 Kavita Jadhav
Kavita Jadhav Taylor S. Cohen
Taylor S. Cohen- Microbiome Discovery, Microbial Sciences, BioPharmaceuticals R&D, AstraZeneca, Gaithersburg, MD, United States
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of disorders, ranging from fatty liver to a more insulin resistant, inflammatory and fibrotic state collectively termed non-alcoholic steatohepatitis (NASH). In the United States, 30%–40% of the adult population has fatty liver and 3%–12% has NASH, making it a major public health concern. Consumption of diets high in fat, obesity and Type II diabetes (T2D) are well-established risk factors; however, there is a growing body of literature suggesting a role for the gut microbiome in the development and progression of NAFLD. The gut microbiota is separated from the body by a monolayer of intestinal epithelial cells (IECs) that line the small intestine and colon. The IEC layer is exposed to luminal contents, participates in selective uptake of nutrients and acts as a barrier to passive paracellular permeability of luminal contents through the expression of tight junctions (TJs) between adjacent IECs. A dysbiotic gut microbiome also leads to decreased gut barrier function by disrupting TJs and the gut vascular barrier (GVB), thus exposing the liver to microbial endotoxins. These endotoxins activate hepatic Toll-like receptors (TLRs), further promoting the progression of fatty liver to a more inflammatory and fibrotic NASH phenotype. This review will summarize major findings pertaining to aforementioned gut-liver interactions and its role in the pathophysiology of NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD), once known as the “un-named” disease, afflicts 80–100 million Americans and is currently the most common cause of chronic liver disease (1). About 20%–30% of NAFLD cases in the United States fall under the more severe category of non-alcoholic steatohepatitis (NASH) (1). With increasing prevalence over the last 20 years, NAFLD presents a burgeoning health problem. Unfortunately no therapies are currently approved for treatment or prevention of NAFLD/NASH. Development of such a therapeutic requires more in depth understanding of this disease, including answers to questions such as: What factors influence progression of steatosis to NASH, to NASH with fibrosis? What predisposes 30% of NAFLD patients to develop NASH? Can we harness pre-disposing factors and other non-invasive methods to accurately predict disease progression?
Pathophysiology of NAFLD
NAFLD covers a wide range of liver morbidities, with accumulation of lipid droplets being its mildest manifestation, and liver failure or cirrhosis being the worst. When the accumulation of lipid droplets exceeds 5% of the total liver weight, an individual may be characterized as having fatty liver, hepatic steatosis, or non-alcoholic fatty liver (NAFL) (1). About 30% of individuals with NAFL progress to NASH which is characterized by inflammation in addition to lipid accumulation (2). About 20% of NASH patients with advanced fibrosis will progress to cirrhosis, which marks an irreversible decline in liver function, in addition to being a risk factor for hepatocellular carcinoma (2).
NAFLD/NASH progression is hypothesized to be due to the combination of insults, termed the two-hit hypothesis (Figure 1) (3). This theory postulates that the “first hit” is the development of fatty liver. The “second hit” is characterized by a multitude of factors including inflammatory cytokines, oxidative stress and/or insulin resistance (IR), although the sequence of these events is unclear (3). Since a two-hit model did not sufficiently explain the complex pathophysiology of NAFLD, a more inclusive theory was proposed, the “multiple hit theory” (4). Fatty liver still remains the first hit, but the complex secondary insults reflect broader metabolic dysfunction that involves crosstalk with other organs central to metabolism such as adipose tissue, pancreas, and gut microbiota (4). However, the multiple hit model is still an oversimplification, and additional factors have yet to be fully explored, with age, obesity, and genetic pre-disposition being just a few of them.
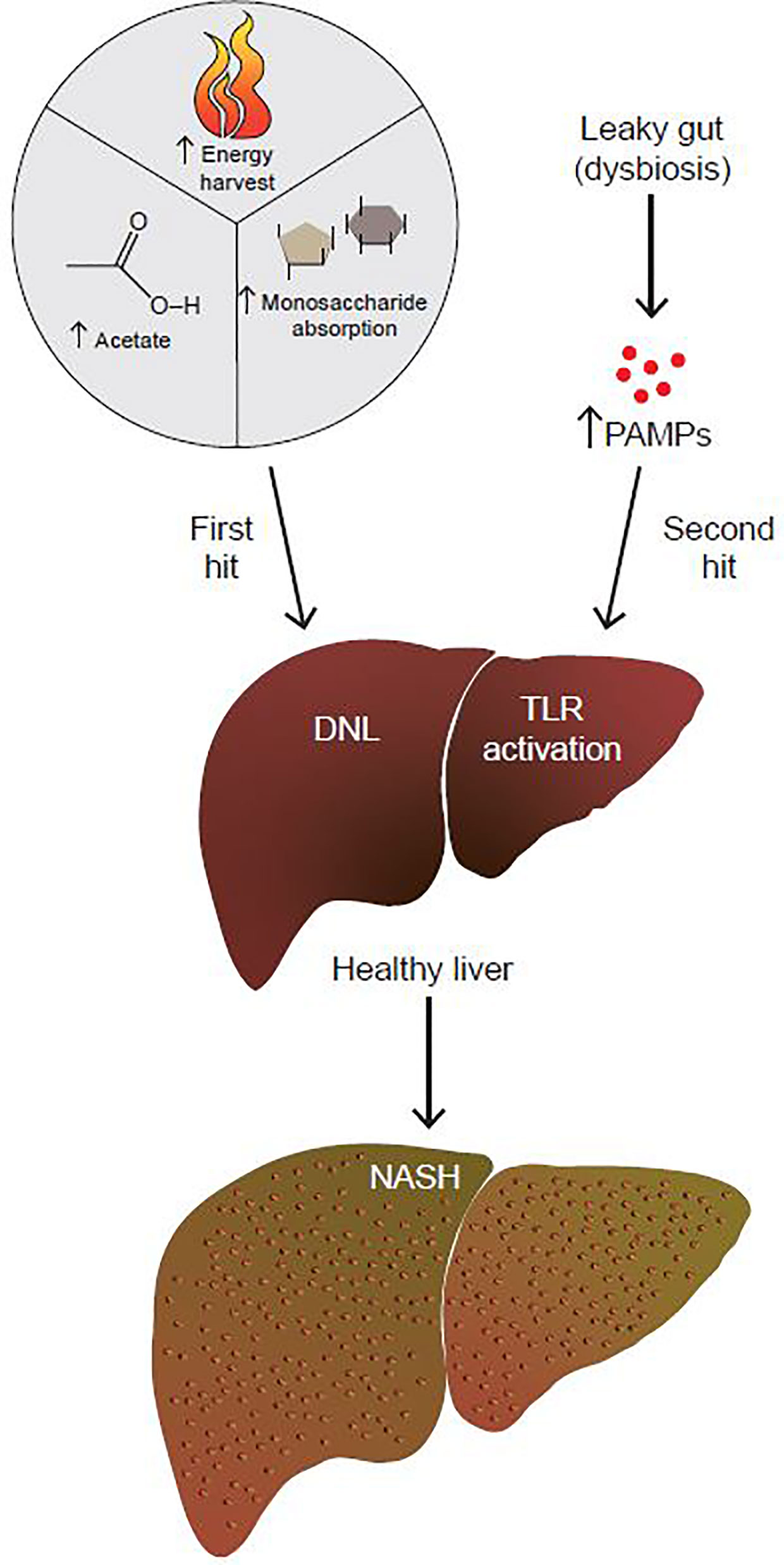
Figure 1 The gut microbiome contributes to both the first and second hits of NAFLD. By increasing energy harvest, monosaccharide absorption, and acetate production, the gut microbiome contributes to the first hit of NAFLD, which is the development of fatty liver. In addition to that, a dysbiotic leaky gut allows for increased passage of PAMPs to the liver. PAMPs activate hepatic TLRs to up-regulate pro-inflammatory and fibrotic pathways. By promoting the development of fatty liver to NASH, the gut microbiome contributes to the second hit of NAFLD.
On top of a complex etiology, tracking progression of NAFLD is an additional challenge. Serum ALT and fibrosis score are surrogate markers to determine liver damage; however, liver biopsy remains the gold standard for diagnosing and characterizing the different stages of NASH (5). Less invasive alternatives, such as ultrasonography and MRI, allow for visualization of fatty liver, but do not evaluate inflammation or accurately assess fibrosis (6). Limited functionality makes these techniques less viable alternatives as disease development/progression indicators. To complement imaging, biomarker research is an active area of the NAFLD/NASH field. The gut microbiome composition or associated metabolites could be one such biomarker, although additional research is needed to confirm the utility of these approaches.
Microbiome and Human NAFLD
Fecal microbiota transplant (FMT) studies have implicated the microbiome and NAFLD development in mice (7, 8). However, no particular microbial signature has emerged in human NAFLD, making it difficult to trace disease development back to any particular cluster of bacterial taxa. Sharpton et al. reviewed the reasons behind discordant results obtained from studies trying to draw correlations between the microbiome and human NAFLD (9). A number of confounders could underlie why no one signature has emerged across multiple studies, including differences in patient age, presence of other metabolic co-morbidities, and geographic location. Differences in handling of stool samples, sequencing and statistical analyses performed also could skew the results of individual studies (9). Additionally, compared to 16S ribosomal RNA sequencing, metagenomic analysis allows for a better understanding of the functional and metabolic potential of the gut microbiome (9).
Despite heterogeneity in specific taxa associated with disease, several cross-sectional studies have shown associations between an unhealthy change in the normal bacterial ecology, also known as dysbiosis, and all stages of NAFLD, including fatty liver, NASH, advanced fibrosis and also cirrhosis and hepatocellular carcinoma (10). Aron-Wisnewsky et al. divided human studies into steatosis to NASH, and NAFLD fibrosis to NASH cirrhosis signatures. In doing so, the authors found significant overlap in microbial signatures in both simple steatosis and NASH (10). In brief, steatosis and NASH patients have increased abundance of Proteobacteria (13.5%) at the phylum level, increased Enterobacteriaceae (12.02%) and decreased Rikenellaceae (0.41% in NASH versus 1.97% in healthy patients) and Ruminococcaceae (7.01% in NASH versus 18.82% in healthy patients) at the family levels, and increased Escherichia (2.36% versus 0.3% in healthy patients), Peptoniphilus (4.1% versus 0.36% in healthy patients) and decreased Anaerosporobacter (1.08% versus 2.02% in healthy patients), Coprococcus (1.03% versus 3.69% in healthy patients), Eubacterium (0.29% versus 1.18% in healthy patients), Faecalibacterium (4.27% versus 8.15% in healthy patients) and more discordant changes in Prevotella at the genera level (6, 11–24). The authors did acknowledge that despite these differences there is widespread divergence in the literature across all levels of taxonomy, with some studies even reporting trends opposite to the ones discussed above (10).
In contrast to simple steatosis, identification of microbial signatures in NASH with fibrosis is less well established, in part due to differences in the threshold for “fibrosis” between studies. For example, some human fibrosis studies have made comparisons between mild to moderate (F0-F2), versus severe fibrosis (F3-F4), while some others have compared no to little fibrosis (F0-F1) to moderate and severe fibrosis (F2-F4), which has created discrepancies in the literature (15, 17). Even then, microbial signatures associated with advanced fibrosis have emerged. In general, advanced fibrosis correlated with increased Gram-negative bacteria, increased Fusobacteria phylum, and decreased Enterobacteriaceae family and Gram-positive bacteria, Firmicutes phylum, Prevotellaceae family, and Prevotella genus (15, 17, 20). One of these studies utilized metagenomic sequencing along with serum metabolomics which allowed the authors to overlay bacterial abundance with pathway and metabolite enrichment data. This approach provided a more holistic microbial profile of patients with mild/moderate fibrosis, and severe fibrosis with NASH (17). While the gut microbiome signature was consistent with previous studies, serum metabolite analysis revealed increased nucleoside metabolism in severe fibrosis and increased amino acid and carbon metabolism related metabolites in mild/moderate fibrosis (16). In terms of pathway enrichment, mild/moderate fibrosis stool samples were enriched in nucleotide and steroid degradation pathways, while severe fibrosis stool samples were enriched in carbon metabolism and detoxification pathways (19). These data suggest the possibility of harnessing the microbiome to differentiate mild/moderate fibrosis from severe fibrosis with NASH. More studies with the same study design and larger cohort sizes are needed to confirm whether these microbiome-derived signatures can truly be used as a diagnostic tool.
Role of the Gut Microbiome in the Development and Progression of NAFLD
Changes in the Gut Microbiome Promote the Development of Fatty Liver
Microbiota and Energy Harvest
The human diet is enriched in all three macronutrients, carbohydrates, protein and fat, with carbohydrates making up a bulk of the standard diet. Dietary carbohydrates come in three forms, polysaccharides, disaccharides, and monosaccharides, as defined by the number of monomeric units. In order to be used as energy sources by the host, poly- and disaccharides must first be broken down to their monosaccharide units. Of all the enzymes required for this hydrolysis to occur, humans only encode amylase which removes monosaccharide units from starch. Other than amylase, the host depends on the gut microbiome to harvest energy from dietary polysaccharides (25). Non-starch polysaccharides such as cellulose or hemicellulose are metabolized by colonic bacteria to generate short chain fatty acids (SCFAs) such as butyrate, acetate, and propionate (25, 26). Analysis of feces originating from germ-free (GF) mice revealed significantly reduced levels of SCFAs in the intestine and cecum when compared to conventional mice, supporting the need for commensal bacteria to metabolize non-digestible carbohydrates to generate SCFAs (27). Microbial-produced monosaccharides and SCFAs are absorbed into the portal vein and serve as substrates for de novo lipogenesis (DNL) in the liver. So far, 130 families of glycoside hydrolases, 22 families of polysaccharide lyases and 16 families of carbohydrate esterases have been discovered, and a vast majority of these are encoded in microbial genomes (28). In addition, metagenomic sequencing of human gut microbiota has uncovered a vast panel of carbohydrate-active enzymes (CAZymes) including hydrolases, lyases and esterases, a great majority of which remain to be characterized (25).
In addition to SCFA generation, another mechanism by which the gut microbiome contributes to energy harvest is by increasing the absorption of dietary monosaccharides across the intestine (29). Conventionally housed mice that were given an oral bolus of glucose showed twice the monosaccharide absorption across the intestine as compared to GF mice (29). Absorbed monosaccharides were then transferred to the portal vein, thereby increasing substrate availability for hepatic DNL (Figure 2).
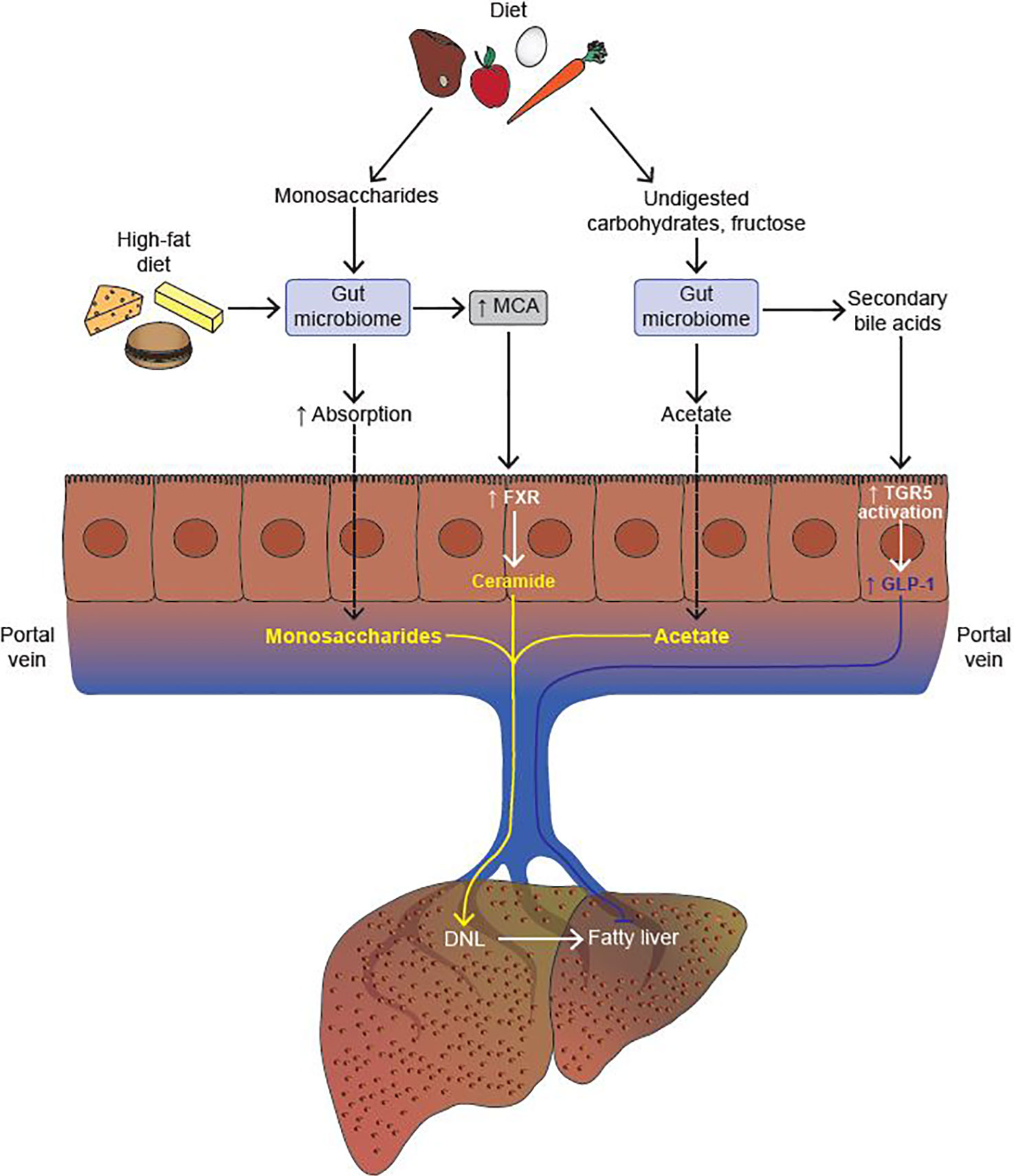
Figure 2 The gut microbiome modulates the development of fatty liver. The gut microbiome increases absorption of monosaccharides from the diet, thereby promoting hepatic de novo lipogenesis (DNL) by increasing substrate availability. Upon consumption of a high fat diet, the gut microbiome increases the production of muricholic acid (MCA) in mice. MCA is a potent activator of intestinal farnesoid X-receptor (FXR), which, in turn, activates the ceramide synthesis pathway in intestinal epithelial cells (IECs). Upon reaching the liver, ceramide promotes the cleavage of Srebp-1c, and thus upregulation of the hepatic DNL program. Undigested carbohydrates and fructose are processed by the gut microbiome to generate acetate, a substrate for hepatic DNL. Lastly, secondary bile acids (BAs) generated by the gut microbiome activate the Takeda G protein-coupled receptor (TGR5) expressed in colonic L cells which, in turn, increases the secretion of glucagon-like peptide 1 (GLP-1). GLP-1 inhibits the development of fatty liver both by driving down DNL, and by increasing fatty acid oxidation in hepatocytes (Activation, indicated in yellow; Inhibition, indicated in blue).
Perhaps, the most direct link between energy harvest, availability of SCFAs and hepatic steatosis was provided in a study that investigated the role of GPR41, a receptor for acetate and propionate (30). Through bomb-calorimetric assays of feces, this study demonstrated that the efficiency of caloric extraction from a polysaccharide rich chow diet was significantly reduced in Gpr41-deficient versus wild-type mice, although the mechanism behind this was unclear. In addition, cecal levels of acetate and propionate were significantly increased in the knockouts, indicating increased excretion of SCFAs. Concomitantly, hepatic triglycerides (TGs) in the Gpr41-deficient mice were significantly reduced (30). The reason behind this phenotype might be that a lack of Gpr41 prevents uptake of dietary polysaccharide-derived SCFAs. Therefore, energy harvest in the absence of SCFA receptors is deemed redundant as this promotes DNL substrates like SCFAs to be excreted in the feces, thereby reducing hepatic steatosis.
From a translational perspective, there are limited human data on any of these mechanisms in fatty liver development. The literature suggests that obese individuals have increased intestinal glucose absorption, but this has not been tied back to the microbiome (31). Monosaccharide transporters might be potential targets, but selection will be a challenge as the GLUT family of transporters alone has 14 members. The SCFA receptors GPR41 and GPR43 show functional divergence when it comes to differentiation of adipocytes, but whether these differences also apply to their role in SCFA uptake is largely unknown (32). In addition, there are no human data on the expression of these receptors in intestinal epithelial cells (IECs) in metabolic disease. Lastly, enrichment of glycoside hydrolase metagenomic signatures in obese mice and humans unable to lose weight on a lifestyle intervention program holds translational promise, but the functional value of these signatures remains to be identified in humans (33, 34). One way to assess functionality would be to measure if microbiome-derived acetate feeds into hepatic DNL as a result of increased expression of glycoside hydrolase in obesity, but this may not be that straight-forward in humans. One potential “fix” for removing glycoside hydrolase-rich microbial populations could be to re-populate the obese gut with FMT from healthy donors. In fact, there currently are clinical trials using this technique to evaluate its impact in NAFLD (ClinicalTrials.gov Identifier: NCT02469272).
Microbiota as a Source of DNL Substrates
Hepatic DNL is the process by which excess carbohydrates in the liver are converted to neutral TGs and stored in lipid droplets. Depending on the energy state of the cell, these TGs are either packaged into very low density lipoprotein (VLDL) particles and secreted out of the liver or are hydrolyzed to undergo β-oxidation. As such, DNL has two components; synthesis of free fatty acids (FFAs) and incorporation of 3 FFAs onto 1 glycerol to form one molecule of tri-acyl glycerol (TAG). Substrates for DNL are sourced from both the host in the form of FFA flux from the adipose tissue, and also microbiome-derived metabolism of carbohydrates and fatty acids from the diet.
Microbial products like SCFAs serve as substrates for hepatic DNL, thereby accelerating the development of fatty liver. Kindt et al. integrated transcriptomic, proteomic, phosphoproteomic, and lipidomic analyses of livers from GF and specific pathogen-free (SPF) mice to provide a comprehensive, multi-OMICS based link between the microbiome and hepatic lipogenesis (35). Presence of microbiota led to a significant increase in desaturation of the FA palmitate by SCD-1, and elongation of the FA γ-linoleic acid by fatty acid elongase (ELOVL)-5. In addition, significant increases were also observed in other TAG-synthesizing enzymes such as fatty acid synthase (FAS), further promoting the development of fatty liver (35). Strikingly, oral gavage of labeled acetate led to its rapid incorporation into newly forming C16 and C18 fatty acids in the livers of SPF mice, further corroborating the idea that SCFAs produced from microbial fermentation of dietary fiber serve as substrates for hepatic DNL (35). While the SCFAs propionate and butyrate have been shown to protect against NAFLD, acetate acts as a substrate for DNL in hepatocytes (36–39) (Figure 2). So while acetate can be assigned as pro-lipogenic, the same does not apply to all SCFAs. Having said that, a recent study by Rau et al. drew correlations between SCFAs and NAFLD severity (40). Thirty-two NAFLD patients were further stratified into non-alcoholic fatty liver (NAFL) and NASH patients. Compared to healthy controls (HCs), there was a 50% increase in fecal acetate and a 100% increase in propionate levels in patients with NAFL, while no difference was observed in butyrate levels (40). A similar trend in acetate and propionate levels was observed when NASH patients were further stratified into mild (F0-F1) and moderate/severe (F2-F4) NASH in comparison to HCs. However, patients with mild NASH had modest but statistically significant higher levels of fecal acetate (~20%) and propionate (~25%) compared to patients with moderate/severe NASH. At the family level, the gut microbiome of NAFLD (NAFL/NASH) patients was enriched in Fusobacteriaceae and Prevotellaceae compared to the gut microbiome of HCs. Both bacterial families are well characterized SCFA-producers, thereby providing a functional link between the microbiome and microbial metabolites (40). There were no differences in bacterial populations at the family level between the mild and moderate/severe NASH groups, but Acadaminococcus and Prevotella were more enriched in moderate/severe NASH (40).
While the above study failed to establish any causal links in the microbiome-SCFA-NAFLD axis, it highlighted some interesting findings. For one, both fecal acetate and propionate are increased in patients with NAFLD versus HCs, but upon closer observation, it is clear that in the NAFL group, acetate levels are three times greater than propionate levels, even though acetate levels went up by only 50%, while propionate went up by 100% when compared to HCs (40). This could mean that while the two SCFAs have opposing roles in DNL, net higher levels of acetate may tip the balance in favor of a pro-lipogenic phenotype. More interestingly, both acetate and propionate levels drop modestly (around 15%) in moderate/severe NASH (F2-F4) in comparison to mild (F0-F1) NASH, which could mean that while more SCFAs are produced in the early stages of NAFL and NASH, this may not be the case when fibrosis becomes more severe. Since it is hard to predict causality in human studies, some future directions for finding a link between SCFAs and NAFLD could be carbon tracing the metabolism of microbiome-derived acetate in mouse models of fatty liver and NASH. Using oral administration of 13C-acetate, the amount of labeled carbons that are incorporated in end products of DNL can be estimated by mass-spectrometry. Another approach would be dual tracing of acetate and propionate in the same mouse models to get a more complete picture of SCFA metabolism in NAFLD. In addition to carbon tracing, another angle would be examining the effect of SCFAs on inhibition of histone deacetylases (HDACs), as it has been demonstrated that SCFAs like butyrate inhibit HDACs to activatethe transcription of activators of fatty acid oxidation such as peroxisome proliferator-activated receptor (PPAR)-α.
While these data are suggestive that gut microbiome derived metabolites contribute to hepatic DNL, they provide an incomplete picture. DNL is also heavily modulated by Srebp-1c and ChREBP transcriptionally, but there are limited data on relative contribution of the transcriptional DNL program versus gut microbiome (41). Indeed, GF mice are resistant to HFD-induced obesity and fatty liver, indicating that complete ablation of the gut microbiome suppresses the transcriptional DNL program through yet unknown mechanisms (42). Conversely, Srebp-1c and ChREBP knockouts are resistant to NAFLD development even in the presence of the microbiome (43, 44). This implies that the absence of the transcriptional DNL program either changes the microbiome composition such that there is less production of DNL substrates, or microbial lipogenic substrates fail to induce DNL by themselves. Either way, crosstalk between the two, or emergence of alternative metabolic pathways in the absence of one or the other remains to be elucidated.
In their recent paper, Zhao et al. set out to tease apart the relative contribution of these two pathways in fructose consumption-mediated increases in hepatic DNL (45). Counter to current dogma surrounding the role of dietary fructose in hepatic steatosis, Zhao et al. demonstrated that dietary fructose is converted to acetate by the gut microbiome, and can be used by the liver as a precursor for DNL (45). Prior to these data, it was believed that once in the hepatocyte, fructose enters the tricarboxylic acid (TCA) cycle and is converted to citrate. ATP citrate lyase (ACLY) converts citrate to acetyl-Coa which serves as a precursor for DNL. By knocking out ACLY, Zhao et al. demonstrated that dietary fructose can still contribute to the NAFLD phenotype by bacterial conversion to acetate, followed by transport to the liver via the portal vein. In the liver, acetate is converted to acetyl-Coa by acetyl-CoA synthetase (ACSS)-2 and is ultimately shunted into the lipogenic pathway. Finally, gene expression of ChREBP-β and other DNL genes is upregulated upon fructose feeding independently of acetyl-CoA metabolism. Collectively, these data indicate dual mechanisms for fructose-mediated hepatic lipogenesis- one via activation of the transcriptional DNL program, and another by providing DNL substrates in the form of microbiome-derived acetate.
Microbiota as a Modulator of Hepatic Lipid Homeostasis via FXR and TGR5
Clues for the role of bile acids (BA) in TG synthesis came in the 1970s when administration of chenodeoxycholic acid (CDCA) for gallstones also resulted in reduced circulating TGs (46). Conversely, patients treated with BA sequestrants were found to have elevated hepatic and serum TGs and VLDL (47). Bile acid synthesis from cholesterol which occurs exclusively in the liver is mediated by two key enzymes- CYP7A1 and CYP8B1, which through a series of reactions catalyze the production of CDCA and cholic acid (CA) respectively (48). CDCA is further converted to α, then β-muricholic acid (β-MCA) in mouse livers and into ursodeoxycholic acid (UDCA) in human livers. These primary BAs are further conjugated in the liver to the amino acids taurine or glycine to generate conjugated BAs such as taurocholic acid (TCA), tauro-alpha/beta-muricholic acid (T-α/β-MCA), etc. (49). Primary BAs are then stored in the gallbladder, wherein they are released upon meal ingestion to facilitate absorption of nutrients across the small intestine (SI). Approximately, 95% of BAs are reabsorbed in the ileum and are acted upon by gut microbiota to undergo de-conjugation by the bacterial enzyme bile acid hydrolase (BSH) and further dehydroxylation by bacterial dehydroxylases to generate the secondary BAs lithocholic acid (LCA) and deoxycholic acid (DCA) from CDCA and CA, respectively (50). Therefore, the gut microbiota plays a key role in maintaining BA composition, and will likely be impacted by any perturbations in microbiome composition.
The Farnesoid X-receptor (FXR) is a ubiquitously expressed nuclear receptor (NR), and plays a particularly important role in gut-liver signaling. Like most NRs, FXR has a N terminal ligand-independent activation function (AF1), a highly conserved DNA-binding domain (DBD), a ligand binding domain (LBD), and finally a C-terminal ligand-dependent activation function (AF2) (51). FXR forms a heterodimer with retinoid X-receptor (RXR), and when there is no ligand binding, the FXR-RXR heterodimer remains bound to FXR responsive elements (FXREs) within the promoters of FXR target genes, bound to co-repressors (52). Upon ligand-induced activation, co-repressors leave the FXR-RXR heterodimer to make way for co-activators, thus upregulating target gene transcription (52). While FXR was initially found to be weakly activated by farnesoid, an intermediate of mevalonate metabolism, it was later found that despite low affinity, BAs potently activate FXR in the order of CDCA > LCA = DCA > CA (53). BA binding to FXR in the intestine leads to the secretion of FGF15 in mice and FGF19 in humans into the hepatic portal vein (54, 55). Upon reaching the liver, FGF15/19 bind to their receptor FGF4 resulting in down-regulation of CYP7A1 and CYP8B1 to stop BA synthesis (54, 55). In this fashion, FXR tightly regulates BA production in the liver.
The role of the beneficial effects of FXR on glucose and lipid metabolism has been studied extensively across many mouse models (56–59). In brief, FXR activation reduces DNL by suppressing the transcription of Srebp-1c (60). It increases TG degradation by inducing the expression of PPARα and fibroblast growth factor (FGF) 21, both activators of fatty acid oxidation (61). Lastly, FXR promotes TG hydrolysis by increasing the expression of apolipoprotein (Apo)-CII which is an activator of lipoprotein lipase (LPL) (59). Taken together, FXR reduces hepatic steatosis by reducing DNL, increasing fatty acid oxidation, and increasing TG clearance.
To elucidate the role of the microbiome in FXR signaling, Jiang et al. treated mice with antibiotics and analyzed changes in BAs and progression of fatty liver (62). Microbiome depletion led to significant increases in the levels of T-β-MCA and TCA, as the bacterial enzyme BSH that catalyzes the conversion of T-β-MCA to MCA is missing in antibiotic treated mice. Increased levels of T-β-MCA inhibits intestinal FXR, which, in turn, reduces the transcription of ceramide synthesis-related genes, resulting in reduced levels of ceramide (62). Since ceramide regulates the cleavage and maturation of the pro-lipogenic Srebp-1c, there is a resultant reduction in HFD-induced hepatic DNL upon antibiotic treatment. Therefore, HFD-feeding leads to increased conversion of T-β-MCA to MCA by the gut microbiome, activation of intestinal FXR, followed by an increase in ceramide synthesis, which upon reaching the liver cleaves Srebp-1c to its active form, thereby increasing hepatic DNL (62) (Figure 1).
Several human studies report that both primary and secondary BAs are elevated in patients with NAFLD (63–65). Part of the explanation for this was the increased abundance of the taurine and glycine de-conjugating bacteria Escherichia and Bilophila, which catalyze the conversion of CA to the secondary BA DCA, which is antagonistic to FXR (63). Due to this inhibition of intestinal FXR, there was reduced secretion of FGF19, thus disrupting the feedback loop and maintaining elevated levels of CYP7A1 and CYP8B1 (63). As a result, there is continued production of BAs in patients with NAFLD, increasing the total primary BA pool size. In another study, NAFLD patients were found to have reduced levels of hepatic FXR, increased cleavage of Srebp1-c, and significantly higher hepatic TGs. Collectively, changes in gut microbiome composition in NAFLD contributes to disrupted primary and secondary BA production, reduced FXR signaling, and resultant fatty liver. Indeed, recent clinical trials have demonstrated that synthetic FXR agonists such as obeticholic acid have a beneficial effect in patients with NASH (66). Additional studies with a larger sample size need to be conducted to validate these findings.
Takeda G protein-coupled Receptor 5 (TGR5) is a G-protein coupled receptor which is less abundant than FXR, but is still highly expressed in the gallbladder, ileum, colon, and on hepatic macrophages known as Kupffer cells (53). As a GPCR, TGR5 activation leads to increase in cyclic AMP levels, thereby increasing the expression of protein kinase A which further mediates downstream effects. BAs activate TGR5 in the order of LCA > DCA > CDCA > CA, implying that TGR5 signaling is strongly associated with the microbiome as both LCA and DCA are products of the microbiome (53). TGR5s key role in the intestine is to facilitate secretion of the incretin hormone GLP-1 from enteroendocrine cells therefore increasing the secretion of insulin from pancreatic beta cells (67). In addition to imparting other metabolic benefits, the administration of GLP-1 agonists in ob/ob mice significantly reduced hepatic steatosis, both by driving down DNL, and up-regulating fatty acid oxidation (68) (Figure 2). Clinical trials with TGR5 agonists are currently underway for the treatment of NASH, and hold promise due to TGR5’s influence on GLP-1 signaling.
Changes in the Gut Microbiome Disrupt Gut Barrier Function
The gut barrier is the first line of defense between intestinal luminal contents and circulation, and mostly consists of the epithelial barrier and the over-laying mucus layer. The epithelial barrier consists of a monolayer of adjacently aligned epithelial cells, a vast majority of which are enterocytes/colonocytes. This layer is also interspersed with four other epithelial cell types—goblet cells, enteroendocrine cells, Paneth cells, and microfold cells (69). Underneath the epithelial cell monolayer is the lamina propria, which houses innate and adaptive immune cells such as T cells, B cells, macrophages, and dendritic cells (70). Finally, under the lamina propria lies a vascular network that eventually converges into the portal vein which, in turn, empties into the liver.
Goblets cells are specialized mucus secreting cells embedded within the epithelial monolayer (71). Secreted mucus is composed of glycosylated mucin proteins that form a gel-like layer and sit above the epithelial monolayer (71). The small intestine (SI) and colon have very distinct physiologies (72). The SI has Immunoglobulin As (IgA) and anti-microbial peptides (AMPs) which are secreted into the mucus layer by plasma cells within the lamina propria, and Paneth cells respectively, making the SI relatively less hospitable for bacterial growth (73, 74). Compared to the SI, the colon has a significantly greater number of goblet cells, and hence more mucus. Unlike the SI, the colon has two layers of mucus, with the bottom layer sitting right above the epithelial monolayer, and is more “tight” in consistency (72). A “loose” mucus layer overlays the bottom layer. This outer mucus layer serves as a habitat for colonic gut microbes (72). Since the colon has fewer Paneth cells, there is less IgA and AMP secretion, which in combination with more mucus production and thickness, makes it a more fertile ground for gut microbes (72). Owing to these differences between the SI and colon, gut microbiome composition varies along the gastrointestinal tract (GI) as well, with more aerobic and facultative anaerobes in the duodenum and jejunum, and more fiber-fermenting, bile acid-metabolizing anaerobes in the colon.
Under the mucus layer lies the intestinal epithelial monolayer. Transport of molecules between the intestinal lumen and the underlying vascular layer is regulated by junctional complexes between epithelial cells within the monolayer (71). The three most important junctional complexes are tight junctions (TJs), adherens junctions (AJs), and gap junctions (75). TJs include proteins like zona occludin-1 (ZO-1), occludin, and members of the claudin family which seal intercellular space. AJs are found below TJs, and along with gap junctions, they help maintain the integrity of the epithelial monolayer and facilitate cell-cell communication. Intracellularly, TJs and AJs are attached to actin and myosin, thereby playing important roles in cytoskeletal dynamics. It should be noted that the gut barrier is not a static organ, but is actually rather dynamic and sensitive to changes occurring in the gut (75).
Since the gut barrier serves to keep intestinal luminal contents from entering the underlying vascular network, any disruption in its integrity leads to a condition called the “leaky gut”. Under certain conditions, expression of TJPs is reduced leading to increased permeability between adjacent epithelial cells. Increased paracellular permeability gives luminal contents access to the underlying lamina propria and vascular network. Leakage of bacterial antigens into the vascular network, portal vein and liver leads to increased hepatic inflammation due to activation of immune signaling (76–78) (Figure 3). Indeed, metabolic diseases are often associated with a loss of intestinal barrier function and an increase in passive transport of microbial pathogen associated molecular patterns (PAMPs) into the body (79) (Figure 3). Emerging evidence suggests a link between a dysfunctional gut barrier and human NAFLD (80–82). A meta-analysis based on five clinical studies demonstrates a linear relationship between increased gut permeability and NAFLD progression, with stronger correlations as disease severity increases (82). Specifically, 39.1% of NAFLD patients had displayed increased intestinal permeability versus only 6.8% of HCs. In addition, it was found that patients with NASH were more likely to have this phenotype with the incidence of gut permeability in this subgroup being 49.2% higher compared to patients with NAFLD as a whole. These data suggest that inflammatory events occurring in the pathophysiology of NASH might be a function of increased gut permeability.
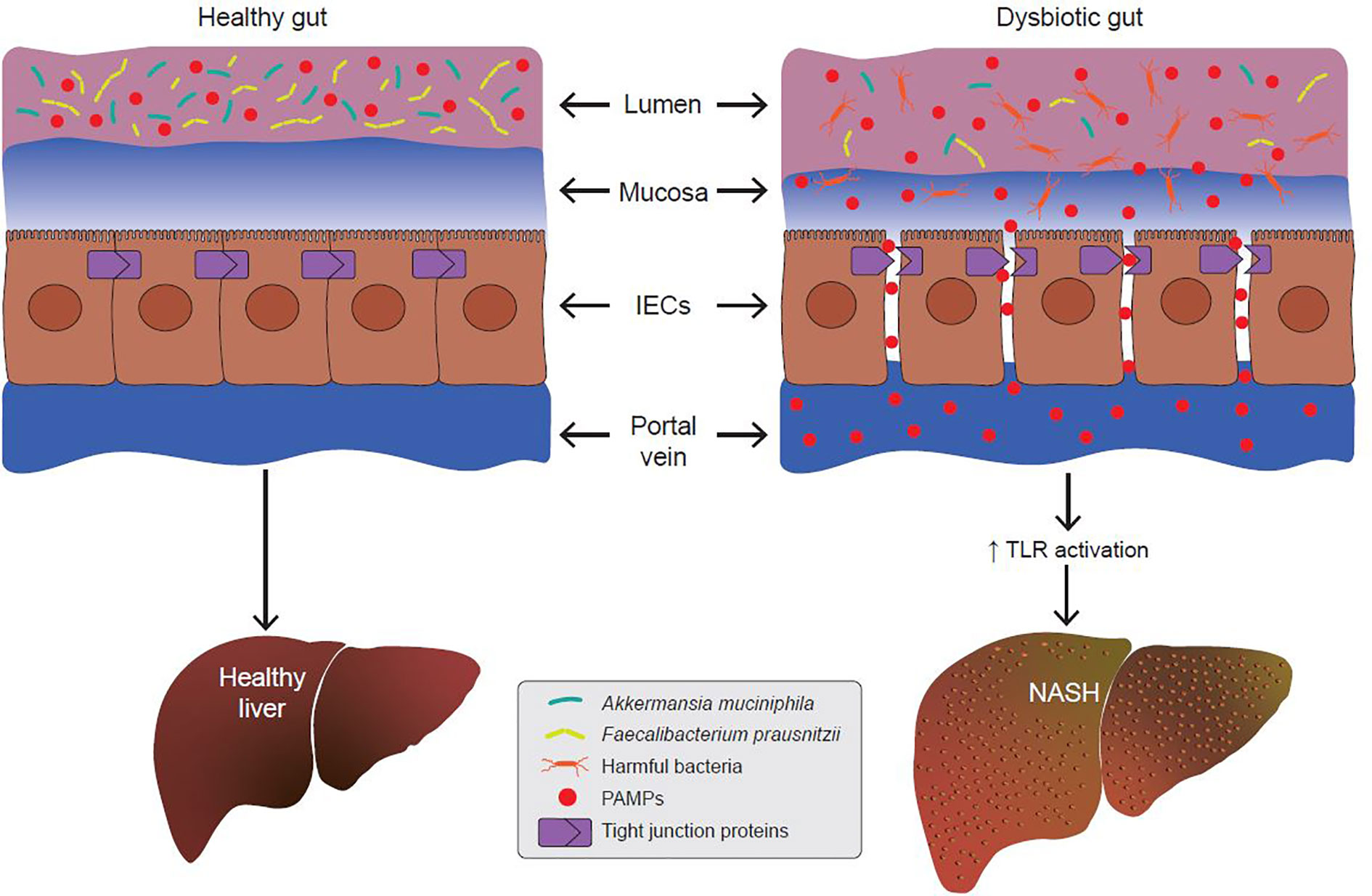
Figure 3 A dysbiotic gut promotes the development of non-alcoholic steatohepatitis (NASH). In metabolic gut dysbiosis, the populations of beneficial microbes like Akkermansia muciniphila and Faecalibacterium prausnitzii decline, and the populations of harmful bacteria increase. Via various mechanisms, this results in disruption of tight junction proteins (TJPs) between adjacent epithelial cells. This allows for increased paracellular passage of pathogen-associated molecular patterns (PAMPs) into the portal vein. PAMPs are endogenous ligands for Toll-like receptors (TLRs), and their binding to hepatic TLRs results in activation of pro-inflammatory and pro-fibrotic cascades which promotes the development of NASH.
To demonstrate that increased intestinal permeability precedes NASH, Mouries et al. showed that intestinal epithelial barrier (IEB) disruption in mice occurs within 48 h of HFD-feeding (83). This was evidenced by reduced expression of ZO-1 and increased bacterial translocation into the ileum and cecum lamina propria. Plasmalemma vesicle-associated protein 1 (PV1), a marker for gut vascular barrier (GVB) damage was unchanged at 48 h. Following 1 week of HFD-feeding, PV1 expression and intestinal permeability were significantly upregulated along all sections of the gut, and stayed that way until the end of the 24-week study. In both 1-week and 6-week HFD-fed mice, disruptions in the IEB and GVB preceded signs of hepatic steatosis and IR, indicating that these are early events in the development of NASH. Impaired GVB is then maintained through development of IR and inflammatory NASH. To further support the hypothesis that a dysbiotic gut disrupts epithelial barrier integrity, when SPF mice were transplanted with fecal matter from control diet and HFD-fed mice, mice receiving FMT from HFD-fed mice had increased adipose mass and expression of PV1, suggesting that HFD-feeding induces dysbiosis, which disrupts the GVB, which, in turn, correlates with increased intestinal blood vessel permeability. Collectively, these data suggest a linear sequence of events- IEB disruption, GVB disruption, IR and hepatic steatosis, and finally NASH (83).
In contrast to the above study, Thaiss et al. absolved the gut microbiome of any culpability in IR-driven gut permeability (84). In addition to showing increased gut permeability in db/db and ob/ob mice, the authors were able to show similar gut barrier dysfunction in STZ-treated mice, therefore demonstrating that IR-driven gut barrier perturbations are associated with, but do not require obesity. To determine the consequence of barrier dysfunction, the authors used a bioluminescent variant of Citrobacter Rodentium to track infection in vivo, which mimics human enteropathogenic E. coli infections. In addition to being hyperglycemic, STZ-treated mice exhibited reduced expression of ZO-1 along with increased gut permeability. Upon receiving C. rodentium, these mice showed increased susceptibility to infection and systemic translocation, enhanced bacterial growth, epithelial adherence and systemic spread. To determine whether these gut dysfunction signals were arising from microbiome alterations upon STZ treatment, FMTs were performed with feces from STZ-untreated and treated mice. No gut barrier dysfunction and associated increase in bacterial infection was seen in mice receiving FMTs from STZ-treated mice, demonstrating that the gut barrier dysfunction observed in these mice is independent of the microbiome. RNA sequencing of IECs revealed global reprogramming of the epithelial transcriptome of STZ-treated mice. In particular, it was found that the transcription of the GLUT2 gene which is responsible for glucose uptake in IECs was significantly upregulated in STZ-treated mice. IEC specific GLUT2 knockouts did not show increased permeability, reduced TJPs or increased susceptibility to infection upon STZ treatment. Taken together, these data suggested that IR-driven gut barrier dysfunction is independent of changes in the gut microbiome, and instead is dependent on GLUT2 dependent signaling in IECs (84).
A key feature that sets the above study apart from the work of Mouries et al. is the animal model used. STZ injection is typically representative of Type 1 (T1D), while ob/ob, db/db, and HFD-feeding models are more representative of Type 2 diabetes (T2D. In humans however, T1D, much like T2D is often accompanied by the presence of metabolic syndrome, thereby making it challenging to investigate microbiome-independent mechanisms behind gut barrier dysfunction in human T1D (85). In conclusion, in mouse models of diabetes, gut barrier dysfunction in T1D is driven by GLUT2 signaling in IECs and in T2D is driven by disruptions in the gut microbiome, and precedes the development of NASH.
Much like NAFLD/NASH, diabetes also has been linked with gut dysbiosis and barrier dysfunction. For example, examination of 345 patients microbiome samples demonstrated a reduction in butyrate producers and increase in opportunistic pathogens in the diabetic microbiome (86). Another study confirmed a significant reduction in the population of Bifidobacteria and Verrucomicrobia (87). Specifically, Akkermansia muciniphila of the Verrucomicrobia phylum has been shown to be significantly reduced in diabetes in both mouse and human studies (87–89). Indeed, oral administration of A. muciniphila resulted in marked improvements in metabolic parameters in genetic and diet-induced models of diabetes, positioning it as a beneficial microbe (89). It is most abundantly found in the loose outer mucus layer of the colon, and uses polysaccharides in the mucus as substrates to generate SCFAs like acetate and propionate (90). A. muciniphila supplementation was reported to restore the colonic mucus layer to its normal thickness in HFD-fed mice, the mechanisms behind which remain unclear (89). In another report, pasteurized A. muciniphila and Amuc_1100, the pili protein in A. muciniphila, were shown to improve gut barrier integrity by upregulating the expression of some TJ proteins (91). Perhaps, most strikingly, A. muciniphila supplementation was shown to significantly reduce circulating LPS levels, which suggests that it lead to improvements in gut barrier integrity (89, 91). In the strongest case yet for using A. muciniphila supplementation as therapy for gut-related and hepatic pathologies, a small exploratory proof-of-concept study was conducted on 40 obese male and female individuals (92). Participants were divided into three groups- placebo, pasteurized A. muciniphila and live A. muciniphila treated groups. At the 3-week end-point, in addition to demonstrating no adverse responses associated with A. muciniphila supplementation, the group receiving pasteurized A. muciniphila had modest, yet statistically significant, reductions in circulating LPS, AST and ALT levels (92). Although more studies with larger patient cohorts are required to confirm these findings, the use of A. muciniphila as a therapeutic agent still holds promise.
Another important function of A. muciniphila is its ability to support the growth of butyrate producing bacteria by a method known as cross-feeding (93). More specifically, in using mucus as a substrate, A. muciniphila produces the SCFA’s acetate and propionate, which are, in turn, utilized by bacteria such as Faecalibacterium prausnitzii, which produce butyrate (93). Much like A. muciniphila, loss of F. prausnitzii also correlates with development of T2D (94) (Figure 2). By producing butyrate, F. prausnitzii enhances mitochondrial function in colonocytes, thereby stabilizing HIF-1α in the gut (95). HIF-1α although considered “bad” in other contexts, has been shown to improve gut barrier integrity through yet unclear mechanisms. In addition to maintaining hypoxic conditions in the gut, butyrate supplementation to colonocyte and epithelial cell lines has led to increased transepithelial resistance (TEER), marking improved barrier function (96). Most importantly, one study found that F. prausnitzii supplementation in HFD-fed mice led to a significant reduction in diet induced steatosis, ALT and AST levels, thereby suggesting that increased butyrate production improves barrier integrity and consequently improves NASH (97).
While the above data paints A. muciniphila as a good player, another study showed that consuming diets depleted of fiber led to significant proliferation of A. muciniphila, correlating with a significant reduction in colonic mucus thickness and a compromised gut barrier (28, 98). While it is unclear why A. muciniphila appears to be a negative component of the microbiome in this study, it is possible that consuming a diet low in fiber deprives A. muciniphila and associated cross-feeders of classic substrates. A known mucus degrader, it is possible that A. muciniphila instead shifts its metabolism to use mucus as a substrate, thereby feeding into a cycle of mucus consumption, reduction in mucus thickness and increased proliferation. Eventually, when mucus consumption exceeds production, a scarcity of substrate availability results, and the population of A. muciniphila declines. It is possible then, that reduced A. muciniphila population size in diabetic patients is a consequence, rather than cause of compromised gut integrity. This might also be the reason why pasteurized forms of A. muciniphila show an improvement in metabolic endpoints, because this form does not have mucus degrading activity. A prospective study where A. muciniphila populations are measured from the onset to full- fledged development of diabetes might be able to answer some of these questions, but until then, the jury is out on the role of A. muciniphila in gut barrier integrity.
Changes in the Gut Microbiome Promote Progression of NAFLD
The previous section elucidates how gut microbiome dysbiosis occurring during metabolic syndromes can alter intestinal biology to make the gut more permeable. This allows passive transport of microbial PAMPs from the intestinal lumen into the portal vein, and eventually the liver (Figure 2). Before diving into how PAMPs contribute to NASH, it is first important to appreciate its pathophysiology. As mentioned, the first step of NAFLD is almost always the development of fatty liver. The second step involves multiple hits like IR, increased gut permeability, inflammation, and reactive oxygen species (ROS) production which leads to the progression of a more inflammatory, fibrotic NASH phenotype. Progression of fatty liver to fibrosis affects all liver cell types (99). While hepatocytes appear injured and undergo a form of cell death termed apoptosis, the resident liver macrophages, Kupffer cells (KCs), start secreting pro-inflammatory chemokines and cytokines. Finally, quiescent stellate cells (SCs), which are the major storage site for retinoids, are activated (99). Activation of stellate cells leads to loss of retinoids and increased expression of signaling receptors including the transforming growth factor β (TGF-β) receptor. Activated SCs proliferate and secrete extracellular matrix proteins to form a fibrous scar, which imparts a “fibrotic” phenotype to NASH (99). PAMP receptors such as the toll-like and nod-like receptors (TLRs and NLRs) are expressed on the cell surface of hepatocytes, Kupffer cells (KCs), and stellate cells (SCs), and are known to contribute to the inflammatory and fibrotic phenotype of NASH (Figure 2). While suppressed in healthy liver, TLR signaling is activated in the presence of pathogenic microorganisms and bacteria-derived molecules. Since other reports have already reviewed the role of TLRs in NAFLD in great detail, this section will briefly highlight some of the major findings (100, 101).
Of all the TLRs, TLR2, -4, -5, and -9 have been shown to contribute to the inflammatory and fibrotic signaling that characterizes NASH. In hepatocytes, LPS binding to TLR4 recruits MyD88, an adapter protein, which, in turn, leads to the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways (102). NF-κB is a transcription factor which upregulates the transcription of pro-inflammatory cytokines including interleukin (IL)-1, 2, 6, and 8 (103). In addition to its role in increasing hepatocyte inflammation, TLR4 plays a role in KC and SC crosstalk. LPS binding to TLR4 in SCs leads to increased production of adhesion molecules and chemokines like vascular cell adhesion protein (VCAM) and methyl-accepting chemotaxis protein (MCP) (104, 105). Adhesion molecules attract KCs and these recruited KCs secrete the pro-fibrogenic TGF-β which binds to TGF-β receptors on SCs (104, 105). This stimulates the secretion of collagen from SCs into hepatocytes, marking the beginning of liver fibrosis. Indeed, KC-specific knockdown of TLR4 in mice on a methionine choline-deficient (MCD) diet led to a significant reduction in hepatic TGs, reduced expression of inflammatory and fibrosis markers, and a resultant reduction in histological markers of NASH (106). Similar findings, demonstrating increased TLR4 mediated signaling contributing to NASH development, have been reported by other groups (106, 107).
In addition to TLR4, other TLRs mentioned in the paragraph above also have been implicated in NAFLD, but there are only a handful of reports elucidating their roles. TLR2 is expressed by HSCs and KCs and is a receptor for bacterial peptidoglycan. To investigate the role of TLR2 in hepatic inflammation, Miura et al. treated KCs with a synthetic TLR2 ligand Pam3CK4, and an endogenous ligand palmitic acid (PA) (108). While priming with Pam3CK4 alone was enough to increase the expression of NLRP3, IL-1β and IL-1α, caspase-1 activity was only induced when KCs were treated with PA after priming with Pam3CK3 first, indicating that both signals were required for activation of the inflammasome complex (108). This further led to the cleavage of the pro-inflammatory cytokines IL-1β and IL-1α to their active form, thereby upregulating hepatic inflammation (108). On the other hand, knock-out of TLR2 has yielded conflicting results in different mouse models, with the more conventional metabolic syndrome models suggesting that loss of TLR2 is protective against NASH (108–110). TLR5 is expressed in hepatocytes, and is a receptor for bacterial flagellin. While the exact role of TLR5 in NAFLD remains unknown, two separate studies have shown that knock-down of TLR5 accelerates hepatic steatosis, susceptibility to liver injury and NASH, thereby assigning it a more protective rather than harmful role (111, 112). Finally, TLR9 is expressed in Kupffer cells, and is a receptor for bacterial DNA. TLR9 activation signals through NF-κB to increase the expression of the cytokine IL-1β from KCs, and induces chemotaxis of macrophages and neutrophils, thereby leading to hepatic steatosis, inflammation and fibrosis (113, 114).
In conclusion, with the exception of TLR5, hepatic TLRs, upon binding by gut bacteria-derived products, set into motion a cascade of inflammatory and fibrotic signals, thereby abetting the progression of fatty liver to NASH.
Discussion
Increasingly, alterations in the gut microbiome have been correlated with NAFLD progression. This remains an important discussion in microbiome research relating to NAFLD for several reasons. Firstly, a unique microbial signature associated with the different phenotypes within NAFLD could serve as a non-invasive tool for accurately determining severity of disease. Secondly, predicting disease progression and prognosis will be easier and less invasive, in comparison to performing a liver biopsy each time an individual comes into the clinic for follow-up. Thirdly, a unique microbial profile in NAFLD overlaid with metagenomic signatures will help predict host metabolic responses, leading to more personalized interventional approaches. Lastly, therapeutically shifting a “disease promoting” microbiome to an “anti-NAFLD/NASH” microbiome remains an attractive strategy for thwarting or reversing the course of NAFLD progression.
As far as host metabolism is concerned, the microbiome may contribute to both hits of NAFLD; first by promoting development of fatty liver via DNL, then through hepatic TLR activation due to dysbiosis (Figure 1). Use of microbial-derived acetate as a substrate for hepatic DNL is a particularly striking finding, as thus far, the hepatic transcriptional lipogenic program alone was thought to play a role in DNL. The studies described in this report identified microbial populations that produce SCFAs, but there are still limited data on specific acetate producers. The modest reduction of fecal acetate levels as fatty liver progresses to NASH also is an important finding because this throws additional light on microbiome-dependent pathophysiology of NAFLD. Do acetate producing microbiome populations decline as NAFLD progresses? What are these populations and could their decline potentially predict onset of fibrosis? Many such outstanding questions remain.
This report also reviewed literature that investigated dysbiosis-induced increases in gut permeability in metabolic syndrome including NAFLD. Interestingly, T2D associated increases in gut permeability were found to be microbiome-dependent, while T1D associated increases in gut permeability relied more on glucose transport pathways in IECs. This is hardly surprising because the pathophysiologies of both are fairly independent, and their gut microbiome signatures are different as well. T2D-associated increases in gut permeability were found to be inversely correlated with A. muciniphila and F. prausnitzii populations. While the literature has painted A. muciniphila as a beneficial microbe, we are of the opinion that its reduced populations are a result, and not cause of T2D induced gut barrier perturbations, given its mucin degrading activity. Given that A. muciniphila cross feeds F. prausnitzii, a decline in the population of the former adversely affects the latter. Since F. prausnitzii is a key butyrate producer, its loss logically leads to compromised gut barrier function. Further research on the full spectrum of functions of both microbes are required before making any firm conclusions about their applicability in human disease.
Lastly, increased barrier permeability leads to leakage of luminal LPS into the portal vein and liver, leading to activation of hepatic TLRs and NASH (Figure 3). Our report described numerous studies that characterized the pro-inflammatory and fibrogenic role of different TLRs in NASH. Targeting circulating LPS and TLRs might be important therapeutic avenues for the treatment of NASH.
As far as translational application of the microbiome in the treatment of NAFLD is concerned, a clinical trial is currently underway that aims at repopulating the gut microbiome of NASH patients via FMT from lean donors (ClinicalTrials.gov Identifier: NCT02469272). The primary end-point of the study at the end of the 12-week FMT period is degree of hepatic steatosis as determined by MRI. The secondary end points are liver function tests and markers of insulin sensitivity. Data from this study will provide preliminary clues on the safety, viability, and efficacy of the use of FMT for the treatment of NASH. Follow-up large-scale studies will be required to truly validate any beneficial findings before FMT is considered as a therapeutic intervention for NASH.
Author Contributions
KJ and TC contributed to the writing of this manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
All authors are employees of AstraZeneca, and may hold stock in AstraZeneca. This work was fully funded by AstraZeneca. The funder has no role in the design, writing or decision to publish this review manuscript.
Acknowledgments
The authors acknowledge Deborah Shuman, Scientific Editor and Communications Specialist for her invaluable contribution in the design of our graphical illustrations.
References
1. Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol (2017) 23:8263–76. doi: 10.3748/wjg.v23.i47.8263
2. DeWeerdt S. Disease progression: Divergent paths. Nature (2017) 551:S92–3. doi: 10.1038/d41586-017-06925-2
3. Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia (2009) 13:9–19.
4. Fang YL, Chen H, Wang CL, Liang L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J Gastroenterol (2018) 24:2974–83. doi: 10.3748/wjg.v24.i27.2974
5. Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD. American Association for the Study of Liver, Liver biopsy. Hepatology (2009) 49:1017–44. doi: 10.1002/hep.22742
6. Loomba R. Role of imaging-based biomarkers in NAFLD: Recent advances in clinical application and future research directions. J Hepatol (2018) 68:296–304. doi: 10.1016/j.jhep.2017.11.028
7. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature (2012) 482:179–85. doi: 10.1038/nature10809
8. Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut (2013) 62:1787–94. doi: 10.1136/gutjnl-2012-303816
9. Sharpton SR, Yong GJM, Terrault NA, Lynch SV. Gut Microbial Metabolism and Nonalcoholic Fatty Liver Disease. Hepatol Commun (2019) 3:29–43. doi: 10.1002/hep4.1284
10. Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol (2020) 17:279–97. doi: 10.1038/s41575-020-0269-9
11. Hoyles L, Fernandez-Real JM, Federici M, Serino M, Abbott J, Charpentier J, et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat Med (2018) 24:1070–80. doi: 10.1038/s41591-018-0061-3
12. Brandl K, Schnabl B. Intestinal microbiota and nonalcoholic steatohepatitis. Curr Opin Gastroenterol (2017) 33:128–33. doi: 10.1097/MOG.0000000000000349
13. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol (2016) 13:412–25. doi: 10.1038/nrgastro.2016.85
14. Wang B, Jiang X, Cao M, Ge J, Bao Q, Tang L, et al. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-alcoholic Fatty Liver Disease. Sci Rep (2016) 6:32002. doi: 10.1038/srep32002
15. Shen F, Zheng RD, Sun XQ, Ding WJ, Wang XY, Fan JG. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int (2017) 16:375–81. doi: 10.1016/S1499-3872(17)60019-5
16. Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol (2013) 11:868–75 e1-3. doi: 10.1016/j.cgh.2013.02.015
17. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab (2017) 25:1054–1062 e5. doi: 10.1016/j.cmet.2017.04.001
18. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology (2013) 57:601–9. doi: 10.1002/hep.26093
19. Del Chierico F, Nobili V, Vernocchi P, Russo A, De Stefanis C, Gnani D, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology (2017) 65:451–64. doi: 10.1002/hep.28572
20. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology (2016) 63:764–75. doi: 10.1002/hep.28356
21. Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology (2013) 58:120–7. doi: 10.1002/hep.26319
22. Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, et al. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol Ecol (2015) 91:1–9. doi: 10.1093/femsec/fiu002
23. Da Silva HE, Teterina A, Comelli EM, Taibi A, Arendt BM, Fischer SE, et al. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci Rep (2018) 8:1466. doi: 10.1038/s41598-018-19753-9
24. Wong VW, Tse CH, Lam TT, Wong GL, Chim AM, Chu WC, et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis–a longitudinal study. PloS One (2013) 8:e62885. doi: 10.1371/journal.pone.0062885
25. Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes (2012) 3:289–306. doi: 10.4161/gmic.19897
26. Holscher HD. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes (2017) 8:172–84. doi: 10.1080/19490976.2017.1290756
27. Hoverstad T, Midtvedt T. Short-chain fatty acids in germfree mice and rats. J Nutr (1986) 116:1772–6. doi: 10.1093/jn/116.9.1772
28. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res (2009) 37:D233–8. doi: 10.1093/nar/gkn663
29. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U.S.A. (2004) 101:15718–23. doi: 10.1073/pnas.0407076101
30. Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U.S.A. (2008) 105:16767–72. doi: 10.1073/pnas.0808567105
31. Nguyen NQ, Debreceni TL, Bambrick JE, Chia B, Wishart J, Deane AM, et al. Accelerated intestinal glucose absorption in morbidly obese humans: relationship to glucose transporters, incretin hormones, and glycemia. J Clin Endocrinol Metab (2015) 100:968–76. doi: 10.1210/jc.2014-3144
32. Ang Z, Ding JL. GPR41 and GPR43 in Obesity and Inflammation - Protective or Causative? Front Immunol (2016) 7:28. doi: 10.3389/fimmu.2016.00028
33. Anderson S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res (1981) 9:3015–27. doi: 10.1093/nar/9.13.3015
34. Muniz Pedrogo DA, Jensen MD, Van Dyke CT, Murray JA, Woods JA, Chen J, et al. Gut Microbial Carbohydrate Metabolism Hinders Weight Loss in Overweight Adults Undergoing Lifestyle Intervention With a Volumetric Diet. Mayo Clin Proc (2018) 93:1104–10. doi: 10.1016/j.mayocp.2018.02.019
35. Kindt A, Liebisch G, Clavel T, Haller D, Hormannsperger G, Yoon H, et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat Commun (2018) 9:3760. doi: 10.1038/s41467-018-05767-4
36. Ye J, Lv L, Wu W, Li Y, Shi D, Fang D, et al. Butyrate Protects Mice Against Methionine-Choline-Deficient Diet-Induced Non-alcoholic Steatohepatitis by Improving Gut Barrier Function, Attenuating Inflammation and Reducing Endotoxin Levels. Front Microbiol (2018) 9:1967. doi: 10.3389/fmicb.2018.01967
37. Endo H, Niioka M, Kobayashi N, Tanaka M, Watanabe T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PloS One (2013) 8:e63388. doi: 10.1371/journal.pone.0063388
38. Nishina PM, Freedland RA. Effects of propionate on lipid biosynthesis in isolated rat hepatocytes. J Nutr (1990) 120:668–73. doi: 10.1093/jn/120.7.668
39. Zambell KL, Fitch MD, Fleming SE. Acetate and butyrate are the major substrates for de novo lipogenesis in rat colonic epithelial cells. J Nutr (2003) 133:3509–15. doi: 10.1093/jn/133.11.3509
40. Rau M, Rehman A, Dittrich M, Groen AK, Hermanns HM, Seyfried F, et al. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. U Eur Gastroenterol J (2018) 6:1496–507. doi: 10.1177/2050640618804444
41. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol (2015) 16:678–89. doi: 10.1038/nrm4074
42. Rabot S, Membrez M, Bruneau A, Gerard P, Harach T, Moser M, et al. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J (2010) 24:4948–59. doi: 10.1096/fj.10-164921
43. Moon YA, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab (2012) 15:240–6. doi: 10.1016/j.cmet.2011.12.017
44. Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes (2006) 55:2159–70. doi: 10.2337/db06-0200
45. Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature (2020) 579:586–91. doi: 10.1038/s41586-020-2101-7
46. Maton PN, Murphy GM, Dowling RH. Ursodeoxycholic acid treatment of gallstones. Dose-response study and possible mechanism of action. Lancet (1977) 2:1297–301. doi: 10.1016/S0140-6736(77)90358-0
47. Shepherd J. Mechanism of action of bile acid sequestrants and other lipid-lowering drugs. Cardiology (1989) 76 Suppl 1:65–71; discussion 71-4. doi: 10.1159/000174548
49. Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev (2003) 83:633–71. doi: 10.1152/physrev.00027.2002
50. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res (2006) 47:241–59. doi: 10.1194/jlr.R500013-JLR200
51. Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocr Rev (1994) 15:391–407. doi: 10.1210/er.15.3.391
52. Wang N, Zou Q, Xu J, Zhang J, Liu J. Ligand binding and heterodimerization with retinoid X receptor alpha (RXRalpha) induce farnesoid X receptor (FXR) conformational changes affecting coactivator binding. J Biol Chem (2018) 293:18180–91. doi: 10.1074/jbc.RA118.004652
53. Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol (2013) 86:1517–24. doi: 10.1016/j.bcp.2013.08.015
54. Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev (2003) 17:1581–91. doi: 10.1101/gad.1083503
55. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab (2005) 2:217–25. doi: 10.1016/j.cmet.2005.09.001
56. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell (2000) 102:731–44. doi: 10.1016/S0092-8674(00)00062-3
57. Lambert G, Amar MJ, Guo G, Brewer HB Jr., Gonzalez FJ, Sinal CJ. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem (2003) 278:2563–70. doi: 10.1074/jbc.M209525200
58. Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem (2000) 43:2971–4. doi: 10.1021/jm0002127
59. Kast HR, Nguyen CM, Sinal CJ, Jones SA, Laffitte BA, Reue K, et al. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol (2001) 15:1720–8. doi: 10.1210/mend.15.10.0712
60. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest (2004) 113:1408–18. doi: 10.1172/JCI21025
61. Cyphert HA, Ge X, Kohan AB, Salati LM, Zhang Y, Hillgartner FB. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem (2012) 287:25123–38. doi: 10.1074/jbc.M112.375907
62. Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest (2015) 125:386–402. doi: 10.1172/JCI76738
63. Jiao N, Baker SS, Chapa-Rodriguez A, Liu W, Nugent CA, Tsompana M, et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut (2018) 67:1881–91. doi: 10.1136/gutjnl-2017-314307
64. Dasarathy S, Yang Y, McCullough AJ, Marczewski S, Bennett C, Kalhan SC. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol (2011) 23:382–8. doi: 10.1097/MEG.0b013e328345c8c7
65. Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L, et al. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PloS One (2016) 11:e0151829. doi: 10.1371/journal.pone.0151829
66. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (2019) 394:2184–96. doi: 10.1016/S0140-6736(19)33041-7
67. Kumar DP, Asgharpour A, Mirshahi F, Park SH, Liu S, Imai Y, et al. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet alpha Cells to Promote Glucose Homeostasis. J Biol Chem (2016) 291:6626–40. doi: 10.1074/jbc.M115.699504
68. Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology (2006) 43:173–81. doi: 10.1002/hep.21006
69. Salim SY, Soderholm JD. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflammation Bowel Dis (2011) 17:362–81. doi: 10.1002/ibd.21403
70. Natividad JM, Verdu EF. Modulation of intestinal barrier by intestinal microbiota: pathological and therapeutic implications. Pharmacol Res (2013) 69:42–51. doi: 10.1016/j.phrs.2012.10.007
71. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol (2009) 9:799–809. doi: 10.1038/nri2653
72. Johansson ME, Hansson GC. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol (2016) 16:639–49. doi: 10.1038/nri.2016.88
73. Mukherjee S, Partch CL, Lehotzky RE, Whitham CV, Chu H, Bevins CL, et al. Regulation of C-type lectin antimicrobial activity by a flexible N-terminal prosegment. J Biol Chem (2009) 284:4881–8. doi: 10.1074/jbc.M808077200
74. Corthesy B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front Immunol (2013) 4:185. doi: 10.3389/fimmu.2013.00185
75. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol (2009) 124:3–20; quiz 21-2. doi: 10.1016/j.jaci.2009.05.038
76. Konturek PC, Harsch IA, Konturek K, Schink M, Zopf Y. Gut-liver axis: How intestinal bacteria affect the liver]. MMW Fortschr Med (2018) 160:11–5. doi: 10.1007/s15006-018-1051-6
77. Yu LC. Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: exploring a common ground hypothesis. J BioMed Sci (2018) 25:79. doi: 10.1186/s12929-018-0483-8
78. Macpherson AJ, Heikenwalder M, Ganal-Vonarburg SC. The Liver at the Nexus of Host-Microbial Interactions. Cell Host Microbe (2016) 20:561–71. doi: 10.1016/j.chom.2016.10.016
79. Assimakopoulos SF, Triantos C, Maroulis I, Gogos C. The Role of the Gut Barrier Function in Health and Disease. Gastroenterol Res (2018) 11:261–3. doi: 10.14740/gr1053w
80. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology (2009) 49:1877–87. doi: 10.1002/hep.22848
81. Volynets V, Kuper MA, Strahl S, Maier IB, Spruss A, Wagnerberger S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci (2012) 57:1932–41. doi: 10.1007/s10620-012-2112-9
82. Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol Gastroenterol Hepatol (2015) 1:222–32. doi: 10.1016/j.jcmgh.2015.01.001
83. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol (2019) 71:1216–28. doi: 10.1016/j.jhep.2019.08.005
84. Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science (2018) 359:1376–83. doi: 10.1126/science.aar3318
85. Jamshidi P, Hasanzadeh S, Tahvildari A, Farsi Y, Arbabi M, Mota JF, et al. Is there any association between gut microbiota and type 1 diabetes? A systematic review. Gut Pathog (2019) 11:49. doi: 10.1186/s13099-019-0332-7
86. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature (2012) 490:55–60. doi: 10.1038/nature11450
87. Zhang X, Shen D, Fang Z, Jie Z, Qiu X, Zhang C, et al. Human gut microbiota changes reveal the progression of glucose intolerance. PloS One (2013) 8:e71108. doi: 10.1371/journal.pone.0071108
88. Greer RL, Dong X, Moraes AC, Zielke RA, Fernandes GR, Peremyslova E, et al. Akkermansia muciniphila mediates negative effects of IFNgamma on glucose metabolism. Nat Commun (2016) 7:13329. doi: 10.1038/ncomms13329
89. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U.S.A. (2013) 110:9066–71. doi: 10.1073/pnas.1219451110
90. Derrien M, Collado MC, Ben-Amor K, Salminen S, de Vos WM. The Mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl Environ Microbiol (2008) 74:1646–8. doi: 10.1128/AEM.01226-07
91. Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med (2017) 23:107–13. doi: 10.1038/nm.4236
92. Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med (2019) 25:1096–103. doi: 10.1038/s41591-019-0495-2
93. Belzer C, Chia LW, Aalvink S, Chamlagain B, Piironen V, Knol J, et al. Microbial Metabolic Networks at the Mucus Layer Lead to Diet-Independent Butyrate and Vitamin B12 Production by Intestinal Symbionts. mBio (2017) 8:e00770-17. doi: 10.1128/mBio.00770-17
94. Gurung M, Li Z, You H, Rodrigues R, Jump DB, Morgun A, et al. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine (2020) 51:102590. doi: 10.1016/j.ebiom.2019.11.051
95. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
96. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr (2009) 139:1619–25. doi: 10.3945/jn.109.104638
97. Munukka E, Rintala A, Toivonen R, Nylund M, Yang B, Takanen A, et al. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J (2017) 11:1667–79. doi: 10.1038/ismej.2017.24
98. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell (2016) 167:1339–1353 e21. doi: 10.1016/j.cell.2016.10.043
99. Brenner DA. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc (2009) 120:361–8.
100. Miura K, Ishioka M, Iijima K. The Roles of the Gut Microbiota and Toll-like Receptors in Obesity and Nonalcoholic Fatty Liver Disease. J Obes Metab Syndr (2017) 26:86–96. doi: 10.7570/jomes.2017.26.2.86
101. Miura K, Ohnishi H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol (2014) 20:7381–91. doi: 10.3748/wjg.v20.i23.7381
102. Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol (2004) 173:7115–9. doi: 10.4049/jimmunol.173.12.7115
103. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
104. Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology (2003) 37:1043–55. doi: 10.1053/jhep.2003.50182
105. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med (2007) 13:1324–32. doi: 10.1038/nm1663
106. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
107. Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology (2009) 50:1094–104. doi: 10.1002/hep.23122
108. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology (2013) 57:577–89. doi: 10.1002/hep.26081
109. Ehses JA, Meier DT, Wueest S, Rytka J, Boller S, Wielinga PY, et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia (2010) 53:1795–806. doi: 10.1007/s00125-010-1747-3
110. Himes RW, Smith CW. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J (2010) 24:731–9. doi: 10.1096/fj.09-141929
111. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science (2010) 328:228–31. doi: 10.1126/science.1179721
112. Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet-Induced Liver Disease. Cell Mol Gastroenterol Hepatol (2016) 2:584–604. doi: 10.1016/j.jcmgh.2016.04.007
113. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology (2010) 139:323–34 e7. doi: 10.1053/j.gastro.2010.03.052
Keywords: microbiome, non-alcoholic steatohepatitis, metabolites, gut barrier, gut permeability, non-alcoholic fatty liver disease
Citation: Jadhav K and Cohen TS (2020) Can You Trust Your Gut? Implicating a Disrupted Intestinal Microbiome in the Progression of NAFLD/NASH. Front. Endocrinol. 11:592157. doi: 10.3389/fendo.2020.592157
Received: 06 August 2020; Accepted: 28 September 2020;
Published: 21 October 2020.
Edited by:
Magdalene K. Montgomery, The University of Melbourne, AustraliaReviewed by:
Amanda Cox, Griffith University, AustraliaBrenna Osborne, University of Copenhagen, Denmark
Copyright © 2020 Jadhav and Cohen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taylor S. Cohen, dGF5bG9yLmNvaGVuQGFzdHJhemVuZWNhLmNvbQ==