, Chen Y-C, Templeman I, James LJ, Betts JA, Trim WV. The effect of hydration status on plasma FGF21 concentrations in humans: A subanalysis of a randomised crossover trial. PLoS One (2020) 15:e0235557–9. doi: 10.1371/journal.pone.0235557
316. He J-L, Zhao M, Xia J-J, Guan J, Liu Y, Wang L-Q, et al. FGF21 ameliorates the neurocontrol of blood pressure in the high fructose-drinking rats. Sci Rep (2016) 6:29582. doi: 10.1038/srep29582
317. Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J, et al. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1-7) Axis in Mice. Cell Metab (2018) 27:1323–37.e5. doi: 10.1016/j.cmet.2018.04.002
318. Shi Y, Wang S, Peng H, Lv Y, Li W, Cheng S, et al. Fibroblast Growth Factor 21 Attenuates Vascular Calcification by Alleviating Endoplasmic Reticulum Stress Mediated Apoptosis in Rats. Int J Biol Sci (2018) 15:138–47. doi: 10.7150/ijbs.28873
319. O’Brown NM, Pfau SJ, Gu C. Bridging barriers: a comparative look at the blood–brain barrier across organisms. Genes Dev (2018) 32:466–78. doi: 10.1101/gad.309823.117
320. Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun (2017) 60:1–12. doi: 10.1016/j.bbi.2016.03.010
321. Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor γ. Proc Natl Acad Sci (2012) 109:3143–8. doi: 10.1073/pnas.1200797109
322. Wang X, Wei W, Krzeszinski JY, Wang Y, Wan Y. A Liver-Bone Endocrine Relay by IGFBP1 Promotes Osteoclastogenesis and Mediates FGF21-Induced Bone Resorption. Cell Metab (2015) 22:811–24. doi: 10.1016/j.cmet.2015.09.010
323. Li X, Stanislaus S, Asuncion F, Niu Q-T, Chinookoswong N, Villasenor K, et al. FGF21 Is Not a Major Mediator for Bone Homeostasis or Metabolic Actions of PPARα and PPARγ Agonists. J Bone Miner Res (2017) 32:834–45. doi: 10.1002/jbmr.2936
324. Guañabens N, Parés A, Alvarez L, Martínez de Osaba MJ, Monegal A, Peris P, et al. Collagen-related markers of bone turnover reflect the severity of liver fibrosis in patients with primary biliary cirrhosis. J Bone Miner Res (1998) 13:731–8. doi: 10.1359/jbmr.1998.13.4.731
325. Veidal SS, Vassiliadis E, Bay-Jensen A-C, Tougas G, Vainer B, Karsdal MA. Procollagen type I N-terminal propeptide (PINP) is a marker for fibrogenesis in bile duct ligation-induced fibrosis in rats. Fibrogenesis Tissue Repair (2010) 3:5–7. doi: 10.1186/1755-1536-3-5
326. Novo Nordisk. R&D Investor Presentation (2020). https://www.novonordisk.com/content/dam/Denmark/HQ/investors/irmaterial/investor_presentations/2020/RD_event_presentation_Final.pdf. [Accessed 30 August 2020].
327. Trujillo J. Safety and tolerability of once-weekly GLP-1 receptor agonists in type 2 diabetes. J Clin Pharm Ther (2020) 45:43–60. doi: 10.1111/jcpt.13225
328. FDA Label for Ocaliva. (2018). https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022341s023lbl.pdf. [Accessed 15 August 2020].
329. Hollander P. Anti-Diabetes and Anti-Obesity Medications: Effects on Weight in People With Diabetes. Diabetes Spectr (2007) 20:159–65. doi: 10.2337/diaspect.20.3.159
Keywords: non-alcoholic steatohepatitis, nonalcoholic fatty liver disease, obesity, insulin resistance, metabolic syndrome, metabolic disease, fibroblast growth factor 21
Citation: Tillman EJ and Rolph T (2020) FGF21: An Emerging Therapeutic Target for Non-Alcoholic Steatohepatitis and Related Metabolic Diseases. Front. Endocrinol. 11:601290. doi: 10.3389/fendo.2020.601290
Received: 31 August 2020; Accepted: 12 November 2020;
Published: 14 December 2020.
Edited by:
Magdalene K. Montgomery, The University of Melbourne, AustraliaReviewed by:
Jörn M. Schattenberg, Johannes Gutenberg University Mainz, GermanyMorichika Konishi, Kobe Pharmaceutical University, Japan
Matthew J. Potthoff, The University of Iowa, United States
Copyright © 2020 Tillman and Rolph. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim Rolph, dGltQGFrZXJvdHguY29t