 Ali A. Hajeyah
Ali A. Hajeyah William J. Griffiths
William J. Griffiths Yuqin Wang2
Yuqin Wang2 Andrew J. Finch
Andrew J. Finch- 1Systems Immunity Research Institute and Division of Infection and Immunity, Cardiff University, Cardiff, United Kingdom
- 2Institute of Life Science, Swansea University Medical School, Swansea, United Kingdom
- 3Centre for Tumour Biology, Barts Cancer Institute, Queen Mary University of London, London, United Kingdom
Enzymatically oxidized lipids are a specific group of biomolecules that function as key signaling mediators and hormones, regulating various cellular and physiological processes from metabolism and cell death to inflammation and the immune response. They are broadly categorized as either polyunsaturated fatty acid (PUFA) containing (free acid oxygenated PUFA “oxylipins”, endocannabinoids, oxidized phospholipids) or cholesterol derivatives (oxysterols, steroid hormones, and bile acids). Their biosynthesis is accomplished by families of enzymes that include lipoxygenases (LOX), cyclooxygenases (COX), cytochrome P450s (CYP), and aldo-keto reductases (AKR). In contrast, non-enzymatically oxidized lipids are produced by uncontrolled oxidation and are broadly considered to be harmful. Here, we provide an overview of the biochemistry and enzymology of LOXs, COXs, CYPs, and AKRs in humans. Next, we present biosynthetic pathways for oxylipins, oxidized phospholipids, oxysterols, bile acids and steroid hormones. Last, we address gaps in knowledge and suggest directions for future work.
Introduction
Lipids are a key component of life. They play essential roles in membrane structure, cell signaling and energy production. Like other biomolecules, they undergo chemical modifications that expand their functional repertoire. One modification that has been steadily gaining attention is lipid oxygenation, with interest being attributed to two factors: advances in analytical methods that allow detection of oxygenated species, and the ever-growing body of literature implicating them in biological processes and disease. “Enzymatically oxidized lipids” constitute a large portion of known oxygenated lipid mediators, being bioactive lipids that are produced locally through specific biosynthetic pathways in response to extracellular stimuli. Those derived from oxygenation of polyunsaturated fatty acids (PUFA) such as arachidonic acid (AA) include prostaglandins (PG), thromboxanes and leukotrienes, which function in modulating inflammation, the immune response, and hemostasis, and are broadly termed “oxylipins” (1, 2). Oxylipins also include multiply oxygenated derivatives of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), of which specific enantiomers have been termed “specialized pro-resolving mediators” (SPM) for their proposed roles in the resolution of inflammation (3). SPMs derived from DHA include D-resolvins, maresins and protectin. On the other hand, E-resolvins are proposed to be generated from EPA but the enzymatic pathways are unclear and require further investigation. Oxylipins also constitute a core functional group on larger lipids including oxidized phospholipids (oxPL), endocannabinoids and cholesteryl esters (CE). Oxidized CEs are conversely associated with atherosclerosis progression (4), whereas enzymatically oxidized phospholipids (eoxPL) are pro-coagulant and promote a variety of innate immune actions in leukocytes and platelets (5).
Enzymatically oxidized lipids are not limited to those containing oxygenated PUFA functional groups. Oxidized derivatives of cholesterol include steroid hormones, bile acids and their oxysterol precursors. Steroid hormones regulate multiple physiological processes including metabolism (e.g., glucose homeostasis by glucocorticoids), water retention, immune function, and development of sex characteristics (6–8). Bile acids, while traditionally known for aiding in fat digestion and bilirubin excretion, are also being increasingly revealed as signaling molecules and metabolic regulators (9, 10). Similarly, accumulating evidence is revealing oxysterols as more than just bile acid intermediates with functions in signaling (11).
Enzymatic lipid oxidation is facilitated by a network of proteins that use PUFAs or sterols as substrates, specifically, lipoxygenase (LOX), cyclooxygenase (COX), and cytochrome P450 (CYP), all of which exist as several isoforms exhibiting broad substrate specificity (12–16). In general, PUFA oxygenation is initiated by COXs, LOXs, and to a lesser extent CYPs. For example, the biosynthesis of PGs is initiated by COXs, whereas the formation of leukotrienes begins with a LOX. Additionally, crossover between COX, LOX, and CYP is proposed to produce various SPMs. On the other hand, cholesterol oxidation is dominated by CYPs. For example, sidechain shortening during steroid hormone synthesis is catalyzed by CYP11A1 (P450scc). Bile acids are synthesized from cholesterol predominantly by two pathways: the neutral pathway starting with an endoplasmic reticulum resident enzyme CYP7A1, and the acidic pathway starting with CYP27A1 in mitochondria (17). CYPs account for many enzymes in both pathways, catalyzing the formation of oxysterols as well as oxygenated PUFAs. Last, aldo-keto reductases (AKR) and hydroxysteroid dehydrogenases (HSD) catalyze key redox reactions in bile acid and steroid hormone biosynthesis.
By contrast, non-enzymatically oxidized lipids are produced through uncontrolled oxidation via free radical mechanisms. This involves the oxidation of lipids by free radicals, followed by chain propagation and ultimately termination. Notably, during oxygenation of PUFA, active site intermediates can escape to react in an uncontrolled manner, leading to some oxidized lipids forming due to non-enzymatic rearrangements of enzymatic pathway intermediates.
Here, we provide an overview of major enzymes involved in lipid metabolism, including LOXs, COXs, CYPs and AKRs. Then we outline the biosynthetic pathways of oxylipins, oxPLs, oxysterols, bile acids and steroid hormones. Finally, we address outstanding questions and suggest directions for future work.
Enzyme Families Involved in the Biosynthesis of Oxidized Lipids
Lipoxygenase (LOX)
Human LOX: Isoforms and Tissue Distribution
Lipoxygenases (LOX) are a family of non-heme iron-containing dioxygenases. They catalyze the stereospecific addition of dioxygen to lipids containing a (1Z,4Z)-pentadiene group producing lipid hydroperoxides. The human genome contains six functional LOX genes, expressed in various tissues (Table 1). LOXs were traditionally named according to their positional specificity for arachidonic acid (AA). However, the latest characterized member eLOX3 has limited lipoxygenase activity (38), while some LOXs show preference for other PUFA. Sequence analysis of human LOXs (using UniProt entries (39)) shows that 12R-LOX (ALOX12B) is evolutionarily closer to 15-LOX-2 (ALOX15B; 48.2% identity) than 12S-LOX (ALOX12; 35.7% identity). Additionally, human 15-LOX-1 (ALOX15) exhibits dual positional specificity which is not reflected in the name (18). The lack of a robust naming system is a common cause of confusion. Thus, the use of gene names alongside enzyme names is recommended (14).
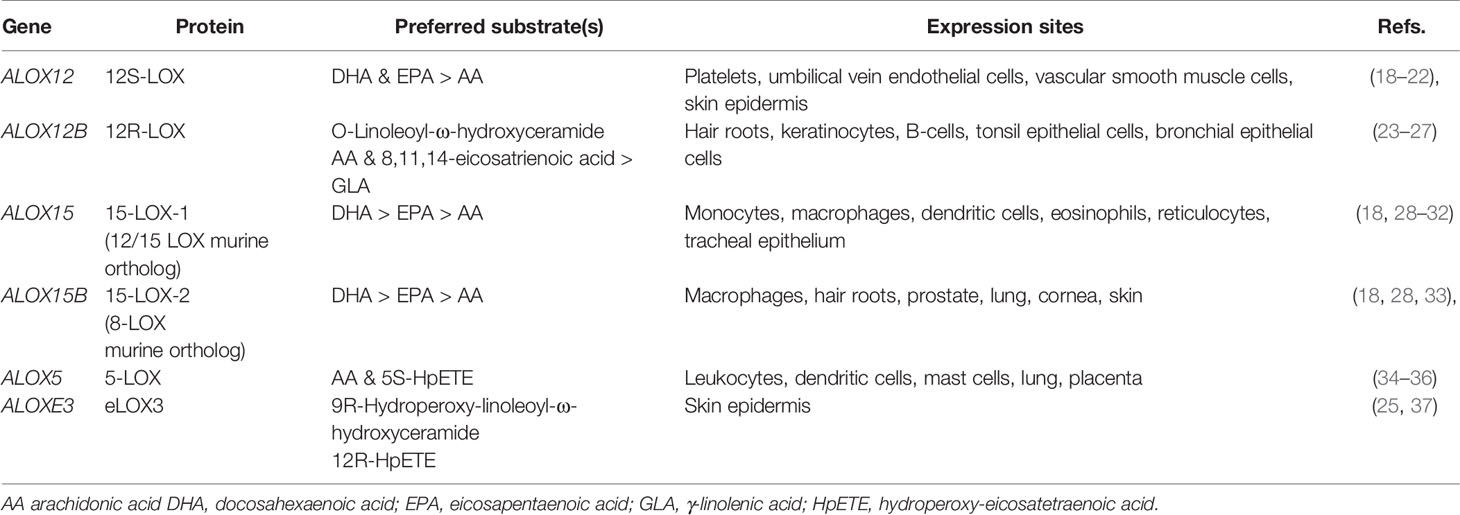
Table 1 Human LOXs: Genes, substrates, and major expression sites.
LOXs are typically constitutively expressed, except for 15-LOX-1 (ALOX15) which is inducible by IL-4 and IL-13 in monocyte-derived macrophages (28). Although 15-LOX-2 (ALOX15B) is constitutively expressed in the same cell type, its expression can be increased by cytokines, hypoxia, and lipopolysaccharide. It is possible that other constitutively expressed LOXs share this property.
Structure and Membrane Association of Mammalian LOXs
There are a limited number of published crystal structures for mammalian LOXs. Available structures of 5- and 15-LOXs show a single polypeptide chain that consists of two domains: a small β-barrel N-terminal domain and a larger α-helix-rich C-terminal domain containing the catalytic non-heme iron (40–42). The coordination positions of the catalytic iron are occupied by three conserved His residues, the carboxyl group of the C-terminus Ile residue, a water molecule and one last variable ligand (water, His, Asn, or Ser). Studies on mammalian LOXs found that the N-terminal domain is not required for catalytic activity, but instead functions in membrane binding and regulation (43, 44). The N-terminal domain of mammalian 5-LOXs (ALOX5) was found to be important for the calcium-dependent translocation from the cytosol or nucleus (depending on cell type) to the nuclear envelope and enzyme activity (45, 46). Similar findings were reported for mammalian 15-LOXs, with the translocation from the cytosol to the cell membrane instead (47, 48). Unlike other LOXs, the activity of 5-LOX requires interaction with partner proteins 5-lipoxygenase activating protein (FLAP), coactosin-like protein (CLP) and cytosolic phospholipase A2 (cPLA2) (49, 50). Additionally, 5-LOX undergoes phosphorylation at multiple sites, regulating its translocation and activity, and is allosterically activated by ATP (51–53).
On the other hand, 15-LOXs do not require accessory proteins for free FA oxygenation. Instead, 15-LOXs form a complex with a small scaffolding protein, phosphatidylethanolamine-binding protein 1 (PEPB1) (54). This complex facilitates 15-LOX activity on PL-esterified PUFA and is proposed to play regulatory roles in ferroptosis along with GPX. PEPB1 has been suggested to direct 15-LOX activity toward PL substrates when free AA is depleted (55). The crystal structure of human 15-LOX-2 (ALOX15B) revealed a hydrophobic loop in the N-terminal domain that is flanked by calcium binding sites, a feature that is absent in 5-LOX (42). Hydrogen-deuterium exchange mass spectrometry showed a decrease in H/D exchange for the hydrophobic loop, supporting a role in membrane hopping (48, 56). Both 5-LOX and 15-LOXs associate with membranes in a calcium-dependent process but they exhibit differences in activity, translocation and binding partners.
Calcium-dependent membrane association has also been shown for platelet 12S-LOX (ALOX12), and more recently, 12R-LOX (ALOX12B), and eLOX3 (19, 57). The activity of 12S-LOX (ALOX12) is thought to be regulated by the availability of its substrates, which are supplied through the action of phospholipases. It is currently unknown whether 12S-LOX is regulated by other mechanisms. Research on the regulation of 12R-LOX and eLOX3 is also limited, although calcium has been shown to increase the activity of mouse 12R-LOX but not eLOX3 (58).
Aside from the catalytic domain of 12S-LOX (ALOX12), no crystal structures are available for 12-LOXs or eLOX3. However, human platelet 12S-LOX (ALOX12) was characterized using small-angle x-ray scattering, showing its occurrence as a dimer in solution (59). This challenged the idea that all human LOXs exist and function as monomers. Human 5-LOX displays full activity as a monomer but forms functional dimers as well, thought to exist in equilibrium (60). Similarly, rabbit 15-LOX-1 (ALOX15) undergoes ligand-induced dimerization in aqueous solutions, and molecular dynamics predicts stable dimers in the presence of substrate fatty acids (61). This supports the idea of monomer-dimer equilibria in LOXs. Human 15-LOX-1 (81.1% sequence identity to rabbit) likely exhibits the same property.
In summary, all six human LOXs associate with membranes in a calcium-dependent manner. 5-LOX requires the help of protein partners for membrane translocation and activity, whereas 15-LOXs only require a partner protein to efficiently catalyze the oxygenation of PL-esterified PUFA. Less is known about the regulation of 12-LOXs and eLOX3. Dimerization and monomer-dimer equilibria have been observed in several LOX isoforms and represent a potential regulatory mechanism. Finally, complete crystal structures have yet to be reported for 12S-LOX, 12R-LOX, and eLOX3. In the case of 12S-LOX, a crystal structure would be beneficial in the rational design of inhibitors, as the enzyme functions in platelet activation.
Reaction Mechanism of LOX
The dioxygenase activity of LOX represents a highly controlled form of lipid peroxidation (Figure 1A). First, stereospecific hydrogen atom removal is carried out by the non-heme ferric iron [Fe(III)]. Fe(III) is not a strong enough oxidizer to abstract hydrogen directly. Thus, a mechanism involving proton-coupled electron transfer (PCET) has been proposed (62) (Figure 1A). In this, the electron is directly transferred to Fe(III) and the proton is picked up by the hydroxide ligand in a concerted mechanism (simultaneously), producing a lipid alkyl radical and ferrous iron [Fe(II)]. This is followed by a rearrangement of the lipid radical into the more stable conjugated diene. After that, dioxygen is introduced onto the opposite side of the removed hydrogen (antarafacially), generating a lipid peroxyl radical. Finally, the lipid peroxyl radical is reduced by Fe(II) and protonated to form a lipid hydroperoxide, through PCET. This reforms Fe(III) for another round of catalysis.
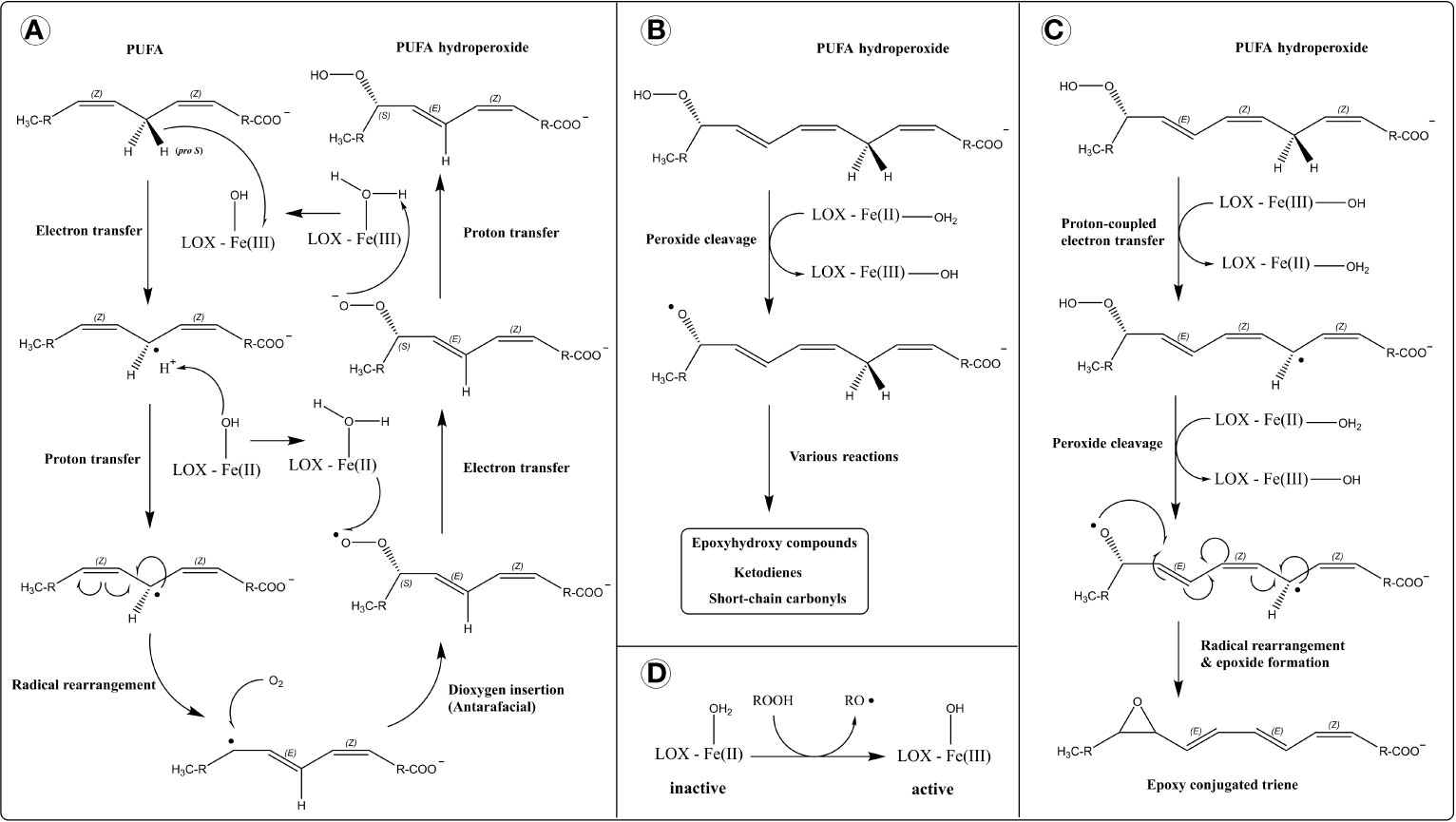
Figure 1 Reaction mechanism of lipoxygenases (LOXs). (A) Dioxygenase activity. Hydrogen atom removal is thought to proceed through a proton-coupled electron transfer mechanism (PCET), in which the transfer of electron and proton occur simultaneously (concerted mechanism), depicted separately for simplicity. (B) Lipohydroperoxidase activity. (C) Leukotriene synthase activity. (D) Activation of LOX by a hydroperoxide.
The stereo and regio-specificity of lipoxygenation are determined by structural features which: (i) accommodate and position the substrate in a specific orientation, and (ii) direct radical rearrangement and oxygen insertion. These features include a U-shaped cavity that accommodates the substrate (63), a migration channel for oxygen (64), a glycine/alanine “switch” that determines stereospecificity by directing oxygen (65), and several amino acid residues that control positional specificity (66, 67). These properties are explored in-depth by Newcomer and Brash (68).
In addition to classical dioxygenase activity, LOXs catalyze other reactions that involve free-radical processes like hydrogen abstraction, homolytic bond cleavage, and radical rearrangement (69). eLOX3 exhibits a hydroperoxide isomerase activity (lipohydroperoxidase activity) which converts hydroperoxides to epoxy-alcohols and ketones (70) (Figure 1B). eLOX3 has restricted dioxygenase activity (38), and its hydroperoxide isomerase activity is considered its primary catalytic reaction. Also, mammalian 5-LOXs and human 15-LOX-1 (ALOX15) possess leukotriene A4 synthase activity which produces epoxides from hydroperoxides (71–74). The mechanism involves the homolytic cleavage of a hydroperoxide into an alkoxy radical and hydrogen atom removal (via PCET) from a bis-allylic methylene carbon generating an alkyl radical. The resulting biradical is stabilized by epoxide formation (Figure 1C).
LOXs are inactive in their basal form with Fe(II) and require activation via oxidation into Fe(III) (Figure 1D). LOX activation is facilitated by hydroperoxides such as their dioxygenase reaction products. Although the proposed LOX catalytic cycle regenerates the ferric iron for another round of catalysis, small amounts of radical intermediates can escape the active site resulting in an incomplete catalytic cycle and requiring repeated activation of the enzyme (formation of Fe(III) by hydroperoxide) for sustained catalysis (69, 75, 76).
Some LOXs exhibit suicide inactivation, a property in which the reaction product rapidly inactivates the enzyme. Human 5-LOX is irreversibly inactivated by both of its products 5-HpETE and leukotriene A4 (77). Similarly, rabbit reticulocyte 15-LOX (human 15-LOX-1 ortholog) is inactivated by its product 15-HpETE, and the mechanism is suggested to be through formation of reactive intermediates that covalently bind the enzyme (78). The dioxygenase reaction and the other two activities (lipohydroperoxidase and leukotriene synthase) have been proposed to inactive LOXs through different mechanisms, but the mechanistic details remain unclear (78).
LOXs Act on a Broad Range of Substrates
LOX isoforms have different substrate preferences. Substrates of mammalian 5-LOXs include AA, its hydroperoxide product 5S-HpETE, as well as epoxy-alcohols derived from EPA and DHA (71, 79). The former two are important in the biosynthesis of leukotrienes, while the latter two are proposed to be precursors of E- and D-series resolvins, respectively (75, 80). 15-LOXs (ALOX15 and ALOX15B) accept AA, EPA, DHA, linoleic acid (LA), and γ-linolenic acid (GLA) as substrates. The preference of human 15-LOX orthologs is: DHA > EPA > AA > GLA > LA (18). One key difference is that 15-LOX-1 (ALOX15) possesses dual positional specificity for some of its substrates (e.g., 12 and 15-lipoxygenase activities for AA), whereas 15-LOX-2 (ALOX15B) exhibits singular specificity (15-lipoxygenase activity) (55). The hydroperoxide isomerase and epoxidase activities of 15-LOX-1 (ALOX15) are proposed to function in the synthesis of eoxins, E-series resolvins, and protectin from their respective precursors. Additionally, human 15-LOX-1 oxygenates PUFA-containing fatty amides and 2-arachidonoyl-glycerol ester (81, 82). Finally, mammalian 15-LOXs can catalyze the oxygenation of PUFA in lysophospholipids (lysoPL), PLs, CEs, and lipoproteins (83–88).
PUFA substrates of platelet 12S-LOX (ALOX12) include AA, EPA, DHA, and dihomo-γ-linolenic acid (DGLA) (18, 89). While no single study has compared all substrates at the same time, one determined the order of preference for the first three substrates to be: DHA > EPA > AA (18). Another found comparable kinetic parameters for EPA, AA, and DGLA as substrates (89). Both observed a singular positional specificity for 12S-LOX. LA, GLA, and α-linolenic acid (ALA) were all found to be poor substrates for 12S-LOX (18, 89, 90). Furthermore, human 12S-LOX possesses lipoxin synthase activity, which converts leukotriene A4 into lipoxins A and B (91). The enzymatic activities of 12S-LOX are also proposed to function in the biosynthesis of hepoxilins and maresins (92, 93). Recently, 12S-LOX was shown to oxygenate 2-AA-lysoPL, proposing alternate pathways for oxylipin and oxPL biosynthesis (94).
Mammalian 12R-LOX (ALOX12B) and eLOX3 (ALOXE3) are co-expressed in skin and their functions are related. 12R-LOX efficiently catalyzes the peroxidation of AA, DGLA and GLA, and less efficiently LA, EPA and DHA (23, 24). Later, 12R-LOX was shown to oxygenate LA esterified to ω-hydroxyacyl-sphingosine more efficiently than free LA (25). The hydroperoxide product of this reaction is further converted into an epoxy-alcohol by the isomerase activity of eLOX3. These two reactions are essential for the proper formation of the water-impermeable barrier in corneocytes (25). Human eLOX3 isomerase activity has also been observed on the 12R- and 12S-hydroperoxides of AA, with the reaction occurring 2-3 times faster for the R-stereoisomer (95). These reactions produce hepoxilins, which play a role in skin inflammation. Curiously, mouse 12R-LOX and eLOX3 metabolize the methyl esters of AA and LA more efficiently than the unmodified FAs (58). However, human and mouse orthologs are known to exhibit differences in substrate preference and regio-specificity (95). Thus, further work is needed to assess the ability of human 12R-LOX to metabolize fatty methyl esters and whether that reaction is biologically relevant.
Several aspects of LOX enzymology remain unresolved. First, the functional implications of suicide inactivation are unknown, that is, the reason why some LOXs have not evolved to resist suicide inactivation. This suggests a biological function for this property, perhaps in regulation. Additionally, the mechanistic details of suicide inactivation are not fully understood. Second, due to the ever-increasing list of LOX substrates, it has been difficult to pinpoint biologically relevant substrates and assign clear functions to all LOX isoforms. For example, the function of 15-LOX-2 (ALOX15B) in macrophages remains unclear, although it has been recently shown to regulate cholesterol levels (96). Unique substrates of each isoform (e.g., CEs for 15-LOXs, lysoPLs for 12S-LOX) might be worth investigating.
Cyclooxygenase (COX)
Human COX: Isoforms and Tissue Distribution
Cyclooxygenases (COXs), also called prostaglandin-endoperoxide synthases and prostaglandin G/H synthases, are heme-containing enzymes that possess both oxygenase and peroxidase activities. There are two COX genes in humans: PTGS1 and PTGS2, which encode for COX-1 and COX-2, respectively. Traditionally, it was thought that COX-1 is constitutively expressed, whereas COX-2 is inducible in response to inflammatory signals. However, several studies suggest constitutive COX-2 expression in the brain, lungs, gut, thymus, kidneys, and blood vessels (97–99). In the vasculature, COX-2 has been demonstrated to be a key source of vascular prostacyclin (100, 101). COX-1 is ubiquitously expressed in the body, and its expression sites include blood vessels, prostate, immune cells (monocytes, T-cells), platelets, stomach, resident inflammatory cells, smooth muscles, and mesothelium of many organs (102–105). On the other hand, COX-2 is inducible in many tissues including prostate, immune cells (T-cells, B-cells, monocytes), and stomach (102–104, 106), but also constitutively expressed in some tissues as previously mentioned.
Structure of COX
The role of COXs as mediators of inflammation and their potential as drug targets were drivers for the elucidation of their structures. The crystal structure of sheep COX-1 has been solved in the presence and absence of various synthetic ligands (107–109). Similarly, there are available structures for mouse COX-2 in the presence and absence of natural and synthetic ligands (13, 110–113), as well as human COX-2 with several inhibitors (114, 115).
Both COX isoforms are homodimers, and this quaternary structure is necessary for enzymatic activity (116). Each monomer consists of three domains: An N-terminal epidermal growth factor (EGF)-like domain, a membrane binding domain, and a large C-terminal catalytic domain. The EGF-like domain is located at the dimer interface and potentially facilitates dimerization. It is also thought to facilitate membrane binding (117). The membrane binding domain consists of four amphipathic α-helices that insert into one face of a membrane bilayer. Both COXs are found on the luminal face of the endoplasmic reticulum (ER) and the inner and outer nuclear membranes (118). However, COX-2 is also found in the Golgi apparatus (119). The catalytic domain (in both isoforms) contains separate oxygenase and peroxidase active sites on opposite sides of the heme cofactor. The oxygenase active site is located towards the membrane binding face at the end of a hydrophobic tunnel that allows substrate entry, whereas the peroxidase active site is in a groove on the opposite face of the enzyme (120).
COXs undergo N-glycosylation at multiple asparagine residues. This facilitates their proper folding, and regulates the turnover of COX-2 by controlling its trafficking between the ER and the Golgi apparatus (119, 121, 122). COX-2 can also be S-nitrosylated by inducible nitric oxide synthase, which has been proposed to enhance its activity (123). In vitro experiments on recombinant human COX-1 and COX-2 demonstrated that both isoforms can be S-nitrosylated by a nitric oxide donor, but only COX-2 showed increased cyclooxygenase activity (124). Circular dichroism data suggest an altered, less dynamic conformation for both isoforms after S-nitrosylation with less α-helices, turns, and random coils, but more β-sheets. The structural change was more pronounced in COX-2 compared to COX-1. These findings provide new insight into the structural dynamics of COXs. The occurrence and biological relevance of this altered COX conformation is unexplored in vivo and would benefit from further investigation.
Although COXs are sequence homodimers, they are considered conformational heterodimers because one monomer functions as the catalytic subunit (Ecat) and the other as an allosteric regulator (Eallo) (125–127). For both COX isoforms, the binding of one heme molecule to Ecat is required for full activity. Several substrate and non-substrate FAs can bind Eallo to regulate the activity of Ecat, and the two COX isoforms exhibit differences in this regard. COX-1 is inhibited by palmitic acid, stearic acid, margaric acid and oleic acid (127), none of which are COX substrates. On the other hand, COX-2 is stimulated by palmitic acid and stearic acid (126). Substrate FAs (like AA and EPA) also bind to Eallo to regulate COX activity, with notable differences in the responses of the two isoforms (reviewed in (128)).
Catalytic Mechanism of COX
COXs possess two enzymatic activities: a dioxygenase activity and a peroxidase activity. The dioxygenase activity is a controlled peroxidation process (129), and the role of the enzyme is to direct hydrogen abstraction and the stereochemistry of formation of intermediates (120). Unlike LOXs where hydrogen abstraction is carried out by a non-heme associated Fe(III), hydrogen abstraction in COX is carried out by a catalytic tyrosyl radical (Figure 2A). The heme group oxidizes a tyrosine residue in the active site into a radical, which then abstracts a hydrogen (stereo- and regio-specifically) from the (1Z,4Z)-pentadiene group of the lipid. Using AA as an example substrate, the 13-pro-S hydrogen is abstracted. The resulting alkyl radical rearranges into a conjugated diene before the addition of dioxygen at C11, generating a peroxyl radical (11R-stereochemistry). Rotation of the peroxyl radical positions the outer oxygen atom in the correct orientation to attack the C9 carbon resulting in an endoperoxide (130). A second cyclization involving C8 and C12 generates a bicyclic ring and a radical at C15. Note that the cyclopentane ring is formed in the trans configuration (131). This differs from the non-enzymatically generated isoprostanes which occur predominantly in the cis configuration (132). Following the second cyclization, dioxygen is added onto the si face of the C15, forming a peroxyl radical (15S-stereochemistry). Hydrogen atom transfer from the active site tyrosine residue forms prostaglandin G2 (PGG2) and regenerates the tyrosyl radical for another round of catalysis.
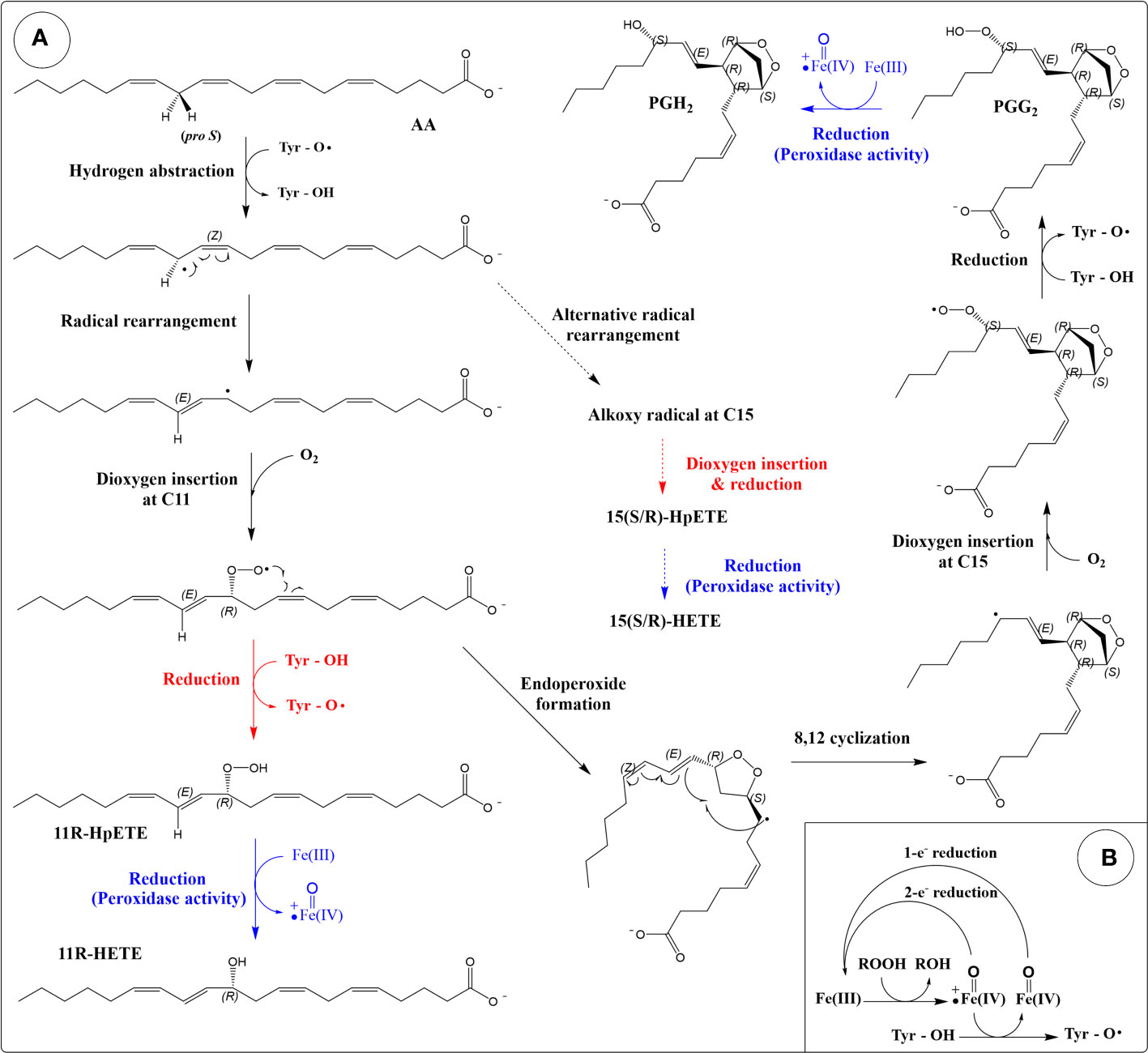
Figure 2 Reaction mechanism of cyclooxygenases (COXs). (A) Production of eicosanoids from arachidonic acid through the dioxygenase and peroxidase activities of COX. The cyclooxygenase reaction is colored black. Peroxidase reactions are coloured blue. Reactions that produce HpETEs are colored red. Side reactions that produce 15-HETEs are depicted by dashed arrows. (B) The peroxidase cycle generates the Tyr radical required for hydrogen abstraction (porphyrin ring of heme not shown).
The peroxidase activity of COX reduces PGG2 into PGH2 (Figure 2A). The mechanism involves a two-electron reduction of the hydroperoxide into an alcohol, with a corresponding oxidation of the heme group into the oxoferryl form (133) (Figure 2B). Regeneration of the heme group can be achieved by two sequential one-electron reductions. The peroxidase activity is required for the dioxygenase activity as the oxidation of the tyrosine into a radical is done by the oxoferryl-porphyrin cation radical generated during the peroxidase reaction cycle (133) (Figure 2B). Suicide inactivation has been described for both the dioxygenase and peroxidase activities of COX (133, 134).
The dioxygenase activity of COX can also result in a lipoxygenase-type reaction, in which one dioxygen molecule is introduced and no endoperoxide formation occurs (135, 136) (Figure 2A). In this case, the peroxyl radical formed after the addition of the oxygen is reduced into a hydroperoxide instead of participating in the cyclization reaction. This reaction represents an incomplete catalytic cycle and occurs as a side product. Using AA as a substrate, this lipoxygenase-type reaction (after reduction by peroxidase) leads to the formation of 11R-HETE (HETE: hydroxy-eicosatetraenoic acid), 15R-HETE and 15S-HETE (minor product compared to 15R-HETE in both isoforms) (137). C11 and C15 are the same oxygen insertion sites in the dioxygenase (cyclooxygenase reaction), and these by-products are thought to arise from alternate conformations of substrate in the active site (138).
Aspirin (acetylsalicylic acid) is an inhibitor of COXs, and its mechanism of action involves the acetylation of a serine residue (Ser530 in COX-2) in the active site. Acetylation of COX-1 leads to complete inhibition (139), whereas the acetylation of COX-2 promotes the lipoxygenase-type reaction and the formation of 15R-HETE (140). It was previously thought that acetylation completely inhibits PG production by COX-2 (cyclooxygenase reaction). However, recent evidence shows that the acetylated enzyme retains that activity (15S-PG formation) but also more favorably forms 15R-PGs (141), although PGs are still minor products compared to 15R-HETE for acetylated-COX-2. These findings are consistent with an earlier study that reported the formation of 15R-PGE2 in COX-2 Ser530 mutants (137). Thus, both acetylation and mutagenesis of Ser530 in COX-2 promote 15R-PG formation.
COX Isoforms Differ in Their Substrate Specificity
COXs catalyze the transformation of AA into PGH2, which is a precursor of other PGs (PGD2, PGE2, PGF2α, PGI2) and thromboxane. These oxygenated AA derivatives are generated through the action of tissue-specific enzymes downstream of COX (discussed in section 3.1) and play roles in inflammation, blood flow regulation and blood clotting through interactions with specific GPCRs (142).
COXs also accept other PUFAs as substrates including DGLA, LA, ALA, EPA, and GLA (143), at least in vitro. An early study determined the efficiency of substrate utilization for human COXs (Kcat/Km) to be AA > DGLA > LA > ALA, with ALA being a poor substrate for COX-1. EPA and GLA are poor substrates for both isoforms but they are better substrates for COX-2 than for COX-1 (143). Later studies have tested other PUFAs like eicosadienoic acid, adrenic acid, docosapentaenoic acid and DHA as COX substrates (127, 144). In general, COX-2 was found to be more efficient than COX-1 for a broader range of PUFAs. That said, AA is the preferred substrate for both isoforms, but COX-2 can oxygenate it at lower concentrations compared to COX-1 due to differences in their allosteric regulation by FAs (145).
COX-2 can also catalyze the 11R-, 15R-, 15S-dioxygenation and bis-oxygenation of 5S-HETE, forming diHETEs in the former three reactions and a di-endoperoxide product in the later reaction. However, acetylation of COX-2 shifts the specificity into favoring 15R-dioxygenation producing 5S,15R-diHETE (146). Additionally, COX-2 can oxygenate AA in complex lipids for example; arachidonoylethanolamide, 2-arachidonoylglycerol (2-AG), and N-arachidonoyl-glycine (147–149). Recently, 2-AA-lysoPL, and ethanolamide derivatives of EPA and DHA were also shown to be COX-2 substrates (88, 150). Oxygenation of AA-lysoPL by COX-2 generates eicosanoid-lysoPL, which can be hydrolyzed to release eicosanoids through intracellular lipases (88). The ability of COX-2 to bind and oxygenate a broader range of substrates compared to COX-1 has been attributed to a larger active site, the orientation of an Arg residue in the substrate binding pocket and amino acid residues lining the tunnel leading to the cyclooxygenase active site (13).
COX isoforms exhibit differences in expression, tissue distribution, allosteric regulation, and substrate specificity. However, some aspects of COX biochemistry are unclear. For example, the physiological functions of COX-2-derived oxygenated endocannabinoids are unclear. Additionally, COX-2 oxygenates AA-lysoPLs into eicosanoid-lysoPLs, which are proposed to function in signaling and as precursors to other mediators (88). However, the metabolism of eicosanoid-lysoPLs requires further investigation.
Cytochrome P450 (CYP)
Human CYPs: Nomenclature and Tissue Distribution
Cytochrome P450s (CYP) are a superfamily of heme-containing monooxygenases that are ubiquitous across all domains of life. There are 57 functional CYP genes in humans, and their products are further divided into 18 families and 41 sub-families based on amino acid sequence (151, 152). A robust, unified nomenclature system has been devised for CYPs, encompassing all known CYPs across living organisms (153, 154). The name includes the root “CYP”, followed by a number for family, a letter for subfamily, and a gene-identifying number for isoforms. Additionally, an asterisk and a number are added at the end to denote alleles (155). Families and subfamilies are based on 40% and 55% amino acid sequence identity, respectively.
CYPs are widely distributed in mammalian tissues (156–158), with a particularly high expression in the liver, brain, kidney and lung. Intracellularly, mammalian CYPs are generally bound to mitochondrial membranes and the endoplasmic reticulum (ER; microsomes when in vitro) (159, 160). There is also evidence of mammalian CYPs in other compartments including the plasma membrane and the nucleus, based on in vitro studies on cultured cells (161–164). Some microsomal CYPs are also targeted into the mitochondria (159). In the human genome, 50 out of the 57 functional CYP genes code for microsomal CYPs, and the remaining seven for mitochondrial isoforms. Some CYPs are inducible by environmental stimuli, whereas others are constitutively expressed (152). Induction of CYP expression by environmental compounds can be through interactions with nuclear receptors, transcriptional regulatory elements, or non-coding RNAs (165).
Two major functions of CYPs are in drug metabolism and lipid metabolism. Here, we focus on lipid metabolism which involves members of most human CYP families. For an extensive review, the reader is referred to Nelson and Nebert (166).
Structure of Mammalian CYPs
Structural characterization was first carried out on bacterial CYPs, which are typically water soluble. On the other hand, mammalian CYPs are membrane-bound, and initial attempts to crystallize them were unsuccessful until a small N-terminal hydrophobic helical segment was replaced with a hydrophilic sequence from a related protein. The first structure to be published was of rabbit CYP2C5, a microsomal CYP isoform that hydroxylates progesterone (167). Comparison of CYP2C5 structure with closely related microbial CYPs showed a similar folding geometry, but unique features of the mammalian enzyme were observed such as a hydrophobic surface that was proposed to play a role in membrane binding (167).
CYPs consist of two domains: a β-sheet-rich N-terminal domain and a larger helix-rich C-terminal catalytic domain. The N-terminal domain in microsomal CYPs contains a transmembrane helix that plays a role in membrane anchoring, but that role is not exclusive as CYPs contain other regions (in the catalytic domain) that bind the membrane. This transmembrane helix is not present in mitochondrial CYPs, and their membrane binding is facilitated by hydrophobic and amphipathic regions on the surface (160, 168). The N-terminus also contains a signal peptide sequence for trafficking into the appropriate compartment, that in the case of mitochondrial CYPs, is cleaved during transport. The catalytic domain contains a deep cavity that houses the heme prosthetic group. A thiolate ion in a conserved cysteine residue occupies the fifth coordination position of heme, and a water molecule occupies the sixth position in the resting state. There is variable flexibility in the active site among CYPs, and a high degree of flexibility has been correlated to substrate promiscuity (169, 170). Residues in helices surrounding the active site heme position the substrate for catalysis. Additionally, in the CYP4 family, a covalent linkage has been identified between the heme group and a conserved glutamic acid residue (171), and this interaction plays a role in substrate positioning for ω-hydroxylation (hydroxylation at the methyl end of FAs) (172).
Other structural features of CYPs include access channels that allow entry of substrates from both the aqueous and membrane compartments and an exit channel for the product (160). Furthermore, oligomerization is well-documented in both microsomal and mitochondrial CYPs, which can be between subunits of the same CYP (homomeric; e.g., CYP2C2) or different CYPs (heteromeric; e.g., CYP2E1/CYP3A4 and CYP1A2/CYP2B4) (173–180). These interactions provide an additional layer of regulation and play a role in substrate specificity.
Enzymology of Mammalian CYPs
CYPs catalyze a wide range of reactions (181, 182), but the most common include C-hydroxylation, heteroatom oxidation, heteroatom dealkylation, epoxidation, and group migration (182). Many reactions of CYPs are dependent on their ability (owing to the thiolate-heme group) to catalyze the scission of dioxygen and incorporate one of the oxygen atoms into the substrate, with the other being reduced to water. These reactions require electron transfer from a donor, NADPH, to the heme iron. However, transfer of electrons from NADPH to CYPs requires protein partners: NADPH cytochrome P450 reductase in the case of microsomal CYPs, and the combined functions of adrenodoxin reductase and adrenodoxin in mitochondrial CYPs. Thus, CYPs form complexes with their redox partners, at least during catalysis. That said, not all CYP reactions require external oxygen or electron donors. Examples are isomerization reactions catalyzed by CYP5A1 (thromboxane synthase) and CYP8A1 (prostacyclin synthase) (183), two enzymes involved in PGH2 metabolism.
A generalized reaction mechanism for CYPs (Figure 3A) contains the following steps: starting with the resting state, substrate recruitment displaces H2O from the 6th (axial) position of the heme Fe(III). This is followed by a 1-electron reduction from a donor forming Fe(II), O2 binding, and another 1-electron reduction resulting in a negatively charged peroxo group. Protonation of this complex twice by nearby H2O or amino acid residues results in the scission of the dioxygen (O-O bond) into an oxoferryl intermediate (compound I) and a water molecule (184). Compound I is thought to be the direct oxidant in many CYP oxidation reactions (185). In hydroxylation reactions, compound I abstracts a hydrogen from an alkyl group in the substrate generating a carbon radical and an iron oxygen complex, which rapidly react together forming a hydroxyl group on the substrate and regenerating the heme iron into the Fe(III) state (Figure 3B). Although this is a generalized reaction scheme for CYPs, the intermediates following O2 binding until compound I have been difficult to characterize due to their instability and the exact electronic structures are not fully assigned (185).
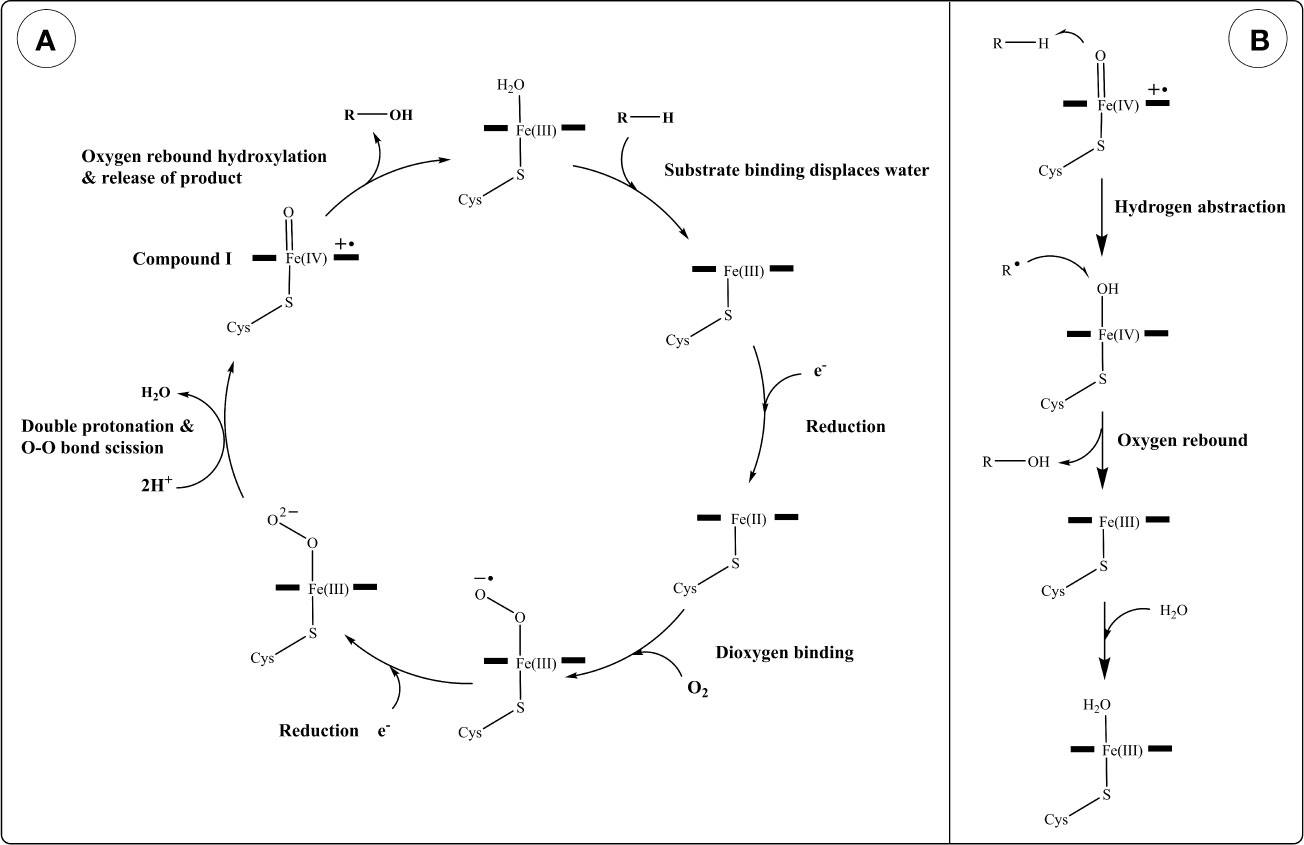
Figure 3 Reaction mechanism of cytochrome P450s (CYPs). (A) Generalized catalytic cycle of CYPs. (B) Hydroxylation of a generic substrate (R–H) by compound I via an oxygen rebound mechanism.
Mammalian CYPs Play Crucial Roles in Oxidative Lipid Metabolism
CYPs are involved in the metabolism of a wide range of lipids including PUFAs and sterols (Table 2). Hydroxylation and epoxidation are two common CYP-mediated reactions for PUFA substrates. CYPs can hydroxylate FAs terminally (ω-hydroxylation) or midchain. Similarly, CYP-mediated epoxidation can occur on various double bonds in PUFA. CYPs exhibit variable preference with respect to the reaction type and positional specificity. CYP1-3 families catalyze epoxidation and hydroxylation reactions for FAs, whereas the CYP4 family favors hydroxylation over epoxidation, specifically ω-hydroxylation (204).
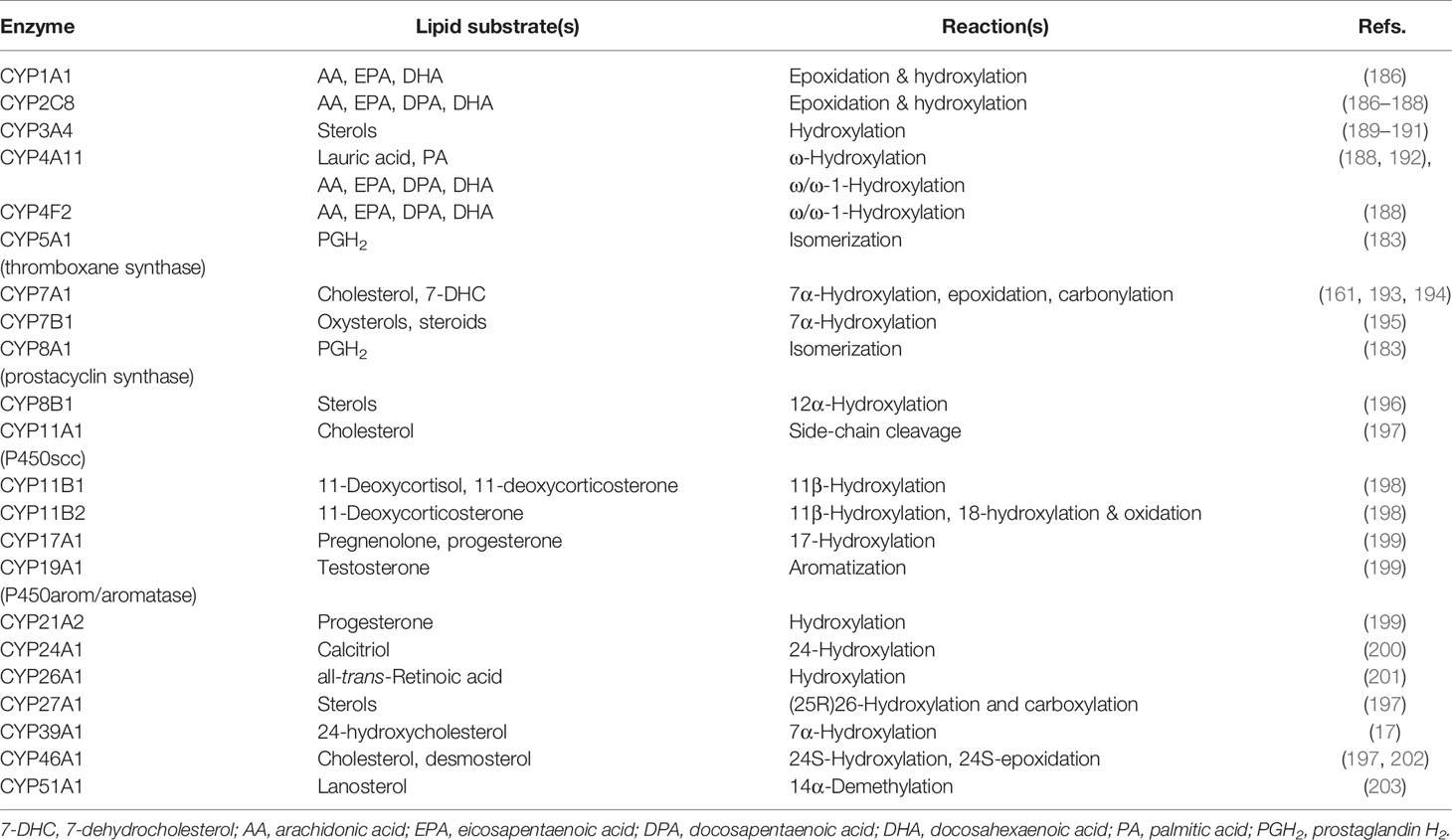
Table 2 Examples of human cytochrome P450s (CYPs) and their involvement in lipid metabolism.
Terminal hydroxylation of AA by CYPs produces 20-hydroxyeicosatetraenoic acid (20-HETE), which functions in blood pressure regulation and water balance (205). Additionally, terminal hydroxylation functions in ω-oxidation (a catabolic pathway for FAs), the degradation of some eicosanoids (206, 207), and the proper formation of the skin permeability barrier (208). Midchain hydroxylation of PUFA produces mono-hydroxylated derivatives (HETEs in the case of AA) with various bioactivities (reviewed in (152)). Unlike LOXs and COXs which oxygenate carbons two bonds away from bis-allylic methylene carbons, CYPs can also oxygenate bis-allylic carbons (producing 7-, 10- and 13-HETE from AA), and various other positions (209–211). However, bis-allylic hydroxylation products are unstable in mildly acidic conditions and rearrange into conjugated dienes (211).
Epoxidation of AA by CYPs produce epoxy-eicosatrienoic acids (EET). However, other PUFA like LA, EPA and DHA also undergo CYP-mediated epoxidation. Epoxy derivatives of PUFAs are implicated in blood pressure regulation and inflammation (15). Their metabolism is undertaken by several epoxide hydrolases including soluble epoxide hydrolase (sEH) and microsomal epoxide hydrolase (mEH) (212). The action of epoxide hydrolases produces dihydroxy derivatives which possess different bioactivities from their epoxide precursors (15).
Arachidonoylethanolamide also undergoes CYP hydroxylation and epoxidation (213, 214). Similarly, CYPs catalyze epoxidation of EPA and DHA ethanolamides, producing compounds with anti-inflammatory and anti-angiogenic effects (215). These lipids can be degraded by both sEH and fatty acid amide hydrolase (FAAH) (215), suggesting a link between the CYP epoxygenase and endocannabinoid pathways. The metabolism and biological roles of these mediators is an active area of research (216).
CYPs catalyze several types of reactions in the metabolism of sterols, some of which involve multiple oxygenation/hydroxylation steps. Demethylation of lanosterol by CYP51A1 is a key reaction in cholesterol biosynthesis and involves three successive oxidations of a methyl group, resulting in its release as formic acid and the introduction of a double bond at the D ring (217). Similarly, the formation of estradiol from testosterone via the action of CYP19A1 (P450arom) involves the elimination of a methyl group by three successive oxidation steps and the introduction of a double bond at the A ring. Additionally, cholesterol side-chain cleavage by CYP11A1 (P450scc) involves two hydroxylation steps on different carbons followed by a C-C bond cleavage (218). This reaction produces pregnenolone which is a key intermediate in the formation of androgens, estrogens, glucocorticoids and mineralocorticoids.
CYP-mediated hydroxylation reactions are ubiquitous in cholesterol metabolism, leading to the formation of oxysterols, steroid hormones and bile acids. Key enzymes in the bile acid biosynthesis pathways are CYP7A1 (cholesterol 7α-hydroxylase), CYP8B1 (sterol 12α-hydroxylase), CYP27A1 (commonly named sterol 27-hydroxylase, but more correctly sterol (25R)26-hydroxylase) and CYP7B1 (oxysterol 7α-hydroxylase) (219, 220). Of these enzymes, CYP27A1 can both hydroxylate and carboxylate the terminal carbon of the sterol side-chain (221). Other enzymes involved in quantitatively minor bile acid biosynthesis pathways are CYP46A1 (cholesterol 24S-hydroxylase) which 24S-hydroxylates cholesterol (222, 223) and CYP3A4 which has 4β- and 25-hydroxylase activity (189, 224). CYP enzymes involved in steroid hormone biosynthesis include CYP11A (P450scc) (225), CYP17A1 (steroid 17α-hydroxylase) (226), CYP21A2 (steroid 21-hydroxylase), CYP11B1 (steroid 11β-hydroxylase), and 11B2 (aldosterone synthase) and CYP19A1 (aromatase) (198).
Thus, CYPs play indispensable roles in the metabolism of both PUFA and cholesterol. However, some human CYPs like CYP2A7 and CYP20A1 have no assigned functions and remain orphan enzymes.
Human Aldo-Keto Reductases (AKRs) and Hydroxysteroid Dehydrogenases (HSDs)
AKRs: Nomenclature, Genes, and Tissue Distribution
Aldo-keto reductases (AKRs) are a superfamily of NADPH-dependent oxidoreductases. They catalyze the reduction of carbonyl groups to alcohols. A nomenclature system has been proposed for AKRs (227). Like CYPs, AKRs are grouped into families and subfamilies based on amino acid sequence. Families have 40% sequence identity, whereas subfamilies are defined by 60% sequence similarity. The nomenclature consists of the root “AKR” followed by a number for family, a letter for subfamily and finally a number as a unique gene identifier. The nomenclature has also been expanded to accommodate multimers (228). The older (trivial) names of AKRs are still heavily in use, which could lead to confusion. Thus, it is advised to follow the nomenclature system proposed by Jez et al. (227).
There are 15 AKRs in the human genome spanning three families and seven subfamilies. Human AKRs have variable expression and accept a wide range of substrates (Table 3). They are involved in the metabolism of sugars, prostaglandins and sterols, as well as the detoxification of carbonyl compounds like lipid peroxidation products. Additionally, members of family 6 (AKR6A3, AKR6A5, and AKR6A9) are constituents of voltage-gated potassium channels (250). All human AKRs have either established or proposed roles in lipid metabolism except for AKR1E2.
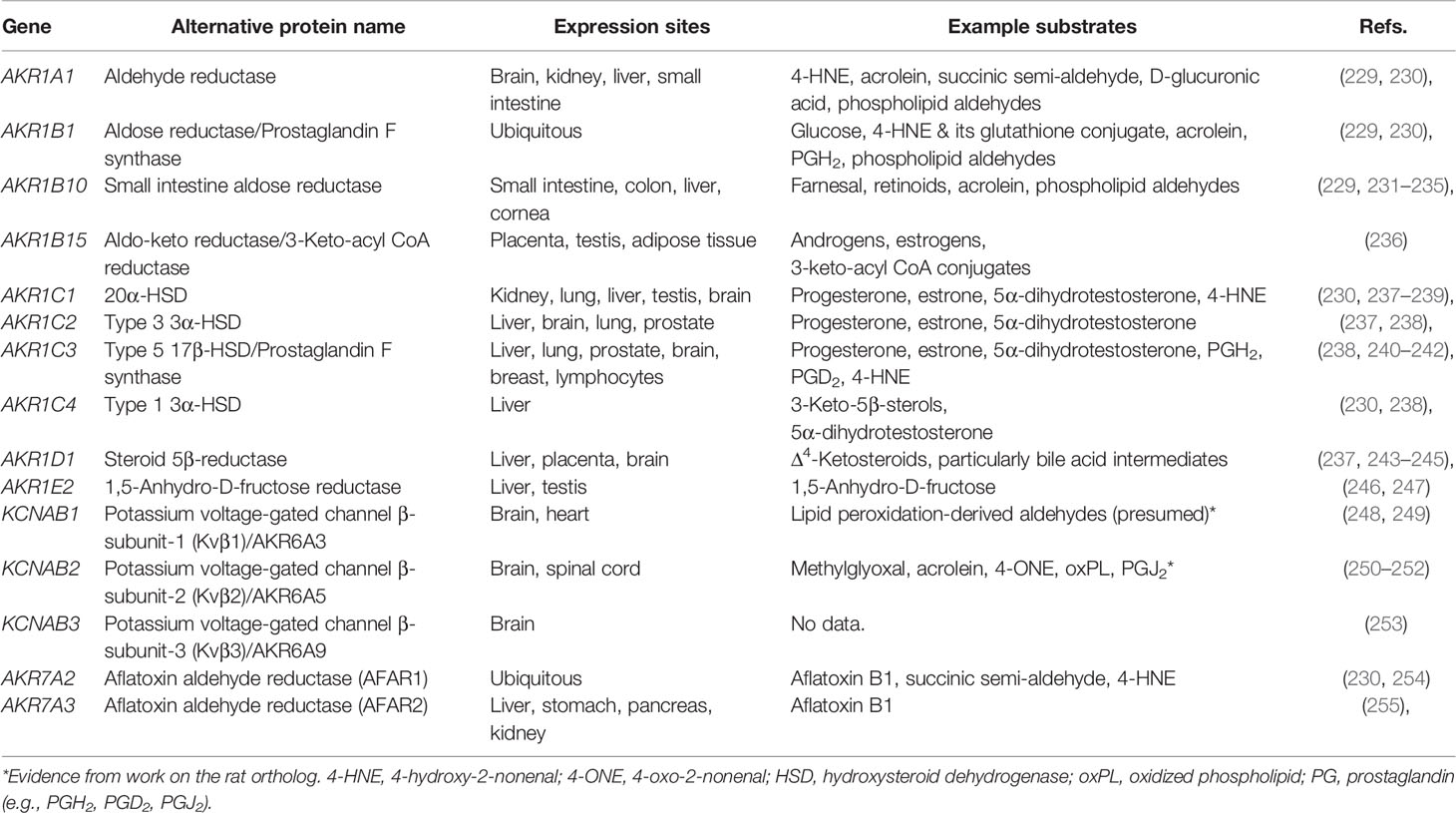
Table 3 Human AKRs: Genes, expression sites, and example substrates.
Human AKRs are generally cytosolic with some exceptions. AKR1B15 is found in mitochondria (236). Members of the AKR1C subfamily in the lung (1C1, 1C2 and 1C3) are also secreted in pulmonary surfactant (256). AKR1B10 is present in lysosomes and is secreted into the intestinal lumen by epithelial cells (257). AKR6 members form a complex with a plasma membrane voltage channel on the cytosolic side. Finally, rat AKR7A2 associates with the Golgi apparatus (258). Human AKR7A2 contains a similar N-terminal amphipathic sequence and likely associates with the Golgi apparatus.
Mammalian AKRs: Structure and Enzymology
Crystal structures have been solved for many mammalian AKRs. They comprise of a triosephosphate isomerase barrel motif (α/β)8 which has alternating α-helices and β-strands repeating eight times. The α-helices surround an internal β-barrel formed by the β-strands. The active site is located at the base of the barrel, in which the substrate and the nicotinamide head group of NADPH (cofactor) are proximally positioned for the reaction, while three flexible loops form the back of the barrel and control substrate specificity (259, 260). The structure also contains two additional helices that are not part of the barrel motif. Family 1 AKRs are monomeric, whereas families 6 and 7 form tetramers and dimers, respectively (258, 261).
AKRs catalyze the stereospecific reduction of carbonyls into alcohols. They can also catalyze the reverse oxidation reaction. However, the reducing direction is generally favored due to the abundance of reduced cofactor in the cellular environment. AKRs can use both NADPH and NADH, but NADPH is the preferred cofactor for most known AKRs as they interact with the phosphoryl group present in NADPH but not NADH (262, 263). The stereospecific nature of the reaction is due to the orientation of the NADPH and the substrate in the active site. The NADPH is bound in the anti-conformation (with respect to the ribose ring) which promotes the transfer of the 4-pro-R hydride (264), and the substrate is positioned perpendicular to the cofactor.
The reaction of AKRs follows an ordered bi-bi mechanism in which the NADPH cofactor binds first and leaves last. The active site of AKRs contains a highly conserved catalytic tetrad of Tyr, Lys, Asp and His. The hydride transfer is facilitated by acid-base catalysis involving the protonation state of Tyr and the other catalytic residues. In the reduction direction, the protonated form of Tyr acts as an acid by participating in proton relay with His (259) (Figure 4A). This polarizes the carbonyl group of the substrate, allowing it to accept a hydride ion from the cofactor. In the oxidation direction, the phenolate form of Tyr (generated through proton relay with Lys and Asp) acts as a base and abstracts a proton from the alcohol group of the substrate, facilitating hydride transfer to the cofactor (259) (Figure 4B).
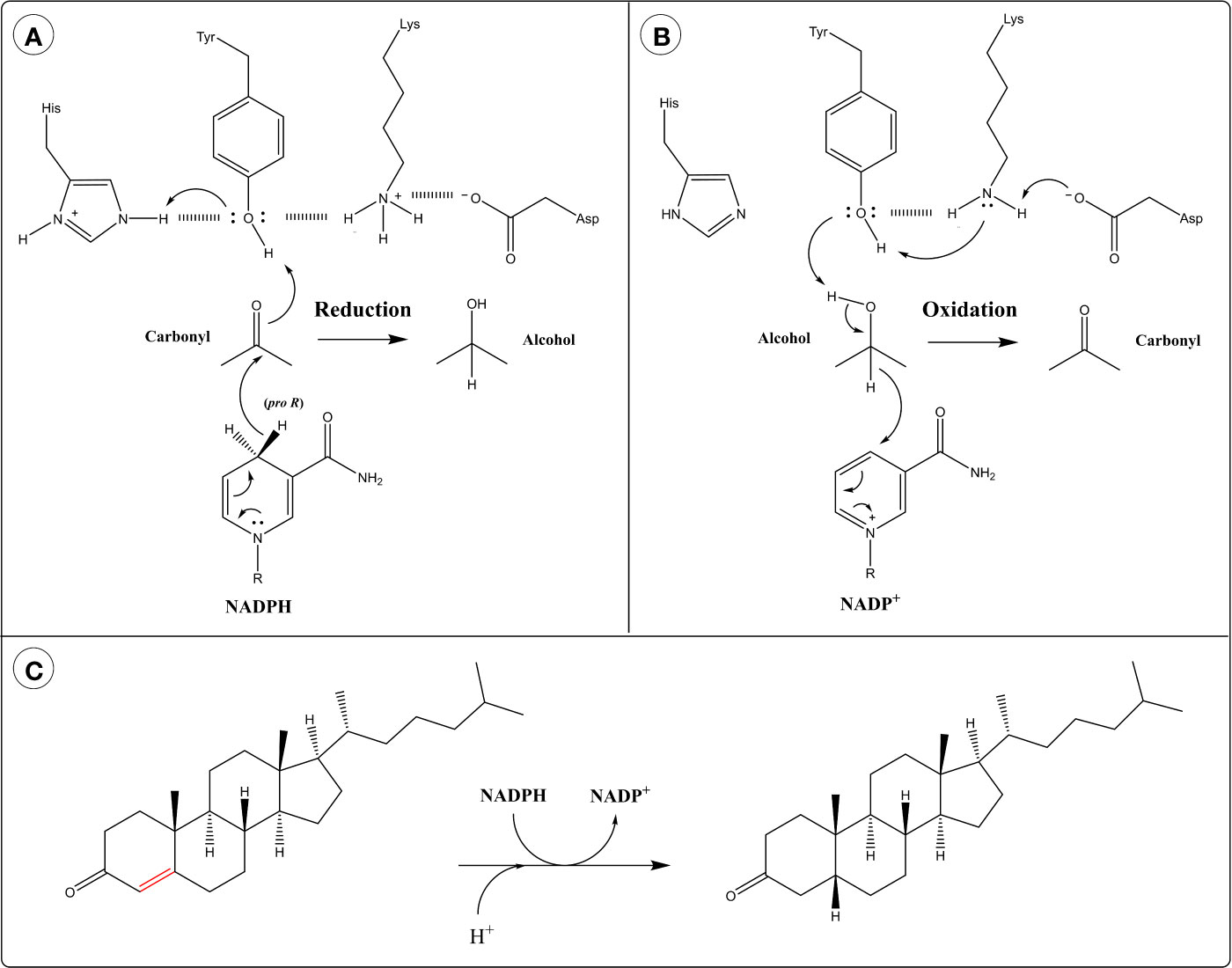
Figure 4 Catalytic mechanism of aldo-keto reductases (AKRs) in the: (A) Reduction direction and (B) Oxidation direction. (C) 5β-Reduction of steroid double bond by AKR1D1.
AKR1D subfamily members catalyze the irreversible 5β-reduction of sterol double bonds into single bonds, instead of the reversible reduction of carbonyls typical of AKRs (Figure 4C). They possess an altered catalytic tetrad with Glu replacing His (265). This substitution allows the substrate to penetrate deeper into the active site pocket, positioning C5 close to the 4-pro-H of NADPH (259). The catalytic Glu residue is thought to act as a superacid, facilitating the enolization of the steroid double bond and allowing hydride transfer to C5 (259, 265).
Human AKR1B1 catalyzes the isomerization of PGH2 to PGD2 in the absence of cofactor, as well as the reduction of PGH2 to PGF2α in the presence of cofactor (266). Curiously, both of these reactions are reported to be facilitated by a catalytic triad of Lys, His and Asp, without the involvement of Tyr in catalysis, although Tyr is still required for the p‐nitrobenzaldehyde reductase activity of the enzyme (266). Thus, both Tyr and His can participate in acid-base catalysis, at least in AKR1B1.
Involvement of Human AKRs in Lipid Metabolism
Human AKRs play roles in the detoxification of reactive carbonyls. Several AKRs metabolize lipid peroxidation-derived aldehydes like acrolein and 4-HNE (Table 3). Additionally, some AKRs (1A1, 1B1, 1B10, 6A5) can reduce oxidized phospholipids, particularly phospholipid aldehydes (229, 251). These activities mitigate the cytotoxicity of reactive carbonyls (240, 267–269), and in the case of family 6 AKRs are also thought to function in redox sensing (251).
Both AKR1B1 and AKR1C3 exhibit prostaglandin F synthase activity (270, 271). AKR1B1 catalyzes the reduction of PGH2 to PGF2α. AKR1C3 catalyzes the same reaction as well as the reversible reduction of PGD2 to 9α,11β-PGF2 (241). Furthermore, AKR1B1 catalyzes the isomerization of PGH2 to PGD2 exclusively in the absence of NADPH as noted previously. However, the biological relevance of this activity is not clear as this isomerization reaction is also catalyzed by two prostaglandin D synthases (272), and the cellular redox status favors the reduction reaction. Both PGF2α and its isomer 9α,11β-PGF2 promote uterine contractions during labor (237).
Six of the 15 human AKRs are involved in sterol/steroid metabolism: AKR1B15, the four members of the AKR1C subfamily (also grouped as hydroxysteroid dehydrogenases; HSDs), and AKR1D1. AKR1B15 catalyzes the 17β-reduction of androgens and estrogens (236). AKR1C1 (20α-HSD) catalyzes the 20α-reduction of progesterone inactivating it (273), whereas AKR1C2 (Type 3 3α-HSD) possesses a 3α-dehydrogenase activity and deactivates 5α-dihydrotesterone (259). AKR1C3 (Type 5 17β-HSD) exhibits 17-ketoreductase activity and produces testosterone and estradiol from their respective precursors.
AKR1D1 and AKR1C4 (Type 1 3α-HSD) catalyze key steps in bile acid biosynthesis. AKR1D1 catalyzes irreversible 5β-reduction of a double bond in 3-ketosterols. This modification changes the geometry of the steroid nucleus from flat to twisted, with a bend in the A/B ring junction. Next, AKR1C4 catalyzes the 3α-reduction of the ketosteroid, resulting in a 3α,5β-configuration, a characteristic feature of bile acids.
Other HSDs Belong to the Short-Chain Dehydrogenase/Reductase (SDR) Family
Some HSDs belong to a different family of enzymes: the short-chain dehydrogenases/reductases (SDR). SDRs exhibit key differences from AKRs in structure, kinetics, catalytic mechanism, and reaction stereochemistry (reviewed in (274)). Of note, HSDs of the SDR family work as either ketosteroid reductases or hydroxysteroid oxidases, depending on their preference for the corresponding forms of NADP(H) or NAD(H). This contrasts with HSDs of the AKR1C family which operate in the reduction direction using (primarily) NADPH (274). Like CYPs and AKRs, a systematic nomenclature has been proposed for SDRs (275). However, the old names (especially for HSDs) remain heavily in use. HSDs of the SDR family have important functions in sterol/steroid metabolism (Table 4).
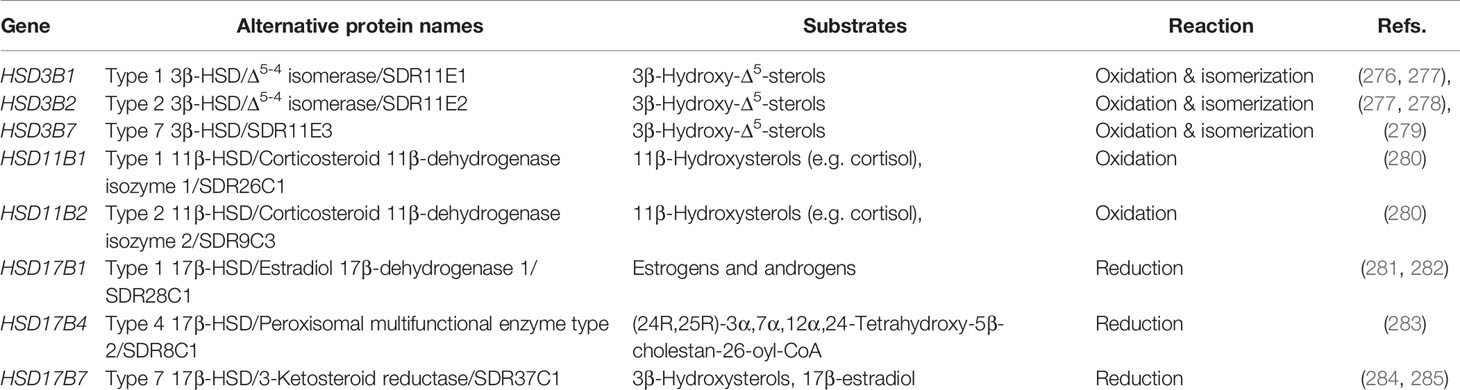
Table 4 Human SDR-HSDs and their substrates/reactions.
Biosynthetic Pathways of Oxidized Lipids
Oxygenated PUFA and Oxidized Phospholipids
Classification of Oxygenated PUFA as Eicosanoids and Docosanoids
The term “oxylipin” refers to oxygenated PUFA derivatives. It encompasses a wide range of oxidized lipids like hydroxy-, epoxy-, oxo-FAs, and endoperoxides. Oxylipins can be classified according to their precursor into eicosanoids (C20) or docosanoids (C22), etc., and this system will be used from here on. Following the classification system of the LIPID MAPS consortium, eicosanoids include prostaglandins, leukotrienes, thromboxanes, lipoxins, hepoxilins, E-resolvins, as well as hydroxy-, hydroperoxy-, epoxy-, and oxo-eicosanoids (286). Likewise, docosanoids include D-resolvins, protectins, maresins, hydroxy-, hydroperoxy-, epoxy-, and oxo-docosanoids. Structures of these lipids are available on the LIPID MAPS database (287).
Here, we focus on the biosynthesis of eicosanoids derived from AA and EPA, and docosanoids derived from DHA, as they are the most studied. Due to the broad substrate specificity of enzymes involved in lipid oxidation, PUFAs with shorter chains or different number of double bonds also undergo similar reactions. Similarly, oxygenation of endocannabinoids occurs on the PUFA moiety and follows the enzymatic mechanisms described here. Further aspects of oxygenated endocannabinoids are discussed in other reviews (216, 288).
Release of PUFA from Membranes for Oxylipin Biosynthesis
Intracellular concentration of free PUFA is tightly regulated through conjugation with coenzyme A (CoA) and subsequent esterification into lysoPL forming PL or shuttling into other pathways (like β-oxidation). PUFAs are abundant in membrane PLs, typically esterified at the sn-2 position. The classical pathway of oxylipin biosynthesis involves the release of PUFA from membrane PL via the action of lipases like phospholipase A2 (PLA2), phospholipase C and diacylglycerol lipase (289, 290). PLA2 enzymes are classified into six types: secreted (sPLA2), cytosolic (cPLA2), calcium-independent (iPLA2), platelet-activating factor acetylhydrolases (PAH-AH), lysosomal (LPLA2), and adipose (AdPLA), and these are further divided into groups and subgroups (291, 292). cPLA2α is localized in the cytosol but translocates to intracellular membranes upon calcium activation, and exhibits selectivity for phospholipids containing AA at the sn-2 position (293, 294). On the other hand, the release of DHA from the membrane can be facilitated by iPLA2β (295, 296). Last, sPLA2 enzymes are involved in the release of AA, EPA and DHA (297, 298). PLA2 enzymes are extensively reviewed by Murakami (299).
Biosynthesis of Eicosanoids
Arachidonic acid is a substrate of eicosanoids produced through the actions of COXs, LOXs, CYPs, and other downstream enzymes (Figure 5A). The most established of these are mono-oxygenation products of AA, DHA, and EPA and classic prostaglandins, leukotrienes, and thromboxane from COXs, LOXs, and CYPs. Most of these were described from the 1950s–90’s in seminal studies, with many conducted at the Karolinska Institute in Stockholm by Sune Bergstrom, Bengt Samuelsson and colleagues. Indeed, the Nobel Prize for Physiology and Medicine was awarded for discovery of prostaglandins and related biologically active substances in 1985 to Bergstrom, Samuelsson and John Vane. More recently, research has focused on characterization of multiply oxygenated PUFAs from AA, EPA, and DHA including several which are proposed to form from transcellular sequential oxygenation by various enzymes. However, for many of these, their specific biosynthetic pathways, enantiomeric composition and biological actions in tissues are not well understood and the levels formed are extremely low in comparison to classic PGs and monohydroxy-oxylipins. Furthermore, their bioactions often require amounts of lipids that are considerably higher than levels detected in cell and tissue samples, and thus could be considered pharmacological.
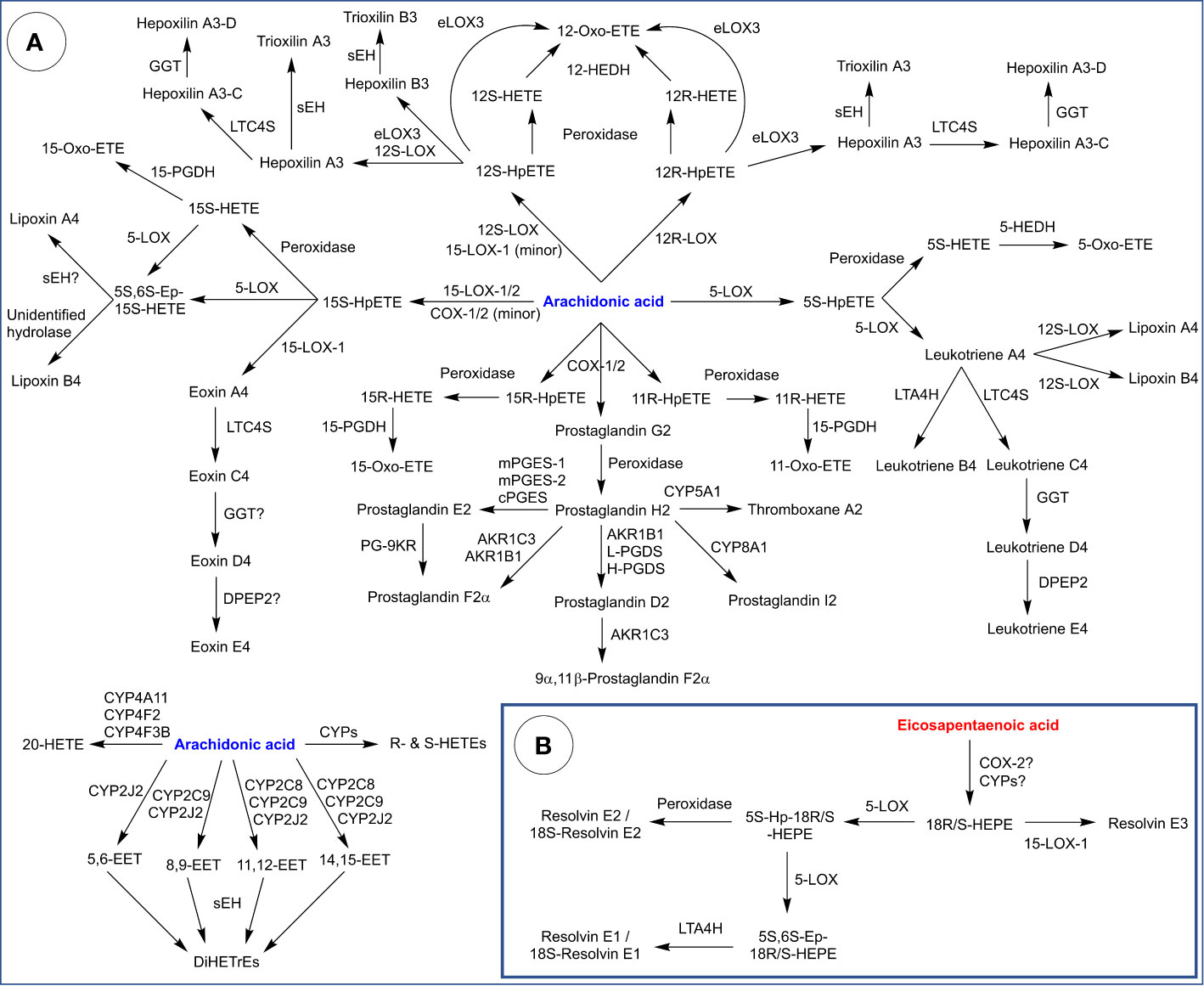
Figure 5 Biosynthesis of eicosanoids derived from: (A) Arachidonic acid and (B) Eicosapentaenoic acid. HETE, hydroxy-eicosatetraenoic acid; Hp, hydroperoxy; Ep, epoxy; LTA4H, leukotriene A4 hydrolase; LTC4S, leukotriene C4 synthase; GGT, γ-glutamyl transpeptidase; DPEP2, dipeptidase 2; sEH, soluble epoxide hydrolase; 12-HEDH, 12-hydroxyeicosanoid dehydrogenase; PGES, prostaglandin E synthase (m: microsomal, c = cytosolic); PGDS (prostaglandin D synthase (L: lipocalin type, H: hematopoietic); 15-PGDH, 15-hydroxyprostaglandin dehydrogenase; PG-9KR, prostaglandin 9-ketoreductase; EET, epoxyeicosatrienoic acid; DiHETrE, dihydroxy-eicosatrienoic acid; HEPE, hydroxy-eicosapenataenoic acid.
COXs convert AA into PGG2 which is reduced into PGH2 (Figure 5A). From there, PGH2 functions as substrate for classic PGs and thromboxane, generated by tissue-specific enzymes. CYP5A1 (thromboxane synthase) is expressed in several cell types and tissues (platelets, macrophages, lung, kidney, liver) and catalyzes the isomerization of PGH2 into thromboxane A2, a potent vasoconstrictor and activator of platelet aggregation (300, 301). CYP5A1 also catalyzes the cleavage PGH2 into malondialdehyde and 12-hydroxyheptadecatrienoic acid. CYP8A1 (prostacyclin synthase) is widely expressed (abundant in ovary, heart, lung, skeletal muscle, and prostate) and catalyzes the isomerization of PGH2 into prostacyclin (PGI2), a vasodilator and inhibitor of platelet aggregation (302).
PGH2 can also be converted into PGD2 through the action of AKR1B1 or PGD synthases, of which there are two isoforms: lipocalin-type PGD synthase (in the central nervous system, male genitalia, heart, cerebrospinal fluid and plasma) and hematopoietic PGD synthase (in antigen-presenting cells, mast cells and megakaryocytes) (266, 303). PGD2 plays roles in the regulation of body temperature, sleep cycle, pain perception and the immune response (304). In the uterus, AKR1C3 catalyzes the reduction of PGD2 into 9α,11β-PGF2, which promotes uterine contractions (237). Similarly, AKR1C3 and AKR1B1 catalyze the reduction of PGH2 into PGF2α, which also promotes uterine contractions (305). Finally, PGH2 can be converted into PGE2 via the action of PGE synthases, of which there are three isoforms: an inducible microsomal isoform (mPGES-1), a constitutive microsomal isoform (mPGES-2) and a constitutive cytosolic isoform (cPGES) (306). PGE2 is abundant in the body and plays a complex role in immunity and inflammation (307). Additionally, PGE2 is reduced into PGF2α by prostaglandin 9-ketoreductase (PG-9KR) (308).
COXs also produce 11R-, 15R-, and 15S-HpETEs as side products, all three of which can be reduced into their corresponding alcohols by peroxidase activity, which are in turn reduced into oxo-ETEs by 15-hydroxyprostaglandin dehydrogenase (15-PGDH). Bioactivities of HETEs and oxo-ETEs are extensively reviewed by Powell and Rokach (309).
5-LOX converts AA into 5S-HpETE (Figure 5A), which is reduced into 5S-HETE through the action of a peroxidase (e.g., GPX) or converted into leukotriene A4 (LTA4) through the leukotriene synthase activity of 5-LOX. Various immune cells (neutrophils, monocytes, platelets) express 5-hydroxyeicosanoid dehydrogenase (5-HEDH) which converts 5S-HETE into 5-oxo-HETE, a potent chemoattractant for eosinophils (310). On the other hand, LTA4 serves as a precursor for LT peptide conjugates (LTC4, D4 and E4) and lipoxins (A and B). LTC4, D4, and E4 are synthesized from LTA4 through LTC4 synthase (a gluthathione-S-transferase), γ-glutamyl transpeptidase (GGT) and dipeptidase 2 (DPEP2). Leukotrienes exhibit pro-inflammatory properties and are implicated in asthma and allergic reactions (311). On the other hand, lipoxins are thought to be synthesized by 12S-LOX through a transcellular pathway which involves interactions between two cell types (312). Lipoxins exhibit anti-inflammatory properties and are proposed to play a role in wound healing and tissue homeostasis (313). Alternatively, LTA4 can be hydrolyzed by leukotriene A4 hydrolase (LTA4H) into LTB4, which attracts neutrophils (314).
15-LOXs converts AA into 15S-HpETE (Figure 5A), which can be reduced into 15S-HETE then oxidized into 15-oxo-ETE. 15S-HpETE and 15S-HETE are also substrates for 5-LOX, which produces a 5S,6S epoxy intermediate that can be hydrolyzed into lipoxins by unidentified hydrolases. Additionally, 15S-HpETE is proposed to be a precursor for eoxin A4 (a 14,15 leukotriene), which is converted into peptide conjugates (eoxin C4, D4, and E4) similar to leukotrienes (315). Conversion of eoxin A4 into C4 is thought to involve LTC4S. Enzymes that catalyze the following two steps leading to eoxin D4 and E4 are unidentified, presumably a GGT isoform and DPEP2. Eoxins are produced in eosinophils, mast cells and airway epithelial cells, and have been shown to possess pro-inflammatory properties (316).
12S-HpETE and 12R-HpETE are produced by 12S-LOX and 12R-LOX, respectively (Figure 5A). These hydroperoxides can be reduced to their corresponding alcohols via a peroxidase, and both alcohols further oxidized into 12-oxo-ETE by 12-hydroxyeicosanoid dehydrogenase (12-HEDH) (309). Both hydroperoxides are also precursors for hepoxilins, which are epoxy-alcohols. eLOX3 in skin converts 12R-HpETE into either hepoxilin A3 or 12-oxo-ETE. On the other hand, 12S-HpETE can be converted into hepoxilin A3 or B3 by either 12S-LOX or eLOX3, but eLOX3 also generates 12-oxo-ETE (70, 317). Hepoxilins A3 and B3 are hydrolyzed by sEH into trioxilins A3 and B3, respectively (318). Hepoxilin A3 can also be conjugated with glutathione via LTC4S producing hepoxilin A3-C, which can be converted into a hepoxilin A3-D by GGT (319). The occurrence of hepoxilin A3-E (cysteinyl conjugate) has also been proposed but not confirmed. Hepoxilin A3 is proposed to regulate mucosal inflammation by recruiting neutrophils across the epithelial junction into the gut lumen (320).
CYPs generate various midchain R- and S-HETEs as well as 20-HETE through hydroxylation of AA (Figure 5A). Alternatively, epoxyeicosatrienoic acids (EET) are produced from AA through epoxidation of its double bonds. Major mammalian CYP epoxygenases include CYP2C8, CYP2C9, and CYP2J2 (15). EETs are further metabolized by sEH (and potentially other epoxide hydrolases) into dihydroxyeicosatrienoic acids (DiHETrE). Note that EPA and other PUFA are also targets of CYP-mediated reactions, which generate the corresponding hydroxy-, epoxy-, and dihydroxy- derivatives.
“E-resolvins”, another class of eicosanoids, are proposed to be generated from EPA (Figure 5B). COX-2 and CYPs have both been proposed to act on EPA to produce 18R-hydroxyeicosapentaenoic acid (18R-HEPE), which is in turn a proposed precursor for lipids termed resolvins E1, E2 and E3 (E-resolvins). It is known that the acetylation of COX-2 shifts its activity into favoring the lipoxygenase-type reaction over the cyclooxygenase activity, and that acetylated COX-2 generates 18R-HEPE from EPA (321). However, acetylated COX-2 is not a physiologically relevant form, and 18R-HEPE could be potentially generated through other means. COX-2 is theoretically capable of generating 18R-HEPE from EPA in a lipoxygenase-type reaction followed by peroxidase activity. However, whether the generation of 18R-HEPE from EPA occurs in vivo and in sufficient amounts without COX-2 acetylation is unknown. Microbial CYPs have also be proposed as sources of 18R-HEPE during infection. E-resolvins are proposed to be produced through a transcellular mechanism involving microbial and mammalian cells (322). Alternatively, mammalian CYPs are also potential sources of 18R-HEPE, although the exact isoforms involved are unknown.
18R-HEPE can be metabolized by 5-LOX into a 5S-hydroperoxy intermediate and further into a 5S,6S-epoxy intermediate (323). The former is proposed to be converted into resolvin E2 by a peroxidase, whereas the latter is proposed to be hydrolyzed into resolvin E1 by LTA4H (324). 18S-analogues of resolvin E1 and E2 have also been described and are proposed to be generated from 18S-HEPE through a similar pathway (Figure 5B), although the enzymatic sources of 18S-HEPE (aside from acetylated COX-2) are also unknown. 18R-HEPE is also proposed to be a precursor to two stereoisomers collectively called resolvin E3 generated by 15-LOX-1 (325). E-resolvins are proposed to exhibit anti-inflammatory effects and promote the resolution of inflammation (325, 326).
Both lipoxins and E-resolvins have been classified as specialized pro-resolving mediators (SPMs), a term which currently describes families of oxygenated PUFA metabolites with proposed roles in the resolution of inflammation and tissue regeneration generated at extremely low levels in biological samples.
Biosynthesis of Docosanoids
Oxygenation of DHA by LOXs and CYPs forms oxygenated docosanoids (hydroxy-, epoxy-, and dihydroxy derivatives). DHA is also the precursor to several classes of multiply oxygenated docosanoids, including additional SPMs which have been named maresins, D-resolvins, and protectins (Figure 6). Maresin biosynthesis has been proposed to start with action of 12S-LOX, generating 14S-hydroperoxy-DHA (14S-HpDoHE) then a 13S,14S-epoxy intermediate. Hydrolysis of this epoxy intermediate by sEH or an unidentified hydrolase is proposed to generate maresin 2 and maresin 1, respectively (93, 327). The biosynthesis of protectin and D-resolvins both begin with 15-LOX-1 which generates a 17S-hydroperoxide (17S-HpDoHE). Further conversion of this hydroperoxide into a 16S,17S-epoxy intermediate by 15-LOX-1 followed by hydrolysis (unidentified hydrolase) is proposed to generate protectin D1 (328). Alternatively, 17S-HpDoHE could be reduced into an alcohol by a peroxidase, which then serves as a precursor for D-resolvins. In this route, 5-LOX is proposed to generate two distinct products hydroperoxides (4S or 7S), which undergo further transformations leading to several D-resolvins (Figure 6). Additionally, epoxide intermediates in the previously described pathways are proposed to undergo glutathione conjugation by LTC4S, generating peptide conjugates of maresin, protectin and D-resolvin named MCTR1, PCTR1, and RCTR1, respectively (CTR = conjugate in tissue regeneration). These conjugates are proposed to be metabolized by GGT and DPEP2, similar to leukotrienes (329).
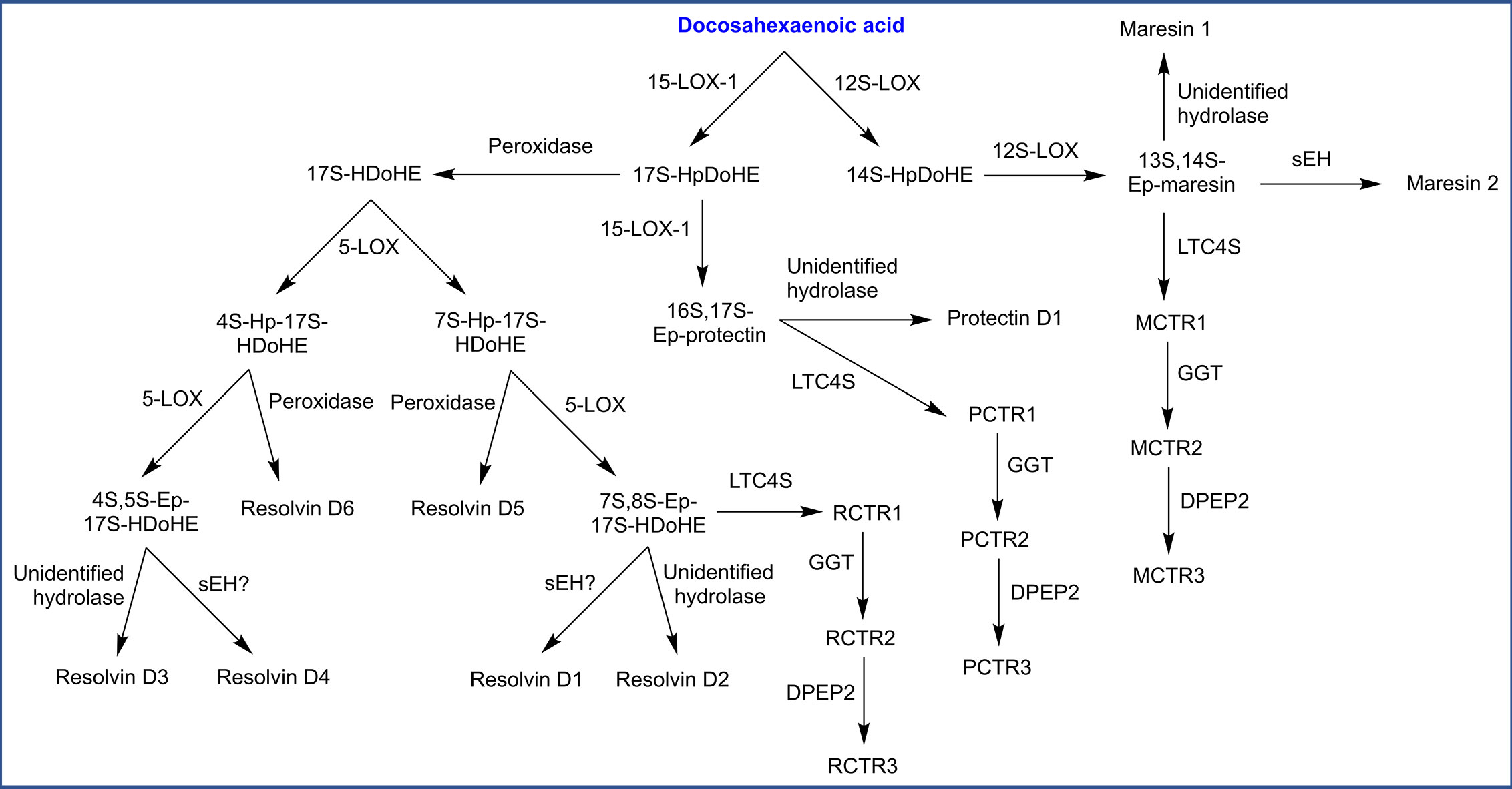
Figure 6 Biosynthesis of specialized pro-resolving mediators (SPMs) derived from docosahexaenoic acid. HDoHE, hydroxy-docosahexaenoic acid; Hp, hydroperoxy; Ep, epoxy; CTR, conjugate in tissue regeneration (M: maresin, P: protectin, R: resolvin); sEH, soluble epoxide hydrolase; LTC4S, leukotriene C4 synthase; GGT, γ-glutamyl transpeptidase; DPEP2, dipeptidase 2.
Oxygenation of Esterified PUFA and Biosynthesis of Oxidized Phospholipids
The classical pathway for the biosynthesis of oxidized phospholipids starts with the release of PUFA from membranes through the action of PLA2. PUFA are then oxygenated by the enzymes discussed earlier. This is followed by acylation of oxygenated PUFA with CoA and esterification into a lysoPL via the action of an sn-2 acyltransferase.
The generation of PL-esterified eicosanoids is well-documented in mammalian cells such as epithelial, endothelial and immune cells (330). HETE-PLs are acutely generated by activated neutrophils, platelets and monocytes (331–333). Similarly, PGD2 and PGE2 generated from COX-1-derived PGH2 in activated platelets are rapidly incorporated into PE-lysoPLs (334). Also, EET-PLs have been detected in rat liver (335). For most of these, the mechanism of formation requires endogenous generation of an oxylipin, which is then rapidly esterified into membrane PL pools (e.g., on a timescale of minutes) via Lands cycle enzymes (336).
15-LOXs also contribute to the formation of oxPL by two other pathways. The first involves direct oxygenation of membrane PLs (331, 337). In the second pathway, 15-LOXs oxygenate the PUFA moiety of CEs, then hydrolysis of oxygenated PUFA from CE liberates oxygenated PUFA which can be esterified to lysoPL (87).
Recent studies found that COX-2, 15-LOX-2, and platelet 12S-LOX can catalyze the oxygenation of 2-AA-lysoPL released by iPLA2γ in response to calcium ionophore stimulation (88, 94). These reactions generate 2-eicosanoid-lysoPLs which are proposed to be a source of free eicosanoids as well as acting as signaling mediators themselves. 2-eicosanoid-lysoPLs can also be converted into oxPLs through the action of sn-1 acyltransferase (338). Cytochrome c has been recently been identified as a plasmalogenase, and is proposed to be a source of 2-eicosanoid-lysoPLs under conditions of oxidative stress (339). These findings describe novel pathways for the biosynthesis of oxPLs (Figure 7). Further research is required to assess the contribution of these pathways to the formation of oxPLs and other mediators in vivo.
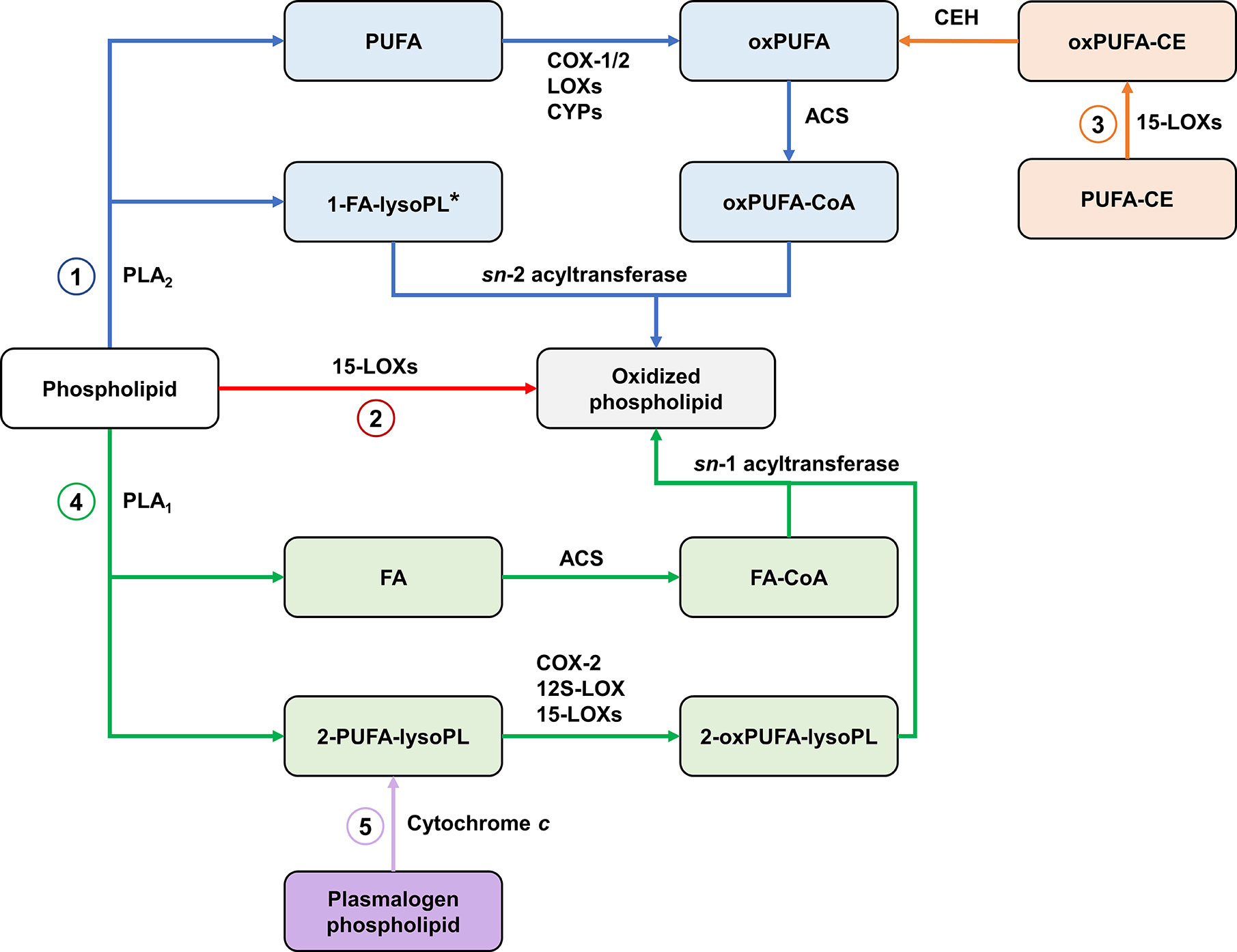
Figure 7 Biosynthetic pathways of oxidized phospholipids. (1) The classical pathway involves the action of PLA2 on membrane phospholipids, releasing sn-2 PUFA which are oxygenated by cyclooxygenases (COXs), lipoxygenases (LOXs), and cytochrome P450s (CYPs) then re-esterified. *Oxygenated PUFA can be also be esterified with plasmalogen lysophospholipids (2) Direct oxygenation of membrane phospholipids by 15-LOXs. (3) 15-LOX-mediated oxygenation of PUFA in cholesteryl esters followed by hydrolysis of oxygenated PUFA provides substrates for the classical pathway. (4) An alternative pathway involves the action of PLA1, forming 2-PUFA-lysophospholipids which are oxygenated by COX-2, 12S-LOX, and 15-LOXs then re-esterified with FA. (5) Cytochrome c releases 2-PUFA-lysophospholipids by cleaving the vinyl ether bond in plasmalogen phospholipids, providing substrates for the alternative pathway. PLA, Phospholipase A; oxPUFA, oxygenated PUFA; ACS, acyl-CoA synthase; CE, cholesteryl ester; CEH, neutral cholesterol ester hydrolase; FA, fatty acid/acyl; lysoPL, lysophospholipid.
Biosynthesis of Oxysterols, Bile Acids, and Steroid Hormones
Oxysterols
Oxysterols are formed in the first steps of cholesterol metabolism: they are oxidized forms of cholesterol and also of its precursors (340). 7α-Hydroxycholesterol (7α-HC) is formed from cholesterol by CYP7A1 and represents the first metabolite in the neutral pathway of bile acid biosynthesis (Figure 8) (219, 220). (25R)26-Hydroxycholesterol (26-HC), more commonly called 27-hydroxycholesterol, and 3β-hydroxycholest-5-en-(25R)26-oic acid (3β-HCA) are both formed from cholesterol by CYP27A1 and are the first members of the acidic or alternative pathway of bile acid biosynthesis (219–221). While CYP7A1 is an endoplasmic reticulum and liver specific protein, CYP27A1 is mitochondrial and expressed in many tissues. CYP46A1 is almost exclusively expressed in neurons, its function is to maintain cholesterol balance in the brain, converting cholesterol from a molecule unable to pass the blood brain barrier to 24S-hydroxycholesterol (24S-HC), a more polar molecule which can cross the barrier (222, 223, 341). CYP11A1, like CYP27A1, is a mitochondrial inner membrane protein. It is highly expressed in steroidogenic tissue and will oxidize cholesterol to pregnenolone in a three step process involving 22R-hydroxycholesterol (22R-HC) and 20R,22R-dihydroxycholesterol as intermediates (20R,22R-diHC) (342, 343) (Figure 9). The pathways of bile acid biosynthesis are discussed in more detail below.
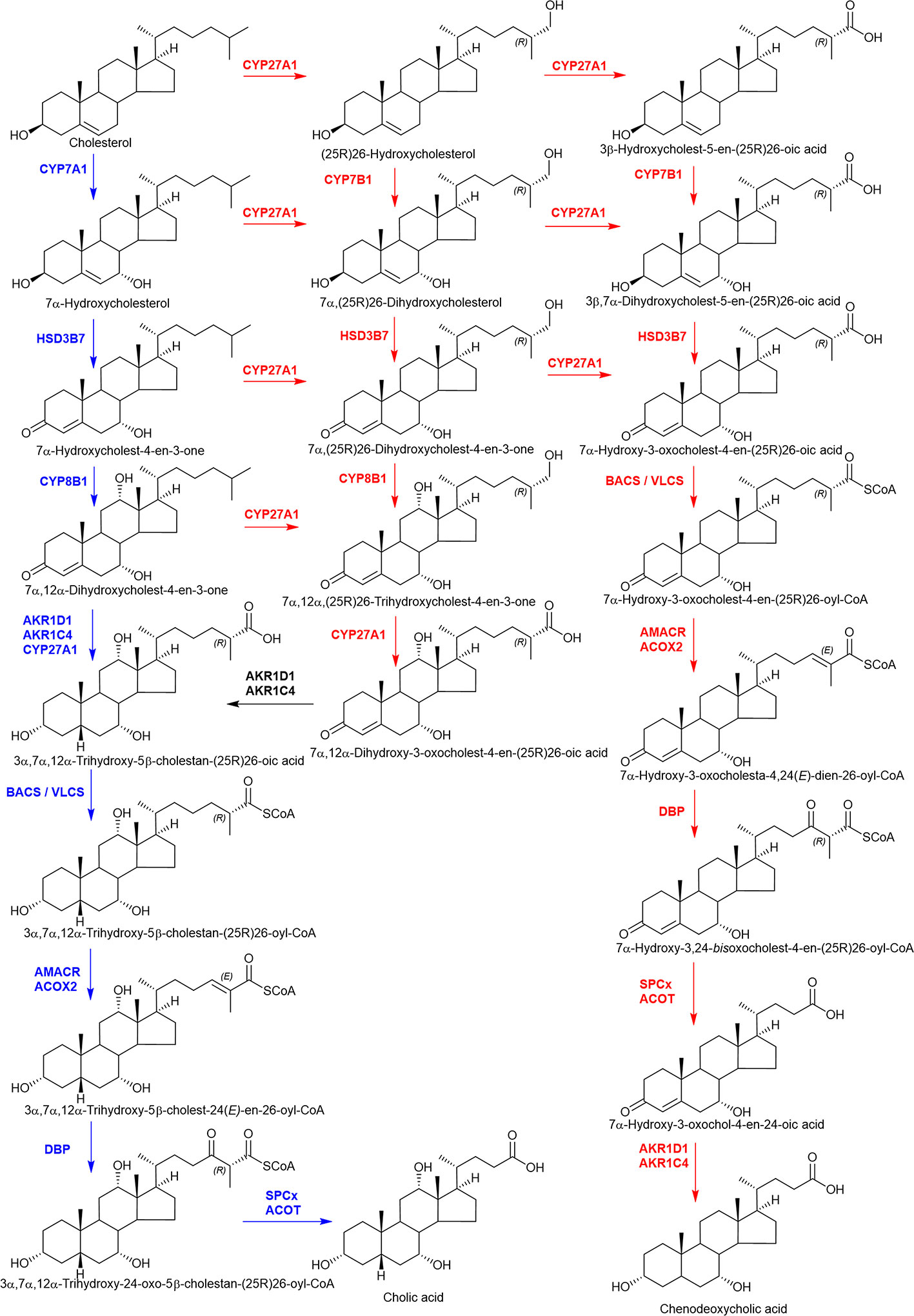
Figure 8 A simplified view of the major bile acid biosynthesis pathways. The “neutral” pathway (highlighted in blue) starts with 7α-hydroxylation of cholesterol by CYP7A1, the “acidic” pathway with (25R)26-hydroxylation then (25R)26-carboxylation of cholesterol by CYP27A1. In the “acidic” pathway (highlighted in red) CYP7B1 is the 7α-hydroxylase.
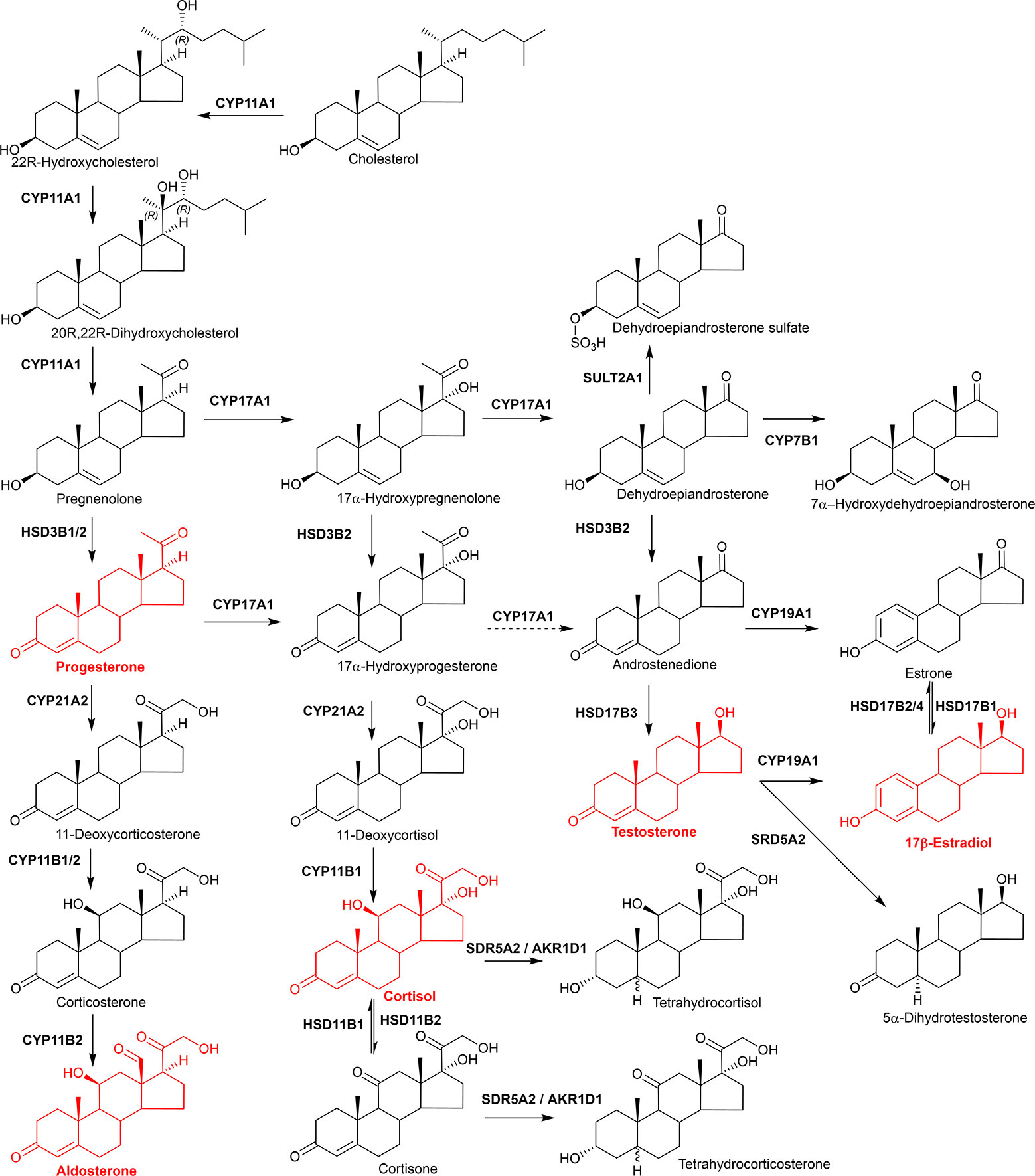
Figure 9 Simplified view of steroid hormone biosynthesis. Highlighted in red are the classical steroid hormones, progesterone, aldosterone, cortisol, testosterone, and 17β-estradiol.
While most “primary” oxysterols are formed from cholesterol in CYP catalyzed reactions, cholesterol 25-hydroxylase (CH25H), the dominating enzyme that generates 25-hydroxycholesterol (25-HC) is an exception, in that is not a CYP, but a member of a family of enzymes that utilize di-iron cofactors to catalyze the hydroxylation (344). CH25H is expressed in activated immune cells and 25-HC has both anti-bacterial and anti-viral activities (345–348). 25-HC is metabolized by CYP7B1 to 7α,25-diHC, a ligand to the GPCR Epstein Barr virus induced gene 2 (EBI2 or GPR183). 7α,25-diHC acts as a chemoattractant to GPR183 expressing immune cells (349, 350). An alternative route to 7α,25-diHC production is through CYP3A4 oxidation of 7α-HC (190), while the same enzyme has also been reported to act as a second cholesterol 25-hydroxylase and also a 4β-hydroxylase of cholesterol (189, 351).
Oxysterols can also be formed from cholesterol precursors in reactions catalyzed by CYP enzymes (202, 352). These reactions may be important in patients suffering from inborn errors of cholesterol biosynthesis such as Smith-Lemli-Opitz syndrome (SLOS, 7-dehydrocholesterol reductase deficiency) and desmosterolosis (3β-hydroxysterol-Δ24-reductase deficiency) or where there is very high expression of sterol hydroxylases, e.g., CYP7A1 in cerebrotendinous xanthomatosis, where CYP27A1 is deficient.
Bile Acid Biosynthesis
There are two quantitatively major and at least five minor pathways of bile acid biosynthesis and all pathways involve multiple oxidation reactions (219, 220, 353–358). Besides CYP enzymes, key oxidation reactions are carried out by hydroxysteroid dehydrogenase (HSD) members of the short chain dehydrogenase/reductase (SDR) family and reductions by aldo-keto reductases (AKR) enzymes. One of the minor pathways that results in the formation of 3β,5α,6β-trihydroxycholan-24-oic acid involves of cholestane-3β,5α,6β-triol which is likely formed via the cholesterol peroxidation product 5,6-epoxycholesterol (219, 354).
While the acidic pathway may be most important in infants (359), at later stages of life the neutral pathway is dominant. This is initiated by CYP7A1 oxidation of cholesterol to generate 7α-HC in the liver. Further oxidation may occur at C-12 by CYP8B1 to generate 7α,12α-dihydroxycholesterol (7α,12α-diHC), which may be preceded or succeeded by oxidation and isomerization of the 3β-hydroxy-5-ene structure to a 3-oxo-4-ene by HSD3B7, giving 7α-hydroxycholest-4-en-3-one (7α-HCO) and 7α,12α-dihydroxycholest-4-en-3-one (7α,12α-diHCO), respectively (Figure 8). A general feature of bile acid biosynthesis is that many of the enzymes involved in the pathways accept multiple substrates resulting in variations in the order of reactions depending on the tissue in which they proceed (360). The next steps involve A-ring reductions which may be succeeded or preceded by (25R)26-hydroxylation and (25R)26-carboxylation to ultimately give 3α,7α,12α-trihydroxy-5β-cholanestan-(25R)26-oic acid. The A-ring reductions are carried out by AKR1D1 and AKR1C4, while CYP27A1 carries out the (25R)26-oxidations. Side-chain shortening of the cholestanoic acid proceeds in the peroxisome through the CoA-thioester formed by bile acid Co-A synthetase (BACS, SLC27A5) or very long chain acyl-CoA synthetase (VLCS, SLC27A2). Following C-25 racemization by α-methylacyl-CoA racemase (AMACR) the next oxidation involves the introduction of Δ24 double bond by the enzyme acyl-CoA oxidase 2 (ACOX2). The Δ24 double bond is then hydrated by D-bifunctional protein (DBP), which then oxidizes the C-24 hydroxy to a C-24 ketone via HSD17B4 activity. The resulting product 3α,7α,12α-trihydroxy-24-oxo-5β-cholestan-(25R)26-oyl-CoA is then oxidized by the enzyme peroxisomal thiolase 2 (SPCx) to the thioester of cholic acid ready for conjugation with glycine, taurine, or hydrolysis to the free acid by peroxisomal acyl-CoA thioesterase (ACOT) (219, 220).
The acidic pathway starts with (25R)26-hydroxylation and (25R)26-carboxylation of cholesterol by CYP27A1 and this and many other steps may proceed extrahepatically (Figure 8). Most of the enzymes involved in the neutral pathway are also involved in the acidic pathway although not in the same order (361). An exception is CYP7A1, which is replaced by CYP7B1 as the 7α-hydroxylase in the acidic pathway. CYP7B1 is expressed in many tissues, not just liver (362, 363), and unlike CYP7A1 uses side-chain oxysterols as its substrate. The acidic pathway mostly generates chenodeoxycholic acid rather than cholic acid, so CYP8B1 has minor involvement. The order of A-ring reduction and side-chain cleavage can be reversed in the acidic pathway with the formation of bile acid intermediates possessing 3-oxo-4-ene or 3β-hydroxy-5-ene functions.
Steroid Hormone Biosynthesis
The classical steroid hormones aldosterone (mineralocorticoid), cortisol (glucocorticoid), testosterone (male sex hormone) and 17β-estradiol (female sex hormone) are all formed from cholesterol through multiple oxidation reactions (Figure 9) (364, 365). Pregnenolone formed via CYP11A1 oxidation of cholesterol represents an intermediate between oxysterol and steroid hormone biosynthesis. It is oxidized by HSD3B2 to progesterone on the pathway to aldosterone or by CYP17A1 to 17α-hydroxypregnenolone on the route to cortisol. Both these pathways use CYP21A2 as a C-21 hydroxylase and CYP11B enzymes as the 11β-hydroxylase. Note, HSD3B1 is the enzyme which generates progesterone from pregnenolone in the placenta, when progesterone acts as the hormone of pregnancy. Further oxidation of 17α-hydroxypregnenolone by CYP17A1 leads to dehydroepiandrosterone on the road to testosterone and17β-estradiol. In this pathway additional oxidations and reductions are carried out by HSD3B2 and HSD17B3, respectively, while CYP19A1 is required to generate estrogens. Besides being synthesized in the adrenal gland and sex organs it is noteworthy that steroids can be synthesized in the brain and are then named neurosteroids (366, 367). A more general term for brain steroids which may be synthesized in the brain or imported from the periphery and exert rapid non-genomic effects is neuroactive steroids. It is beyond the scope of this review to discuss steroid hormone biosynthesis and metabolism in greater detail and the reader is directed to the excellent reviews of Shackleton and colleagues (364, 368–370).
Concluding Remarks
Enzymatically oxidized lipids are derivatives of PUFA or cholesterol with critical functions in cellular and physiological processes as signaling mediators and hormones. Their biosynthesis is highly regulated and carried out by enzymes that include LOXs, COXs, CYPs, and AKRs. Advances in our understanding of these enzymes have led to the discovery of novel lipid mediators and their biosynthetic routes. Many functional aspects of these enzymes and their products remain unclear, requiring further investigation. These include elucidating the function of 15-LOX-2 (ALOX15B) in macrophages, the regulatory mechanisms of 12-LOXs and eLOX3, the mechanistic details of transcellular biosynthesis, the origin of 18-HEPE required for E-resolvin biosynthesis, the biological functions of orphan CYPs, and the bioactivities/functions of oxygenated endocannabinoids. The relative physiological importance of some multiply oxygenated PUFA mediators, which are generated in extremely low amounts, also needs to be further clarified. This is also true for multiply oxygenated derivatives of cholesterol.
Research on oxygenated PUFA and oxysterols has been carried out largely in parallel, and both areas have benefited greatly from advances in analytical methods. However, there are still major questions to be answered in terms of their proposed roles in human disease including atherosclerosis and neurodegeneration. Here, we presented biosynthetic pathways of oxygenated PUFA and oxysterols, highlighting their (known) functions to show the diversity of products but also to draw connections between the two groups. An integrated approach encompassing the analysis of both lipid groups could be useful in examining the etiology of disease. Despite their involvement in the progression of disease such as atherosclerosis and neurodegenerative disease, the two lipid groups are rarely analyzed together. We propose that oxygenated PUFA and oxysterols are more connected than previously thought, especially as 15-LOX is being increasingly recognized as a regulator of cholesterol metabolism. The immune and nervous systems are both of particular interest as major sites of cholesterol metabolism and 15-LOX expression. Further work on the enzymology of 15-LOX with cholesteryl substrates could lead to the discovery of novel oxidized lipids. For example, 15-LOX oxygenation of PUFA esters of oxysterols could generate novel lipids that link both classes directly in cell types that possess the enzymatic machinery for oxysterol and oxylipin biosyntheses (e.g., macrophages). An alternative biosynthetic route could be through the esterification of oxylipins with free oxysterols. That said, the detection of these proposed molecules could be challenging due to low abundance and sensitivity to alkaline conditions commonly used in analysis. Finally, the substrate promiscuity of many of the enzymes involved in production of oxidized lipids provides a technical challenge for dissection of (patho)physiological function of specific oxidized lipids. Clearly, there is plenty of scope for ongoing exploration of the molecular, cellular, and physiological functions of oxidized lipids.
Author Contributions
AH planned the manuscript. AH, WG, and YW wrote the manuscript with input from AF and VO. All authors contributed to the article and approved the submitted version.
Acknowledgments
AAH acknowledges funding from Kuwait University and support from the Kuwait Cultural Office (KCO) in London. Work in Swansea was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC, grant numbers BB/N015932/1 to WJG and BB/L001942/1 to YW). AJF is supported by funding from Barts Charity. VBO is a Royal Society Wolfson Merit Award Holder and acknowledges funding from the Wellcome Trust for LIPID MAPS (203014/Z/16/Z).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
4-HNE, 4-hydroxy-2-nonenal; AA, arachidonic acid; AKR, aldo-keto reductase; ALA, α-linolenic acid; COX, cyclooxygenase; CE, cholesteryl ester; CYP, cytochrome P450; DGLA, dihomo-γ-linolenic acid; DHA, docosahexaenoic acid; DPEP2, dipeptidase 2; EET, epoxy-eicosatrienoic acid; EPA, eicosapentaenoic acid; GGT, γ-glutamyl transpeptidase; GLA, γ-linolenic acid; GPCR, G protein-coupled receptor; HETE, hydroxy-eicosatetraenoic acid; HpETE, hydroperoxy-eicosatetraenoic acid; HSD, hydroxysteroid dehydrogenase; LA, linoleic acid; LOX, lipoxygenase; LTA4, leukotriene A4; lysoPL, lysophospholipid; oxPL, oxidized phospholipids; PG, prostaglandin; PL, phospholipid; PUFA, polyunsaturated fatty acid; SDR, short-chain dehydrogenase/reductase; SPM, specialized pro-resolving mediator.
References
1. Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest (2001) 108(1):15–23. doi: 10.1172/JCI13416
2. Slatter DA, Percy CL, Allen-Redpath K, Gajsiewicz JM, Brooks NJ, Clayton A, et al. Enzymatically oxidized phospholipids restore thrombin generation in coagulation factor deficiencies. JCI Insight (2018) 3(6):e98459. doi: 10.1172/jci.insight.98459
3. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 40(3):315–27. doi: 10.1016/j.immuni.2014.02.009
4. Choi SH, Sviridov D, Miller YI. Oxidized cholesteryl esters and inflammation. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862(4):393–7. doi: 10.1016/j.bbalip.2016.06.020
5. Lauder SN, Allen-Redpath K, Slatter DA, Aldrovandi M, O’Connor A, Farewell D, et al. Networks of enzymatically oxidized membrane lipids support calcium-dependent coagulation factor binding to maintain hemostasis. Sci Signal (2017) 10(507):eaan2787. doi: 10.1126/scisignal.aan2787
6. Stachenfeld NS. Sex hormone effects on body fluid regulation. Exerc Sport Sci Rev (2008) 36(3):152–9. doi: 10.1097/JES.0b013e31817be928
7. Kuo T, McQueen A, Chen TC, Wang JC. Regulation of Glucose Homeostasis by Glucocorticoids. Adv Exp Med Biol (2015) 872:99–126. doi: 10.1007/978-1-4939-2895-8_5
8. Bereshchenko O, Bruscoli S, Riccardi C. Glucocorticoids, Sex Hormones, and Immunity. Front Immunol (2018) 9:1332(1332):1332. doi: 10.3389/fimmu.2018.01332
9. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev (2009) 89(1):147–91. doi: 10.1152/physrev.00010.2008
10. Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology (2012) 55(1):267–76. doi: 10.1002/hep.24681
11. Griffiths WJ, Wang Y. Oxysterol research: a brief review. Biochem Soc Trans (2019) 47(2):517–26. doi: 10.1042/BST20180135
12. Garavito RM, DeWitt DL. The cyclooxygenase isoforms: structural insights into the conversion of arachidonic acid to prostaglandins. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (1999) 1441(2-3):278–87. doi: 10.1016/s1388-1981(99)00147-x
13. Vecchio AJ, Orlando BJ, Nandagiri R, Malkowski MG. Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J Biol Chem (2012) 287(29):24619–30. doi: 10.1074/jbc.M112.372243
14. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta (2015) 1851(4):308–30. doi: 10.1016/j.bbalip.2014.10.002
15. Spector AA, Kim HY. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim Biophys Acta (2015) 1851(4):356–65. doi: 10.1016/j.bbalip.2014.07.020
16. Mikulska-Ruminska K, Shrivastava I, Krieger J, Zhang S, Li H, Bayir H, et al. Characterization of Differential Dynamics, Specificity, and Allostery of Lipoxygenase Family Members. J Chem Inf Model (2019) 59(5):2496–508. doi: 10.1021/acs.jcim.9b00006
17. Pandak WM, Kakiyama G. The acidic pathway of bile acid synthesis: Not just an alternative pathway(). Liver Res (2019) 3(2):88–98. doi: 10.1016/j.livres.2019.05.001
18. Kutzner L, Goloshchapova K, Heydeck D, Stehling S, Kuhn H, Schebb NH. Mammalian ALOX15 orthologs exhsibit pronounced dual positional specificity with docosahexaenoic acid. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862(7):666–75. doi: 10.1016/j.bbalip.2017.04.001
19. Simard-Bisson C, Parent LA, Moulin VJ, Fruteau de Laclos B. Characterization of Epidermal Lipoxygenase Expression in Normal Human Skin and Tissue-Engineered Skin Substitutes. J Histochem Cytochem (2018) 66(11):813–24. doi: 10.1369/0022155418788117
20. Funk CD, Funk LB, FitzGerald GA, Samuelsson B. Characterization of human 12-lipoxygenase genes. Proc Natl Acad Sci U S A (1992) 89(9):3962–6. doi: 10.1073/pnas.89.9.3962
21. Chen XS, Brash AR, Funk CD. Purification and characterization of recombinant histidine-tagged human platelet 12-lipoxygenase expressed in a baculovirus/insect cell system. Eur J Biochem (1993) 214(3):845–52. doi: 10.1111/j.1432-1033.1993.tb17988.x
22. Weisinger G, Grafi-Cohen M, Hirsh M, Knoll E, Sharon O, Many A, et al. 12S-Lipoxygenase is necessary for human vascular smooth muscle cell survival. Exp Cell Res (2013) 319(10):1586–93. doi: 10.1016/j.yexcr.2013.04.001
23. Boeglin WE, Kim RB, Brash AR. A 12R-lipoxygenase in human skin: mechanistic evidence, molecular cloning, and expression. Proc Natl Acad Sci U.S.A. (1998) 95(12):6744–9. doi: 10.1073/pnas.95.12.6744
24. Sun D, McDonnell M, Chen XS, Lakkis MM, Li H, Isaacs SN, et al. Human 12(R)-lipoxygenase and the mouse ortholog. Molecular cloning, expression, and gene chromosomal assignment. J Biol Chem (1998) 273(50):33540–7. doi: 10.1074/jbc.273.50.33540
25. Zheng Y, Yin H, Boeglin WE, Elias PM, Crumrine D, Beier DR, et al. Lipoxygenases mediate the effect of essential fatty acid in skin barrier formation: a proposed role in releasing omega-hydroxyceramide for construction of the corneocyte lipid envelope. J Biol Chem (2011) 286(27):24046–56. doi: 10.1074/jbc.M111.251496
26. Schneider C, Keeney DS, Boeglin WE, Brash AR. Detection and cellular localization of 12R-lipoxygenase in human tonsils. Arch Biochem Biophys (2001) 386(2):268–74. doi: 10.1006/abbi.2000.2217
27. Garcia-Verdugo I, BenMohamed F, Tattermusch S, Leduc D, Charpigny G, Chignard M, et al. A role for 12R-lipoxygenase in MUC5AC expression by respiratory epithelial cells. Eur Respir J (2012) 40(3):714–23. doi: 10.1183/09031936.00023111
28. Wuest SJ, Crucet M, Gemperle C, Loretz C, Hersberger M. Expression and regulation of 12/15-lipoxygenases in human primary macrophages. Atherosclerosis (2012) 225(1):121–7. doi: 10.1016/j.atherosclerosis.2012.07.022
29. Turk J, Maas RL, Brash AR, Roberts LJ,2, Oates JA. Arachidonic acid 15-lipoxygenase products from human eosinophils. J Biol Chem (1982) 257(12):7068–76.
30. Sigal E, Grunberger D, Highland E, Gross C, Dixon RA, Craik CS. Expression of cloned human reticulocyte 15-lipoxygenase and immunological evidence that 15-lipoxygenases of different cell types are related. J Biol Chem (1990) 265(9):5113–20.
31. Conrad DJ, Kuhn H, Mulkins M, Highland E, Sigal E. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci U S A (1992) 89(1):217–21. doi: 10.1073/pnas.89.1.217
32. Spanbroek R, Hildner M, Kohler A, Muller A, Zintl F, Kuhn H, et al. IL-4 determines eicosanoid formation in dendritic cells by down-regulation of 5-lipoxygenase and up-regulation of 15-lipoxygenase 1 expression. Proc Natl Acad Sci U.S.A. (2001) 98(9):5152–7. doi: 10.1073/pnas.091076998
33. Brash AR, Boeglin WE, Chang MS. Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci U S A (1997) 94(12):6148–52. doi: 10.1073/pnas.94.12.6148
34. Matsumoto T, Funk CD, Radmark O, Hoog JO, Jornvall H, Samuelsson B. Molecular cloning and amino acid sequence of human 5-lipoxygenase. Proc Natl Acad Sci U S A (1988) 85(1):26–30. doi: 10.1073/pnas.85.1.26
35. Colamorea T, Di Paola R, Macchia F, Guerrese MC, Tursi A, Butterfield JH, et al. 5-Lipoxygenase upregulation by dexamethasone in human mast cells. Biochem Biophys Res Commun (1999) 265(3):617–24. doi: 10.1006/bbrc.1999.1732
36. Spanbroek R, Hildner M, Steinhilber D, Fusenig N, Yoneda K, Radmark O, et al. 5-lipoxygenase expression in dendritic cells generated from CD34(+) hematopoietic progenitors and in lymphoid organs. Blood (2000) 96(12):3857–65. doi: 10.1182/blood.V96.12.3857
37. Krieg P, Marks F, Furstenberger G. A gene cluster encoding human epidermis-type lipoxygenases at chromosome 17p13.1: cloning, physical mapping, and expression. Genomics (2001) 73(3):323–30. doi: 10.1006/geno.2001.6519
38. Zheng Y, Brash AR. Dioxygenase activity of epidermal lipoxygenase-3 unveiled: typical and atypical features of its catalytic activity with natural and synthetic polyunsaturated fatty acids. J Biol Chem (2010) 285(51):39866–75. doi: 10.1074/jbc.M110.155374
39. UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res (2019) 47(D1):D506–D15. doi: 10.1093/nar/gky1049
40. Choi J, Chon JK, Kim S, Shin W. Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins (2008) 70(3):1023–32. doi: 10.1002/prot.21590
41. Gilbert NC, Bartlett SG, Waight MT, Neau DB, Boeglin WE, Brash AR, et al. The structure of human 5-lipoxygenase. Science (2011) 331(6014):217–9. doi: 10.1126/science.1197203
42. Kobe MJ, Neau DB, Mitchell CE, Bartlett SG, Newcomer ME. The structure of human 15-lipoxygenase-2 with a substrate mimic. J Biol Chem (2014) 289(12):8562–9. doi: 10.1074/jbc.M113.543777
43. Walther M, Anton M, Wiedmann M, Fletterick R, Kuhn H. The N-terminal domain of the reticulocyte-type 15-lipoxygenase is not essential for enzymatic activity but contains determinants for membrane binding. J Biol Chem (2002) 277(30):27360–6. doi: 10.1074/jbc.M203234200
44. Walther M, Hofheinz K, Vogel R, Roffeis J, Kuhn H. The N-terminal beta-barrel domain of mammalian lipoxygenases including mouse 5-lipoxygenase is not essential for catalytic activity and membrane binding but exhibits regulatory functions. Arch Biochem Biophys (2011) 516(1):1–9. doi: 10.1016/j.abb.2011.09.004
45. Hammarberg T, Provost P, Persson B, Radmark O. The N-terminal domain of 5-lipoxygenase binds calcium and mediates calcium stimulation of enzyme activity. J Biol Chem (2000) 275(49):38787–93. doi: 10.1074/jbc.M006136200
46. Chen XS, Funk CD. The N-terminal “beta-barrel” domain of 5-lipoxygenase is essential for nuclear membrane translocation. J Biol Chem (2001) 276(1):811–8. doi: 10.1074/jbc.M008203200
47. Brinckmann R, Schnurr K, Heydeck D, Rosenbach T, Kolde G, K̈hn H. Membrane Translocation of 15-Lipoxygenase in Hematopoietic Cells Is Calcium-Dependent and Activates the Oxygenase Activity of the Enzyme. Blood (1998) 91(1):64–74. doi: 10.1182/blood.V91.1.64
48. Bender G, Schexnaydre EE, Murphy RC, Uhlson C, Newcomer ME. Membrane-dependent Activities of Human 15-LOX-2 and Its Murine Counterpart: IMPLICATIONS FOR MURINE MODELS OF ATHEROSCLEROSIS. J Biol Chem (2016) 291(37):19413–24. doi: 10.1074/jbc.M116.741454
49. Basavarajappa D, Wan M, Lukic A, Steinhilber D, Samuelsson B, Radmark O. Roles of coactosin-like protein (CLP) and 5-lipoxygenase-activating protein (FLAP) in cellular leukotriene biosynthesis. Proc Natl Acad Sci U S A (2014) 111(31):11371–6. doi: 10.1073/pnas.1410983111
50. Hafner AK, Gerstmeier J, Hornig M, George S, Ball AK, Schroder M, et al. Characterization of the interaction of human 5-lipoxygenase with its activating protein FLAP. Biochim Biophys Acta (2015) 1851(11):1465–72. doi: 10.1016/j.bbalip.2015.08.010
51. Luo M, Jones SM, Peters-Golden M, Brock TG. Nuclear localization of 5-lipoxygenase as a determinant of leukotriene B4 synthetic capacity. Proc Natl Acad Sci U S A (2003) 100(21):12165–70. doi: 10.1073/pnas.2133253100
52. Sun QY, Zhou HH, Mao XY. Emerging Roles of 5-Lipoxygenase Phosphorylation in Inflammation and Cell Death. Oxid Med Cell Longev (2019) 2019:2749173. doi: 10.1155/2019/2749173
53. Smyrniotis CJ, Barbour SR, Xia Z, Hixon MS, Holman TR. ATP allosterically activates the human 5-lipoxygenase molecular mechanism of arachidonic acid and 5(S)-hydroperoxy-6(E),8(Z),11(Z),14(Z)-eicosatetraenoic acid. Biochemistry (2014) 53(27):4407–19. doi: 10.1021/bi401621d
54. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell (2017) 171(3):628–41 e26. doi: 10.1016/j.cell.2017.09.044
55. Snodgrass RG, Brune B. Regulation and Functions of 15-Lipoxygenases in Human Macrophages. Front Pharmacol (2019) 10:719:719. doi: 10.3389/fphar.2019.00719
56. Droege KD, Keithly ME, Sanders CR, Armstrong RN, Thompson MK. Structural Dynamics of 15-Lipoxygenase-2 via Hydrogen-Deuterium Exchange. Biochemistry (2017) 56(38):5065–74. doi: 10.1021/acs.biochem.7b00559
57. Hagmann W, Kagawa D, Renaud C, Honn KV. Activity and protein distribution of 12-lipoxygenase in HEL cells: Induction of membrane-association by phorbol ester TPA, modulation of activity by glutathione and 13-HPODE, and Ca2+-dependent translocation to membranes. Prostaglandins (1993) 46(6):471–7. doi: 10.1016/0090-6980(93)90066-g
58. Siebert M, Krieg P, Lehmann WD, Marks F, Furstenberger G. Enzymic characterization of epidermis-derived 12-lipoxygenase isoenzymes. Biochem J (2001) 355(Pt 1):97–104. doi: 10.1042/0264-6021:3550097
59. Aleem AM, Jankun J, Dignam JD, Walther M, Kuhn H, Svergun DI, et al. Human platelet 12-lipoxygenase, new findings about its activity, membrane binding and low-resolution structure. J Mol Biol (2008) 376(1):193–209. doi: 10.1016/j.jmb.2007.11.086
60. Hafner AK, Cernescu M, Hofmann B, Ermisch M, Hornig M, Metzner J, et al. Dimerization of human 5-lipoxygenase. Biol Chem (2011) 392(12):1097–111. doi: 10.1515/BC.2011.200
61. Ivanov I, Shang W, Toledo L, Masgrau L, Svergun DI, Stehling S, et al. Ligand-induced formation of transient dimers of mammalian 12/15-lipoxygenase: a key to allosteric behavior of this class of enzymes? Proteins (2012) 80(3):703–12. doi: 10.1002/prot.23227
62. Lehnert N, Solomon EI. Density-functional investigation on the mechanism of H-atom abstraction by lipoxygenase. J Biol Inorg Chem (2003) 8(3):294–305. doi: 10.1007/s00775-002-0415-6
63. Neau DB, Gilbert NC, Bartlett SG, Boeglin W, Brash AR, Newcomer ME. The 1.85 A structure of an 8R-lipoxygenase suggests a general model for lipoxygenase product specificity. Biochemistry (2009) 48(33):7906–15. doi: 10.1021/bi900084m
64. Collazo L, Klinman JP. Control of the Position of Oxygen Delivery in Soybean Lipoxygenase-1 by Amino Acid Side Chains within a Gas Migration Channel. J Biol Chem (2016) 291(17):9052–9. doi: 10.1074/jbc.M115.709154
65. Coffa G, Brash AR, Samuelsson B. A single active site residue directs oxygenation stereospecificity in lipoxygenases: Stereocontrol is linked to the position of oxygenation. Proc Natl Acad Sci U States America (2004) 101(44):15579–84. doi: 10.1073/pnas.0406727101
66. Schwarz K, Walther M, Anton M, Gerth C, Feussner I, Kuhn H. Structural basis for lipoxygenase specificity. Conversion of the human leukocyte 5-lipoxygenase to a 15-lipoxygenating enzyme species by site-directed mutagenesis. J Biol Chem (2001) 276(1):773–9. doi: 10.1074/jbc.M005114200
67. Walther M, Ivanov I, Myagkova G, Kuhn H. Alterations of lipoxygenase specificity by targeted substrate modification and site-directed mutagenesis. Chem Biol (2001) 8(8):779–90. doi: 10.1016/s1074-5521(01)00050-3
68. Newcomer ME, Brash AR. The structural basis for specificity in lipoxygenase catalysis. Protein Sci (2015) 24(3):298–309. doi: 10.1002/pro.2626
69. Ivanov I, Kuhn H, Heydeck D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene (2015) 573(1):1–32. doi: 10.1016/j.gene.2015.07.073
70. Yu Z, Schneider C, Boeglin WE, Marnett LJ, Brash AR. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc Natl Acad Sci U S A (2003) 100(16):9162–7. doi: 10.1073/pnas.1633612100
71. Rouzer CA, Matsumoto T, Samuelsson B. Single protein from human leukocytes possesses 5-lipoxygenase and leukotriene A4 synthase activities. Proc Natl Acad Sci U S A (1986) 83(4):857–61. doi: 10.1073/pnas.83.4.857
72. Shimizu T, Izumi T, Seyama Y, Tadokoro K, Radmark O, Samuelsson B. Characterization of leukotriene A4 synthase from murine mast cells: evidence for its identity to arachidonate 5-lipoxygenase. Proc Natl Acad Sci U S A (1986) 83(12):4175–9. doi: 10.1073/pnas.83.12.4175
73. Ueda N, Yamamoto S, Oates JA, Brash AR. Stereoselective hydrogen abstraction in leukotriene A4 synthesis by purified 5-lipoxygenase of porcine leukocytes. Prostaglandins (1986) 32(1):43–8. doi: 10.1016/0090-6980(86)90141-3
74. MacMillan DK, Hill E, Sala A, Sigal E, Shuman T, Henson PM, et al. Eosinophil 15-lipoxygenase is a leukotriene A4 synthase. J Biol Chem (1994) 269(43):26663–8.
75. Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem (2003) 278(17):14677–87. doi: 10.1074/jbc.M300218200
76. Kuhn H, Wiesner R, Stender H, Schewe T, Lankin VZ, Nekrasov A, et al. Requirement of monohydroperoxy fatty acids for the oxygenation of 15LS-HETE by reticulocyte lipoxygenase. FEBS Lett (1986) 203(2):247–52. doi: 10.1016/0014-5793(86)80752-9
77. Lepley RA, Fitzpatrick FA. Irreversible inactivation of 5-lipoxygenase by leukotriene A4. Characterization of product inactivation with purified enzyme and intact leukocytes. J Biol Chem (1994) 269(4):2627–31.
78. Wiesner R, Suzuki H, Walther M, Yamamoto S, Kuhn H. Suicidal inactivation of the rabbit 15-lipoxygenase by 15S-HpETE is paralleled by covalent modification of active site peptides. Free Radical Biol Med (2003) 34(3):304–15. doi: 10.1016/s0891-5849(02)01244-3
79. Kohli P, Levy BD. Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol (2009) 158(4):960–71. doi: 10.1111/j.1476-5381.2009.00290.x
80. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci U.S.A. (2005) 102(21):7671–6. doi: 10.1073/pnas.0409271102
81. Forsell PK, Brunnstrom A, Johannesson M, Claesson HE. Metabolism of anandamide into eoxamides by 15-lipoxygenase-1 and glutathione transferases. Lipids (2012) 47(8):781–91. doi: 10.1007/s11745-012-3684-z
82. Kim HY, Spector AA. Synaptamide, endocannabinoid-like derivative of docosahexaenoic acid with cannabinoid-independent function. Prostaglandins Leukot Essent Fatty Acids (2013) 88(1):121–5. doi: 10.1016/j.plefa.2012.08.002
83. Schewe T, Halangk W, Hiebsch C, Rapoport SM. A lipoxygenase in rabbit reticulocytes which attacks phospholipids and intact mitochondria. FEBS Lett (1975) 60(1):149–52. doi: 10.1016/0014-5793(75)80439-x
84. Belkner J, Wiesner R, Rathman J, Barnett J, Sigal E, Kuhn H. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur J Biochem (1993) 213(1):251–61. doi: 10.1111/j.1432-1033.1993.tb17755.x
85. Kuhn H, Barnett J, Grunberger D, Baecker P, Chow J, Nguyen B, et al. Overexpression, purification and characterization of human recombinant 15-lipoxygenase. Biochim Biophys Acta (1993) 1169(1):80–9. doi: 10.1016/0005-2760(93)90085-n
86. Huang LS, Kang JS, Kim MR, Sok DE. Oxygenation of arachidonoyl lysophospholipids by lipoxygenases from soybean, porcine leukocyte, or rabbit reticulocyte. J Agric Food Chem (2008) 56(4):1224–32. doi: 10.1021/jf073016i
87. Hutchins PM, Murphy RC. Cholesteryl ester acyl oxidation and remodeling in murine macrophages: formation of oxidized phosphatidylcholine. J Lipid Res (2012) 53(8):1588–97. doi: 10.1194/jlr.M026799
88. Liu X, Moon SH, Jenkins CM, Sims HF, Gross RW. Cyclooxygenase-2 Mediated Oxidation of 2-Arachidonoyl-Lysophospholipids Identifies Unknown Lipid Signaling Pathways. Cell Chem Biol (2016) 23(10):1217–27. doi: 10.1016/j.chembiol.2016.08.009
89. Ikei KN, Yeung J, Apopa PL, Ceja J, Vesci J, Holman TR, et al. Investigations of human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation. J Lipid Res (2012) 53(12):2546–59. doi: 10.1194/jlr.M026385
90. Hada T, Ueda N, Takahashi Y, Yamamoto S. Catalytic properties of human platelet 12-lipoxygenase as compared with the enzymes of other origins. Biochim Biophys Acta (BBA) - Lipids Lipid Metab (1991) 1083(1):89–93. doi: 10.1016/0005-2760(91)90128-5
91. Romano M, Chen XS, Takahashi Y, Yamamoto S, Funk CD, Serhan CN. Lipoxin synthase activity of human platelet 12-lipoxygenase. Biochem J (1993) 296( Pt 1):127–33. doi: 10.1042/bj2960127
92. Anton R, Vila L. Stereoselective biosynthesis of hepoxilin B3 in human epidermis. J Invest Dermatol (2000) 114(3):554–9. doi: 10.1046/j.1523-1747.2000.00903.x
93. Deng B, Wang CW, Arnardottir HH, Li Y, Cheng CY, Dalli J, et al. Maresin biosynthesis and identification of maresin 2, a new anti-inflammatory and pro-resolving mediator from human macrophages. PloS One (2014) 9(7):e102362. doi: 10.1371/journal.pone.0102362
94. Liu X, Sims HF, Jenkins CM, Guan S, Dilthey BG, Gross RW. 12-LOX catalyzes the oxidation of 2-arachidonoyl-lysolipids in platelets generating eicosanoid-lysolipids that are attenuated by iPLA2gamma knockout. J Biol Chem (2020) 295(16):5307–20. doi: 10.1074/jbc.RA119.012296
95. Yu Z, Schneider C, Boeglin WE, Brash AR. Human and mouse eLOX3 have distinct substrate specificities: implications for their linkage with lipoxygenases in skin. Arch Biochem Biophys (2006) 455(2):188–96. doi: 10.1016/j.abb.2006.09.002
96. Snodgrass RG, Zezina E, Namgaladze D, Gupta S, Angioni C, Geisslinger G, et al. A Novel Function for 15-Lipoxygenases in Cholesterol Homeostasis and CCL17 Production in Human Macrophages. Front Immunol (2018) 9:1906:1906. doi: 10.3389/fimmu.2018.01906
97. Therland KL, Stubbe J, Thiesson HC, Ottosen PD, Walter S, Sorensen GL, et al. Cycloxygenase-2 is expressed in vasculature of normal and ischemic adult human kidney and is colocalized with vascular prostaglandin E2 EP4 receptors. J Am Soc Nephrol (2004) 15(5):1189–98. doi: 10.1097/01.asn.0000124673.79934.24
98. Kirkby NS, Zaiss AK, Urquhart P, Jiao J, Austin PJ, Al-Yamani M, et al. LC-MS/MS confirms that COX-1 drives vascular prostacyclin whilst gene expression pattern reveals non-vascular sites of COX-2 expression. PloS One (2013) 8(7):e69524. doi: 10.1371/journal.pone.0069524
99. Kirkby NS, Chan MV, Zaiss AK, Garcia-Vaz E, Jiao J, Berglund LM, et al. Systematic study of constitutive cyclooxygenase-2 expression: Role of NF-kappaB and NFAT transcriptional pathways. Proc Natl Acad Sci U S A (2016) 113(2):434–9. doi: 10.1073/pnas.1517642113
100. Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Investigation (2006) 116(1):4–15. doi: 10.1172/JCI27291
101. Tang SY, Monslow J, Todd L, Lawson J, Puré E, FitzGerald GA. Cyclooxygenase-2 in endothelial and vascular smooth muscle cells restrains atherogenesis in hyperlipidemic mice. Circulation (2014) 129(17):1761–9. doi: 10.1161/circulationaha.113.007913
102. Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A (1992) 89(16):7384–8. doi: 10.1073/pnas.89.16.7384
103. O’Neill GP, Ford-Hutchinson AW. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett (1993) 330(2):156–60. doi: 10.1016/0014-5793(93)80263-t
104. Pablos JL, Santiago B, Carreira PE, Galindo M, Gomez-Reino JJ. Cyclooxygenase-1 and -2 are expressed by human T cells. Clin Exp Immunol (1999) 115(1):86–90. doi: 10.1046/j.1365-2249.1999.00780.x
105. Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. Cyclooxygenase in normal human tissues–is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med (2009) 13(9B):3753–63. doi: 10.1111/j.1582-4934.2008.00430.x
106. Ryan EP, Pollack SJ, Murant TI, Bernstein SH, Felgar RE, Phipps RP. Activated Human B Lymphocytes Express Cyclooxygenase-2 and Cyclooxygenase Inhibitors Attenuate Ab Production. J Immunol (2005) 174(8):5134.2–. doi: 10.4049/jimmunol.174.8.5134-a
107. Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature (1994) 367(6460):243–9. doi: 10.1038/367243a0
108. Rimon G, Sidhu RS, Lauver DA, Lee JY, Sharma NP, Yuan C, et al. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc Natl Acad Sci U.S.A. (2010) 107(1):28–33. doi: 10.1073/pnas.0909765106
109. Sidhu RS, Lee JY, Yuan C, Smith WL. Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry (2010) 49(33):7069–79. doi: 10.1021/bi1003298
110. Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature (1996) 384(6610):644–8. doi: 10.1038/384644a0
111. Rowlinson SW, Kiefer JR, Prusakiewicz JJ, Pawlitz JL, Kozak KR, Kalgutkar AS, et al. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem (2003) 278(46):45763–9. doi: 10.1074/jbc.M305481200
112. Vecchio AJ, Malkowski MG. The structural basis of endocannabinoid oxygenation by cyclooxygenase-2. J Biol Chem (2011) 286(23):20736–45. doi: 10.1074/jbc.M111.230367
113. Xu S, Hermanson DJ, Banerjee S, Ghebreselasie K, Clayton GM, Garavito RM, et al. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding Network. J Biol Chem (2014) 289(10):6799–808. doi: 10.1074/jbc.M113.517987
114. Lucido MJ, Orlando BJ, Vecchio AJ, Malkowski MG. Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry. Biochemistry (2016) 55(8):1226–38. doi: 10.1021/acs.biochem.5b01378
115. Orlando BJ, Malkowski MG. Substrate-selective Inhibition of Cyclooxygeanse-2 by Fenamic Acid Derivatives Is Dependent on Peroxide Tone. J Biol Chem (2016) 291(29):15069–81. doi: 10.1074/jbc.M116.725713
116. Xiao G, Chen W, Kulmacz RJ. Comparison of structural stabilities of prostaglandin H synthase-1 and -2. J Biol Chem (1998) 273(12):6801–11. doi: 10.1074/jbc.273.12.6801
117. Michael Garavito R, Malkowski MG, DeWitt DL. The structures of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediators (2002) 68-69:129–52. doi: 10.1016/s0090-6980(02)00026-6
118. Spencer AG, Woods JW, Arakawa T, Singer II, Smith WL. Subcellular localization of prostaglandin endoperoxide H synthases-1 and -2 by immunoelectron microscopy. J Biol Chem (1998) 273(16):9886–93. doi: 10.1074/jbc.273.16.9886
119. Yuan C, Smith WL. A cyclooxygenase-2-dependent prostaglandin E2 biosynthetic system in the Golgi apparatus. J Biol Chem (2015) 290(9):5606–20. doi: 10.1074/jbc.M114.632463
120. Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res (2009) 50 Suppl(Suppl):S29–34. doi: 10.1194/jlr.R800042-JLR200
121. Otto JC, DeWitt DL, Smith WL. N-glycosylation of prostaglandin endoperoxide synthases-1 and -2 and their orientations in the endoplasmic reticulum. J Biol Chem (1993) 268(24):18234–42.
122. Sevigny MB, Li CF, Alas M, Hughes-Fulford M. Glycosylation regulates turnover of cyclooxygenase-2. FEBS Lett (2006) 580(28-29):6533–6. doi: 10.1016/j.febslet.2006.10.073
123. Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science (2005) 310(5756):1966–70. doi: 10.1126/science.1119407
124. Qiao J, Ma L, Roth J, Li Y, Liu Y. Kinetic basis for the activation of human cyclooxygenase-2 rather than cyclooxygenase-1 by nitric oxide. Org Biomol Chem (2018) 16(5):765–70. doi: 10.1039/c7ob02992f
125. Yuan C, Rieke CJ, Rimon G, Wingerd BA, Smith WL. Partnering between monomers of cyclooxygenase-2 homodimers. Proc Natl Acad Sci U.S.A. (2006) 103(16):6142–7. doi: 10.1073/pnas.0601805103
126. Dong L, Vecchio AJ, Sharma NP, Jurban BJ, Malkowski MG, Smith WL. Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J Biol Chem (2011) 286(21):19035–46. doi: 10.1074/jbc.M111.231969
127. Zou H, Yuan C, Dong L, Sidhu RS, Hong YH, Kuklev DV, et al. Human cyclooxygenase-1 activity and its responses to COX inhibitors are allosterically regulated by nonsubstrate fatty acids. J Lipid Res (2012) 53(7):1336–47. doi: 10.1194/jlr.M026856
128. Smith WL, Malkowski MG. Interactions of fatty acids, nonsteroidal anti-inflammatory drugs, and coxibs with the catalytic and allosteric subunits of cyclooxygenases-1 and -2. J Biol Chem (2019) 294(5):1697–705. doi: 10.1074/jbc.TM118.006295
129. Marnett LJ. Cyclooxygenase mechanisms. Curr Opin Chem Biol (2000) 4(5):545–52. doi: 10.1016/s1367-5931(00)00130-7
130. Silva PJ, Fernandes PA, Ramos MJ. A theoretical study of radical-only and combined radical/carbocationic mechanisms of arachidonic acid cyclooxygenation by prostaglandin H synthase. Theor Chem Acc: Theory Computation Model (Theoretica Chim Acta) (2003) 110(5):345–51. doi: 10.1007/s00214-003-0476-9
131. Cebrián-Prats A, González-Lafont À, Lluch JM. Unraveling the Molecular Details of the Complete Mechanism That Governs the Synthesis of Prostaglandin G2 Catalyzed by Cyclooxygenase-2. ACS Omega (2019) 4(1):2063–74. doi: 10.1021/acsomega.8b03575
132. O’Connor DE, Mihelich ED, Coleman MC. Stereochemical course of the autooxidative cyclization of lipid hydroperoxides to prostaglandin-like bicyclic endoperoxides. J Am Chem Soc (1984) 106(12):3577–84. doi: 10.1021/ja00324a028
133. Rouzer CA, Marnett LJ. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem Rev (2003) 103(6):2239–304. doi: 10.1021/cr000068x
134. Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Med (2002) 68-69:115–28. doi: 10.1016/s0090-6980(02)00025-4
135. Bailey JM, Bryant RW, Whiting J, Salata K. Characterization of 11-HETE and 15-HETE, together with prostacyclin, as major products of the cyclooxygenase pathway in cultured rat aorta smooth muscle cells. J Lipid Res (1983) 24(11):1419–28.
136. Setty BN, Stuart MJ, Walenga RW. Formation of 11-hydroxyeicosatetraenoic acid and 15-hydroxyeicosatetraenoic acid in human umbilical arteries is catalyzed by cyclooxygenase. Biochim Biophys Acta (1985) 833(3):484–94. doi: 10.1016/0005-2760(85)90106-7
137. Schneider C, Boeglin WE, Prusakiewicz JJ, Rowlinson SW, Marnett LJ, Samel N, et al. Control of prostaglandin stereochemistry at the 15-carbon by cyclooxygenases-1 and -2. A critical role for serine 530 and valine 349. J Biol Chem (2002) 277(1):478–85. doi: 10.1074/jbc.M107471200
138. Thuresson ED, Lakkides KM, Smith WL. Different catalytically competent arrangements of arachidonic acid within the cyclooxygenase active site of prostaglandin endoperoxide H synthase-1 lead to the formation of different oxygenated products. J Biol Chem (2000) 275(12):8501–7. doi: 10.1074/jbc.275.12.8501
139. Loll PJ, Picot D, Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol (1995) 2(8):637–43. doi: 10.1038/nsb0895-637
140. Lecomte M, Laneuville O, Ji C, DeWitt DL, Smith WL. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J Biol Chem (1994) 269(18):13207–15.
141. Gimenez-Bastida JA, Boeglin WE, Boutaud O, Malkowski MG, Schneider C. Residual cyclooxygenase activity of aspirin-acetylated COX-2 forms 15 R-prostaglandins that inhibit platelet aggregation. FASEB J (2019) 33(1):1033–41. doi: 10.1096/fj.201801018R
142. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449
143. Laneuville O, Breuer DK, Xu N, Huang ZH, Gage DA, Watson JT, et al. Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2. Formation of 12-hydroxy-(9Z, 13E/Z, 15Z)- octadecatrienoic acids from alpha-linolenic acid. J Biol Chem (1995) 270(33):19330–6. doi: 10.1074/jbc.270.33.19330
144. Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, et al. Cyclooxygenase Allosterism, Fatty Acid-mediated Cross-talk between Monomers of Cyclooxygenase Homodimers. J Biol Chem (2009) 284(15):10046–55. doi: 10.1074/jbc.M808634200
145. Dong L, Zou H, Yuan C, Hong YH, Kuklev DV, Smith WL. Different Fatty Acids Compete with Arachidonic Acid for Binding to the Allosteric or Catalytic Subunits of Cyclooxygenases to Regulate Prostanoid Synthesis. J Biol Chem (2016) 291(8):4069–78. doi: 10.1074/jbc.M115.698001
146. Mulugeta S, Suzuki T, Hernandez NT, Griesser M, Boeglin WE, Schneider C. Identification and absolute configuration of dihydroxy-arachidonic acids formed by oxygenation of 5S-HETE by native and aspirin-acetylated COX-2. J Lipid Res (2010) 51(3):575–85. doi: 10.1194/jlr.M001719
147. Yu M, Ives D, Ramesha CS. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem (1997) 272(34):21181–6. doi: 10.1074/jbc.272.34.21181
148. Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem (2000) 275(43):33744–9. doi: 10.1074/jbc.M007088200
149. Prusakiewicz JJ, Kingsley PJ, Kozak KR, Marnett LJ. Selective oxygenation of N-arachidonylglycine by cyclooxygenase-2. Biochem Biophys Res Commun (2002) 296(3):612–7. doi: 10.1016/s0006-291x(02)00915-4
150. de Bus I, Zuilhof H, Witkamp R, Balvers M, Albada B. Novel COX-2 products of n-3 polyunsaturated fatty acid-ethanolamine-conjugates identified in RAW264.7 macrophages. J Lipid Res (2019) 60(11):1829–40. doi: 10.1194/jlr.M094235
151. Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics (2004) 14(1):1–18. doi: 10.1097/00008571-200401000-00001
152. Nebert DW, Wikvall K, Miller WL. Human cytochromes P450 in health and disease. Philos Trans R Soc Lond B Biol Sci (2013) 368(1612):20120431. doi: 10.1098/rstb.2012.0431
153. Nebert DW, Adesnik M, Coon MJ, Estabrook RW, Gonzalez FJ, Guengerich FP, et al. The P450 gene superfamily: recommended nomenclature. DNA (1987) 6(1):1–11. doi: 10.1089/dna.1987.6.1
154. Nelson DR. The cytochrome p450 homepage. Hum Genomics (2009) 4(1):59–65. doi: 10.1186/1479-7364-4-1-59
155. Sim SC, Ingelman-Sundberg M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: a peer-reviewed database of CYP variants and their associated effects. Hum Genomics (2010) 4(4):278–81. doi: 10.1186/1479-7364-4-4-278
156. Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci (2001) 58(5-6):737–47. doi: 10.1007/pl00000897
157. Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi (2003) 123(5):369–75. doi: 10.1248/yakushi.123.369
158. Renaud HJ, Cui JY, Khan M, Klaassen CD. Tissue distribution and gender-divergent expression of 78 cytochrome P450 mRNAs in mice. Toxicol Sci (2011) 124(2):261–77. doi: 10.1093/toxsci/kfr240
159. Sangar MC, Bansal S, Avadhani NG. Bimodal targeting of microsomal cytochrome P450s to mitochondria: implications in drug metabolism and toxicity. Expert Opin Drug Metab Toxicol (2010) 6(10):1231–51. doi: 10.1517/17425255.2010.503955
160. Srejber M, Navratilova V, Paloncyova M, Bazgier V, Berka K, Anzenbacher P, et al. Membrane-attached mammalian cytochromes P450: An overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J Inorg Biochem (2018) 183:117–36. doi: 10.1016/j.jinorgbio.2018.03.002
161. Erickson SK, Bosterling B. Cholesterol 7 alpha-hydroxylase from human liver: partial purification and reconstruction into defined phospholipid-cholesterol vesicles. J Lipid Res (1981) 22(5):872–6.
162. Loeper J, Descatoire V, Maurice M, Beaune P, Belghiti J, Houssin D, et al. Cytochromes P-450 in human hepatocyte plasma membrane: Recognition by several autoantibodies. Gastroenterology (1993) 104(1):203–16. doi: 10.1016/0016-5085(93)90853-5
163. Neve EP, Ingelman-Sundberg M. Molecular basis for the transport of cytochrome P450 2E1 to the plasma membrane. J Biol Chem (2000) 275(22):17130–5. doi: 10.1074/jbc.M000957200
164. Leung YK, Lau KM, Mobley J, Jiang Z, Ho SM. Overexpression of cytochrome P450 1A1 and its novel spliced variant in ovarian cancer cells: alternative subcellular enzyme compartmentation may contribute to carcinogenesis. Cancer Res (2005) 65(9):3726–34. doi: 10.1158/0008-5472.CAN-04-3771
165. Li D, Tolleson WH, Yu D, Chen S, Guo L, Xiao W, et al. Regulation of cytochrome P450 expression by microRNAs and long noncoding RNAs: Epigenetic mechanisms in environmental toxicology and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev (2019) 37(3):180–214. doi: 10.1080/10590501.2019.1639481
166. Nelson DR, Nebert DW. Cytochrome P450 (CYP) Gene Superfamily. eLS. Chichester: John Wiley & Sons, Ltd (Ed (2018) p. 1–19. doi: 10.1002/9780470015902.a0005667.pub3
167. Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell (2000) 5(1):121–31. doi: 10.1016/s1097-2765(00)80408-6
168. Annalora AJ, Goodin DB, Hong WX, Zhang Q, Johnson EF, Stout CD. Crystal structure of CYP24A1, a mitochondrial cytochrome P450 involved in vitamin D metabolism. J Mol Biol (2010) 396(2):441–51. doi: 10.1016/j.jmb.2009.11.057
169. Pochapsky TC, Kazanis S, Dang M. Conformational plasticity and structure/function relationships in cytochromes P450. Antioxid Redox Signal (2010) 13(8):1273–96. doi: 10.1089/ars.2010.3109
170. Ohkura K, Kawaguchi Y, Watanabe Y, Masubuchi Y, Shinohara Y, Hori H. Flexible structure of cytochrome P450: promiscuity of ligand binding in the CYP3A4 heme pocket. Anticancer Res (2009) 29(3):935–42.
171. Hoch U, Ortiz De Montellano PR. Covalently linked heme in cytochrome p4504a fatty acid hydroxylases. J Biol Chem (2001) 276(14):11339–46. doi: 10.1074/jbc.M009969200
172. Hsu MH, Baer BR, Rettie AE, Johnson EF. The Crystal Structure of Cytochrome P450 4B1 (CYP4B1) Monooxygenase Complexed with Octane Discloses Several Structural Adaptations for omega-Hydroxylation. J Biol Chem (2017) 292(13):5610–21. doi: 10.1074/jbc.M117.775494
173. Schwarz D, Chernogolov A, Kisselev P. Complex formation in vesicle-reconstituted mitochondrial cytochrome P450 systems (CYP11A1 and CYP11B1) as evidenced by rotational diffusion experiments using EPR and ST-EPR. Biochemistry (1999) 38(29):9456–64. doi: 10.1021/bi990584l
174. Szczesna-Skorupa E, Mallah B, Kemper B. Fluorescence resonance energy transfer analysis of cytochromes P450 2C2 and 2E1 molecular interactions in living cells. J Biol Chem (2003) 278(33):31269–76. doi: 10.1074/jbc.M301489200
175. Hu G, Johnson EF, Kemper B. CYP2C8 exists as a dimer in natural membranes. Drug Metab Dispos (2010) 38(11):1976–83. doi: 10.1124/dmd.110.034942
176. Li B, Yau P, Kemper B. Identification of cytochrome P450 2C2 protein complexes in mouse liver. Proteomics (2011) 11(16):3359–68. doi: 10.1002/pmic.201100001
177. Kandel SE, Lampe JN. Role of protein-protein interactions in cytochrome P450-mediated drug metabolism and toxicity. Chem Res Toxicol (2014) 27(9):1474–86. doi: 10.1021/tx500203s
178. Davydov DR, Davydova NY, Sineva EV, Halpert JR. Interactions among cytochromes P450 in microsomal membranes: oligomerization of cytochromes P450 3A4, 3A5, and 2E1 and its functional consequences. J Biol Chem (2015) 290(6):3850–64. doi: 10.1074/jbc.M114.615443
179. Connick JP, Reed JR, Backes WL. Characterization of Interactions Among CYP1A2, CYP2B4, and NADPH-cytochrome P450 Reductase: Identification of Specific Protein Complexes. Drug Metab Dispos (2018) 46(3):197–203. doi: 10.1124/dmd.117.078642
180. Zhang H, Yokom AL, Cheng S, Su M, Hollenberg PF, Southworth DR, et al. The full-length cytochrome P450 enzyme CYP102A1 dimerizes at its reductase domains and has flexible heme domains for efficient catalysis. J Biol Chem (2018) 293(20):7727–36. doi: 10.1074/jbc.RA117.000600
181. Isin EM, Guengerich FP. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta (2007) 1770(3):314–29. doi: 10.1016/j.bbagen.2006.07.003
182. Lamb DC, Waterman MR. Unusual properties of the cytochrome P450 superfamily. Philos Trans R Soc Lond B Biol Sci (2013) 368(1612):20120434. doi: 10.1098/rstb.2012.0434
183. Hecker M, Ullrich V. On the mechanism of prostacyclin and thromboxane A2 biosynthesis. J Biol Chem (1989) 264(1):141–50.
184. Cook DJ, Finnigan JD, Cook K, Black GW, Charnock SJ. Chapter Five - Cytochromes P450: History, Classes, Catalytic Mechanism, and Industrial Application. In: Christov CZ, editor. Advances in Protein Chemistry and Structural Biology. Academic Press (2016). p. 105–26. doi: 10.1016/bs.apcsb.2016.07.003
185. Guengerich FP. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal (2018) 8(12):10964–76. doi: 10.1021/acscatal.8b03401
186. Fer M, Dreano Y, Lucas D, Corcos L, Salaun JP, Berthou F, et al. Metabolism of eicosapentaenoic and docosahexaenoic acids by recombinant human cytochromes P450. Arch Biochem Biophys (2008) 471(2):116–25. doi: 10.1016/j.abb.2008.01.002
187. Barbosa-Sicard E, Markovic M, Honeck H, Christ B, Muller DN, Schunck WH. Eicosapentaenoic acid metabolism by cytochrome P450 enzymes of the CYP2C subfamily. Biochem Biophys Res Commun (2005) 329(4):1275–81. doi: 10.1016/j.bbrc.2005.02.103
188. Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}-3 fatty acids. J Biol Chem (2010) 285(43):32720–33. doi: 10.1074/jbc.M110.118406
189. Honda A, Miyazaki T, Ikegami T, Iwamoto J, Maeda T, Hirayama T, et al. Cholesterol 25-hydroxylation activity of CYP3A. J Lipid Res (2011) 52(8):1509–16. doi: 10.1194/jlr.M014084
190. Griffiths WJ, Crick PJ, Meljon A, Theofilopoulos S, Abdel-Khalik J, Yutuc E, et al. Additional pathways of sterol metabolism: Evidence from analysis of Cyp27a1-/- mouse brain and plasma. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(2):191–211. doi: 10.1016/j.bbalip.2018.11.006
191. Bodin K, Andersson U, Rystedt E, Ellis E, Norlin M, Pikuleva I, et al. Metabolism of 4 beta -hydroxycholesterol in humans. J Biol Chem (2002) 277(35):31534–40. doi: 10.1074/jbc.M201712200
192. Kawashima H, Naganuma T, Kusunose E, Kono T, Yasumoto R, Sugimura K, et al. Human fatty acid omega-hydroxylase, CYP4A11: determination of complete genomic sequence and characterization of purified recombinant protein. Arch Biochem Biophys (2000) 378(2):333–9. doi: 10.1006/abbi.2000.1831
193. Shinkyo R, Xu L, Tallman KA, Cheng Q, Porter NA, Guengerich FP. Conversion of 7-dehydrocholesterol to 7-ketocholesterol is catalyzed by human cytochrome P450 7A1 and occurs by direct oxidation without an epoxide intermediate. J Biol Chem (2011) 286(38):33021–8. doi: 10.1074/jbc.M111.282434
194. Bjorkhem I, Diczfalusy U, Lovgren-Sandblom A, Starck L, Jonsson M, Tallman K, et al. On the formation of 7-ketocholesterol from 7-dehydrocholesterol in patients with CTX and SLO. J Lipid Res (2014) 55(6):1165–72. doi: 10.1194/jlr.P048603
195. Schwarz M, Lund EG, Lathe R, Bjorkhem I, Russell DW. Identification and characterization of a mouse oxysterol 7alpha-hydroxylase cDNA. J Biol Chem (1997) 272(38):23995–4001. doi: 10.1074/jbc.272.38.23995
196. Gafvels M, Olin M, Chowdhary BP, Raudsepp T, Andersson U, Persson B, et al. Structure and chromosomal assignment of the sterol 12alpha-hydroxylase gene (CYP8B1) in human and mouse: eukaryotic cytochrome P-450 gene devoid of introns. Genomics (1999) 56(2):184–96. doi: 10.1006/geno.1998.5606
197. Pikuleva IA. Cholesterol-metabolizing cytochromes P450. Drug Metab Dispos (2006) 34(4):513–20. doi: 10.1124/dmd.105.008789
198. Coulter CL, Jaffe RB. Functional maturation of the primate fetal adrenal in vivo: 3. Specific zonal localization and developmental regulation of CYP21A2 (P450c21) and CYP11B1/CYP11B2 (P450c11/aldosterone synthase) lead to integrated concept of zonal and temporal steroid biosynthesis. Endocrinology (1998) 139(12):5144–50. doi: 10.1210/endo.139.12.6333
199. Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev (2004) 25(6):947–70. doi: 10.1210/er.2003-0030
200. Omdahl JL, Morris HA, May BK. Hydroxylase enzymes of the vitamin D pathway: expression, function, and regulation. Annu Rev Nutr (2002) 22(1):139–66. doi: 10.1146/annurev.nutr.22.120501.150216
201. White JA, Guo YD, Baetz K, Beckett-Jones B, Bonasoro J, Hsu KE, et al. Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J Biol Chem (1996) 271(47):29922–7. doi: 10.1074/jbc.271.47.29922
202. Goyal S, Xiao Y, Porter NA, Xu L, Guengerich FP. Oxidation of 7-dehydrocholesterol and desmosterol by human cytochrome P450 46A1. J Lipid Res (2014) 55(9):1933–43. doi: 10.1194/jlr.M051508
203. Lepesheva GI, Waterman MR. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta (2007) 1770(3):467–77. doi: 10.1016/j.bbagen.2006.07.018
204. Fer M, Corcos L, Dreano Y, Plee-Gautier E, Salaun JP, Berthou F, et al. Cytochromes P450 from family 4 are the main omega hydroxylating enzymes in humans: CYP4F3B is the prominent player in PUFA metabolism. J Lipid Res (2008) 49(11):2379–89. doi: 10.1194/jlr.M800199-JLR200
205. Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev (2014) 22(1):1–12. doi: 10.1097/CRD.0b013e3182961659
206. Kikuta Y, Kusunose E, Kusunose M. Prostaglandin and leukotriene omega-hydroxylases. Prostaglandins Other Lipid Med (2002) 68-69:345–62. doi: 10.1016/s0090-6980(02)00039-4
207. Hardwick JP. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem Pharmacol (2008) 75(12):2263–75. doi: 10.1016/j.bcp.2008.03.004
208. Ohno Y, Nakamichi S, Ohkuni A, Kamiyama N, Naoe A, Tsujimura H, et al. Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc Natl Acad Sci U.S.A. (2015) 112(25):7707–12. doi: 10.1073/pnas.1503491112
209. Laethem RM, Balazy M, Falck JR, Laethem CL, Koop DR. Formation of 19(S)-, 19(R)-, and 18(R)-hydroxyeicosatetraenoic acids by alcohol-inducible cytochrome P450 2E1. J Biol Chem (1993) 268(17):12912–8.
210. Oliw EH. bis-Allylic hydroxylation of linoleic acid and arachidonic acid by human hepatic monooxygenases. Biochim Biophys Acta (1993) 1166(2-3):258–63. doi: 10.1016/0005-2760(93)90106-j
211. Brash AR, Boeglin WE, Capdevila JH, Yeola S, Blair IA. 7-HETE, 10-HETE, and 13-HETE are major products of NADPH-dependent arachidonic acid metabolism in rat liver microsomes: analysis of their stereochemistry, and the stereochemistry of their acid-catalyzed rearrangement. Arch Biochem Biophys (1995) 321(2):485–92. doi: 10.1006/abbi.1995.1421
212. Morisseau C. Role of epoxide hydrolases in lipid metabolism. Biochimie (2013) 95(1):91–5. doi: 10.1016/j.biochi.2012.06.011
213. Snider NT, Kornilov AM, Kent UM, Hollenberg PF. Anandamide metabolism by human liver and kidney microsomal cytochrome p450 enzymes to form hydroxyeicosatetraenoic and epoxyeicosatrienoic acid ethanolamides. J Pharmacol Exp Ther (2007) 321(2):590–7. doi: 10.1124/jpet.107.119321
214. Sridar C, Snider NT, Hollenberg PF. Anandamide oxidation by wild-type and polymorphically expressed CYP2B6 and CYP2D6. Drug Metab Dispos (2011) 39(5):782–8. doi: 10.1124/dmd.110.036707
215. McDougle DR, Watson JE, Abdeen AA, Adili R, Caputo MP, Krapf JE, et al. Anti-inflammatory ω-3 endocannabinoid epoxides. Proc Natl Acad Sci (2017) 114(30):E6034–E43. doi: 10.1073/pnas.1610325114
216. Watson JE, Kim JS, Das A. Emerging class of omega-3 fatty acid endocannabinoids & their derivatives. Prostaglandins Other Lipid Med (2019) 143:106337. doi: 10.1016/j.prostaglandins.2019.106337
217. Shyadehi AZ, Lamb DC, Kelly SL, Kelly DE, Schunck WH, Wright JN, et al. The mechanism of the acyl-carbon bond cleavage reaction catalyzed by recombinant sterol 14 alpha-demethylase of Candida albicans (other names are: lanosterol 14 alpha-demethylase, P-45014DM, and CYP51). J Biol Chem (1996) 271(21):12445–50. doi: 10.1074/jbc.271.21.12445
218. Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, Park HW. Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc Natl Acad Sci U S A (2011) 108(25):10139–43. doi: 10.1073/pnas.1019441108
219. Griffiths WJ, Wang Y. Oxysterols as lipid mediators: Their biosynthetic genes, enzymes and metabolites. Prostaglandins Other Lipid Med (2020) 147:106381. doi: 10.1016/j.prostaglandins.2019.106381
220. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem (2003) 72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712
221. Cali JJ, Russell DW. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J Biol Chem (1991) 266(12):7774–8.
222. Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A (1999) 96(13):7238–43. doi: 10.1073/pnas.96.13.7238
223. Mast N, Norcross R, Andersson U, Shou M, Nakayama K, Bjorkhem I, et al. Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry (2003) 42(48):14284–92. doi: 10.1021/bi035512f
224. Bodin K, Andersson U, Rystedt E, Ellis E, Norlin M, Pikuleva I, et al. Metabolism of 4 beta -hydroxycholesterol in humans. J Biol Chem (2002) 277(35):31534–40. doi: 10.1074/jbc.M201712200
225. Tuckey RC, Cameron KJ. Side-chain specificities of human and bovine cytochromes P-450scc. Eur J Biochem (1993) 217(1):209–15. doi: 10.1111/j.1432-1033.1993.tb18235.x
226. Petrunak EM, DeVore NM, Porubsky PR, Scott EE. Structures of human steroidogenic cytochrome P450 17A1 with substrates. J Biol Chem (2014) 289(47):32952–64. doi: 10.1074/jbc.M114.610998
227. Jez JM, Flynn TG, Penning TM. A new nomenclature for the aldo-keto reductase superfamily. Biochem Pharmacol (1997) 54(6):639–47. doi: 10.1016/s0006-2952(97)84253-0
228. Hyndman D, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chem Biol Interact (2003) 143-144:621–31. doi: 10.1016/s0009-2797(02)00193-x
229. Spite M, Baba SP, Ahmed Y, Barski OA, Nijhawan K, Petrash JM, et al. Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem J (2007) 405(1):95–105. doi: 10.1042/BJ20061743
230. O’Connor T, Ireland LS, Harrison DJ, Hayes JD. Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem J (1999) 343 Pt 2(Pt 2):487–504. doi: 10.1042/bj3430487
231. Cao D, Fan ST, Chung SS. Identification and characterization of a novel human aldose reductase-like gene. J Biol Chem (1998) 273(19):11429–35. doi: 10.1074/jbc.273.19.11429
232. Endo S, Matsunaga T, Mamiya H, Ohta C, Soda M, Kitade Y, et al. Kinetic studies of AKR1B10, human aldose reductase-like protein: endogenous substrates and inhibition by steroids. Arch Biochem Biophys (2009) 487(1):1–9. doi: 10.1016/j.abb.2009.05.009
233. Huang SP, Palla S, Ruzycki P, Varma RA, Harter T, Reddy GB, et al. Aldo-keto reductases in the eye. J Ophthalmol (2010) 2010:521204. doi: 10.1155/2010/521204
234. Ruiz FX, Porte S, Pares X, Farres J. Biological role of aldo-keto reductases in retinoic Acid biosynthesis and signaling. Front Pharmacol (2012) 3:58:58. doi: 10.3389/fphar.2012.00058
235. Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev (2008) 40(4):553–624. doi: 10.1080/03602530802431439
236. Weber S, Salabei JK, Moller G, Kremmer E, Bhatnagar A, Adamski J, et al. Aldo-keto Reductase 1B15 (AKR1B15): a mitochondrial human aldo-keto reductase with activity toward steroids and 3-keto-acyl-CoA conjugates. J Biol Chem (2015) 290(10):6531–45. doi: 10.1074/jbc.M114.610121
237. Byrns MC. Role of aldo-keto reductase enzymes in mediating the timing of parturition. Front Pharmacol (2011) 2:92:92. doi: 10.3389/fphar.2011.00092
238. Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, et al. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J (2000) 351(Pt 1):67–77. doi: 10.1042/0264-6021:3510067
239. Burczynski ME, Sridhar GR, Palackal NT, Penning TM. The reactive oxygen species–and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the alpha,beta-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. J Biol Chem (2001) 276(4):2890–7. doi: 10.1074/jbc.M006655200
240. Lyon RC, Li D, McGarvie G, Ellis EM. Aldo-keto reductases mediate constitutive and inducible protection against aldehyde toxicity in human neuroblastoma SH-SY5Y cells. Neurochem Int (2013) 62(1):113–21. doi: 10.1016/j.neuint.2012.10.007
241. Suzuki-Yamamoto T, Nishizawa M, Fukui M, Okuda-Ashitaka E, Nakajima T, Ito S, et al. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett (1999) 462(3):335–40. doi: 10.1016/s0014-5793(99)01551-3
242. Penning TM. Aldo-Keto Reductase (AKR) 1C3 inhibitors: a patent review. Expert Opin Ther Pat (2017) 27(12):1329–40. doi: 10.1080/13543776.2017.1379503
243. Kondo KH, Kai MH, Setoguchi Y, Eggertsen G, Sjoblom P, Setoguchi T, et al. Cloning and expression of cDNA of human delta 4-3-oxosteroid 5 beta-reductase and substrate specificity of the expressed enzyme. Eur J Biochem (1994) 219(1-2):357–63. doi: 10.1111/j.1432-1033.1994.tb19947.x
244. Chen M, Drury JE, Penning TM. Substrate specificity and inhibitor analyses of human steroid 5beta-reductase (AKR1D1). Steroids (2011) 76(5):484–90. doi: 10.1016/j.steroids.2011.01.003
245. Nikolaou N, Gathercole LL, Marchand L, Althari S, Dempster NJ, Green CJ, et al. AKR1D1 is a novel regulator of metabolic phenotype in human hepatocytes and is dysregulated in non-alcoholic fatty liver disease. Metabolism (2019) 99:67–80. doi: 10.1016/j.metabol.2019.153947
246. Sakuma M, Kametani S, Akanuma H. Purification and some properties of a hepatic NADPH-dependent reductase that specifically acts on 1,5-anhydro-D-fructose. J Biochem (1998) 123(1):189–93. doi: 10.1093/oxfordjournals.jbchem.a021909
247. Azuma Y, Nishinaka T, Ushijima S, Soh J, Katsuyama M, Lu HP, et al. Characterization of htAKR, a novel gene product in the aldo-keto reductase family specifically expressed in human testis. Mol Hum Reprod (2004) 10(7):527–33. doi: 10.1093/molehr/gah062
248. England SK, Uebele VN, Shear H, Kodali J, Bennett PB, Tamkun MM. Characterization of a voltage-gated K+ channel beta subunit expressed in human heart. Proc Natl Acad Sci U S A (1995) 92(14):6309–13. doi: 10.1073/pnas.92.14.6309
249. Pan Y, Weng J, Cao Y, Bhosle RC, Zhou M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvbeta1. J Biol Chem (2008) 283(13):8634–42. doi: 10.1074/jbc.M709304200
250. Tipparaju SM, Barski OA, Srivastava S, Bhatnagar A. Catalytic mechanism and substrate specificity of the beta-subunit of the voltage-gated potassium channel. Biochemistry (2008) 47(34):8840–54. doi: 10.1021/bi800301b
251. Xie Z, Barski OA, Cai J, Bhatnagar A, Tipparaju SM. Catalytic reduction of carbonyl groups in oxidized PAPC by Kvbeta2 (AKR6). Chem Biol Interact (2011) 191(1-3):255–60. doi: 10.1016/j.cbi.2011.01.032
252. Rasband MN, Trimmer JS. Subunit composition and novel localization of K+ channels in spinal cord. J Comp Neurol (2001) 429(1):166–76. doi: 10.1002/1096-9861(20000101)429:1<166::aid-cne13>3.0.co;2-y
253. Leicher T, Bahring R, Isbrandt D, Pongs O. Coexpression of the KCNA3B gene product with Kv1.5 leads to a novel A-type potassium channel. J Biol Chem (1998) 273(52):35095–101. doi: 10.1074/jbc.273.52.35095
254. Ireland LS, Harrison DJ, Neal GE, Hayes JD. Molecular cloning, expression and catalytic activity of a human AKR7 member of the aldo-keto reductase superfamily: evidence that the major 2-carboxybenzaldehyde reductase from human liver is a homologue of rat aflatoxin B1-aldehyde reductase. Biochem J (1998) 332( Pt 1):21–34. doi: 10.1042/bj3320021
255. Knight LP, Primiano T, Groopman JD, Kensler TW, Sutter TR. cDNA cloning, expression and activity of a second human aflatoxin B1-metabolizing member of the aldo-keto reductase superfamily, AKR7A3. Carcinogenesis (1999) 20(7):1215–23. doi: 10.1093/carcin/20.7.1215
256. Matsunaga T, Haga M, Watanabe G, Shinoda Y, Endo S, Kajiwara Y, et al. 9,10-Phenanthrenequinone promotes secretion of pulmonary aldo-keto reductases with surfactant. Cell Tissue Res (2012) 347(2):407–17. doi: 10.1007/s00441-011-1304-5
257. Luo DX, Huang MC, Ma J, Gao Z, Liao DF, Cao D. Aldo-keto reductase family 1, member B10 is secreted through a lysosome-mediated non-classical pathway. Biochem J (2011) 438(1):71–80. doi: 10.1042/BJ20110111
258. Kelly VP, Sherratt PJ, Crouch DH, Hayes JD. Novel homodimeric and heterodimeric rat gamma-hydroxybutyrate synthases that associate with the Golgi apparatus define a distinct subclass of aldo-keto reductase 7 family proteins. Biochem J (2002) 366(Pt 3):847–61. doi: 10.1042/BJ20020342
259. Penning TM, Wangtrakuldee P, Auchus RJ. Structural and Functional Biology of Aldo-Keto Reductase Steroid-Transforming Enzymes. Endocr Rev (2019) 40(2):447–75. doi: 10.1210/er.2018-00089
260. Ma H, Penning TM. Conversion of mammalian 3alpha-hydroxysteroid dehydrogenase to 20alpha-hydroxysteroid dehydrogenase using loop chimeras: changing specificity from androgens to progestins. Proc Natl Acad Sci U S A (1999) 96(20):11161–6. doi: 10.1073/pnas.96.20.11161
261. Gulbis JM, Mann S, MacKinnon R. Structure of a voltage-dependent K+ channel beta subunit. Cell (1999) 97(7):943–52. doi: 10.1016/s0092-8674(00)80805-3
262. Liu SQ, Jin H, Zacarias A, Srivastava S, Bhatnagar A. Binding of pyridine coenzymes to the beta-subunit of the voltage sensitive potassium channels. Chem Biol Interact (2001) 130-132(1-3):955–62. doi: 10.1016/s0009-2797(00)00248-9
263. Di Luccio E, Elling RA, Wilson DK. Identification of a novel NADH-specific aldo-keto reductase using sequence and structural homologies. Biochem J (2006) 400(1):105–14. doi: 10.1042/BJ20060660
264. Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J (1997) 326Pt 3)(Pt 3):625–36. doi: 10.1042/bj3260625
265. Di Costanzo L, Drury JE, Christianson DW, Penning TM. Structure and catalytic mechanism of human steroid 5beta-reductase (AKR1D1). Mol Cell Endocrinol (2009) 301(1-2):191–8. doi: 10.1016/j.mce.2008.09.013
266. Nagata N, Kusakari Y, Fukunishi Y, Inoue T, Urade Y. Catalytic mechanism of the primary human prostaglandin F2alpha synthase, aldo-keto reductase 1B1–prostaglandin D2 synthase activity in the absence of NADP(H). FEBS J (2011) 278(8):1288–98. doi: 10.1111/j.1742-4658.2011.08049.x
267. Wang C, Yan R, Luo D, Watabe K, Liao DF, Cao D. Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J Biol Chem (2009) 284(39):26742–8. doi: 10.1074/jbc.M109.022897
268. Li D, Ferrari M, Ellis EM. Human aldo-keto reductase AKR7A2 protects against the cytotoxicity and mutagenicity of reactive aldehydes and lowers intracellular reactive oxygen species in hamster V79-4 cells. Chem Biol Interact (2012) 195(1):25–34. doi: 10.1016/j.cbi.2011.09.007
269. Li D, Ellis EM. Inducible protection of human astrocytoma 1321N1 cells against hydrogen peroxide and aldehyde toxicity by 7-hydroxycoumarin is associated with the upregulation of aldo-keto reductases. Neurotoxicology (2012) 33(5):1368–74. doi: 10.1016/j.neuro.2012.08.015
270. Kabututu Z, Manin M, Pointud JC, Maruyama T, Nagata N, Lambert S, et al. Prostaglandin F2alpha synthase activities of aldo-keto reductase 1B1, 1B3 and 1B7. J Biochem (2009) 145(2):161–8. doi: 10.1093/jb/mvn152
271. Komoto J, Yamada T, Watanabe K, Takusagawa F. Crystal structure of human prostaglandin F synthase (AKR1C3). Biochemistry (2004) 43(8):2188–98. doi: 10.1021/bi036046x
272. Zhou Y, Shaw N, Li Y, Zhao Y, Zhang R, Liu ZJ. Structure-function analysis of human l-prostaglandin D synthase bound with fatty acid molecules. FASEB J (2010) 24(12):4668–77. doi: 10.1096/fj.10-164863
273. Rizner TL, Smuc T, Rupreht R, Sinkovec J, Penning TM. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol Cell Endocrinol (2006) 248(1-2):126–35. doi: 10.1016/j.mce.2005.10.009
274. Penning TM. Human hydroxysteroid dehydrogenases and pre-receptor regulation: insights into inhibitor design and evaluation. J Steroid Biochem Mol Biol (2011) 125(1-2):46–56. doi: 10.1016/j.jsbmb.2011.01.009
275. Persson B, Kallberg Y, Bray JE, Bruford E, Dellaporta SL, Favia AD, et al. (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem Biol Interact (2009) 178(1-3):94–8. doi: 10.1016/j.cbi.2008.10.040
276. Dumont M, Luu-The V, Dupont E, Pelletier G, Labrie F. Characterization, expression, and immunohistochemical localization of 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4 isomerase in human skin. J Invest Dermatol (1992) 99(4):415–21. doi: 10.1111/1523-1747.ep12616131
277. Labrie F, Simard J, Luu-The V, Belanger A, Pelletier G. Structure, function and tissue-specific gene expression of 3beta-hydroxysteroid dehydrogenase/5-ene-4-ene isomerase enzymes in classical and peripheral intracrine steroidogenic tissues. J Steroid Biochem Mol Biol (1992) 43(8):805–26. doi: 10.1016/0960-0760(92)90308-6
278. Rheaume E, Lachance Y, Zhao HF, Breton N, Dumont M, de Launoit Y, et al. Structure and expression of a new complementary DNA encoding the almost exclusive 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4-isomerase in human adrenals and gonads. Mol Endocrinol (1991) 5(8):1147–57. doi: 10.1210/mend-5-8-1147
279. Schwarz M, Wright AC, Davis DL, Nazer H, Bjorkhem I, Russell DW. The bile acid synthetic gene 3beta-hydroxy-Delta(5)-C(27)-steroid oxidoreductase is mutated in progressive intrahepatic cholestasis. J Clin Invest (2000) 106(9):1175–84. doi: 10.1172/JCI10902
280. Odermatt A, Arnold P, Stauffer A, Frey BM, Frey FJ. The N-terminal anchor sequences of 11beta-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J Biol Chem (1999) 274(40):28762–70. doi: 10.1074/jbc.274.40.28762
281. Luu The V, Labrie C, Zhao HF, Couet J, Lachance Y, Simard J, et al. Characterization of cDNAs for human estradiol 17 beta-dehydrogenase and assignment of the gene to chromosome 17: evidence of two mRNA species with distinct 5’-termini in human placenta. Mol Endocrinol (1989) 3(8):1301–9. doi: 10.1210/mend-3-8-1301
282. He W, Gauri M, Li T, Wang R, Lin SX. Current knowledge of the multifunctional 17beta-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene (2016) 588(1):54–61. doi: 10.1016/j.gene.2016.04.031
283. van Grunsven EG, van Berkel E, Ijlst L, Vreken P, de Klerk JB, Adamski J, et al. Peroxisomal D-hydroxyacyl-CoA dehydrogenase deficiency: resolution of the enzyme defect and its molecular basis in bifunctional protein deficiency. Proc Natl Acad Sci U.S.A. (1998) 95(5):2128–33. doi: 10.1073/pnas.95.5.2128
284. Nokelainen P, Peltoketo H, Vihko R, Vihko P. Expression cloning of a novel estrogenic mouse 17 beta-hydroxysteroid dehydrogenase/17-ketosteroid reductase (m17HSD7), previously described as a prolactin receptor-associated protein (PRAP) in rat. Mol Endocrinol (1998) 12(7):1048–59. doi: 10.1210/mend.12.7.0134
285. Marijanovic Z, Laubner D, Moller G, Gege C, Husen B, Adamski J, et al. Closing the gap: identification of human 3-ketosteroid reductase, the last unknown enzyme of mammalian cholesterol biosynthesis. Mol Endocrinol (2003) 17(9):1715–25. doi: 10.1210/me.2002-0436
286. Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CR, Shimizu T, et al. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res (2009) 50 Suppl(Suppl):S9–14. doi: 10.1194/jlr.R800095-JLR200
287. Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res (2007) 35(Database issue):D527–32. doi: 10.1093/nar/gkl838
288. Urquhart P, Nicolaou A, Woodward DF. Endocannabinoids and their oxygenation by cyclo-oxygenases, lipoxygenases and other oxygenases. Biochim Biophys Acta (2015) 1851(4):366–76. doi: 10.1016/j.bbalip.2014.12.015
289. Abraham RT, McKinney MM, Forray C, Shipley GD, Handwerger BS. Stimulation of arachidonic acid release and eicosanoid biosynthesis in an interleukin 2-dependent T cell line. J Immunopharmacol (1986) 8(2):165–204. doi: 10.3109/08923978609028614
290. Tang X, Edwards EM, Holmes BB, Falck JR, Campbell WB. Role of phospholipase C and diacylglyceride lipase pathway in arachidonic acid release and acetylcholine-induced vascular relaxation in rabbit aorta. Am J Physiol Heart Circ Physiol (2006) 290(1):H37–45. doi: 10.1152/ajpheart.00491.2005
291. Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev (2011) 111(10):6130–85. doi: 10.1021/cr200085w
292. Vasquez AM, Mouchlis VD, Dennis EA. Review of four major distinct types of human phospholipase A2. Adv Biol Regul (2018) 67:212–8. doi: 10.1016/j.jbior.2017.10.009
293. Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, et al. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell (1991) 65(6):1043–51. doi: 10.1016/0092-8674(91)90556-e
294. Hirabayashi T, Murayama T, Shimizu T. Regulatory mechanism and physiological role of cytosolic phospholipase A2. Biol Pharm Bull (2004) 27(8):1168–73. doi: 10.1248/bpb.27.1168
295. Strokin M, Sergeeva M, Reiser G. Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and Ca2+. Br J Pharmacol (2003) 139(5):1014–22. doi: 10.1038/sj.bjp.0705326
296. Cheon Y, Kim HW, Igarashi M, Modi HR, Chang L, Ma K, et al. Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A(2)-VIA (iPLA(2)beta)-knockout mice. Biochim Biophys Acta (2012) 1821(9):1278–86. doi: 10.1016/j.bbalip.2012.02.003
297. Fonteh AN, Bass DA, Marshall LA, Seeds M, Samet JM, Chilton FH. Evidence that secretory phospholipase A2 plays a role in arachidonic acid release and eicosanoid biosynthesis by mast cells. J Immunol (1994) 152(11):5438–46.
298. Murase R, Sato H, Yamamoto K, Ushida A, Nishito Y, Ikeda K, et al. Group X Secreted Phospholipase A2 Releases omega3 Polyunsaturated Fatty Acids, Suppresses Colitis, and Promotes Sperm Fertility. J Biol Chem (2016) 291(13):6895–911. doi: 10.1074/jbc.M116.715672
299. Murakami M. Lipoquality control by phospholipase A2 enzymes. Proc Jpn Acad Ser B Phys Biol Sci (2017) 93(9):677–702. doi: 10.2183/pjab.93.043
300. Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature (1976) 261(5561):558–60. doi: 10.1038/261558a0
301. Nusing R, Sauter G, Fehr P, Durmuller U, Kasper M, Gudat F, et al. Localization of thromboxane synthase in human tissues by monoclonal antibody Tu 300. Virchows Arch A Pathol Anat Histopathol (1992) 421(3):249–54. doi: 10.1007/BF01611182
302. Miyata A, Hara S, Yokoyama C, Inoue H, Ullrich V, Tanabe T. Molecular cloning and expression of human prostacyclin synthase. Biochem Biophys Res Commun (1994) 200(3):1728–34. doi: 10.1006/bbrc.1994.1652
303. Urade Y, Hayaishi O. Prostaglandin D synthase: structure and function. Vitam Horm (2000) 58:89–120. doi: 10.1016/s0083-6729(00)58022-4
304. Joo M, Sadikot RT. PGD synthase and PGD2 in immune resposne. Mediators Inflamm (2012) 2012:503128. doi: 10.1155/2012/503128
305. Ziganshin AU, Zefirova JT, Zefirova TP, Ziganshina LE, Hoyle CH, Burnstock G. Potentiation of uterine effects of prostaglandin F2{alpha} by adenosine 5’-triphosphate. Obstet Gynecol (2005) 105(6):1429–36. doi: 10.1097/01.AOG.0000161314.50808.dd
306. Sampey AV, Monrad S, Crofford LJ. Microsomal prostaglandin E synthase-1: the inducible synthase for prostaglandin E2. Arthritis Res Ther (2005) 7(3):114–7. doi: 10.1186/ar1748
307. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol (2015) 15(8):511–23. doi: 10.1038/nri3859
308. Schieber A, Frank RW, Ghisla S. Purification and properties of prostaglandin 9-ketoreductase from pig and human kidney. Identity with human carbonyl reductase. Eur J Biochem (1992) 206(2):491–502. doi: 10.1111/j.1432-1033.1992.tb16952.x
309. Powell WS, Rokach J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim Biophys Acta (2015) 1851(4):340–55. doi: 10.1016/j.bbalip.2014.10.008
310. Powell WS, Gravel S, Halwani F. 5-oxo-6,8,11,14-eicosatetraenoic acid is a potent stimulator of L-selectin shedding, surface expression of CD11b, actin polymerization, and calcium mobilization in human eosinophils. Am J Respir Cell Mol Biol (1999) 20(1):163–70. doi: 10.1165/ajrcmb.20.1.3141
311. Hallstrand TS, Henderson WR Jr. An update on the role of leukotrienes in asthma. Curr Opin Allergy Clin Immunol (2010) 10(1):60–6. doi: 10.1097/ACI.0b013e32833489c3
312. Capra V, Rovati GE, Mangano P, Buccellati C, Murphy RC, Sala A. Transcellular biosynthesis of eicosanoid lipid mediators. Biochim Biophys Acta (2015) 1851(4):377–82. doi: 10.1016/j.bbalip.2014.09.002
313. Chandrasekharan JA, Sharma-Walia N. Lipoxins: nature’s way to resolve inflammation. J Inflammation Res (2015) 8:181–92. doi: 10.2147/JIR.S90380
314. Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest (1989) 84(5):1609–19. doi: 10.1172/JCI114338
315. Feltenmark S, Gautam N, Brunnstrom A, Griffiths W, Backman L, Edenius C, et al. Eoxins are proinflammatory arachidonic acid metabolites produced via the 15-lipoxygenase-1 pathway in human eosinophils and mast cells. Proc Natl Acad Sci U.S.A. (2008) 105(2):680–5. doi: 10.1073/pnas.0710127105
316. Claesson HE. On the biosynthesis and biological role of eoxins and 15-lipoxygenase-1 in airway inflammation and Hodgkin lymphoma. Prostaglandins Other Lipid Med (2009) 89(3-4):120–5. doi: 10.1016/j.prostaglandins.2008.12.003
317. Nigam S, Patabhiraman S, Ciccoli R, Ishdorj G, Schwarz K, Petrucev B, et al. The rat leukocyte-type 12-lipoxygenase exhibits an intrinsic hepoxilin A3 synthase activity. J Biol Chem (2004) 279(28):29023–30. doi: 10.1074/jbc.M307576200
318. Cronin A, Decker M, Arand M. Mammalian soluble epoxide hydrolase is identical to liver hepoxilin hydrolase. J Lipid Res (2011) 52(4):712–9. doi: 10.1194/jlr.M009639
319. Pace-Asciak CR, Laneuville O, Su WG, Corey EJ, Gurevich N, Wu P, et al. A glutathione conjugate of hepoxilin A3: formation and action in the rat central nervous system. Proc Natl Acad Sci U.S.A. (1990) 87(8):3037–41. doi: 10.1073/pnas.87.8.3037
320. Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, et al. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc Natl Acad Sci U.S.A. (2004) 101(19):7421–6. doi: 10.1073/pnas.0400832101
321. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med (2000) 192(8):1197–204. doi: 10.1084/jem.192.8.1197
322. Arita M, Clish CB, Serhan CN. The contributions of aspirin and microbial oxygenase to the biosynthesis of anti-inflammatory resolvins: novel oxygenase products from omega-3 polyunsaturated fatty acids. Biochem Biophys Res Commun (2005) 338(1):149–57. doi: 10.1016/j.bbrc.2005.07.181
323. Tjonahen E, Oh SF, Siegelman J, Elangovan S, Percarpio KB, Hong S, et al. Resolvin E2: identification and anti-inflammatory actions: pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem Biol (2006) 13(11):1193–202. doi: 10.1016/j.chembiol.2006.09.011
324. Oh SF, Vickery TW, Serhan CN. Chiral lipidomics of E-series resolvins: aspirin and the biosynthesis of novel mediators. Biochim Biophys Acta (2011) 1811(11):737–47. doi: 10.1016/j.bbalip.2011.06.007
325. Isobe Y, Arita M, Matsueda S, Iwamoto R, Fujihara T, Nakanishi H, et al. Identification and structure determination of novel anti-inflammatory mediator resolvin E3, 17,18-dihydroxyeicosapentaenoic acid. J Biol Chem (2012) 287(13):10525–34. doi: 10.1074/jbc.M112.340612
326. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest (2018) 128(7):2657–69. doi: 10.1172/JCI97943
327. Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggstrom JZ, Petasis NA, et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J (2013) 27(7):2573–83. doi: 10.1096/fj.13-227728
328. Aursnes M, Tungen JE, Colas RA, Vlasakov I, Dalli J, Serhan CN, et al. Synthesis of the 16S,17S-Epoxyprotectin Intermediate in the Biosynthesis of Protectins by Human Macrophages. J Nat Prod (2015) 78(12):2924–31. doi: 10.1021/acs.jnatprod.5b00574
329. Jouvene CC, Shay AE, Soens MA, Norris PC, Haeggstrom JZ, Serhan CN. Biosynthetic metabolomes of cysteinyl-containing immunoresolvents. FASEB J (2019) 33(12):13794–807. doi: 10.1096/fj.201902003R
330. Hammond VJ, O’Donnell VB. Esterified eicosanoids: generation, characterization and function. Biochim Biophys Acta (2012) 1818(10):2403–12. doi: 10.1016/j.bbamem.2011.12.013
331. Maskrey BH, Bermudez-Fajardo A, Morgan AH, Stewart-Jones E, Dioszeghy V, Taylor GW, et al. Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J Biol Chem (2007) 282(28):20151–63. doi: 10.1074/jbc.M611776200
332. Morgan LT, Thomas CP, Kuhn H, O’Donnell VB. Thrombin-activated human platelets acutely generate oxidized docosahexaenoic-acid-containing phospholipids via 12-lipoxygenase. Biochem J (2010) 431(1):141–8. doi: 10.1042/BJ20100415
333. Thomas CP, Morgan LT, Maskrey BH, Murphy RC, Kuhn H, Hazen SL, et al. Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J Biol Chem (2010) 285(10):6891–903. doi: 10.1074/jbc.M109.078428
334. Aldrovandi M, Hammond VJ, Podmore H, Hornshaw M, Clark SR, Marnett LJ, et al. Human platelets generate phospholipid-esterified prostaglandins via cyclooxygenase-1 that are inhibited by low dose aspirin supplementation. J Lipid Res (2013) 54(11):3085–97. doi: 10.1194/jlr.M041533
335. Karara A, Dishman E, Falck JR, Capdevila JH. Endogenous epoxyeicosatrienoyl-phospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. J Biol Chem (1991) 266(12):7561–9.
336. O’Donnell VB, Aldrovandi M, Murphy RC, Krönke G. Enzymatically oxidized phospholipids assume center stage as essential regulators of innate immunity and cell death. Sci Signaling (2019) 12(574):eaau2293. doi: 10.1126/scisignal.aau2293
337. Morgan AH, Dioszeghy V, Maskrey BH, Thomas CP, Clark SR, Mathie SA, et al. Phosphatidylethanolamine-esterified eicosanoids in the mouse: tissue localization and inflammation-dependent formation in Th-2 disease. J Biol Chem (2009) 284(32):21185–91. doi: 10.1074/jbc.M109.021634
338. Liu GY, Moon SH, Jenkins CM, Sims HF, Guan S, Gross RW. Synthesis of oxidized phospholipids by sn-1 acyltransferase using 2-15-HETE lysophospholipids. J Biol Chem (2019) 294(26):10146–59. doi: 10.1074/jbc.RA119.008766
339. Jenkins CM, Yang K, Liu G, Moon SH, Dilthey BG, Gross RW. Cytochrome c is an oxidative stress-activated plasmalogenase that cleaves plasmenylcholine and plasmenylethanolamine at the sn-1 vinyl ether linkage. J Biol Chem (2018) 293(22):8693–709. doi: 10.1074/jbc.RA117.001629
340. Schroepfer GJ Jr. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev (2000) 80(1):361–554. doi: 10.1152/physrev.2000.80.1.361
341. Bjorkhem I, Andersson U, Ellis E, Alvelius G, Ellegard L, Diczfalusy U, et al. From brain to bile. Evidence that conjugation and omega-hydroxylation are important for elimination of 24S-hydroxycholesterol (cerebrosterol) in humans. J Biol Chem (2001) 276(40):37004–10. doi: 10.1074/jbc.M103828200
342. Tuckey RC, Cameron KJ. Catalytic properties of cytochrome P-450scc purified from the human placenta: comparison to bovine cytochrome P-450scc. Biochim Biophys Acta (1993) 1163(2):185–94. doi: 10.1016/0167-4838(93)90180-y
343. Mast N, Annalora AJ, Lodowski DT, Palczewski K, Stout CD, Pikuleva IA. Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J Biol Chem (2011) 286(7):5607–13. doi: 10.1074/jbc.M110.188433
344. Lund EG, Kerr TA, Sakai J, Li WP, Russell DW. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem (1998) 273(51):34316–27. doi: 10.1074/jbc.273.51.34316
345. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity (2013) 38(1):106–18. doi: 10.1016/j.immuni.2012.11.004
346. Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A (2009) 106(39):16764–9. doi: 10.1073/pnas.0909142106
347. McDonald JG, Russell DW. Editorial: 25-Hydroxycholesterol: a new life in immunology. J Leukoc Biol (2010) 88(6):1071–2. doi: 10.1189/jlb.0710418
348. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity (2013) 38(1):92–105. doi: 10.1016/j.immuni.2012.11.005
349. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature (2011) 475(7357):524–7. doi: 10.1038/nature10280
350. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct B-cell migration through EBI2. Nature (2011) 475(7357):519–23. doi: 10.1038/nature10226
351. Bodin K, Bretillon L, Aden Y, Bertilsson L, Broome U, Einarsson C, et al. Antiepileptic drugs increase plasma levels of 4beta-hydroxycholesterol in humans: evidence for involvement of cytochrome P450 3A4. J Biol Chem (2001) 276(42):38685–9. doi: 10.1074/jbc.M105127200
352. Heverin M, Meaney S, Brafman A, Shafir M, Olin M, Shafaati M, et al. Studies on the cholesterol-free mouse: strong activation of LXR-regulated hepatic genes when replacing cholesterol with desmosterol. Arterioscler Thromb Vasc Biol (2007) 27(10):2191–7. doi: 10.1161/ATVBAHA.107.149823
353. Pandak WM, Kakiyama G. The acidic pathway of bile acid synthesis: Not just an alternative pathway(☆). Liver Res (2019) 3(2):88–98. doi: 10.1016/j.livres.2019.05.001
354. Griffiths WJ, Yutuc E, Abdel-Khalik J, Crick PJ, Hearn T, Dickson A, et al. Metabolism of non-enzymatically derived oxysterols: Clues from sterol metabolic disorders. Free Radic Biol Med (2019) 144:124–33. doi: 10.1016/j.freeradbiomed.2019.04.020
355. Abdel-Khalik J, Crick PJ, Yutuc E, DeBarber AE, Duell PB, Steiner RD, et al. Identification of 7alpha,24-dihydroxy-3-oxocholest-4-en-26-oic and 7alpha,25-dihydroxy-3-oxocholest-4-en-26-oic acids in human cerebrospinal fluid and plasma. Biochimie (2018) 153:86–98. doi: 10.1016/j.biochi.2018.06.020
356. Vaz FM, Ferdinandusse S. Bile acid analysis in human disorders of bile acid biosynthesis. Mol Aspects Med (2017) 56:10–24. doi: 10.1016/j.mam.2017.03.003
357. Mazzacuva F, Mills P, Mills K, Camuzeaux S, Gissen P, Nicoli ER, et al. Identification of novel bile acids as biomarkers for the early diagnosis of Niemann-Pick C disease. FEBS Lett (2016) 590(11):1651–62. doi: 10.1002/1873-3468.12196
358. Jiang X, Sidhu R, Mydock-McGrane L, Hsu FF, Covey DF, Scherrer DE, et al. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci Transl Med (2016) 8(337):337ra63. doi: 10.1126/scitranslmed.aaf2326
359. Setchell KD, Schwarz M, O’Connell NC, Lund EG, Davis DL, Lathe R, et al. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J Clin Invest (1998) 102(9):1690–703. doi: 10.1172/JCI2962
360. Sjövall J, Griffiths WJ, Setchell KD, Mano N, Goto J. Analysis of Bile Acids. In: Makin H, Gower D, editors. Steroid Analysis. Dordrecht: Springer (2010). p. 837–966. doi: 10.1023/b135931_10
361. Axelson M, Sjovall J. Potential bile acid precursors in plasma–possible indicators of biosynthetic pathways to cholic and chenodeoxycholic acids in man. J Steroid Biochem (1990) 36(6):631–40. doi: 10.1016/0022-4731(90)90182-R
362. Rose KA, Stapleton G, Dott K, Kieny MP, Best R, Schwarz M, et al. Cyp7b, a novel brain cytochrome P450, catalyzes the synthesis of neurosteroids 7alpha-hydroxy dehydroepiandrosterone and 7alpha-hydroxy pregnenolone. Proc Natl Acad Sci U.S.A. (1997) 94(10):4925–30. doi: 10.1073/pnas.94.10.4925
363. Schwarz M, Lund EG, Russell DW. Two 7 alpha-hydroxylase enzymes in bile acid biosynthesis. Curr Opin Lipidol (1998) 9(2):113–8. doi: 10.1097/00041433-199804000-00006
364. Shackleton CH. Role of a disordered steroid metabolome in the elucidation of sterol and steroid biosynthesis. Lipids (2012) 47(1):1–12. doi: 10.1007/s11745-011-3605-6
365. Andrew R. Clinical measurement of steroid metabolism. Best Pract Res Clin Endocrinol Metab (2001) 15(1):1–16. doi: 10.1053/beem.2001.0116
366. Baulieu EE, Robel P, Schumacher M. Neurosteroids: beginning of the story. Int Rev Neurobiol (2001) 46:1–32. doi: 10.1016/s0074-7742(01)46057-0
367. Schumacher M, Guennoun R, Mattern C, Oudinet JP, Labombarda F, De Nicola AF, et al. Analytical challenges for measuring steroid responses to stress, neurodegeneration and injury in the central nervous system. Steroids (2015) 103:42–57. doi: 10.1016/j.steroids.2015.08.013
368. Storbeck KH, Schiffer L, Baranowski ES, Chortis V, Prete A, Barnard L, et al. Steroid Metabolome Analysis in Disorders of Adrenal Steroid Biosynthesis and Metabolism. Endocr Rev (2019) 40(6):1605–25. doi: 10.1210/er.2018-00262
369. Schiffer L, Barnard L, Baranowski ES, Gilligan LC, Taylor AE, Arlt W, et al. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: A comprehensive review. J Steroid Biochem Mol Biol (2019) 194:105439. doi: 10.1016/j.jsbmb.2019.105439
370. Krone N, Hughes BA, Lavery GG, Stewart PM, Arlt W, Shackleton CH. Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS). J Steroid Biochem Mol Biol (2010) 121(3-5):496–504. doi: 10.1016/j.jsbmb.2010.04.010
Keywords: biosynthesis of oxidized lipids, lipoxygenase (LOX), cyclooxygenase (COX), cytochrome P450, aldo-keto reductase (AKR), oxylipins, oxidized phospholipids, sterols and steroid hormones
Citation: Hajeyah AA, Griffiths WJ, Wang Y, Finch AJ and O’Donnell VB (2020) The Biosynthesis of Enzymatically Oxidized Lipids. Front. Endocrinol. 11:591819. doi: 10.3389/fendo.2020.591819
Received: 05 August 2020; Accepted: 26 October 2020;
Published: 19 November 2020.
Edited by:
Maria Fedorova, Leipzig University, GermanyReviewed by:
Consolato Sergi, University of Alberta Hospital, CanadaKatja Hummitzsch, University of Adelaide, Australia
Copyright © 2020 Hajeyah, Griffiths, Wang, Finch and O’Donnell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali A. Hajeyah, aGFqZXlhaGFhQGNhcmRpZmYuYWMudWs=