 L. Kořínková1†
L. Kořínková1† Lenka Maletínská
Lenka Maletínská- 1Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Prague, Czechia
- 2Institute of Physiology, Czech Academy of Sciences, Prague, Czechia
Obesity, diabetes, insulin resistance, sedentary lifestyle, and Western diet are the key factors underlying non-alcoholic fatty liver disease (NAFLD), one of the most common liver diseases in developed countries. In many cases, NAFLD further progresses to non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and to hepatocellular carcinoma. The hepatic lipotoxicity and non-liver factors, such as adipose tissue inflammation and gastrointestinal imbalances were linked to evolution of NAFLD. Nowadays, the degree of adipose tissue inflammation was shown to directly correlate with the severity of NAFLD. Consumption of higher caloric intake is increasingly emerging as a fuel of metabolic inflammation not only in obesity-related disorders but also NAFLD. However, multiple causes of NAFLD are the reason why the mechanisms of NAFLD progression to NASH are still not well understood. In this review, we explore the role of food intake regulating peptides in NAFLD and NASH mouse models. Leptin, an anorexigenic peptide, is involved in hepatic metabolism, and has an effect on NAFLD experimental models. Glucagon-like peptide-1 (GLP-1), another anorexigenic peptide, and GLP-1 receptor agonists (GLP-1R), represent potential therapeutic agents to prevent NAFLD progression to NASH. On the other hand, the deletion of ghrelin, an orexigenic peptide, prevents age-associated hepatic steatosis in mice. Because of the increasing incidence of NAFLD and NASH worldwide, the selection of appropriate animal models is important to clarify aspects of pathogenesis and progression in this field.
Introduction
Chronic liver diseases represent a major global health problem (1). In addition to genetic factors, various other stimuli, such as diet, metabolic diseases, etc. can alter liver function, especially the intracellular accumulation of lipids in hepatocytes. If these stimuli act for a sufficient time period, steatosis can induce inflammation resulting in non-alcoholic steatohepatitis (NASH). NASH, an extremely advanced form of non-alcoholic fatty liver disease (NAFLD), is defined as hepatic steatosis with inflammation and hepatocyte injury. NASH can eventually lead to advanced fibrosis, liver cirrhosis and liver failure (Figure 1). Over the past 20 years, the incidence of NAFLD has more than doubled, and it is now one of the most common liver diseases in Western countries (1). In the United States (US), the rates of prevalence of hepatic steatosis and NAFLD were estimated to 24.13%; however, it can vary by the ethnicity. It is reported that the highest prevalence is in the Hispanic Americans, followed by Americans of Europe descent and then African Americans (2). Moreover, only 3-5% of biopsy-proven NASH in the US population has been convincingly shown to progress to cirrhosis, liver failure and hepatocellular carcinoma (3) and NASH-associated cirrhosis is currently the third most frequent reason for liver transplantation. The epidemiological data from individual states of Europe suggested that approximately one-quarter of the European population is affected by NAFLD (2). As expected, the prevalence of NAFLD is substantially increased with type 2 diabetes mellitus (T2DM) and with increased body mass index, as demonstrated across Europe (2).
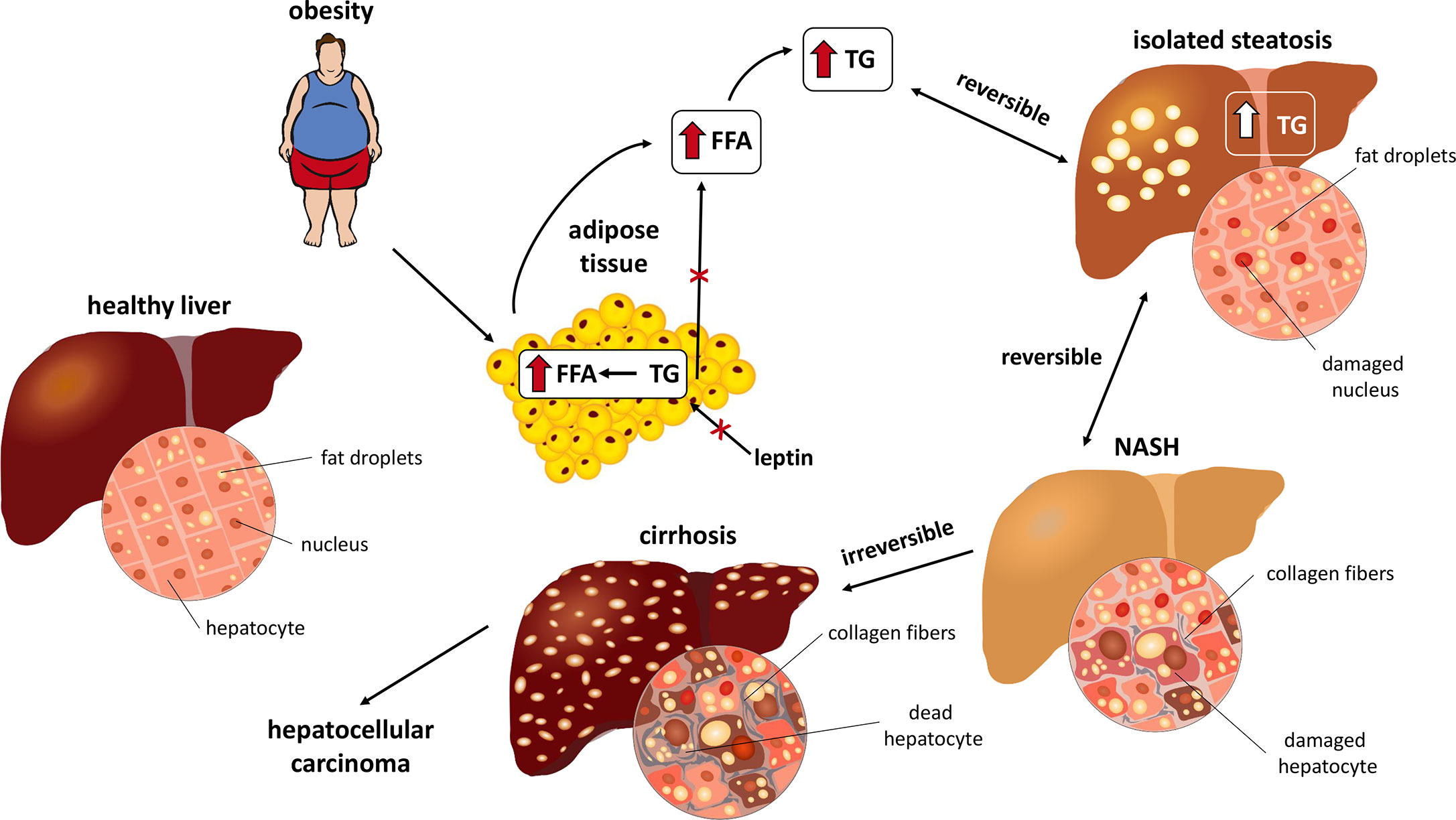
Figure 1 Stages of NASH.
The pathogenesis of NAFLD and NASH appears to be multifactorial and many mechanisms have been proposed as possible causes of fatty liver infiltration (4). The association of steatosis with a number of different clinical conditions has been suggested. Common metabolic diseases such as central obesity, T2DM and hyperlipidemia are well-established risk factors and have been associated with both benign liver steatosis and progressive NASH (Figure 1). Moreover, it has recently been proposed that metabolic syndrome may play a causal role in the pathogenesis of NASH. Management of both NAFLD and NASH has recently become a major challenge to healthcare systems, and many different interventions have been proposed (5). In this case, one may speculate that the treatment of obesity might reduce or stop the development of NASH. The treatment of obesity is mainly related to weight reduction and improvement of eating habits. All patients with NAFLD, whether obese or normal weight, should be informed that a healthy diet has many benefits in addition to weight reduction. They should reduce the added sugar to a minimum and also minimize unhealthy fast eating and, conversely, increase fiber intake. An increase in physical activity should also be recommended. It is likely that there will not be only one right approach for all patients with NAFLD, it will be necessary to adapt the diet individually, including the inclusion of n-3 fatty acids, foods with higher monounsaturated fatty acids, fruits, vegetables and reducing the intake of saturated fats or simple carbohydrates (6, 7). Recently, it was proposed that food intake regulating peptides play a significant role in obesity regulation and may have the potential to be a drug for obesity treatment (8–11). Nevertheless, to elucidate the detailed pathophysiological mechanisms of NAFLD and NASH, appropriate experimental models need to be used. Because there are many causes of human steatohepatitis pathology, it is difficult to establish a universal experimental model. Thus, several genetic, nutritional (diet-induced) and other mouse, rat, and rabbit models have been established (12–15). Models based on overnutrition with adipose tissue enlargement and resulting metabolic complications, particularly insulin resistance, may be most useful to investigate critical etiopathogenic factors. Not only environmental factors, but also genetics play a role in the development and progression of NAFLD, as reviewed by (16). At least four genetic variants that play a role in lipid metabolism in the liver are robustly associated with the development and progression of NAFLD in humans. Patatin-like phospholipase domain-containing protein 3 (PNPLA3), involved in lipid droplet remodeling, is the most robust and replicable genetic variant associated with NAFLD. Transmembrane 6 superfamily member 2 (TM6SF2) is involved in very low-density lipoprotein (VLDL) secretion. Other genes causing the development of NASH are membrane bound O-acyltransferase domain-containing 7 (MBOAT7) and glucokinase regulator (GCKR). Additionally, other genetic variants involved in the regulation of lipid metabolism, inflammation, insulin signaling, oxidative stress and fibrogenesis in NAFLD progression have been studied (16).
This review explores the role of food intake regulating peptides in the pathology of NAFLD and NASH. Special attention will be paid to several anorexigenic peptides, such as leptin, glucagon-like peptide-1 (GLP-1), and to the orexigenic peptide ghrelin. Because anorexigenic peptides are currently promising substances in the treatment of obesity, they may also play an important role in the future treatment of liver pathology. On the other hand, the inhibition of the orexigenic peptide ghrelin could prevent age-associated hepatic steatosis. Another aim of our review is the critical view of experimental models for the study of NAFLD and NASH.
Features of NASH
The individual markers for NASH, such as biochemical markers and inflammatory or fibrogenic factors, improve our understanding of disease pathogenesis and allow therapies to be developed. Biochemical markers measured in plasma or the liver, such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST), reflect nonspecific hepatocellular damage. Aminotransferase levels can be increased two to four times the upper normal limit in NASH. Other biochemical markers such as triglyceride (TG) and cholesterol levels are measured in plasma and from liver biopsies. A high content of liver hydroxyproline (originating mostly from collagen) was also observed in obese leptin‐deficient (ob/ob) mice, in both obesity and NASH mouse models (17). On the other hand, inflammatory cytokines and chemokines such as tumor necrosis factor α (TNFα), interleukin (IL)-6, chemokine CC ligand-2 (CCL2) and another inflammation marker, C-reactive protein (CRP), are among the main markers of NASH (18–20). Moreover, liver biopsy is used to verify or diagnose the stage of NASH and to monitor histopathological changes in NASH. To monitor these histopathological changes, several histological scoring systems, including the NAFLD activity score (NAS) and the fibrosis scoring system, were established. The NAS system evaluates the severity of macrovesicular and microvesicular steatosis, hepatocellular ballooning or lobular inflammation. Fibrosis is scored in 7 grades with the Laennec scoring system, in which 0 indicates no fibrosis; 1, minimal fibrosis; 2, mild fibrosis; 3, moderate fibrosis; 4A, cirrhosis, mild, definite, or probable; 4B, moderate cirrhosis; and 4C, severe cirrhosis (21). However, noninvasive approaches such as magnetic resonance spectroscopy provide a sensitive method to detect hepatic steatosis (22). Other inflammatory and fibrotic markers discovered by liver mRNA sequencing include the inflammatory marker cluster of differentiation 68 (CD68), which is highly expressed in macrophages, the cluster of differentiation molecule 11b (CD11b) marker expressed in Kupffer cells (also known as stellate macrophages located in the liver) and fibrosis markers, such as collagen type 1 α 1 chain (col1a1), collagen type 3 α 1 chain (col3a1) and α-smooth muscle actin (α-SMA) (17, 21, 23, 24).
Mouse Models of NASH
Various mouse models that mirror both the pathophysiology and the histopathology of NAFLD/NASH have been developed to elucidate the progression of NAFLD to NASH and its link to metabolic syndrome. Dietary approaches including a high-fat diet (HFD) and atherogenic and methionine- and choline-deficient (MCD) diets with many variations produce different severity of disease and are widely used (Table 1) (25). Other commonly used models are mice with genetic manipulations that allow to study different lipid pathways or glucose metabolism during the development of steatosis or fibrosis (Table 1). Toxin-induced models are not very commonly used; however, these models also develop some features of NAFLD and NASH with connection to metabolic syndrome (Table 1).
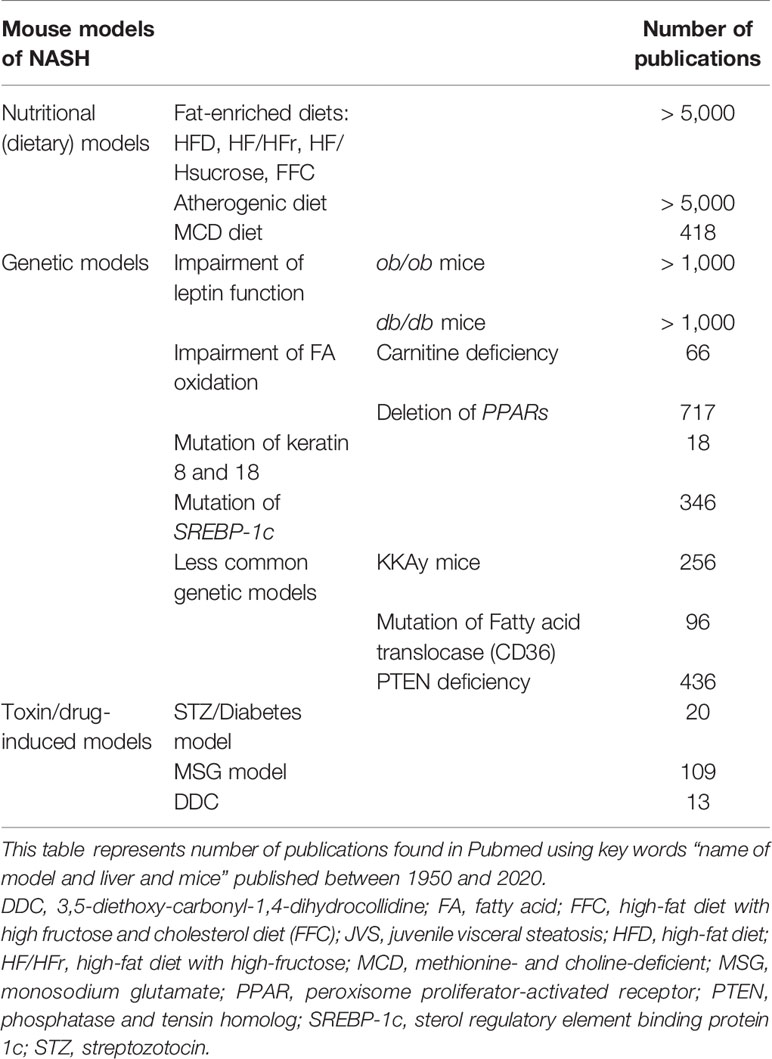
Table 1 Number of publications in mouse models of NASH.
Nutritional (Dietary) Models
High-Fat Diet
Dietary models, including obesogenic and nutrient-deficient models, can effectively trigger the development of NAFLD/NASH. Obesogenic models imitate overnutrition and a sedentary lifestyle leading to overweight or obesity in humans; therefore, the use of diet-induced obesity (DIO) models represents the natural development of NASH. The classic HFD contains 45%–75% of total calories from fat, without any nutrient deficiencies, and represents the most commonly used model of obesity in rodents (Table 2) (62). Mice fed this diet develop obesity with an increase in adiposity and metabolic syndrome (63, 64) and display severe liver steatosis with micro- and macrovesicular lipid accumulation and increased total liver TG but without marks of fibrosis (26–28). However, the length of diet administration, the content and type of fat used, the sex, species, and genetic background of the model can play roles in adiposity and subsequent NAFLD development.
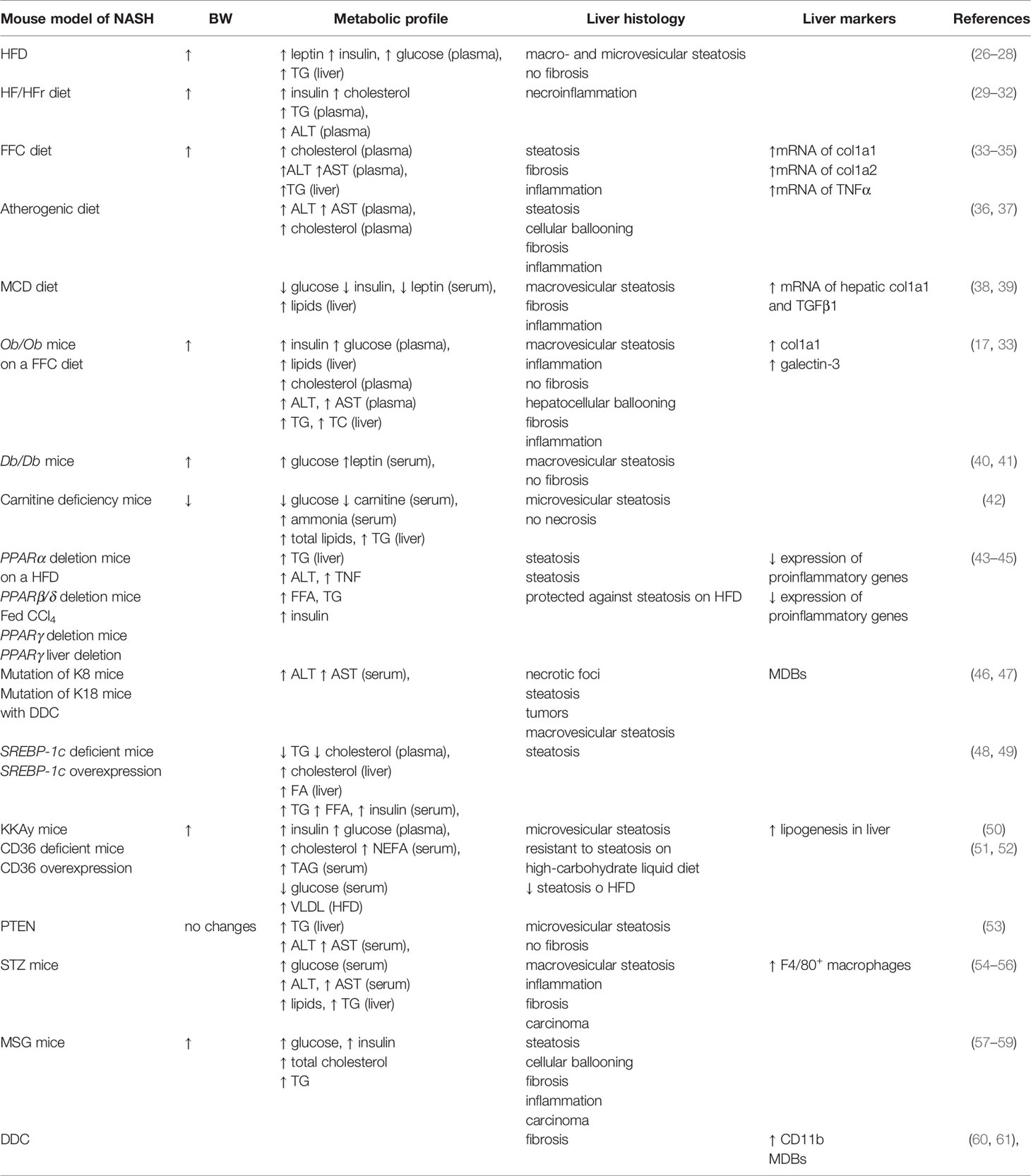
Table 2 Mouse model and features of NAFLD/NASH.
High-Fat Diet With High-Fructose/Sucrose
To increase hepatic fibrosis and preserve steatosis, HFD with high-fructose or high-sucrose (HF/HFr or HF/Hsucrose) consumption can be used; moreover, this approach has been linked to the development of NASH (Table 2) (65, 66). The metabolism of fructose differs from that of glucose. Hepatic metabolism of fructose favors de novo lipogenesis, and fructose stimulates the synthesis of TG and free fatty acids (FFAs). Fructose overconsumption is related to the obesity epidemic (67). In addition, high fructose consumption and increased fat intake is reported to be a risk factor for the development of NAFLD (29, 68). Fructose consumption, even in the absence of obesity, causes serious changes in the liver, such as dyslipidemia, insulin resistance, and NAFLD, with areas of inflammation (30). Compared to lean controls, male mice fed a high-fat, high-fructose (HFr) diet alone or combined with a high-fat (HF/HFr) diet had increased plasma cholesterol and TG and increased homeostasis model assessment for insulin resistance (HOMA-IR). Nevertheless, a dramatic increase in body mass was observed only in the HFD and HF/HFr groups and not in the HFr group. All three groups displayed features of NAFLD, with increased lipogenesis mediated by sterol regulatory element binding protein 1c (SREBP-1c), peroxisome proliferator-activated receptor γ (PPARγ) and reduced peroxisome proliferator-activated receptor α (PPARα). Inflammation was observed in the HFr and HF/HFr groups, leading to the development of NASH (30). It was reported that mice fed a high-fat, high-carbohydrate diet and a 55% fructose and 45% sucrose solution for 16 weeks developed obesity and a NASH-like phenotype with significant fibrosis (31). Mice fed a high-fat, high-sucrose diet developed severe hepatic steatosis with low-grade inflammation and fibrosis and increased inflammatory and fibrosis marker expression (32).
Mice fed a high fat, high-fructose and high-cholesterol (FFC) diet demonstrated increased body weight, plasma cholesterol, ALT, and AST and hepatic TG. Mice presented steatosis with an NAS score of 3 and fibrosis with a score of 1–3 and increased expression of fibrillary collagens such as col1a1 and collagen type 1 α 2 chain (col1a2) (33–35, 69). These mice also exhibited leukocyte infiltration in the liver and high expression of monocyte chemotactic protein-1 and TNFα (34).
The consumption of a HFD alone or a HFD enriched with other mentioned components greatly mimics features of human obesity and steatosis. However, only low-grade fibrosis and inflammation develop with the consumption of different types of HFDs. Therefore, the addition of fructose or cholesterol could enhance all features of NASH.
Atherogenic (High-Cholesterol, High-Cholate) Diet
An atherogenic diet enriched with cholesterol and cholic acid (or sodium cholate) has been widely used to study atherosclerosis and coronary heart disease (Table 2). A diet supplemented with 1.0% cholesterol and 0.3% sodium cholate increased body weight and led to elevated AST, ALT and cholesterol in Wistar rats. The liver displayed features of increased steatosis, hepatic necrosis, macrophage infiltration and hepatic fibrosis (36). An atherogenic diet induced dyslipidemia, lipid peroxidation and stellate cell activation leading to precirrhotic steatohepatitis after 24 weeks on the diet, and in contrast to HFD, cellular ballooning, one of the features of NASH, was observed. An atherogenic diet increased the expression of genes for fatty acid (FA) synthesis, oxidative stress, inflammation, and fibrogenesis, which were further accelerated by the addition of a HFD but did not change body weight. This model suggests the critical role of lipids in causing oxidative stress and insulin resistance leading to steatohepatitis (37). Hypercholesterolemia should be considered a risk factor for hepatic fibrosis, and it could be enhanced to develop all metabolic features when a high content of cholesterol is combined with a high content of fat in the diet.
Methionine- and Choline-Deficient Diet
Nutrient-deficient models with low contents or lacking certain nutrients could also be used for the development of NAFLD/NASH features. The MCD diet contains high sucrose (40%) and fat (10%) but lacks methionine and choline, which are essential for hepatic β-oxidation and VLDL production (Table 2). Rats fed an MCD diet develop macrovesicular steatosis, inflammation, and hepatic fibrosis (70). In mice, the MCD diet caused liver injury, which is associated with hepatic microsomal lipid peroxidation. Livers revealed macrosteatosis and inflammation, together with perivenular and pericellular fibrosis (71). In different strains and animal models, the severity of NASH induced by an MCD diet differed; however, C57BL/6 mice developed the most inflammation and necrosis and best approximated the histological features of NASH (72). Although the MCD diet causes severe inflammation, oxidative stress, mitochondrial damage, apoptosis, and fibrogenesis, the metabolic profile of the model is the opposite of that of typical human NASH (73).
Specifically, mice fed an MCD diet show weight loss, decreased fasted glucose, no insulin resistance and low insulin and leptin levels (38, 39). On the other hand, the main advantage of the MCD diet is that it is easy to obtain and use.
Genetic Models
Impairment of Leptin Function
Ob/ob Mice With Leptin Deficiency
Ob/ob mice carry a spontaneous mutation in the leptin gene (74) but do not have impaired leptin receptors. Ob/ob mice develop severe obesity due to hyperphagia and reduced energy expenditure (Table 2). Hyperinsulinemia, insulin resistance and hyperglycemia develop before obesity occurs (75, 76). Leptin-deficient mice are predisposed to develop severe steatohepatitis with macrovesicular steatosis; however, when maintained on a standard chow diet, they do not develop fibrosis (35). Ob/ob mice are protected from fibrosis because leptin is a mediator of hepatic fibrosis during chronic toxic liver injury (77). Nevertheless, leptin-deficient mice maintained on an FFC diet are more susceptible to developing features of NASH, such as steatohepatitis and fibrosis than wild-type C57BL/6J mice (33). Generally, it is believed that additional stimuli, such as an MCD diet or toxin exposure, must be added to develop inflammation and fibrosis, but interestingly, ob/ob mice fed a MCD diet develop moderate lobular inflammation and no detectable fibrosis (40, 77).
Ob/ob mice on an FFC diet displayed strong features of NASH with fibrosis, elevated plasma ALT, AST and total cholesterol, and high expression of col1a1, a marker of fibrosis, and galectin-3, a marker of inflammation (17).
Db/db Mice With Leptin Receptor Mutation
Db/db mice have a spontaneous mutation in the leptin receptor, and therefore, they are resistant to the effects of leptin (78, 79). These mice are hyperphagic, and early onset morbid obesity occurs with severe T2DM, insulin resistance, hyperglycemia and hyperleptinemia (Table 2) (80, 81). Db/db mice have macrovesicular hepatic steatosis, but they have only modestly increased liver TG compared to the control mice and do not spontaneously develop steatohepatitis or fibrosis. When db/db mice are exposed to a MCD diet, they exhibit increased inflammation and serum ALT compared with db/db mice fed a control diet and even ob/ob mice. Moreover, db/db mice fed a MCD diet revealed pericellular fibrosis (40). Interestingly, hepatic steatosis and insulin resistance, as well as body weight and adiposity, were reversed to the level of control mice in db/db mice by caloric restriction, a condition under which mice receive a restricted amount of food (2 g/day) (41).
Impairment of FA Oxidation
FA oxidation takes place in three cellular organelles: mitochondria, peroxisomes and the endoplasmic reticulum (ER) (microsomes). Mitochondrial β‐oxidation is the main route for the metabolism of FAs under normal physiological conditions (82). Deficiencies of the enzymes involved in FA oxidation have been recognized as important causes of the pathogenesis of macrovesicular and microvesicular hepatic steatosis (Table 2).
Carnitine Deficiency Leading to Juvenile Visceral Steatosis Mice
Juvenile visceral steatosis (JVS) mice have a systemic carnitine deficiency caused by mutation of the organic cation/carnitine transporter 2 gene, which encodes a plasma membrane carnitine transporter (Table 2) (42, 83). JVS mice develop severe lipid accumulation in the liver from an early age, with microvesicular swelling of hepatocytes, hypoglycemia, high levels of ammonia in serum, and growth retardation (42, 83, 84). This model can represent a model to examine changes in FA metabolism in the liver.
Deletion of Peroxisome Proliferator-Activated Receptors
Peroxisome proliferator-activated receptors (PPARs) belong to the nuclear receptor transcription factor family. This class comprises three PPAR isoforms, namely, α, β/δ, and γ, which are expressed in various tissues; PPARs play a role in the transcriptional regulation of glucose and lipid metabolism (Table 2) (85, 86).
PPARα is ubiquitous, but it is most highly expressed in the liver. It has a critical role in the regulation of FA uptake, β-oxidation, ketogenesis, bile acid synthesis, and TG turnover (85). PPARα was suggested to have an anti-inflammatory role in the liver and in adipose tissue. Mice lacking PPARα are more susceptible to the negative effects of a HFD, and feeding them a HFD results in increased steatosis and oxidative stress and inflammation markers compared to their controls (43).
PPARβ/δ is highly expressed in skeletal muscle, adipose tissue, and skin. PPARβ/δ is also expressed in the liver, hepatocytes, Kupffer cells and hepatic stellate cells, suggesting a potential role in inflammation and fibrosis (85). A study with PPARβ/δ-deficient mice suggested that PPARβ/δ is hepatoprotective against chemically induced hepatotoxicity, such as carbon tetrachloride (CCl4), by downregulating the expression of proinflammatory genes (87).
PPARγ is highly expressed in adipose tissue, where it controls adipocyte differentiation, adipogenesis, and lipid metabolism, but the expression of PPARγ in the liver is very low (85). Mice with adipose tissue-specific PPARγ deficiency have increased plasma FFA and TG, fatty liver, and enhanced hepatic gluconeogenesis (88). Moreover, these mice were significantly more susceptible to HFD-induced steatosis, hyperinsulinemia, and insulin resistance than their controls. Ob/ob mice with PPARγ deficiency in the liver had increased serum TG and FFA and decreased mRNA expression of hepatic lipogenic genes compared with their controls. A deficiency in hepatic PPARγ further aggravated the severity of diabetes in ob/ob mice due to decreased insulin sensitivity in muscle and fat. Hepatic PPARγ plays a critical role in the regulation of TG content and in the homeostasis of blood glucose and insulin resistance in steatotic diabetic mice (89).
Models with PPAR deficiencies are used to study the role of PPARs in glucose and lipid metabolism, but studies on their role in the development of NASH features could be considered rather uncommon.
Mutation of Keratin 8 and Keratin 18
Keratins belong to a large family of intermediate filaments, and they are expressed in epithelial cells as specific keratin pairs. Keratin 8 (K8) and keratin 18 (K18) are expressed mainly in adult hepatocytes, and mutations of this K8/K18 pair can lead to various liver diseases (Table 2) (90–92). Mice expressing mutant K8/18 represent an animal model for human chronic hepatitis and for studying the tissue-specific function of K8/18 (93). The increased frequency of keratin K8/K18 variants in NAFLD patients was previously reported (94). Keratin phosphorylation, transamidation and glycosylation (95) are involved in rearranging the keratin cytoskeleton into cytoplasmic inclusions, known as Mallory-Denk bodies (MDBs), found in specific liver diseases such as alcoholic hepatitis and cirrhosis (90).
Mutation of Sterol Regulatory Element Binding Protein-1c
SREBP plays a key role in lipid homeostasis by regulating the expression of genes involved in lipid synthesis (96, 97). SREBP-1c is transcriptionally controlled by various factors, mainly insulin and glucose, and regulates hepatic glucose and lipid metabolism (Table 2) (98). In cultured hepatocytes, insulin and glucose activate the transcription of the SREBP-1c gene, whereas glucagon has an inhibitory effect (99, 100). SREBP-1c expression was positively correlated with fatty acid synthase (FASN) expression, and higher levels were found in the liver in NAFLD models than in controls (101). The SREBP-1 gene has a role in the genetic predisposition of metabolic diseases such as obesity, T2DM, and dyslipidemia (102).
SREBP-1c deficiency led to the reduced expression of enzymes involved in FA and TG synthesis. SREBP-1c KO mice had reduced total plasma TG and cholesterol. In contrast, the liver cholesterol content was higher in SREBP-1c KO mice than in WT mice (48). Mice with liver-specific overexpression of human SREBP-1c (alb-SREBP-1c) developed hepatic lipid accumulation featuring a fatty liver by the age of 24 weeks. Moreover, liver-specific over-expression of human SREBP-1c (alb-SREBP-1c) mice had increased liver FA levels, serum TG and FFA, and insulin levels, indicating insulin resistance (49).
Less Common Genetic Models
KKAy Mice
Yellow KKAy mice carry the yellow obese gene (Ay) and they develop spontaneous obesity due to hyperphagia with increased adiposity and diabetic symptoms. KKAy mice are characterized by insulin resistance with increased blood glucose, circulating insulin levels and increased lipogenesis in the liver and in adipose tissue (Table 2) (103). The livers of KKAy mice had microvesicular steatosis and an increased degree of hepatic steatosis (50). KKAy mice represent a model that closely resembles obesity and T2DM in humans, who develop several metabolic diseases therefore, this model is valuable for the development of potential therapeutic strategies not only for diabetes (104).
Deficiency or Overexpression of Fatty Acid Translocase (CD36)
Fatty acid translocase, or cluster of differentiation 36 (CD36), is a transmembrane glycoprotein that facilitates lipid transport (105). The circulating serum level of CD36 is increased in NAFLD and correlates with the histological grade of steatosis intrahepatic lipids, ALT and TG (106, 107). CD36-deficient mice showed a significant increase in fasting levels of cholesterol, non-esterified fatty acid (NEFA) and TG with lower fasting serum glucose levels (Table 2) (51). CD36 KO mice are resistant to hepatic steatosis when fed a high-carbohydrate liquid diet, and they do not develop alcoholic steatosis when chronically fed alcohol (108). CD36 was suggested to be a protective metabolic sensor in the liver under lipid overload and metabolic stress (52).
Mutation of Phosphatase and Tensin Homolog in the Liver
Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene mutated in many human cancers but has multiple roles in organisms. Its expression is reduced or absent in almost half of hepatoma patients (109, 110). The hepatocyte-specific null mutation of PTEN in mice (PTEN-HEP-KO) showed marked hepatomegaly and steatohepatitis with TG accumulation, a phenotype similar to that observed in human NASH (Table 1). PTEN deficiency in hepatocytes led to steatosis through increased FA uptake and de novo lipogenesis (53).
Toxin/Drug-Induced Models
Streptozotocin Diabetes Model
Streptozotocin (STZ) is a broad-spectrum antibiotic with antitumor, oncogenic, and diabetogenic properties (111). Multiple small intraperitoneal injections of STZ (40 mg/kg) in mice produce pancreatic insulitis, with the selective destruction of pancreatic β cells and diabetes mellitus (112), causing insulin deficiency, hyperglycemia, polydipsia, and polyuria, all of which mimic human type 1 diabetes mellitus (Table 2) (113). Hepatic changes, including lipid peroxidation, mitochondrial swelling, peroxisome proliferation and hepatocyte proliferation inhibition, occur before STZ induces hyperglycemia (114). In the subacute phase, intraperitoneal injection of STZ resulted in an increase in serum glucose and in serum and liver lipids and a decrease in liver glycogen (54). Neonatal male mice exposed to low-dose STZ developed liver steatosis with diabetes after one week on a HFD. With continuous HFD, neonatal STZ mice develop NASH pathology with decreased hepatic fat deposits and increased lobular inflammation and fibrosis. At older ages, mice develop hepatocellular carcinoma (55). Neonatal STZ mice demonstrated focal liver lesions and hepatocellular carcinoma. Hematoxylin-eosin staining showed macrovesicular steatosis, lobular inflammation, hepatocellular ballooning and mild fibroblast proliferation by silver impregnation (56). This model represents a model of NASH linked to diabetes and hepatocellular carcinoma without the accumulation of fat and the development of steatosis.
Monosodium Glutamate Model
Subcutaneous injections of monosodium glutamate (MSG) into newborn mice cause acute neuronal necrosis mainly in the arcuate nucleus (ARC) of the hypothalamus. MSG-treated mice display stunted growth due to impaired growth hormone production (115), marked obesity, and female sterility, but they are rather hypophagic (116). MSG mice had an 8 times higher fat-to-body mass ratio and developed hyperglycemia and hyperinsulinemia (Table 2) (117–119), which was more pronounced in males than in females. The obesity-related changes in the feeding behavior of the MSG-treated mice are possibly the result of missing leptin and insulin receptors in ARCs and consequent altered neuropeptide signaling (120). The injection of MSG in ICR mice leads to the development of significant inflammation, central obesity, and T2DM. Compared with control mice, MSG-ICR mice had increased concentrations of glucose, insulin, total cholesterol, and TG (57). MSG-ICR mice developed NAFLD and NASH-like pathology and had steatohepatitis and dysplastic nodular lesions within the fibrotic liver (121). A different strain, MSG-DIAR (ddY, Institute for Animal Reproduction, Japan) mice, revealed a similar pattern of T2DM and macrovesicular steatosis, lobular inflammation with neutrophils, and ballooning degeneration. At an older age, they developed cellular structures mimicking human hepatocellular carcinoma (58). The crucial metabolic window for studying pathophysiological events involved in NAFLD/NASH progression is considered at 4–6 months of age, the age at which MSG-treated mice also have peripheral insulin resistance (59). The MSG model develops features of NASH with obesity and steatosis, high TG, and insulin resistance; however, this model is not used frequently.
Porphyrinogen Agent as Inducer of NASH-Like Liver Lesions
Feeding with 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) leads to chronic xenobiotic-induced cholangiopathy in mice (Table 2). DDC feeding results in significantly increased number of CD11b-positive cells. Moreover, mice fed DDC have a biliary type of liver fibrosis (60). DDC triggered the formation of MDBs and with that, the expression of keratins 8/18 was elevated (61).
Role of Anorexigenic Peptides in NASH
Anorexigenic peptides, of both hypothalamic and gut origin, play important roles in the pathology of NAFLD and NASH. The most studied peripheral gut hormones include cholecystokinin, leptin, amylin, GLP-1, oxyntomodulin, and bombesin, and the main hypothalamic anorexigenic peptides include cocaine- and amphetamine-regulated transcript peptide, α-melanocyte-stimulating hormone, corticotropin-releasing factor and prolactin- releasing peptide. This review is focused mainly on leptin and GLP-1 as they are the best known anorexigenic peptides involved in NAFLD and NASH pathogenesis (122, 123).
Leptin
Leptin, a product of the ob gene, is a hormone secreted by white adipose tissue that acts as a major regulator of food intake and energy homeostasis. Because leptin receptors are found in the brain and in many peripheral tissues, leptin triggers many biological effects and has the potential to affect a wide range of diseases (124–127). Obese individuals usually have high plasma leptin concentrations, and this hyperleptinemia leads to leptin resistance (128). Mutations in the ob gene are rarely responsible for obesity in humans, but several animal models with ob gene mutations exist (73).
The binding of leptin to its receptor activates Janus kinase 2 (JAK2) and phosphorylates specific tyrosine residues of the receptor and downstream proteins, including signal transducer and activation of transcription (STAT3), Src homology region 2 (SH2)-containing protein tyrosine phosphatase 2 (SHP2), insulin receptor substrate 2 (IRS2), and phosphatidylinositol 3-kinase (PI3K), which regulate the transcription of genes involved in food intake and lipid metabolism. Leptin activates 5′ adenosine monophosphate-activated protein kinase (AMPK) and decreases acetyl-CoA carboxylase (ACC) activity in skeletal muscle while increasing mitochondrial β-oxidation. Another aspect of the metabolic activity of leptin is the inhibition of hepatic stearoyl-CoA desaturase-1 (SCD-1) activity, which regulates lipoprotein metabolism and energy consumption. On the other hand, leptin inactivates AMPK, increases ACC activity and decreases food intake in the hypothalamus (126).
Role of Leptin in NAFLD/NASH Progression to Fibrosis
Ten years ago, hepatic lipotoxicity and non-hepatic factors such as adipose tissue inflammation and gastrointestinal imbalance were linked to evolution of NAFLD (129). Nowadays, the degree of adipose tissue inflammation was shown to directly correlate with the severity of NAFLD. Consumption of higher caloric intake is increasingly emerging as a fuel for metabolic inflammation not only in obesity-related disorders but also NAFLD. Gut microbiome in NAFLD is gaining importance too (130). Characteristic features of NAFLD progression include imbalance in FA metabolism, cytokine dysregulation, and oxidative metabolic stress. Oxidative stress is mainly characterized by the excessive production of reactive oxygen species by three main mechanisms, namely, lipid peroxidation, cytokine induction, and Fas ligand (type II transmembrane protein belonging to the TNF family), and induces the progression from steatosis to steatohepatitis and to fibrosis (131). In NAFLD progression, oxidative stress causes the induction of purinergic receptor X7 expression at both the mRNA and protein levels in inflammatory cells, but the detailed mechanisms are not yet clear (132).
Circulating leptin levels primarily reflect energy stores in the body but also indicate acute changes in caloric intake. Leptin appears to exert a dual action in experimental NAFLD models; it protects against liver steatosis, at least in the early stages of the disease, but it also acts as an inflammatory and fibrogenic mediator when the disease persists or continues (133). An important role of leptin is to reduce the deposition of TG in adipocytes and at the same time to limit the storage of TG in nonadipose tissues, including the liver, and thus protect them from lipotoxicity and lipoapoptosis (Figure 2) (134). Leptin prevents liver steatosis in animal models by affecting both lipid and glucose metabolism. Under normal conditions, leptin suppresses hepatic glucose production and hepatic lipogenesis (135). Chronic central administration of leptin reduces the expression of hepatic lipogenic genes and reduces TG content by stimulating hepatic sympathetic activity; this function requires PI3K signaling because the leptin-mediated impairment of PI3K signaling leads to hepatic steatosis without inducing obesity (Figure 2) (133). For lipid accumulation in the liver, glycerol is needed. A decrease in glycerol availability might be involved in the beneficial effects of leptin on NAFLD. Integral membrane aquaglycerolporins (AQP3, AQP7, AQP9, and AQP10) form a channel across the cell membrane and thus facilitate glycerol transport. The main gateway facilitating the release of glycerol from adipocytes is AQP7; however, AQP3, AQP9, and AQP10 also aid in glycerol efflux from fat depots (Figure 2). Subsequently, circulating plasma glycerol is transferred to hepatocytes by hepatic-specific AQP9, where glycerol kinase catalyzes the initial step for its conversion to glucose (gluconeogenesis) or to TG in ob/ob mice (136). More specifically, leptin inhibits hepatic de novo lipogenesis while stimulating FA oxidation, thereby reducing lipid content in isolated livers (133, 137, 138). The steatosis observed in ob/ob mice suggests that leptin is taken up by the fatty liver, indirectly owing to central nerve pathways and directly by hepatic AMPK activation. However, the regulation of glucose production in the liver by leptin but not insulin requires hepatic AMPKα2 activity (131). On the other hand, leptin may promote hepatic fibrogenesis through the upregulation of transforming growth factor β (TGF-β) in Kupffer cells and sinusoidal endothelial cells. Leptin prevents the upregulation of col1a1 mRNA, a change associated with the fibrotic process in the liver. Fibrosis develops in the absence of leptin in ob/ob mice; therefore, liver fibrosis depends on the presence of leptin in chronic liver damage (77, 139). In Kupffer cells, leptin induces cluster of differentiation 14 (CD14) expression by activating STAT3 (140). This results in a hepatic hypersensitive response and progression from simple steatosis or liver inflammation to fibrosis (Figure 2). This points to possible therapeutic approaches for NASH by targeting leptin-dependent STAT3 and CD14 signaling. During liver fibrosis, the role of leptin and its functioning receptors, in particular in activated hematopoietic stem cells (HSCs), has been demonstrated. Increases in leptin-associated col1a2 gene expression and in leptin-enhanced col1a2 gene promoter activity were observed by ribonuclease protection analysis in vitro (141). Furthermore, leptin enhances platelet-derived growth factor-dependent proliferative responses in HSCs, most likely through actions involving the PI3K/Akt pathway (140, 141).
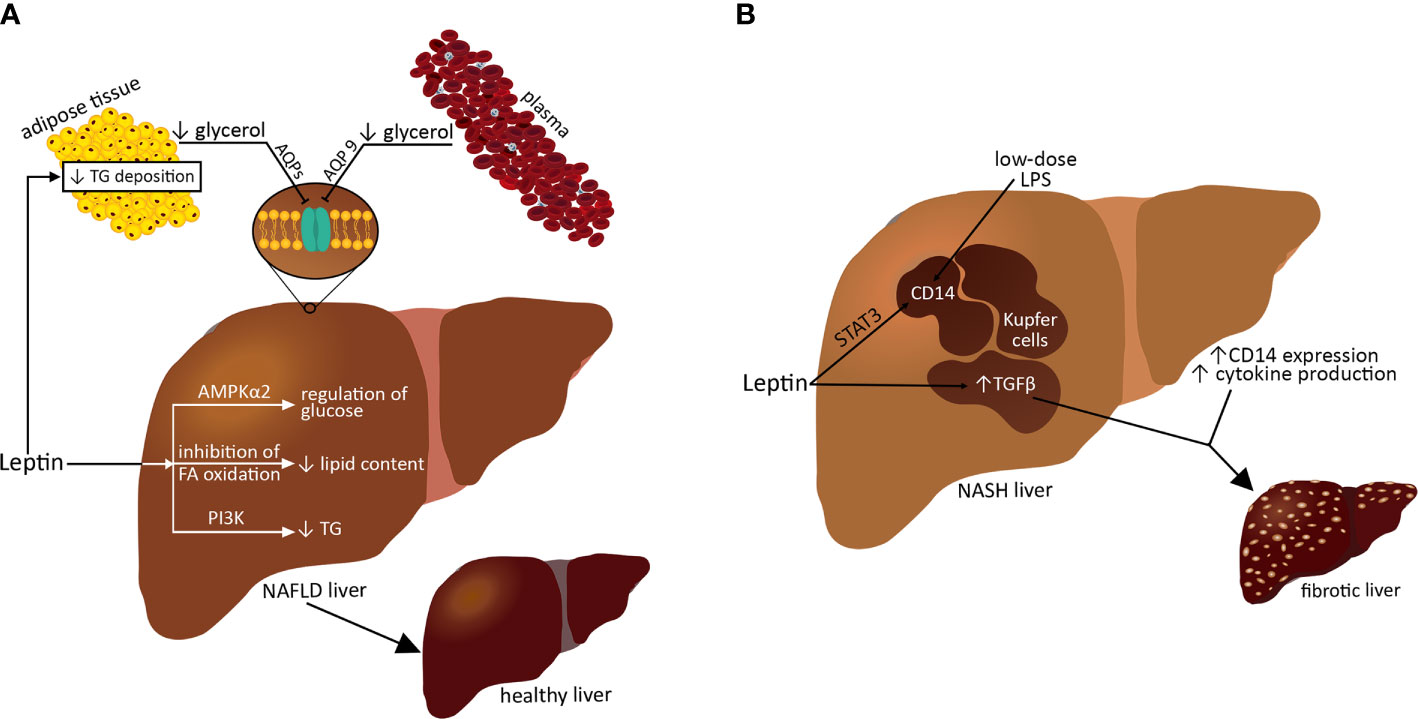
Figure 2 Role of leptin in NAFDL/NASH progression.
In conclusion, it seems that in the initial stages of the disease, leptin may protect against hepatic steatosis. However, when the disease progresses, leptin may act as an inflammatory and fibrogenic agent. Leptin deficiency can lead to hepatic steatosis, and excess leptin can promote hepatitis and fibrosis.
GLP-1
The gut incretin hormones glucose-dependent insulinotropic peptide (GIP) and GLP-1 are secreted after the oral administration of nutrients and stimulate insulin secretion, which leads to a decrease in glucose concentration (142, 143). GLP-1 slows gastric motility, enhances satiety, reduces appetite and energy intake, and suppresses postprandial glucagon secretion (Figure 3) (144, 145). Glucagon-like peptide-1 receptor (GLP-1R; also termed secretin-like receptor) belongs to the G protein-coupled receptor family B. GLP-1R mRNA is found in the pancreatic islets, lungs, hypothalamus, hippocampus, cerebral cortex, brainstem, kidney, stomach, intestine, skin, and heart of rodents and humans (146, 147).
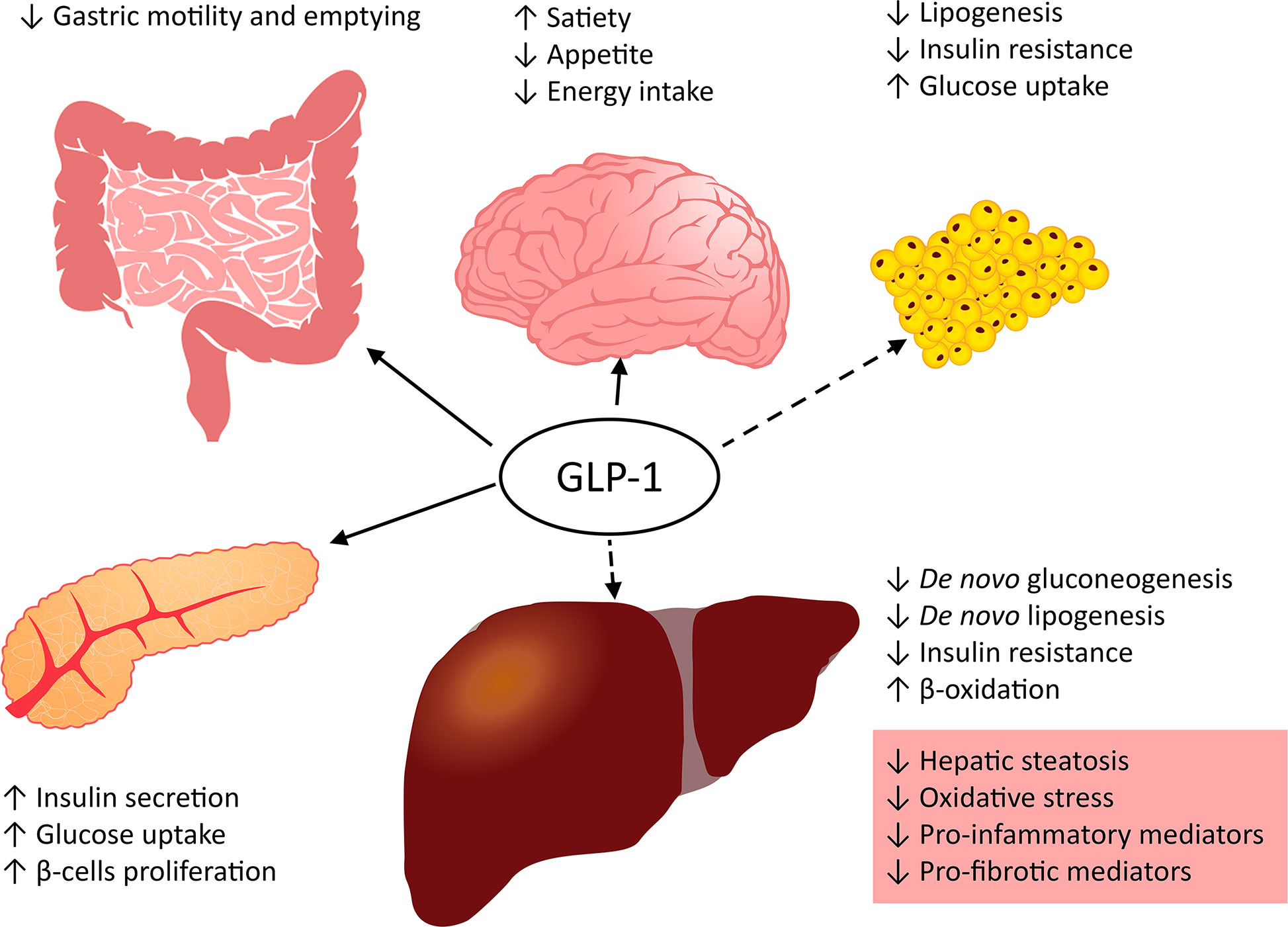
Figure 3 Role of GLP-1 in metabolism.
GLP-1–Activated Pathways in Hepatic Lipid Metabolism
GLP-1R agonists stimulate the production of cAMP through adenylate cyclase, and cAMP activates protein kinase A (PKA) and exchange protein activated by cAMP (Epac) (148, 149). PKA and Epac induce insulin gene transcription and secretion (150). Activation of cAMP in turn activates PKA, which leads to the phosphorylation of AMPK (151). In addition, cAMP activation stimulates the epidermal growth factor receptor, pointing to the activation of the PI3K/Akt signaling pathway. These actions suppress the expression of genes that have a key role in the stimulation of insulin secretion affects the repression of hepatic gluconeogenesis and lipogenesis and reduce postprandial plasma glucose levels by elevating insulin-regulated glucose uptake (Figure 3) (148, 149). Insulin or ER stimulation is required for release SREBP-1c, which activates the expression of the lipogenic genes ACC and fatty acid synthase (FASN) and enhances glycolytic flux. The expression of lipogenic genes requires the activation of the transcription factors SREBP-1c and carbohydrate response element binding protein (ChREBP). GLP-1R agonists reduce the expression of both ChREBP and SREBP-1c genes and inhibit de novo lipogenesis in the liver (98, 152).
PPAR transcription factors are also involved in the regulation of lipid metabolism. GLP-1 analogs may have a key role in increasing the expression of PPARα and suppressing PPARγ genes (153, 154).
The Role of GLP-1 in NAFLD/NASH Progression to Fibrosis
GLP-1R agonists are approved for the treatment of T2DM, and they have great potential in the treatment of NAFLD and NASH. GLP-1R agonists enhance insulin secretion and improve glucose tolerance, which leads to a decrease in lipogenesis de novo and enhances hepatic FA oxidation and lipid export (Figure 3). Moreover, receptor activation leads to the central regulation of satiety and decreases in appetite and energy intake. GLP-1R agonists alleviate metabolic inflammation and NASH by suppressing the expression of inflammatory genes such as TNFα, IL-6, and nuclear factor NF-kappa-B (NFκB) (155, 156). Many studies in mice show improvement and the possibility of preventing hepatic steatosis using different GLP-1R agonists (35, 157–161). GLP-1R agonists directly affect Kupffer cell function and reduce the influx of macrophages into the liver (162). Together with dipeptidyl-peptidase 4 (DPP4) inhibitors, they decrease not only proinflammatory but also profibrotic mediators. Decreasing profibrotic mediators in mice occurs via the reduction of TGF-β gene expression and other profibrotic mediators through the stimulation of cAMP production (151, 159, 163). Epac and PKA activation by cAMP leads to a reduction in col1, col3, and angiotensin II expression and to smad (TGF-β superfamily member) pathway inhibition (164, 165).
GLP-1R Agonists in Experimental Models
Despite the fact that food intake-regulating peptides have great potential in the treatment of metabolic diseases and NAFLD/NASH, there is very little known about their effects on current mouse models of NASH. Only GLP-1R agonists, primarily used as antidiabetic/antiobesity drugs, are used as potential agents for NAFLD/NASH treatment, as demonstrated in several studies with mouse models. The GLP-1R agonists exenatide, liraglutide and semaglutide are already used in clinical practice for the treatment of T2DM and obesity. The available mouse models of NASH address different aspects of the disease, leading to various clinical utilities in drug discovery. The advantages and limitations of current in vivo mouse models of NASH in view of different targets for NASH treatment are summarized in the review by Hansen et al. (23). The GLP-1R agonist Exendin-4 was originally isolated from the saliva of the Gila monster. Because of the change in the amino sequence at position 8, Exendin-4 has a longer half-life and more stable interaction with DPP4 than GLP-1 (148, 157). Treatment with Exendin-4 (synthetic form is called exenatide) reduced serum glucose levels and body weight and improved serum ALT in ob/ob mice. Exendin-4 also has a positive effect on hepatocyte lipid metabolism. Treatment significantly decreased hepatic lipid content and thiobarbituric acid reactive substance concentration, which is an important parameter of hepatic oxidative stress in NASH and NAFLD. Moreover, Exedin-4 significantly reduced the mRNA levels of SREBP-1c and SCD-1, parameters regulating de novo lipogenesis in the liver, and increased the level of PPARα mRNA in ob/ob mice (157). The results from the Trevaskis et al. study in ob/ob and C57BL/6 mice on a HFD support the therapeutic potential of the exendin-4 analog AC3174 in NASH and NAFLD treatment. The analog AC3174 diminished plasma TG and ALT levels and lipid accumulation in the liver and attenuated fibrosis (35). In db/db mice fed an MCD diet, exendin-4 treatment attenuated hepatic steatosis, TG and FFA content, oxidative stress and hepatic inflammation (166).
Several studies in mice have shown that treatment with liraglutide, a long-acting palmitoylated analog of GLP-1, alleviates hepatic steatosis and has great potential to inhibit NASH or NAFLD (160, 161, 167, 168). In two different NASH mouse obesity models, an atherogenic diet model and ob/ob mice, chronic treatment with liraglutide reduced body weight, lowered steatosis scores and inhibited fibrosis (through a decreased col1a1) (17). In male C57BL/6J mice fed a western diet, liraglutide significantly improved insulin sensitivity and prevented NASH pathology. Liraglutide improved lipid flux between liver and adipose tissue, downregulated genes regulating de novo lipogenesis and increased the expression of genes associated with β-oxidation, FA uptake and VLDL transport (167). The administration of liraglutide in male C57BL/6 mice fed a HFD reduced ALT and AST serum levels and inhibited NOD-like receptor family pyrin-containing 3 inflammasome gene expression, which has a critical role in NAFLD pathogenesis (161). Another study using (C/EBP) homologous protein (CHOP) C57BL/6 KO mice fed an FFC diet significantly attenuated hepatic steatosis after 4 weeks of liraglutide treatment in WT mice and showed that CHOPs play an important role in the protection of hepatocytes from diet-induced ER stress (160).
GLP-1R and glucagon receptor (GLP-1R/GR) dual agonists also have potential in the treatment of NAFLD. DIO C57BL/6 mice treated with GLP-1R/GR dual agonists showed improved glucose metabolism and reduced food intake and weight loss. In the liver, decreases in acetyl-CoA and malonyl-CoA were observed, and ketogenesis was increased (169). Antiobesity effects were not the only effects observed in C57BL/6J mice fed a HFD treated with GLP-1R/GR dual agonists. This study showed reduced mRNA expression of hepatic SREBP-1c and SCD-1 and increased PPARα expression. Reduced levels of the inflammation factors TNFα and IL-6 in plasma and of TGF-ß, monocyte chemoattractant protein-1, matrix metalloproteinase-9 and TNFα in the liver inhibited the development of NASH and NAFLD (159). Moreover, a GLP-1R and GIP receptor dual agonist attenuated NASH in C57BL/6J mice fed an atherogenic diet, significantly decreased body and liver weight, decreased liver TG and improved NAS, showing synergistic action compared to monotherapy with GLP-1R agonist or GIP (170).
GHRELIN
Orexigenic peptides such as ghrelin, neuropeptide Y, agouti-related protein, and orexins play an important role in the mechanism of food intake (171). Ghrelin peptide is released from the stomach as the endogenous ligand for the growth hormone secretagogue receptor (GHSR), which stimulates growth hormone (GH) release from the anterior pituitary gland. Ghrelin regulates food intake, adiposity, body weight, glucose metabolism, taste sensation, sleep modulation, brown fat thermogenesis, stress and anxiety responses, muscle atrophy, gut motility, gastric acid secretion, and cardiovascular function (172–174).
Ghrelin is the only known peptide with an attached FA. The octanoylation of ghrelin is catalyzed by ghrelin-O-acyltransferase. This modification is essential for the biological activity of ghrelin (175). Ghrelin without the acyl group (des-acyl ghrelin) is biologically inactive (101, 176). Des-acyl ghrelin is also present at significant levels in both blood and stomach, but des-acyl ghrelin can neither bind to GHSR nor exhibit GH release (177).
Ghrelin-Activated Pathways in Hepatic Lipid Metabolism
Ghrelin plays a complex role in hepatic metabolism and liver diseases. On the one hand, ghrelin induces adiposity in the liver; on the other hand, ghrelin elicits a protective effect against inflammation and fibrosis. Currently, there is no clear explanation for opposite actions of ghrelin in these pathologies (178).
Central ghrelin administration leads to lipid storage in the liver, which is independent of GH. Ghrelin promotes energy storage to minimize negative effects in periods of food shortage (179).
Ghrelin has a direct peripheral effect on lipogenesis in hepatocytes. Ghrelin activates GHSR in hepatocytes, which leads to increased TG synthesis, by increasing the expression of lipogenesis-related genes in hepatocytes. These effects are mediated by mammalian target of rapamycin (mTOR) – PPARγ signaling pathway activation (180). The activation of this pathway is independent of the central stimulation of energy intake in the hypothalamus, where activated AMPK mediates the orexigenic action of ghrelin. Ghrelin stimulates the activity of AMPK in the hypothalamus but inhibits AMPK activity in the liver and adipose tissue, resulting in increased lipogenesis (181).
Other studies have shown that the tumor protein p53 is crucial for the stimulation of lipid storage in fat and the liver by ghrelin. Lack of p53 abolishes the stimulation of lipid storage induced by administered ghrelin (182).
In the gastrointestinal tract, ghrelin has potent anti-inflammatory properties. Exogenous ghrelin pretreatment augmented the release of the anti-inflammatory cytokine interleukin IL-10 (183) and inhibited the production of various pro-inflammatory cytokines, such as IL-1β, IL-6, IL-8, and TNFα (184). Furthermore, the hepatoprotective effect of ghrelin is caused by inhibition of apoptosis and proliferation stimulation in various cell types (185, 186).
The Role of Ghrelin in NAFLD/NASH Progression to Fibrosis
The activation of the gastric ghrelin-brain axis is essential to maintain biological homeostasis. The liver damage signal caused by FA infiltration is sent to the brain and stomach via autonomic nerve connections, which causes an increase in ghrelin release. These signals could slow down the progression of NAFLD. Impairment of this appetite control is necessary for NASH pathology (187).
Low levels of acylated ghrelin in plasma are found in NASH (188, 189). Moreover, decreased plasma ghrelin correlates with increased immunoglobulin production that is often observed in patients with chronic liver disease (190) and correlates with liver inflammation (191). The most likely reason for the low ghrelin levels in NAFLD patients is insulin resistance. Low ghrelin levels are observed in several diseases characterized by insulin resistance, including severe obesity (192), acromegaly (193), hypogonadism (194), and polycystic ovary syndrome (195). Nevertheless, the molecular mechanisms remain elusive (196).
Patients with NASH had a twofold higher concentration of des-acyl ghrelin compared with healthy humans. Des-acyl ghrelin also correlated with ALT, AST, TG levels, fasting glucose, MDBs, and portal fibrosis, which are strongly associated with the occurrence of NASH (197).
Oxidative stress and inflammation are key factors in the development of NAFLD/NASH. Thus, the impairment of these processes could revert the development of NASH. The administration of ghrelin during and after NAFLD development reduced inflammation, apoptosis, and oxidative stress and improved lipid metabolism in the rat liver (198). In addition to the protective effects of ghrelin mentioned above, ghrelin also exerts antifibrotic and hepatoprotective effects in the injured livers of rodents (199). Antifibrotic effects can also be seen in other tissues, such as the heart (200) and colon (201).
Exogenous and endogenous ghrelin regulates fibrogenesis in mice and humans (199). Antifibrotic effects are caused by several mechanisms. Ghrelin protects hepatocytes from cell death by reducing inflammatory cells, decreasing apoptosis, and increasing the activation of hepatoprotective signaling pathways such as Akt phosphorylation. Ghrelin modulates inflammation by downregulating the NFκB pathway (202). The wound-healing response to injury is caused by decreasing oxidative stress in livers (199). Ghrelin also reduces profibrogenic cytokine TGF-β1 and p-Smad3 expression levels that are involved in increased deposition of fibronectin, col1 and α-SMA in liver fibrosis. Ghrelin suppresses autophagy, thus reducing available energy from intracellular lipid degradation (202).
Pharmacological Therapies for NAFLD/NASH
NAFLD is associated with obesity, T2DM, dyslipidemia, and metabolic syndrome. Weight loss through dietary changes and lifestyle modifications is now the only proven effective therapy for patients with NAFLD/NASH. Nevertheless, these approaches are not sufficient for the treatment of fibrosis and even cirrhosis. Pharmaceutical companies are developing new drugs for the treatment of NASH, but no drugs have yet been approved (203, 204). The pathophysiology of NAFLD is very complex and associated with different features, such as lipotoxicity, inflammatory cytokines, apoptosis, and insulin resistance. Therefore, drugs to treat NASH could target these features (203).
In patients with T2DM, the prevalence of NAFLD is 75% (205); therefore, the use of antidiabetic drugs to improve insulin resistance could be one approach for treatment. Metformin is an insulin sensitizer used as a major therapy for T2DM because of its low cost, body weight-lowering effect and safety profile (203). Nevertheless, metformin was reported to not significantly improve liver histology and therefore is not recommended in the treatment of NASH (206).
Pioglitazone belongs to the thiazolidinedione family, agonists of PPARγ, and improves glucose and lipid metabolism. Treatment with pioglitazone improved insulin sensitivity, plasma ALT and AST levels and liver steatosis, inflammation, and ballooning (206, 207). Pioglitazone causes some side effects, such as body weight gain, possible bladder cancer and bone loss in women (206). Nevertheless, risks and benefits should be considered, as pioglitazone improves liver histology in patients with and without T2DM with NASH.
GLP-1R agonists and DPP4 inhibitors were also investigated as possible therapeutics for NASH treatment. Liraglutide, a stable GLP-1 analog, improved steatosis and hepatocyte ballooning and slowed fibrosis progression (205, 208). On the other hand, DPP4 inhibitors delay the quick inactivation of GLP-1 in plasma. Sitagliptin was unable to improve the fibrosis score or NAS or to reduce liver fat after 24 weeks of therapy (209, 210).
Conclusions
A large number of mouse models could be used to study NASH pathogenesis and its possible treatment. However, some approaches do not coincide with human NALFD. Currently, NASH is associated with the development of prediabetes and metabolic syndrome with elevated ALT, AST, cholesterol, and FFA levels.
Genetic models represent advantages in the time required and development of concrete metabolic features associated with NAFLD. However, these mutations are very rare in humans. Moreover, some of these models fail to induce the metabolic comorbidities typically observed in humans with NASH, such as insulin resistance, obesity and dyslipidemia.
Regarding nutritional models, a large number of different approaches, with variable fat content, glucose/fructose enrichment or other substance additions or deficiencies, create complicated decisions for researchers. The crucial advantage of nutritional models is the ability to mimic human NAFLD, both pathophysiologically and phenotypically. In contrast, a longer period is necessary for the development of NAFLD, and a lesser degree of pathology is observed. Nevertheless, this issue could be overcome by the use of high glucose/fructose levels or increased cholesterol levels in the diet.
Finally, regarding chemically induced models, toxic agents are not pathophysiologically related to NASH disease. However, those models could be used to enhance fibrosis and cirrhosis leading to liver failure.
All these models should be used with caution, and their use should be limited to clearly defining liver-specific research and to studying the pathologic features of human NAFLD/NASH.
It seems that anorexigenic and orexigenic peptides are involved in the pathology of NAFLD and NASH. Leptin may have a potential dual action in NAFLD and NASH. Leptin may protect the liver from hepatic steatosis at the initial stage of the disease but also acts as an inflammatory and fibrogenic marker when the disease progresses. Leptin deficiency can lead to hepatic steatosis, and excess leptin can promote hepatitis and fibrosis. The efficacy of NASH treatment with anorexigenic leptin is questionable, similar to potential treatment with orexigenic ghrelin. Ghrelin induces adiposity in the liver, but also ghrelin elicits a protective effect against inflammation and fibrosis.
Pharmaceutical companies are developing new drugs for the treatment of NAFLD and NASH; however, no drugs have been approved yet. Because NAFLD is associated with obesity, T2DM, dyslipidemia, and metabolic syndrome, weight loss through dietary changes and lifestyle modifications is now the only proven effective therapy for patients with NAFLD/NASH. Nevertheless, these approaches are not sufficient for the treatment of fibrosis and even cirrhosis.
Recently, GLP-1R agonists used primarily as antidiabetic or antiobesity drugs have shown the greatest potential in the possible treatment of NASH. GLP-1R agonists enhance insulin secretion and improve glucose tolerance, which leads to a decrease in lipogenesis de novo and enhances hepatic FA oxidation and lipid export. GLP-1R agonists alleviate metabolic inflammation and NASH by suppressing the expression of inflammatory genes. Nevertheless, more attention should be paid to the potential role of other anorexigenic and/or orexigenic peptides in the pathophysiology of NAFLD and NASH in the future, especially in relation to the treatment of obesity and T2DM.
Author Contributions
LK and LM coordinated writting of the manuscript, LK, JK, VP, LM, AK, and LC wrote the manuscript. BZ, JK, and LM revised and completed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Grant Agency of the Czech Republic (Grant No. 18-10591S).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACC, acetyl-CoA carboxylase; Alb-SREBP-1c, liver-specific over-expression of human SREBP-1c; ALT, alanine aminotransferase; AMPK, 5’ adenosine monophosphate-activated protein kinase; AQPs, aquaglyceroporins; ARC, nucleus arcuatus; AST, aspartate aminotransferase; Ay, yellow obese gene; CC, chemokine; CCL, chemokine ligand-2; CCl4, carbon tetrachloride; CD11b, cluster of differentiation 11B; CD14, cluster of differentiation 14; CD36, cluster of differentiation 36; CD68, cluster of differentiation 68; CHOP, C/EBP homologous protein; ChREBP, carbohydrate response element binding protein; Col1a1, collagen type 1 α 1 chain; Col1a2, collagen type 1 α 2 chain; Col3a1, collagen type 3 α 1 chain; CRP, C-reactive protein; DDC, 3,5-dietoxy-carbonyl-1,4-dihydrocollidine; DIAR, ddY, Institute for Animal Reproduction; DIO, diet-induced obesity; DPP4, dipeptidyl peptidase-4; Epac, exchange protein activated by cAMP; ER, endoplasmatic reticulum; FA, fatty acid; FASN, fatty acid synthase; FFA, free fatty acid; FFC, high fructose and cholesterol; GCKR, glucokinase regulator; GH, growth hormone; GHSR, growth hormone secretagogue receptor; GIP, glucose-dependent insulinotropic peptide; GLP-1, glucagon-like peptide 1; GLP-1R, glucagon-like peptide 1 receptor; GLP-1R/GR, GLP-1R and glucagon receptor dual agonists; HF/HFr, high-fat high-fructose; HFD, high-fat diet; HFr, high-fructose; HOMA-IR, homeostasis model assessment for insulin resistance; HSCs, hematopoetic stem cells; IL, interleukin; IRS2, insulin receptor substrate 2; JAK2, Janus kinase 2; JVS, juvenile visceral steatosis; K18, keratin 18; K8, keratin 8; MBOAT7, membrane bound O-acyltransferase domain-containing 7; MCD, methionine- and choline-deficient; MDBs, Mallory-Denk bodies; MSG, monosodium glutamate; mTOR, mammalian target of rapamycin; NAFLD, non-alcoholic fatty liver disease; NAS, NAFLD activity score; NASH, non-alcoholic steatohepatitis; NEFA, non-esterified fatty acid; NFκB, nuclear factor NF-kappa-B; p53, tumor protein; PI3K, phosphatidylinositol 3-kinase; PKA, protein kinase A; PNPLA3, patatin-like phospholipase domain-containing protein 3; PPARs, peroxisome proliferator-activated receptors; PTEN, phosphatase and tensin homolog; PTEN-HEP-KO, hepatocyte-specific null mutation of PTEN in mice; SCD-1, stearoyl-CoA desaturase-1; SHP2, Src homology region 2 (SH2)-containing protein tyrosine phosphatase 2; Smad, TGF-β superfamily member; SREBP-1c, sterol regulatory element binding protein 1c; STAT, signal transducer and activation of transcription; STZ, streptozotocin; T2DM, type 2 diabetes mellitus; TG, triglyceride; TGF-β, transforming growth factor β; TM6SF2, transmembrane 6 superfamily member 2; TNFα, tumor necrosis factor α; US, United States; VLDL, very low-density lipoprotein; α-SMA, alpha-smooth muscle actin.
References
1. Marcellin P, Kutala BK. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int (2018) 38 Suppl 1:2–6. doi: 10.1111/liv.13682
2. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of nafld and nash: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
3. Younossi ZM. Review article: Current management of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment Pharmacol Ther (2008) 28:2–12. doi: 10.1111/j.1365-2036.2008.03710.x
4. Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: Pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol (2013) 28 Suppl 1:68–76. doi: 10.1111/jgh.12212
5. Del Ben M, Polimeni L, Baratta F, Pastori D, Loffredo L, Angelico F. Modern approach to the clinical management of non-alcoholic fatty liver disease. World J Gastroenterol (2014) 20:8341–50. doi: 10.3748/wjg.v20.i26.8341
6. Finelli C, Tarantino G. Is there any consensus as to what diet or lifestyle approach is the right one for nafld patients? J Gastrointestin Liver Dis (2012) 21:293–302.
7. Jordão Candido C, Silva Figueiredo P, Del Ciampo Silva R, Candeloro Portugal L, Augusto Dos Santos Jaques J, Alves de Almeida J, et al. Protective effect of α-linolenic acid on non-alcoholic hepatic steatosis and interleukin-6 and -10 in wistar rats. Nutrients (2019) 12:1–15. doi: 10.3390/nu12010009
8. Brandt SJ, Kleinert M, Tschop MH, Muller TD. Are peptide conjugates the golden therapy against obesity? J Endocrinol (2018) 238:R109–R19. doi: 10.1530/JOE-18-0264
9. Mikulaskova B, Maletinska L, Zicha J, Kunes J. The role of food intake regulating peptides in cardiovascular regulation. Mol Cell Endocrinol (2016) 436:78–92. doi: 10.1016/j.mce.2016.07.021
10. Müller TD, Clemmensen C, Finan B, DiMarchi RD, Tschöp MH. Anti-obesity therapy: From rainbow pills to polyagonists. Pharmacol Rev (2018) 70:712–46. doi: 10.1124/pr.117.014803
11. Williams DM, Nawaz A, Evans M. Drug therapy in obesity: A review of current and emerging treatments. Diabetes Ther (2020) 11:1199–216. doi: 10.1007/s13300-020-00816-y
12. Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical models for studying nash-driven hcc: How useful are they? Cell Metab (2019) 29:18–26. doi: 10.1016/j.cmet.2018.10.012
13. Imajo K, Yoneda M, Kessoku T, Ogawa Y, Maeda S, Sumida Y, et al. Rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Int J Mol Sci (2013) 14:21833–57. doi: 10.3390/ijms141121833
14. Nevzorova YA, Boyer-Diaz Z, Cubero FJ, Gracia-Sancho J. Animal models for liver disease - a practical approach for translational research. J Hepatol (2020) 73:423–40. doi: 10.1016/j.jhep.2020.04.011
15. Sanches SC, Ramalho LN, Augusto MJ, da Silva DM, Ramalho FS. Nonalcoholic steatohepatitis: A search for factual animal models. BioMed Res Int (2015) 2015:574832. doi: 10.1155/2015/574832
16. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of nafld and nash: Clinical impact. J Hepatol (2018) 68:268–79. doi: 10.1016/j.jhep.2017.09.003
17. Tolbol KS, Kristiansen MN, Hansen HH, Veidal SS, Rigbolt KT, Gillum MP, et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J Gastroenterol (2018) 24:179–94. doi: 10.3748/wjg.v24.i2.179
18. Neuman MG, Cohen LB, Nanau RM. Biomarkers in nonalcoholic fatty liver disease. Can J Gastroenterol Hepatol (2014) 28:607–18. doi: 10.1155/2014/757929
19. Roth JD, Feigh M, Veidal SS, Fensholdt LK, Rigbolt KT, Hansen HH, et al. Int-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis. World J Gastroenterol (2018) 24:195–210. doi: 10.3748/wjg.v24.i2.195
20. Schuppan D, Surabattula R, Wang XY. Determinants of fibrosis progression and regression in nash. J Hepatol (2018) 68:238–50. doi: 10.1016/j.jhep.2017.11.012
21. Kim MY, Cho MY, Baik SK, Park HJ, Jeon HK, Im CK, et al. Histological subclassification of cirrhosis using the laennec fibrosis scoring system correlates with clinical stage and grade of portal hypertension. J Hepatol (2011) 55:1004–09. doi: 10.1016/j.jhep.2011.02.012
22. Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab (2005) 288:E462–E68. doi: 10.1152/ajpendo.00064.2004
23. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discovery Today (2017) 22:1707–18. doi: 10.1016/j.drudis.2017.06.007
24. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (2005) 41:1313–21. doi: 10.1002/hep.20701
25. Radhakrishnan S, Ke JY, Pellizzon MA. Targeted nutrient modifications in purified diets differentially affect nonalcoholic fatty liver disease and metabolic disease development in rodent models. Curr Dev Nutr (2020) 4:nzaa078. doi: 10.1093/cdn/nzaa078
26. Lo L, McLennan SV, Williams PF, Bonner J, Chowdhury S, McCaughan GW, et al. Diabetes is a progression factor for hepatic fibrosis in a high fat fed mouse obesity model of non-alcoholic steatohepatitis. J Hepatol (2011) 55:435–44. doi: 10.1016/j.jhep.2010.10.039
27. Lanthier N, Molendi-Coste O, Cani PD, van Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J (2011) 25:4301–11. doi: 10.1096/fj.11-189472
28. Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, et al. Fast food diet mouse: Novel small animal model of nash with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol (2011) 301:G825–34. doi: 10.1152/ajpgi.00145.2011
29. Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology (2010) 51:1961–71. doi: 10.1002/hep.23535
30. Schultz A, Neil D, Aguila MB, Mandarim-de-Lacerda CA. Hepatic adverse effects of fructose consumption independent of overweight/obesity. Int J Mol Sci (2013) 14:21873–86. doi: 10.3390/ijms141121873
31. Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology (2010) 52:934–44. doi: 10.1002/hep.23797
32. Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology (2013) 58:1632–43. doi: 10.1002/hep.26594
33. Kristiansen MN, Veidal SS, Rigbolt KT, Tolbol KS, Roth JD, Jelsing J, et al. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J Hepatol (2016) 8:673–84. doi: 10.4254/wjh.v8.i16.673
34. Kawashita E, Ishihara K, Nomoto M, Taniguchi M, Akiba S. A comparative analysis of hepatic pathological phenotypes in c57bl/6j and c57bl/6n mouse strains in non-alcoholic steatohepatitis models. Sci Rep (2019) 9:204. doi: 10.1038/s41598-018-36862-7
35. Trevaskis JL, Griffin PS, Wittmer C, Neuschwander-Tetri BA, Brunt EM, Dolman CS, et al. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol (2012) 302:G762–72. doi: 10.1152/ajpgi.00476.2011
36. Jeong WI, Jeong DH, Do SH, Kim YK, Park HY, Kwon OD, et al. Mild hepatic fibrosis in cholesterol and sodium cholate diet-fed rats. J Vet Med Sci (2005) 67:235–42. doi: 10.1292/jvms.67.235
37. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology (2007) 46:1392–403. doi: 10.1002/hep.21874
38. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol (2004) 40:47–51. doi: 10.1016/j.jhep.2003.09.020
39. Leclercq IA, Lebrun VA, Starkel P, Horsmans YJ. Intrahepatic insulin resistance in a murine model of steatohepatitis: Effect of ppargamma agonist pioglitazone. Lab Invest (2007) 87:56–65. doi: 10.1038/labinvest.3700489
40. Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, et al. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: Role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol (2004) 287:G1035–43. doi: 10.1152/ajpgi.00199.2004
41. Kim KE, Jung Y, Min S, Nam M, Heo RW, Jeon BT, et al. Caloric restriction of db/db mice reverts hepatic steatosis and body weight with divergent hepatic metabolism. Sci Rep (2016) 6:30111. doi: 10.1038/srep30111
42. Kuwajima M, Kono N, Horiuchi M, Imamura Y, Ono A, Inui Y, et al. Animal model of systemic carnitine deficiency: Analysis in c3h-h-2 degrees strain of mouse associated with juvenile visceral steatosis. Biochem Biophys Res Commun (1991) 174:1090–4. doi: 10.1016/0006-291x(91)91532-h
43. Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. Pparalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr (2011) 141:603–10. doi: 10.3945/jn.110.135210
44. Shan W, Palkar PS, Murray IA, McDevitt EI, Kennett MJ, Kang BH, et al. Ligand activation of peroxisome proliferator-activated receptor beta/delta (pparbeta/delta) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol Sci (2008) 105:418–28. doi: 10.1093/toxsci/kfn142
45. Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, et al. Role for ppargamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J (2011) 25:2538–50. doi: 10.1096/fj.10-173716
46. Loranger A, Duclos S, Grenier A, Price J, Wilson-Heiner M, Baribault H, et al. Simple epithelium keratins are required for maintenance of hepatocyte integrity. Am J Pathol (1997) 151:1673–83.
47. Bettermann K, Mehta AK, Hofer EM, Wohlrab C, Golob-Schwarzl N, Svendova V, et al. Keratin 18-deficiency results in steatohepatitis and liver tumors in old mice: A model of steatohepatitis-associated liver carcinogenesis. Oncotarget (2016) 7:73309–22. doi: 10.18632/oncotarget.12325
48. Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver x receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem (2002) 277:9520–8. doi: 10.1074/jbc.M111421200
49. Knebel B, Haas J, Hartwig S, Jacob S, Kollmer C, Nitzgen U, et al. Liver-specific expression of transcriptionally active srebp-1c is associated with fatty liver and increased visceral fat mass. PloS One (2012) 7:e31812. doi: 10.1371/journal.pone.0031812
50. Zhu W, Chen S, Li Z, Zhao X, Li W, Sun Y, et al. Effects and mechanisms of resveratrol on the amelioration of oxidative stress and hepatic steatosis in kkay mice. Nutr Metab (Lond) (2014) 11:35. doi: 10.1186/1743-7075-11-35
51. Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, et al. A null mutation in murine cd36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem (1999) 274:19055–62. doi: 10.1074/jbc.274.27.19055
52. Garbacz WG, Lu P, Miller TM, Poloyac SM, Eyre NS, Mayrhofer G, et al. Hepatic overexpression of cd36 improves glycogen homeostasis and attenuates high-fat diet-induced hepatic steatosis and insulin resistance. Mol Cell Biol (2016) 36:2715–27. doi: 10.1128/MCB.00138-16
53. Peyrou M, Bourgoin L, Poher AL, Altirriba J, Maeder C, Caillon A, et al. Hepatic pten deficiency improves muscle insulin sensitivity and decreases adiposity in mice. J Hepatol (2015) 62:421–9. doi: 10.1016/j.jhep.2014.09.012
54. Kume E, Ohmachi Y, Itagaki S, Tamura K, Doi K. Hepatic changes of mice in the subacute phase of streptozotocin (sz)-induced diabetes. Exp Toxicol Pathol (1994) 46:368–74. doi: 10.1016/s0940-2993(11)80119-3
55. Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol (2013) 46:141–52. doi: 10.1007/s00795-013-0016-1
56. Akai H, Kiryu S, Ohta Y, Yasaka K, Nakano Y, Inoue Y, et al. The natural history of streptozotocin-stimulated non-alcoholic steatohepatitis mice followed by gd-eob-dtpa-enhanced mri: Comparison with simple steatosis mice. Magn Reson Imaging (2017) 38:123–28. doi: 10.1016/j.mri.2016.12.027
57. Nagata M, Suzuki W, Iizuka S, Tabuchi M, Maruyama H, Takeda S, et al. Type 2 diabetes mellitus in obese mouse model induced by monosodium glutamate. Exp Anim (2006) 55:109–15. doi: 10.1538/expanim.55.109
58. Tsuneyama K, Nishida T, Baba H, Taira S, Fujimoto M, Nomoto K, et al. Neonatal monosodium glutamate treatment causes obesity, diabetes, and macrovesicular steatohepatitis with liver nodules in diar mice. J Gastroenterol Hepatol (2014) 29:1736–43. doi: 10.1111/jgh.12610
59. Coelho CFF, Franca LM, Nascimento JR, Dos Santos AM, Azevedo-Santos APS, Nascimento FRF, et al. Early onset and progression of non-alcoholic fatty liver disease in young monosodium l-glutamate-induced obese mice. J Dev Orig Health Dis (2019) 10:188–95. doi: 10.1017/S2040174418000284
60. Fickert P, Stoger U, Fuchsbichler A, Moustafa T, Marschall HU, Weiglein AH, et al. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol (2007) 171:525–36. doi: 10.2353/ajpath.2007.061133
61. Stumptner C, Fuchsbichler A, Lehner M, Zatloukal K, Denk H. Sequence of events in the assembly of mallory body components in mouse liver: Clues to the pathogenesis and significance of mallory body formation. J Hepatol (2001) 34:665–75. doi: 10.1016/s0168-8278(00)00099-4
62. Speakman JR. Use of high-fat diets to study rodent obesity as a model of human obesity. Int J Obes (Lond) (2019) 43:1491–92. doi: 10.1038/s41366-019-0363-7
63. Maletinska L, Nagelova V, Ticha A, Zemenova J, Pirnik Z, Holubova M, et al. Novel lipidized analogs of prolactin-releasing peptide have prolonged half-lives and exert anti-obesity effects after peripheral administration. Int J Obes (Lond) (2015) 39:986–93. doi: 10.1038/ijo.2015.28
64. Shi H, Akunuru S, Bierman JC, Hodge KM, Mitchell MC, Foster MT, et al. Diet-induced obese mice are leptin insufficient after weight reduction. Obesity (Silver Spring) (2009) 17:1702–9. doi: 10.1038/oby.2009.106
65. Lustig RH, Schmidt LA, Brindis CD. Public health: The toxic truth about sugar. Nature (2012) 482:27–9. doi: 10.1038/482027a
66. Nomura K, Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem (2012) 23:203–8. doi: 10.1016/j.jnutbio.2011.09.006
67. Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr (2004) 79:537–43. doi: 10.1093/ajcn/79.4.537
68. Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol (2008) 48:993–9. doi: 10.1016/j.jhep.2008.02.011
69. Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol (2013) 305:G483–95. doi: 10.1152/ajpgi.00079.2013
70. Weltman MD, Farrell GC, Liddle C. Increased hepatocyte cyp2e1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology (1996) 111:1645–53. doi: 10.1016/s0016-5085(96)70028-8
71. Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. Cyp2e1 and cyp4a as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest (2000) 105:1067–75. doi: 10.1172/JCI8814
72. Kirsch R, Clarkson V, Shephard EG, Marais DA, Jaffer MA, Woodburne VE, et al. Rodent nutritional model of non-alcoholic steatohepatitis: Species, strain and sex difference studies. J Gastroenterol Hepatol (2003) 18:1272–82. doi: 10.1046/j.1440-1746.2003.03198.x
73. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol (2012) 18:2300–8. doi: 10.3748/wjg.v18.i19.2300
74. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science (1995) 269:540–3. doi: 10.1126/science.7624776
75. Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr Diabetes Rev (2014) 10:131–45. doi: 10.2174/1573399810666140508121012
76. Lindstrom P. The physiology of obese-hyperglycemic mice [ob/ob mice]. ScientificWorldJournal (2007) 7:666–85. doi: 10.1100/tsw.2007.117
77. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol (2002) 37:206–13. doi: 10.1016/S0168-8278(02)00102-2
78. Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell (1996) 84:491–5. doi: 10.1016/s0092-8674(00)81294-5
79. Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature (1996) 379:632–5. doi: 10.1038/379632a0
80. Campfield LA, Smith FJ, Burn P. The ob protein (leptin) pathway–a link between adipose tissue mass and central neural networks. Horm Metab Res (1996) 28:619–32. doi: 10.1055/s-2007-979867
81. Roesler WJ, Pugazhenthi S, Khandelwal RL. Hepatic glycogen metabolism in the db/db mouse. Mol Cell Biochem (1990) 92:99–106. doi: 10.1007/BF00218127
82. Rao MS, Reddy JK. Peroxisomal beta-oxidation and steatohepatitis. Semin Liver Dis (2001) 21:43–55. doi: 10.1055/s-2001-12928
83. Koizumi T, Nikaido H, Hayakawa J, Nonomura A, Yoneda T. Infantile disease with microvesicular fatty infiltration of viscera spontaneously occurring in the c3h-h-2(0) strain of mouse with similarities to reye’s syndrome. Lab Anim (1988) 22:83–7. doi: 10.1258/002367788780746511
84. Narama I, Ozaki K, Matsuura T, Ono A, Sei M, Shima K, et al. Heterogeneity of histopathologic features in the congenitally carnitine-deficient juvenile visceral steatosis (jvs) mouse. Biomed Res (1997) 18:247–55. doi: 10.2220/biomedres.18.247
85. Liss KH, Finck BN. Ppars and nonalcoholic fatty liver disease. Biochimie (2017) 136:65–74. doi: 10.1016/j.biochi.2016.11.009
86. Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim Biophys Acta (2016) 1859:1083–99. doi: 10.1016/j.bbagrm.2016.03.002
87. Shan W, Nicol CJ, Ito S, Bility MT, Kennett MJ, Ward JM, et al. Peroxisome proliferator-activated receptor-beta/delta protects against chemically induced liver toxicity in mice. Hepatology (2008) 47:225–35. doi: 10.1002/hep.21925
88. He W, Barak Y, Hevener A, Olson P, Liao D, Le J, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A (2003) 100:15712–7. doi: 10.1073/pnas.2536828100
89. Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liver-specific disruption of ppargamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest (2003) 111:737–47. doi: 10.1172/JCI17223
90. Ku NO, Strnad P, Zhong BH, Tao GZ, Omary MB. Keratins let liver live: Mutations predispose to liver disease and crosslinking generates mallory-denk bodies. Hepatology (2007) 46:1639–49. doi: 10.1002/hep.21976
91. Ku NO, Strnad P, Bantel H, Omary MB. Keratins: Biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology (2016) 64:966–76. doi: 10.1002/hep.28493
92. Omary MB, Ku NO, Strnad P, Hanada S. Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest (2009) 119:1794–805. doi: 10.1172/JCI37762
93. Baribault H, Penner J, Iozzo RV, Wilson-Heiner M. Colorectal hyperplasia and inflammation in keratin 8-deficient fvb/n mice. Genes Dev (1994) 8:2964–73. doi: 10.1101/gad.8.24.2964
94. Li R, Liao XH, Ye JZ, Li MR, Wu YQ, Hu X, et al. Association of keratin 8/18 variants with non-alcoholic fatty liver disease and insulin resistance in chinese patients: A case-control study. World J Gastroenterol (2017) 23:4047–53. doi: 10.3748/wjg.v23.i22.4047
95. Ku NO, Toivola DM, Strnad P, Omary MB. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat Cell Biol (2010) 12:876–85. doi: 10.1038/ncb2091
96. Moslehi A, Hamidi-Zad Z. Role of srebps in liver diseases: A mini-review. J Clin Transl Hepatol (2018) 6:332–38. doi: 10.14218/JCTH.2017.00061
97. Horton JD, Goldstein JL, Brown MS. Srebps: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109:1125–31. doi: 10.1172/JCI15593
98. Ferre P, Foufelle F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor srebp-1c. Diabetes Obes Metab (2010) 12 Suppl 2:83–92. doi: 10.1111/j.1463-1326.2010.01275.x
99. Foretz M, Guichard C, Ferre P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci U S A (1999) 96:12737–42. doi: 10.1073/pnas.96.22.12737
100. Hasty AH, Shimano H, Yahagi N, Amemiya-Kudo M, Perrey S, Yoshikawa T, et al. Sterol regulatory element-binding protein-1 is regulated by glucose at the transcriptional level. J Biol Chem (2000) 275:31069–77. doi: 10.1074/jbc.M003335200
101. Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, et al. Srebp-1c, regulated by the insulin and ampk signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med (2008) 21:507–11. doi: 10.3892/ijmm.21.4.507
102. Eberle D, Clement K, Meyre D, Sahbatou M, Vaxillaire M, Le Gall A, et al. Srebf-1 gene polymorphisms are associated with obesity and type 2 diabetes in french obese and diabetic cohorts. Diabetes (2004) 53:2153–7. doi: 10.2337/diabetes.53.8.2153
103. Iwatsuka H, Shino A, Suzuoki Z. General survey of diabetic features of yellow kk mice. Endocrinol Jpn (1970) 17:23–35. doi: 10.1507/endocrj1954.17.23
104. Liu P, Feng T, Zuo X, Wang X, Luo J, Li N, et al. A novel sirt1 activator e6155 improves insulin sensitivity in type 2 diabetic kkay mice. Biochem Biophys Res Commun (2018) 498:633–39. doi: 10.1016/j.bbrc.2018.03.034
105. Park YM. Cd36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med (2014) 46:e99. doi: 10.1038/emm.2014.38
106. Heeboll S, Poulsen MK, Ornstrup MJ, Kjaer TN, Pedersen SB, Nielsen S, et al. Circulating scd36 levels in patients with non-alcoholic fatty liver disease and controls. Int J Obes (Lond) (2017) 41:262–67. doi: 10.1038/ijo.2016.223
107. Garcia-Monzon C, Lo Iacono O, Crespo J, Romero-Gomez M, Garcia-Samaniego J, Fernandez-Bermejo M, et al. Increased soluble cd36 is linked to advanced steatosis in nonalcoholic fatty liver disease. Eur J Clin Invest (2014) 44:65–73. doi: 10.1111/eci.12192
108. Clugston RD, Yuen JJ, Hu Y, Abumrad NA, Berk PD, Goldberg IJ, et al. Cd36-deficient mice are resistant to alcohol- and high-carbohydrate-induced hepatic steatosis. J Lipid Res (2014) 55:239–46. doi: 10.1194/jlr.M041863
109. Peyrou M, Bourgoin L, Foti M. Pten in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and cancer. Dig Dis (2010) 28:236–46. doi: 10.1159/000282095
110. Vinciguerra M, Foti M. Pten at the crossroad of metabolic diseases and cancer in the liver. Ann Hepatol (2008) 7:192–9. doi: 10.1016/S1665-2681(19)31848-4
111. Wu KK, Huan Y. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol (2008). Chapter 5:Unit 5 47. doi:10.1002/0471141755.ph0547s40. doi: 10.1002/0471141755.ph0547s40
112. Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: New model of diabetes mellitus. Science (1976) 193:415–7. doi: 10.1126/science.180605
113. Kolb H. Mouse models of insulin dependent diabetes: Low-dose streptozocin-induced diabetes and nonobese diabetic (nod) mice. Diabetes Metab Rev (1987) 3:751–78. doi: 10.1002/dmr.5610030308
114. Kume E, Fujimura H, Matsuki N, Ito M, Aruga C, Toriumi W, et al. Hepatic changes in the acute phase of streptozotocin (sz)-induced diabetes in mice. Exp Toxicol Pathol (2004) 55:467–80. doi: 10.1078/0940-2993-00351
115. Tamura H, Kamegai J, Shimizu T, Ishii S, Sugihara H, Oikawa S. Ghrelin stimulates gh but not food intake in arcuate nucleus ablated rats. Endocrinology (2002) 143:3268–75. doi: 10.1210/en.2002-220268
116. Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science (1969) 164:719–21. doi: 10.1126/science.164.3880.719
117. Matyskova R, Maletinska L, Maixnerova J, Pirnik Z, Kiss A, Zelezna B. Comparison of the obesity phenotypes related to monosodium glutamate effect on arcuate nucleus and/or the high fat diet feeding in c57bl/6 and nmri mice. Physiol Res (2008) 57:727–34.
118. Pelantova H, Bartova S, Anyz J, Holubova M, Zelezna B, Maletinska L, et al. Metabolomic profiling of urinary changes in mice with monosodium glutamate-induced obesity. Anal Bioanal Chem (2016) 408:567–78. doi: 10.1007/s00216-015-9133-0
119. Yamamoto T, Matsuo S, Ueshima Y, Inoue F, Kinugasa A, Sawada T. Plasma levels of insulin-like growth factor-i are reduced at one week of age in monosodium l-glutamate-treated mice. Endocr J (1993) 40:461–5. doi: 10.1507/endocrj.40.461
120. Maletínská L, Toma RS, Pirnik Z, Kiss A, Slaninová J, Haluzík M, et al. Effect of cholecystokinin on feeding is attenuated in monosodium glutamate obese mice. Regul Pept (2006) 136:58–63. doi: 10.1016/j.regpep.2006.04.020
121. Nakanishi Y, Tsuneyama K, Fujimoto M, Salunga TL, Nomoto K, An JL, et al. Monosodium glutamate (msg): A villain and promoter of liver inflammation and dysplasia. J Autoimmun (2008) 30:42–50. doi: 10.1016/j.jaut.2007.11.016
122. Sobrino Crespo C, Perianes Cachero A, Puebla Jiménez L, Barrios V, Arilla Ferreiro E. Peptides and food intake. Front Endocrinol (2014) 5:58. doi: 10.3389/fendo.2014.00058
123. Tarantino G, Balsano C. Gastrointestinal peptides and nonalcoholic fatty liver disease. Curr Opin Endocrinol Diabetes Obes (2020) 27:11–5. doi: 10.1097/MED.0000000000000514
124. Clement K, Garner C, Hager J, Philippi A, LeDuc C, Carey A, et al. Indication for linkage of the human ob gene region with extreme obesity. Diabetes (1996) 45:687–90. doi: 10.2337/diab.45.5.687
125. Reed DR, Ding Y, Xu W, Cather C, Green ED, Price RA. Extreme obesity may be linked to markers flanking the human <em<ob</em< gene. Diabetes (1996) 45:691. doi: 10.2337/diab.45.5.691
126. Zhang F, Chen Y, Heiman M, Dimarchi R. Leptin: Structure, function and biology. Vitam Horm (2005) 71:345–72. doi: 10.1016/S0083-6729(05)71012-8
127. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi: 10.1038/372425a0
128. Enriori PJ, Evans AE, Sinnayah P, Cowley MA. Leptin resistance and obesity. Obesity (Silver Spring) (2006) 14 Suppl 5:254S–58S. doi: 10.1038/oby.2006.319
129. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology (2010) 52:1836–46. doi: 10.1002/hep.24001
130. Tilg H, Adolph TE, Moschen AR. Multiple parallel hits hypothesis in nafld – revisited after a decade. Hepatology (2020). doi: 10.1002/hep.31518
131. Procaccini C, Galgani M, De Rosa V, Carbone F, La Rocca C, Ranucci G, et al. Leptin: The prototypic adipocytokine and its role in nafld. Curr Pharm Des (2010) 16:1902–12. doi: 10.2174/138161210791208884
132. Chandrashekaran V, Das S, Seth RK, Dattaroy D, Alhasson F, Michelotti G, et al. Purinergic receptor x7 mediates leptin induced glut4 function in stellate cells in nonalcoholic steatohepatitis. Biochim Biophys Acta (2016) 1862:32–45. doi: 10.1016/j.bbadis.2015.10.009
133. Polyzos SA, Kountouras J, Mantzoros CS. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism (2015) 64:60–78. doi: 10.1016/j.metabol.2014.10.012
134. Unger RH, Zhou YT, Orci L. Regulation of fatty acid homeostasis in cells: Novel role of leptin. Proc Natl Acad Sci U S A (1999) 96:2327–32. doi: 10.1073/pnas.96.5.2327
135. Moon HS, Dalamaga M, Kim SY, Polyzos SA, Hamnvik OP, Magkos F, et al. Leptin’s role in lipodystrophic and nonlipodystrophic insulin-resistant and diabetic individuals. Endocr Rev (2013) 34:377–412. doi: 10.1210/er.2012-1053
136. Rodriguez A, Moreno NR, Balaguer I, Mendez-Gimenez L, Becerril S, Catalan V, et al. Leptin administration restores the altered adipose and hepatic expression of aquaglyceroporins improving the non-alcoholic fatty liver of ob/ob mice. Sci Rep (2015) 5:12067. doi: 10.1038/srep12067
137. Cohen SM, Werrmann JG, Tota MR. 13c nmr study of the effects of leptin treatment on kinetics of hepatic intermediary metabolism. Proc Natl Acad Sci (1998) 95:7385–90. doi: 10.1073/pnas.95.13.7385
138. Lee Y, Yu X, Gonzales F, Mangelsdorf DJ, Wang M-Y, Richardson C, et al. Pparα is necessary for the lipopenic action of hyperleptinemia on white adipose and liver tissue. Proc Natl Acad Sci (2002) 99:11848–53. doi: 10.1073/pnas.182420899
139. Ikejima K, Okumura K, Kon K, Takei Y, Sato N. Role of adipocytokines in hepatic fibrogenesis. J Gastroenterol Hepatol (2007) 22:S87–92. doi: 10.1111/j.1440-1746.2007.04961.x
140. Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab (2012) 16:44–54. doi: 10.1016/j.cmet.2012.05.012
141. Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology (2002) 35:762–71. doi: 10.1053/jhep.2002.32029
142. Herrmann C, Goke R, Richter G, Fehmann HC, Arnold R, Goke B. Glucagon-like peptide-1 and glucose-dependent insulin-releasing polypeptide plasma levels in response to nutrients. Digestion (1995) 56:117–26. doi: 10.1159/000201231
143. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: A physiological incretin in man. Lancet (1987) 2:1300–4. doi: 10.1016/s0140-6736(87)91194-9
144. Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest (1998) 101:515–20. doi: 10.1172/JCI990
145. Stoffers DA, Kieffer TJ, Hussain MA, Drucker DJ, Bonner-Weir S, Habener JF, et al. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein idx-1 and increase islet size in mouse pancreas. Diabetes (2000) 49:741–8. doi: 10.2337/diabetes.49.5.741
146. Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology (1996) 137:2968–78. doi: 10.1210/endo.137.7.8770921
147. Hällbrink M, Holmqvist T, Olsson M, Östenson C-G, Efendic S, Langel Ü. Different domains in the third intracellular loop of the glp-1 receptor are responsible for gαs and gαi/gαo activation. Biochim Biophys Acta (BBA) - Protein Struct Molecular Enzymol (2001) 1546:79–86. doi: 10.1016/S0167-4838(00)00270-3
148. Baggio LL, Drucker DJ. Biology of incretins: Glp-1 and gip. Gastroenterology (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
149. Buteau J. Glp-1 receptor signaling: Effects on pancreatic beta-cell proliferation and survival. Diabetes Metab (2008) 34 Suppl 2:S73–7. doi: 10.1016/S1262-3636(08)73398-6
150. Graaf C, Donnelly D, Wootten D, Lau J, Sexton PM, Miller LJ, et al. Glucagon-like peptide-1 and its class b g protein-coupled receptors: A long march to therapeutic successes. Pharmacol Rev (2016) 68:954–1013. doi: 10.1124/pr.115.011395
151. Ben-Shlomo S, Zvibel I, Shnell M, Shlomai A, Chepurko E, Halpern Z, et al. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of amp-activated protein kinase. J Hepatol (2011) 54:1214–23. doi: 10.1016/j.jhep.2010.09.032
152. Bifari F, Manfrini R, Dei Cas M, Berra C, Siano M, Zuin M, et al. Multiple target tissue effects of glp-1 analogues on non-alcoholic fatty liver disease (nafld) and non-alcoholic steatohepatitis (nash). Pharmacol Res (2018) 137:219–29. doi: 10.1016/j.phrs.2018.09.025
153. Lee J, Hong SW, Rhee EJ, Lee WY. Glp-1 receptor agonist and non-alcoholic fatty liver disease. Diabetes Metab J (2012) 36:262–7. doi: 10.4093/dmj.2012.36.4.262
154. Rui L. Energy metabolism in the liver. Compr Physiol (2014) 4:177–97. doi: 10.1002/cphy.c130024
155. Lee YS, Jun HS. Anti-inflammatory effects of glp-1-based therapies beyond glucose control. Mediators Inflammation (2016) 2016:3094642. doi: 10.1155/2016/3094642
156. Seghieri M, Christensen AS, Andersen A, Solini A, Knop FK, Vilsboll T. Future perspectives on glp-1 receptor agonists and glp-1/glucagon receptor co-agonists in the treatment of nafld. Front Endocrinol (Lausanne) (2018) 9:649. doi: 10.3389/fendo.2018.00649
157. Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (glp-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology (2006) 43:173–81. doi: 10.1002/hep.21006
158. Kawakubo M, Tanaka M, Ochi K, Watanabe A, Saka-Tanaka M, Kanamori Y, et al. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis-associated liver fibrosis and tumor development in mice independently of its anti-diabetic effects. Sci Rep (2020) 10:983. doi: 10.1038/s41598-020-57935-6
159. Patel V, Joharapurkar A, Kshirsagar S, Sutariya B, Patel M, Patel H, et al. Coagonist of glp-1 and glucagon receptor ameliorates development of non-alcoholic fatty liver disease. Cardiovasc Hematol Agents Med Chem (2018) 16:35–43. doi: 10.2174/1871525716666180118152158
160. Rahman K, Liu Y, Kumar P, Smith T, Thorn NE, Farris AB, et al. C/ebp homologous protein modulates liraglutide-mediated attenuation of non-alcoholic steatohepatitis. Lab Invest (2016) 96:895–908. doi: 10.1038/labinvest.2016.61
161. Zhu W, Feng PP, He K, Li SW, Gong JP. Liraglutide protects non-alcoholic fatty liver disease via inhibiting nlrp3 inflammasome activation in a mouse model induced by high-fat diet. Biochem Biophys Res Commun (2018) 505:523–29. doi: 10.1016/j.bbrc.2018.09.134
162. Petit JM, Verges B. Glp-1 receptor agonists in nafld. Diabetes Metab (2017) 43 Suppl 1:2S28–33. doi: 10.1016/S1262-3636(17)30070-8
163. Choi SH, Leem J, Park S, Lee CK, Park KG, Lee IK. Gemigliptin ameliorates western-diet-induced metabolic syndrome in mice. Can J Physiol Pharmacol (2017) 95:129–39. doi: 10.1139/cjpp-2016-0026
164. Insel PA, Murray F, Yokoyama U, Romano S, Yun H, Brown L, et al. Camp and epac in the regulation of tissue fibrosis. Br J Pharmacol (2012) 166:447–56. doi: 10.1111/j.1476-5381.2012.01847.x
165. Wahlang B, McClain C, Barve S, Gobejishvili L. Role of camp and phosphodiesterase signaling in liver health and disease. Cell Signal (2018) 49:105–15. doi: 10.1016/j.cellsig.2018.06.005
166. Yamamoto T, Nakade Y, Yamauchi T, Kobayashi Y, Ishii N, Ohashi T, et al. Glucagon-like peptide-1 analogue prevents nonalcoholic steatohepatitis in non-obese mice. World J Gastroenterol (2016) 22:2512–23. doi: 10.3748/wjg.v22.i8.2512
167. Mells JE, Fu PP, Sharma S, Olson D, Cheng L, Handy JA, et al. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in c57bl/6j mice fed a western diet. Am J Physiol Gastrointest Liver Physiol (2012) 302:G225–35. doi: 10.1152/ajpgi.00274.2011
168. Moreira GV, Azevedo FF, Ribeiro LM, Santos A, Guadagnini D, Gama P, et al. Liraglutide modulates gut microbiota and reduces nafld in obese mice. J Nutr Biochem (2018) 62:143–54. doi: 10.1016/j.jnutbio.2018.07.009
169. Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes (2009) 58:2258–66. doi: 10.2337/db09-0278
170. Kannt A, Madsen AN, Kammermeier C, Elvert R, Klöckener T, Bossart M, et al. Incretin combination therapy for the treatment of non-alcoholic steatohepatitis. Diabetes Obesity Metab (2020). doi: 10.1111/dom.14035
171. Patel CA, Acharya SR. Energy homeostasis and obesity: The therapeutic role of anorexigenic and orexigenic peptide. Int J Pept Res Ther (2018) 25:919–32. doi: 10.1007/s10989-018-9740-7
172. Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans**this work was supported in part by grants-in-aid from the ministry of education, science, sports, and culture, japan, and the ministry of health and welfare, japan (to m.N.). Endocrinology (2000) 141:4255–61. doi: 10.1210/endo.141.11.7757
173. Muller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, et al. Ghrelin. Mol Metab (2015) 4:437–60. doi: 10.1016/j.molmet.2015.03.005
174. Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature (2000) 407:908–13. doi: 10.1038/35038090
175. Sato T, Nakamura Y, Shiimura Y, Ohgusu H, Kangawa K, Kojima M. Structure, regulation and function of ghrelin. J Biochem (2012) 151:119–28. doi: 10.1093/jb/mvr134
176. Maletinska L, Pychova M, Holubova M, Blechova M, Demianova Z, Elbert T, et al. Characterization of new stable ghrelin analogs with prolonged orexigenic potency. J Pharmacol Exp Ther (2012) 340:781–6. doi: 10.1124/jpet.111.185371
177. Sato T, Ida T, Nakamura Y, Shiimura Y, Kangawa K, Kojima M. Physiological roles of ghrelin on obesity. Obes Res Clin Pract (2014) 8:e405–13. doi: 10.1016/j.orcp.2013.10.002
178. Quinones M, Ferno J, Al-Massadi O. Ghrelin and liver disease. Rev Endocr Metab Disord (2020) 21:45–56. doi: 10.1007/s11154-019-09528-6
179. Sangiao-Alvarellos S, Vazquez MJ, Varela L, Nogueiras R, Saha AK, Cordido F, et al. Central ghrelin regulates peripheral lipid metabolism in a growth hormone-independent fashion. Endocrinology (2009) 150:4562–74. doi: 10.1210/en.2009-0482
180. Li Z, Xu G, Qin Y, Zhang C, Tang H, Yin Y, et al. Ghrelin promotes hepatic lipogenesis by activation of mtor-ppargamma signaling pathway. Proc Natl Acad Sci U S A (2014) 111:13163–8. doi: 10.1073/pnas.1411571111
181. Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via amp-activated protein kinase. J Biol Chem (2005) 280:25196–201. doi: 10.1074/jbc.C500175200
182. Porteiro B, Diaz-Ruiz A, Martinez G, Senra A, Vidal A, Serrano M, et al. Ghrelin requires p53 to stimulate lipid storage in fat and liver. Endocrinology (2013) 154:3671–9. doi: 10.1210/en.2013-1176
183. Waseem T, Duxbury M, Ito H, Ashley SW, Robinson MK. Exogenous ghrelin modulates release of pro-inflammatory and anti-inflammatory cytokines in lps-stimulated macrophages through distinct signaling pathways. Surgery (2008) 143:334–42. doi: 10.1016/j.surg.2007.09.039
184. Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and t cells. J Clin Invest (2004) 114:57–66. doi: 10.1172/JCI21134
185. Kim MS, Yoon CY, Jang PG, Park YJ, Shin CS, Park HS, et al. The mitogenic and antiapoptotic actions of ghrelin in 3t3-l1 adipocytes. Mol Endocrinol (2004) 18:2291–301. doi: 10.1210/me.2003-0459
186. Nanzer AM, Khalaf S, Mozid AM, Fowkes RC, Patel MV, Burrin JM, et al. Ghrelin exerts a proliferative effect on a rat pituitary somatotroph cell line via the mitogen-activated protein kinase pathway. Eur J Endocrinol (2004) 151:233–40. doi: 10.1530/eje.0.1510233
187. Nagoya T, Kamimura K, Inoue R, Ko M, Owaki T, Niwa Y, et al. Ghrelin-insulin-like growth factor-1 axis is activated via autonomic neural circuits in the non-alcoholic fatty liver disease. Neurogastroenterol Motil (2020) 32:e13799. doi: 10.1111/nmo.13799
188. Sajjad A, Mottershead M, Syn WK, Jones R, Smith S, Nwokolo CU. Ciprofloxacin suppresses bacterial overgrowth, increases fasting insulin but does not correct low acylated ghrelin concentration in non-alcoholic steatohepatitis. Aliment Pharmacol Ther (2005) 22:291–9. doi: 10.1111/j.1365-2036.2005.02562.x
189. Yalniz M, Bahcecioglu IH, Ataseven H, Ustundag B, Ilhan F, Poyrazoglu OK, et al. Serum adipokine and ghrelin levels in nonalcoholic steatohepatitis. Mediators Inflammation (2006) 2006:34295. doi: 10.1155/MI/2006/34295
190. Okamatsu Y, Matsuda K, Hiramoto I, Tani H, Kimura K, Yada Y, et al. Ghrelin and leptin modulate immunity and liver function in overweight children. Pediatr Int (2009) 51:9–13. doi: 10.1111/j.1442-200X.2008.02647.x
191. Machado MV, Coutinho J, Carepa F, Costa A, Proenca H, Cortez-Pinto H. How adiponectin, leptin, and ghrelin orchestrate together and correlate with the severity of nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol (2012) 24:1166–72. doi: 10.1097/MEG.0b013e32835609b0
192. Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes (2001) 50:707–9. doi: 10.2337/diabetes.50.4.707
193. Cappiello V, Ronchi C, Morpurgo PS, Epaminonda P, Arosio M, Beck-Peccoz P, et al. Circulating ghrelin levels in basal conditions and during glucose tolerance test in acromegalic patients. Eur J Endocrinol (2002) 147:189–94. doi: 10.1530/eje.0.1470189
194. Pagotto U, Gambineri A, Pelusi C, Genghini S, Cacciari M, Otto B, et al. Testosterone replacement therapy restores normal ghrelin in hypogonadal men. J Clin Endocrinol Metab (2003) 88:4139–43. doi: 10.1210/jc.2003-030554
195. Pagotto U, Gambineri A, Vicennati V, Heiman ML, Tschop M, Pasquali R. Plasma ghrelin, obesity, and the polycystic ovary syndrome: Correlation with insulin resistance and androgen levels. J Clin Endocrinol Metab (2002) 87:5625–9. doi: 10.1210/jc.2002-020776
196. Vestergaard ET, Jessen N, Moller N, Jorgensen JO. Acyl ghrelin induces insulin resistance independently of gh, cortisol, and free fatty acids. Sci Rep (2017) 7:42706. doi: 10.1038/srep42706
197. Estep M, Abawi M, Jarrar M, Wang L, Stepanova M, Elariny H, et al. Association of obestatin, ghrelin, and inflammatory cytokines in obese patients with non-alcoholic fatty liver disease. Obes Surg (2011) 21:1750–7. doi: 10.1007/s11695-011-0475-1
198. Li Y, Hai J, Li L, Chen X, Peng H, Cao M, et al. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine (2013) 43:376–86. doi: 10.1007/s12020-012-9761-5
199. Moreno M, Chaves JF, Sancho-Bru P, Ramalho F, Ramalho LN, Mansego ML, et al. Ghrelin attenuates hepatocellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humans. Hepatology (2010) 51:974–85. doi: 10.1002/hep.23421
200. Chang L, Ren Y, Liu X, Li WG, Yang J, Geng B, et al. Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J Cardiovasc Pharmacol (2004) 43:165–70. doi: 10.1097/00005344-200402000-00001
201. Gonzalez-Rey E, Chorny A, Delgado M. Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology (2006) 130:1707–20. doi: 10.1053/j.gastro.2006.01.041
202. Mao Y, Zhang S, Yu F, Li H, Guo C, Fan X. Ghrelin attenuates liver fibrosis through regulation of tgf-beta1 expression and autophagy. Int J Mol Sci (2015) 16:21911–30. doi: 10.3390/ijms160921911
203. Sumida Y, Yoneda M. Current and future pharmacological therapies for nafld/nash. J Gastroenterol (2018) 53:362–76. doi: 10.1007/s00535-017-1415-1
204. Yoon IC, Eun JR. Pharmacologic therapy for nonalcoholic steatohepatitis focusing on pathophysiology. Yeungnam Univ J Med (2019) 36:67–77. doi: 10.12701/yujm.2019.00171
205. Kothari S, Dhami-Shah H, Shah SR. Antidiabetic drugs and statins in nonalcoholic fatty liver disease. J Clin Exp Hepatol (2019) 9:723–30. doi: 10.1016/j.jceh.2019.06.003
206. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the american association for the study of liver diseases. Hepatology (2018) 67:328–57. doi: 10.1002/hep.29367
207. Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med (2006) 355:2297–307. doi: 10.1056/NEJMoa060326
208. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (lean): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet (2016) 387:679–90. doi: 10.1016/S0140-6736(15)00803-X
209. Joy TR, McKenzie CA, Tirona RG, Summers K, Seney S, Chakrabarti S, et al. Sitagliptin in patients with non-alcoholic steatohepatitis: A randomized, placebo-controlled trial. World J Gastroenterol (2017) 23:141–50. doi: 10.3748/wjg.v23.i1.141
Keywords: peptides, leptin, glucagon-like peptide-1, ghrelin, non-alcoholic steatohepatitis
Citation: Kořínková L, Pražienková V, Černá L, Karnošová A, Železná B, Kuneš J and Maletínská L (2020) Pathophysiology of NAFLD and NASH in Experimental Models: The Role of Food Intake Regulating Peptides. Front. Endocrinol. 11:597583. doi: 10.3389/fendo.2020.597583
Received: 21 August 2020; Accepted: 28 October 2020;
Published: 26 November 2020.
Edited by:
Magdalene K. Montgomery, The University of Melbourne, AustraliaReviewed by:
Carmine Finelli, Ospedale Cav. R. Apicella – ASL Napoli 3 Sud, ItalyXiang Zhang, The Chinese University of Hong Kong, China
Copyright © 2020 Kořínková, Pražienková, Černá, Karnošová, Železná, Kuneš and Maletínská. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lenka Maletínská, maletin@uochb.cas.cz
†These authors have contributed equally to this work