 Bohan Lv1†
Bohan Lv1† Xueli Bao2†Ping Li2Juan Lian1Yanxiang Wu1Tian An1Jing Zhang3Xiuyan Yang1Tingye Wang1Jiajian Zhu1Yuanyuan Hu1Guangjian Jiang1*Sihua Gao1*
Xueli Bao2†Ping Li2Juan Lian1Yanxiang Wu1Tian An1Jing Zhang3Xiuyan Yang1Tingye Wang1Jiajian Zhu1Yuanyuan Hu1Guangjian Jiang1*Sihua Gao1*- 1Traditional Chinese Medicine School, Beijing University of Chinese Medicine, Beijing, China
- 2Third Affiliated Hospital, Beijing University of Chinese Medicine, Beijing, China
- 3Department of Endocrinology, Tangshan People's Hospital, Tangshan, China
Purpose: The purpose of this study is to explore the differences in transcriptome expression profiles between healthy subjects and type 2 diabetes mellitus patients with thirst and fatigue (D-T2DM) and, in addition, to investigate the possible role of noncoding ribonucleic acids (RNAs) in the pathogenesis of D-T2DM.
Methods: We constructed the expression profiles of RNAs by RNA sequencing in the peripheral blood of D-T2DM patients and healthy subjects and analyzed differentially expressed RNAs.
Results: Compared with healthy subjects, a total of 469 mRNAs, 776 long non-coding RNAs (lncRNAs), and 21 circular RNAs (circRNAs) were differentially expressed in D-T2DM patients. Furthermore, several genes associated with insulin resistance, inflammation, and mitochondrial dysfunction were identified within the differentially expressed mRNAs. Differentially expressed lncRNAs were primarily involved in biological processes associated with immune responses. In addition, differentially expressed circRNAs may target miRNAs associated with glucose metabolism and mitochondrial function.
Conclusions: Our results may bring a new perspective on differential RNA expression involved in the pathogenesis of D-T2DM and promote the development of novel treatments for this disease.
Introduction
In today's world, with the improvement of living conditions and the change of lifestyle, more and more diabetic patients are diagnosed. The number of people with diabetes is expected to reach 700 million by 2045 (1). In traditional Chinese medicine, diabetes is divided into different types according to the clinical symptoms such as fever, fatigue, sputum, and urinary frequency (2). The “dual deficiency of Qi and Yin syndrome” is a common type of diabetes, and the main symptoms of this type are dry mouth and throat and weakness with general fatigue. Similarly, a proportion of patients with T2DM are accompanied with thirst and fatigue in modern medicine (3–7). Therefore, the diagnosis and treatment of this type of T2DM (D-T2DM) have become an important issue in clinical work.
Non-coding ribonucleic acids (ncRNAs) are transcripts that cannot be translated into proteins, but are generally considered to have a role in regulating protein expression (8). ncRNAs are divided into long non-coding RNAs (lncRNAs) and circular loops RNAs (circRNAs) according to their morphology (9). lncRNAs are transcripts longer than 200 nucleotides that have been implicated in diverse biological functions, such as transcriptional and posttranscriptional regulation and chromatin modification (10–12). CircRNAs are a type of circular ncRNA discovered recently and attracted wide attention (13). In our previous research, ncRNAs have been shown to play a key role in the pathogenesis of T2DM and other endocrine diseases (14, 15). However, the role of ncRNAs in D-T2DM development remains unclear. Therefore, comprehensive detection and analysis of ncRNAs in the development of D-T2DM are critical for the prevention and treatment of D-T2DM.
In this study, we used RNA sequencing technology to construct RNA expression profiles in peripheral blood of patients with D-T2DM. Differentially expressed mRNAs, lncRNAs, and circRNAs were detected, and their functions were predicted by bioinformatics analysis, in order to discover new targets and provide assistance for the treatment of D-T2DM.
Materials and Methods
Ethics Statement and Information of Subjects
This research was approved by the Ethics Committee of Beijing University of Chinese Medicine (BUCM) (2017BZHYLL0105). All subjects, enrolled from the Affiliated Hospital of BUCM, agreed to participate in the study after fully understanding the purpose and procedure of the experiment. Full inclusion and exclusion criteria are listed in Table 1. The subjects were divided into two groups: D-T2DM group (ID: QYD1, QYD2, QYD3, QYD4, QYD5, and QYD6) and the healthy subjects as the control group (ID: LZC001, LZC002, LZC003, LZC004, LZC005, and LZC006). Subsequently, the fasting venous blood of all subjects were obtained and stored at −80°C until analysis.
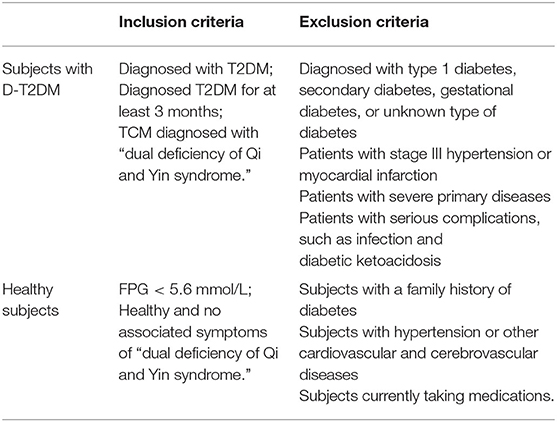
Table 1. Inclusion and exclusion criteria.
Total RNA Extraction, Library Construction, and Illumina Sequencing
According to the previous experimental methods, we extracted the total RNA and constructed the RNA library (16). Briefly, the total RNA was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and tested for purity using the NanoPhotometer spectrophotometer (IMPLEN, CA, USA). In addition, the concentration and integrity of RNA were assessed using the Qubit 2.0 fluorometer (Life Technologies, CA, USA) and the Agilent 2100 bioanalyzer (Agilent Technologies, CA, USA), respectively. Next, Ribo-Zero rRNA Removal Kit (Epicentre, USA) was used to remove ribosomal RNA (rRNA). RNA-sequencing libraries were prepared using the rRNA-depleted RNA by NEBnext ultra RNA library prep kit following the manufacturer's instruction. After that, the quality of libraries was determined using the Agilent 2100 System (NanoDrop ND-1000) and accurately quantified by quantitative real-time polymerase chain reaction. Lastly, the libraries were pooled according to the requirements of effective concentration and data volume and then sequenced on Illumina Hiseq 2000 platform.
Quality Control and Mapping
Raw data were processed through in-house Perl scripts. In this step, the raw data were cleaned by removing reads containing adaptors, contaminants, and low-quality reads. All clean data have been submitted to Sequence Read Archive with accession number SRP274496. Additionally, the Q20, Q30, and GC content were calculated to estimate the quality of clean reads. Next, the clean reads were aligned to the reference genome (GRCh37/hg19) using STAR (v2.5.1b).
Quantitative Analysis of Genes and ncRNAs
The FPKM of transcripts and ncRNAs were calculated by Cuffdiff (v2.1.1) (17). FPKM refers to the number of fragments per kilobase length from a gene per million fragments mapped. It considers the effect of both sequencing depth and gene length on fragments count.
GO and KEGG Pathway Annotation
In order to understand the biological function of differentially expressed genes, we performed enrichment analysis by topGO software. Based on the newest KEGG database, the pathway analysis was performed to determine the significant pathway of the differential genes. Fisher test was used for enrichment analysis, and those with P < 0.05 were significantly enriched.
Interaction Network Analysis
Cytoscape v2.8.2 software (http://www.cytoscape.org/) was used to construct the lncRNA–mRNA and circRNA–miRNA regulatory network based on the differentially expressed gene data in the blood between D-T2DM and healthy subjects.
Statistical Analysis
This study used GraphPad Prism 7 (GraphPad Software, CA) and SPSS software (version 20.0) for the statistical evaluations. The results are expressed as mean ± SEM. Statistical differences were determined by Student independent t-test, and the significance was accepted at P < 0.05. Each experiment was repeated for three technical replicates.
Results
Clinical Characteristics of the Participants
In this study, six D-T2DM patients and six healthy subjects were enrolled. All patients fulfilled the diagnostic criteria for D-T2DM. There was no significant difference in age between the D-T2DM and control groups. The characteristics of all subjects are shown in Table 2. Compared with the control group, the D-T2DM group exhibited significant increases in FPG and TG levels.
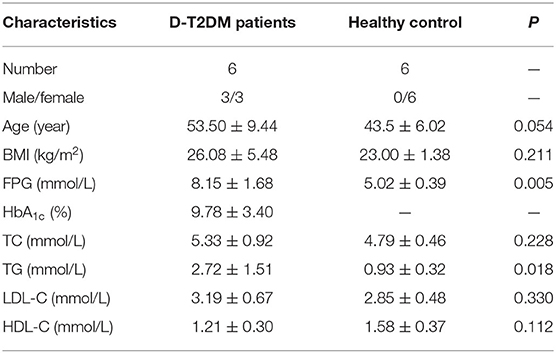
Table 2. Characteristics of study subjects.
Quality Assessments and Mapping Results
To construct expression profiles of mRNAs and ncRNAs of D-T2DM and healthy control, transcriptome data sets were generated by RNA-seq. Subsequently, quality control and mapping analysis were performed for the sequencing output (Supplementary Tables 1, 2). The base percentage of Q20 was >95.29%, Q30 was >88.92%, and the average mapping rate of the 12 samples was 95.21%. These results could indicate the high quality of transcriptome sequencing data with suitable mapping.
Differentially Expressed mRNAs, lncRNAs, and circRNAs
Sequencing technique was used to detect differentially expressed ncRNAs in the peripheral blood of D-T2DM patients. In Figure 1, the results showed that a total of 469 differentially expressed mRNAs (341 up- and 128 down-regulated), 776 differentially expressed lncRNAs (688 up- and 88 down-regulated), and 21 differentially expressed circRNAs (5 up- and 16 down-regulated) were detected in D-T2DM patients compared with healthy control (fold change >1.5, P < 0.05). Tables 3–5 list the top 10 up- and down-regulated mRNAs, lncRNAs, and circRNAs, respectively. Furthermore, Figures 1A–F shows the volcano plot, cluster map of differentially expressed mRNAs, lncRNAs, and circRNAs, respectively. Among these genes, LRRC19, GCNT3, and CKMT2 were associated with the pathogenesis of T2DM. Furthermore, mRNAs involved in regulation of mitochondrial function and lipid metabolism were significantly expressed, such as MT-ND1, MT-ND2, and OSBL6.
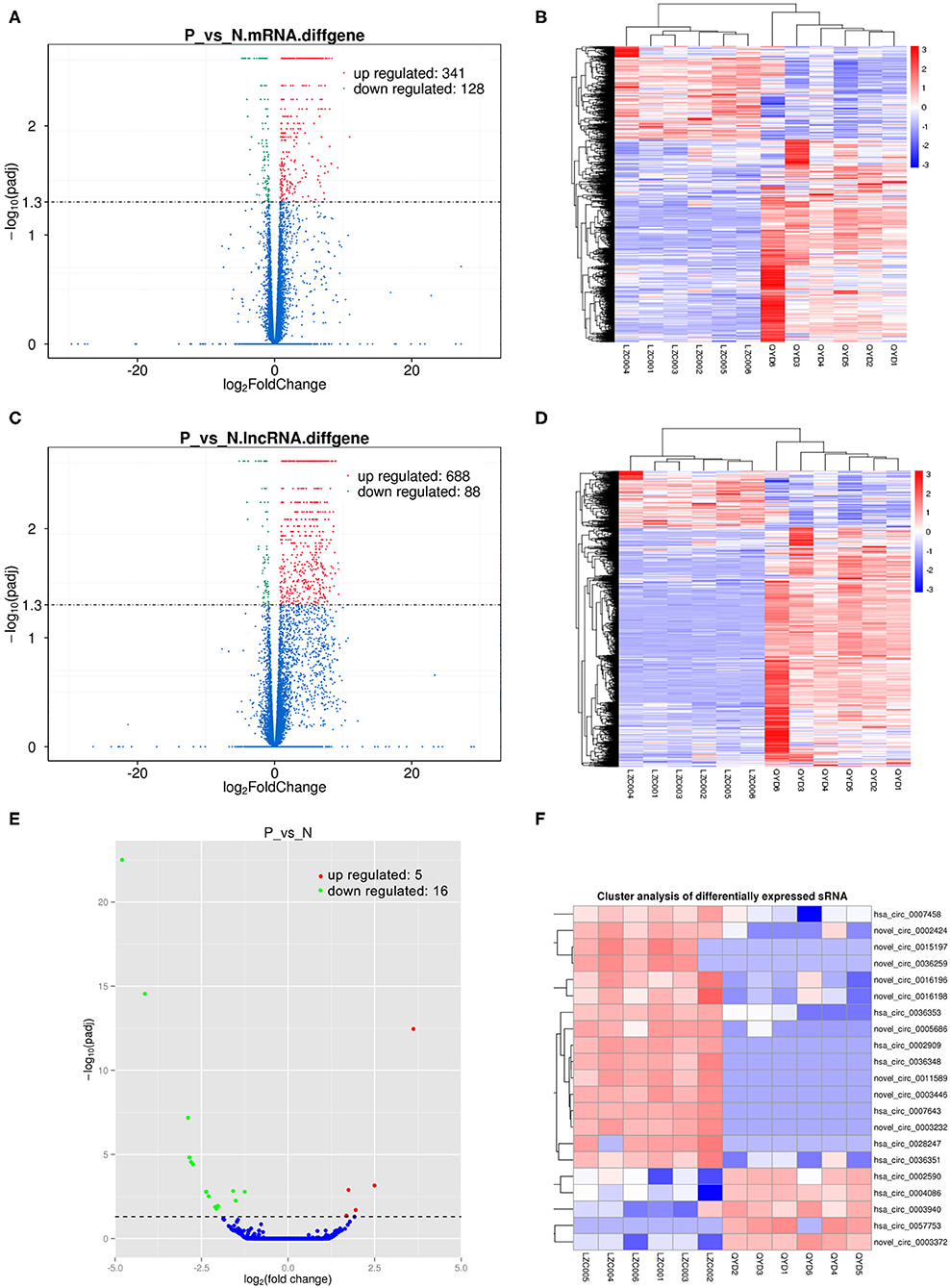
Figure 1. The expression profiling changes of RNAs in peripheral blood sample of D-T2DM patients. The volcano plot (A), cluster map (B) of differentially expressed mRNAs. The volcano plot (C) and cluster map (D) of differentially expressed lncRNAs. The volcano plot (E) and cluster map (F) of differentially expressed circRNAs.
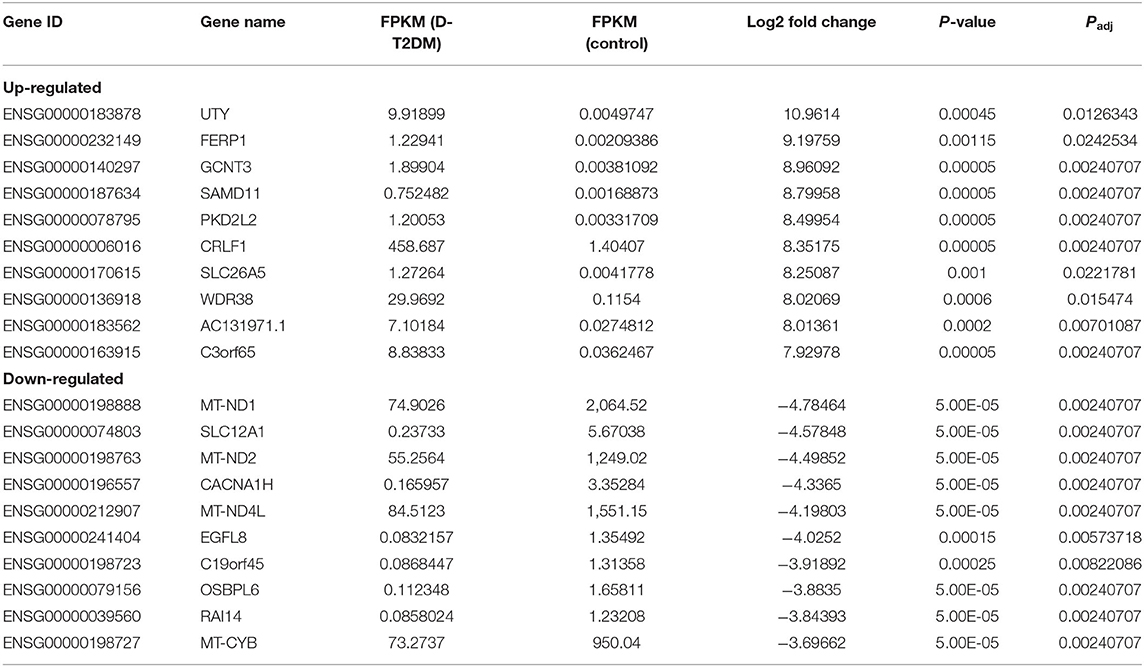
Table 3. The top 10 up-regulated and down-regulated mRNAs.
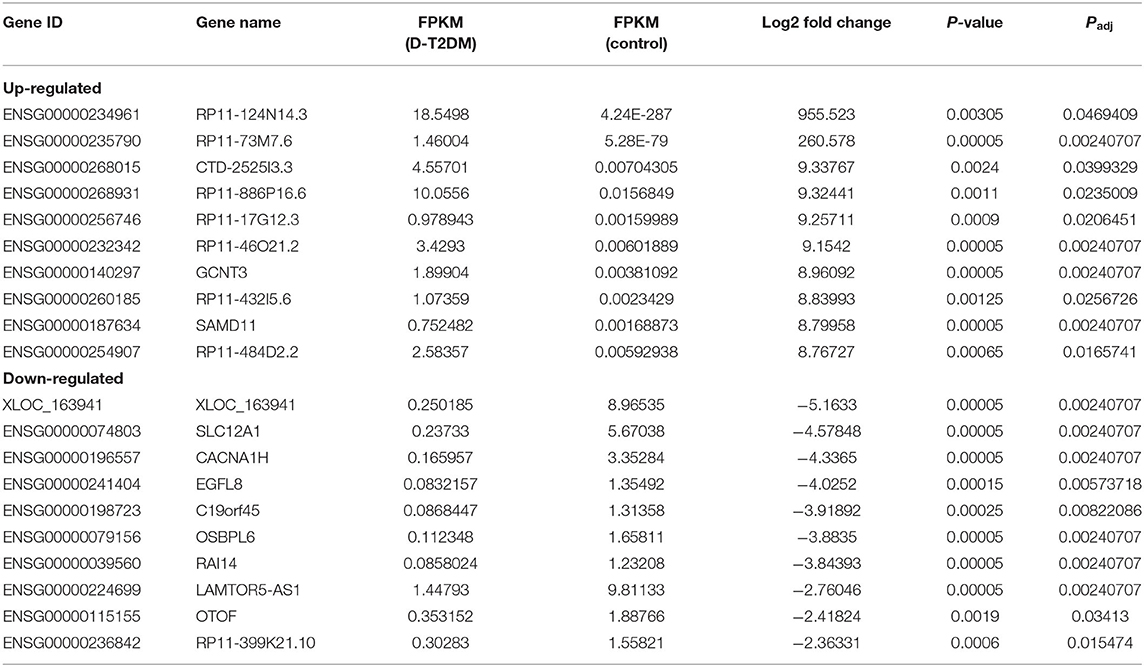
Table 4. The top 10 up-regulated and down-regulated lncRNAs.
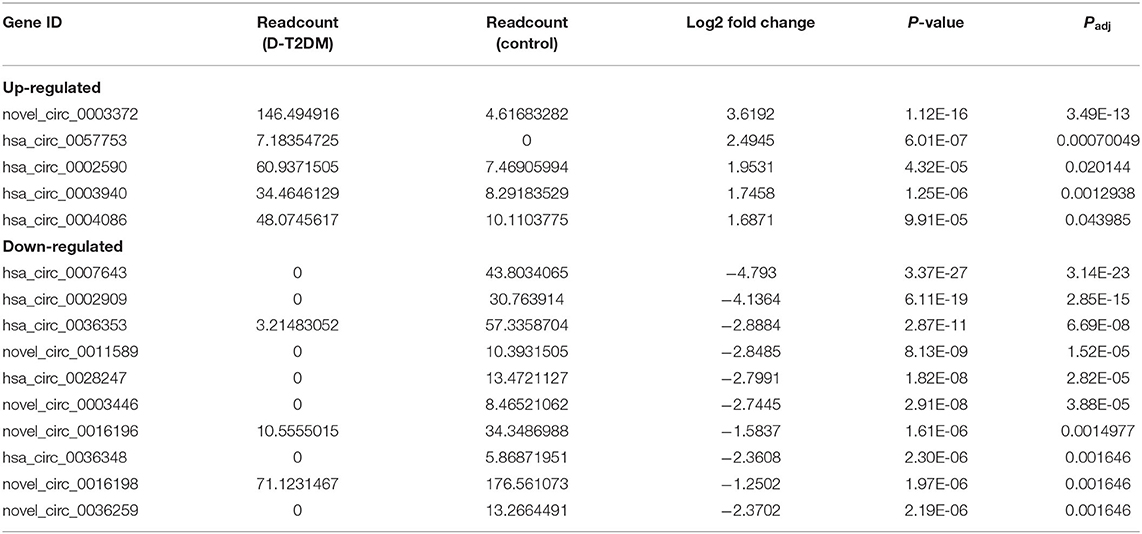
Table 5. The top 10 up-regulated and down-regulated circRNAs.
lncRNA Target Gene Prediction
lncRNA may perform its function by regulating genes. Therefore, we predicted the biological function of lncRNA by its colocated and coexpressed genes. We set the threshold of the colocated genes to 100 kb upstream and downstream of lncRNA; mRNA gene with an absolute value of Pearson correlation coefficient >0.95 was defined as lncRNA coexpressed mRNAs (Supplementary Tables 3, 4).
GO and KEGG Enrichment Analysis of Differentially Expressed RNAs in D-T2DM
We performed functional enrichment analysis of differentially expressed RNAs in bioinformatics databases, including GO and KEGG analysis. As shown in Figure 2A, GO analysis of differentially expressed mRNAs revealed that the most significantly enriched biological process (BP) were disruption of cells of other organism and killing of cells of other organism, and the most significantly enriched cellular component (CC) were actin cytoskeleton, cytosolic ribosome, and secretory granule. The GO analysis of differentially expressed lncRNAs showed that the most significantly enriched BPs were adaptive immune response, positive regulation of lymphocyte activation, and response to virus, and the most noteworthy enriched CCs were MHC class II protein complex, cytosolic part, and MHC protein complex. The most significantly enriched molecular functions (MFs) were MHC protein binding, tumor necrosis factor receptor binding, and antigen binding (Figure 2C). GO analysis of differentially expressed circRNAs revealed that the most significantly enriched BPs were biological regulation, CC organization or biogenesis, and cellular process, and the most notable enrichment of CCs were cell, cell junction, and cell part. The most significantly enriched MFs were binding, catalytic activity, and channel regulator activity (Figure 3A).
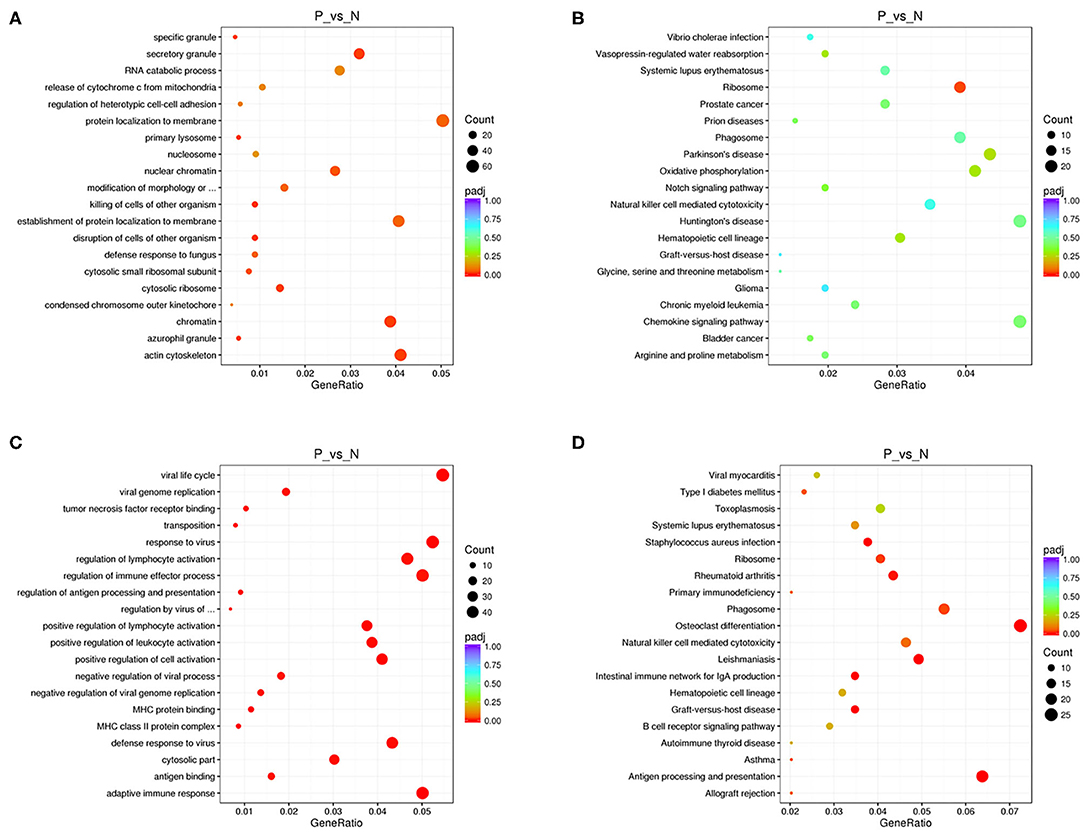
Figure 2. GO and KEGG analysis of mRNAs and lncRNAs in peripheral blood sample of D-T2DM patients. The scatterplots of GO enrichment results of differentially expressed mRNAs (A) and lncRNAs (C). The scatterplots of KEGG pathway analysis of differentially expressed mRNAs (B) and lncRNAs (D). The degree of GO and KEGG enrichment is assessed by enrichment of factors, Q-values, and number of genes. When the rich factor is larger, the Q-value is closer to zero, and the more the number of genes, the more significant the enrichment.
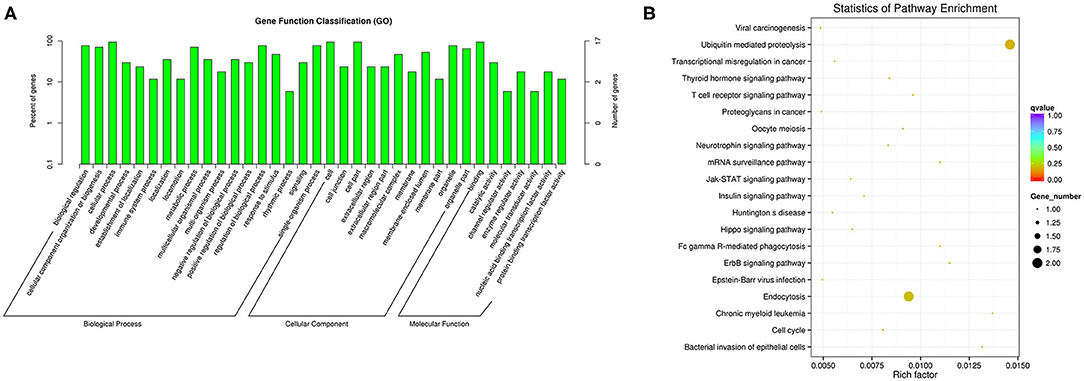
Figure 3. GO (A) and KEGG (B) analysis of circRNAs in peripheral blood sample of D-T2DM patients.
KEGG analysis of differentially expressed mRNAs revealed that the most significantly enriched pathway in D-T2DM pathogenesis was ribosome (Figure 2B). The colocated mRNAs of differentially expressed lncRNAs were significantly enriched in antigen processing and presentation, osteoclast differentiation, and leishmaniasis (Figure 2D). Furthermore, differentially expressed circRNAs-derived genes were mostly involved in the ubiquitin-mediated proteolysis (Figure 3B).
Protein–Protein Interaction Network of lncRNA Colocated mRNA Corresponding Genes
We extracted the interaction relationship of differential gene sets to build a network forms the STRING Protein Interaction Database (http://string-db.org/) and then imported them into Cytoscape software for visual editing (Figure 4). As shown in Table 6, we listed the top 10 BP terms enriched for the genes involved in the protein–protein interaction network. UBA52 had the highest degree of network connectivity and enriched in the BP terms such as immune system process, regulation of immune response, and regulation of immune system process.

Figure 4. Protein interaction network of DELncRNA colocated mRNA corresponding genes.
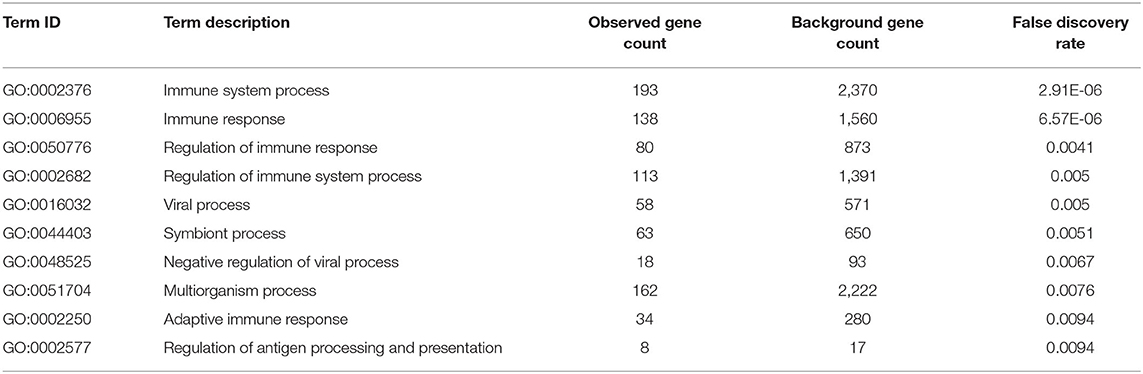
Table 6. Top 10 enriched biological process terms for the genes involved in the protein–protein interaction network.
Regulatory Network of circRNA and miRNA
Using differentially expressed circRNA as the center and miRNA as the target, we constructed the circRNA–miRNA regulatory network of up-regulated and down-regulated circRNAs, respectively (Figure 5). We found that 47, 38, 48, and 51 miRNA sites can be combined with up-regulated novel_circ_0003372, hsa_circ_0002590, hsa_circ_0003940, and hsa_circ_0004086, respectively; 70, 76, 80, 41, 22, 63, and 30 miRNA sites associated with down-regulated hsa_circ_0036353, novel_circ_0005686, novel_circ_0002424, hsa_circ_0036351, novel_circ_0016196, hsa_circ_0007458, and novel_circ_0016198, respectively. Among the results, MiR-877-3p was associated with T2DM and mitochondrial function and combined with novel_circ_0016196, novel_circ_0016198, and novel_circ_0005686. circRNA has_circ_0002590 and novel_circ_000372 regulated the expression of MiR-149-5p, which could regulate the insulin secretion.
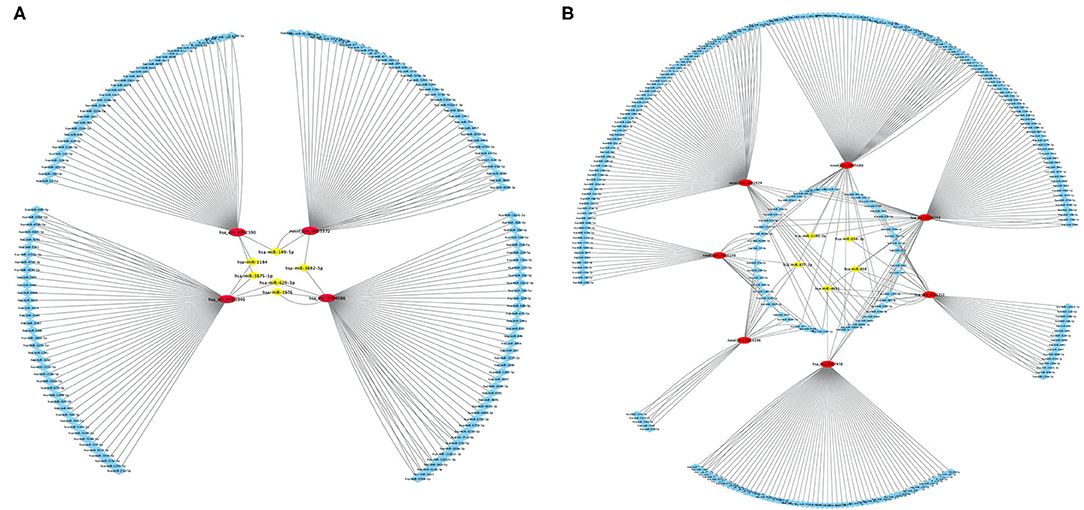
Figure 5. CircRNA–miRNA regulatory network analysis. (A) The circRNA–miRNA regulatory network analysis of up-regulated circRNA. (B) The circRNA–miRNA regulatory network analysis of down-regulated circRNA. Squares represent miRNAs; circles represent circRNAs.
Discussion
In this study, we used sequencing technology to determine the significantly differentially expressed mRNAs, lncRNAs, and circRNAs in the peripheral blood of patients with D-T2DM. Afterward, we performed GO and KEGG pathway analysis on these differentially expressed RNAs to predict their potential biological functions. In addition, we also constructed protein–protein interaction network and circRNA–miRNA regulatory network. Our results indicated that ncRNAs may play a role in the pathogenesis of D-T2DM and provide some potential targets for the treatment of D-T2DM.
The sequencing results showed that there were 469 mRNAs, 776 lncRNAs, and 21 circRNAs significantly dysregulated in D-T2DM patients compared with healthy subjects. In up-regulated mRNAs, we have found some targets related to the pathogenesis of T2DM, such as LRRC19, GCNT3, and CKMT2. Among them, LRRC19 could activate nuclear factor κB (NF-κB) and induce expression of pro-inflammatory cytokines (18). GCNT3 was also related to inflammation (19–21). Inflammation can lead to a cluster of chronic metabolic disorders such as insulin resistance, obesity, type 2 diabetes, and cardiovascular disease (22–24). In addition, previous studies have revealed that NF-κB and its target inflammatory factor genes such as IL-1 and IL-6 played a key role in the development of insulin resistance and T2DM (25–27). CKMT2, creatine kinase, mitochondrial 2, was an effective modulator of ATP synthase–coupled respiration (28). Among down-regulated mRNAs, there were some mitochondrial genes, for instance, MT-ND1, MT-ND2, MT-ND5, and MT-ND4L. These mitochondrial genes were associated with fatty acid metabolism, mitochondrial oxidative phosphorylation, mitochondrial energy transduction, and the diabetes mellitus pathogenesis (29–31). Mitochondria are the most essential energy production organelles, supplying energy for cell metabolism in the form of ATP (32). In traditional Chinese medicine, fatigue is a major symptom of D-T2DM patients. Meanwhile, the hallmark symptom of mitochondrial dysfunction is fatigue (33). Therefore, mitochondrial dysfunction may play a role in the pathogenesis of D-T2DM. Furthermore, OSBPL6 was positively correlated with plasma levels of high-density lipoprotein cholesterol (34). SLC12A1 may play a role in glucose-induced insulin secretion (35). In general, the results indicated that the pathogenesis of D-T2DM may be related to the glucose and lipid metabolism disorders, occurrence of inflammation, and mitochondrial dysfunction.
At present, the mechanism of interaction between lncRNA and protein-coding genes is not clear. We predicted the biological function of lncRNA through its colocated and coexpressed protein-coding genes. Among the differentially expressed lncRNAs, lncRNA-SLC12A1, lncRNA-OSBPL6, and lncRNA-GCNT3 were colocated with SLC12A1, OSBPL6, and GCNT3, respectively. Therefore, these lncRNAs may play a role by regulating the genes related to glucose metabolism, lipid metabolism, and inflammation. Afterward, this study used GO and KEGG pathway analyses to analyze the biological function and pathways of lncRNAs-related genes in the peripheral blood of patients with D-T2DM. The results showed that the most enriched GO term was adaptive immune response. Previous studies have demonstrated that adaptive immune factors was recognized as important etiological components in the development of insulin resistance (36). In our future work, the specific role of lncRNAs predicted by GO and KEGG analysis in the pathogenesis D-T2DM needs to be further studied. Protein–protein interaction network represents the interaction of the protein products of the lncRNA colocated genes and was used to predict the biological function of differentially expressed lncRNAs. The results showed that the proteins were mainly enriched in BPs related to immune response. In addition, UBA52 had the highest degree of connectivity, and previous study suggested that UBA52 may be implicated in the diabetic nephropathy (37). Therefore, differentially expressed lncRNAs appear to function in the pathogenesis of D-T2DM by regulating immune response.
In order to discover the molecular mechanism of circRNAs in D-T2DM, we constructed the circRNA–miRNA regulatory network based on the sequencing data. In our results, MiR-877-3p was combined with down-regulated circRNA novel_circ_0016196, novel_circ_0016198, and novel_circ_0005686. Xie et al. (38) found that MiR-877-3p was deregulated in type 2 diabetic kidney disease. Another study revealed that MiR-877-3p exerts its effects via the Blc-2–mediated mitochondrial apoptotic pathway (39). CircRNAs generally act as an miRNA sponge to regulate gene expression. Therefore, the down-regulation of these circRNAs may improve the function of MiR-877-3p and thus mediate mitochondrial apoptosis. On the other hand, up-regulated circRNA has_circ_0002590 and novel_circ_000372 were associated with MiR-149-5p. The overexpression of MiR-149-5p could ameliorate the high glucose–induced injury in human umbilical vein endothelial cells, whereas the inhibition of MiR-149-5p could suppress cell viability, induce cell apoptosis, and inhibit insulin secretion (40, 41). In this research, the up-regulated circRNAs competitively bound with MiR-149-5p to inhibit its function. In general, circRNAs may play a role in the pathogenesis of D-T2DM by regulating the function of MiR-877-3p and MiR-149-5p, but the specific mechanisms need further research.
In this study, we have performed a comprehensive transcriptome analysis in D-T2DM patients' peripheral blood, revealing the contribution of epigenetic changes to D-T2DM. However, the present study has several limitations. First, in order to better understand the mechanism of occurrence and development of D-T2DM, it is important to compare D-T2DM with other types of T2DM, such as a milder form of T2DM. This will be the main part of our future study. Next, the major targets of insulin actions are skeletal muscle, liver, and adipose tissue rather than the blood. Therefore, our results can explain the pathogenesis of D-T2DM only partially. We anticipate the transcriptome profile of typical insulin-targeted tissues of D-T2DM patients will be investigated in the future. In addition, because peripheral blood also includes multiple cell populations and bioactive substances, further studies are required to characterize a more precise role of ncRNAs in peripheral blood of D-T2DM patients. Our results indicate that ncRNAs may exert their biological functions in the pathogenesis of D-T2DM by interacting with each other or related proteins genes. However, the mechanism of ncRNAs is complicated; thus, we will further study these predicted ncRNAs and their involved signaling pathways. Finally, it will help to reveal the internal mechanism and therapeutic targets of D-T2DM.
In conclusion, we used sequencing analysis to study the expression profile of RNAs in the peripheral blood of D-T2DM and healthy subjects. Differentially expressed mRNAs, lncRNAs, and circRNAs were screened, and their potential biological functions were predicted by bioinformatics analysis. These results may bring new perspectives on the pathogenesis of D-T2DM and promote the development of new therapeutic approaches targeting RNAs.
Data Availability Statement
All clean data have been submitted to Sequence Read Archive with accession number SRP274496.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Beijing University of Chinese Medicine (BUCM) (2017BZHYLL0105). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
SG and GJ designed the experiments. BL and XB wrote the manuscript. PL and TA interpreted data and revised the manuscript. JL, YW, JZha, and XY performed experiments. TA, TW, JZhu, and YH performed data collection and analysis. All authors reviewed the manuscript and agreed to the publication of this article.
Funding
This work was supported by Grants from the Qihuang Scholars National Leading Talents Supporting Plan in Traditional Chinese Medicine (10400633210005) and the National Natural Science Foundation of China (NSFC 81774171).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.558344/full#supplementary-material
References
1. International Diabetes Federation. IDF Diabetes Atlas. 9th ed. Brussels: Belgium Int Diabetes Fed 2019. (2019).
2. Guo J, Chen H, Song J, Wang J, Zhao L, Tong X. Syndrome differentiation of diabetes by the traditional Chinese medicine according to evidence-based medicine and expert consensus opinion. Evid Based Complement Alternat Med. (2014) 2014:492193. doi: 10.1155/2014/492193
3. Kamoi K, Ishibashi M, Yamaji T. Thirst and plasma levels of vasopressin, angiotensin II and atrial natriuretic peptide in patients with non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract. (1991) 11:195–202. doi: 10.1016/S0168-8227(05)80033-6
4. Thompson CJ, Davis SN, Butler PC, Charlton JA, Baylis PH. Osmoregulation of thirst and vasopressin secretion in insulin-dependent diabetes mellitus. Clin Sci. (1988) 74:599–606. doi: 10.1042/cs0740599
5. Morley JE, Malmstrom TK, Rodriguez-Mañas L, Sinclair AJ. Frailty, sarcopenia and diabetes. J Am Med Dir Assoc. (2014) 15:853–9. doi: 10.1016/j.jamda.2014.10.001
6. Yanase T, Yanagita I, Muta K, Nawata H. Frailty in elderly diabetes patients. Endocr J. (2018) 65:1–11. doi: 10.1507/endocrj.EJ17-0390
7. Sinclair AJ, Abdelhafiz AH, Rodríguez-Mañas L. Frailty and sarcopenia - newly emerging and high impact complications of diabetes. J Diabetes Complications. (2017) 31:1465–73. doi: 10.1016/j.jdiacomp.2017.05.003
8. Hon C-C, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJL, Gough J, et al. An atlas of human long non-coding RNAs with accurate 5' ends. Nature. (2017) 543:199–204. doi: 10.1038/nature21374
9. Chen L-L. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. (2016) 17:205–11. doi: 10.1038/nrm.2015.32
10. Tung M-C, Wen Y-C, Wang S-S, Lin Y-W, Chow J-M, Yang S-F, et al. Impact of long non-coding RNA HOTAIR genetic variants on the susceptibility and clinicopathologic characteristics of patients with urothelial cell carcinoma. J Clin Med. (2019) 8:282. doi: 10.3390/jcm8030282
11. Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. (2016) 17:756–70. doi: 10.1038/nrm.2016.126
12. Marchese FP, Huarte M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics. (2014) 9:21–6. doi: 10.4161/epi.27472
13. Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. (2013) 19:141–57. doi: 10.1261/rna.035667.112
14. An T, Zhang J, Ma Y, Lian J, Wu YX, Lv BH, et al. Relationships of non-coding RNA with diabetes and depression. Sci Rep. (2019) 9:10707. doi: 10.1038/s41598-019-47077-9
15. Jiang G, Ma Y, An T, Pan Y, Mo F, Zhao D, et al. Relationships of circular RNA with diabetes and depression. Sci Rep. (2017) 7:7285. doi: 10.1038/s41598-017-07931-0
16. An T, He Z-C, Zhang X-Q, Li J, Chen A-L, Tan F, et al. Baduanjin exerts anti-diabetic and anti-depression effects by regulating the expression of mRNA, lncRNA, and circRNA. Chinese Med. (2019) 14:3. doi: 10.1186/s13020-019-0225-1
17. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. (2010) 28:511–5. doi: 10.1038/nbt.1621
18. Chai L, Dai L, Che Y, Xu J, Liu G, Zhang Z, et al. LRRC19, a novel member of the leucine-rich repeat protein family, activates NF-kappaB and induces expression of pro-inflammatory cytokines. Biochem Biophys Res Commun. (2009) 388:543–8. doi: 10.1016/j.bbrc.2009.08.043
19. Yonezawa S, Nakamura A, Horinouchi M, Sato E. The expression of several types of mucin is related to the biological behavior of pancreatic neoplasms. J Hepatobiliary Pancreat Surg. (2002) 9:328–41. doi: 10.1007/s005340200037
20. Kim GE, Bae H-I, Park H-U, Kuan S-F, Crawley SC, Ho JJL, et al. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology. (2002) 123:1052–60. doi: 10.1053/gast.2002.36018
21. Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. (2008) 321:1801–6. doi: 10.1126/science.1164368
22. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
23. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
24. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006) 116:1793–801. doi: 10.1172/JCI29069
25. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. (2005) 11:183–90. doi: 10.1038/nm1166
26. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. (2001) 293:1673–7. doi: 10.1126/science.1061620
27. Arkan MC, Hevener AL, Greten FR, Maeda S, Li Z-W, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. (2005) 11:191–8. doi: 10.1038/nm1185
28. Müller S, Balaz M, Stefanicka P, Varga L, Amri E-Z, Ukropec J, et al. Proteomic analysis of human brown adipose tissue reveals utilization of coupled and uncoupled energy expenditure pathways. Sci Rep. (2016) 6:30030. doi: 10.1038/srep30030
29. Deng G, Zhou X, Pang Z, Liu S, Xie Y. Study on mitochondrial function of ND1 gene with 3316 G–>A mutation in human diabetes. Zhonghua Yi Xue Za Zhi. (2009) 89:2822–6.
30. McKenzie M, Duchen MR. Impaired cellular bioenergetics causes mitochondrial calcium handling defects in MT-ND5 mutant cybrids. PLoS ONE. (2016) 11:e0154371. doi: 10.1371/journal.pone.0154371
31. Bai M, Chen H, Ding D, Song R, Lin J, Zhang Y, et al. MicroRNA-214 promotes chronic kidney disease by disrupting mitochondrial oxidative phosphorylation. Kidney Int. (2019) 95:1389–404. doi: 10.1016/j.kint.2018.12.028
32. Zhang D, Liu Z-X, Choi CS, Tian L, Kibbey R, Dong J, et al. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci USA. (2007) 104:17075–80. doi: 10.1073/pnas.0707060104
33. Filler K, Lyon D, Bennett J, McCain N, Elswick R, Lukkahatai N, et al. Association of mitochondrial dysfunction and fatigue: a review of the literature. BBA Clin. (2014) 1:12–23. doi: 10.1016/j.bbacli.2014.04.001
34. Ouimet M, Hennessy EJ, van Solingen C, Koelwyn GJ, Hussein MA, Ramkhelawon B, et al. miRNA Targeting of Oxysterol-Binding Protein-Like 6 Regulates Cholesterol Trafficking and Efflux. Arterioscler Thromb Vasc Biol. (2016) 36:942–51. doi: 10.1161/ATVBAHA.116.307282
35. Alshahrani S, Alvarez-Leefmans FJ, Di Fulvio M. Expression of the Slc12a1 gene in pancreatic β-cells: molecular characterization and in silico analysis. Cell Physiol Biochem. (2012) 30:95–112. doi: 10.1159/000339050
36. Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab. (2009) 94:3171–82. doi: 10.1210/jc.2008-2534
37. Dihazi H, Müller GA, Lindner S, Meyer M, Asif AR, Oellerich M, et al. Characterization of diabetic nephropathy by urinary proteomic analysis: identification of a processed ubiquitin form as a differentially excreted protein in diabetic nephropathy patients. Clin Chem. (2007) 53:1636–45. doi: 10.1373/clinchem.2007.088260
38. Xie Y, Jia Y, Cuihua X, Hu F, Xue M, Xue Y. Urinary exosomal MicroRNA profiling in incipient type 2 diabetic kidney disease. J Diabetes Res. (2017) 2017:6978984. doi: 10.1155/2017/6978984
39. Li X, Luo S, Zhang J, Yuan Y, Jiang W, Zhu H, et al. lncRNA H19 alleviated myocardial I/RI via suppressing miR-877-3p/Bcl-2-mediated mitochondrial apoptosis. Mol Ther Nucleic Acids. (2019) 17:297–309. doi: 10.1016/j.omtn.2019.05.031
40. Yuan J, Chen M, Xu Q, Liang J, Chen R, Xiao Y, et al. Effect of the diabetic environment on the expression of MiRNAs in endothelial cells: Mir-149-5p restoration ameliorates the high glucose-induced expression of TNF-α and ER stress markers. Cell Physiol Biochem. (2017) 43:120–35. doi: 10.1159/000480330
Keywords: circRNAs, lncRNAs, miRNA, T2DM, RNA sequencing
Citation: Lv B, Bao X, Li P, Lian J, Wu Y, An T, Zhang J, Yang X, Wang T, Zhu J, Hu Y, Jiang G and Gao S (2020) Transcriptome Sequencing Analysis of Peripheral Blood of Type 2 Diabetes Mellitus Patients With Thirst and Fatigue. Front. Endocrinol. 11:558344. doi: 10.3389/fendo.2020.558344
Received: 04 May 2020; Accepted: 04 September 2020;
Published: 09 November 2020.
Edited by:
Marcus M. Seldin, University of California, Irvine, United StatesReviewed by:
Emily Louise Clark, University of Edinburgh, United KingdomDaniela Patrizia Foti, University of Catanzaro, Italy
Copyright © 2020 Lv, Bao, Li, Lian, Wu, An, Zhang, Yang, Wang, Zhu, Hu, Jiang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangjian Jiang, Z3VhbmdqaWFuamlhbmdAc2luYS5jb20=; Sihua Gao, c2lodWFnYW9wYXBlckBzaW5hLmNvbQ==
†These authors have contributed equally to this work