 Manuel A. Riquelme
Manuel A. Riquelme Eduardo R. Cardenas
Eduardo R. Cardenas Jean X. Jiang
Jean X. Jiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 14 October 2020
Sec. Bone Research
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.567844
This article is part of the Research Topic Bone Inside-Out and Outside-In Signals: Control of Body Homeostasis View all 21 articles
Bone is the most frequent site of breast cancer and prostate cancer metastasis, and one of the most common sites of metastasis for many solid tumors. Once cancer cells colonize in the bone, it imposes a major clinical challenge for the treatment of the disease, and fatality rates increase drastically. Bone, the largest organ in the body, provides a fertile microenvironment enriched with nutrients, growth factors and hormones, a generous reward for cancer cells. Dependent on cancer type, cancer cells can cause osteoblastic (bone forming) or osteolytic lesions to promote the net resorption and/or release of growth factors from the bone extracellular matrix. These processes activate a “vicious cycle”, leading to disruption of bone integrity and promoting cancer cell growth and migration. Cancer cells influence the bone microenvironment favoring their colonization and growth. In order to metastasize to the bone, cancer cells must first migrate from the site of origin, and once established within the bone, they must overcome the dormant inducing effects of resident cells. If successful, cancer cells can then colonize and continually disrupt bone homeostasis that is primarily maintained by osteocytes, the most abundant bone cell type. For example, it has been shown that exercise induces osteocytes to release anabolic factors that inhibit osteoclast resorptive activity, promote dormancy and the release of anti-cancer factors that inhibit breast cancer cell metastasis. In this review, we will summarize recent research findings and provide mechanistic insights related to the role of osteocytes in osteolytic metastasis.
The bone is a mineralized tissue highly regulated to adapt and meet the diverse needs of the host relative to physical demand, hormones, metabolic state, and environmental stimulation. Bone remodeling involves three major bone cell types; osteoblasts (bone forming cells) and osteoclasts (bone resorbing cells) that function in maintaining the structural balance, and the osteocytes that function in bone remodeling in response to environmental and mechanical signals and stimuli (1). The osteocyte, which is the most abundant cell type (~95%) in the bone, is the primary cell responsible for bone remodeling and homeostasis. Embedded inside the bone mineral matrix, osteocytes are connected and able to sense and respond coordinately to environmental cues, such as hormones, physical stress, and mechanical loading and unloading. These properties allow osteocytes to modulate the bone microenvironment by promoting the release of factors that regulate bone formation or resorption with respect to demands. Disease and aging can disrupt bone homeostasis, create structural defects, and alter the bone macro- and microenvironment, ultimately leading to cancer cells colonizing within the tissue (2). The bone, along with the liver and lung, is one of the most frequent sites of cancer metastasis (2). Bone metastasis is an unfortunate outcome of many solid tumors, typically breast, lung, prostate, thyroid, renal carcinoma, melanoma, gastrointestinal tumors, and head and neck cancers (2, 3). Tumors that originate in the bone represent a small fraction of diagnosed cancers. Originating from cells found in bone tissue of osteosarcomas, these cancers are of transformed osteoblastic lineage, and occur most often in adolescents (4, 5). Other cancers, such as multiple myeloma arise from the bone marrow, but do not come from mesenchymal lineage. Approximately 80% of bone lesions and tumors originate in the bone marrow as multiple myeloma (3, 6, 7). The process of other cancers metastasizing to the bone is as complicated as one would expect considering that it is not one single cancer type that favors bone metastasis. This article primarily focuses on cancer bone metastasis from cancer not originating from the bone. Bone metastasis greatly affects the quality of life of patients, causing complications, such as pain, nerve root or spine cord compression, vertebral or peripheral fractures, hypercalcemia, and bone marrow infiltration that lead to cytopenia (3, 8).
The reasons why tumor cells metastasize to the bone are poorly understood. Bone tissue provides an ideal microenvironment for metastatic tumor cells. Bone marrow endothelium, adipocytes, and the immune response all participate in maintaining bone homeostasis in ways that are only partially understood. Although solid tumor metastasis to the bone is common, not all cancers preferably metastasize to the bone. Thus, disseminated tumor cells homing to the bone may be a targeted, and/or the microenvironment found in the bone, including cellular, hormonal or otherwise is not suitable for the growth of certain cancer types (8, 9). Interestingly, highly vascularized bone containing red bone marrow and cancellous bone (e.g. pelvis and long bones) are common sites of metastasis (rarely hand and foot bones) (3). It has been shown that primary tumors from near and distal regions of the body organize and make ready premetastatic niches. For instance, myeloid cells can be recruited from the bone marrow by tumor-derived exosomes that release a plethora of soluble factors, including, proteins, enzymes, and small nucleic acids, which are capable of homing in circulating tumor cells to the newly forming metastatic niche (10).
αvβ3 integrins (acting as cell surface adhesion receptors) have been found to play a key role in mediating the metastatic MDA-MB-231 and Chines Hamster Ovary tumor cells into the bone (11, 12). Metastatic cancer cells are attracted and retained in the bone marrow through the sensing and signaling of chemokines, for example, the C-X-C motif chemokine ligand 12 (CXCL12), which is expressed in bone marrow stromal cells, attracts tumor cells overexpressing the C-X-C chemokine receptor type 4 (CXCR4) (13). In another study by Cox et al., lysyl oxidase (LOX) was identified in hypoxic ER-negative breast tumor cells to play a key role in preparing the bone metastatic niche. LOX induces osteoclastogenesis independent of RANKL, disrupts bone homeostasis, ultimately leading to the formation of premetastatic bone lesions (14). Moreover, high LOX activity has been clinically associated with increased collagen cross-linking, fibrosis, and elevated risk of cancer metastasis (15). LOX, secreted by primary tumor cells, is responsible for catalyzing the cross-linking of both collagen and elastin, which increases matrix stiffness, alignment, and total ECM volume. The increase of ECM stiffness facilitates the activation of integrins and augments Rho-generated cytoskeletal tension promoting focal adhesion formation and cell motility (14, 15).
Adaptive immune cells also play a role in setting up the bone metastatic niche. Immune-competent mice orthotopically injected with metastatic 4T1 breast cancer cells are shown to have increased osteoclastogenesis; this induces the pre-metastatic osteolytic niche required for colony formation. It is further shown that the primary tumor environment promotes the differentiation of helper T cells (CD4+), and the tumor-specific Th17 cells expressing RANKL, which stimulates osteoclast activation and induces osteolytic bone lesions, ultimately promoting breast cancer colonization in the bone (16).
The seemingly self-perpetuating metastatic growth to bone has been described as a ‘vicious cycle’. In an intricate process inside the bone, tumor cells secrete osteoclastogenic factors (e.g., IL-1, IL-6, IL-11, PDGF, MIP1α, TNF, M-CFS, RANKL, and PTHrP) that help stimulate the recruitment and activity of osteoclast, key players in the formation of osteolytic lesions (17). This process disrupts bone homeostasis and induces the release of growth factors, including, activin, transforming growth factor β (TGFβ), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) from the bone mineral matrix. In turn, these released factors promote tumor cell growth and increase further bone resorption (Figure 1, step ①) (18). This feedback loop, or ‘vicious cycle’, increases the incidence of metastatic lesions in the bone and eventually leads to related ailments, e.g. bone fractures, and high levels of blood calcium (hypercalcemia).
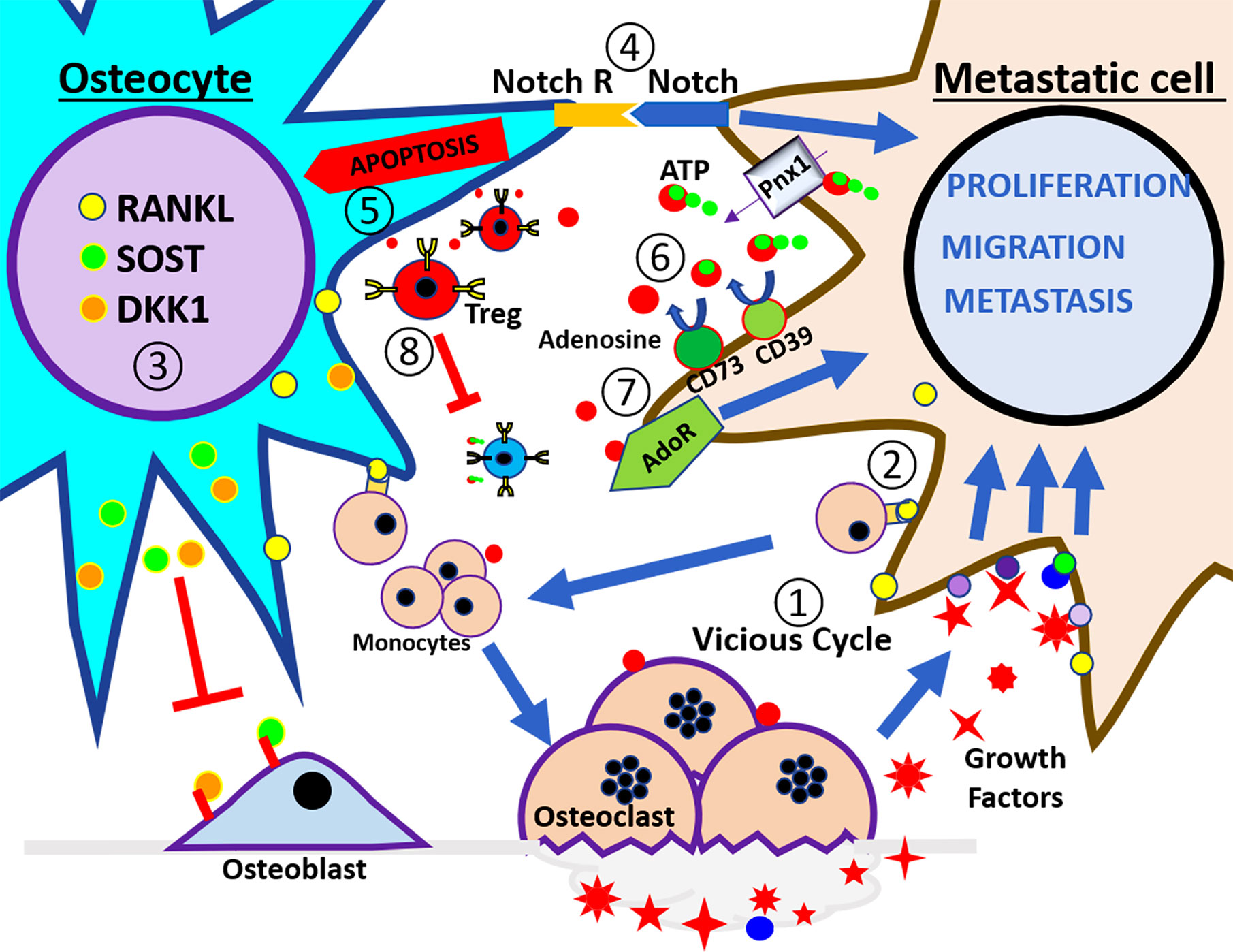
Figure 1 Schematic illustration of tumor microenvironment in the bone. At the left side of the panel is the osteocyte and the right side is the breast cancer cell. These cells interact in a bone catabolic environment. The numbers indicate the steps of events that may happen during breast metastasis described in the review. ① The vicious cycle; cancer cells interact with monocytes to increase the osteoclast number and activity in order to release growth factors embedded in the bone. ② RANKL expressed by osteocytes and cancer cells increases the recruitment of monocytes and stimulates the osteoclast differentiation. ③ Under this condition osteocytes increase the release of Sost and DKK1 that inhibit osteoblast activity and increase the expression of RANKL. ④ Notch signaling pathway induces apoptosis in the osteocytes and increases the proliferation of cancer cells. ⑤ Osteocytes apoptosis signals promote osteoclasts for bone resorption. ⑥ eATP is hydrolyzed to adenosine through CD39/CD73 enzyme activation, and generated adenosine activates adenosine receptor in cancer cells, leading to increased proliferation, migration, and metastasis. ⑦ Extracellular adenosine increases osteoclast activity, ⑧ also promotes Treg activity, and increases immune tolerance.
As mentioned, an important player in the vicious cycle is osteoclasts, large bone resorbing multinucleated cells originating from the fusion of bone marrow-derived monocytes/macrophages. Activated osteoclasts adhere to bone surfaces, forming an acting ring that covers a space in which bone demineralizing enzymes and proteases are secreted. Key players in osteoclast differentiation include adenosine nucleotides, receptor activator of nuclear factor κ-B ligand (RANKL), macrophage colony-stimulating factor (M-CSF), and other molecules (19), which are principally generated from nearby osteoblasts, osteocytes, and immune cells (20). Osteoclast generation and activation is achieved directly, or indirectly by RANKL production by neighboring cells, or by bone trophic tumor cells. These activities are eventually used by tumor derived cells to create the bone niche, leading to further osteoclastogenesis and bone resorption. The mechanistic comprehension of bone turnover in tumor growth has led to the clinical use of osteoclast inhibiting bisphosphonates, and Denosumab (anti-RANKL antibody) in patients with bone metastasis, and has become the standard of care to improve quality of life by limiting bone turnover (Figure 1, step ②) (21). In addition to molecules directly involved in bone resorption, other factors involved in bone resorption include interleukins-6 and 11 (22), parathyroid hormone-related peptide (PTHrP) (23, 24), soluble intercellular adhesion molecule 1 (ICAM-1) (25), Wnt molecules (26, 27), macrophage-stimulating protein (MSP) (28), and extracellular adenosine (Figure 1, step ③) (29).
Although crucial to bone metastasis and creating the metastatic niche, osteoclasts are not the only cell type to participate in bone metastasis, and osteoblasts also play a vital role. Osteoblasts participate in matrix mineralization, which provides strength (hardness) to the bone (30). Osteoblasts are derived from skeletal bone marrow stromal cells that differentiate into preosteoblasts and secrete numerous factors, including RANKL that directly impact osteoclastogenesis (Figure 1, step ③) (30). These cells eventually differentiate into mature osteoblasts, which secrete the mineral matrix proteins and mineralize bone (30). Some osteoblasts may become bone lining cells or become embedded in lacunae where they differentiate into fully mature mechanosensing osteocytes (1, 30). While studies show osteoclasts can induce tumor proliferation by releasing growth factors stored within the mineral matrix, osteoblast activity has been associated with tumor cell growth and tumor cell dormancy. Preosteoblasts and osteoblasts express tumor-promoting osteoprotegerin (OPG) (31), hepatocyte growth factor (HGF), and secrete connective tissue growth factor (CTGF) and TGFβ (22). Furthermore, osteoblasts express IL-6, which increases osteoclastogenesis, and has been shown to drive proliferation of multiple myeloma plasma cells (24, 32).
Tumor–osteoblast interactions have been shown to be critical in establishing bone metastasis (33, 34). Circulating (prostate) metastatic cells have been shown to have an affinity for the bone endosteal surface where they interact with osteoblasts through annexin2/annexin2 receptor interactions (33, 34). These micro-metastases are formed in regions of new bone formation, where differentiating and actively mineralizing osteoblasts are located. Furthermore, osteoblast and breast tumor interaction is shown to require adherent junction formation for tumor cell proliferation (9), supporting the notion that factors produced during osteogenesis promote cancer proliferation. Disseminated cancer cells must also compete for the endosteal surface of the bone, a niche occupied by non-proliferating long-term hematopoietic stem cells (LT-HSCs) (Figure 2, step ①). The mechanisms of cell cycle arrest of breast cancer cells once established in the endosteal niche are likely the same mechanisms that induce the non-proliferating status of long-term resident hematopoietic stem cells, unfortunately, only to later escape dormancy and proliferate (35, 36). In one study using a 3D co-culture model (osteoblast/breast cancer), it was identified that the addition of bone remodeling cytokines, tumor necrosis factor (TNF)-α and interleukin (IL)-1β and tumor necrosis factor resulted in increased proliferation in the breast cancer cell line MDA-MB-231 BMRS1 (37). The inhibition of TNF-α and IL-1β downstream targets, cycloxygenase (COX) and PGE2 receptors, resulted in decreased cancer cell proliferation (37). Moreover, osteoblasts and osteocytes also secrete leukemia inhibitory factor (LIF), and the activation of LIF receptors present in breast cancer cells is shown to maintain them in a dormant state. Loss of LIFR resulted in decreased expression of genes associated with cell dormancy. LIFR knockdown increased cancer cell migration and invasion, proliferation, and osteoclastogenesis. Interestingly, overexpression of PTHrP also decreased LIFR signaling. (38).
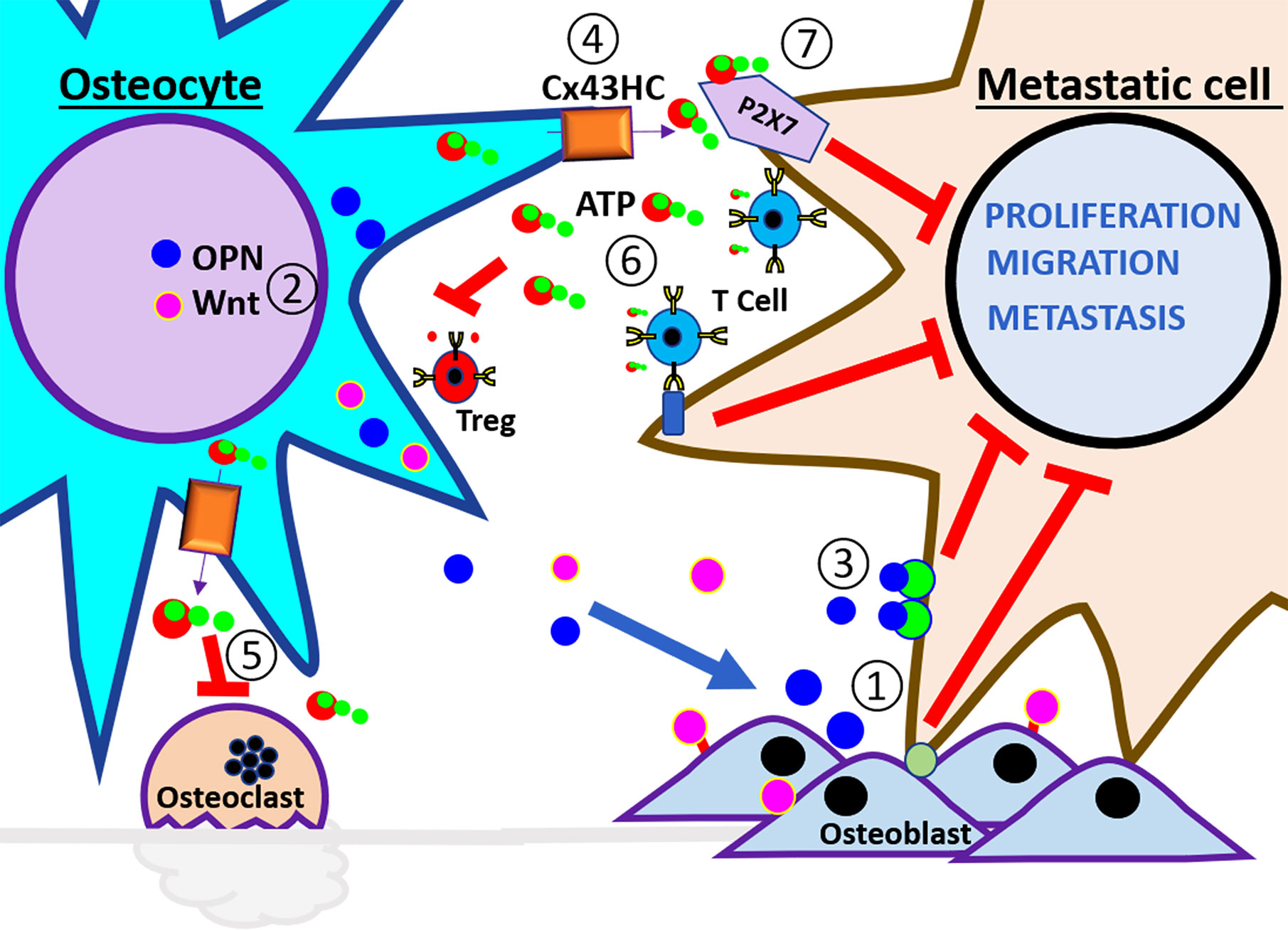
Figure 2 Schematic illustration of anti-tumor microenvironment in the bone. At the left side of the panel are the osteocytes and the right side are the breast cancer cells. These cells interact in a healthy bone environment. The numbers indicate the steps of events that may happen during breast metastasis described in the review. ① The interaction of metastatic cells with osteoblast promotes dormancy. ② Physiological level of mechanical loading stimulates osteocyte release of anabolic factors, Wnt, and OPN, to increase osteoblast differentiation, activity, and bone strength. ③ High concentration of OPN reduces EMT in metastatic cells. ④ Mechanical loading increases opening of Cx43 hemichannels and the release of ATP. ⑤ eATP inhibits osteoclast activity and ⑥ also inhibits Treg formation and stimulates immune surveillance. ⑦ eATP activates P2X receptor and reduces the proliferation, migration, and metastatic potential of cancer cells.
An early study by Kobayashi et al. found bone stromal cells induced dormancy by the release of bone morphogenic protein 7 (BMP7) and activation of prostate BMP receptor 2. BMP7-treated prostate cancer cells resulted in activated p38 MAPK, increased expression of p21 and the metastasis suppressor gene, NDRG1 (N-myc downstreamregulated gene 1) (39). Key studies have given further insight into the mechanisms in which osteoblasts may induce dormancy in disseminated tumor cells (40, 41). In another study, osteoblast conditioned media increased cellular quiescence of prostate cancer cells. TGF-β2 and growth differentiation factor (GDF)10 were identified as osteoblast secretory factors that induced quiescence in several prostate cancer cell lines. The binding of these factors to the TGF-βRIII receptor expressed in prostate cancer cell lines activated (phosphorylation at Thr180/Tyr182) p38 mitogen activated protein kinase (MAPK). Activated p38-MAPK phosphorylation of downstream target retinoblastoma protein (Rb) resulted in the inhibition of cancer cell-cycle progression (40). In another study of prostate cancer metastasis to the bone, Yumoto et al. identified osteoblast-derived ligand growth arrest specific 6 (GAS6) and the tumoral tyrosine kinase receptor Axl as required for the TGF-β2-induced response towards prostate cancer cell dormancy (41). Multiple myeloma cells have also been shown to be affected by the bone microenvironment. These cells can occupy the endosteal niche, remain dormant, and escape therapies that largely target dividing cells. The interaction of multiple myeloma cells with cells of osteoblastic lineage along the endosteal bone surface was associated with single, non-dividing tumor cells. Interestingly, dormant myeloma cells that were insensitive to melphalan, a chemotherapeutic agent, could be reactivated upon osteoclast activation with the soluble form of RANKL (42).
The bone is home to the hematopoietic system, and it integrates an assortment of systemic physiological signals. Bone homeostasis is affected directly or indirectly by many pathological conditions, including diabetes, gastrointestinal diseases, physical stress, etc. (43–48). One example is the increased risk of cancer and tumor growth under inflammatory conditions (32, 49) or the propensity of cells metastasizing to fractures sites (50). Similar correlations have been associated with surgical procedures (51–53), which intrinsically induce trauma, inflammation, and an increase in innate immune cells needed for tissue repair. These responses have been shown to promote conditions conducive to metastatic growth at non-surgical sites (51, 53).
Bone, like other organs, changes with age, which includes an accumulation of senescent cells, such as, osteoblasts, and resident bone cells harboring genetic mutations (54–56). The loss of bone density with age is well known. Equally concerning is how the accumulated damage to cells and their genetic makeup, caused by environmental toxins, or byproducts of cellular respiration, can result in an environment primed for tumor growth and metastasis (57). How does an increase in senescent bone cells affect certain cancers metastasizing to the bone, or dormant cells in the bone being released from dormancy? As discussed here and shown in numerous studies, senescent osteoblasts can promote osteoclastogenesis, which leads to increased metastasis, dissemination, and metastatic growth of cancer in the bone. This supports and explains the possible mechanisms in which elder cancer patients in remission with dormant cancer cells in the bone often relapse. A possible underlying reason is that cancer cells can take residence in the bone through the contribution of senescent cells accumulated over time (42, 58). An important detail to keep in mind is that most experimental studies have not used aged animal models (59, 60). Studies delving into how age affects metastasis are in dire need.
Bone impacts metastasis in unexpected and sometimes complex manners. Mechanical loading inhibits secondary growth and osteolytic capability of metastatic tumors in nude mice by modulating osteoblastic/osteoclastic activities and communication between osteocytes and tumor cells (61). Osteocytic release of osteopontin (OPN) (Figure 2, step ②), a secreted phosphoprotein with a high avidity to bone mineral matrix, has been reported to induce activators of the EMT process (62). Interestingly, lower OPN levels (0.1 to 0.5 μg/ml) induce EMT markers. Low levels of mechanical loading (1 N) are shown to increase the expression and secretion of OPN (Fan, 2020 #82). The increase in OPN in turn inhibits the expression of TGF-β in osteocytes, increases the adhesion of tumoral cells, thus possibly inhibiting growth and migration by anchoring tumor cells at the primary site. This bone microenvironmental condition is pro-mesenchymal to epithelial transition (MET) reducing the aggressiveness and allowing the settlement of secondary tumor (Figure 2, step ③) (63). Similar intensities of mechanical loading inhibiting tumor growth in nude mice have been previously reported (61). As studies have shown, high loading intensity of the bone enhances breast cancer cell malignancy; therefore, the extent of mechanical loading should be carefully monitored (63). Taken together, studies indicate osteocytes are an important player in providing the ‘soil’ for bone metastasis/progression as well as associated skeletal diseases (27, 64–66).
The extensive lacuna–canaliculi network allows osteocytes to directly communicate with one another. It also allows for osteocytes to respond to local and distant signals, including mechanical (bone stress) or biological (paracrine and endocrine) (1, 67). Osteocytes control bone remodeling through regulation of bone-forming and bone-destroying cells. During bone demineralization, osteocytes decrease osteoblast differentiation and function through secreted factors, including the Wnt signaling antagonists sclerostin and Dickkopf Wnt signaling pathway inhibitor 1 (DKK1) (67). Osteocytes are the main sclerostin producer in the bone, and this protein inhibits the association of Wnt ligands with their receptor in osteocytes and osteoblast. Therefore, the key bone formation inhibitor, sclerostin, has been given much attention as a targeted therapeutic approach for low bone density (66, 68). Bone RANKL is primarily produced by osteocytes (Figure 1, step ③). RANKL promotes monocyte differentiation into bone resorbing osteoclasts. Neutralization of RANKL with the antibody Denosumab is currently in use to reduce fracture incidence in low bone density, bone metastasis, and rare bone cancers (Figure 3, step ①) (69, 70). The Denosumab Clinical trial (ABCSG-18) showed that postmenopausal breast cancer patients under aromatase inhibitor treatment had a significant latency of apparition of bone fracture with Denosumab as an adjuvant (71). This was also true in metastatic breast cancers and new primary malignancies.
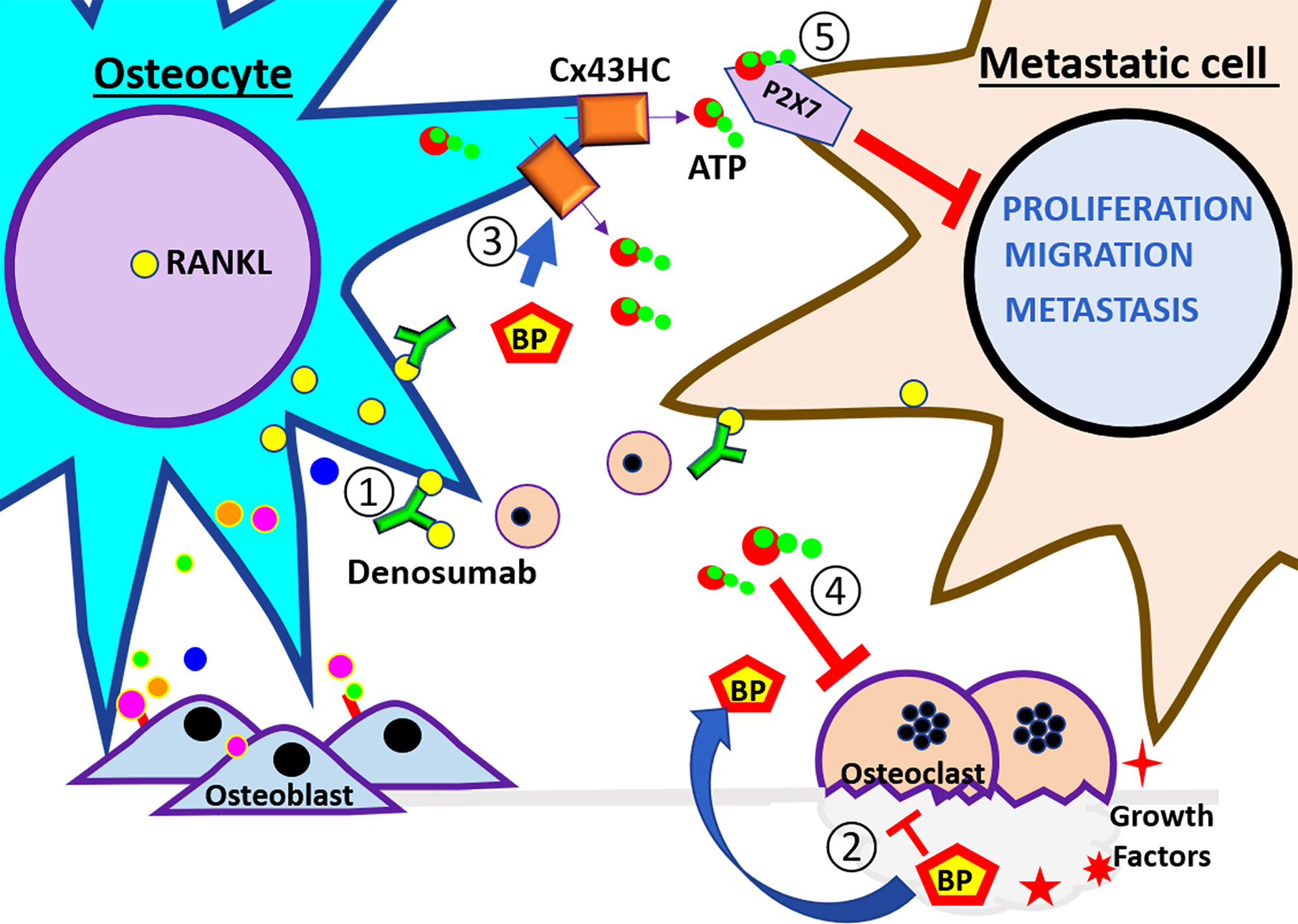
Figure 3 Schematic illustration of therapeutic agents used to treat bone metastasis. At the left side of the panel are the osteocytes and the right side are the breast cancer cells. These cells are subjected to the therapeutic treatment under the bone environment. The numbers indicate the steps of events that may happen during breast metastasis described in the review. ① Denosumab, a RANKL neutralizing antibody, binds RankL expressed by osteocytes and cancer cells, and inhibits the recruitment of monocytes and osteoclast differentiation. ② Bisphosphonates (BPs) inhibit osteoclast activity. Also, ③ BP increases osteocyte survival and induces opening of Cx43 hemichannels and the release of ATP. ④ eATP inhibits osteoclast activity. ⑤ eATP activates P2X receptor and reduces the proliferation, migration, and metastatic potential of cancer cells. The effects of the pharmacological agents reduce the activity of the vicious cycle.
In contrast, in a clinical trial (D-CARE study) of patients with stage II/III breast cancer, Denosumab treatment did not improve bone metastasis free survival. Denosumab did increase the incidence of osteonecrosis of the jaw (5 versus <1%) and hypocalcaemia (7 versus 4%) in comparison with placebo (72). Although the above mentioned trials do not corroborate Denosumab having a beneficial effect on survival, the results obtained from these two trials have a significant difference. The D-care trial patient profile was of early stage high risk breast cancer patients, while the ABCSC-18 study focused on early stage low risk breast cancer patients. It is also important to note the trials were conducted with different Denosumab schedules and endpoints (70).
As mentioned above, bones inevitably age and undergo numerous changes, including bone loss, osteocyte apoptosis, and increased oxidative stress. Osteocyte apoptosis is a key stimulus that triggers bone resorption (Figure 1, step ⑤) (73, 74). The slowed production of sex hormones that comes with aging promotes osteoclast activity (75, 76), osteocyte apoptosis (73), elevated oxidative stress (76, 77), and a reduction in osteoblast function (78). Thus, reduction in sex hormones culminates in bone fragility and bone loss. This bone destructive environment is further enhanced by a decline in immune surveillance and increased fat formation; this disturbs the balance of critical osteoclastogenic proteins, RANKL, and OPG, towards bone destruction (79). The AZURE phase 3 clinical trial for post-menopausal women was designed to study the effects of adjuvant zoledronic acid treatment in early high risk breast cancer patients. The AZURE trial showed that with treatment, incidence of bone metastasis was reduced. Critical to our understanding, this benefit was restricted to postmenopausal women and those under ovarian suppression treatments with hormone-receptor-positive breast cancer. This observation highlights how critical it is to understand the differences between old and young bones in metastasis (80, 81).
Emerging studies indicate how osteocytes could have a positive impact on tumor growth, motility, and survival, a scenario leading to poor outcomes in cancer patients. The interaction between prostate cancer cells and osteocytes induces the osteocytic production and release growth-derived factor 15 (GDF15) promoting prostate cancer cell proliferation, migration, and invasion of prostate cell in the bone (82). MLO-Y4 osteocytes stimulated by hydrostatic pressure, similar to what is observed in bone metastasis, increased the viability of prostate, breast, and lung cancer cell lines. Hydrostatic pressure also improved motile and invasive capacity through increased expression of chemokine (C-C motif) ligand 5 (CCL5, also known as RANTES) and MMPs (83). Likewise, high intensity shear stress increased survival and migration in the MDA-MBA-231 breast cancer cell line (63, 84). However, these studies also show how intensity of load imparts opposing effects on survival and migration. This underscores the importance of osteocytes in the modulation of the bone microenvironment with regard to tumor progression particularly with respect to the relationship between the magnitude of mechanical stimulation and breast cancer cell apoptosis or migration (84).
Increased osteocyte apoptosis within lytic bone lesions has been found in patients with multiple myeloma (85). During the progression of multiple myeloma, osteocytes directly interact with multiple myeloma cells, which stimulate osteocytes to produce sclerostin and RANKL. This results in the recruitment of osteoclast precursors and a reduction of Wnt signaling, leading to the inhibition of osteoblast differentiation (Figure 1, step ③). Concomitantly, cell to cell interactions reduce osteocyte viability due to apoptosis triggered Notch signaling and sustained by multiple myeloma derived TNFα. Furthermore, Notch signaling interaction increases the proliferation of multiple myeloma by increasing cyclin D1 RNA levels and accelerating cell proliferation (Figure 1, step ④) (27). This highlights how osteocytes play a constant integrative role of endocrine, paracrine, and mechanical signals, and the output of those signals results in bone formation or resorption responses. The complexity of such integration makes it particularly difficult to predict the impact of osteocytes on cancer cells metastasized to the bone.
The skeleton is a dynamic organ that responds to physical stress by promoting bone remodeling, which includes the addition and removal of bone. Although several resident bone cells are involved in mechanosensing, osteocytes are regarded as the major mechanosensory cell within the bone (67, 86). The long dendritic processes of osteocytes form gap junction channels composed primarily by gap junction proteins (connexins). These gap junction networks connect not only neighboring osteocytes, but also cells on the bone surface, including osteoblasts and osteoclasts (1, 67). The mechanosensing osteocytes form connexin 43 (Cx43) hemichannels (half a gap junction channel) which allows for the communication between the internal environment of the cell and its extracellular environment. Gap junction channels are involved in the global regulation and fine tuning of bone formation and resorption as it was evidenced by the altered levels of serum remodeling markers N-terminal propeptide of type I procollagen and C-terminal telopeptide of type I collagen, respectively (87). Osteocyte hemichannels likely play a predominant role in its response to mechanical stimulation; given that bone and osteocytes are constantly subjected to mechanical stimuli as a result of physical movement, gravity, and blood circulation. This is evident by the major impact that Cx43 hemichannels have on the expression of OPG and RANKL, and osteocyte viability, which are essential for bone integrity and longevity (87, 88). Concordantly, the anti-apoptotic effect of bisphosphonates on osteoblasts and osteocytes has been shown to be through regulation of Cx43 hemichannels (Figure 3, step ③) (89). Bisphosphonates are the gold standard for therapy for bone diseases in cancer patients (6, 80) as they inhibit osteoclast activity and prevent bone loss induced by cancer cells, thus reducing fracture risk (21, 28). In vivo, osteocyte Cx43 hemichannel activity is an important mediator of the growth inhibitory effects of bisphosphonates in breast cancer (65). In vitro and in vivo studies by our group further underscore the key role of Cx43 in mediating the tumor inhibitory effects of bisphosphonates. Bisphosphonate conditioned media from osteocytes inhibited breast cancer cell growth (MDA-MB-231), migration, and invasion. These effects were abrogated by treating with Cx43 hemichannel specific blocking antibody (65). Moreover, mice with impaired Cx43 gap junctions and hemichannels showed significantly increased tumor burden and a reduced effect of bisphosphonates on tumor growth compared to mice with impaired Cx43 gap junction channel function or wild type (65). This implies Cx43 hemichannels in osteocytes are responsive to bisphosphonates, thus making Cx43 a promising novel drug target for the treatment of breast cancer metastasis to the bone. In a previous study, we have shown that these effects are mediated by adenosine triphosphate (ATP) released by osteocyte Cx43 hemichannel opening (Figure 2, steps ④ and ⑦) (Figure 3, steps ③ and ④) (90). These important findings highlight that more work is needed to determine exactly how osteocytes impact various cancers metastatic potential, and if these cells can be targeted to stop bone metastasis.
As a response to tissue damage and cellular stress, cells, including osteocytes, secrete/release ATP to the extracellular space (Figure 2, step ④) (65, 91, 92). The intracellular concentration of ATP is ~3–10 mM, and the extracellular ATP (eATP) is about 10 nM. The big difference between intracellular and extracellular ATP is from active ATP degradation through ectonucleotidases in the extracellular compartment (93). The presence of eATP has been shown to inhibit the growth of pancreatic, colon, prostate, breast, liver, ovarian, colorectal, esophageal, melanoma, and leukemia (94). Intravenous ATP used in clinical trials of patients with pre-terminal lung cancer, showed an increase in survival rate and had a beneficial effect on weight and muscle strength (95–97). Multiple studies show an anticancer action of eATP or eATP analogs by binding to P2 purinergic receptors (90, 98). eATP is rapidly degraded to adenosine, a well-known tumorigenic factor (92, 93, 99). Additionally, eATP or P2 receptor agonist decreases osteoclast activity and bone resorption (Figure 2, step ⑤) (100), reduces the number of T regulatory lymphocytes (Tregs) (101), and prolongs the activity of T lymphocytes (Figure 2, step ⑥) (102). However, ATP metabolites through P1 purinergic receptor activation also mediate pro-tumorigenic effects in prostate and breast cancer cells (90, 103). This suggests that ATP and/or ATP metabolite balance plays a key role in the tumor microenvironment. The solid tumor microenvironment is usually hypoxic and/or inflammatory, and the extracellular concentration of nucleotides (ATP/adenosine) is higher in comparison to normal tissue (92, 103, 104). In this hypoxic/inflammatory microenvironment, adenosine promotes cancer cell migration and chemotaxis in breast cancer and melanoma cells, along with an increase in osteoclastic activity and bone resorption (Figure 1, ⑥ and ⑦) (29). This microenvironment also results in poor immune surveillance with high lymphocytic tolerance (Figure 1, steps ⑥and ⑧) (92, 105, 106). The main pathway leading to high extracellular adenosine levels is the hydrolysis of eATP by a family of enzymes known as ectonucleotidases, such as CD39 and CD73, which hydrolyze ATP and ADP to AMP, and AMP further to adenosine (Figure 1, step ⑥) (93, 105). CD73 has been associated with a pro-metastatic phenotype in breast cancer, and CD73 knockdown leads to suppression of breast cancer cell growth, migration, and invasion both in vivo and in vitro (105). Therefore, we must practice caution since the function of eATP on tumorigenesis could largely depend on the activity of ecto-ATPases in the tissue.
Adenosine and ATP bind to specific purinergic receptors at the cell surface, which are divided into P1 receptors, with adenosine as the main ligand, and P2 receptors, with ATP and ADP as the main agonists. P1 receptors have four subtypes: A1, A2a, A2b, and A3. There are two major P2 receptor subtypes, seven P2X, and eight P2Y subtypes (92, 107). The presence of P2Y subtypes has been shown to play important roles in cell survival under mechanical stress, although the role of specific P2X subtypes remains unclear. It has been suggested that autocrine activation of breast cancer P2X7 receptors participates in the activation of cell proliferation, cancer cell process elongation, and further ATP release (99). The tumor microenvironment rich in ATP is shown to either promote or inhibit cell migration, enough to activate multiple P2 receptors, but not enough to induce cell death trough P2X7 activation. The study by Zhou et al. provides clues to clarify the mixed effects of ATP. They showed that the addition of a non-hydrolysable P2X receptor agonist resulted in an inhibitory effect on cancer cell migration and growth. This inhibitory effect was dependent on P2X7 activation (Figure 2, step ⑦) (Figure 3, step ⑤) (90). A2A receptor activation, on the other hand, resulted in a stimulatory effect on breast cancer cell migration and growth (Figure 1, step ⑦) (90). Cancer cell-specific expression of P1 receptor subtypes has been reported. for example, in breast cancer. A2b receptors are absent on ER-positive MCF-7 cells, whereas MDA-MB-231 cells express very high levels of A2b (90). In order to understand and establish strategies to control tumor growth and metastasis, it is very important to evaluate the presence and expression levels of the specific P1 or P2 receptor subtypes in cancer cells. These are likely key factors determining if a cellular response to adenosine nucleotides will be elicited.
The osteocyte is a key player modulating the bone cancer microenvironment. Metastatic cancer cells have shown the potential to utilize osteocyte signaling by turning the bone microenvironment osteoclastogenic and transforming osteocytes into pro-tumorigenic cells. Moreover, osteocytic overproduction of Wnt inhibitors contributes to the suppression of bone formation. In addition, metastatic cancer cells colonized in the bone reduce osteocyte viability, resulting in reduced cell capacity to maintain bone homeostasis. However, the bone forming ability of osteocytes is related with an anti-resorptive microenvironment. This condition reduces osteocyte apoptosis, enhances Cx43 hemichannel activity, increases bone strength, and reduces osteoclast recruitment and activity. Taken together, these osteoclast activities will inhibit the overall activation of metastatic dormant cells, and tumor growth, and motility. Although we have just started a fascinating journey to understand the functional relationship between cancer and osteocytes, targeting osteocyte signaling pathways and molecular messengers has already shown to have a positive impact on preventing/improving bone pathologies associated with cancers. These results offer encouraging and supportive ideas of a targeted approach on osteocytes that reside in the cancer niche. These studies may offer guidance and a map to develop new therapies for cancers metastasized to bone, or the prevention thereof.
In conclusion, osteocytes play a large role as gate keepers of the bone and bone homeostasis. By having a broader knowledge on how osteocytes influence cancer cells, osteoblasts, and osteoclasts, we may improve and increase pharmacological strategies to help keep the bone healthy and free of cancer. Further work is needed to uncover key events and players that coordinate the communication between cancer cells and bone cells. The goal is to identify target points that can disrupt the initial and key steps of bone metastasis. Osteocytes may prove to be a key ally in combating cancer cell progression in the bone.
All authors contributed to the article and approved the submitted version.
JJ was supported by the US National Institutes of Health grant CA196214, US Department of Defense grant BC161273, and Welch Foundation grant AQ-1507.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Buenrostro D, Mulcrone PL, Owens P, Sterling JA. The Bone Microenvironment: a Fertile Soil for Tumor Growth. Curr Qsteoporos Rep (2016) 14:151–8. doi: 10.1007/s11914-016-0315-2
3. Chin H, Kim J. Bone Metastasis: Concise Overview. Federal practitioner : for the health care professionals of the VA. DoD PHS (2015) 32:24–30.
4. Moore DD, Luu HH. Osteosarcoma. Cancer Treat Res (2014) 162:65–92. doi: 10.1007/978-3-319-07323-1_4
5. de Azevedo JWV, de Medeiros Fernandes TAA, Fernandes JV Jr., de Azevedo JCV, Lanza DCF, Bezerra CM, et al. Biology and pathogenesis of human osteosarcoma. Oncol Lett (2020) 19:1099–116. doi: 10.3892/ol.2019.11229
6. Terpos E, Berenson J, Raje N, Roodman GD. Management of bone disease in multiple myeloma. Expert Rev Hematol (2014) 7:113–25. doi: 10.1586/17474086.2013.874943
7. Minnie SA, Hill GR. Immunotherapy of multiple myeloma. J Clin Invest (2020) 130:1565–75. doi: 10.1172/JCI129205
8. Hernandez RK, Wade SW, Reich A, Pirolli M, Liede A, Lyman GH. Incidence of bone metastases in patients with solid tumors: analysis of oncology electronic medical records in the United States. BMC Cancer (2018) 18:44. doi: 10.1186/s12885-017-3922-0
9. Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell (2015) 27:193–210. doi: 10.1016/j.ccell.2014.11.017
10. Liu Y, Cao X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
11. Kwakwa KA, Sterling JA. Integrin alphavbeta3 Signaling in Tumor-Induced Bone Disease. Cancers (2017) 9. doi: 10.3390/cancers9070084
12. Pecheur I, Peyruchaud O, Serre CM, Guglielmi J, Voland C, Bourre F, et al. Integrin alpha(v)beta3 expression confers on tumor cells a greater propensity to metastasize to bone. FASEB J (2002) 16:1266–8. doi: 10.1096/fj.01-0911fje
13. Weidle UH, Birzele F, Kollmorgen G, Ruger R. Molecular Mechanisms of Bone Metastasis. Cancer Genomics Proteomics (2016) 13:1–12. doi: 10.21873/cgp.20004
14. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature (2015) 522:106–10. doi: 10.1038/nature14492
15. Walker C, Mojares E, Del Río Hernández A. Role of Extracellular Matrix in Development and Cancer Progression. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19103028
16. Monteiro AC, Leal AC, Gonçalves-Silva T, Mercadante AC, Kestelman F, Chaves SB, et al. T cells induce pre-metastatic osteolytic disease and help bone metastases establishment in a mouse model of metastatic breast cancer. PloS One (2013) 8:e68171.doi: 10.1371/journal.pone.0068171
17. Wu MY, Li CJ, Yiang GT, Cheng YL, Tsai AP, Hou YT, et al. Molecular Regulation of Bone Metastasis Pathogenesis. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2018) 46:1423–38. doi: 10.1159/000489184
18. Chen YC, Sosnoski DM, Mastro AM. Breast cancer metastasis to the bone: mechanisms of bone loss. Breast Cancer Res BCR (2010) 12:215. doi: 10.1186/bcr2781
19. Pellegatti P, Falzoni S, Donvito G, Lemaire I, Di Virgilio F. P2X7 receptor drives osteoclast fusion by increasing the extracellular adenosine concentration. FASEB J (2011) 25:1264–74. doi: 10.1096/fj.10-169854
20. Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev (2008) 29:155–92. doi: 10.1210/er.2007-0014
21. Gralow J, Tripathy D. Managing metastatic bone pain: the role of bisphosphonates. J Pain Symptom Manage (2007) 33:462–72. doi: 10.1016/j.jpainsymman.2007.01.001
22. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell (2003) 3:537–49. doi: 10.1016/S1535-6108(03)00132-6
23. Soki FN, Park SI, McCauley LK. The multifaceted actions of PTHrP in skeletal metastasis. Future Oncol (Lond Engl) (2012) 8:803–17. doi: 10.2217/fon.12.76
24. Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer (2011) 11:411–25. doi: 10.1038/nrc3055
25. Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell (2013) 24:542–56. doi: 10.1016/j.ccr.2013.09.008
26. Colombo M, Thummler K, Mirandola L, Garavelli S, Todoerti K, Apicella L, et al. Notch signaling drives multiple myeloma induced osteoclastogenesis. Oncotarget (2014) 5:10393–406. doi: 10.18632/oncotarget.2084
27. Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res (2016) 76:1089–100. doi: 10.1158/0008-5472.CAN-15-1703
28. Andrade K, Fornetti J, Zhao L, Miller SC, Randall RL, Anderson N, et al. RON kinase: A target for treatment of cancer-induced bone destruction and osteoporosis. Sci Trans Med (2017) 9. doi: 10.1126/scitranslmed.aai9338
29. Knowles HJ. The Adenosine A2B Receptor Drives Osteoclast-Mediated Bone Resorption in Hypoxic Microenvironments. Cells (2019) 8. doi: 10.3390/cells8060624
30. Dirckx N, Moorer MC, Clemens TL, Riddle RC. The role of osteoblasts in energy homeostasis. Nat Rev Endocrinol (2019) 15:651–65. doi: 10.1038/s41574-019-0246-y
31. Weichhaus M, Chung ST, Connelly L. Osteoprotegerin in breast cancer: beyond bone remodeling. Mol Cancer (2015) 14:117. doi: 10.1186/s12943-015-0390-5
32. Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manage Res (2011) 3:177–89. doi: 10.2147/CMAR.S18101
33. Shiozawa Y, Havens AM, Jung Y, Ziegler AM, Pedersen EA, Wang J, et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem (2008) 105:370–80. doi: 10.1002/jcb.21835
34. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest (2011) 121:1298–312. doi: 10.1172/JCI43414
35. Capulli M, Hristova D, Valbret Z, Carys K, Arjan R, Maurizi A, et al. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br J Cancer (2019) 121:157–71. doi: 10.1038/s41416-019-0501-y
36. Widner DB, Park SH, Eber MR, Shiozawa Y. Interactions Between Disseminated Tumor Cells and Bone Marrow Stromal Cells Regulate Tumor Dormancy. Curr Qsteoporos Rep (2018) 16:596–602. doi: 10.1007/s11914-018-0471-7
37. Sosnoski DM, Norgard RJ, Grove CD, Foster SJ, Mastro AM. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin Exp Metastasis (2015) 32:335–44. doi: 10.1007/s10585-015-9710-9
38. Johnson RW, Finger EC, Olcina MM, Vilalta M, Aguilera T, Miao Y, et al. Erratum: Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol (2016) 18:1260. doi: 10.1038/ncb3433
39. Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med (2011) 208:2641–55. doi: 10.1084/jem.20110840
40. Yu-Lee LY, Yu G, Lee YC, Lin SC, Pan J, Pan T, et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFβRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res (2018) 78:2911–24. doi: 10.1158/0008-5472.CAN-17-1051
41. Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, et al. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci Rep (2016) 6:36520. doi: 10.1038/srep36520
42. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun (2015) 6:8983. doi: 10.1038/ncomms9983
43. Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology (2007) 148:198–205. doi: 10.1210/en.2006-1006
44. Burghardt AJ, Issever AS, Schwartz AV, Davis KA, Masharani U, Majumdar S, et al. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab (2010) 95:5045–55. doi: 10.1210/jc.2010-0226
45. Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes–a meta-analysis. Osteoporos Int J established as result cooperation between Eur Foundation Osteoporos Natl Osteoporos Foundation USA (2007) 18:427–44. doi: 10.1007/s00198-006-0253-4
46. Zong JC, Wang X, Zhou X, Wang C, Chen L, Yin LJ, et al. Gut-derived serotonin induced by depression promotes breast cancer bone metastasis through the RUNX2/PTHrP/RANKL pathway in mice. Oncol Rep (2016) 35:739–48. doi: 10.3892/or.2015.4430
47. Campbell JP, Karolak MR, Ma Y, Perrien DS, Masood-Campbell SK, Penner NL, et al. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PloS Biol (2012) 10:e1001363. doi: 10.1371/journal.pbio.1001363
48. Katz S, Weinerman S. Osteoporosis and gastrointestinal disease. Gastroenterol Hepatol (2010) 6:506–17.
49. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (Lond Engl) (2001) 357:539–45. doi: 10.1016/S0140-6736(00)04046-0
50. Lee JY, Murphy SM, Scanlon EF. Effect of trauma on implantation of metastatic tumor in bone in mice. J Surg Oncol (1994) 56:178–84. doi: 10.1002/jso.2930560311
51. Alieva M, van Rheenen J, Broekman MLD. Potential impact of invasive surgical procedures on primary tumor growth and metastasis. Clin Exp Metastasis (2018) 35:319–31. doi: 10.1007/s10585-018-9896-8
52. Demicheli R, Retsky MW, Hrushesky WJ, Baum M, Gukas ID. The effects of surgery on tumor growth: a century of investigations. Ann Oncol (2008) 19:1821–8. doi: 10.1093/annonc/mdn386
53. Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Trans Med (2018) 10. doi: 10.1126/scitranslmed.aan3464
54. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8:729–40. doi: 10.1038/nrm2233
55. Alspach E, Fu Y, Stewart SA. Senescence and the pro-tumorigenic stroma. Crit Rev Oncogenesis (2013) 18:549–58. doi: 10.1615/CritRevOncog.2014010630
56. Luo X, Fu Y, Loza AJ, Murali B, Leahy KM, Ruhland MK, et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep (2016) 14:82–92. doi: 10.1016/j.celrep.2015.12.016
57. Li Y, Terauchi M, Vikulina T, Roser-Page S, Weitzmann MN. B Cell Production of Both OPG and RANKL is Significantly Increased in Aged Mice. Open Bone J (2014) 6:8–17. doi: 10.2174/1876525401406010008
58. Ottewell PD, Wang N, Brown HK, Reeves KJ, Fowles CA, Croucher PI, et al. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin Cancer Res (2014) 20:2922–32. doi: 10.1158/1078-0432.CCR-13-1246
59. Schosserer M, Grillari J, Breitenbach M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front Oncol (2017) 7:278. doi: 10.3389/fonc.2017.00278
60. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol (2019) 21:94–101. doi: 10.1038/s41556-018-0249-2
61. Lynch ME, Brooks D, Mohanan S, Lee MJ, Polamraju P, Dent K, et al. In vivo tibial compression decreases osteolysis and tumor formation in a human metastatic breast cancer model. J Bone Mineral Res (2013) 28:2357–67. doi: 10.1002/jbmr.1966
62. Kothari AN, Arffa ML, Chang V, Blackwell RH, Syn WK, Zhang J, et al. Osteopontin-A Master Regulator of Epithelial-Mesenchymal Transition. J Clin Med (2016) 5. doi: 10.3390/jcm5040039
63. Fan Y, Jalali A, Chen A, Zhao X, Liu S, Teli M, et al. Skeletal loading regulates breast cancer-associated osteolysis in a loading intensity-dependent fashion. Bone Res (2020) 8:9. doi: 10.1038/s41413-020-0083-6
64. Delgado-Calle J, Bellido T, Roodman GD. Role of osteocytes in multiple myeloma bone disease. Curr Opin Support Palliative Care (2014) 8:407–13. doi: 10.1097/SPC.0000000000000090
65. Zhou JZ, Riquelme MA, Gu S, Kar R, Gao X, Sun L, et al. Osteocytic connexin hemichannels suppress breast cancer growth and bone metastasis. Oncogene (2016) 35:5597–607. doi: 10.1038/onc.2016.101
66. McDonald MM, Delgado-Calle J. Sclerostin: an Emerging Target for the Treatment of Cancer-Induced Bone Disease. Curr Qsteoporos Rep (2017) 15:532–41. doi: 10.1007/s11914-017-0403-y
67. Robling AG, Bonewald LF. The Osteocyte: New Insights. Annu Rev Physiol (2020) 82:485–506. doi: 10.1146/annurev-physiol-021119-034332
68. Delgado-Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone (2017) 96:29–37. doi: 10.1016/j.bone.2016.10.007
69. Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PloS One (2015) 10:e0138189. doi: 10.1371/journal.pone.0138189
70. D’Oronzo S, Silvestris E, Paradiso A, Cives M, Tucci M. Role of Bone Targeting Agents in the Prevention of Bone Metastases from Breast Cancer. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21083022
71. Gnant M, Pfeiler G, Steger GG, Egle D, Greil R, Fitzal F, et al. Adjuvant denosumab in postmenopausal patients with hormone receptor-positive breast cancer (ABCSG-18): disease-free survival results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2019) 20:339–51. doi: 10.1016/S1470-2045(18)30862-3
72. Coleman R, Finkelstein DM, Barrios C, Martin M, Iwata H, Hegg R, et al. Adjuvant denosumab in early breast cancer (D-CARE): an international, multicentre, randomised, controlled, phase 3 trial. Lancet Oncol (2020) 21:60–72. doi: 10.1016/S1470-2045(19)30687-4
73. Plotkin LI, Gortazar AR, Davis HM, Condon KW, Gabilondo H, Maycas M, et al. Inhibition of osteocyte apoptosis prevents the increase in osteocytic receptor activator of nuclear factor kappaB ligand (RANKL) but does not stop bone resorption or the loss of bone induced by unloading. J Biol Chem (2015) 290:18934–42. doi: 10.1074/jbc.M115.642090
74. McCutcheon S, Majeska RJ, Spray DC, Schaffler MB, Vazquez M. Apoptotic Osteocytes Induce RANKL Production in Bystanders via Purinergic Signaling and Activation of Pannexin Channels. J Bone Mineral Res (2020) 35:966–77. doi: 10.1002/jbmr.3954
75. Kameda T, Mano H, Yuasa T, Mori Y, Miyazawa K, Shiokawa M, et al. Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J Exp Med (1997) 186:489–95. doi: 10.1084/jem.186.4.489
76. Ma L, Hua R, Tian Y, Cheng H, Fajardo RJ, Pearson JJ, et al. Connexin 43 hemichannels protect bone loss during estrogen deficiency. Bone Res (2019) 7:11. doi: 10.1038/s41413-019-0050-2
77. Li J, Karim MA, Che H, Geng Q, Miao D. Deletion of p16 prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Am J Trans Res (2020) 12:672–83.
78. Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell (2001) 104:719–30. doi: 10.1016/S0092-8674(01)00268-9
79. Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem (2014) 289:16699–710. doi: 10.1074/jbc.M114.547919
80. Coleman R, Cameron D, Dodwell D, Bell R, Wilson C, Rathbone E, et al. Adjuvant zoledronic acid in patients with early breast cancer: final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. Lancet Oncol (2014) 15:997–1006. doi: 10.1016/S1470-2045(14)70302-X
81. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Adjuvant bisphosphonate treatment in early breast cancer: meta-analyses of individual patient data from randomised trials. Lancet (Lond Engl) (2015) 386:1353–61. doi: 10.1016/S0140-6736(15)60908-4
82. Wang W, Yang X, Dai J, Lu Y, Zhang J, Keller ET. Prostate cancer promotes a vicious cycle of bone metastasis progression through inducing osteocytes to secrete GDF15 that stimulates prostate cancer growth and invasion. Oncogene (2019) 38:4540–59. doi: 10.1038/s41388-019-0736-3
83. Sottnik JL, Dai J, Zhang H, Campbell B, Keller ET. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res (2015) 75:2151–8. doi: 10.1158/0008-5472.CAN-14-2493
84. Ma YV, Lam C, Dalmia S, Gao P, Young J, Middleton K, et al. Mechanical regulation of breast cancer migration and apoptosis via direct and indirect osteocyte signaling. J Cell Biochem (2018) 119:5665–75. doi: 10.1002/jcb.26745
85. Giuliani N, Ferretti M, Bolzoni M, Storti P, Lazzaretti M, Dalla Palma B, et al. Increased osteocyte death in multiple myeloma patients: role in myeloma-induced osteoclast formation. Leukemia (2012) 26:1391–401. doi: 10.1038/leu.2011.381
86. Riquelme MA, Cardenas ER, Xu H, Jiang JX. The Role of Connexin Channels in the Response of Mechanical Loading and Unloading of Bone. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21031146
87. Xu H, Gu S, Riquelme MA, Burra S, Callaway D, Cheng H, et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. J Bone Mineral Res (2015) 30:436–48. doi: 10.1002/jbmr.2374
88. Kar R, Riquelme MA, Werner S, Jiang JX. Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J Bone Mineral Res (2013) 28:1611–21. doi: 10.1002/jbmr.1917
89. Bellido T, Plotkin LI. Novel actions of bisphosphonates in bone: preservation of osteoblast and osteocyte viability. Bone (2011) 49:50–5. doi: 10.1016/j.bone.2010.08.008
90. Zhou JZ, Riquelme MA, Gao X, Ellies LG, Sun LZ, Jiang JX. Differential impact of adenosine nucleotides released by osteocytes on breast cancer growth and bone metastasis. Oncogene (2015) 34:1831–42. doi: 10.1038/onc.2014.113
91. Genetos DC, Kephart CJ, Zhang Y, Yellowley CE, Donahue HJ. Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J Cell Physiol (2007) 212:207–14. doi: 10.1002/jcp.21021
92. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
93. Feng LL, Cai YQ, Zhu MC, Xing LJ, Wang X. The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int (2020) 20:110. doi: 10.1186/s12935-020-01195-x
94. Rapaport E, Fishman RF, Gercel C. Growth inhibition of human tumor cells in soft-agar cultures by treatment with low levels of adenosine 5’-triphosphate. Cancer Res (1983) 43:4402–6.
95. Beijer S, Hupperets PS, van den Borne BE, Eussen SR, van Henten AM, van den Beuken-van Everdingen M, et al. Effect of adenosine 5’-triphosphate infusions on the nutritional status and survival of preterminal cancer patients. Anti-cancer Drugs (2009) 20:625–33. doi: 10.1097/CAD.0b013e32832d4f22
96. Agteresch HJ, Burgers SA, van der Gaast A, Wilson JH, Dagnelie PC. Randomized clinical trial of adenosine 5’-triphosphate on tumor growth and survival in advanced lung cancer patients. Anti-cancer Drugs (2003) 14:639–44. doi: 10.1097/00001813-200309000-00009
97. Agteresch HJ, Dagnelie PC, van der Gaast A, Stijnen T, Wilson JH. Randomized clinical trial of adenosine 5’-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Institute (2000) 92:321–8. doi: 10.1093/jnci/92.4.321
98. White N, Burnstock G. P2 receptors and cancer. Trends Pharmacol Sci (2006) 27:211–7. doi: 10.1016/j.tips.2006.02.004
99. Furlow PW, Zhang S, Soong TD, Halberg N, Goodarzi H, Mangrum C, et al. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat Cell Biol (2015) 17:943–52. doi: 10.1038/ncb3194
100. Miyazaki T, Iwasawa M, Nakashima T, Mori S, Shigemoto K, Nakamura H, et al. Intracellular and extracellular ATP coordinately regulate the inverse correlation between osteoclast survival and bone resorption. J Biol Chem (2012) 287:37808–23. doi: 10.1074/jbc.M112.385369
101. Schenk U, Frascoli M, Proietti M, Geffers R, Traggiai E, Buer J, et al. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signaling (2011) 4:ra12. doi: 10.1126/scisignal.2001270
102. Canale FP, Ramello MC, Nunez N, Araujo Furlan CL, Bossio SN, Gorosito Serran M, et al. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8(+) T Cells. Cancer Res (2018) 78:115–28. doi: 10.1158/0008-5472.CAN-16-2684
103. Kazemi MH, Raoofi Mohseni S, Hojjat-Farsangi M, Anvari E, Ghalamfarsa G, Mohammadi H, et al. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J Cell Physiol (2018) 233:2032–57. doi: 10.1002/jcp.25873
104. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PloS One (2008) 3:e2599. doi: 10.1371/journal.pone.0002599
105. Allard D, Allard B, Stagg J. On the mechanism of anti-CD39 immune checkpoint therapy. J Immunotherapy Cancer (2020) 8. doi: 10.1136/jitc-2019-000186
106. Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell (2018) 22:445–53.e5. doi: 10.1016/j.stem.2018.01.017
Keywords: osteocyte, bone, cancer, metastasis, microenvironment
Citation: Riquelme MA, Cardenas ER and Jiang JX (2020) Osteocytes and Bone Metastasis. Front. Endocrinol. 11:567844. doi: 10.3389/fendo.2020.567844
Received: 30 May 2020; Accepted: 24 September 2020;
Published: 14 October 2020.
Edited by:
Lilian Irene Plotkin, Indiana University Bloomington, United StatesReviewed by:
Gabriel M. Pagnotti, Stony Brook University, United StatesCopyright © 2020 Riquelme, Cardenas and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean X. Jiang, amlhbmdqQHV0aHNjc2EuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.