 Pengyue Xiang
Pengyue Xiang Qiuxia Wu1
Qiuxia Wu1 Huajie Zou
Huajie Zou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 07 October 2020
Sec. Pituitary Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.579436
This article is part of the Research Topic Refractory Pituitary Adenoma—Current Challenges and Emerging Treatments View all 17 articles
Background: Autoimmune hypophysitis (AH) is a primary autoimmune inflammatory disorder of the pituitary gland, which usually presents as a mass in the sella turcica. Systemic lupus erythematosus (SLE) is another inflammatory disorder in which the immune system attacks healthy cells and tissues throughout the body. Although both diseases are autoimmune disorders, they rarely coexist, and the relationship between them is unclear.
Case Report: A 66-year-old man was evaluated at the endocrinology clinic because of worsening fatigue, anorexia, drowsiness, and leg oedema. Examination revealed alertness impairment and lower limb oedema. Laboratory tests showed anterior pituitary hypofunction. The treatment approach, with glucocorticoids and immunosuppressive agents, resulted in long-term remission of symptoms of hypopituitarism and hyponatraemia.
Conclusions: Our case demonstrates a potential association between AH and SLE. AH may need to be considered in the evaluation of SLE patients with headache, hyperprolactinemia, a pituitary mass, and hypopituitarism.
Autoimmune hypophysitis (AH), also known as lymphocytic hypophysitis, is a rare inflammatory disorder that can affect the anterior, posterior or both pituitary lobes (1). Most cases have been diagnosed in women during late pregnancy or the postpartum period (2). Systemic lupus erythematosus (SLE) is a condition where the immune system attacks healthy cells as well as tissues around the body (3). AH is rarely associated with rheumatic diseases, and only 1.3% of AH cases have previously been described to be associated with SLE (4). Here, we describe a case of AH and SLE in a 66-year-old man presenting with fatigue, anorexia, drowsiness, and leg oedema, and we summarize all case reports of AH with SLE to provide some evidence of the pathogenesis, diagnosis and treatments.
A 66-year-old man with a history of diabetes and percutaneous transluminal coronary intervention (PCI) was evaluated at the endocrinology clinic because of worsening fatigue, anorexia, drowsiness, and leg oedema.
The patient had been followed up by one of us (P X) for 5 years because of non-insulin-dependent diabetes mellitus (NIDDM). Three years ago, the patient was diagnosed with cardiovascular disease (CVD) and treated with percutaneous coronary intervention (PCI) in the Department of Cardiology. Five months before presentation, the patient was readmitted to the Department of Cardiology because of fatigue, anorexia, drowsiness, and leg oedema. His temperature was 36.5°C, heart rate was 72 beats/min, and blood pressure was 135/85 mmHg. At the Department of Cardiology, the white-cell and differential counts and blood levels of erythrocyte sedimentation rate (ESR), high-sensitivity C-reactive protein, thyroid stimulating hormone (TSH), and free T3 (FT3) were normal; the level of free T4 (FT4) was decreased; and levels of glycated haemoglobin A1c (HbA1c), triglycerides, and brain natriuretic peptide (BNP) were elevated. The blood sodium level was 131.7 mmol/L [normal range (NR), 137 to 147]. During hospitalization, he received treatment with laxatives due to constipation, and his blood sodium level decreased to 110.3 mmol/L. Therefore, treatment with 3% NaCl was initiated, with obvious relief in symptoms. The patient was discharged when the sodium level reached 130.1 mmol/L.
At the last visit in the endocrinology clinic, the patient complained that these symptoms had worsened during the past week. He reported no headache, dizziness, polydipsia, polyuria, or vomiting. Examination revealed alertness impairment and lower limb oedema. No trochlear nerve palsy, oculomotor nerve paralysis, or visual field defects were detected during examination. As shown in Table 1, laboratory tests showed anterior pituitary hypofunction. The FT3 (1.93 pg/ml; NR, 2.27 to 4.22) and FT4 (0.63 ng/dl; NR, 0.9 to 1.76) decreased without a corresponding increase in TSH (3.00 uIU/ml; NR, 0.55 to 4.78), suggesting central hypothyroidism. Follicle-stimulating hormone (FSH), luteinizing hormone (LH), and testosterone levels were all below the sensitivity of the assay, consistent with hypogonadotropic hypogonadism. Laboratory examination also showed low sodium (124 mmol/L; NR, 137 to 147) and chloride (89 mmol/L; NR, 99 to 110) and low morning cortisol (0.49 µg/dl; NR, 4.26 to 24.85) without a corresponding increase in corticotrophin (ACTH 8.14 pg/ml, NR 7.2–63.4), suggesting central hypercortisolism. The insulin-like growth factor 1 (IGF-1) level was decreased (53 ng/ml; NR, 69-211), and the serum growth hormone (GH) level was normal. Interestingly, prolactin (PRL) was slightly elevated (26.6 ng/ml; NR, 3.6 to 16.3), which occurs in approximately 18% of AH patients (5) and can decrease LH and FSH levels. Test results for rheumatoid factor (RF), antinuclear antibody (ANA), anti-Sm antibody, anti-Sjögren’s syndrome B (anti-SSB), anti-double-stranded DNA antibodies (anti-dsDNA), anti-nRNP/Sm, and anticardiolipin antibody-IgG (ACL-IgG) were positive. Test results for other relevant antibodies were negative (Table 1). The levels of all tumour markers were normal. The magnetic resonance imaging (MRI) scan of the pituitary gland revealed an empty sella (Figure 1). No abnormality was observed in the adrenal MRI.
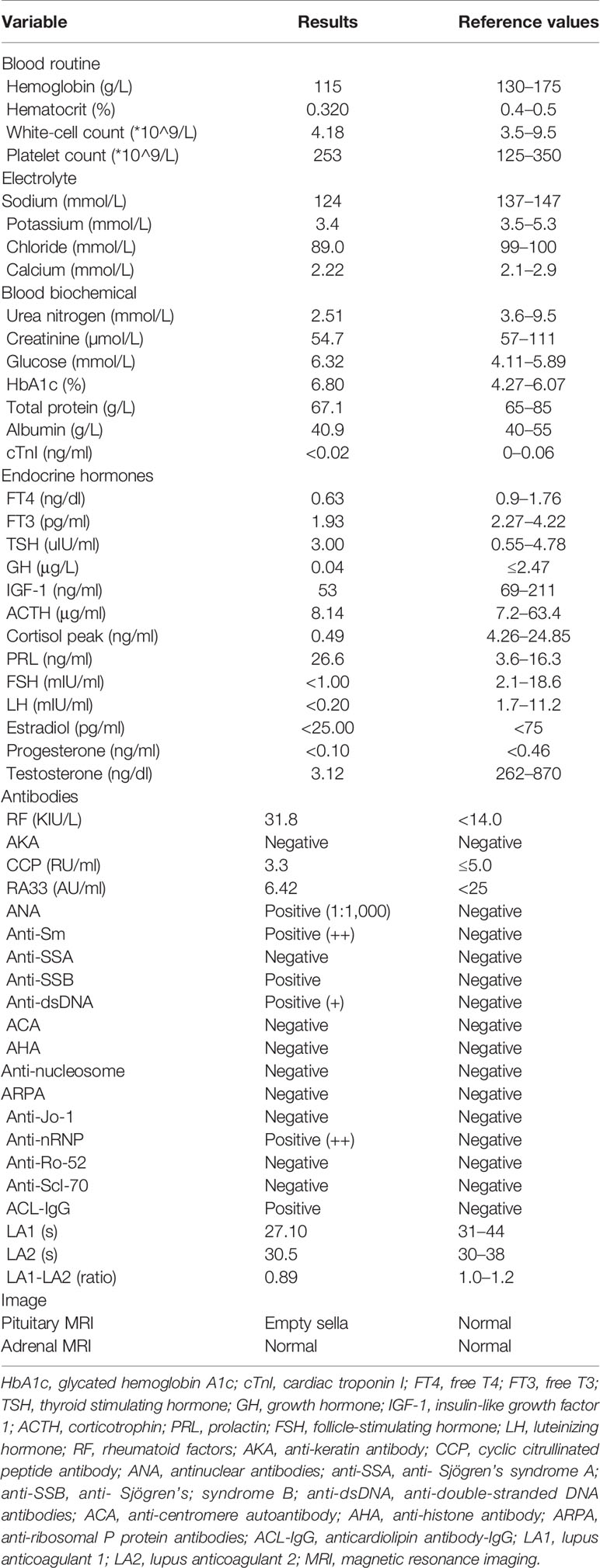
Table 1 Laboratory findings.
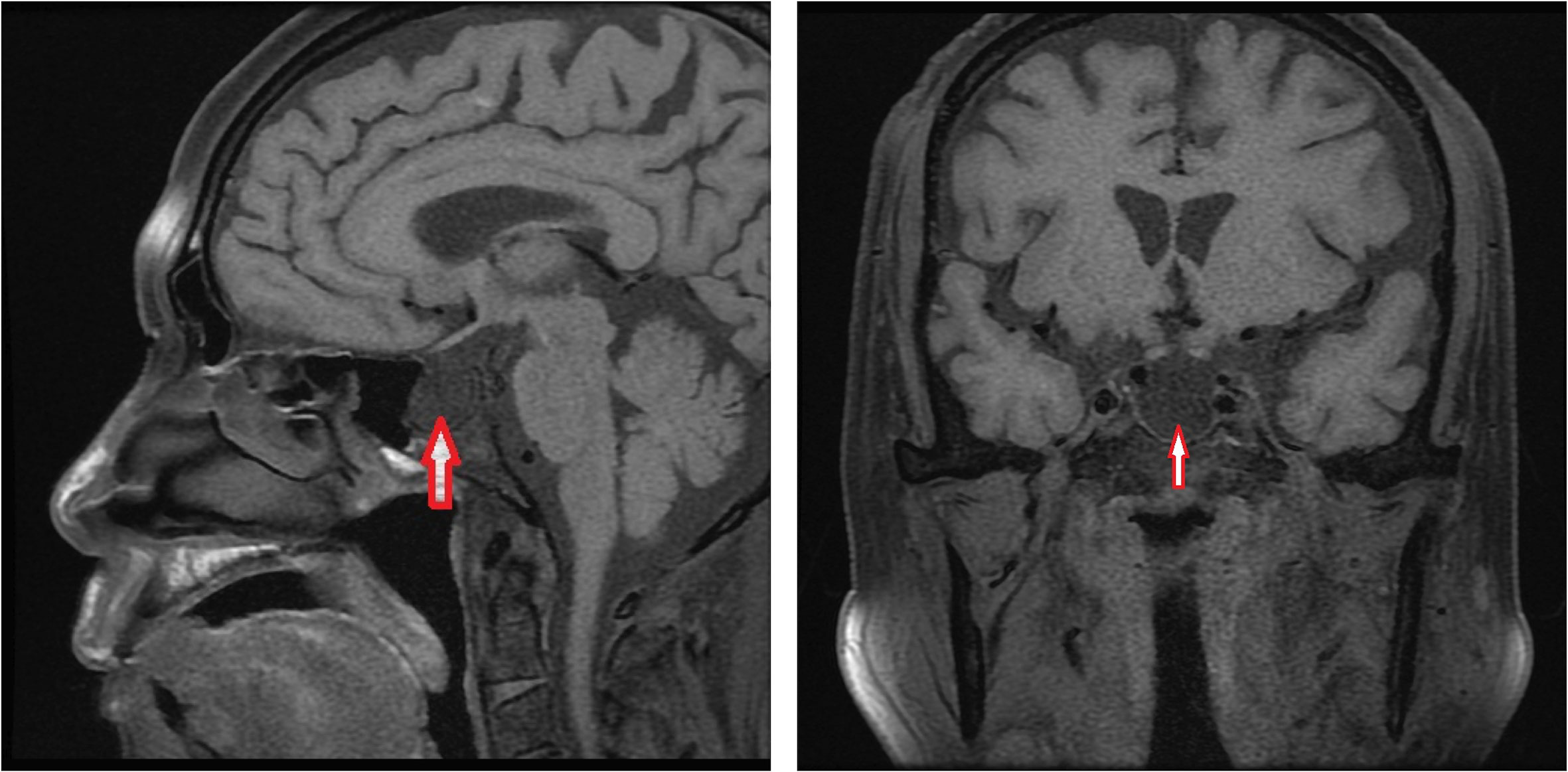
Figure 1 Brain MRI.
Based on the above, the patient was diagnosed with AH, anterior pituitary hypofunction, SLE, NIDDM, and CVD. According to the clinical guidelines (6, 7), the patient was treated with 30 mg/d prednisone, 80 mg/d testosterone undecanoate, and 50 µg/d levothyroxine for the treatment of anterior pituitary hypofunction; 0.4 g/d hydroxychloroquine for the treatment of SLE; 22 U/d premixed human insulin for the treatment of NIDDM; and 75 mg/d clopidogrel, 10 mg/d atorvastatin calcium tablets, 50 mg/d captopril and 100 mg/d metoprolol for the treatment of CVD. Three days after the treatment, the sodium level had risen to 134 mmol/L, and the patient’s symptoms resolved gradually. Then, the patient was discharged with prescriptions for oral administration on the 11th hospital day. The dosage of prednisone was decreased after one month, with a monthly decrease of 5 mg until it was maintained at 10 mg/d. Two months after discharge, the patient reported no fatigue, anorexia, drowsiness, or leg oedema, and the patient’s sodium level was normal at the follow-up visit.
We conducted a literature search with the terms “autoimmune hypophysitis” or “lymphocytic hypophysitis” and “systemic lupus erythematosus” in the PubMed, Embase, and CNKI databases for case reports of AH with SLE from inception to August 10, 2020. Then, manual searching was conducted according to the references of relevant acquired articles. Five cases of AH with SLE were reported (2, 8–11). Unfortunately, we did not obtain the complete publication of the first case reported by Hasegawa et al. (8). We summarized the remaining 4 cases in Table 2 (2, 9–11).
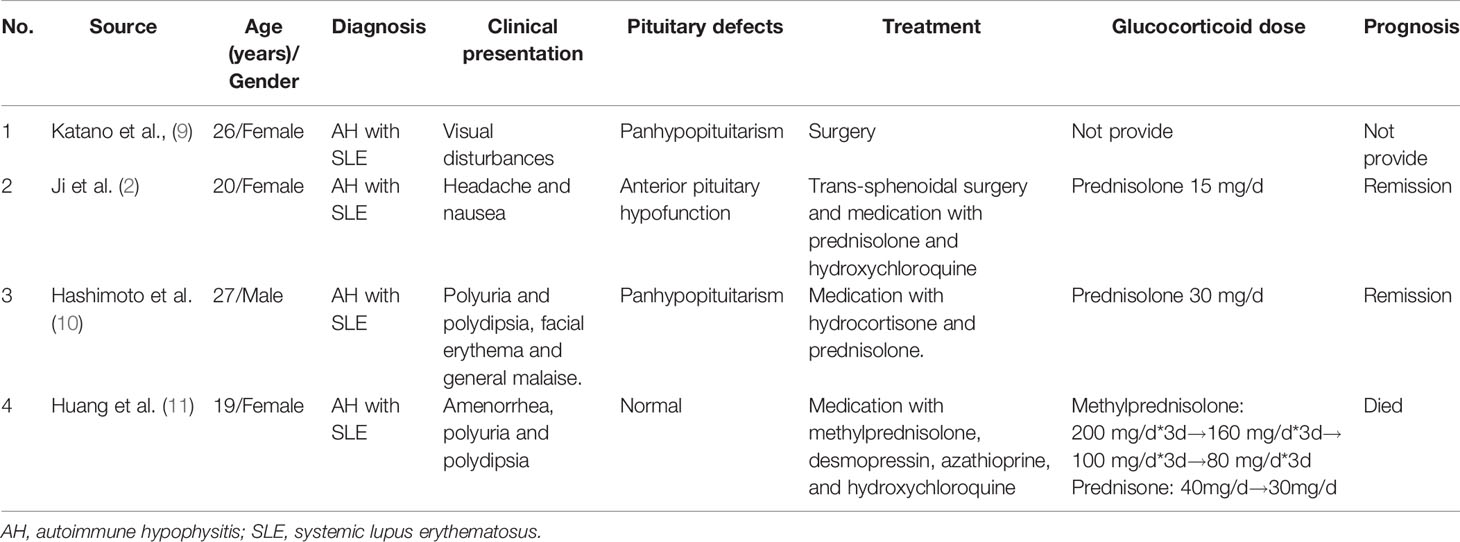
Table 2 Reported cases of AH associated with SLE.
In the four cases of AH with SLE, only one patient was male, and the remaining patients were female. The onset age ranged from 19 to 27 years. Three of them were SLE patients who then developed visual impairment, headache, nausea, polyuria, or polydipsia. One patient was previously diagnosed with diabetes insipidus and then presented with facial erythema and general malaise. Most of the patients were diagnosed based on clinical and imaging features. Two patients underwent surgery, and postoperative pathological examination confirmed the diagnosis. Three patients were treated with glucocorticoid medication. One patient received hydroxychloroquine and azathioprine for the treatment of SLE. Three patients achieved remission, but one died of severe hyponatraemia.
AH is related to a disruption of tissue organization and frequently leads to glandular malfunction, which generally manifests as a mass within the sella turcica, with headache and/or visual disturbances (5). Pituitary enlargement is often secondary to infiltration and oedema. The progression of AH includes remission, spontaneous or pharmacological resolution of the inflammation, or progressive diffuse destruction with glandular atrophy for fibrotic replacement, thus resulting in varying degrees of pituitary dysfunction (1) and symptoms of hypopituitarism, including low ACTH, low TSH, low gonadotropins, or low prolactin (5). Currently, a definitive diagnosis of AH can be based only on pathological examination of a pituitary biopsy sample after invasive surgical intervention. For patients with glandular atrophy, as in the present case, clinicians should make a non-invasive diagnosis through clinical and imaging features and then treat with conservative management with glucocorticoids and other anti-inflammatory and immunosuppressive agents (methotrexate, azathioprine) (12), which aims to reduce inflammation and restore pituitary function.
SLE is a chronic autoimmune disease in which the immune system attacks healthy cells and tissues throughout the body, and the most frequently targeted organs are the kidneys, skin, lungs, brain, and heart (13). AH and SLE are both autoimmune diseases, but we are not aware of any histologically proven description or mechanism that links AH with SLE. It is still unclear whether the pituitary is a target of SLE. Our case demonstrates a potential association between AH and SLE. AH may need to be considered in the evaluation of SLE patients with headache, hyperprolactinemia, a pituitary mass, and hypopituitarism.
In conclusion, we report the case of a 66-year-old man with AH and SLE who presented with hypopituitarism and hyponatraemia. The diagnosis was made mainly based on clinical manifestations, laboratory assessment, and imaging studies. The treatment approach, with glucocorticoids and immunosuppressive agents, resulted in long-term remission of symptoms of hypopituitarism and hyponatraemia.
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HJZ and PX collected data and wrote the manuscript. QW, HZ, and CL provided suggestion during the diagnosis and treatment in this case. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Falorni A, Minarelli V, Bartoloni E, Alunno A, Gerli R. Diagnosis and classification of autoimmune hypophysitis. Autoimmun Rev (2014) 13:412–6. doi: 10.1016/j.autrev.2014.01.021
2. Ji JD, Lee SY, Choi SJ, Lee YH, Song GG. Lymphocytic hypophysitis in a patient with systemic lupus erythematosus. Clin Exp Rheumato (2000) 78–80. doi: 10.1186/ar132
3. Kiriakidou M, Cotton D, Taichman D, Williams S. Systemic lupus erythematosus. Ann Intern Med (2013) 159:ITC4–1. doi: 10.7326/0003-4819-159-7-201310010-01004
4. da Silva BSP, Bonin C, Bueno CBF, Glezer A, Bronstein MD, Carvalho JF. Primary antiphospholipid syndrome and panhypopituitarism: a unique presentation. Acta Reumatol Port (2012) 37:272–5. doi: 10.1186/1471-2474-13-117
5. Caturegli P, Lupi I, Landek-Salgado M, Kimura H, Rose NR. Pituitary autoimmunity: 30 years later. Autoimmun Rev (2008) 7:631–7. doi: 10.1016/j.autrev.2008.04.016
6. Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. Update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis (2019) 78:736–45. doi: 10.1136/annrheumdis-2019-215089
7. Takagi H, Iwama S, Sugimura Y, Takahashi Y, Oki Y, Akamizu T, et al. Diagnosis and treatment of autoimmune and IgG4-related hypophysitis: Clinical guidelines of the Japan endocrine society. Endocr J (2020) 67:373–8. doi: 10.1507/endocrj.EJ19-0569
8. Hasegawa Y, Matsumoto M, Kamimura AYM. A case of systematic lupus erythematosus with autoimmune hypophysitis. Nihon Naika Gakkai Zasshi (1993) 82:582–3. doi: 10.2169/naika.82.582
9. Katano H, Umemura A, Kamiya K, Kanal H, Yamada K. Visual disturbance by lymphocytic hypophysitis in a non-pregnant woman with systemic lupus erythematosus. Lupus (1998) 7:554–6. doi: 10.1191/096120398678920514
10. Hashimoto K, Asaba K, Tamura K, Takao T, Nakamura T. A case of lymphocytic infundibuloneurohypophysitis associated with systemic lupus erythematosus. Endocr J (2002) 49:605–10. doi: 10.1507/endocrj.49.605
11. Huang A, Liu Y, Zhang N, Zhang L, Wang Z, Su X, et al. A case of systemic lupus erythematosus with hypophysitis as the first manifestation. Chin J Rheumatol (2013) 17:285–6. doi: 10.3760/cma.j.issn.1007-7480.2013.04.018
12. Xu C, Ricciuti A, Caturegli P, Keene CD, Kargi AY. Autoimmune lymphocytic hypophysitis in association with autoimmune eye disease and sequential treatment with infliximab and rituximab. Pituitary (2015) 18:441–7. doi: 10.1007/s11102-014-0592-7
Keywords: autoimmune hypophysitis, systemic lupus erythematosus, anterior pituitary hypofunction, autoimmune disease, sella turcica
Citation: Xiang P, Wu Q, Zhang H, Luo C and Zou H (2020) Autoimmune Hypophysitis With Systemic Lupus Erythematosus: A Case Report and Literature Review. Front. Endocrinol. 11:579436. doi: 10.3389/fendo.2020.579436
Received: 02 July 2020; Accepted: 14 September 2020;
Published: 07 October 2020.
Edited by:
Renzhi Wang, Peking Union Medical College Hospital (CAMS), ChinaReviewed by:
Leila Warszawski, Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione, BrazilCopyright © 2020 Xiang, Wu, Zhang, Luo and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huajie Zou, aGp6b3UwNDA2QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.