 Vassos Neocleous1,2†
Vassos Neocleous1,2† Pavlos Fanis1,2†
Pavlos Fanis1,2† Meropi Toumba1,3
Meropi Toumba1,3 George A. Tanteles2,4
George A. Tanteles2,4 Melpo Schiza1,2
Melpo Schiza1,2 Feride Cinarli1,2
Feride Cinarli1,2 Nicolas C. Nicolaides5,6
Nicolas C. Nicolaides5,6 Anastasis Oulas2,7
Anastasis Oulas2,7 George M. Spyrou2,7
George M. Spyrou2,7 Christos S. Mantzoros8,9
Christos S. Mantzoros8,9 Dimitrios Vlachakis10,11,12
Dimitrios Vlachakis10,11,12 Nicos Skordis1,13,14*
Nicos Skordis1,13,14* Leonidas A. Phylactou1,2*
Leonidas A. Phylactou1,2*- 1Department of Molecular Genetics, Function and Therapy, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
- 2Cyprus School of Molecular Medicine, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
- 3Pediatric Endocrine Clinic, IASIS Hospital, Paphos, Cyprus
- 4Clinical Genetics Department, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
- 5Division of Endocrinology, Diabetes and Metabolism, First Department of Pediatrics, National and Kapodistrian University of Athens Medical School, “Aghia Sophia” Childrens Hospital, Athens, Greece
- 6Division of Endocrinology and Metabolism, Biomedical Research Foundation of the Academy of Athens, Athens, Greece
- 7Bioinformatics ERA Chair, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
- 8Division of Endocrinology, Diabetes and Metabolism, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
- 9Section of Endocrinology, Diabetes and Metabolism, Boston VA Healthcare System, Boston, MA, United States
- 10Laboratory of Genetics, Department of Biotechnology, School of Food, Biotechnology and Development, Agricultural University of Athens, Athens, Greece
- 11Lab of Molecular Endocrinology, Center of Clinical, Experimental Surgery and Translational Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece
- 12Department of Informatics, Faculty of Natural and Mathematical Sciences, King's College London, London, United Kingdom
- 13Division of Pediatric Endocrinology, Paedi Center for Specialized Pediatrics, Nicosia, Cyprus
- 14St George's, University of London Medical School at the University of Nicosia, Nicosia, Cyprus
Background: Congenital hypogonadotropic hypogonadism (CHH) is a rare genetic disease caused by Gonadotropin-Releasing Hormone (GnRH) deficiency. So far a limited number of variants in several genes have been associated with the pathogenesis of the disease. In this original research and review manuscript the retrospective analysis of known variants in ANOS1 (KAL1), RNF216, WDR11, FGFR1, CHD7, and POLR3A genes is described, along with novel variants identified in patients with CHH by the present study.
Methods: Seven GnRH deficient unrelated Cypriot patients underwent whole exome sequencing (WES) by Next Generation Sequencing (NGS). The identified novel variants were initially examined by in silico computational algorithms and structural analysis of their predicted pathogenicity at the protein level was confirmed.
Results: In four non-related GnRH males, a novel X-linked pathogenic variant in ANOS1 gene, two novel autosomal dominant (AD) probably pathogenic variants in WDR11 and FGFR1 genes and one rare AD probably pathogenic variant in CHD7 gene were identified. A rare autosomal recessive (AR) variant in the SRA1 gene was identified in homozygosity in a female patient, whilst two other male patients were also, respectively, found to carry novel or previously reported rare pathogenic variants in more than one genes; FGFR1/POLR3A and SRA1/RNF216.
Conclusion: This report embraces the description of novel and previously reported rare pathogenic variants in a series of genes known to be implicated in the biological development of CHH. Notably, patients with CHH can harbor pathogenic rare variants in more than one gene which raises the hypothesis of locus-locus interactions providing evidence for digenic inheritance. The identification of such aberrations by NGS can be very informative for the management and future planning of these patients.
Introduction
Congenital hypogonadotropic hypogonadism (CHH) is a rare disorder that is mainly caused by gonadotropin releasing hormone (GnRH) deficiency and characterized by delayed sexual development and infertility in both males and females (1–6). The pulsatile secretion of the decapeptide GnRH from the hypothalamus into the hypophyseal-portal vessels exerts control in the synthesis and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH) in the anterior pituitary gland (7, 8). In CHH, the GnRH secretion and/or action is impaired and as a consequence patients with the disorder exhibit low levels of gonadotropins, low sex steroids, absent, incomplete or delayed puberty and subsequently hypogonadotropic hypogonadism (HH) (1, 9, 10). The prevalence of CHH is estimated to be 1:8,000 male and 1:40,000 female live births with slightly fewer than 50% of cases suffering from hyposmia or anosmia (10–12). CHH is divided into two subtypes, which include of congenital normosmic isolated hypogonadotropic hypogonadism (HH) and anosmic HH or Kallmann syndrome (KS) (13, 14). Current research regarding the pathophysiology of CHH provides evidence that genetic abnormalities play a key role in the development of the disease and is estimated that a genetic cause is apparent in almost 50% of CHH cases (1, 6). Up-to-date there have been reported more than 60 putative loci for CHH, 17 of which have been linked with KS (1, 6, 13–16).
Over the last few years with the use of the high throughput next generation sequencing (NGS) the number of genes shown to be responsible for causing CHH/KS has radically increased (15, 17). Therefore, the purpose of the present study was to determine the genetic involvement in a series of clinically diagnosed with CHH/KS Cypriot patients.
Materials and Methods
Patients
A total of seven (six males and one female) unrelated Cypriot patients with CHH/KS were included in the present study and underwent whole exome sequencing by NGS. Clinical criteria included the absence or incomplete development of secondary sexual characteristics after the age of 16 years in females and 18 years in males. The biochemical criteria included low levels of basal and GnRH stimulated gonadotrophins (LH, FSH) as well as low levels of sex steroids (testosterone in males and estradiol in females). MRI scans were performed for all patients, with the exception of patients 2 and 6. Five male patients and one female exhibited isolated hypogonadotropic hypogonadism. Only one patient, a 72-year-old male, had KS with anosmia. Written, informed consent was obtained from all seven adult individuals that participate in the study for the publication of any potentially identifiable images or data included in this article. The study was approved by the Cyprus National Bioethics Committee and all methods were performed in accordance with the relevant guidelines and regulations.
Genetic Analysis
Genomic DNA was isolated from peripheral blood using the Gentra Puregene Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. The DNA concentration and purity was measured using the Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Prior to library preparation for whole exome sequencing (WES) genomic DNA was quantified using the Qubit dsDNA BR Assay Kit (Invitrogen, Life Technologies, Eugene, OR, USA) on a Qubit® 2.0 Fluorometer (Invitrogen, Life Technologies, Eugene, OR, USA). WES was performed by using the TruSeq Exome Kit (Illumina Inc., San Diego, CA, USA) with paired-end 150 bp reads. NGS was performed using the NextSeq 500/550 High Output Kit v2.5 (150 Cycles) on an NextSeq500 system (Illumina Inc., San Diego, CA, USA). The FastQC quality control tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to evaluate the quality of the WES procedure. The mean target coverage for the exome was 70.88X. Specifically, 10X coverage was achieved for 99.22% of the nucleotides, 20X coverage for 87.68% of the nucleotides and 30X coverage for 79.35% of the nucleotides, indicating that the WES reaction was of sufficiently high quality for subsequent analysis.
Variant Analysis
The fastqc data obtained by WES were processed using an in-house bioinformatics pipeline. Briefly, all variants were inputted into the VarApp Browser and filtered. VarApp is a graphical user interface, which supports GEMINI (18). Variants in selected genes with biological involvement in the GnRH neuronal system and CHH (Supplementary Table 1) were further analyzed using the Qualimap v2.2.1 tool (19) to calculate the target coverage. Mean target coverage was >20X for 93.2% of the selected genes and >30X for 89% of the selected genes (Supplementary Table 1). Variants in these genes were additionally filtered using the VarApp Browser for minor allele frequencies of <1% in public databases such as 1000 genomes, ExAC browser and Exome Sequencing Project (ESP). Moreover, variants were filtered and selected according to their impact such as frameshift, splice acceptor, splice donor, start lost, stop gained, stop lost, inframe deletion, inframe insertion, missense, protein altering and splice region. In addition, variants were filtered by the VarApp Browser for their pathogenicity by two in silico tools, SIFT and Polyphen2. Population-specific data from an in-house WES library composed of 51 randomly selected samples of Cypriot origin were used to evaluate the potential disease-causing variants. All variants identified were confirmed by Sanger sequencing. When genetic material of relatives was obtainable, familial segregation was performed (Table 2, Supplementary Figure 5). For the cases where two potentially pathogenic variants were identified in an individual, we employed the ORVAL platform for predicting pathogenicity due to digenicity (20). Finally, the variants were categorized for their pathogenicity using the standards and guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (21).
In silico Analysis of the Single Nucleotide Variants
In silico prediction on protein function of the pathogenicity effect of the different amino acid substitutions identified by NGS and confirmed with Sanger analysis was performed by the PredictSNP tool using the default settings (22). The PredictSNP tool evaluates the pathogenicity of a variant by using seven different protein functionality prediction tools: MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, SNAP, and PANTHER.
Molecular Modeling and Homology Modeling of the Mutated Genes
By using MOE (Chemical Computing Group, MOE, v2014.0901, www.chemcomp.com) homology modeling was attempted in all seven genes that carried the missense variants identified in the present study. The selection of template crystal structures for homology modeling was based on the primary sequence identity and the crystal resolution. The MOE homology model method is separated into four main steps. First, a primary fragment geometry specification. Second the insertion and deletions task. The third step is the loop selection and the side-chain packing and the last step is the final model selection and refinement. The template selection was as follows: for PROP1 the 3A01 PDB file was used, for SRA1 the 4NBO PDB file was used, for RNF216 the 5l1V PDB file was used, for FGFR1 the 1CVS PDB file was used and for POLR3A the 5FJ8 PDB file was used. All models were handled, verified and visualized using the Drugster suite (23).
Model Optimization
Energy minimization for all four models was done in MOE initially using the Amber99 (24) force-field implemented into the same package, up to a root mean square deviation (RMSd) gradient of 0.0001 to remove the geometrical strain. The models were subsequently solvated with simple point charge (SPC) water using the truncated octahedron box extending to 7 Å from the model, and molecular dynamics was performed at 300 K, 1 atm with 2 fs step size for a total of 10 ns, using the NVT ensemble in a canonical environment (NVT stands for Number of atoms, Volume and Temperature that remain constant throughout the calculation). The results of the molecular dynamics simulation were collected into a database by MOE for further analysis.
Results
Genetic Findings
All seven patients were sequenced by WES. The clinical, biochemical and genetic characteristics are summarized in Table 1. A total of nine variants were identified in genes that are known to be linked with the development of CHH/KS (Table 2). Seven of these variants were novel and two were previously reported. The novel X-linked p.Gln82* in the ANOS1 (KAL1) gene was found in patient 1, a 28-year-old CHH male with pubertal absence, cryptorchidism and micropenis (Table 1, Figure 1). The novel WDR11 p.Leu244Pro variant is probably pathogenic and is inherited in an autosomal dominant fashion (AD). This variant was identified in patient 2, a 72-year-old male with KS and associated clinical characteristics of anosmia, cryptorchidism and micropenis. Patient 2 first sought medical advice at the age of 40 years and since then remains a patient of our clinic. Molecular diagnosis was only possible 32 years later (Table 1, Figure 2). The previously reported AR, probably pathogenic p.Ile179Thr variant in the SRA1 gene was identified in heterozygosity in patient 3, a 19-year-old male with partial hypogonadism and upper limb defects (Table 1, Figure 3). In addition, the novel p.Asp792Asn in the RNF216 gene was also identified in heterozygosity in the same patient (Figures 3, 8, Supplementary Figure 1). Evaluation by the ORVAL platform for digenicity predicted this novel variant to have a neutral effect (Supplementary Figure 6). However, familial segregation data and in silico structural models indicated a digenic mode of inheritance (Figures 3, 8, Supplementary Figure 5). Variants in the SRA1 and RNF216 genes have been associated with effects on the CHH phenotype. Thus, the presence in patient 3 of pathogenic, heterozygous variants in SRA1 and RNF216 genes could potentially be another example of digenic inheritance for the development of CHH (28).
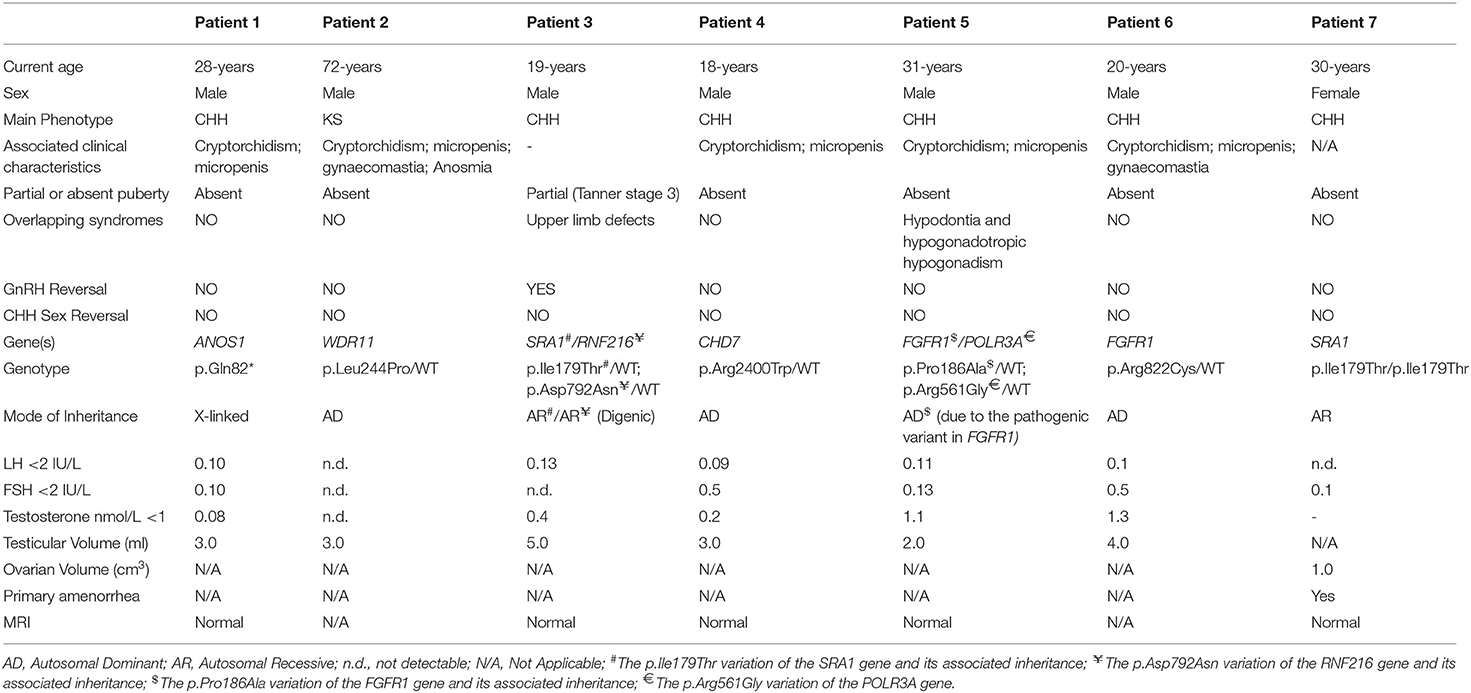
Table 1. Clinical and biochemical characteristics of the patients with CHH.
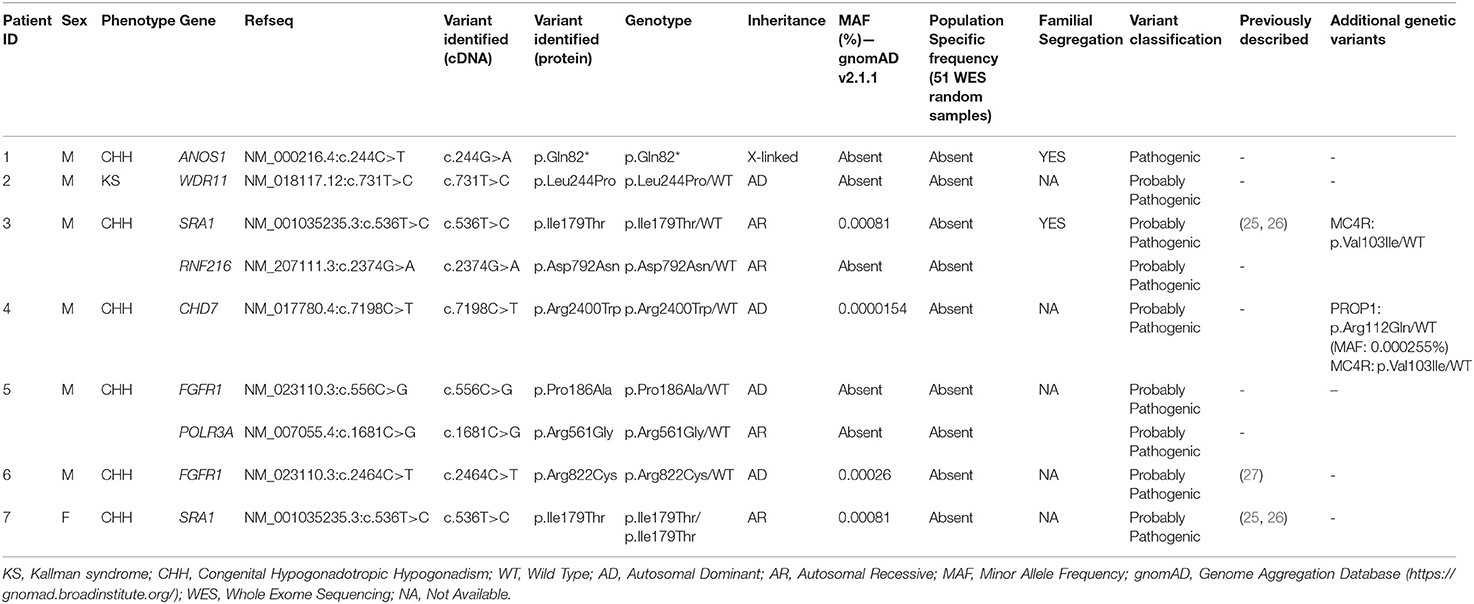
Table 2. Variants identified in the seven non-related patients.
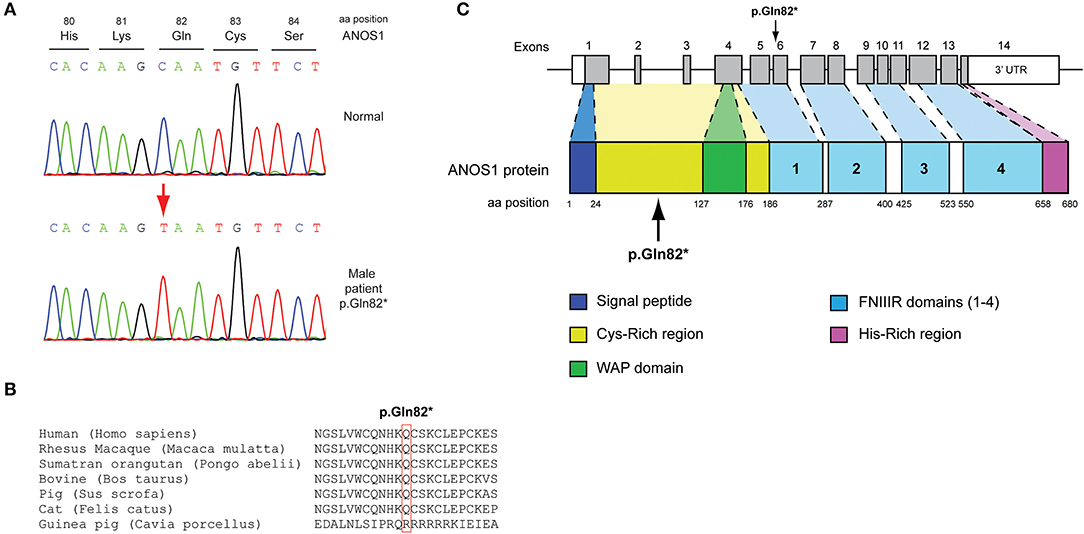
Figure 1. Identification of a novel p.Gln82* ANOS1 mutation. (A) Sequence electropherogram of the novel p.Gln82* ANOS1 mutation. (B) Multiple sequence alignment of the amino acid at position 82 of the ANOS1 protein from various species. The conserved glutamine amino acid at position 82 is indicated by red color. (C) Schematic representation of the ANOS1 gene and protein of a male patient identified with the novel p.Gln82* non-sense pathogenic variant. WAP, whey acidic protein; FNIII, fibronectin type III.
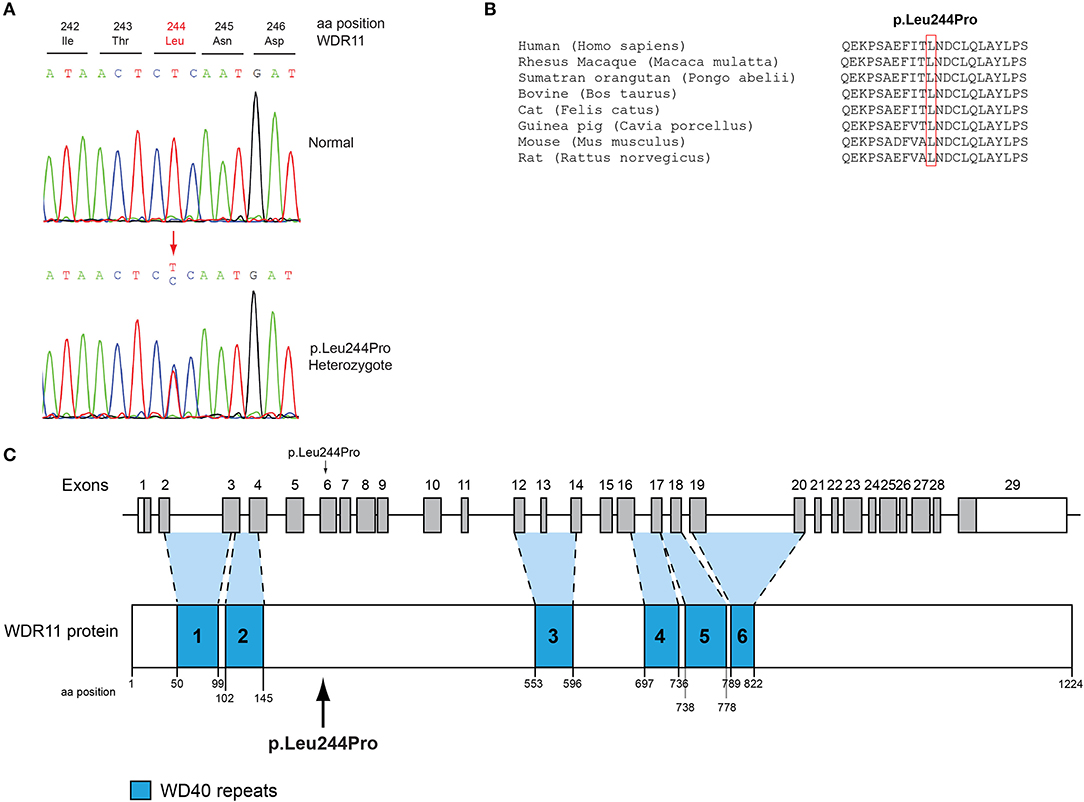
Figure 2. Identification of a novel p.Leu244Pro WDR11 variant. (A) Sequence electropherogram of the novel WDR11 p.Leu244Pro variant. (B) Multiple sequence alignment of the amino acid at position 244 of the WDR11 protein from various species. The conserved leucine amino acid at position 244 is indicated by red color. (C) Schematic representation of the WDR11 gene and protein of a male patient identified with the novel p.Leu244Pro variant.
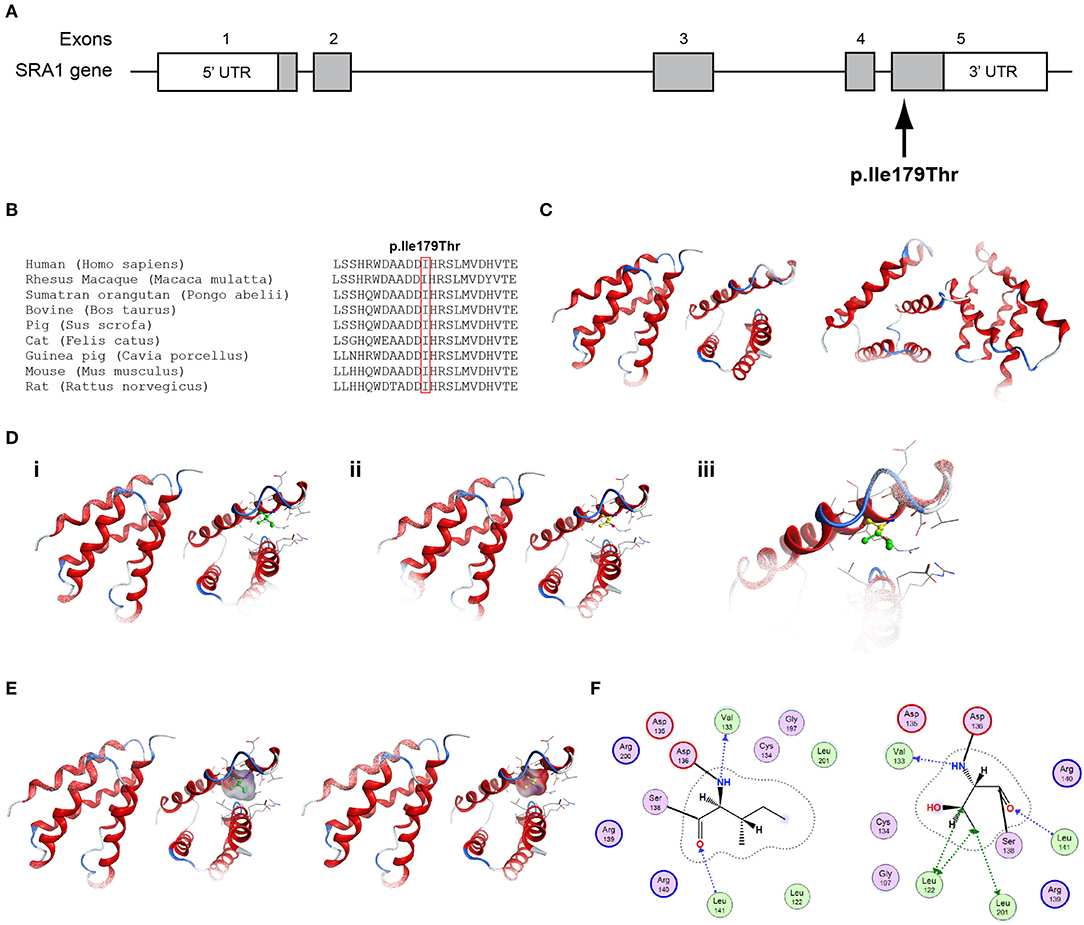
Figure 3. Identification of the p.Ile179Thr SRA1 variant. (A) Schematic representation of the SRA1 gene. The known SRA1 p.Ile179Thr variant identified in a male patient in the heterozygote state and in a female patient in the homozygous state is indicated. (B) Multiple sequence alignment of the amino acid at position 179 of the SRA1 protein from various species. The conserved isoleucine amino acid at position 179 is indicated by red color. (C) The homology model for SRA1. (D) Design of the wild type (i) and mutant model (ii) of SRA1. (iii) Wild type and mutant models superposed. (E) Electrostatic surface calculated and drawn for the wild type and mutant models. (F) 2D interaction diagram for wild type and mutant models.
Patient 4, an 18-year-old male with CHH and the associated clinical characteristics of cryptorchidism and micropenis, was found to carry the novel AD p.Arg2400Trp variant in the CHD7 gene (Table 1, Figure 4). Patient 5, a 31-year-old male with CHH and associated clinical characteristics of cryptorchidism and micropenis, was found to carry the novel AD p.Pro186Ala in the FGFR1 gene (Table 1, Figure 5). The previously reported AD p.Arg822Cys also in the FGFR1 gene was found in patient 6, a 20-year-old male with CHH and associated clinical characteristics of cryptorchidism, micropenis and gynaecomastia (Table 1, Figure 5, Supplementary Figure 2). The known p.Ile179Thr variant in the SRA1 gene was also identified in the homozygous state in patient five, a 30-year-old female with CHH and primary amenorrhea (Figure 3, Supplementary Figure 1). Furthermore, patient five also carried the novel p.Arg561Gly variant in the POLR3A gene (Figure 6, Supplementary Figure 3). The ORVAL platform was used to evaluate the pathogenicity of the variants regarding digenic inheritance, with respect to which they were shown to be positive (Supplementary Figure 6). Such variants when inherited in the AR form are usually associated with Pol III-related hypomyelinating leukodystrophies and not with CHH. Patients with Pol III-related leukodystrophies may have various clinical characteristics including ataxia, delayed dentition, hypomyelination, hypodontia, and hypogonadotropic hypogonadism. Patient 5, in addition to CHH, also developed a mild hypomyelinating leukodystrophy phenotype, which is likely associated with the heterozygous condition found in the POLR3A gene.
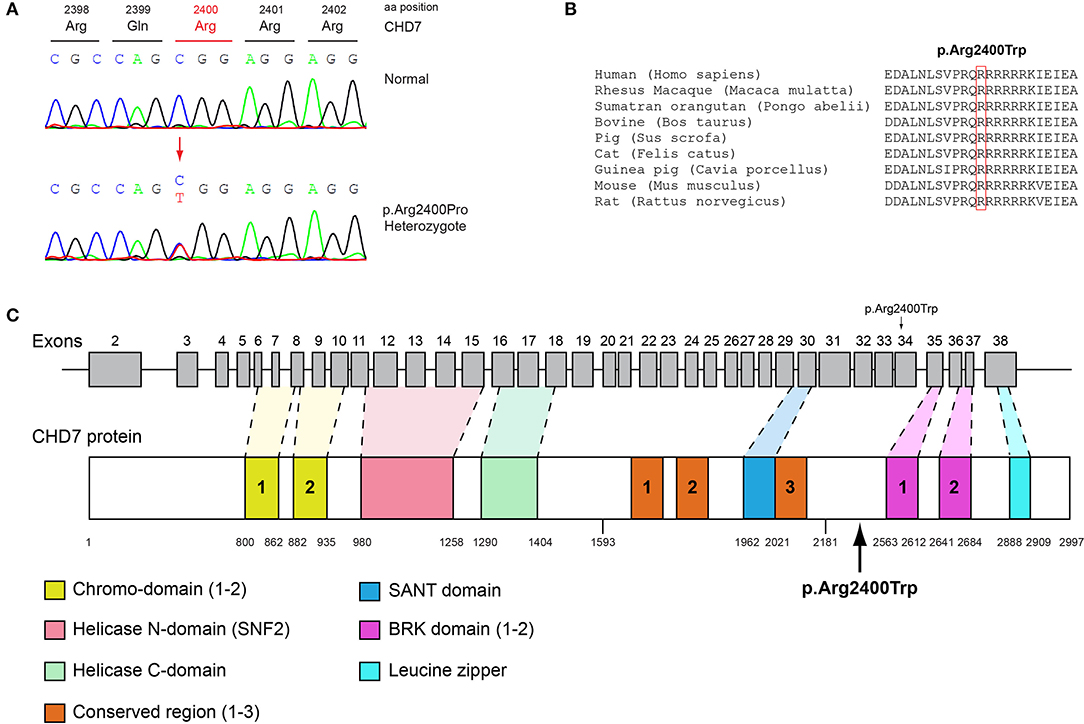
Figure 4. Identification of the p.Arg2400Trp CHD7 variant. (A) Sequence electropherogram of a male patient identified with the novel CHD7 p.Arg2400Trp variant. (B) Multiple sequence alignment of the amino acid at position 2,400 of the CHD7 protein from various species. The conserved arginine amino acid at position 2,400 is indicated by red color. (C) Schematic representation of the CHD7 gene and protein of a male patient identified with the novel p.Arg2400Trp variant.
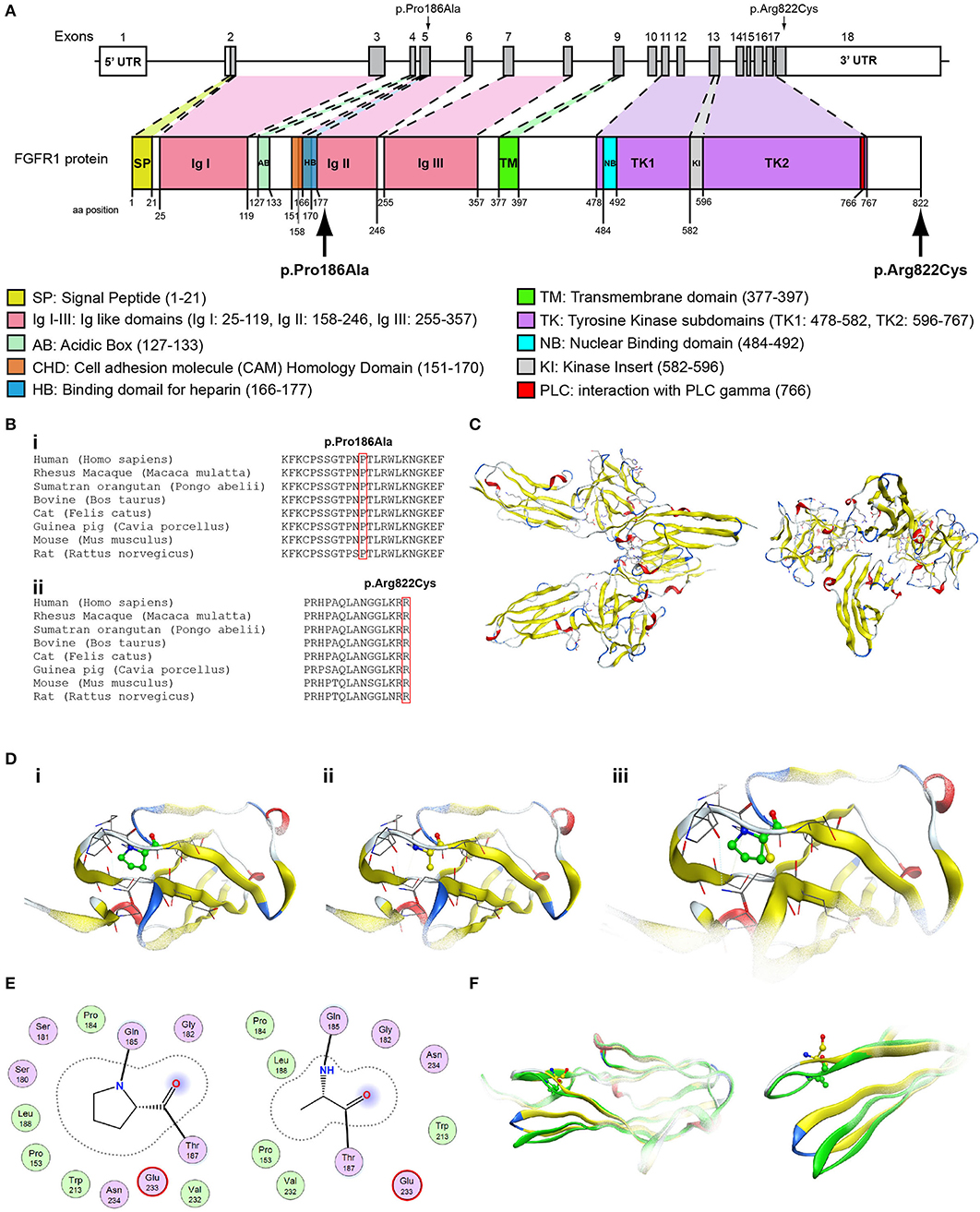
Figure 5. Identification of the p.Pro186Ala and p.Arg822Cys FGFR1 variants. (A) Schematic representation of the FGFR1 gene and protein. The p.Pro186Ala and p.Arg822Cys FGFR1 variants are indicated with arrows. (B) Multiple sequence alignment of the amino acids at position 186 (i) and 822 (ii) of the FGFR1 protein from various species. The conserved amino acids at positions 186 and 822 are indicated by red color. (C) The homology model of the FGFR1. (D) Design of the wildtype (i) and p.Pro186Ala mutant model (ii); the wildtype and p.Pro186Ala mutant models superposed (iii). (E) 2D interaction diagram for wildtype and p.Pro186Ala mutant models. (F) Conformational change induced upon variant. Wildtype is showing in green ribbon, while p.Pro186Ala mutant is showing in yellow ribbon.
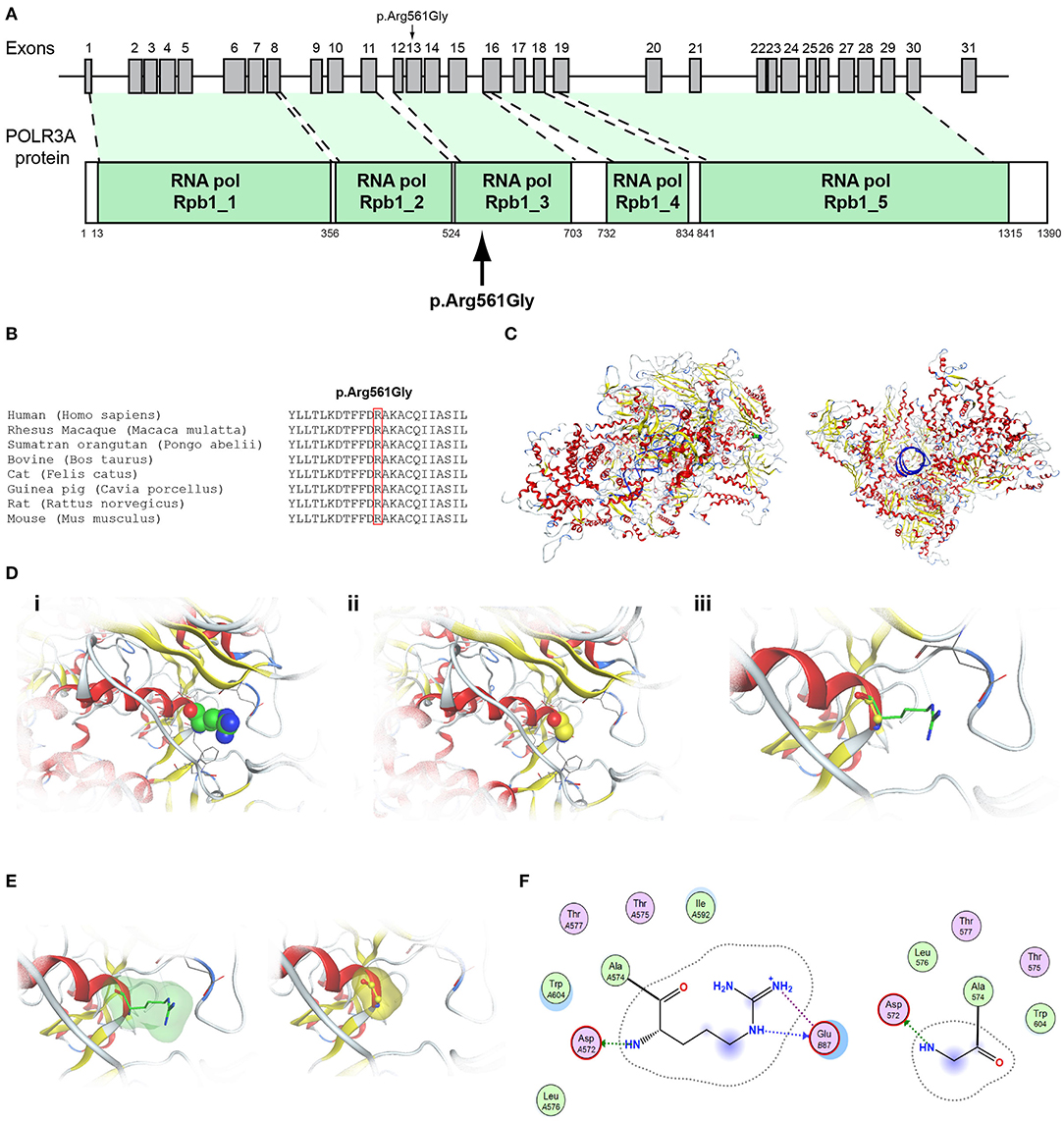
Figure 6. Identification of the p.Arg561Gly POLR3A variant. (A) Schematic representation of the POLR3A gene and protein. The p.Arg561Gly POLR3A variant is indicated with arrows. (B) Multiple sequence alignment of the amino acid at position 561 of the POLR3A protein from various species. The conserved arginine amino acid at position 561 is indicated by red color. (C) The homology model for POLR3A. (D) Design of the wild type (i) and mutant model (ii) of POLR3A. (iii) Wild type and mutant models superposed. (E) Electrostatic surface calculated and drawn for the wild type and mutant models. (F) 2D interaction diagram for wild type and mutant models.
It should also be noted that 3 of our patients were found to have other variants as well. The known p.Val103Ile variation of the MC4R gene, which has been linked to obesity, was found in heterozygocity in two of the male patients (the 18 and the 19-year-old males, Patients two and four). Both the patients were obese, with BMI above +2SDS and both developed insulin resistance. In addition the novel variant p.Arg112gGln in the PROP1 gene was found in the heterozygous state also in patient 4, an 18-year-old male (Figure 7, Supplementary Figure 4). Such variants have been reported to be associated with combined pituitary hormone deficiency. Our patient also had central hypothyroidism and he is currently on treatment with thyroxin.
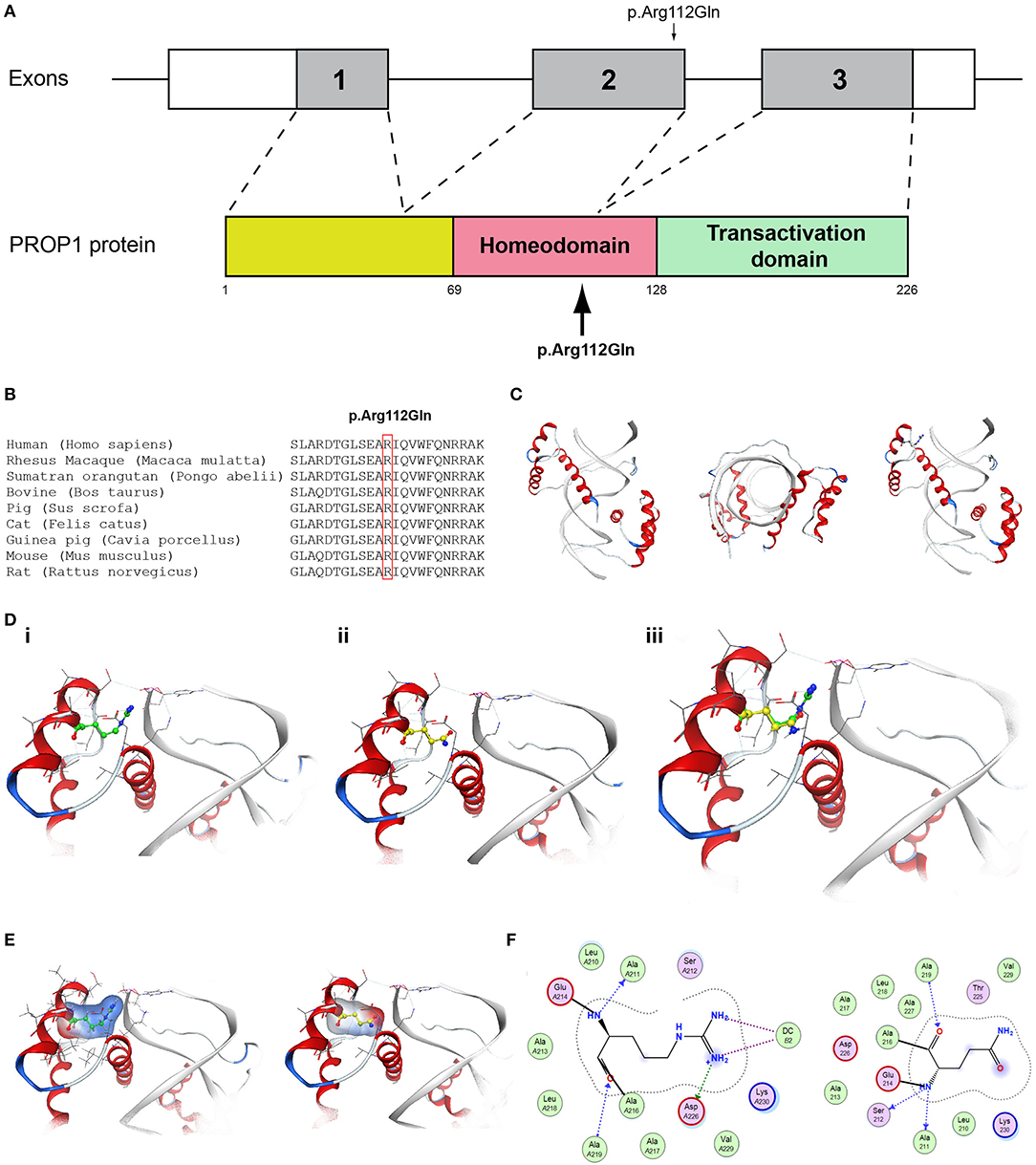
Figure 7. Identification of the p.Arg112Gln PROP1 variant. (A) Schematic representation of the PROP1 gene and protein. The p.Arg112Gln PROP1 variant is indicated with arrows. (B) Multiple sequence alignment of the amino acid at position 112 of the PROP1 protein from various species. The conserved arginine amino acid at position 112 is indicated by red color. (C) The homology model for PROP1. (D) Design of the wild type (i) and mutant model (ii) of PROP1. (iii) Wild type and mutant models superposed. (E) Electrostatic surface calculated and drawn for the wild type and mutant models. (F) 2D interaction diagram for wild type and mutant models.
The novel non-sense pathogenic variant p.Gln82* in the ANOS1 gene, which was identified in a 28-year-old male, encodes a premature termination codon. It is expected to yield a truncated ANOS1 protein, missing the whey acidic protein (WAP)-like protease inhibitor domain and the four fibronectin type III (FN[III]) domains (Figure 1). The missense variants, identified by WES and confirmed by Sanger sequencing, were predicted to be deleterious by at least two prediction tools using the PredictSNP consensus classifier (22) (Table 3). The PredictSNP consensus classifier evaluates the pathogenicity of a variant by using seven different prediction tools: MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, SNAP, and PANTHER. Furthermore, the identified variants were absent from the population-specific data in an in-house database composed of 51 random samples of Cypriot origin. Familial segregation was available for two patients, patient 1 and patient 3 (Supplementary Figure 5, Table 2). For patient one the identified pathogenic variant followed the X-liked mode of inheritance and for patient 3, variants followed the digenic mode of inheritance (Supplementary Figure 5).
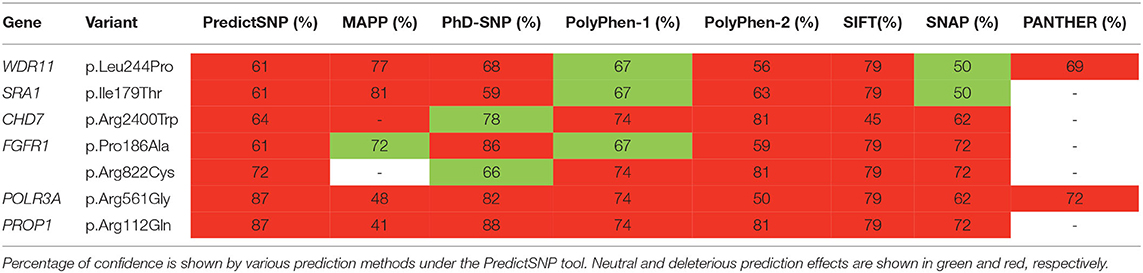
Table 3. Prediction analysis of the variants identified.
Conserved Protein Sequences Among Species
Protein alignment analyses of all identified pathogenic and probably pathogenic variants including the p.Gln82* of the ANOS1 gene, the p.Leu244Pro of the WDR11 gene, the p.Asp792Asn of the RNF216 gene, the p.Arg2400Trp of the CHD7 gene, the p.Pro186Ala and p.Arg822Cys of the FGFR1 gene, the p.Ile179Thr of the SRA1 gene, the p.Arg561Gly of the POLR3A gene and the p.Arg112gGln of the PROP1 gene showed high amino acid conservation among different species (Figures 1B, 2B, 3B, 4B, 5B, 6B, 7B, 8C). Therefore, the newly and previously discovered variant sites of the above genes were probably located in a vital region of the coding genes that might affect the corresponding proteins mechanistically and/or structurally.
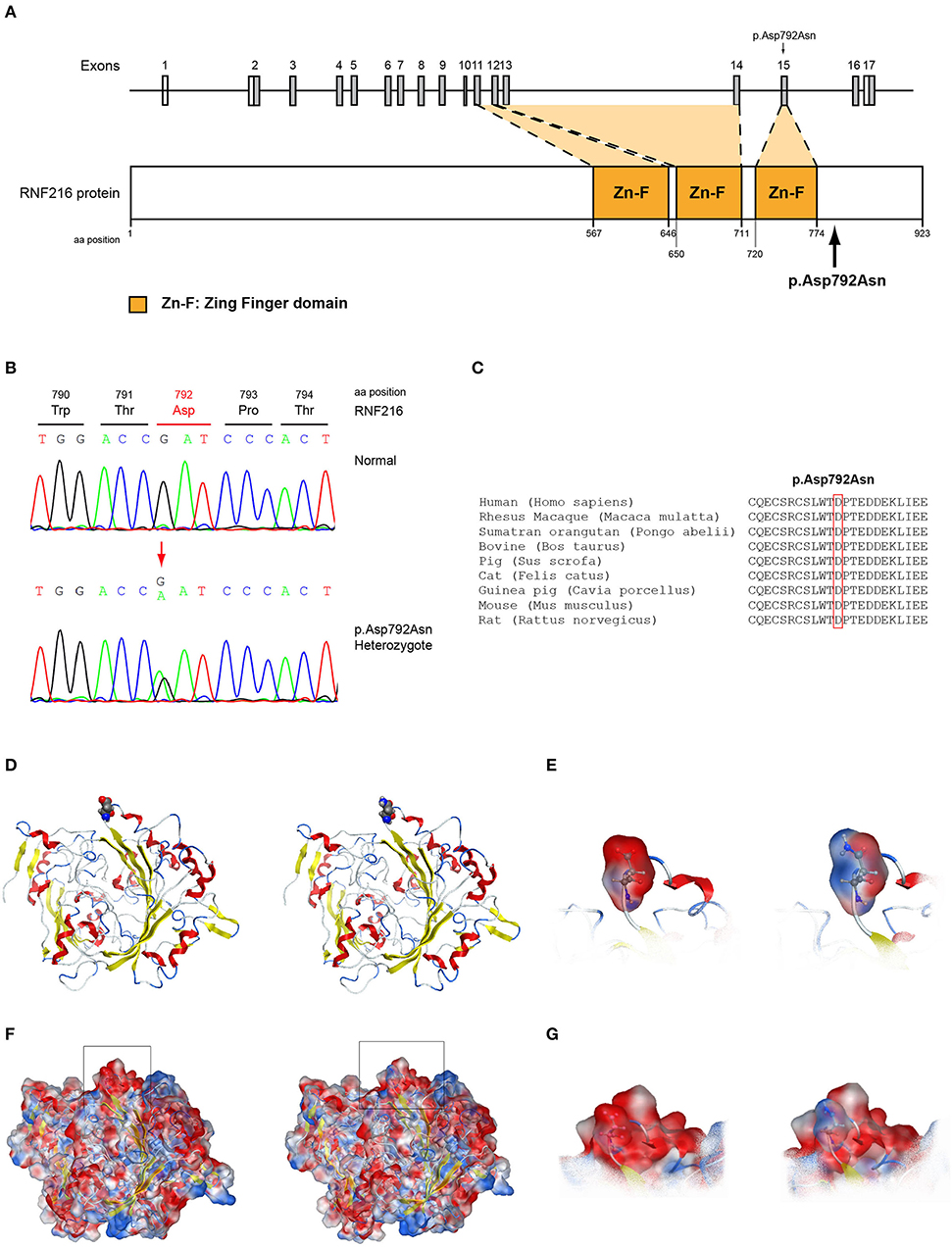
Figure 8. Identification of the p.Asp792Asn RNF216 variant. (A) Schematic representation of the RNF216 gene and protein of a male patient identified with the novel p.Asp792Asn variant. (B) Sequence electropherogram of a male patient identified with the novel RNF216 p.Asp792Asn variant. (C) Multiple sequence alignment of the amino acid at position 792 of the RNF216 protein from various species. The conserved aspartic acid amino acid at position 792 is indicated by red color. (D) The p.Asp792Asn RNF216 variant is indicated with spacefill atoms. (E) The electrostatic potential surface calculated and drawn for the wild type and mutant residues at the 792 position of RNF216. (F) The electrostatic potential surface calculated and drawn for the whole protein. In squares the regions presented in (G). (G) The electrostatic potential surface calculated and drawn for the adjacent to the 792 position residues.
Effect of the Mutated PROP1, SRA1, FGFR1, POLR3A, and RNF216 at the Related Protein Structure
The selection of template crystal structures for homology modeling was based on the primary sequence identity and crystal resolution models were obtained for the PROP1, SRA1, FGFR1, POLR3A, and RNF216. Unfortunately, no crystal resolution models were obtained for the CHD7 and WDR11 proteins.
The models for the SRA1, FGFR1, POLR3A, PROP1, and RNF216 were designed using means of homology modeling (Figures 3C, 5C, 6C, 7C, 8D). Each one of the recorded variants were induced in silico and each model was energetically optimized via Energy minimizations and Molecular Dynamics [Figures 3D (i-ii), 5D (i-ii), 6D (i-ii), 7D (i-ii)]. The wild type and variant models were subsequently superposed and the variant residues were inevitably superposed too [Figures 3D (iii), 5D (iii), 6D (iii), 7D (iii)]. The electrostatic surface was calculated for each wild type and variant set for the SRA1, POLR3A, and PROP1 models (Figures 3E, 6E, 7E) and finally the 2D interaction diagram was drawn for each pair to visualize all bonding and conformation changes induced upon by the variants (Figures 3F, 5E, 6F, 7F). Since the FGFR1 variant involved the replacement of a proline residue the conformational change in the 3D conformation of the beta sheet formation is showed in Figure 5F.
More specifically, the p.Arg112Gln variant in the PROP1 results in a significant physicochemical change in the 112 position. The bulkier and positively charged arginine residue is now replaced by a smaller polar and uncharged glutamine residue. Inevitably the interaction to the DNA molecule that was also included in the model is now lost, as well as a strong stabilizing H-bond to a nearby negatively charged aspartic acid residue. It is evident from the 2D interaction map that the mutant PROP1 has lost its potential to interact with DNA and that would unavoidably alter its molecular and cellular function (Figure 7). Moving on, the p.Ile179Thr variant of the SRA1 also changes significantly the physicochemical profile of the amino acid at 179 position. Isoleucine is a non-polar aliphatic residue, whereas threonine is a polar residue. This electrostatic change results in the establishment of two new bonds with two nearby leucine residues. This fixates the local three helical bundle conformation more strongly, thus making the 3D structural arrangement adjacent to the 179 position more compact and conformationally rigid compared to the wild type (Figure 3). Likewise, the p.Pro186Ala variant in the FGFR1 has a significant conformational impact. The proline residue is a very special amino acid that bears a cyclic side chain. This gives this residue a unique property of inducing a kink in the 3D conformational arrangement of the protein. Removing it, and replacing it with an alanine residue resulted in a 2.5 Angstrom shift outwards of the beta sheet it is located in and that consequently led to a 4.5 Angstrom shift of the neighboring antiparallel beta sheet formation too. Such significant changes in structural arrangements are bound to change the mechanics and function of the FGFR1 protein (Figure 5). Lastly, the p.Arg561Gly in the POLR3A is a fine example of a variant change from a very bulky and positively charged residue to a small amino acid. From the 2D interaction diagram for this residue we can deduce that only the arginine amino acid is long enough to reach the nearby positively charged aspartic acid residue and to establish with the latter strong H-bonds. The glycine residue is not just small but it also misses the essential amino-groups that are required to establish H-bonding to the glutamic acid (Figure 6). The p.Asp792Asn variant according to the homology model of RNF216 is exposed to the solvent. It is located on a hairpin like loop, linking a beta-sheet and an a-helix in an antiparallel fashion (Figure 8D). Therefore, there is high probability that this could be an interacting part of the RNF216 protein, judging from the rotamer of this residue, which is pointing outwards. In this direction, we modeled the electrostatic potential of the 792 residue position (Figure 8E), of the adjacent residues within a 7 Angstrom radius (Figure 8G) and of the whole RNF216 protein (Figure 8F). It was found that the variation of aspartic acid to asparagine changes significantly the electrostatic potential and nature of the 792 position due to the extra NH2 moiety on the asparagine amino acid. That, coupled with the fact that all amino acids around the 792 position are negatively charged (red color–Figure 8G, left) is very significant as with the introduction of the asparagine residue a positively charged group is now introduced. Taking the abovementioned facts into consideration, we propose that the p.Asp792Asn variant is very significant as the positively charged moiety that is introduced could disrupt the conserved negatively charged region of RNF216, thus leading to a considerable change in the physicochemical and electrostatic profile of that domain that would inevitably affect its binding/interaction potential and would probably change its functional properties (e.g., loss of recognition and even interaction).
Discussion
The present study investigated by high-throughput whole exome sequencing the genetic impact in patients with CHH. The seven patients of Cypriot origin with CHH/KS were identified with variants in genes linked with this phenotype: ANOS1, SRA1, CHD7, WDR11, FGFR1, RNF216, and POLR3A. A total of seven novel and two rare previously reported variants were identified in the patients of the current study and were found as novel or very rare in the ExAC population database (29). All these variants were also predicted to be pathogenic by at least two computational programs (22, 30–36). Our results once more confirmed the genetic complexity of CHH and the roles that exemplify a series of pleiotropic genes during development (13, 37, 38). More specifically, we identified the novel X-linked hemizygous truncated p.Gln82* pathogenic variant in the ANOS1 gene in a 28-year-old male with CHH (Table 1). Patients with sporadic KS/CHH due to ANOS1 gene defects have been correlated with the phenotype of right renal agenesis/dysgenesis, thus provide evidence for the X-linked mode of inheritance and offering the opportunity for genetic counseling (12, 39–44). Approximately 10–20% of males with KS carry ANOS1 variants or intragenic microdeletions (38). The majority of the X-linked KS variants cause alteration of splicing, frameshift or stop codons leading to synthesis of truncated anosmin (45). Nonsense variants and full deletions in the ANOS1 gene are the most pathogenic and lead to a truncated and absent anosmin protein, respectively (37). Two of our normosmic male adult patients have been identified with the novel AD p.Pro186Ala and the previously reported p.Arg822Cys (46) variants in the FGFR1 gene. Both of these patients were also characterized by delayed puberty during adolescence and later CHH. In a similar fashion with ANOS1, the expression of the FGFR1 gene is also generated in the apparent olfactory bulbs and loss-of-function variants cause a form of KS with autosomal dominant inheritance (4–24, 28–50). FGFR1 is a cell surface membrane receptor that possesses tyrosine kinase activity and mediates fibroblast growth factor signaling (51). Patients with variants in the FGFR1 gene present also various congenital anomalies that are not associated with the reproductive system and are often associated with kidney and tooth differentiation, ear and palate morphogenesis and the development of cortico-spinal axonal tracts (52). Notably, patient five, the 31-year-old male patient with CHH identified with the novel FGFR1 p.Pro186Ala also shared in heterozygosity the novel POLR3A p.Arg561Gly missense variant. This finding adds to the already known spectrum of phenotypes resulting from POLR3A and POLR3B variants. POLR3A and POLR3B can be also associated to neurological or dental anomalies and isolated hypogonadotropic hypogonadism (53).
Patient 4 in addition to the novel AD CHD7 p.Arg2400Trp variant also carried the known MC4R p.Val103Ile variant implicated in BMI and the novel p.Arg112gGln in the PROP1 gene. Various studies have described PROP1 gene variants as responsible for causing combined pituitary hormone deficiency (54–56). Heterozygous autosomal dominant loss-of-function variants in the CHD7 gene are the major causal factor of CHARGE syndrome (57, 58), in addition to the fact that CHD7 variants have been also been reported in patients with isolated CHH (59–61). Several reports have also linked PROP1 variants with gonadotroph function that progressively declines and clinically patients with such variants may manifest a shortage of pubertal development, i.e., failure to enter or complete puberty (62, 63). There are several reports of spontaneous puberty with a posterior decline of gonadotrophic function that have been linked to p.Arg112Ter, p.Arg120Cys, p.Phe88Ser, and c.150delA PROP1 variants (64–67). Since the PROP1 gene is involved in the anterior pituitary, cell lineage specification variants could behave as an additive factor in the development of CHH when co-inherited with variants from genes involved in normal gonadotroph function. Such could be the case with the 18-year-old CHH patient of the present study identified with the novel CHD7 p.Arg2400Trp and the novel p.Arg112gGln variant in the PROP1 gene.
In the present study, a 72-year-old anosmic KS patient originally sought medical advice at the age of 40 in our clinic. Since then, he remains a patient of our clinic and at the age of 72-years he was identified with the novel AD p.Leu244Pro in the WDR11 gene. WDR11 has been implicated in CHH and KS, human developmental genetic disorders defined by delayed puberty and infertility (68, 69). Several reports in CHH patients with and without anosmia identified in heterozygosity variants in the WDR11 gene (68). WDR11 is expressed in several adult organs including the brain and the gonads. Comprehensive analysis of the mouse brain displayed WDR11 expression in the GnRH neuronal migratory location including nasal cavity and cribriform plate area in E12.5 mouse embryo as well as the median eminence in the adult brain, showing co-localization with GnRH. Furthermore, WDR11 is expressed all over the developing and adult olfactory bulb (OB) and its WD domains are important for β-propeller formation and protein-protein interaction (70). In addition, WDR11 interacts with EMX1, a homeodomain transcription factor involved in the development of olfactory neurons, and missense variants diminish or eliminate this interaction (68). Therefore, it is highly likely that the impaired pubertal development in these patients results from a deficiency of productive WDR11 protein interaction.
Interestingly, two out of the seven CHH patients in our cohort, a 30-year-old female (Patient 7) and a 19-year-old (Patient 3) male were both identified with variants in the SRA1 gene. More specifically, the 30-year-old female carried in homozygosity the previously reported p.Ile179Thr variant in the SRA1 gene (25). The 19-year-old male also carried this same SRA1 variant in heterozygosity together with the novel p.Asp792Asn variant in the RNF216 gene. As reported by Kotan et al. (25), the variant p.Ile179Thr was reported only once in one independent Turkish family with IHH/delayed puberty and its severity was supported by functional studies. Using a mutant SRA1 construct, reduced co-activation of ligand-dependent activity of the estrogen receptor alpha was demonstrated (25).
The variant p.Ile179Thr was not found in 51 Cypriots used as controls for the purposes of the present study and was reported with an allele frequency of 0.00081 in GnomAD v2.1.1. Therefore, most likely, a hot spot exists for this specific variant in the greater Eastern Mediterranean region, suggesting a founder effect phenomenon, which has been also seen for other rare endocrine conditions in this area (71).
The SRA1 is a steroid receptor RNA activator that has been shown to positively regulate the activity of the androgen receptor and the estrogen receptor (72, 73). In recent years only a few studies linked the SRA1 gene as responsible for causing CHH when patients inherit pathogenic variants in the AR form (25, 26).
The concepts of incomplete penetrance and variable expressivity have been notified in such cases as the 19-year-old patient where digenic variants are observed. Digenic variants account for variable phenotypes in idiopathic hypogonadotropic hypogonadism and other disorders and several recent and older reports identified such conditions (17, 40, 74, 75). The existence of digenic and oligogenic inheritance in CHH is quite common, with about 20% of CHH cases reported to share at least two causative variants that could result in a disease phenotype (1, 15). The most appropriate way to examine the possibility of low penetrance and variable expressivity of CHH genes is by concurrently carrying out targeted genetic analyses, or preferably by performing WES on the probands and available relatives so as to establish digenic or oligogenic transmission.
Review of the Literature
Congenital hypogonadotropic hypogonadism (CHH) is a rare disorder of sexual maturation characterized by GnRH deficiency with low sex steroid levels associated with low levels of LH and FSH. CHH may be caused by variants in numerous genes and recent studies shed light on the complexity of CHH genetics (6, 15, 76). Over the recent few years, genetic evaluation of patients with inherited diseases, including CHH, has increasingly utilized massive parallel sequencing by next-generation sequencing (NGS), that allows the concurrent investigation of thousands of genes (1, 15, 77). At this scale of analysis, NGS is inexpensive and rapid compared to the traditional Sanger sequencing and is increasingly being used in medical practice. NGS has certainly facilitated CHH genetic diagnoses and aided healthcare professionals to provide reliable and informed genetic counseling for patients with CHH. The crucial challenge regarding NGS concerns the identification of true oligogenism in circumstances involving several rare variants which do not have a clear phenotypic effect and are identified by coincidence. Such a challenge also concerns the identification of genes underlying CHH pathogenesis and which are likewise reported to act in an oligogenic context (78). Since the discovery of ANOS1 (79), more than sixty genes have been reported to underlie CHH and were previously considered to be inherited in the AD form (6). Herein, we review six of these genes: ANOS1, FGFR1, CHD7, WDR11, RNF216, and POLR3A, since novel variants in these genes have been identified in our cohort of patients under investigation.
ANOS1 (KAL1) Gene Variants Causing X-Linked Recessive KS/CHH
ANOS1 was the first gene linked to Kallmann syndrome (KS) and since the early nineties when the first reports demonstrated variants with an X-linked mode of inheritance (80–83), many others followed throughout the years (6, 84–88). KS occurs more frequently in males than in females, with an estimated prevalence of 1 in 30,000 males and 1 in 120,000 females (12). Patients with KS associated with ANOS1 pathogenic variants usually exhibit anosmia accompanied with CHH (12, 14, 85, 86). Fewer patients with pathogenic variants in ANOS1 are either anosmic or hyposmic and have been reported to exhibit other signs, such as mirror movements and renal agenesis, but they do not always co-segregate with the variant recognized in a given family (85, 89–91). According to the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) more than 150 ANOS1 pathogenic variants have been reported as the causative factor in KS patients. Most of these pathogenic variants mainly consist of nucleotide deletions or insertions and to a lesser extend of variants that involve amino acid missense substitutions (88, 92, 93). The ANOS1 gene encodes anosmin, a protein which plays a significant role in the embryogenesis of brain, kidneys, respiratory and digestive systems (92, 94). Anosmin, as an extracellular matrix protein binds to the cell membrane and stimulates the development of the olfactory system and behaves as an axonal guidance for the GnRH neurons, the olfactory cells and the Purkinje cerebellum neurons (95). Monogenic loss-of-function pathogenic variants in ANOS1 gene have been estimated to account for 4–10% of KS/CHH cases and has been principally studied in many reported cohorts (12, 41–44, 88, 96–99).
Regarding the reproductive phenotype, male KS patients with ANOS1 variants display a complete penetrance of CHH and their pre- and postnatal gonadotropin deficit is severe with a high frequency of micropenis, cryptorchidism and complete absence of gonadal development (15, 16).
FGFR1 Gene Variants Causing KS/CHH
The presence of variants in the FGFR1 gene is another important cause of KS and was the first gene to be identified as an AD form of the disease (49, 100). More than 140 loss-of-function mutations in the FGFR1 gene have been reported with missense, non-sense and frameshift defects being the most frequent (101). Less frequent autosomal gene deletions have also been reported in patients with CHH and KS (102, 103). FGFR1 is considered to be a pleiotropic gene that can display different roles during development and variants found in it can cause CHH with or without anosmia (49, 100). Genotype-phenotype correlations in patients with AD variants in the FGFR1 gene demonstrated some clinical features linked with KS, such as loss of nasal cartilage, hearing deficit and anomalies of the limbs (6, 93, 101). The function of FGFR1 in the normal development of the olfactory bulb proposes the link of anosmia with GnRH deficiency in the FGFR1-mutated patients (104). Phenotype analysis proposes that FGFR1 elaborates in the normal migration of GnRH fetal neurons, but this is not entirely clear-cut as a considerable proportion of FGFR1-mutated patients have normosmic GnRH deficiency (15). Regarding the reproductive phenotype of male patients with FGFR1 variants, the penetrance of CHH and GnRH deficiency is variable and ranges from profound to partial puberty and even to reversal (1, 99, 105, 106).
Several groups have reported patients harboring FGFR1 variants linked to non-reproductive signs. Patients with FGFR1 mutations have been reported to suffer from health conditions such as 8p11 myeloproliferative syndrome (107, 108), encephalocraniocutaneous lipomatosis (109, 110), Hartsfield syndrome, a rare condition characterized by holoprosencephaly, which is an abnormality of brain development (111, 112), osteoglophonic dysplasia, a condition characterized by abnormal bone growth that leads to craniofacial abnormalities and dwarfism (113, 114) and Pfeiffer syndrome, which is characterized by craniosynostosis (115, 116). Somatic pathogenic variants involving the FGFR1 gene have also been reported in several types of cancers, including the lung, breast, esophagous, oral cavity and brain tumors (101, 117–119). Taking into consideration the genotypic and phenotypic heterogeneity that is observed in patients with FGFR1 variants and the fact that their prevalence is not clearly established makes genetic counseling rather complicated.
CHD7 Gene Variants Causing CHARGE Syndrome and CHH/KS
CHD7 is the gene that codes for the chromodomain helicase DNA binding protein 7 and variants found in the AD form were first reported as the genetic cause in a series of patients with CHARGE (coloboma, heart defect, atresia choanae, growth retardation, genital abnormality, and ear abnormality) syndrome (57, 120). CHARGE syndrome occurs in approximately 1 in 8,500 to 10,000 new-borns and up-to-date more than 600 CHD7 variants in the AD form have been associated with the disorder (57, 121–124). Several other studies of families carrying CHD7 mutations in the AD form also demonstrated a broad phenotypic variability and linked more than 50 of them with KS and congenital hypogonadotropic hypogonadism (59, 60, 69, 125). It has been estimated that inherited and de novo CHD7 mutations account for 5–10 percent of all cases of KS and an accountable number of these patients exhibit mild form features of CHARGE syndrome, such as abnormally shaped ears, hearing loss, hare lip/cleft palate and cardiac abnormalities (60, 61, 69, 121, 126).
WDR11 Gene Variants Causing CHH/KS
WDR11 is a member of the WD-repeat containing protein family and comprises of twelve conserved domains of approximately 40 amino acids (68). The WDR11 gene is located in the chromosome 10q25-26 region and is expressed in various human organs including the brain, ear, lung, heart, kidney and the gonads (70). WDR11 is a scaffolding protein that is involved in multiple of cellular proceedings, including cell cycle progression, signal transduction, apoptosis and gene regulation (70). Kim et al. first reported that when mutated WDR11 is linked with idiopathic HH and KS (68). Since the initial report by Kim et al. (68), a few others followed and linked the WDR11 gene with different pathogenic variants in male patients without anosmia and CHH (127, 128). Recently, the WDR11 gene has also been shown to be involved in the Hedgehog (Hh) signaling pathway which is important for the normal ciliogenesis and when mutated can be the causal factor of KS and HH (68, 70). Another recent report by Sutani et al. (129) linked WDR11 as another causative gene for coloboma, cardiac anomaly and growth retardation in the 10q26 deletion syndrome.
RNF216 Gene Variants Linked to Gordon Holmes Syndrome
The RNF216 protein is a cytoplasmic protein which interacts with the serine/threonine protein kinase i.e., the receptor-interacting protein (RIP). Particular zinc finger domains of the RNF216 protein are necessary for its interaction with RIP and for the inhibition of TNF- and IL1-induced NF-kappa B activation pathways (130, 131). Additionally, the RNF216 protein plays a role in the ubiquitin-proteasome system for the break-down and degradation of unwanted proteins. Specifically, this protein functions as an E3 ubiquitin ligase (132). Variants in the RNF216 gene have been linked with hypogonadotropic hypogonadism, ataxia and dementia (28). More explicitly digenic homozygous variants in RNF216 and OTUD4, which encode a ubiquitin E3 ligase and a deubiquitinase, respectively, were identified in three affected siblings in a consanguineous family (28). Several other recent studies also reported variants in the RNF216 gene as a result of consanguinity to cause Gordon Holmes syndrome, a rare disorder characterized by diminished production of hormones leading to hypogonadotropic hypogonadism and difficulty in the coordination of movements i.e., cerebellar ataxia (133–136). These recent findings regarding the RNF216 gene associate the disorderly ubiquitination to neurodegeneration and reproductive dysfunction in combination with functional studies to reveal specific genetic interactions that cause disease.
POLR3A Gene Variants Associated With Hypomyelinating Leukodystrophy and HH
The POLR3A gene provides instructions for the production of the largest subunit of RNA polymerase III which is the enzyme involved in the RNA synthesis (137). The gene is located in chromosome 10q22.3 and variants inherited in the AR form have been initially reported in French-Canadian families with hypomyelinating leukodystrophy (138). Interestingly, these families were mapped to the same locus as leukodystrophy with oligodontia and demonstrated clinical and radiological overlap with patients with hypomyelination, hypodontia and hypogonadotropic hypogonadism syndrome (138). Several other recent studies that followed also reported variants in the POLR3A gene as being responsible for causing hypomyelination, hypodontia and hypogonadotropic hypogonadism, thus establishing a series of POLR3A gene variants to be associated with polymerase III-related leukodystrophy (139–144). It is estimated that 30–40% of patients with leukodystrophies remain without a molecular diagnosis (138, 141). The existence of mild and overlapping hypomyelinating leukodystrophy phenotypes could be attributed to heterozygous variants found in the POLR3A gene as a result of an abnormal enzymatic function of the RNA Pol III catalytic subunit. The role of heterozygosity in POLR3A in the overall pathogenesis of CHH is not well-established, and the possibility of a synergistic effect between these variants and variants identified in other genes cannot be excluded. Additionally, POLR3A gene could also be speculated to be a phenocopy gene due to the observed variability of phenotypes, therefore, patients and family members identified with mutations in this gene should be re-evaluated for understated and previously unrecognized clinical signs.
Conclusion
GnRH deficiency has been recognized both clinically and genetically as a heterogeneous disease with a range of different reproductive phenotypes including of congenital GnRH deficiency with anosmia (KS) and congenital GnRH deficiency with normal olfaction (normosmic CHH). The present study/review discusses the involvement of known and novel variants in patients with CHH/KS and adds up to the ontogeny of GnRH deficiency.
Moreover, this study provides new genetic findings and reinforces the significance of the use of NGS technology for the accurate molecular diagnosis and treatment of this rare condition.
Data Availability Statement
The datasets from this study can be found in DRYAD repository (https://datadryad.org) under accession number doi: 10.5061/dryad.1vhhmgqqj Link: https://datadryad.org/stash/share/uhb7bIQj8JDLq9TKTtb51pF7W1xprCFi58lt7vcRxU4.
Ethics Statement
The studies involving human participants were reviewed and approved by The Cyprus National Ethics Committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written, informed consent was obtained from all seven adult individuals that participate in the study for the publication of any potentially identifiable images or data included in this article.
Author Contributions
VN, PF, MT, NS, and LP contributed to the conception, design, and interpretation of the project. MS, FC, AO, GS, and DV contributed to the experimental part and data analysis of the project. NN, GT, and CM contributed to drafting or revising intellectual content of the manuscript. VN, NS, and LP had primary responsibility for final content. All authors read and approved the final manuscript.
Funding
AO and GS were supported by the European Commission Research Executive Agency Grant BIORISE (No. 669026), under the Spreading Excellence, Widening Participation, Science with and for Society Framework. The six (VN, PF, MT, GT, NS, LP) authors of this publication are members of the European Reference Network on Rare Endocrine Conditions: Project ID No. 739543 https://endo-ern.eu/about/reference-centres/. This work was supported by the A. G. Leventis Foundation & the RCB Bank Ltd.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.00626/full#supplementary-material
Supplementary Figure 1. Sequence electropherograms of the novel SRA1 p.Ile179Thr variant in heterozygous and homozygous state. The variant is indicated by a red arrow.
Supplementary Figure 2. Sequence electropherograms of the (A) p.Pro186Ala and (B) p.Arg822Cys FGFR1 variants in heterozygous state. The variants are indicated by red arrows.
Supplementary Figure 3. Sequence electropherogram of the POLR3A p.Arg561Gly variant in heterozygous state. The variant is indicated by a red arrow.
Supplementary Figure 4. Sequence electropherogram of the novel PROP1 p.Arg112Gln variant in heterozygous state. The variant is indicated by a red arrow.
Supplementary Figure 5. Family pedigrees and inheritance patterns of the (A) ANOS1:p.Gln82* mutation in patient 1 and the (B) SRA1:p.Ile179Thr, RNF216:p.Asp792Asn variations in patient 3.
Supplementary Figure 6. Predicted pathogenicity of the due to digenicity by ORVAL platform. (A) S-plot and corresponding table with score values of the results for the FGFR1/POLR3A digenic combination. (B) S-plot and corresponding table with score values of the results for the RNF216/SRA1 digenic combination. Black arrows indicate the disease-causing position of the digenic combination on the plot. Y-axis, Support Score: percentage of individual predictors agreeing on the disease-causing class of the digenic combination. X-axis, Classification Score: median probability among all predictors that the digenic combination is disease-causing. The higher the scores, the more confident the predictor is for the disease-causing class.
Supplementary Table 1. Genes with biological involvement in the GnRH neuronal system and CHH selected for variant analysis of the WES. For each gene the corresponding genetic and phenotypic information is indicated. The mean target coverage of the target region for each gene is designated in a separate column.
References
1. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2015) 11:547–64. doi: 10.1038/nrendo.2015.112
2. Dwyer AA, Raivio T, Pitteloud N. Gonadotrophin replacement for induction of fertility in hypogonadal men. Best Pract Res Clin Endocrinol Metab. (2015) 29:91–103. doi: 10.1016/j.beem.2014.10.005
3. Bouvattier C, Maione L, Bouligand J, Dode C, Guiochon-Mantel A, Young J. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nat Rev Endocrinol. (2011) 8:172–82. doi: 10.1038/nrendo.2011.164
4. Shoham Z, Smith H, Yeko T, O'Brien F, Hemsey G, O'Dea L. Recombinant LH (lutropin alfa) for the treatment of hypogonadotrophic women with profound LH deficiency: a randomized, double-blind, placebo-controlled, proof-of-efficacy study. Clin Endocrinol (Oxf). (2008) 69:471–8. doi: 10.1111/j.1365-2265.2008.03299.x
5. Seminara SB, Hayes FJ, Crowley WF Jr. Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations. Endocr Rev. (1998) 19:521–39. doi: 10.1210/edrv.19.5.0344
6. Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamaki J, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev. (2019) 40:669–710. doi: 10.1210/er.2018-00116
7. Wierman ME, Pawlowski JE, Allen MP, Xu M, Linseman DA, Nielsen-Preiss S. Molecular mechanisms of gonadotropin-releasing hormone neuronal migration. Trends Endocrinol Metab. (2004) 15:96–102. doi: 10.1016/j.tem.2004.02.003
8. Farr OM, Mantzoros CS. Old and new tools to study human brain physiology: Current state, future directions and implications for metabolic regulation. Metabolism. (2019) 99:iii-viii. doi: 10.1016/j.metabol.2019.153957
9. Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med. (2012) 366:443–53. doi: 10.1056/NEJMcp1109290
10. Bonomi M, Vezzoli V, Krausz C, Guizzardi F, Vezzani S, Simoni M, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol. (2018) 178:23–32. doi: 10.1530/EJE-17-0065
11. Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. (2009) 5:569–76. doi: 10.1038/nrendo.2009.177
12. Laitinen EM, Vaaralahti K, Tommiska J, Eklund E, Tervaniemi M, Valanne L, et al. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis. (2011) 6:41. doi: 10.1186/1750-1172-6-41
13. Kim SH. Congenital hypogonadotropic hypogonadism and kallmann syndrome: past, present, and future. Endocrinol Metab. (2015) 30:456–66. doi: 10.3803/EnM.2015.30.4.456
14. Stamou MI, Georgopoulos NA. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism. (2018) 86:124–34. doi: 10.1016/j.metabol.2017.10.012
15. Maione L, Dwyer AA, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, et al. GENETICS IN ENDOCRINOLOGY: genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. (2018) 178:R55–R80. doi: 10.1530/EJE-17-0749
16. Mason KA, Schoelwer MJ, Rogol AD. Androgens during infancy, childhood, and adolescence: physiology and use in clinical practice. Endocr Rev. (2020) 41:421–56. doi: 10.1210/endrev/bnaa003
17. Amato LGL, Montenegro LR, Lerario AM, Jorge AAL, Guerra Junior G, Schnoll C, et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Europ J Endocrinol. (2019) 181:103–19. doi: 10.1530/EJE-18-0764
18. Delafontaine J, Masselot A, Liechti R, Kuznetsov Xenarios I, Pradervand S Varapp: a reactive web-application for variants filtering bioRxiv. (2016). doi: 10.1101/060806
19. Okonechnikov K, Conesa A, Garcia-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. (2016) 32:292–4. doi: 10.1093/bioinformatics/btv566
20. Papadimitriou S, Gazzo A, Versbraegen N, Nachtegael C, Aerts J, Moreau Y, et al. Predicting disease-causing variant combinations. Proc Natl Acad Sci USA. (2019) 116:11878–87. doi: 10.1101/520353
21. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
22. Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comp Biol. (2014) 10:e1003440. doi: 10.1371/journal.pcbi.1003440
23. Vlachakis D, Tsagrasoulis D, Megalooikonomou V, Kossida S. Introducing Drugster: a comprehensive and fully integrated drug design, lead and structure optimization toolkit. Bioinformatics. (2013) 29:126–8. doi: 10.1093/bioinformatics/bts637
24. Wang J CP, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. (2000) 21:1049–71. doi: 10.1002/1096-987X(200009)21:12<1049::AID-JCC3>3.0.CO;2-F
25. Kotan LD, Cooper C, Darcan S, Carr IM, Ozen S, Yan Y, et al. Idiopathic hypogonadotropic hypogonadism caused by inactivating mutations in SRA1. J Clin Res Pediatr Endocrinol. (2016) 8:125–34. doi: 10.4274/jcrpe.3248
26. Kotan LD, Isik E, Turan I, Mengen E, Akkus G, Tastan M, et al. Prevalence and associated phenotypes of PLXNA1 variants in normosmic and anosmic idiopathic hypogonadotropic hypogonadism. Clin Genetics. (2019) 95:320–4. doi: 10.1111/cge.13482
27. Dode C, Hardelin JP. Kallmann syndrome. Europ J Hum Genet. (2009) 17:139–46. doi: 10.1038/ejhg.2008.206
28. Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. (2013) 368:1992–2003. doi: 10.1056/NEJMoa1215993
29. ExAC. Exome Aggregation Consortium. Available online at: http://exac.broadinstitute.org/ (2019).
30. Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. (2005) 15:978–86. doi: 10.1101/gr.3804205
31. Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. (2006) 22:2729–34. doi: 10.1093/bioinformatics/btl423
32. Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. (2002) 30:3894–900. doi: 10.1093/nar/gkf493
33. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
34. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. (2012) 40:W452–7. doi: 10.1093/nar/gks539
35. Leskovec J, Sosic R. SNAP: a general purpose network analysis and graph mining library. ACM Trans Intell Syst Technol. (2016) 8:1. doi: 10.1145/2898361
36. Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. (2019) 47:D419–D26. doi: 10.1093/nar/gky1038
37. Lima Amato LG, Latronico AC, Gontijo Silveira LF. Molecular and genetic aspects of congenital isolated hypogonadotropic hypogonadism. Endocrinol Metab Clinics NA. (2017) 46:283–303. doi: 10.1016/j.ecl.2017.01.010
38. Vezzoli V, Duminuco P, Bassi I, Guizzardi F, Persani L, Bonomi M. The complex genetic basis of congenital hypogonadotropic hypogonadism. Minerva Endocrinol. (2016) 41:223–39.
39. Georgopoulos NA, Koika V, Galli-Tsinopoulou A, Spiliotis BE, Adonakis G, Keramida MK, et al. Renal dysgenesis and KAL1 gene defects in patients with sporadic Kallmann syndrome. Fertil Steril. (2007) 88:1311–7. doi: 10.1016/j.fertnstert.2006.12.044
40. Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA. (2010) 107:15140–4. doi: 10.1073/pnas.1009622107
41. Hanchate NK, Giacobini P, Lhuillier P, Parkash J, Espy C, Fouveaut C, et al. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. (2012) 8:e1002896. doi: 10.1371/journal.pgen.1002896
42. Basaran Y, Bolu E, Unal HU, Sagkan RI, Taslipinar A, Ozgurtas T, et al. Multiplex ligation dependent probe amplification analysis of KAL1, GNRH1, GNRHR, PROK2 and PROKR2 in male patients with idiopathic hypogonadotropic hypogonadism. Endokrynol Polska. (2013) 64:285–92. doi: 10.5603/EP.2013.0007
43. Stamou MI, Varnavas P, Plummer L, Koika V, Georgopoulos NA. Next-generation sequencing refines the genetic architecture of Greek GnRH-deficient patients. Endocr Connect. (2019) 8:468–80. doi: 10.1530/EC-19-0010
44. Tommiska J, Kansakoski J, Christiansen P, Jorgensen N, Lawaetz JG, Juul A, et al. Genetics of congenital hypogonadotropic hypogonadism in Denmark. Eur J Med Genet. (2014) 57:345–8. doi: 10.1016/j.ejmg.2014.04.002
46. Dode C, Fouveaut C, Mortier G, Janssens S, Bertherat J, Mahoudeau J, et al. Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Hum Mutat. (2007) 28:97–8. doi: 10.1002/humu.9470
47. Hardelin JP, Julliard AK, Moniot B, Soussi-Yanicostas N, Verney C, Schwanzel-Fukuda M, et al. Anosmin-1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis: implications for the developmental anomalies of X chromosome-linked Kallmann syndrome. Dev Dynam. (1999) 215:26–44. doi: 10.1002/(SICI)1097-0177(199905)215:1<26::AID-DVDY4>3.0.CO;2-D
48. Hebert JM, Lin M, Partanen J, Rossant J, McConnell SK. FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development. (2003) 130:1101–11. doi: 10.1242/dev.00334
49. Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Gen. (2003) 33:463–5. doi: 10.1038/ng1122
50. Albuisson J, Pecheux C, Carel JC, Lacombe D, Leheup B, Lapuzina P, et al. Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2). Hum Mutat. (2005) 25:98–9. doi: 10.1002/humu.9298
51. Hardelin JP, Dode C. The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sexual Dev. (2008) 2:181–93. doi: 10.1159/000152034
52. Howard SR, Dunkel L. Delayed puberty-phenotypic diversity, molecular genetic mechanisms, and recent discoveries. Endocr Rev. (2019) 40:1285–317. doi: 10.1210/er.2018-00248
53. Richards MR, Plummer L, Chan YM, Lippincott MF, Quinton R, Kumanov P, et al. Phenotypic spectrum of POLR3B mutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J Med Genet. (2017) 54:19–25. doi: 10.1136/jmedgenet-2016-104064
54. Wu W, Cogan JD, Pfaffle RW, Dasen JS, Frisch H, O'Connell SM, et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. (1998) 18:147–9. doi: 10.1038/ng0298-147
55. Kelberman D, Turton JP, Woods KS, Mehta A, Al-Khawari M, Greening J, et al. Molecular analysis of novel PROP1 mutations associated with combined pituitary hormone deficiency (CPHD). Clin Endocrinol (Oxf). (2009) 70:96–103. doi: 10.1111/j.1365-2265.2008.03326.x
56. Correa FA, Nakaguma M, Madeira JLO, Nishi MY, Abrao MG, Jorge AAL, et al. Combined pituitary hormone deficiency caused by PROP1 mutations: update 20 years post-discovery. Arch Endocrinol Metab. (2019) 63:167–74. doi: 10.20945/2359-3997000000139
57. Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. (2004) 36:955–7. doi: 10.1038/ng1407
58. Balasubramanian R, Crowley WF, Jr. Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am J Med Genet Part C Sem Med Genet. (2017) 175:507–15. doi: 10.1002/ajmg.c.31585
59. Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. (2008) 83:511–9. doi: 10.1016/j.ajhg.2008.09.005
60. Balasubramanian R, Choi JH, Francescatto L, Willer J, Horton ER, Asimacopoulos EP, et al. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc Natl Acad Sci USA. (2014) 111:17953–8. doi: 10.1073/pnas.1417438111
61. Marcos S, Sarfati J, Leroy C, Fouveaut C, Parent P, Metz C, et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J Clin Endocrinol Metab. (2014) 99:E2138–43. doi: 10.1210/jc.2014-2110
62. Deladoey J, Fluck C, Buyukgebiz A, Kuhlmann BV, Eble A, Hindmarsh PC, et al. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. (1999) 84:1645–50. doi: 10.1210/jc.84.5.1645
63. Pavel ME, Hensen J, Pfaffle R, Hahn EG, Dorr HG. Long-term follow-up of childhood-onset hypopituitarism in patients with the PROP-1 gene mutation. Horm Res. (2003) 60:168–73. doi: 10.1159/000073228
64. Avbelj Stefanija M, Kotnik P, Bratanic N, Zerjav Tansek M, Bertok S, Bratina N, et al. Novel mutations in HESX1 and PROP1 genes in combined pituitary hormone deficiency. Horm Res Paediatr. (2015) 84:153–8. doi: 10.1159/000433468
65. Osorio MG, Kopp P, Marui S, Latronico AC, Mendonca BB, Arnhold IJ. Combined pituitary hormone deficiency caused by a novel mutation of a highly conserved residue (F88S) in the homeodomain of PROP-1. J Clin Endocrinol Metab. (2000) 85:2779–85. doi: 10.1210/jc.85.8.2779
66. Fluck C, Deladoey J, Rutishauser K, Eble A, Marti U, Wu W, et al. Phenotypic variability in familial combined pituitary hormone deficiency caused by a PROP1 gene mutation resulting in the substitution of Arg–>Cys at codon 120 (R120C). J Clin Endocrinol Metab. (1998) 83:3727–34. doi: 10.1210/jc.83.10.3727
67. Riepe FG, Partsch CJ, Blankenstein O, Monig H, Pfaffle RW, Sippell WG. Longitudinal imaging reveals pituitary enlargement preceding hypoplasia in two brothers with combined pituitary hormone deficiency attributable to PROP1 mutation. J Clin Endocrinol Metab. (2001) 86:4353–7. doi: 10.1210/jcem.86.9.7828
68. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. (2010) 87:465–79. doi: 10.1016/j.ajhg.2010.08.018
69. Kim HG, Layman LC. The role of CHD7 and the newly identified WDR11 gene in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Cell Endocrinol. (2011) 346:74–83. doi: 10.1016/j.mce.2011.07.013
70. Kim YJ, Osborn DP, Lee JY, Araki M, Araki K, Mohun T, et al. WDR11-mediated Hedgehog signalling defects underlie a new ciliopathy related to Kallmann syndrome. EMBO Rep. (2018) 19:269–89. doi: 10.15252/embr.201744632
71. Shammas C, Neocleous V, Toumba M, Costi C, Phedonos AA, Efstathiou E, et al. Overview of genetic defects in endocrinopathies in the island of Cyprus; evidence of a founder effect. Genet Test Mol Biomarkers. (2012) 16:1073–9. doi: 10.1089/gtmb.2011.0381
72. Kawashima H, Takano H, Sugita S, Takahara Y, Sugimura K, Nakatani T. A novel steroid receptor co-activator protein (SRAP) as an alternative form of steroid receptor RNA-activator gene: expression in prostate cancer cells and enhancement of androgen receptor activity. Biochem J. (2003) 369(Pt 1):163–71. doi: 10.1042/bj20020743
73. Chooniedass-Kothari S, Hamedani MK, Auge C, Wang X, Carascossa S, Yan Y, et al. The steroid receptor RNA activator protein is recruited to promoter regions and acts as a transcriptional repressor. FEBS Lett. (2010) 584:2218–24. doi: 10.1016/j.febslet.2010.04.022
74. Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. (2007) 117:457–63. doi: 10.1172/JCI29884
75. Neocleous V, Byrou S, Toumba M, Costi C, Shammas C, Kyriakou C, et al. Evidence of digenic inheritance in autoinflammation-associated genes. J Genet. (2016) 95:761–6. doi: 10.1007/s12041-016-0691-5
76. Stamou MI, Cox KH, Crowley WF Jr. Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the “-omics” era. Endocr Rev. (2016) 2016:4–22. doi: 10.1210/er.2015-1045.2016.1.test
77. Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, et al. Exome sequencing and the management of neurometabolic disorders. N Engl J Med. (2016) 374:2246–55. doi: 10.1056/NEJMoa1515792
78. Cangiano B, Swee DS, Quinton R, Bonomi M. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. (2020) doi: 10.1007/s00439-020-02147-1
79. Hardelin JP, Levilliers J, del Castillo I, Cohen-Salmon M, Legouis R, Blanchard S, et al. X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc Natl Acad Sci USA. (1992) 89:8190–4. doi: 10.1073/pnas.89.17.8190
80. Legouis R, Hardelin JP, Levilliers J, Claverie JM, Compain S, Wunderle V, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. (1991) 67:423–35. doi: 10.1016/0092-8674(91)90193-3
81. Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. (1991) 353:529–36. doi: 10.1038/353529a0
82. Bick D, Franco B, Sherins RJ, Heye B, Pike L, Crawford J, et al. Brief report: intragenic deletion of the KALIG-1 gene in Kallmann's syndrome. N Engl J Med. (1992) 326:1752–5. doi: 10.1056/NEJM199206253262606
83. Hardelin JP, Levilliers J, Young J, Pholsena M, Legouis R, Kirk J, et al. Xp22.3 deletions in isolated familial Kallmann's syndrome. J Clin Endocrinol Metab. (1993) 76:827–31. doi: 10.1210/jcem.76.4.8473391
84. Georgopoulos NA, Pralong FP, Seidman CE, Seidman JG, Crowley WF Jr. Genetic heterogeneity evidenced by low incidence of KAL-1 gene mutations in sporadic cases of gonadotropin-releasing hormone deficiency. J Clin Endocrinol Metab. (1997) 82:213–7. doi: 10.1210/jc.82.1.213
85. Dode C, Hardelin JP. Clinical genetics of Kallmann syndrome. Ann Endocrinol (Paris). (2010) 71:149–57. doi: 10.1016/j.ando.2010.02.005
86. Goncalves CI, Fonseca F, Borges T, Cunha F, Lemos MC. Expanding the genetic spectrum of ANOS1 mutations in patients with congenital hypogonadotropic hypogonadism. Hum Reprod. (2017) 32:704–11. doi: 10.1093/humrep/dew354
87. Nie M, Xu H, Chen R, Mao J, Wang X, Xiong S, et al. Analysis of genetic and clinical characteristics of a Chinese Kallmann syndrome cohort with ANOS1 mutations. Eur J Endocrinol. (2017) 177:389–98. doi: 10.1530/EJE-17-0335
88. Gach A, Pinkier I, Szarras-Czapnik M, Sakowicz A, Jakubowski L. Expanding the mutational spectrum of monogenic hypogonadotropic hypogonadism: novel mutations in ANOS1 and FGFR1 genes. Reprod Biol Endocrinol. (2020) 18:8. doi: 10.1186/s12958-020-0568-6
89. Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, de Zoysa PA, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf). (2001) 55:163–74. doi: 10.1046/j.1365-2265.2001.01277.x
90. Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, et al. Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab. (2013) 98:E943–53. doi: 10.1210/jc.2012-4116
91. Madhu SV, Kant S, Holla VV, Arora R, Rathi S. Unusual presentation of Kallmannn syndrome with contiguous gene deletion in three siblings of a family. Indian J Endocrinol Metab. (2012) 16 (Suppl. 2):S326–8. doi: 10.4103/2230-8210.104077
92. Tsai PS, Gill JC. Mechanisms of disease: insights into X-linked and autosomal-dominant Kallmann syndrome. Nat Clin Pract Endocrinol Metab. (2006) 2:160–71. doi: 10.1038/ncpendmet0119
93. Maione L, Pala G, Bouvattier C, Trabado S, Papadakis G, Chanson P, et al. Congenital hypogonadotropic hypogonadism/Kallmann syndrome is associated with statural gain in both men and women: a monocentric study. Eur J Endocrinol. (2020) 182:185. doi: 10.1530/EJE-19-0537
94. Kaprara A, Huhtaniemi IT. The hypothalamus-pituitary-gonad axis: tales of mice and men. Metabolism. (2018) 86:3–17. doi: 10.1016/j.metabol.2017.11.018
95. Soussi-Yanicostas N, de Castro F, Julliard AK, Perfettini I, Chedotal A, Petit C. Anosmin-1, defective in the X-linked form of Kallmann syndrome, promotes axonal branch formation from olfactory bulb output neurons. Cell. (2002) 109:217–28. doi: 10.1016/S0092-8674(02)00713-4
96. Oliveira LM, Seminara SB, Beranova M, Hayes FJ, Valkenburgh SB, Schipani E, et al. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. (2001) 86:1532–8. doi: 10.1210/jcem.86.4.7420
97. Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, Kawakita S, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab. (2004) 89:1079–88. doi: 10.1210/jc.2003-030476
98. Trarbach EB, Costa EM, Versiani B, de Castro M, Baptista MT, Garmes HM, et al. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J Clin Endocrinol Metab. (2006) 91:4006–12. doi: 10.1210/jc.2005-2793
99. Salenave S, Chanson P, Bry H, Pugeat M, Cabrol S, Carel JC, et al. Kallmann's syndrome: a comparison of the reproductive phenotypes in men carrying KAL1 and FGFR1/KAL2 mutations. J Clin Endocrinol Metab. (2008) 93:758–63. doi: 10.1210/jc.2007-1168
100. Pitteloud N, Acierno JS Jr., Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. (2006) 103:6281–6. doi: 10.1073/pnas.0600962103
101. Villanueva C, de Roux N. FGFR1 mutations in Kallmann syndrome. Front Horm Res. (2010) 39:51–61. doi: 10.1159/000312693
102. Trarbach EB, Teles MG, Costa EM, Abreu AP, Garmes HM, Guerra G, et al. Screening of autosomal gene deletions in patients with hypogonadotropic hypogonadism using multiplex ligation-dependent probe amplification: detection of a hemizygosis for the fibroblast growth factor receptor 1. Clin Endocrinol (Oxf). (2010) 72:371–6. doi: 10.1111/j.1365-2265.2009.03642.x
103. Pedersen-White JR, Chorich LP, Bick DP, Sherins RJ, Layman LC. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Hum Reprod. (2008) 14:367–70. doi: 10.1093/molehr/gan027
104. Valdes-Socin H, Rubio Almanza M, Tome Fernandez-Ladreda M, Debray FG, Bours V, Beckers A. Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front Endocrinol. (2014) 5:109. doi: 10.3389/fendo.2014.00109
105. Dwyer AA, Raivio T, Pitteloud N. Management of endocrine disease: reversible hypogonadotropic hypogonadism. Eur J Endocrinol. (2016) 174:R267–74. doi: 10.1530/EJE-15-1033
106. Pitteloud N, Meysing A, Quinton R, Acierno JS Jr, Dwyer AA, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. (2006) 254–255:60–9. doi: 10.1016/j.mce.2006.04.021
107. Dong S, Kang S, Gu TL, Kardar S, Fu H, Lonial S, et al. 14-3-3 Integrates prosurvival signals mediated by the AKT and MAPK pathways in ZNF198-FGFR1-transformed hematopoietic cells. Blood. (2007) 110:360–9. doi: 10.1182/blood-2006-12-065615
108. Goradia A, Bayerl M, Cornfield D. The 8p11 myeloproliferative syndrome: review of literature and an illustrative case report. Int J Clin Exp Pathol. (2008) 1:448–56.
109. Bennett JT, Tan TY, Alcantara D, Tetrault M, Timms AE, Jensen D, et al. Mosaic activating mutations in FGFR1 cause encephalocraniocutaneous lipomatosis. Am J Hum Genet. (2016) 98:579–87. doi: 10.1016/j.ajhg.2016.02.006
110. Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet. (2009) 46:721–9. doi: 10.1136/jmg.2009.066068
111. Dhamija R, Babovic-Vuksanovic D. Hartsfield syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle, WA: University of Washington, Seattle (1993).
112. Hong S, Hu P, Marino J, Hufnagel SB, Hopkin RJ, Toromanovic A, et al. Dominant-negative kinase domain mutations in FGFR1 can explain the clinical severity of Hartsfield syndrome. Hum Mol Genet. (2016) 25:1912–22. doi: 10.1093/hmg/ddw064
113. White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. (2005) 76:361–7. doi: 10.1086/427956
114. Sow AJ, Ramli R, Latiff ZA, Ichikawa S, Gray AK, Nordin R, et al. Osteoglophonic dysplasia: a 'common' mutation in a rare disease. Clin Genet. (2010) 78:197–8. doi: 10.1111/j.1399-0004.2010.01382.x
115. Cornejo-Roldan LR, Roessler E, Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum Genet. (1999) 104:425–31. doi: 10.1007/s004390050979
116. Chokdeemboon C, Mahatumarat C, Rojvachiranonda N, Tongkobpetch S, Suphapeetiporn K, Shotelersuk V. FGFR1 and FGFR2 mutations in Pfeiffer syndrome. J Craniofac Surg. (2013) 24:150–2. doi: 10.1097/SCS.0b013e3182646454
117. Balasubramanian R, Crowley WF Jr. Isolated gonadotropin-releasing hormone (GnRH) deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle, WA: University of Washington, Seattle (1993).
118. Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res BCR. (2007) 9:R23. doi: 10.1186/bcr1665
119. Jiang T, Gao G, Fan G, Li M, Zhou C. FGFR1 amplification in lung squamous cell carcinoma: a systematic review with meta-analysis. Lung Cancer. (2015) 87:1–7. doi: 10.1016/j.lungcan.2014.11.009
120. Pinto G, Abadie V, Mesnage R, Blustajn J, Cabrol S, Amiel J, et al. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. (2005) 90:5621–6. doi: 10.1210/jc.2004-2474
121. Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. (2011) 48:334–42. doi: 10.1136/jmg.2010.087106
122. Kong F, Martin DM. Atopic disorders in CHARGE syndrome: a retrospective study and literature review. Eur J Med Genet. (2018) 61:225–9. doi: 10.1016/j.ejmg.2017.11.019
123. Hsu P, Ma A, Wilson M, Williams G, Curotta J, Munns CF, et al. CHARGE syndrome: a review. J Paediatr Child Health. (2014) 50:504–11. doi: 10.1111/jpc.12497
124. Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. (2016) 170A:344–54. doi: 10.1002/ajmg.a.37435
125. Bergman JE, de Ronde W, Jongmans MC, Wolffenbuttel BH, Drop SL, Hermus A, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab. (2012) 97:E858–62. doi: 10.1210/jc.2011-2652
126. Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome–the clinical overlap with CHARGE syndrome. Clin Genet. (2009) 75:65–71. doi: 10.1111/j.1399-0004.2008.01107.x
127. Mao J, Chen R, Wu X, Liu Z, Zheng J, Wang X, et al. Association of the extra-gonadal manifestations with different pathogenic gene mutations in male patients with congenital hypogonadotropic hypogonadism. Zhonghua Yi Xue Za Zhi. (2015) 95:3424–7.
128. Ayers KL, Bouty A, Robevska G, van den Bergen JA, Juniarto AZ, Listyasari NA, et al. Variants in congenital hypogonadotrophic hypogonadism genes identified in an Indonesian cohort of 46,XY under-virilised boys. Hum Genom. (2017) 11:1. doi: 10.1186/s40246-017-0098-2
129. Sutani A, Shima H, Hijikata A, Hosokawa S, Katoh-Fukui Y, Takasawa K, et al. WDR11 is another causative gene for coloboma, cardiac anomaly and growth retardation in 10q26 deletion syndrome. Eur J Med Genet. (2020) 63:103626. doi: 10.1016/j.ejmg.2019.01.016
130. van der Reijden BA, Erpelinck-Verschueren CA, Lowenberg B, Jansen JH. TRIADs: a new class of proteins with a novel cysteine-rich signature. Protein Sci. (1999) 8:1557–61. doi: 10.1110/ps.8.7.1557
131. Chen D, Li X, Zhai Z, Shu HB. A novel zinc finger protein interacts with receptor-interacting protein (RIP) and inhibits tumor necrosis factor (TNF)- and IL1-induced NF-kappa B activation. J Biol Chem. (2002) 277:15985–91. doi: 10.1074/jbc.M108675200
132. Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. (2004) 5:495–502. doi: 10.1038/ni1066
133. Alqwaifly M, Bohlega S. Ataxia and hypogonadotropic hypogonadism with intrafamilial variability caused by RNF216 mutation. Neurol Int. (2016) 8:6444. doi: 10.4081/ni.2016.6444
134. Husain N, Yuan Q, Yen YC, Pletnikova O, Sally DQ, Worley P, et al. TRIAD3/RNF216 mutations associated with Gordon Holmes syndrome lead to synaptic and cognitive impairments via Arc misregulation. Aging Cell. (2017) 16:281–92. doi: 10.1111/acel.12551
135. Santens P, Van Damme T, Steyaert W, Willaert A, Sablonniere B, De Paepe A, et al. RNF216 mutations as a novel cause of autosomal recessive Huntington-like disorder. Neurology. (2015) 84:1760–6. doi: 10.1212/WNL.0000000000001521
136. Sawyer SL, Schwartzentruber J, Beaulieu CL, Dyment D, Smith A, Warman Chardon J, et al. Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum Mutat. (2014) 35:45–9. doi: 10.1002/humu.22451
137. Sepehri S, Hernandez N. The largest subunit of human RNA polymerase III is closely related to the largest subunit of yeast and trypanosome RNA polymerase III. Genome Res. (1997) 7:1006–19. doi: 10.1101/gr.7.10.1006
138. Bernard G, Chouery E, Putorti ML, Tetreault M, Takanohashi A, Carosso G, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. (2011) 89:415–23. doi: 10.1016/j.ajhg.2011.07.014
139. Cayami FK, La Piana R, van Spaendonk RM, Nickel M, Bley A, Guerrero K, et al. POLR3A and POLR3B mutations in unclassified hypomyelination. Neuropediatrics. (2015) 46:221–8. doi: 10.1055/s-0035-1550148
140. Daoud H, Tetreault M, Gibson W, Guerrero K, Cohen A, Gburek-Augustat J, et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet. (2013) 50:194–7. doi: 10.1136/jmedgenet-2012-101357
141. Potic A, Brais B, Choquet K, Schiffmann R, Bernard G. 4H syndrome with late-onset growth hormone deficiency caused by POLR3A mutations. Arch Neurol. (2012) 69:920–3. doi: 10.1001/archneurol.2011.1963
142. La Piana R, Cayami FK, Tran LT, Guerrero K, van Spaendonk R, Ounap K, et al. Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology. (2016) 86:1622–6. doi: 10.1212/WNL.0000000000002612
143. Khalifa M, Naffaa L. Exome sequencing reveals a novel WDR45 frameshift mutation and inherited POLR3A heterozygous variants in a female with a complex phenotype and mixed brain MRI findings. Eur J Med Genet. (2015) 58:381–6. doi: 10.1016/j.ejmg.2015.05.009
Keywords: GnRH, hypogonadotropic hypogonadism, genes, digenic inheritance, next generation sequencing
Citation: Neocleous V, Fanis P, Toumba M, Tanteles GA, Schiza M, Cinarli F, Nicolaides NC, Oulas A, Spyrou GM, Mantzoros CS, Vlachakis D, Skordis N and Phylactou LA (2020) GnRH Deficient Patients With Congenital Hypogonadotropic Hypogonadism: Novel Genetic Findings in ANOS1, RNF216, WDR11, FGFR1, CHD7, and POLR3A Genes in a Case Series and Review of the Literature. Front. Endocrinol. 11:626. doi: 10.3389/fendo.2020.00626
Received: 10 April 2020; Accepted: 31 July 2020;
Published: 28 August 2020.
Edited by:
Rosario Pivonello, University of Naples Federico II, ItalyReviewed by:
Biagio Cangiano, University of Milan, ItalyFrancesca Cioppi, University of Florence, Italy
Copyright © 2020 Neocleous, Fanis, Toumba, Tanteles, Schiza, Cinarli, Nicolaides, Oulas, Spyrou, Mantzoros, Vlachakis, Skordis and Phylactou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicos Skordis, bmljb3Nza29yZGlzQHBhZWRpLm9yZy5jeQ==; Leonidas A. Phylactou, bGFwaHlsYWNAY2luZy5hYy5jeQ==
†These authors have contributed equally to this work and share first authorship