 Jorge A. Berlanga-Acosta1,2
Jorge A. Berlanga-Acosta1,2 Gerardo E. Guillén-Nieto1,2
Gerardo E. Guillén-Nieto1,2 Nadia Rodríguez-Rodríguez2
Nadia Rodríguez-Rodríguez2 Yssel Mendoza-Mari2
Yssel Mendoza-Mari2 Maria Luisa Bringas-Vega1,3Jorge O. Berlanga-Saez4
Maria Luisa Bringas-Vega1,3Jorge O. Berlanga-Saez4 Diana García del Barco Herrera2Indira Martinez-Jimenez2Sandra Hernandez-Gutierrez5
Diana García del Barco Herrera2Indira Martinez-Jimenez2Sandra Hernandez-Gutierrez5 Pedro A. Valdés-Sosa1,3*
Pedro A. Valdés-Sosa1,3*- 1The Clinical Hospital Chengdu Brain Sciences Institute, University of Electronic Science and Technology of China, Chengdu, China
- 2Tissue Repair, Wound Healing and Cytoprotection Research Group, Biomedical Research Direction, Center for Genetic Engineering and Biotechnology, Playa, Cuba
- 3Cuban Neurosciences Center, Playa, Cuba
- 4Applied Mathematics Department, Institute of Mathematics, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 5Department of Surgery, Hospital Salvador Allende, Havana, Cuba
Diabetes is constantly increasing at a rate that outpaces genetic variation and approaches to pandemic magnitude. Skin cells physiology and the cutaneous healing response are progressively undermined in diabetes which predisposes to lower limb ulceration, recidivism, and subsequent lower extremities amputation as a frightened complication. The molecular operators whereby diabetes reduces tissues resilience and hampers the repair mechanisms remain elusive. We have accrued the notion that diabetic environment embraces preconditioning factors that definitively propel premature cellular senescence, and that ulcer cells senescence impair the healing response. Hyperglycemia/oxidative stress/mitochondrial and DNA damage may act as major drivers sculpturing the senescent phenotype. We review here historical and recent evidences that substantiate the hypothesis that diabetic foot ulcers healing trajectory, is definitively impinged by a self-expanding and self-perpetuative senescent cells society that drives wound chronicity. This society may be fostered by a diabetic archetypal secretome that induces replicative senescence in dermal fibroblasts, endothelial cells, and keratinocytes. Mesenchymal stem cells are also susceptible to major diabetic senescence drivers, which accounts for the inability of these cells to appropriately assist in diabetics wound healing. Thus, the use of autologous stem cells has not translated in significant clinical outcomes. Novel and multifaceted therapeutic approaches are required to pharmacologically mitigate the diabetic cellular senescence operators and reduce the secondary multi-organs complications. The senescent cells society and its adjunctive secretome could be an ideal local target to manipulate diabetic ulcers and prevent wound chronification and acute recidivism. This futuristic goal demands harnessing the diabetic wound chronicity epigenomic signature.
Introduction
Diabetes mellitus (DM) is a heterogeneous metabolic disease characterized by chronic hyperglycemia resulting from defects in insulin secretion, insulin action, or both (1). Despite the genetic underpinnings of the diseases, the prevalence of both type 1 and type 2 diabetes (T1DM and T2DM) is globally increasing at a rate that outpaces genetic variation and approaches to pandemic magnitude (2, 3). Like other age-related chronic diseases, diabetes is essentially impinged by the convergence of basic aging mechanisms that underlie age-related tissue dysfunction (4, 5).
Although more than 300 theories have emerged over the years to explain the intrinsic molecular and evolutionary drivers behind organismal aging (6); the onset of cellular senescence seems to act as a foundational pillar for multi-organs and organismal aging (6–9).
An intense debate has existed so far addressing whether senescence precedes or follows the onset of perpetual inflammation and insulin resistance (IR) (10). Irrespective to “who-precedes-who,” diabetic patients experience an obvious accelerated aging process that increases their susceptibility to morbidity and earlier mortality (10). Hence, diabetes-affected patients have a significantly shorter life expectancy than non-diabetic individuals (11, 12), while this life expectancy reduction is largely dependent on diabetes duration (2, 13).
Aside from the reduced number of fatalities caused by acute diabetic complications, the major clinical challenge of diabetes is the progressive and expansive morbidity and mortality resulting from the long-term secondary complications (14–16). Within the constellation of diabetic complications, the delayed and poorness in triggering and progressing along a physiological repair response is of major clinical significance (17, 18). As in most chronic wounds, the diabetic wound is frequently distinguished by its chronicity and by the asynchrony of the healing phases within a specific wound niche (17, 19, 20).
Diabetes undermines skin cells physiology and progressively intoxicates the dermal layer by the accumulation of advanced glycation end products (AGEs) and free radicals derivatives (21–23). Accordingly, most if not all of the events encompassed within the cutaneous healing process including hemostasis, inflammation, matrix deposition, angiogenesis, contraction, remodeling, and re-epithelialization are somewhat buffeted by diabetes (20, 24). Aside from the impaired healing response, a parallel dark arista of diabetic ulcer pathology is the high rate of ulcer recidivism after the primary wound closure (25, 26). In line with this fact, a classic study reveals that roughly 40% of patients have a recurrence within 1 year after ulcer healing. Thus, the epithelized ulcers are considered as being in remission rather than being healed (10). Additionally, an underappreciated risk of diabetic foot ulcers (DFU) recidivism is its ability to recur or “metastize” at anatomical locations distinct from the primary occurrence, often involving the contralateral limb which entails the risk of subsequent amputation within 5 years following a primary amputation (25, 27).
The synergy between compromised healing machinery and the predisposition to peripheral tissues infection nurture the alarming figures of lower extremity amputations (28). Thus, DFU has historically remained as the worldwide leading factor for non-traumatic lower extremities amputations (29, 30). These evidences support the David's Armstrong “cancer analogy” concept. The concept itself highlights that the 5 year mortality rates associated to foot ulceration and lower extremity amputation, surpasses those registered for five common cancers (31). Subsequent studies have further enriched the notion that lower extremity amputation reduces life expectancy in diabetics (32); and that ulcer severity is a more significant predictor of subsequent mortality than coronary and peripheral arterial disease, or stroke (33).
While healing diabetic wounds is instrumental for amputation prevention, there exists the underlying need to identify the molecular factors driving the secondary diabetic complication of impaired healing (20, 34–36). Hyperglycemia is the major systemic risk factor for the onset and progression of diabetic complications (37, 38); and mechanistically speaking, it is endowed with a proximal position in the diabetic biochemical environment where numerous endogenous stressors drive cellular senescence (7). Chronic low-grade inflammation and an increased burden of senescent cells are hallmarks of aging in diabetic subjects (10). Hyperglycemia per se is known to act as a senescence-promoting factor for cultured cells (39, 40), and steadily precipitates organs complications and functional demise by different mechanistic pathways (10, 41–44).
The notion that cellular senescence is an imperceptible underlying force in the pathogenesis of wound chronicity and ulcers recurrence has been accrued for years (45–48). Consequently, we suggest that diabetes-associated wound healing failure and reduced tissue resilience are clinical translations of an “entrenched” wound senescent cells society, with self-perpetuating and propagating abilities. Subsequent to the enlightening opinion article by Sahin and DePinho (49), which postulates a rational model depicting how mitochondria is intersected by different pro-senescence pathways—We decided to review the current knowledge on cellular senescence as a putative founding pillar for diabetic wound healing impairment. The search strategy involved Medline/Pubmed and other reference data sources as Google Scholar, Scielo, Bioline International (www.bioline.org.br), etc; introducing key words as: chronic wounds+senescent cells, diabetes+senescence, DFU, mitochondria, telomeres erosion, replicative senescence+chronic wounds, fibroblasts+proliferative arrest. In first instance, we delineated the major molecular factors invoked as diabetes-associated senescence drivers in a systemic level and attempted to extrapolate the impact of these hallmarks for the wound microenvironment. The contemporary evidences describing the participation of granulation tissue senescent cells and the impact of diabetic-senescence drivers in mesenchymal stem cells biology were reviewed. All the articles reviewed were limited to English language and with no date restriction. Thus, classic articles dated on the 70's and 80's are referenced.
Diabetes and Cellular Senescence. Brief Mechanistic Overview
Conceptually, aging is defined as an evolutionary process with a time-dependent functional decline that affects all higher organisms. Since cellular senescence contributes to organismal demise aging is associated to morbidity and mortality. Aging therefore involves different interdependent hallmarks on cellular, molecular, and organism levels (50, 51). Senescence is a cellular program that induces proliferative arrest accompanied by morphological modifications, metabolic reprogramming, implementation of a complex proinflammatory spreadable secretome, increased autophagy, apoptosis resistance, and epigenetic reprograming (52–54).
Diabetic environment is overwhelmed of multiple cellular senescence-contributing factors that ultimately translate in organismal aging with well-known nosogenic consequences (7, 41, 55). Diabetes is therefore representative of the model that “organisms accumulate modified macromolecules” and that these macromolecules increase over time and interacts with proteins and tissues, inducing structural changes generating the so called “damage accumulation” (56, 57). As elegantly described by Palmer et al., the fundamental aging mechanisms broadly fall into the following categories: (1) macromolecular dysfunction, (2) sterile inflammation, (3) progenitor cell dysfunction, and (4) cellular senescence (58). Of note, these four mechanisms are represented in diabetes and reciprocally link aging and diabetes (58, 59). Accordingly, many aging hallmarks appear earlier or are overexpressed in T2DM, including the “inflammaging” as other forms of cellular stress: oxidative stress, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress (10, 58, 60).
Hyperglycemia which also comprises the oscillation of the “trivial” blood glucose levels, known as hyperglycemia transient spikes, is a major risk factor for a variety of pathologies and clinical and surgical complications (61, 62), including poor wound healing and wound infections (63–65). Hyperglycemia is also the proximal trigger for all the downstream molecular derangements in diabetes including cellular senescence (66, 67). The excessive generation of ROS and the expansive oxidative attack to multicellular targets including mitochondrial structures, generate an interdependent and perpetuative loop. Mitochondrial and genomic DNA damages including or not telomeric regions trigger different effector pathways that provoke cell senescence and proliferative arrest (Figure 1) (10, 68–72).
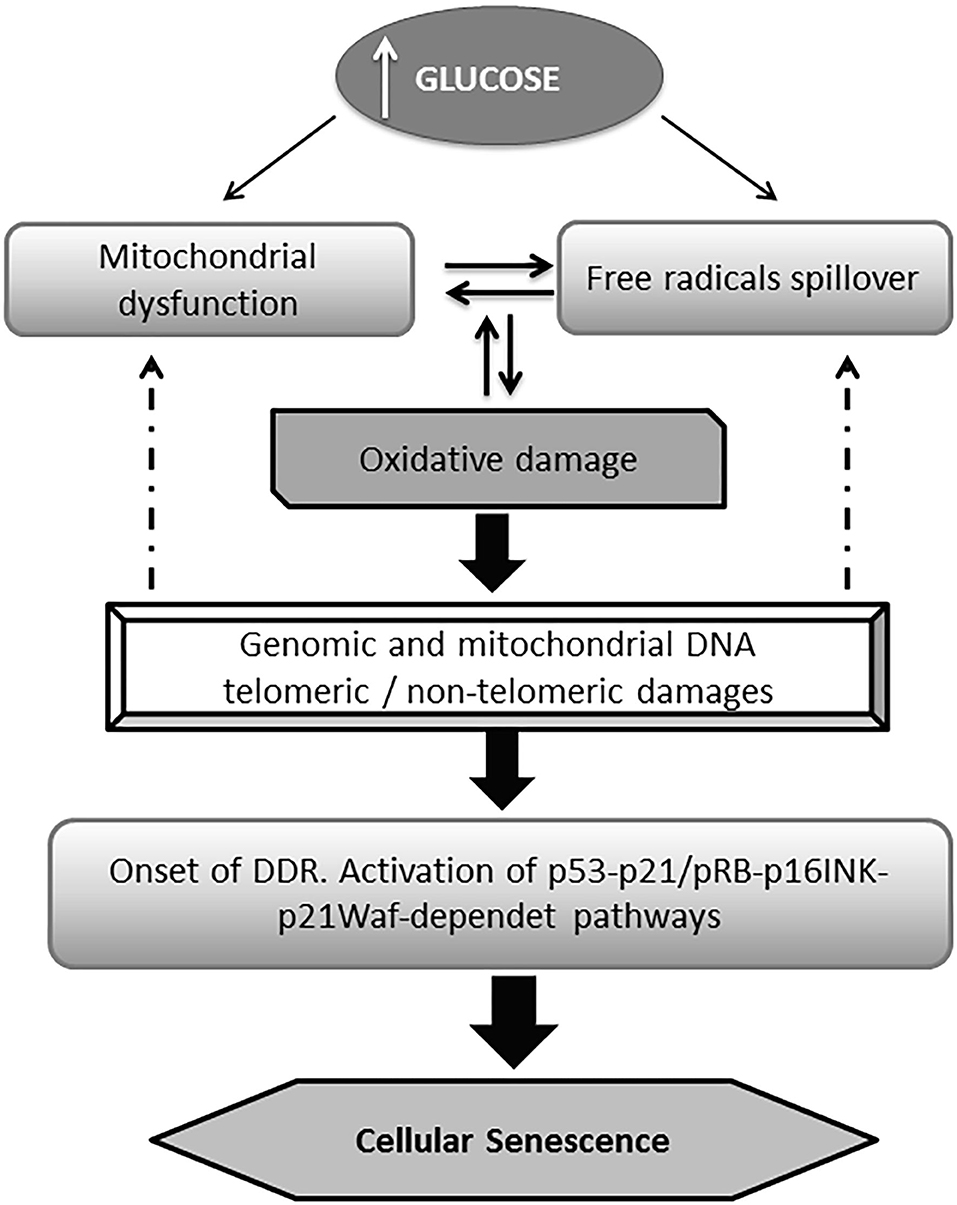
Figure 1. General pathway of glucose-induced senescence. Multiple evidences converge to indicate that high glucose drives premature senescence acting as a proximal trigger for a myriad of subsequent biochemical derangements. Hyperglycemia is ensued by an excessive mitochondrial production of reactive oxygen species (ROS) which in turn generates the typical pro-oxidative environment of diabetes. Alike, mitochondrial dysfunction derives from the ROS-mediated attack to its DNA, and the structural damage to respiratory chain proteins which generates a destructive vicious circuitry. Senescent burden can be multiplied in neighboring cells via the local oxidative milieu. The DNA chemical damage (telomeric and non-telomeric) induced by the impact of ROS is followed by the activation of the DNA damage responses (DDR) pathway; which in turn activates the p53 and/or the p16INK4A pathways. The later facilitates the accumulation of phosphorylated pRb eventually accounting for cell senescence program orchestration.
From the molecular point of view, two major basic operators have been described for the onset of the cellular senescence phenotype derived from the impact of a high glucose burden: (1) p53-p21 and (2) pRb-p16INK4a-p21Waf1 (73–75). Moreover, convergent lines of evidence point to the role played by p53 in the regulation of cell cycle arrest and senescence (50, 76, 77). It has been recently shown that telomeric damage activates a p53-dependent program which leads to mitochondrial failure via the repression of peroxisome proliferator-activated receptor gamma co-activator 1α (PGC-1α) gene expression (78). PGC-1α is a master regulator of mitochondrial biogenesis and function, including oxidative phosphorylation and (ROS) detoxification (79). Other cognate proteins involved in mitochondrial biogenesis and function are also impaired upon the orchestration of the DDR, which ultimately amplifies mitochondrial functional impairment (Figure 2).
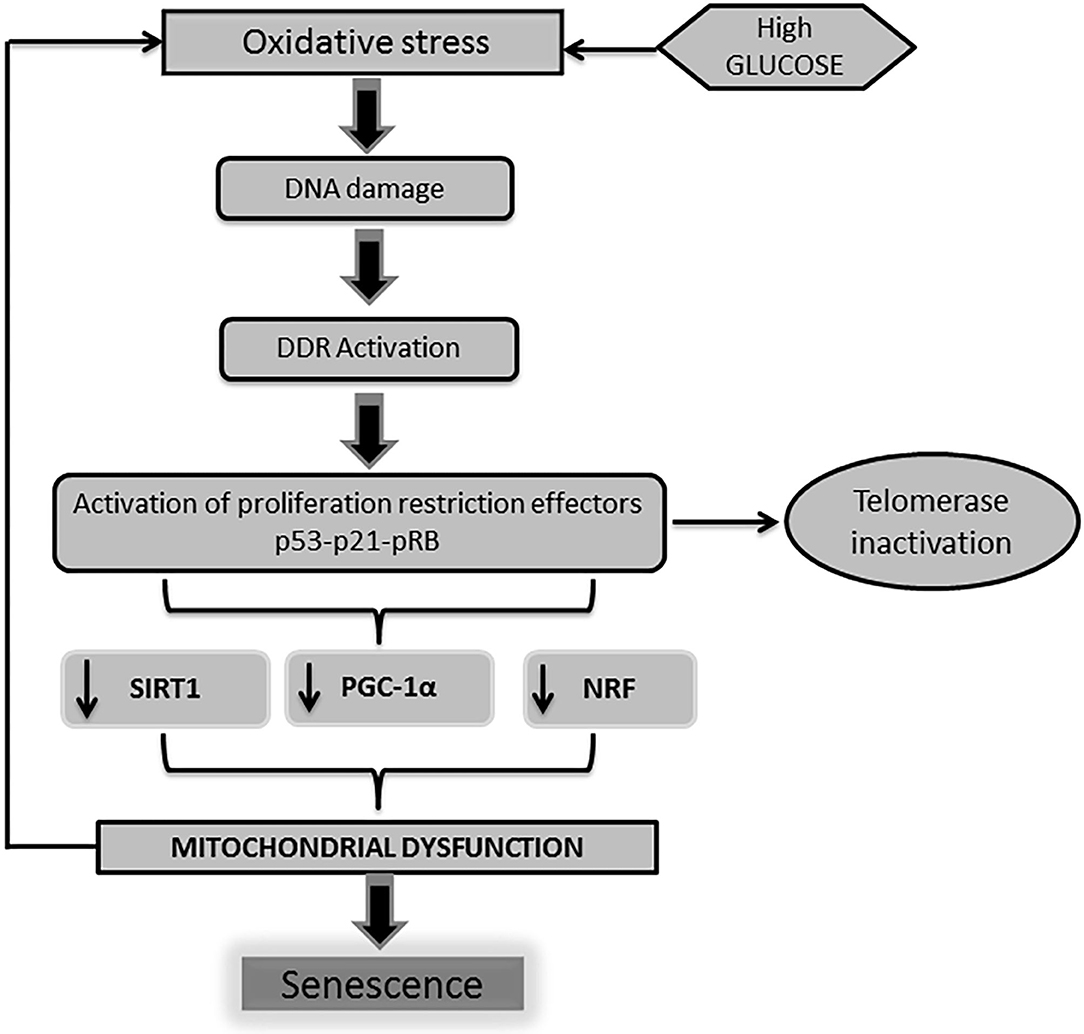
Figure 2. DNA damage is linked to mitochondrial physiology. As described earlier, high glucose levels trigger a cascade of events leading to the activation of professional inhibitors of cell cycle proliferation as p53-p21-pRB. The activation of p53 by DNA damage response (DDR) impacts on mitochondrial physiology and energy metabolism. Telomeric shortening negatively influences mitochondrial homeostasis. This mitochondrial failure mostly derives from the p53-mediated repression of peroxisome proliferator-activated receptor gamma co-activator 1α/β gene (PGC-1α/β) expression. Of note, PGC-1 is largely involved in mitochondrial biogenesis and function, including oxidative phosphorylation and reactive oxygen species (ROS) detoxification. Along with PGC-1 activity abrogation, other mitochondrial biogenesis regulators as nuclear respiratory factor (NRF) and SIRT-1 are also impaired. Here again, pathogenic vicious circles are established leading and amplification of cells senescence as a final outcome.
Likewise, the retinoblastoma protein (pRb) plays a central role toward the onset of senescence. The cell cycle inhibitors p16INK4a and p21Waf1 cooperate to keep pRB in its active hypophosphorylated/growth inhibitory state (80, 81). Of note, nuclear overexpression of p53 phosphorylated on serine-15 and p21Cip have been detected in fibroblasts derived from reluctant-to-heal DFU, supporting the notion that granulation tissue fibroblasts-proliferative arrest, promote cellular senescence and fibrovascular stagnancy (82). Similarly, cultured diabetic cutaneous fibroblasts also exhibit an exaggerated activation of the p53/p21-dependent pathways along with a significant increase in senescence-associated β-galactosidase (SA-β-Gal) activity and phospho-γ-histone H2AX (pH2AX) level, as indicative of cellular senescence and impaired wound healing (83). Outspoken morphological and biochemical alterations in diabetic ulcer and diabetic intact skin-derived fibroblasts are described, which are all suggestive of impaired proliferative capability (84–86). A seminal study by Vande Berg et al. confirmed the existence of senescent fibroblasts in pressure ulcers, which exhibit limited proliferative capability (87). Finally, senescent cells are readily distinguished in addition to their blunted proliferative activity (88, 89), by their larger size and flattened morphology, and an altered gene expression including upregulation of SA-β-Gal and pro-inflammatory chemokines and cytokines (90, 91).
The Senescence Messenger Organ, Metabolic Derangements, and Oxidative Stress on Wound Cells Biology
A singular and distinctive marker for senescent cells is the onset of a pro-inflammatory program identified as senescence-associated secretory phenotype (SASP) (92, 93). This “messenger organ” is able to reinforce the cellular proliferative arrest, and spread different types of senescence messages integrated in the senescence-messaging secretome (SMS) (94). SASP ingredients are mostly constituted by interleukins, chemokines, growth factors, secreted proteases, and secreted insoluble proteins/extracellular matrix (ECM) components (92, 95, 96). Hyperglycemia-induced senescence is associated to a typical diabetic SASP. Reciprocally, SASP further enhances insulin resistance (97), amplifies diabetes-related endovascular and tissue inflammation, and ultimately disseminate a senescence message that impairs diabetic wound matrix accumulation (44). This archetypical SASP transforms healthy cells into aged cells, and perpetuates the residence and the turnover of the senescence cells society and of inflammation- polarized macrophages (44, 98–102) (Figure 3).
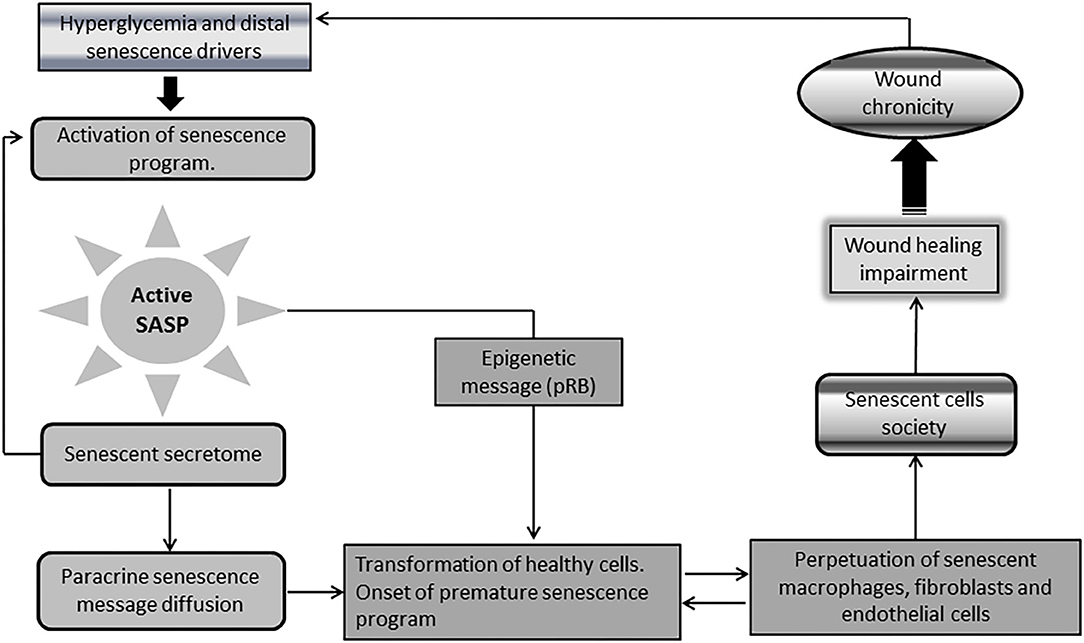
Figure 3. Chronic wounds and the society of senescent cells. A singular and distinctive marker for senescence cells is the onset of a pro-inflammatory program identified as senescence-associated secretory phenotype (SASP). Diabetic senescent cells SASP are typically endowed with inflammatory effectors and matrix degradative enzymes. The paracrine release of this secretome accounts for the diffusion of a progeroid message and ultimately engenders a threshold of senescent cells within the wound via transformation of normal neighbor cells into senescent cells. SASP also spread the epigenetic signature ingredients of senescence which further contributes to expand and perpetuate the so called “senescence cells society” Accordingly, irreversibility of senescence is largely dependent upon pRB epigenetic events. It is therefore plausible that epigenetic mechanisms contribute to shape a long-lasting epigenetic memory for SASP, and the perdurability of the senescent society. The society integrates within the wound bed to M1-polarized macrophages, as arrested fibroblasts, and endothelial cells that “contaminate” keratinocytes leading edge, thus impairing wound closure and promoting wound chronicity. Given that the diabetic chronic wound is constant source of inflammatory mediators and ROS, insulin resistance is amplified leading to more hyperglycemia. This is a major perpetuative pathogenic circuitry. These factors nurture the existence and perdurability of the.
By the contrary, SASP attenuation prolongs health span and lifespan of diabetic mice and slows diabetes progression and/or the development of its different complications (44). Of relevance for the tissue repair process, SASP cocktail contains growth factors and extracellular matrix-remodeling proteases with important functions across the different overlapping phases of the healing process (103). Via the paracrine activity of its soluble senescence messages, SASP may disrupt tissues homeostasis provoking a variety of age-related pathologies and disabilities (95, 104). Surprisingly, two well-reputed growth factors implicated in wound fibro-angiogenic response: transforming growth factor β1 (TGF-β1) and vascular endothelial growth factor (VEGF), are involved in the remote transformation of healthy to senescent cells (105, 106).
Although there is relative scarcity of information regarding the identification or quantification of senescent cells-extracellular vesicles cargo (95); important findings have revealed the link between extracellular miRNAs and the development and progression of T2DM complications. About 40 circulating miRNAs are significantly deregulated and implicated in diabetic endothelial dysfunction, inflammation, cellular senescence (107), and in the pathogenesis of diabetic impaired wound healing (108, 109). Diabetic wounds are typically associated with a persistent inflammatory infiltration that involves accumulation of senescent macrophages. These cells are key effectors for the clearance of senescent cells, and their absence or dormancy may favor a SASP with sustained inflammatory message (48). In line with this, it has been observed that aged and diabetic wounds populated with senescent macrophages produce a CXCR2-enriched SASP. Importantly, CXCR2 is an inflammatory chemokine that drives primary human dermal fibroblasts toward a pro-fibrotic and senescent phenotype, via the paracrine induction of nuclear p21 (53). Furthermore, clinical samples of diabetic wounds exhibit a protracted population of M1 phenotype polarized senescent macrophages, driven by the expressions of NLRP3, caspase1, and IL-1 (110). Taken together these evidences incite to reinforce that: (a) both diabetic and aged wounds are chronically infiltrated by inflammatory mononuclear cells, (b) that diabetes seems to disrupt the physiological fine tuning for macrophages turnover and polarization, (c) these population of inflammation-polarized macrophages (M1) contribute to impaired healing, become senescent, and impose senescence to other granulation tissue cells via an inflammation loaded-SASP (111–114). Once again, the pathogenic synergistic link between chronic inflammation and cellular senescence, toward the implementation of a pathological wound repair is further strengthened.
On the opposite extreme, an elegant study by Birgit Ritschka et al. uncovered the benefits of a transient exposure to SASP by inducing cell plasticity, stemness, and regeneration. Thus, it was anticipated that transient induction of a senescent phenotype produces a “wound healing-promoting SASP” (92). For the scenario of an acute cutaneous healing in which ordinarily senescent fibroblasts and endothelial cells appear few days after skin injury, the SASP-derived platelet growth factor (PDGF)-AA stimulates wound closure via myofibroblast differentiation. Accordingly, there are circumstances in which SASP may exercise a positive role in normal wound repair (115). Likewise, the acute exposure of human dermal fibroblast to hydrogen peroxide successfully induced fibroblasts senescence and an archetypal SASP, which stimulated keratinocytes migration and myofibroblast transition via increased secretion of growth factors including different isoforms of PDGF, TGF-β, and VEGF. Again, this finding indicates that a population of senescent fibroblast from acute wounds, may positively modulate the wound healing process (116).
Chronic diabetic wounds show marked differences not only compared to acute non-diabetic wounds, but also between both chronic healing, and non-healing diabetic wounds (117–119). Given that senescent cells may have diverse and context-dependent effects on tissue, the existence of two different types of SASP is predictable. One associated to acutely induced and physiological healing wounds, and the concomitant to chronic wounds—as diabetic foot ulcers in which the secretome is basically inflammatory and anti-proliferative. Therefore, SASP is a dynamic and malleable organ whose phenotype appears to act in a tissue-specific manner, within a temporary window, and vectoring particular chemical and epigenetic signatures (120–122).
In the realm of cellular metabolism, it is known that metabolic dysfunction is a powerful organismal driving force for aging, and a senescence hallmark (123) with meaningful repercussion in diabetes (59, 124). Diabetes per se is a metabolic disease in its basic origin, but that progress along with a continuous deterioration of proximal metabolic regulators such as PGC-1α, SIRT 1, and mTOR (78, 79, 125–132). PGC-1 plays an essential role as a master metabolic regulator as mitochondrial biogenesis and function, oxidative phosphorylation, and ROS detoxification (79). PGC-1α axis failure contributes to glucose intolerance, mitochondrial bankruptcy, systemic chronic inflammation, amplified pro-oxidative environment, and ultimately to organismal aging (133, 134). SIRT1 is a prominent member of a family of NAD-dependent enzymes that inhibits transcriptional factors, such as p300, NF-κB, P38MAPK, Histone 3, MMP-9, FOXO3a, and most importantly to p53 (135). SIRT1 exhibit numerous and diverse biological functions including the regulation of energy homeostasis, cell cycle, inflammation, oxidative stress threshold, ER stress-related apoptosis, and ultimately a senescence-preventive factor (136, 137). Conclusively, both PGC-1 and SIRT1 alone or in physiological synergy are key modulators of molecules and pathways involved in cellular senescence. This anti-senescence guardian role is played by different interconnected mechanisms, that protect and generates mitochondria, reduce tissue inflammation, and combat oxidative and nitrosilative stress (138–142).
Recent evidences document that SIRT1 activation accelerates and improves the wound healing response in diabetic mice by promoting angiogenesis, via an angio-protective effect against oxidative stress injury (143). In line with this, the SIRT 1 agonist Resveratrol has shown to reduce the hyperglycemia-triggered endothelial dysfunction, to stimulate angiogenesis and consequently wound closure in db/db mice. Again SIRT 1 axis activation reduces oxidative stress and favors downstream pro-angiogenic pathways (144). Furthermore, two anti-aging proxies like metformin and resveratrol have proved to stimulate wound healing in aged animals, by preventing age-related AMPK deactivation, and attenuating age-associated angiogenic inhibition (145). These experiments confirm the pro-senescent driving effect of ROS-oxidative stress and their negative impact for wound healing biology, especially in diabetic organisms (143, 146–149).
Mitochondria, routinely identified as the powerhouse of the cell are the major generator of members of the ROS family such as superoxide anion, hydroxyl and peroxyl radicals, and hydrogen peroxide (150–153). Furthermore, ROS/oxidative stress/mitochondrial dysfunction and inflammation behave as an indissoluble, interconnected nosogenic unit that links organismal aging, T2DM, and poor wound healing (73, 154–158). Converging evidences demonstrate that high levels of ROS disrupt the normal healing process (159–161) and constitute a hallmark in chronic wounds including recalcitrant DFU (147, 159, 162). In this scenario ROS overproduction is associated to the upregulation of cell cycle inhibitors as to positivity for the SA-β gal marker. The two most studied cell cycle proliferation inhibitors the p53-p21 and the p16-Rb pathways are responsive to ROS-induced telomere erosion, and to non-telomeric DNA damage (SIPS) via oxidative stress (7, 163). Accordingly, oxidative stress-influenced events translate in a collection of wound cells senescence attributes as proliferative arrest, fibroblasts reduced migration, imperfect and poor angiogenesis, amplified apoptosis of granulation tissue productive cells, extended inflammatory polarization, and ultimately wound chronicity (164–166).
Glycolysis is enhanced as cells undergo replicative senescence. Glycolytic enzymes are a major target of oxidation and modification by AGEs, and ROS products during replicative senescence (167). This accounts for a fall of ATP and GTP intracellular levels (167). Mechanistically, enhanced glycolysis contributes to the development of a senescent phenotype (168, 169); as it is the case for the diabetic abnormal increase in glycolysis which seems to drive vascular aging (170). Experiments from two decades ago illustrated that normal cutaneous fibroblasts became arrested and reluctant to proliferate, when they were exposed T2DM-wound fibroblasts conditioned media. This proliferative arrest was associated to decreased DNA content, and was proportional to L-lactate production and incorporation of D-glucose in the media (171, 172).
Conclusively, diabetic metabolic and biochemical derangements as glycolysis are drivers for the onset of cellular senescence and organismal aging. Metabolic dysfunctions trigger a wave of molecular derangements where glucooxidative and nitrosilative stress, inflammatory, and cellular senescence, intersect and cooperate for the implementation of a wound chronicity profile.
Cellular Senescence in Diabetic Sweet Environment
The role of cellular senescence within the complex pathogenic realm of T2DM dates back to the 70's and 80's of last century. The inspiring theory stated the existence of a kind of a pre-senescent state in fibroblast from pre-diabetic subjects which appear to predetermine the wound healing failure (45). This “pre-senescent state” notion has remained effective and validated nowadays (173), and has been expanded to extracutaneous cells (60). Aside from the impact generated by the exposure to glucose spikes which enforce pro-senescent and apoptogenic signatures in most skin cells (45), and even cerebral cells populations (174); these germane studies allow to infer that a persistent heritable abnormality, related to a precocious cellular senescence is present in mesenchymal tissues of genetically predisposed subjects to suffer of diabetes (45). The forces that “erase” from the core of the cell the intrinsic mechanisms to migrate and proliferate within a physiological repair response remain unclear (175–177). Nevertheless, local fibroblasts senescence has been invoked as a major wound healing deterrent factor (53, 178, 179); particularly when a senescent cells threshold is reached per tissue area (180, 181).
The skin is the preferential target organ for extrinsic and intrinsic aging driving forces which account for keratinocytes and fibroblasts to precocious decline (182). Skin fibroblast is a mesenchymal-derived cell with a pivotal role in wound repair. It is difficult to conceive granulation tissue creation, contraction, and basement membrane elaboration in a mammal devoid of this type of cells (183, 184). Cutaneous fibroblasts are sensitive cells, impacted by a variety of stressors which may modify their biological behavior and ultimately the wound healing course (185, 186). High glucose levels and their adjacent biochemical derangements act as a major skin cells pro-senescent factor (7, 187, 188). Even in non-diabetic subjects, high glucose levels are associated to an apparent cutaneous advanced age (189). Proliferative refractoriness, torpid migration capabilities, susceptibility to apoptosis, and limited secretion of extracellular matrix constituents, are hallmarks of diabetics' cutaneous fibroblasts (165, 190). These hallmarks have been historically reproduced in vitro when healthy donor-derived cells, are exposed to high glucose burden stress (63, 171, 191, 192). These fibroblasts adverse behavior is over-represented in DFU, and represent active pieces within the pathogenic puzzle of wound chronicity (17, 193, 194). Classic studies by Miriam Loots and co-workers illustrated that fibroblasts isolated from the ulcer bed of T2DM-donor patients, exhibit a diminished proliferative capacity and an abnormal morphology (84). This observation became paradigmatic when converging studies reproducibly showed that human cutaneous, renal, and periodontal fibroblasts cultured under “hyperglycemic” conditions, expressed a variety of premature cellular senescence markers including inhibition of spontaneous and growth factors-stimulated proliferation (20, 195–198).
Influential studies of late 70's indicated that high glucose concentrations directly or indirectly damage cell's DNA, induce telomeric attrition and other forms of DNA damage in cultured cells (199), These findings have been recently substantiated including the toxic effects of AGEs (200, 201). The early in vitro models founded the classic thesis of a “point-of-no-return,” beyond which hyperglycemia resulted in irreversible progression to premature senescence (202). Form those years it was also known that cutaneous fibroblasts derived from insulin-dependent or insulin-independent diabetic patients, exhibit abnormal replicative capacity in vitro and that senescence, was far more precocious than in non-diabetic control subjects (203). Another sequence of enlightening in vitro experiments documented that diabetic' cutaneous fibroblasts exhibited impoverished synthetic and secreting capabilities (192, 204). By the contrary, glucose restriction to cultured human diploid fibroblasts inhibited the onset of premature senescence, and extended mean survival days and lifespan (205). It was later shown that when normal subjects-derived fibroblasts were challenged with the conditioned media from T2DM counterparts, proliferation was inhibited in a dose-dependent manner. Accordingly, this proliferative arrest turned a “reproducible and transmissible trait” which nurtured our hypothesis on the existence of a “senescence memory-transmissible factor” (88).
Premature cellular senescence has also been observed in endothelial cells exposed to a “diabetic environment” recreated by exposure to glycated collagen I. Although these cells did not show telomeric dysfunction, the process appeared presided by an increased expression of p14 and p53 consequent to an excessive oxidative stress (39, 206). Similarly, adipocytes subjected to glucose oscillations elicit an oxidative environment that damaged telomeres, and increased the presence of p53, p21, as pro-inflammatory cytokines (207). High glucose also impairs cell migration. Fibroblasts from diabetic mice migrate 75% less than those from normoglycemic controls, and display a defective response to hypoxia, a condition commonly present in chronic wounds (208, 209). Furthermore, skin keratinocytes exposed to high glucose also exhibit abnormal cellular morphology, insulin resistance, and decreased proliferation (66, 210). Taken together these experimental data support the clinical statement that abnormal levels of glycated hemoglobin and fasting glucose, are associated to poor ulcers healing and higher figures of lower extremity amputation in diabetics (211). Conclusively, fibroblast, endothelial cells, and keratinocytes –the most important cells for skin injury repair– are sensitive to hyperglycemia and oxidative stress, which act as the main steering factors toward senescence (202, 212).
Wound Cells-Senescence Induction and Maintenance
Acute senescence seems to be a programmed process that is triggered in response to discrete stressors. It is established with fast kinetics and normally contributes to tissue homeostasis and repair (115). As described earlier, this type of acute injury-induced senescent response is endowed with its own SASP/secretome with wound healing promoting activity (115, 213). In contrast, chronic senescence may result from long-term unscheduled damage, and it is often associated with detrimental processes such as the creation of a stable society of senescent cells (90, 214).
Senescence maintenance poises as a relevant and futuristic target given its clinical implications for novel diagnostic procedures and therapeutic approaches. As most biological process, the acquisition of a senescent phenotype may be a progressively escalating, multistep event (215), what suggests that the molecular operators eliciting senescence are not necessarily those involved in its perpetuation. Accordingly, the chronic senescent phenotype is dictated by epigenetic mechanisms that ensure its long survival within the wound bed. The existence of a clinical correlation between a quantitative in vitro senescent cells population and a time-to-healing, support the notion of a perpetuated senescence society (216, 217). Mathematical simulation models have in silico predicted the dynamics of a senescent cells society. It is concluded that within a week time period a cell has an 83.22% possibility of entering a pre-senescent state if senescence soluble instructions are received from a neighbor senescent cell (218). This suggests the existence of a continuous population turnover with a stable senescent phenotype.
Diabetic ulcer cells are chronically exposed to major stressors that sculpt wound cells phenotype. In this respect, the local impact of a metabolically active SASP is likely to be pathogenically crucial via its SMS. A living, active SASP secretome would guarantee a constant pro-senescent cells threshold toward the establishment of a chronicity phenotype. In support to this observation is the fact that cells isolated from chronic wounds, become incompetent to upregulate those chemokines and cytokines involved in chronic inflammation resolution (219). Of note, cultured senescent fibroblasts in addition to be proliferative incompetent, decrease the synthesis and drive the degradation of matrix components through the expression of SASP ingredients (196, 220). This behavior suggests the persistence and dominance of the senescent imprint even after separated from the donor organism. These elements together again emphasize on the hypothesis of an existing “senescence phenotype memory.” This alleged memory may justify why wound bed cells subjected to “ideal” tissue culture conditions recreate the same behavioral traits as when in the donor's organism (165). In line with this we and other groups, have accumulated evidences supporting that wound senescent cells are able to reproduce and transmit their original morphological and functional phenotypic traits to their progeny.
1) Young granulation tissue of neuropathic diabetic ulcers reproduces a collection of senescent vascular defects observed in distant, intact, dermal vessel that take years-long for evolvement. The driving forces behind the “inheritance” of these progeroid morphological traits are likely associated to an epigenetic senescence signature (221).
2) Similarly, diabetic granulation tissue although being a “de novo” and short-lived “welding tissue,” its cells exhibit a sort of genetic or epigenetic imprinting for the deranged expression of glucose metabolism-related genes, which are implicated in insulin resistance, cellular senescence, and T2DM pathophysiology (221).
3) The in vivo proliferative arrest shown by ischemic ulcers fibroblasts is maintained even when cultured under “ideal physiological” conditions. This in vitro proliferative arrest coincides with the transcriptional upregulation of p53 mRNA, and its nuclear translocation with phosphorylation on serine 15. Ultimately these cells exhibited a conspicuous expression of p21Cip along with a marked under-expression of cyclin D1. Taken together, these findings describe molecular ingredients of senescence (82).
4) Classic studies by other groups also document the involvement of senescent phenotype fibroblasts derived from pressure ulcers in the mechanism of wound chronification. Accordingly, although these cells are cultured under standard conditions and remain viable, still they exhibit proliferative arrest as in vivo, being unable to complete DNA synthesis (87). Remarkably, elimination of pressure/ischemia stressors does not guarantee the resumption of a physiological healing trajectory (178).
5) Paradigmatic evidence on the existence of a senescent perpetuation program within the ulcer is the need for frequent sharp debridement (178). This procedure is ultimately addressed to remove proliferative senescent cells which hamper wound closure (178, 222). In this scenario Stojadinovic and co-workers demonstrated that c-myc overexpression is implicated in keratinocytes proliferative and migration arrest (223).
In conclusion, senescent phenotypic traits are transmitted and reproduced to descendent cells within the diabetic ulcer bed, and do not seem to fade away along successive generations' of in vitro passages.
Diabetics' Stem Cells are not Exempted From Senescence
Mesenchymal stem cells (MSCs) are multipotent cells with a fibroblast-like morphology that are considered a lifelong cellular reservoir to ensure the continuous generation, replacement, and restitution of multiple tissue lineages (224–226). MSCs regenerative properties are mediated by their ability to infiltrate and engraft injured areas, where they reduce inflammation, promote angiogenesis, prevent apoptosis, improve scar formation, and mediate tissue remodeling via the paracrine secretion of chemokines and growth factors (227, 228). These pluripotent stem cells are capable of reprogramming into differentiated phenotypes participating in regenerative and reparative programs of most tissues and organs (229–234).
Within the realm of impaired diabetic wound healing, MSCs allogenic transplantation has demonstrated significant healing improvement at both experimental (235–237), and clinical scenarios (238–240). It is controversial however whether autologous MSCs regenerative and pro-survival capabilities, are conserved within diabetic organisms given the complex, invasive, and “metastazing” pathophysiology of this disease (241, 242). Diabetic animals show a lower number of circulating MSCs with deteriorated proliferation and survival capabilities. Furthermore, these cells also exhibit an impaired recruitment and insufficient engraftment response within diabetic wounds (241, 243). Similarly, diabetic-associated MSCs dysfunctionalities are described in humans. Diabetic individuals show lower levels of CD34–/CD133+/KDR+ endothelial progenitor cells (EPC) counts along with higher apoptotic rates of circulating EPC (244). Ultimately, MSCs population is affected at every physiological respect by the cytotoxic diabetic environment (245, 246), which turns it prone to replicative senescence and limits its multipotency expansion ability (69, 247, 248).
Converging evidences conclusively demonstrate how toxic high glucose burden may be for survival, differentiation plasticity, and regenerative competence for different stem cells lineages (249–253). MSCs are markedly susceptible to hyperglycemia-induced ROS/oxidative stress-mediated senescence (245, 254). The perpetuative vicious circle integrated by hyperglycemia/mitochondrial dysfunction/oxidative stress appears again as a master driver to MSCs senescence (255, 256). In this scenario, ROS/oxidative stress activate a p53 program with the ensued cells proliferative arrest as described for mitotic differentiated stem cells (257). Thus, as for other cells stirpes, redox imbalance is a key factor in imposing a premature senescence program (249, 256). Accordingly, the diabetic pro-oxidative environment is a major contributing factor for premature MSCs senescence and functional demise (246). Ultimately, MSCs are victims like other somatic cells of the hyperglycemia-determined oxidative stress with the ensued mitochondrial and genomic DNA damage, the onset of a pathogenic SASP, and the paracrine inflammatory reaction (256, 258, 259). The continuous surge of cytotoxic and pro-inflammatory mediators of the diabetic environment, render MSCs numerical and functionally deficitary for the repair process of diabetic ulcers (260). As summarized in Figure 4, MSCs do not escape from the entangled and interconnected mechanisms that drive cellular senescence in diabetes.
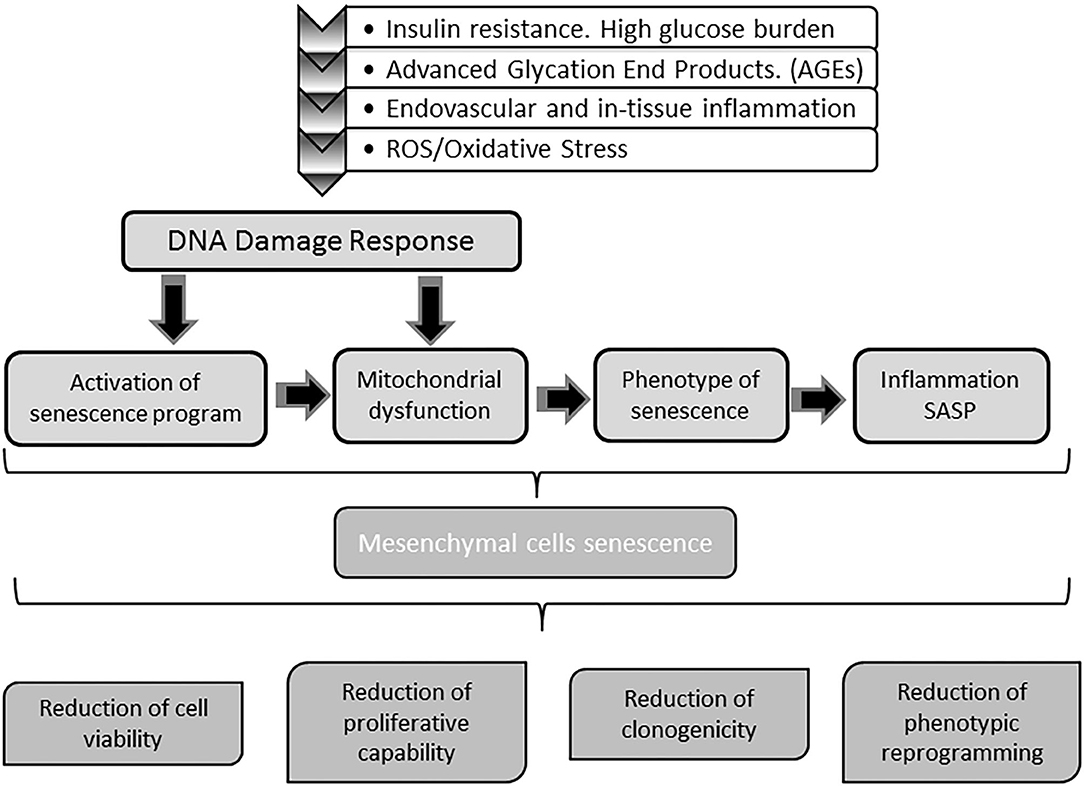
Figure 4. Impact of diabetes on stem cells physiology. Mesenchymal stem cells (MSC) are also targeted by the molecular gluco-oxidative hostile environment of diabetes. The mechanisms and pathways whereby MSC become senescent are the same senescent stressors buffeting differentiated somatic cells. Senescence of MSC impairs stem cells physiology and indirectly that of somatic cells in both mitotic and post-mitotic populations. Upon senescence, stem cells circulating pool is reduced and their ability to engraft injured tissues is also compromised. Concurrently, the capabilities of these cells to assist in wound repair as to participate in epithelial cells populations turnover is also impaired. Globally speaking, with diabetes MSC senescence the tissue's resilience based on the self-renewal potential are undermined.
Concluding Remarks
Diabetes is a pre-conditioning and powerful driver of organismal aging. The biological foundations of aging primarily involve cellular senescence and diabetes is plethoric in senescence-driving factors. Definitively, high glucose concentrations are the proximal trigger for all the subsequent and long lasting molecular by-products, which turn diabetes an archetypal disease of “aging hallmarks.”
Cellular senescence is considered today as intimately related to diabetes complications progression, which are a major cause for poor health span and decreased lifespan. Cellular senescence is also a major steering factor for diabetic poor wound healing response. Pathogenically speaking, fibroblasts, endothelial cells, and keratinocytes are targeted by a broad range of diabetes-related pro-senescence factors. Most interestingly, these cells become and remain stubbornly refractory to migrate, proliferate, and secrete matrix ingredients both in vivo and in vitro—For the later, suggesting the existence of a sculptured epigenomic memory that along with the wound local senescence secretome creates the long-lasting senescent cells society within the wound. This society with its active pro-aging secretome contributes to wound chronicity perpetuation. Diabetic wounds chronicity phenotype is also impinged by two major integrated and cooperative forces: “metabolic memory” and “senescence memory” —both endowed with the ability to manipulate cells behavior. A frightened stamp of diabetic foot wounds is their recurrences short time after re-epithelialization. We deem that the episode of this in situ re-ulceration is an illustrative reflection of the consequence of a long-subsisting senescence society with imprinted “memories” and an active diabetic SASP secretome.
Both bone marrow and tissue niches of MSCs become prematurely senescent under the pressure of the diabetes biochemical environment. These cells are extremely sensitive to glycoxidation products as to oxidative stress, which reduces the population of biological competent cells for epithelial organs self-renewal, for tissue maintenance, and ultimately to actively participate in tissue injury repair, especially in post-mitotic organs.
All together these molecular and cellular senescence traits underlie the diabetic's organismal illnesses and tissues vulnerabilities. Although this has been a long-sought goal we deem that diabetes still waits for a pharmacological re-interpretation. It is likely that establishing “lines of treatments” beyond glucose-lowering medications and improving insulin sensitivity, will translate in great therapeutic value for reducing, and even preventing diabetic complications including the torpid healing phenotype. These “lines of treatments” may represent a cluster of tactics addressed to control cellular senescence and ultimately organismal aging. Alluring targets could be:
1) The molecular basis of diabetic metabolic and senescence memories and their epigenetic signatures for selective pharmacological manipulation.
2) Pharmacological compounds with the ability to selectively modify the histone landscape in a complication and tissue-specific manner.
3) Mitochondria are a sensitive, appealing, and unexplored therapeutic target field. Pharmacological tools aimed to preserve the axis telomeres-mitochondrial function and accordingly to regulate its biogenesis could be promising. “Mitochondrio-therapy” could not only revolutionize diabetes, but expand to cancer and neurodegenerative diseases.
4) Finally, the identification of therapeutic interventions acting as selective senolytics is justified.
Harnessing and tempering diabetic cellular senescence in an appropriate therapeutic opportunity window, will be of paramount clinical and societal significance.
Author Contributions
JB-A, GG-N, and PV-S contributed equally to the initial project conception. JB-A wrote the initial draft of the manuscript. DG, YM-M, NR-R, IM-J, SH-G, and JB-S contributed to analysis and interpretation of all the references and data related to this MS. MB-V was in charge of the preparation of the manuscript and the whole editorial process. JB-A and JB-S contributed equally to data collection. All authors contributed to the interpretation, critical review, and approved the final version of the manuscript.
Funding
The participation of JB-A, GG-N, MB-V, and PV-S in this research was funded by the International Academician Station for Global Precision Medicine and the Project number Y03111023901014005 at the University of the Electronic Sciences and Technology of China UESTC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to the Joint China-Cuba Lab for Frontiers Research in Translational Neurotechnology, subordinated to the University of Electronic Sciences and Technology and the Cuban Neuroscience Center for the support to this work. Also to the dedicated physicians and patients from the Hospital Salvador Allende, La Habana Cuba, where most of the clinical work of this research has been conducted.
References
1. Kerner W, Bruckel J, German Diabetes A. Definition, classification and diagnosis of diabetes mellitus. Exp Clin Endocrinol Diabetes. (2014) 122:384–6. doi: 10.1055/s-0034-1366278
2. Skyler JS, Bakris GL, Bonifacio E, Darsow T, Eckel RH, Groop L, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes. (2017) 66:241–55. doi: 10.2337/db16-0806
3. Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys Ther. (2008) 88:1254–64. doi: 10.2522/ptj.20080020
4. Longo M, Bellastella G, Maiorino MI, Meier JJ, Esposito K, Giugliano D. Diabetes and aging: from treatment goals to pharmacologic therapy. Front Endocrinol. (2019) 10:45. doi: 10.3389/fendo.2019.00045
5. Schwartz SS, Epstein S, Corkey BE, Grant SFA, Gavin Iii JR, Aguilar RB, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. (2017) 28:645–55. doi: 10.1016/j.tem.2017.05.005
6. Pomatto LCD, Davies KJA. Adaptive homeostasis and the free radical theory of ageing. Free Radic Biol Med. (2018) 124:420–30. doi: 10.1016/j.freeradbiomed.2018.06.016
7. Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL. Cellular senescence in Type 2 diabetes: a therapeutic opportunity. Diabetes. (2015) 64:2289. doi: 10.2337/db14-1820
8. Warraich U-e-A, Hussain F, Kayani HUR. Aging - oxidative stress, antioxidants and computational modeling. Heliyon. (2020) 6:e04107. doi: 10.1016/j.heliyon.2020.e04107
9. Vaiserman A, Lushchak O. Developmental origins of type 2 diabetes: focus on epigenetics. Ageing Res Rev. (2019) 55:100957. doi: 10.1016/j.arr.2019.100957
10. Prattichizzo F, de Nigris V, La Sala L, Procopio AD, Olivieri F, Ceriello A. “Inflammaging” as a druggable target: a senescence-associated secretory phenotype-centered view of Type 2 diabetes. Oxid Med Cell Longev. (2016) 2016:1810327. doi: 10.1155/2016/1810327
11. Dale AC, Nilsen TI, Vatten L, Midthjell K, Wiseth R. Diabetes mellitus and risk of fatal ischaemic heart disease by gender: 18 years follow-up of 74,914 individuals in the HUNT 1 study. Eur Heart J. (2007) 28:2924–9. doi: 10.1093/eurheartj/ehm447
12. Huang ES, Laiteerapong N, Liu JY, John PM, Moffet HH, Karter AJ. Rates of complications and mortality in older patients with diabetes mellitus: the diabetes and aging study. JAMA Intern Med. (2014) 174:251–8. doi: 10.1001/jamainternmed.2013.12956
13. Leal J, Gray AM, Clarke PM. Development of life-expectancy tables for people with type 2 diabetes. Eur Heart J. (2009) 30:834–9. doi: 10.1093/eurheartj/ehn567
14. LeRoith D, Biessels GJ, Braithwaite SS, Casanueva FF, Draznin B, Halter JB, et al. Treatment of diabetes in older adults: an endocrine society* clinical practice guideline. J Clin Endocrinol Metab. (2019) 104:1520–74. doi: 10.1210/jc.2019-00198
15. Wu T, Qiao S, Shi C, Wang S, Ji G. Metabolomics window into diabetic complications. J Diabetes Invest. (2018) 9:244–55. doi: 10.1111/jdi.12723
16. Ahlqvist E, van Zuydam NR, Groop LC, McCarthy MI. The genetics of diabetic complications. Nat Rev Nephrol. (2015) 11:277–87. doi: 10.1038/nrneph.2015.37
17. Davis FM, Kimball A, Boniakowski A, Gallagher K. Dysfunctional wound healing in diabetic foot ulcers: new crossroads. Curr Diab Rep. (2018) 18:2. doi: 10.1007/s11892-018-0970-z
18. Frykberg RG, Banks J. Challenges in the treatment of chronic wounds. Adv Wound Care. (2015) 4:560–82. doi: 10.1089/wound.2015.0635
19. Gomes A, Teixeira C, Ferraz R, Prudencio C, Gomes P. Wound-healing peptides for treatment of chronic diabetic foot ulcers and other infected skin injuries. Molecules. (2017) 22:1743. doi: 10.3390/molecules22101743
20. Berlanga-Acosta J, Fernández-Montequín J, Valdés-Pérez C, Savigne-Gutiérrez W, Mendoza-Marí Y, García-Ojalvo A, et al. Diabetic foot ulcers and epidermal growth factor: revisiting the local delivery route for a successful outcome. Biomed Res Int. (2017) 2017:2923759. doi: 10.1155/2017/2923759
21. Argyropoulos AJ, Robichaud P, Balimunkwe RM, Fisher GJ, Hammerberg C, Yan Y, et al. Alterations of dermal connective tissue collagen in diabetes: molecular basis of aged-appearing skin. PLoS ONE. (2016) 11:e0153806. doi: 10.1371/journal.pone.0153806
22. Fournet M, Bonte F, Desmouliere A. Glycation damage: a possible hub for major pathophysiological disorders and aging. Aging Dis. (2018) 9:880–900. doi: 10.14336/AD.2017.1121
23. Yamagishi S, Maeda S, Matsui T, Ueda S, Fukami K, Okuda S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim Biophys Acta. (2012) 1820:663–71. doi: 10.1016/j.bbagen.2011.03.014
24. Perez-Favila A, Martinez-Fierro ML, Rodriguez-Lazalde JG, Cid-Baez MA, Zamudio-Osuna MJ, Martinez-Blanco MDR, et al. Current therapeutic strategies in diabetic foot ulcers. Medicina. (2019) 55:714. doi: 10.3390/medicina55110714
25. Petersen BJ, Rothenberg GM, Lakhani PJ, Zhou M, Linders DR, Bloom JD, et al. Ulcer metastasis? Anatomical locations of recurrence for patients in diabetic foot remission. J Foot Ankle Res. (2020) 13:1. doi: 10.1186/s13047-020-0369-3
26. Huang ZH, Li SQ, Kou Y, Huang L, Yu T, Hu A. Risk factors for the recurrence of diabetic foot ulcers among diabetic patients: a meta-analysis. Int Wound J. (2019) 16:1373–82. doi: 10.1111/iwj.13200
27. Orneholm H, Apelqvist J, Larsson J, Eneroth M. Recurrent and other new foot ulcers after healed plantar forefoot diabetic ulcer. Wound Repair Regen. (2017) 25:309–15. doi: 10.1111/wrr.12522
28. Blakytny R, Jude E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med. (2006) 23:594–608. doi: 10.1111/j.1464-5491.2006.01773.x
29. Armstrong DG, Boulton AJM, Bus SA. Diabetic foot ulcers and their recurrence. N Engl J Med. (2017) 376:2367–75. doi: 10.1056/NEJMra1615439
30. Bus SA, Lavery LA, Monteiro-Soares M, Rasmussen A, Raspovic A, Sacco ICN, et al. Guidelines on the prevention of foot ulcers in persons with diabetes (IWGDF 2019 update). Diabetes Metab Res Rev. (2020) 36(Suppl. 1):e3269. doi: 10.1002/dmrr.3269
31. Armstrong D, Wrobel J, Robbins J. Guest editorial: are diabetes-related wounds and amputations worse than cancer? Int Wound J. (2008) 4:286–7. doi: 10.1111/j.1742-481X.2007.00392.x
32. Martins-Mendes D, Monteiro-Soares M, Boyko EJ, Ribeiro M, Barata P, Lima J, et al. The independent contribution of diabetic foot ulcer on lower extremity amputation and mortality risk. J Diabetes Complications. (2014) 28:632–8. doi: 10.1016/j.jdiacomp.2014.04.011
33. Brennan MB, Hess TM, Bartle B, Cooper JM, Kang J, Huang ES, et al. Diabetic foot ulcer severity predicts mortality among veterans with type 2 diabetes. J Diabetes Complications. (2017) 31:556–61. doi: 10.1016/j.jdiacomp.2016.11.020
34. den Dekker A, Davis FM, Kunkel SL, Gallagher KA. Targeting epigenetic mechanisms in diabetic wound healing. Transl Res. (2019) 204:39–50. doi: 10.1016/j.trsl.2018.10.001
35. Okonkwo UA, Chen L, Ma D, Haywood VA, Barakat M, Urao N, et al. Compromised angiogenesis and vascular integrity in impaired diabetic wound healing. PLoS ONE. (2020) 15:e0231962. doi: 10.1371/journal.pone.0231962
36. Barman PK, Koh TJ. Macrophage dysregulation and impaired skin wound healing in diabetes. Front Cell Dev Biol. (2020) 8:528. doi: 10.3389/fcell.2020.00528
37. Ma RC. Genetics of cardiovascular and renal complications in diabetes. J Diabetes Invest. (2016) 7:139–54. doi: 10.1111/jdi.12391
38. Blair M. Diabetes mellitus review. Urol Nurs. (2016) 36:27–36. doi: 10.7257/1053-816X.2016.36.1.27
39. Chen J, Brodsky SV, Goligorsky DM, Hampel DJ, Li H, Gross SS, et al. Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circ Res. (2002) 90:1290–8. doi: 10.1161/01.RES.0000022161.42655.98
40. Li Y, Tollefsbol TO. p16(INK4a) suppression by glucose restriction contributes to human cellular lifespan extension through SIRT1-mediated epigenetic and genetic mechanisms. PLoS ONE. (2011) 6:e17421. doi: 10.1371/journal.pone.0017421
41. Verzola D, Gandolfo MT, Gaetani G, Ferraris A, Mangerini R, Ferrario F, et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol. (2008) 295:F1563–73. doi: 10.1152/ajprenal.90302.2008
42. Meza CA, La Favor JD, Kim D-H, Hickner RC. Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int J Mol Sci. (2019) 20:3775. doi: 10.3390/ijms20153775
43. Murray CE, Coleman CM. Impact of diabetes mellitus on bone health. Int J Mol Sci. (2019) 20:4873. doi: 10.3390/ijms20194873
44. Prattichizzo F, de Nigris V, Mancuso E, Spiga R, Giuliani A, Matacchione G, et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. (2018) 15:170–81. doi: 10.1016/j.redox.2017.12.001
45. Berlanga-Acosta J, Armstrong DG, Schultz GS, Herrera-Martinez L. Chronic wounds with emphasis in diabetic foot ulcers. Biomed Res Int. (2014) 2014:890352. doi: 10.1155/2014/890352
46. Da Silva-Alvarez S, Guerra-Varela J, Sobrido-Camean D, Quelle A, Barreiro-Iglesias A, Sanchez L, et al. Cell senescence contributes to tissue regeneration in zebrafish. Aging Cell. (2020) 19:e13052. doi: 10.1111/acel.13052
47. Song P, An J, Zou MH. Immune clearance of senescent cells to combat ageing and chronic diseases. Cells. (2020) 9:671. doi: 10.3390/cells9030671
48. Wang Z, Shi C. Cellular senescence is a promising target for chronic wounds: a comprehensive review. Burns Trauma. (2020) 8:tkaa021-tkaa. doi: 10.1093/burnst/tkaa021
49. Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. (2010) 464:520–8. doi: 10.1038/nature08982
50. Hohn A, Weber D, Jung T, Ott C, Hugo M, Kochlik B, et al. Happily (n)ever after: aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. (2017) 11:482–501. doi: 10.1016/j.redox.2016.12.001
52. McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. (2018) 217:65–77. doi: 10.1083/jcb.201708092
53. Wilkinson HN, Clowes C, Banyard KL, Matteuci P, Mace KA, Hardman MJ. Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. J Invest Dermatol. (2019) 139:1171–81.e6. doi: 10.1016/j.jid.2019.01.005
54. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
55. Rosso A, Balsamo A, Gambino R, Dentelli P, Falcioni R, Cassader M, et al. p53 Mediates the accelerated onset of senescence of endothelial progenitor cells in diabetes. J Biol Chem. (2006) 281:4339–47. doi: 10.1074/jbc.M509293200
56. Moldogazieva NT, Mokhosoev IM, Mel'nikova TI, Porozov YB, Terentiev AA. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid Med Cell Longev. (2019) 2019:3085756. doi: 10.1155/2019/3085756
57. Ogrodnik M, Salmonowicz H, Gladyshev VN. Integrating cellular senescence with the concept of damage accumulation in aging: relevance for clearance of senescent cells. Aging Cell. (2019) 18:e12841. doi: 10.1111/acel.12841
58. Palmer AK, Gustafson B, Kirkland JL, Smith U. Cellular senescence: at the nexus between ageing and diabetes. Diabetologia. (2019) 62:1835–41. doi: 10.1007/s00125-019-4934-x
59. Stout MB, Justice JN, Nicklas BJ, Kirkland JL. Physiological aging: links among adipose tissue dysfunction, diabetes, and frailty. Physiology. (2017) 32:9–19. doi: 10.1152/physiol.00012.2016
60. Giovos G, Yavropoulou MP, Yovos JG. The role of cellular senescence in diabetes mellitus and osteoporosis: molecular pathways and potential interventions. Hormones. (2019) 18:339–51. doi: 10.1007/s42000-019-00132-6
61. Mouri M, Badireddy M. Hyperglycemia. StatPearls. Treasure Island, FL: StatPearls Publishing Copyright © 2020 StatPearls Publishing LLC (2020).
62. Chia CW, Egan JM, Ferrucci L. Age-related changes in glucose metabolism, hyperglycemia, and cardiovascular risk. Circ Res. (2018) 123:886–904. doi: 10.1161/CIRCRESAHA.118.312806
63. Hu SC, Lan CE. High-glucose environment disturbs the physiologic functions of keratinocytes: focusing on diabetic wound healing. J Dermatol Sci. (2016) 84:121–7. doi: 10.1016/j.jdermsci.2016.07.008
64. Kruse CR, Singh M, Sorensen JA, Eriksson E, Nuutila K. The effect of local hyperglycemia on skin cells in vitro and on wound healing in euglycemic rats. J Surg Res. (2016) 206:418–26. doi: 10.1016/j.jss.2016.08.060
65. Rodriguez-Buitrago A, Basem A, Okwumabua E, Enata N, Evans A, Pennings J, et al. Hyperglycemia as a risk factor for postoperative early wound infection after bicondylar tibial plateau fractures: determining a predictive model based on four methods. Injury. (2019) 50:2097–102. doi: 10.1016/j.injury.2019.07.028
66. Spravchikov N, Sizyakov G, Gartsbein M, Accili D, Tennenbaum T, Wertheimer E. Glucose effects on skin keratinocytes: implications for diabetes skin complications. Diabetes. (2001) 50:1627–35. doi: 10.2337/diabetes.50.7.1627
67. Lee PG, Halter JB. The pathophysiology of hyperglycemia in older adults: clinical considerations. Diabetes Care. (2017) 40:444–52. doi: 10.2337/dc16-1732
68. Ksiazek K, Korybalska K, Jorres A, Witowski J. Accelerated senescence of human peritoneal mesothelial cells exposed to high glucose: the role of TGF-beta1. Lab Invest. (2007) 87:345–56. doi: 10.1038/labinvest.3700519
69. Stolzing A, Coleman N, Scutt A. Glucose-induced replicative senescence in mesenchymal stem cells. Rejuvenation Res. (2006) 9:31–5. doi: 10.1089/rej.2006.9.31
70. Chang TC, Hsu MF, Wu KK. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PLoS ONE. (2015) 10:e0126537. doi: 10.1371/journal.pone.0126537
71. Aluksanasuwan S, Plumworasawat S, Malaitad T, Chaiyarit S, Thongboonkerd V. High glucose induces phosphorylation and oxidation of mitochondrial proteins in renal tubular cells: a proteomics approach. Sci Rep. (2020) 10:5843. doi: 10.1038/s41598-020-62665-w
72. Pu J, Zhu S, Zhou D, Zhao L, Yin M, Wang Z, et al. Propofol alleviates apoptosis induced by chronic high glucose exposure via regulation of HIF-1alpha in H9c2 cells. Oxid Med Cell Longev. (2019) 2019:4824035. doi: 10.1155/2019/4824035
73. Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am J Physiol Endocrinol Metab. (2019) 316:E268–E85. doi: 10.1152/ajpendo.00314.2018
74. Pinti M, Cevenini E, Nasi M, de Biasi S, Salvioli S, Monti D, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur J Immunol. (2014) 44:1552–62. doi: 10.1002/eji.201343921
75. Sahin E, dePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. (2012) 13:397–404. doi: 10.1038/nrm3352
76. Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules. (2020) 10:420. doi: 10.3390/biom10030420
77. Fujita K. p53 isoforms in cellular senescence- and ageing-associated biological and physiological functions. Int J Mol Sci. (2019) 20:6023. doi: 10.3390/ijms20236023
78. Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. (2011) 470:359–65. doi: 10.1038/nature09787
79. Rius-Perez S, Torres-Cuevas I, Millan I, Ortega AL, Perez S. PGC-1alpha, inflammation, and oxidative stress: an integrative view in metabolism. Oxid Med Cell Longev. (2020) 2020:1452696. doi: 10.1155/2020/1452696
80. Becker T, Haferkamp S. Molecular mechanisms of cellular senescence. In: Wang Z, Inuzuka H, editors. Senescence and Senescence-Related Disorders. London: IntechOpen (2013). doi: 10.5772/54120
81. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. doi: 10.1038/nrm3823
82. Berlanga-Acosta J, Mendoza-Mari Y, Martinez MD, Valdes-Perez C, Ojalvo AG, Armstrong DG. Expression of cell proliferation cycle negative regulators in fibroblasts of an ischemic diabetic foot ulcer. a clinical case report. Int Wound J. (2013) 10:232–6. doi: 10.1111/j.1742-481X.2012.12000.x
83. Bitar MS, Abdel-Halim SM, Al-Mulla F. Caveolin-1/PTRF upregulation constitutes a mechanism for mediating p53-induced cellular senescence: implications for evidence-based therapy of delayed wound healing in diabetes. Am J Physiol Endocrinol Metab. (2013) 305:E951–63. doi: 10.1152/ajpendo.00189.2013
84. Loots MA, Lamme EN, Mekkes JR, Bos JD, Middelkoop E. Cultured fibroblasts from chronic diabetic wounds on the lower extremity (non-insulin-dependent diabetes mellitus) show disturbed proliferation. Arch Dermatol Res. (1999) 291:93–9. doi: 10.1007/s004030050389
85. Raizada MK, Tan G, Fellows RE. Fibroblastic cultures from the diabetic db/db mouse. Demonstration of decreased insulin receptors and impaired responses to insulin. J Biol Chem. (1980) 255:9149–55.
86. Arquilla ER, Weringer EJ, Nakajo M. Wound healing: a model for the study of diabetic angiopathy. Diabetes. (1976) 25(Suppl. 2):811–9.
87. Vande Berg JS, Rudolph R, Hollan C, Haywood-Reid PL. Fibroblast senescence in pressure ulcers. Wound Repair Regen. (1998) 6:38–49. doi: 10.1046/j.1524-475X.1998.60107.x
88. Berlanga-Acosta J, Mendoza Y, García Ojalvo A, Fernández Mayola M, Guillen G. Epidermal growth factor therapy impact on scar tissue resilience of diabetic lower limbs ulcers-An enlightening hypothesis. J Diabetes Metab. (2018) 9:1000798. doi: 10.4172/2155-6156.1000798
89. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. (2018) 28:436–53. doi: 10.1016/j.tcb.2018.02.001
90. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. (2018) 128:1238–46. doi: 10.1172/JCI95148
91. Anerillas C, Abdelmohsen K, Gorospe M. Regulation of senescence traits by MAPKs. GeroScience. (2020) 42:397–408. doi: 10.1007/s11357-020-00183-3
92. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. (2017) 31:172–83. doi: 10.1101/gad.290635.116
93. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. (2017) 21:21–8. doi: 10.1016/j.ebiom.2017.04.013
94. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. (2009) 9:81–94. doi: 10.1038/nrc2560
95. Basisty N, Kale A, Patel S, Campisi J, Schilling B. The power of proteomics to monitor senescence-associated secretory phenotypes and beyond: toward clinical applications. Expert Rev Proteomics. (2020) 17:297–308. doi: 10.1080/14789450.2020.1766976
96. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
97. Palmer AK, Kirkland JL. Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp Gerontol. (2016) 86:97–105. doi: 10.1016/j.exger.2016.02.013
98. Donath MY. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia. (2016) 59:679–82. doi: 10.1007/s00125-016-3873-z
99. de Rekeneire N, Peila R, Ding J, Colbert LH, Visser M, Shorr RI, et al. Diabetes, hyperglycemia, and inflammation in older individuals: the health, aging and body composition study. Diabetes Care. (2006) 29:1902–8. doi: 10.2337/dc05-2327
100. Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes. (2008) 57:3090–8. doi: 10.2337/db08-0564
101. Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. (2003) 52:1256–64. doi: 10.2337/diabetes.52.5.1256
102. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. (2017) 8:1960. doi: 10.3389/fimmu.2017.01960
103. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
104. Ferrucci L, Gonzalez-Freire M, Fabbri E, Simonsick E, Tanaka T, Moore Z, et al. Measuring biological aging in humans: a quest. Aging Cell. (2020) 19:e13080. doi: 10.1111/acel.13080
105. Balducci L, Ershler WB. Cancer and ageing: a nexus at several levels. Nat Rev Cancer. (2005) 5:655–62. doi: 10.1038/nrc1675
106. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. (2013) 15:978–90. doi: 10.1038/ncb2784
107. Prattichizzo F, Giuliani A, De Nigris V, Pujadas G, Ceka A, La Sala L, et al. Extracellular microRNAs and endothelial hyperglycaemic memory: a therapeutic opportunity? Diabetes Obes Metab. (2016) 18:855–67. doi: 10.1111/dom.12688
108. Rafehi H, El-Osta A, Karagiannis TC. Genetic and epigenetic events in diabetic wound healing. Int Wound J. (2011) 8:12–21. doi: 10.1111/j.1742-481X.2010.00745.x
109. Jhamb S, Vangaveti VN, Malabu UH. Genetic and molecular basis of diabetic foot ulcers: clinical review. J Tissue Viability. (2016) 25:229–36. doi: 10.1016/j.jtv.2016.06.005
110. Zhang X, Dai J, Li L, Chen H, Chai Y. NLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. J Diabetes Res. (2017) 2017:5281358. doi: 10.1155/2017/5281358
111. Morey M, O'Gaora P, Pandit A, Helary C. Hyperglycemia acts in synergy with hypoxia to maintain the pro-inflammatory phenotype of macrophages. PLoS ONE. (2019) 14:e0220577. doi: 10.1371/journal.pone.0220577
112. Makrantonaki E, Wlaschek M, Scharffetter-Kochanek K. Pathogenesis of wound healing disorders in the elderly. J Dtsch Dermatol Ges. (2017) 15:255–75. doi: 10.1111/ddg.13199
113. Zhao R, Liang H, Clarke E, Jackson C, Xue M. Inflammation in chronic wounds. Int J Mol Sci. (2016) 17:2085. doi: 10.3390/ijms17122085
114. Lau EYM, Carroll EC, Callender LA, Hood GA, Berryman V, Pattrick M, et al. Type 2 diabetes is associated with the accumulation of senescent T cells. Clin Exp Immunol. (2019) 197:205–13. doi: 10.1111/cei.13344
115. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. (2014) 31:722–33. doi: 10.1016/j.devcel.2014.11.012
116. Hou J, Kim S. Possible role of ginsenoside Rb1 in skin wound healing via regulating senescent skin dermal fibroblast. Biochem Biophys Res Commun. (2018) 499:381–8. doi: 10.1016/j.bbrc.2018.03.170
117. Strang H, Kaul A, Parikh U, Masri L, Saravanan S, Li H, et al. Role of cytokines and chemokines in wound healing. In: Bagchi D, Das A, Roy S, editors. Wound Healing, Tissue Repair, and Regeneration in Diabetes. Cambridge, MA: Academic Press (2020) p. 197–235. doi: 10.1016/B978-0-12-816413-6.00011-3
118. Schultz G, Acosta JB, Cowan L, Stechmiller J. “Linking the advanced glycation endproducts/receptor for advanced glycation endproducts pathway in diabetics with inflammation and topical antiinflammatory treatments of chronic wounds,” in Advances in Wound Care, Vol 1. New Rochelle, NY: Mary Ann Liebert, Inc., publishers (2010). p. 248–52.
119. Acosta JB, del Barco DG, Vera DC, Savigne W, Lopez-Saura P, Guillen Nieto G, et al. The pro-inflammatory environment in recalcitrant diabetic foot wounds. Int Wound J. (2008) 5:530–9. doi: 10.1111/j.1742-481X.2008.00457.x
120. Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. (2019) 38:e100492. doi: 10.15252/embj.2018100492
121. Moreno-Blas D, Gorostieta-Salas E, Pommer-Alba A, Mucino-Hernandez G, Geronimo-Olvera C, Maciel-Baron LA, et al. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging. (2019) 11:6175–98. doi: 10.18632/aging.102181
122. Karin O, Agrawal A, Porat Z, Krizhanovsky V, Alon U. Senescent cell turnover slows with age providing an explanation for the Gompertz law. Nat Commun. (2019) 10:5495. doi: 10.1038/s41467-019-13192-4
123. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
124. Guzik TJ, Cosentino F. Epigenetics and immunometabolism in diabetes and aging. Antioxid Redox Signal. (2018) 29:257–74. doi: 10.1089/ars.2017.7299
125. Wenz T. Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion. (2013) 13:134–42. doi: 10.1016/j.mito.2013.01.006
126. Berlanga-Acosta J, Iglesias-Marichal I, Rodriguez-Rodriguez N, Mendoza-Mari Y, Garcia-Ojalvo A, Fernandez-Mayola M, et al. Review: insulin resistance and mitochondrial dysfunction following severe burn injury. Peptides. (2020) 126:170269. doi: 10.1016/j.peptides.2020.170269
127. Yaribeygi H, Sathyapalan T, Atkin SL, Sahebkar A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid Med Cell Longev. (2020) 2020:8609213. doi: 10.1155/2020/8609213
128. Wu Y, Chen L, Wang Y, Li W, Lin Y, Yu D, et al. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-kappaB mediated inflammatory responses in osteoarthritis development. Sci Rep. (2015) 5:17602. doi: 10.1038/srep17602
129. Xiong S, Patrushev N, Forouzandeh F, Hilenski L, Alexander RW. PGC-1alpha modulates telomere function and DNA damage in protecting against aging-related chronic diseases. Cell Rep. (2015) 12:1391–9. doi: 10.1016/j.celrep.2015.07.047
130. Su KH, Dai C. mTORC1 senses stresses: coupling stress to proteostasis. Bioessays. (2017) 39:1600268,. doi: 10.1002/bies.201600268
131. Ong PS, Wang LZ, Dai X, Tseng SH, Loo SJ, Sethi G. Judicious toggling of mTOR activity to combat insulin resistance and cancer: current evidence and perspectives. Front Pharmacol. (2016) 7:395. doi: 10.3389/fphar.2016.00395
132. Cosentino C, Mostoslavsky R. Metabolism, longevity and epigenetics. Cell Mol Life Sci. (2013) 70:1525–41. doi: 10.1007/s00018-013-1295-3
133. Anderson R, Prolla T. PGC-1α in aging and anti-aging interventions. Biochim et Biophys Acta (BBA) - Gen Subjects. (2009) 1790:1059–66. doi: 10.1016/j.bbagen.2009.04.005
134. Sczelecki S, Besse-Patin A, Abboud A, Kleiner S, Laznik-Bogoslavski D, Wrann CD, et al. Loss of Pgc-1alpha expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. Am J Physiol Endocrinol Metab. (2014) 306:E157–67. doi: 10.1152/ajpendo.00578.2013
135. Karbasforooshan H, Karimi G. The role of SIRT1 in diabetic cardiomyopathy. Biomed Pharmacother. (2017) 90:386–92. doi: 10.1016/j.biopha.2017.03.056
136. Sosnowska B, Mazidi M, Penson P, Gluba-Brzozka A, Rysz J, Banach M. The sirtuin family members SIRT1, SIRT3 and SIRT6: Their role in vascular biology and atherogenesis. Atherosclerosis. (2017) 265:275–82. doi: 10.1016/j.atherosclerosis.2017.08.027
137. Hsu Y-J, Hsu S-C, Hsu C-P, Chen Y-H, Chang Y-L, Sadoshima J, et al. Sirtuin 1 protects the aging heart from contractile dysfunction mediated through the inhibition of endoplasmic reticulum stress-mediated apoptosis in cardiac-specific Sirtuin 1 knockout mouse model. Int J Cardiol. (2017) 228:543–52. doi: 10.1016/j.ijcard.2016.11.247
138. Kitada M, Ogura Y, Koya D. The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging. (2016) 8:2290–307. doi: 10.18632/aging.101068
139. Karbasforooshan H, Karimi G. The role of SIRT1 in diabetic retinopathy. Biomed Pharmacother. (2018) 97:190–4. doi: 10.1016/j.biopha.2017.10.075
140. Wang J, Li S, Wang J, Wu F, Chen Y, Zhang H, et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging. (2020) 12:650–71. doi: 10.18632/aging.102647
141. Yuan Y, Cruzat VF, Newsholme P, Cheng J, Chen Y, Lu Y. Regulation of SIRT1 in aging: roles in mitochondrial function and biogenesis. Mech Ageing Dev. (2016) 155:10–21. doi: 10.1016/j.mad.2016.02.003
142. Bu X, Wu D, Lu X, Yang L, Xu X, Wang J, et al. Role of SIRT1/PGC-1α in mitochondrial oxidative stress in autistic spectrum disorder. Neuropsychiatr Dis Treat. (2017) 13:1633–45. doi: 10.2147/NDT.S129081
143. Li X, Wu G, Han F, Wang K, Bai X, Jia Y, et al. SIRT1 activation promotes angiogenesis in diabetic wounds by protecting endothelial cells against oxidative stress. Arch Biochem Biophys. (2019) 661:117–24. doi: 10.1016/j.abb.2018.11.016
144. Huang X, Sun J, Chen G, Niu C, Wang Y, Zhao C, et al. Resveratrol promotes diabetic wound healing via SIRT1-FOXO1-c-Myc signaling pathway-mediated angiogenesis. Front Pharmacol. (2019) 10:421. doi: 10.3389/fphar.2019.00421
145. Zhao P, Sui BD, Liu N, Lv YJ, Zheng CX, Lu YB, et al. Anti-aging pharmacology in cutaneous wound healing: effects of metformin, resveratrol, and rapamycin by local application. Aging Cell. (2017) 16:1083–93. doi: 10.1111/acel.12635
146. Ojalvo AG, Acosta JB, Marí YM, Mayola MF, Pérez CV, Gutiérrez WS, et al. Healing enhancement of diabetic wounds by locally infiltrated epidermal growth factor is associated with systemic oxidative stress reduction. Int Wound J. (2017) 14:214–25. doi: 10.1111/iwj.12592
147. Dietrich I, Braga GA, de Melo FG, da Costa Silva Silva ACC. The diabetic foot as a proxy for cardiovascular events and mortality review. Curr Atheroscler Rep. (2017) 19:44. doi: 10.1007/s11883-017-0680-z
148. Izadi M, Kheirjou R, Mohammadpour R, Aliyoldashi MH, Moghadam SJ, Khorvash F, et al. Efficacy of comprehensive ozone therapy in diabetic foot ulcer healing. Diabetes Metab Syndrome. (2019) 13:822–5. doi: 10.1016/j.dsx.2018.11.060
149. Zheng Y, Wang X, Ji S, Tian S, Wu H, Luo P, et al. Mepenzolate bromide promotes diabetic wound healing by modulating inflammation and oxidative stress. Am J Transl Res. (2016) 8:2738–47.
150. Fazakerley DJ, Minard AY, Krycer JR, Thomas KC, Stockli J, Harney DJ, et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J Biol Chem. (2018) 293:7315–28. doi: 10.1074/jbc.RA117.001254
151. Hurrle S, Hsu WH. The etiology of oxidative stress in insulin resistance. Biomed J. (2017) 40:257–62. doi: 10.1016/j.bj.2017.06.007
152. Mello T, Zanieri F, Ceni E, Galli A. Oxidative stress in the healthy and wounded hepatocyte: a cellular organelles perspective. Oxid Med Cell Longev. (2016) 2016:8327410. doi: 10.1155/2016/8327410
153. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1066–77. doi: 10.1016/j.bbadis.2016.11.010
154. Liu L, Trimarchi JR, Smith PJ, Keefe DL. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell. (2002) 1:40–6. doi: 10.1046/j.1474-9728.2002.00004.x
155. Palmieri B, Vadala M, Laurino C. Review of the molecular mechanisms in wound healing: new therapeutic targets? J Wound Care. (2017) 26:765–75. doi: 10.12968/jowc.2017.26.12.765
156. Picard M, McEwen BS, Epel ES, Sandi C. An energetic view of stress: focus on mitochondria. Front Neuroendocrinol. (2018) 49:72–85. doi: 10.1016/j.yfrne.2018.01.001
157. Rocha M, Apostolova N, Diaz-Rua R, Muntane J, Victor VM. Mitochondria and T2D: role of autophagy, ER stress, and inflammasome. Trends Endocrinol Metab. (2020). doi: 10.1016/j.tem.2020.03.004. [Epub ahead of print].
158. Nisr RB, Shah DS, Ganley IG, Hundal HS. Proinflammatory NFkB signalling promotes mitochondrial dysfunction in skeletal muscle in response to cellular fuel overloading. Cell Mol Life Sci. (2019) 76:4887–904. doi: 10.1007/s00018-019-03148-8
159. Bolajoko E, Akinosun O, Khine A. Hyperglycemia-induced oxidative stress in the development of diabetic foot ulcers. In: Preedy VR, editor. Diabetes: Oxidative Stress and Dietary Antioxidants. 2nd ed. Academic Press (2020). p. 35–48. doi: 10.1016/B978-0-12-815776-3.00004-8
160. Dunnill C, Patton T, Brennan J, Barrett J, Dryden M, Cooke J, et al. Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int Wound J. (2017) 14:89–96. doi: 10.1111/iwj.12557
161. Kunkemoeller B, Kyriakides TR. Redox signaling in diabetic wound healing regulates extracellular matrix deposition. Antioxid Redox Signal. (2017) 27:823–38. doi: 10.1089/ars.2017.7263
162. Nouvong A, Ambrus AM, Zhang ER, Hultman L, Coller HA. Reactive oxygen species and bacterial biofilms in diabetic wound healing. Physiol Genomics. (2016) 48:889–96. doi: 10.1152/physiolgenomics.00066.2016
163. Feng C, Liu H, Yang M, Zhang Y, Huang B, Zhou Y. Disc cell senescence in intervertebral disc degeneration: causes and molecular pathways. Cell Cycle. (2016) 15:1674–84. doi: 10.1080/15384101.2016.1152433
164. Berlanga-Acosta J, Mendoza-Marí Y, Fernández-Mayola M, García AF, Valdés-Pérez C, Savigne-Gutiérrez W, et al. Torpid diabetic wound healing: evidence on the role of epigenetic forces. Int J Diabetes Clin Res. (2015) 2:1. doi: 10.23937/2377-3634/1410020
165. Berlanga-Acosta J, Schultz GS, Lopez-Mola E, Guillen-Nieto G, Garcia-Siverio M, Herrera-Martinez L. Glucose toxic effects on granulation tissue productive cells: the diabetics' impaired healing. Biomed Res Int. (2013) 2013:256043. doi: 10.1155/2013/256043
166. Chang M. Restructuring of the extracellular matrix in diabetic wounds and healing: a perspective. Pharmacol Res. (2016) 107:243–8. doi: 10.1016/j.phrs.2016.03.008
167. Baraibar MA, Hyzewicz J, Rogowska-Wrzesinska A, Bulteau A-L, Prip-Buus C, Butler-Browne G, et al. Impaired energy metabolism of senescent muscle satellite cells is associated with oxidative modifications of glycolytic enzymes. Aging. (2016) 8:3375–89. doi: 10.18632/aging.101126
168. Birch J, Passos JF. Targeting the SASP to combat ageing: Mitochondria as possible intracellular allies? Bioessays. (2017) 39:1600235. doi: 10.1002/bies.201600235
169. Wiley CD, Campisi J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. (2016) 23:1013–21. doi: 10.1016/j.cmet.2016.05.010
170. Rabbani N, Thornalley PJ. Hexokinase-2 glycolytic overload in diabetes and ischemia-reperfusion injury. Trends Endocrinol Metab. (2019) 30:419–31. doi: 10.1016/j.tem.2019.04.011
171. Hehenberger K, Heilborn JD, Brismar K, Hansson A. Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of l-lactate. WoundRepair Regen. (1998) 6:135–41. doi: 10.1046/j.1524-475X.1998.60207.x
172. Sabbatinelli J, Prattichizzo F, Olivieri F, Procopio AD, Rippo MR, Giuliani A. Where metabolism meets senescence: focus on endothelial cells. Front Physiol. (2019) 10:1523. doi: 10.3389/fphys.2019.01523
173. Choi EJ, Kil IS, Cho EG. Extracellular vesicles derived from senescent fibroblasts attenuate the dermal effect on keratinocyte differentiation. Int J Mol Sci. (2020) 21:1022. doi: 10.3390/ijms21031022
174. Quincozes-Santos A, Bobermin LD, de Assis AM, Gonçalves C-A, Souza DO. Fluctuations in glucose levels induce glial toxicity with glutamatergic, oxidative and inflammatory implications. Biochim et Biophys Acta (BBA) - Mol Basis Dis. (2017) 1863:1–14. doi: 10.1016/j.bbadis.2016.09.013
175. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. (2014) 6:265sr6. doi: 10.1126/scitranslmed.3009337
176. Owlarn S, Klenner F, Schmidt D, Rabert F, Tomasso A, Reuter H, et al. Generic wound signals initiate regeneration in missing-tissue contexts. Nat Commun. (2017) 8:2282. doi: 10.1038/s41467-017-02338-x
177. Marshall CD, Hu MS, Leavitt T, Barnes LA, Lorenz HP, Longaker MT. Cutaneous scarring: basic science, current treatments, and future directions. Adv Wound Care. (2018) 7:29–45. doi: 10.1089/wound.2016.0696
178. Mulder GD, vande Berg JS. Cellular senescence and matrix metalloproteinase activity in chronic wounds. Relevance to debridement and new technologies. J Am Podiatr Med Assoc. (2002) 92:34–7. doi: 10.7547/87507315-92-1-34
179. Henderson EA. The potential effect of fibroblast senescence on wound healing and the chronic wound environment. J Wound Care. (2006) 15:315–8. doi: 10.12968/jowc.2006.15.7.26932
180. Dekker P, Maier AB, van Heemst D, de Koning-Treurniet C, Blom J, Dirks RW, et al. Stress-induced responses of human skin fibroblasts in vitro reflect human longevity. Aging Cell. (2009) 8:595–603. doi: 10.1111/j.1474-9726.2009.00506.x
181. Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. (2007) 128:36–44. doi: 10.1016/j.mad.2006.11.008
182. Gruber F, Kremslehner C, Eckhart L, Tschachler E. Cell aging and cellular senescence in skin aging - recent advances in fibroblast and keratinocyte biology. Exp Gerontol. (2020) 130:110780. doi: 10.1016/j.exger.2019.110780
183. Werner S, Krieg T, Smola H. Keratinocyte-fibroblast interactions in wound healing. J Invest Dermatol. (2007) 127:998–1008. doi: 10.1038/sj.jid.5700786
184. Stunova A, Vistejnova L. Dermal fibroblasts—A heterogeneous population with regulatory function in wound healing. Cytokine Growth Factor Rev. (2018) 39:137–50. doi: 10.1016/j.cytogfr.2018.01.003
185. Xue SN, Lei J, Lin DZ, Yang C, Yan L. Changes in biological behaviors of rat dermal fibroblasts induced by high expression of MMP9. World J Emerg Med. (2014) 5:139–43. doi: 10.5847/wjem.j.issn.1920-8642.2014.02.011
186. desJardins-Park HE, Foster DS, Longaker MT. Fibroblasts and wound healing: an update. Regen Med. (2018) 13:491–5. doi: 10.2217/rme-2018-0073
187. Zhang D, Lu H, Chen Z, Wang Y, Lin J, Xu S, et al. High glucose induces the aging of mesenchymal stem cells via Akt/mTOR signaling. Mol Med Rep. (2017) 16:1685–90. doi: 10.3892/mmr.2017.6832
188. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. (2020) 16:263–75. doi: 10.1038/s41574-020-0335-y
189. Noordam R, Gunn DA, Tomlin CC, Maier AB, Mooijaart SP, Slagboom PE, et al. High serum glucose levels are associated with a higher perceived age. Age. (2013) 35:189–95. doi: 10.1007/s11357-011-9339-9
190. Maione AG, Brudno Y, Stojadinovic O, Park LK, Smith A, Tellechea A, et al. Three-Dimensional human tissue models that incorporate diabetic foot ulcer-derived fibroblasts mimic in vivo features of chronic wounds. Tissue Eng Part C: Methods. (2014) 21:499–508. doi: 10.1089/ten.tec.2014.0414
191. Wall SJ, Sampson MJ, Levell N, Murphy G. Elevated matrix metalloproteinase-2 and−3 production from human diabetic dermal fibroblasts. Br J Dermatol. (2003) 149:13–6. doi: 10.1046/j.1365-2133.2003.05262.x
192. Soydas T, Yaprak Sarac E, Cinar S, Dogan S, Solakoglu S, Tuncdemir M, et al. The protective effects of metformin in an in vitro model of aging 3T3 fibroblast under the high glucose conditions. J Physiol Biochem. (2018) 74:273–81. doi: 10.1007/s13105-018-0613-5
193. Patel S, Srivastava S, Singh MR, Singh D. Mechanistic insight into diabetic wounds: pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed Pharmacother. (2019) 112:108615. doi: 10.1016/j.biopha.2019.108615
194. Wilkinson HN, Hardman MJ. Wound senescence: a functional link between diabetes and ageing? Exp Dermatol. (2020) doi: 10.1111/exd.14082. [Epub ahead of print].
195. Hehenberger K, Hansson A. High glucose-induced growth factor resistance in human fibroblasts can be reversed by antioxidants and protein kinase C-inhibitors. Cell Biochem Function. (1997) 15:197–201. doi: 10.1002/(SICI)1099-0844(199709)15:3<197::AID-CBF740=3.0.CO;2-7
196. Lerman OZ, Galiano RD, Armour M, Levine JP, Gurtner GC. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am J Pathol. (2003) 162:303–12. doi: 10.1016/S0002-9440(10)63821-7
197. Kim HS, Park JW, Yeo SI, Choi BJ, Suh JY. Effects of high glucose on cellular activity of periodontal ligament cells in vitro. Diabetes Res Clin Pract. (2006) 74:41–7. doi: 10.1016/j.diabres.2006.03.034
198. Maione AG, Smith A, Kashpur O, Yanez V, Knight E, Mooney DJ, et al. Altered ECM deposition by diabetic foot ulcer-derived fibroblasts implicates fibronectin in chronic wound repair. Wound Repair Regen. (2016) 24:630–43. doi: 10.1111/wrr.12437
199. Goldstein S, Moerman EJ, Soeldner JS, Gleason RE, Barnett DM. Diabetes mellitus and genetic prediabetes. decreased replicative capacity of cultured skin fibroblasts. J Clin Invest. (1979) 63:358–70. doi: 10.1172/JCI109311
200. Sutanto SSI, McLennan SV, Keech AC, Twigg SM. Shortening of telomere length by metabolic factors in diabetes: protective effects of fenofibrate. J Cell Commun Signal. (2019) 13:523–30. doi: 10.1007/s12079-019-00521-x
201. Nagy T, Fisi V, Frank D, Kátai E, Nagy Z, Miseta A. Hyperglycemia-induced aberrant cell proliferation; a metabolic challenge mediated by protein O-GlcNAc modification. Cells. (2019) 8:999. doi: 10.3390/cells8090999
202. Blazer S, Khankin E, Segev Y, Ofir R, Yalon-Hacohen M, Kra-Oz Z, et al. High glucose-induced replicative senescence: point of no return and effect of telomerase. Biochem Biophys Res Commun. (2002) 296:93–101. doi: 10.1016/S0006-291X(02)00818-5
203. Goldstein S, Moerman EJ, Soeldner JS, Gleason RE, Barnett DM. Chronologic and physiologic age affect replicative life-span of fibroblasts from diabetic, prediabetic, and normal donors. Science. (1978) 199:781–2. doi: 10.1126/science.622567
204. Rowe DW, Starman BJ, Fujimoto WY, Williams RH. Abnormalities in proliferation and protein synthesis in skin fibroblast cultures from patients with diabetes mellitus. Diabetes. (1977) 26:284–90. doi: 10.2337/diabetes.26.4.284
205. Jin J, Zhang T. Effects of glucose restriction on replicative senescence of human diploid fibroblasts IMR-90. Cell Physiol Biochem. (2013) 31:718–27. doi: 10.1159/000350090
206. Zhang S, Duan E. Fighting against skin aging: the way from bench to bedside. Cell Transplant. (2018) 27:729–38. doi: 10.1177/0963689717725755
207. Monickaraj F, Aravind S, Nandhini P, Prabu P, Sathishkumar C, Mohan V, et al. Accelerated fat cell aging links oxidative stress and insulin resistance in adipocytes. J Biosci. (2013) 38:113–22. doi: 10.1007/s12038-012-9289-0
208. Almeida ME, Monteiro KS, Kato EE, Sampaio SC, Braga TT, Camara NO, et al. Hyperglycemia reduces integrin subunits alpha v and alpha 5 on the surface of dermal fibroblasts contributing to deficient migration. Mol Cell Biochem. (2016) 421:19–28. doi: 10.1007/s11010-016-2780-4
209. Lamers ML, Almeida ME, Vicente-Manzanares M, Horwitz AF, Santos MF. High glucose-mediated oxidative stress impairs cell migration. PLoS ONE. (2011) 6:e22865. doi: 10.1371/journal.pone.0022865
210. Rizwan H, Pal S, Sabnam S, Pal A. High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sci. (2020) 241:117148. doi: 10.1016/j.lfs.2019.117148
211. Lane KL, Abusamaan MS, Voss BF, Thurber EG, Al-Hajri N, Gopakumar S, et al. Glycemic control and diabetic foot ulcer outcomes: a systematic review and meta-analysis of observational studies. J Diabetes Complications. (2020) 2020:107638. doi: 10.1016/j.jdiacomp.2020.107638
212. Buranasin P, Mizutani K, Iwasaki K, Pawaputanon Na Mahasarakham C, Kido D, Takeda K, et al. High glucose-induced oxidative stress impairs proliferation and migration of human gingival fibroblasts. PLoS ONE. (2018) 13:e0201855. doi: 10.1371/journal.pone.0201855
213. Serrano M. Senescence helps regeneration. Dev Cell. (2014) 31:671–2. doi: 10.1016/j.devcel.2014.12.007
214. Ito Y, Hoare M, Narita M. Spatial and temporal control of senescence. Trends Cell Biol. (2017) 27:820–32. doi: 10.1016/j.tcb.2017.07.004
215. van Deursen JM. The role of senescent cells in ageing. Nature. (2014) 509:439–46. doi: 10.1038/nature13193
216. Stanley A, Osler T. Senescence and the healing rates of venous ulcers. J Vasc Surg. (2001) 33:1206–11. doi: 10.1067/mva.2001.115379
217. Clark RA. Oxidative stress and “senescent” fibroblasts in non-healing wounds as potential therapeutic targets. J Invest Dermatol. (2008) 128:2361–4. doi: 10.1038/jid.2008.257
218. Martinez Guimera A, Welsh C, Dalle Pezze P, Fullard N, Nelson G, Roger MF, et al. Systems modelling ageing: from single senescent cells to simple multi-cellular models. Essays Biochem. (2017) 61:369–77. doi: 10.1042/EBC20160087
219. Galkowska H, Wojewodzka U, Olszewski WL. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen. (2006) 14:558–65. doi: 10.1111/j.1743-6109.2006.00155.x
220. Rybinski B, Franco-Barraza J, Cukierman E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol Genomics. (2014) 46:223–44. doi: 10.1152/physiolgenomics.00158.2013
221. Mendoza-Marí Y, Valdés-Pérez C, Rodríguez-Corrales E, Suárez-Alba J, García-Ojalvo A, Guillén-Nieto G, et al. Histological and transcriptional expression differences between diabetic foot and pressure ulcers. J Diabetes Metab. (2013) 4:1000296. doi: 10.4172/2155-6156.1000296
222. Zacur H, Kirsner RS. Debridement: rationale and therapeutic options. Wounds. (2002) 14(2 Suppl. E):2E−7E.
223. Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, et al. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol. (2005) 167:59–69. doi: 10.1016/S0002-9440(10)62953-7
224. Kamal MM, Kassem DH. Therapeutic potential of wharton's jelly mesenchymal stem cells for diabetes: achievements and challenges. Front Cell Dev Biol. (2020) 8:16. doi: 10.3389/fcell.2020.00016
225. Andrzejewska A, Lukomska B, Janowski M. Concise review: mesenchymal stem cells: from roots to boost. Stem Cells. (2019) 37:855–64. doi: 10.1002/stem.3016
226. Saeedi P, Halabian R, Imani Fooladi AA. A revealing review of mesenchymal stem cells therapy, clinical perspectives and modification strategies. Stem Cell Investig. (2019) 6:34. doi: 10.21037/sci.2019.08.11
227. Saheli M, Bayat M, Ganji R, Hendudari F, Kheirjou R, Pakzad M, et al. Human mesenchymal stem cells-conditioned medium improves diabetic wound healing mainly through modulating fibroblast behaviors. Arch Dermatol Res. (2020) 312:325–36. doi: 10.1007/s00403-019-02016-6
228. Ankrum J, Karp JM. Mesenchymal stem cell therapy: two steps forward, one step back. Trends Mol Med. (2010) 16:203–9. doi: 10.1016/j.molmed.2010.02.005
229. Li H, Fu X. Mechanisms of action of mesenchymal stem cells in cutaneous wound repair and regeneration. Cell Tissue Res. (2012) 348:371–7. doi: 10.1007/s00441-012-1393-9
230. Jiang XY, Lu DB, Chen B. Progress in stem cell therapy for the diabetic foot. Diabetes Res Clin Pract. (2012) 97:43–50. doi: 10.1016/j.diabres.2011.12.011
231. Peired AJ, Sisti A, Romagnani P. Mesenchymal stem cell-based therapy for kidney disease: a review of clinical evidence. Stem Cells Int. (2016) 2016:4798639. doi: 10.1155/2016/4798639
232. Xu S, Liu C, Ji H-L. Concise review: therapeutic potential of the mesenchymal stem cell derived secretome and extracellular vesicles for radiation-induced lung injury: progress and hypotheses. Stem Cells Transl Med. (2019) 8:344–54. doi: 10.1002/sctm.18-0038
233. Freitag J, Bates D, Boyd R, Shah K, Barnard A, Huguenin L, et al. Mesenchymal stem cell therapy in the treatment of osteoarthritis: reparative pathways, safety and efficacy - a review. BMC Musculoskelet Disord. (2016) 17:230. doi: 10.1186/s12891-016-1085-9
234. Khan S, Mafi P, Mafi R, Khan W. A systematic review of mesenchymal stem cells in spinal cord injury, intervertebral disc repair and spinal fusion. Curr Stem Cell Res Ther. (2018) 13:316–23. doi: 10.2174/1574888X11666170907120030
235. Asai J, Takenaka H, Ii M, Asahi M, Kishimoto S, Katoh N, et al. Topical application of ex vivo expanded endothelial progenitor cells promotes vascularisation and wound healing in diabetic mice. Int Wound J. (2013) 10:527–33. doi: 10.1111/j.1742-481X.2012.01010.x
236. O'Loughlin A, Kulkarni M, Creane M, Vaughan EE, Mooney E, Shaw G, et al. Topical administration of allogeneic mesenchymal stromal cells seeded in a collagen scaffold augments wound healing and increases angiogenesis in the diabetic rabbit ulcer. Diabetes. (2013) 62:2588–94. doi: 10.2337/db12-1822
237. Wan J, Xia L, Liang W, Liu Y, Cai Q. Transplantation of bone marrow-derived mesenchymal stem cells promotes delayed wound healing in diabetic rats. J Diabetes Res. (2013) 2013:647107. doi: 10.1155/2013/647107
238. Cao Y, Gang X, Sun C, Wang G. Mesenchymal stem cells improve healing of diabetic foot ulcer. J Diabetes Res. (2017) 2017:9328347. doi: 10.1155/2017/9328347
239. Dash SN, Dash NR, Guru B, Mohapatra PC. Towards reaching the target: clinical application of mesenchymal stem cells for diabetic foot ulcers. Rejuvenation Res. (2014) 17:40–53. doi: 10.1089/rej.2013.1467
240. Jackson WM, Nesti LJ, Tuan RS. Concise review: clinical translation of wound healing therapies based on mesenchymal stem cells. Stem Cells Transl Med. (2012) 1:44–50. doi: 10.5966/sctm.2011-0024
241. Shin L, Peterson DA. Impaired therapeutic capacity of autologous stem cells in a model of type 2 diabetes. Stem Cells Transl Med. (2012) 1:125–35. doi: 10.5966/sctm.2012-0031
242. Inoue O, Usui S, Takashima S-i, Nomura A, Yamaguchi K, Takeda Y, et al. Diabetes impairs the angiogenic capacity of human adipose-derived stem cells by reducing the CD271+ subpopulation in adipose tissue. Biochem Biophys Res Commun. (2019) 517:369–75. doi: 10.1016/j.bbrc.2019.07.081
243. Orlandi A, Chavakis E, Seeger F, Tjwa M, Zeiher AM, Dimmeler S. Long-term diabetes impairs repopulation of hematopoietic progenitor cells and dysregulates the cytokine expression in the bone marrow microenvironment in mice. Basic Res Cardiol. (2010) 105:703–12. doi: 10.1007/s00395-010-0109-0
244. Jung C, Rafnsson A, Shemyakin A, Bohm F, Pernow J. Different subpopulations of endothelial progenitor cells and circulating apoptotic progenitor cells in patients with vascular disease and diabetes. Int J Cardiol. (2010) 143:368–72. doi: 10.1016/j.ijcard.2009.03.075
245. Fijany A, Sayadi LR, Khoshab N, Banyard DA, Shaterian A, Alexander M, et al. Mesenchymal stem cell dysfunction in diabetes. Mol Biol Rep. (2019) 46:1459–75. doi: 10.1007/s11033-018-4516-x
246. Cassidy FC, Shortiss C, Murphy CG, Kearns SR, Curtin W, de Buitléir C, et al. Impact of Type 2 diabetes mellitus on human bone marrow stromal cell number and phenotypic characteristics. Int J Mol Sci. (2020) 21:2476. doi: 10.3390/ijms21072476
247. Goodell MA, Rando TA. Stem cells and healthy aging. Science. (2015) 350:1199–204. doi: 10.1126/science.aab3388
248. Li Y, Wu Q, Wang Y, Li L, Bu H, Bao J. Senescence of mesenchymal stem cells (Review). Int J Mol Med. (2017) 39:775–82. doi: 10.3892/ijmm.2017.2912
249. Neri S, Borzi RM. Molecular mechanisms contributing to mesenchymal stromal cell aging. Biomolecules. (2020) 10:340. doi: 10.3390/biom10020340
250. Yang G, Jia Y, Li C, Cheng Q, Yue W, Pei X. Hyperglycemic stress impairs the stemness capacity of kidney stem cells in rats. PLoS ONE. (2015) 10:e0139607. doi: 10.1371/journal.pone.0139607
251. Dienelt A, zur Nieden NI. Hyperglycemia impairs skeletogenesis from embryonic stem cells by affecting osteoblast and osteoclast differentiation. Stem Cells Dev. (2011) 20:465–74. doi: 10.1089/scd.2010.0205
252. Xu W, Yu Wl, Gao H, Wang L, Liu Xp, Wang Yg. Effects of high glucose and high lipid on proliferation and apoptosis of human umbilical cord mensenchymal stem cells. Chin J Tissue Eng Res. (2012) 16:5001–5. doi: 10.3969/j.issn.2095-4344.2012.27.012
253. Krankel N, Adams V, Linke A, Gielen S, Erbs S, Lenk K, et al. Hyperglycemia reduces survival and impairs function of circulating blood-derived progenitor cells. Arterioscler Thromb Vasc Biol. (2005) 25:698–703. doi: 10.1161/01.ATV.0000156401.04325.8f
254. Denu RA, Hematti P. Effects of oxidative stress on mesenchymal stem cell biology. Oxid Med Cell Longevity. (2016) 2016:2989076. doi: 10.1155/2016/2989076
255. Kornicka K, Houston J, Marycz K. Dysfunction of mesenchymal stem cells isolated from metabolic syndrome and Type 2 diabetic patients as result of oxidative stress and autophagy may limit their potential therapeutic use. Stem Cell Rev Rep. (2018) 14:337–45. doi: 10.1007/s12015-018-9809-x
256. Vono R, Jover Garcia E, Spinetti G, Madeddu P. Oxidative stress in mesenchymal stem cell senescence: regulation by coding and noncoding RNAs. Antioxid Redox Signal. (2017) 29:864–79. doi: 10.1089/ars.2017.7294
257. Fu X, Wu S, Li B, Xu Y, Liu J. Functions of p53 in pluripotent stem cells. Protein Cell. (2020) 11:71–8. doi: 10.1007/s13238-019-00665-x
258. Ko KI, Coimbra LS, Tian C, Alblowi J, Kayal RA, Einhorn TA, et al. Diabetes reduces mesenchymal stem cells in fracture healing through a TNFalpha-mediated mechanism. Diabetologia. (2015) 58:633–42. doi: 10.1007/s00125-014-3470-y
259. van de Vyver M, Niesler C, Myburgh KH, Ferris WF. Delayed wound healing and dysregulation of IL6/STAT3 signalling in MSCs derived from pre-diabetic obese mice. Mol Cell Endocrinol. (2016) 426:1–10. doi: 10.1016/j.mce.2016.02.003
Keywords: senescence, proliferative senescence, chronic wounds, diabetic ulcers, aging
Citation: Berlanga-Acosta JA, Guillén-Nieto GE, Rodríguez-Rodríguez N, Mendoza-Mari Y, Bringas-Vega ML, Berlanga-Saez JO, García del Barco Herrera D, Martinez-Jimenez I, Hernandez-Gutierrez S and Valdés-Sosa PA (2020) Cellular Senescence as the Pathogenic Hub of Diabetes-Related Wound Chronicity. Front. Endocrinol. 11:573032. doi: 10.3389/fendo.2020.573032
Received: 15 June 2020; Accepted: 13 August 2020;
Published: 16 September 2020.
Edited by:
Ioanna Eleftheriadou, National and Kapodistrian University of Athens, GreeceReviewed by:
Vinod Tiwari, Indian Institute of Technology (BHU), IndiaFederico Biscetti, Catholic University of the Sacred Heart, Italy
Copyright © 2020 Berlanga-Acosta, Guillén-Nieto, Rodríguez-Rodríguez, Mendoza-Mari, Bringas-Vega, Berlanga-Saez, García del Barco Herrera, Martinez-Jimenez, Hernandez-Gutierrez and Valdés-Sosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro A. Valdés-Sosa, cGVkcm8udmFsZGVzQG5ldXJvaW5mb3JtYXRpY3MtY29sbGFib3JhdG9yeS5vcmc=