 Eline Constance Brombacher
Eline Constance Brombacher Bart Everts
Bart Everts- Department of Parasitology, Leiden University Medical Center, Leiden, Netherlands
Nutrients are required for growth and survival of all cells, but are also crucially involved in cell fate determination of many cell types, including immune cells. There is a growing appreciation that the metabolic micro-environment also plays a major role in shaping the functional properties of dendritic cells (DCs). Under pathological conditions nutrient availability can range from a very restricted supply, such as seen in a tumor micro-environment, to an overabundance of nutrients found in for example obese adipose tissue. In this review we will discuss what is currently known about the metabolic requirements for DC differentiation and immunogenicity and compare that to how function and fate of DCs under pathological conditions can be affected by alterations in environmental levels of carbohydrates, lipids and amino acids as well as by other metabolic cues, including availability of oxygen, redox homeostasis and lactate levels. Many of these insights have been generated using in vitro model systems, which have revealed highly diverse effects of different metabolic cues on DC function. However, they also stress the importance of shifting toward more physiologically relevant experimental settings to be able to fully delineate the role of the metabolic surroundings in its full complexity in shaping the functional properties of DCs in health and disease.
Introduction
DCs are crucial for the regulation of immunity and tolerance during infections as well as during immune homeostasis in steady state, by governing the activation and maintenance of different CD4+ T helper subsets and CD8+ cytotoxic T cell responses. DCs are a heterogenic population of cells that comprise several lineages, including conventional DCs (cDCs), plasmacytoid DCs (pDCs), Langerhans cells (LCs), and inflammatory monocyte-derived DCs (infDCs) (1). Two main lineages can be identified within the cDCs, IRF8-dependent cDC1s and IRF4-dependent cDC2s (2). These different DC subsets have specialized largely non-redundant functions in priming and regulation of T cell responses (2). For instance, cDC1s and cDC2s are considered to be the most potent activators of T cells, that are particularly well-equipped to prime CD8+ and CD4+ T cell responses, respectively. pDCs, on the other hand, play an important role in the viral defense by producing large amounts of type I interferons. infDCs develop from monocytes during inflammation and have been described to have a role in innate inflammatory responses as well as in T cell priming (3). Given the scarcity of DCs in vivo, several in vitro experimental models have been developed to study DC biology. In this respect monocyte-derived DCs (moDCs), obtained from monocyte cultures supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 are a widely used model to study human DC biology and metabolism (4). moDCs share phenotypical and functional similarities with cDCs, but closely resemble infDCs at the transcriptional level (5, 6). In vitro generated murine DCs are commonly derived from GM-CSF-stimulated bone marrow (BMDCs), which bear resemblance to antigen presenting monocyte-derived murine inflammatory DCs. Flt3L-stimulated bone marrow cultures (Flt3L-DCs) give rise to equivalents of multiple steady state splenic DC subsets, including cDC1s, cDC2s, and pDCs (7–9). Regulation of the functional properties of DCs is dictated by their ontogeny, but also strongly influenced by their micro-environment. While the effects of danger signals, cytokines and chemokines are extensively reviewed elsewhere, we will focus on the role of metabolic cues in tuning DC function (10–12).
Over the last decade it has become increasingly clear that immune cell activation, proliferation, fate and function are closely linked to, and dependent on activation of specific metabolic pathways (13). Since these metabolic shifts are largely fueled and dependent on nutrients present in the immune cell niche it is becoming evident that the metabolic micro-environment is a vital factor in shaping the outcome of an immune response (14, 15). This review describes the latest insights into the nutritional requirements for DCs to drive an effective immune response and how altered nutrient availability in diseased states may affect the immunogenic or tolerogenic properties of DCs. In this context we will particularly focus on nutrient-limiting and excessive nutrient conditions as found in cancer and diabetes, respectively. Overall we here aim to provide an overview of how the metabolic micro-environment affects the functional properties of DCs.
Metabolic Demands of DCs During an Immune Response
During homeostasis DCs reside in peripheral tissues in a relatively quiescent state. However, upon sensing of changes in this homeostatic state, either due to invading pathogens or tissue-derived inflammatory signals, DCs undergo a well-defined activation program that involves increased capturing and processing of antigens for presentation in major histocompatibility complex I (MHC-I) and MHC-II and the induction of expression of chemokine receptors, cytokines, and costimulatory molecules. This enables DCs to traffic, via tissue-draining lymphatic vessels, to T cell zones of secondary lymphoid organs to efficiently prime and control effector T cell responses. Here, we discuss how the availability of carbohydrates (glucose), amino acids and lipids influences these functions of DCs during an immune response.
Glucose
A well-described phenomenon in immune cells, including DCs, is the switch from catabolic metabolism, characterized by fatty acid oxidation and mitochondrial respiration (Figure 1A) to anabolic metabolism, with enhanced glycolytic rates and lowered oxidative phosphorylation following activation (Figure 1B). BMDCs increase glycolytic rates within minutes after stimulation with LPS (TLR4), Poly(I:C) (TLR3), and CPG (TLR9) and also for moDCs a rapid glycolytic increase has been observed upon LPS stimulation (16–20). Likewise, human myeloid CD1c+ DCs and human pDCs show a similar metabolic reprogramming upon pRNA (TLR7/8) stimulation or viral infection (TLR7), respectively (21, 22). Although this points toward increased glucose utilization to fuel glycolysis as a conserved metabolic response to TLR stimulation by DCs, the function of this metabolic reprogramming and fate of glucose-derived carbons can change over time after activation, as discussed below.
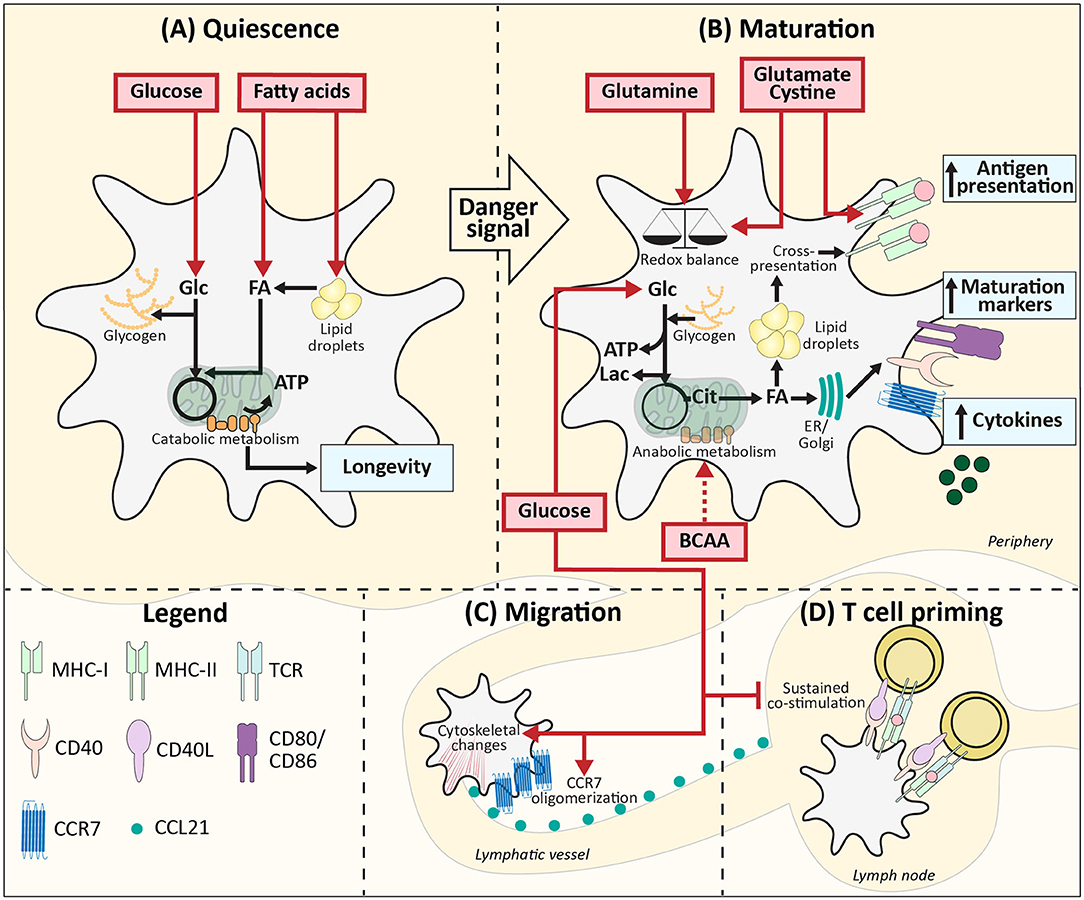
Figure 1. Metabolic demands of conventional DCs during homeostasis. (A) Quiescent DCs in peripheral tissues require, glucose and lipids as fuel for mitochondrial ATP generation and to build up intracellular storage of lipids and glycogen. (B) After TLR ligation uptake of glucose and BCAAs increases and together with glucose derived from glycogen this supports the switch from catabolic to anabolic metabolism, which is required for DC maturation. During this process, lipid bodies support cross-presentation. Glutamine, cysteine, and glutamate promote DC activation via maintaining redox homeostasis and promoting antigen presentation. (C) Glucose also promotes migration via stimulating CCR7 oligomerization and inducing cytoskeletal changes. (D) In the lymph nodes local glucose availability may be reduced due to glucose consumption by T cells, which may allow for more sustained expression of costimulatory markers and thereby more potent T cell priming. Red boxes: nutrients. Blue boxes: Functional consequences. Dotted arrow: presumed mechanism. ER, Endoplasmatic reticulum; Golgi, Golgi apparatus.
For LPS-induced BMDCs the switch to enhanced glycolysis is essential, given that inhibition of glycolysis using 2-deoxyglucose (2-DG) interferes with activation and their capacity to induce T cell proliferation in vitro and in vivo (16, 23). Correspondingly, changing glucose concentrations in culture media from 10 to 0 mM diminished the upregulation of co-stimulatory markers CD40 and CD86, and production of p40, subunit of IL-12 and IL-23 in LPS-stimulated murine BMDCs (16). In the presence of glucose, LPS stimulation enhances glycolysis via activation of the PI3K/Akt pathway and promotes loss of mitochondrial respiration via stabilization of Hypoxia-inducible factor 1-alpha (HIF1α) and expression of Inducible nitric oxide synthase (iNOS) in BMDCs (16, 17, 24). iNOS is involved in production of nitric oxide (NO) which in an autocrine manner poisons oxidative phosphorylation (17, 24). These TLR-induced events are largely absent in BMDCs activated by LPS in the absence of glucose, due to impaired mTOR activation, thereby further establishing the crucial role for glucose in supporting TLR-induced DC activation (Figure 1B). Mechanistically, BMDCs require glucose as a carbon source for fatty acid synthesis (FAS) to support ER and Golgi membrane expansion that is needed to accommodate the increased demands for protein translation during activation (23). To this end, glucose is metabolized in the TCA cycle to citrate, which is used as a carbon substrate for FAS (Figure 1B). In contrast to this anabolic role that glucose-derived carbon plays during BMDC activation, once activated, glucose is used in glycolysis by BMDCs purely for bioenergetic purposes (e.g., synthesis of ATP) to compensate for loss of OXPHOS due to the autocrine effects of NO derived from iNOS that poisons the ETC (17). Apart from direct usage of extracellular glucose, BMDCs and moDCs can also utilize intracellular glycogen stores to fuel glycolysis. These glycogen stores are formed prior to TLR stimulation and essential for activation and subsequent T cell stimulation in both moDCs and BMDCs by directly contributing to FAS (Figures 1A,B) (19).
Upon TLR-ligation, DCs upregulate CCR7 and are attracted toward lymphatic vessels that secrete CCR7 ligand CCL21 (25). In BMDCs the presence of glucose in the medium and subsequent activation of glycolysis are required for CCR7 oligomerization and cytoskeletal changes that support the mobility of DCs. Correspondingly, glucose depletion reduced migration of splenic DCs toward CCL21 ex vivo and 2-DG administration in an experimental model of allergic asthma, reduced migration of CD11c+MHCIIhi DCs to the lung (26). Similarly, BMDCs pulsed ex vivo with OVA plus LPS in the presence of 2-DG, displayed impaired migration toward skin-draining lymph nodes following subcutaneous injection. Altogether this points toward an important role for glucose in DC migration in vivo (Figure 1C) (16).
Interestingly, there are indications that the role of glucose in priming of T cell responses by DCs changes once the cells interact with T cells. It has been shown that the T cell-priming capacity of BMDCs declines after 24 h following TLR stimulation, which was associated with a reduction in expression of costimulatory molecules (27). However, this reduction in expression was prevented when 8 h after stimulation glucose was replaced with galactose, a carbohydrate that cannot be efficiently used in glycolysis. These data indicate that after the initial need for glucose, sustained high glycolytic rates repress BMDC activation (Figure 1D). Hence a local glucose-limiting micro-environment when DCs interact with T cells, may actually support T cell priming and an active immune response (24). Interestingly, both in vitro and in vivo data suggest that reduction of glucose availability may occur naturally during T cell priming in lymph nodes, due to scavenging of glucose by T cells that are being activated by DCs (24).
Many studies addressing the role of glycolysis, use glucose analog 2-DG to mimic glucose starvation. 2-DG can reduce mitochondrial metabolism independently of reducing glycolysis (28). Moreover, treatment of LPS-activated moDCs with 2-DG has been documented to result in ER-induced upregulation of IL-23 expression, while glucose depletion did not. These data indicate that the effects of 2-DG are not always connected to glycolysis and warrant caution when interpreting data from studies that have used 2-DG to interrogate the role of glycolysis in DC biology (29). In addition, most of the studies described above have been performed in BMDCs, in which iNOS plays a major role in the suppression of mitochondrial respiration in response to TLR stimulation. Most other DC subsets, including moDCs and primary human DCs, do not express iNOS (30). Hence, the role of oxidative phosphorylation during an immune response in human DCs may be underestimated and in physiological settings DCs may rely less on glucose as predicted based on BMDC experiments. Thus far, it has not been addressed whether other nutritional carbohydrates shape DC function, but it is worth investigating, given that an immunosuppressive role for D-mannose was found in T cells (31). In conclusion, there is clear evidence that many aspects of DC activation are dependent on availability of glucose in the micro-environment. Glucose initiates metabolic reprogramming required for activation and boosts DC migration toward lymph nodes, while during T cell interaction glucose may have an immunosuppressive effect on DCs.
Amino Acids
Amino acids are important for fueling mitochondrial respiration, for protein synthesis, as well as acting as a source of carbon and nitrogen for the synthesis of various other macromolecules. There are clear indications that amino acids in the environment of DCs play an important role in regulating their function. For example, lowering the supraphysiological amino acid concentrations commonly found in standard culture media to ones found in human plasma, increased the efficiency of moDC differentiation (32, 33). Conversely, moDCs in media containing imbalanced amino acid concentrations, as found in plasma of liver cirrhosis patients, showed impaired expression of maturation markers, secretion of IL-12 and migratory capacity in response to LPS stimulation, compared to moDCs stimulated in control medium (32, 33). The amino acid imbalance interfered with mitochondrial metabolism of immature DCs, causing a reduction in ATP levels and an increase in glucose consumption, which could not be further increased by LPS stimulation. This together supports the idea that several aspects of DC biology, including differentiation, activation and core metabolism, are sensitive to changes in amino acid concentrations in their environment (33).
In addition to the aforementioned work implicating amino acids in general in regulating DC function, there are several studies that have specifically interrogated the role of individual amino acids in this context. LPS stimulation of moDCs has been shown to enhance the uptake of aspartate, cystine, glutamate, and branched chain amino acids (BCAAs) valine, leucine, and isoleucine (33). Depletion of BCAAs and in particular valine from culture media of moDCs impairs maturation upon LPS stimulation, characterized by lowered CD83 levels and decreased ability to induce T cell proliferation. Additionally, mTOR signaling was impaired, which raises the possibility that BCAAs may affect DC maturation through modulation of metabolism via the mTOR pathway (Figure 1B) (34). Of note, as the above mentioned studies were performed in serum-free media with high glucose concentrations (25 mM), the relevance of these results under more physiological settings remains to be established. BCAAs are also important for maturation of moDCs stimulated with TLR7/8 ligand protamine-RNA (pRNA). In contrast to LPS stimulation, pRNA ligation boosts fatty acid oxidation (FAO)-dependent mitochondrial respiration and high intracellular levels of BCAAs are required to induce moDC maturation via FAO (20). BCAA leucine may play a key role in supporting FAO, as leucine can promote mitochondrial biogenesis (35). LPS treatment also increases uptake of glutamate and cystine in moDCs and inhibition of the cystine/glutamate antiporter in these cells reduced glutathione synthesis, but did not change the expression of maturation markers. Nevertheless, treating murine splenic DCs with an cystine/glutamate antiporter inhibitor resulted in lowered antigen presentation to both class I and class II MHC-restricted T cells (Figure 1B) (36). Hence, cystine and glutamate may be crucial metabolites for DC maturation via their role in redox homeostasis and antigen presentation.
As mentioned before, FAS is upregulated in BMDCs following LPS-stimulation. In cancer cells glutamine can contribute to lipogenesis via NADPH production that takes place when glutamine is metabolized to lactate or when glutamine is converted to citrate, facilitated by reductive carboxylation (37, 38). However, depleting glutamine (from 2 to 0 mM) from BMDC culture media did not affect CD40 and CD86 levels and inhibition of glutaminolysis had no effect on the metabolic alterations 6 h after LPS stimulation (19, 23). Interestingly, in BMDCs stimulated with TLR7/8 ligand imiquimod, glutamine deprivation, or disruption of glutaminolysis enhanced mitochondrial reactive oxygen species (ROS) production and subsequently IL-23 expression. This may suggest that glutamine, by supporting NADPH production, may contribute to scavenging of ROS in BMDCs, rather than to FAS-dependent activation (Figure 1B) (39). In addition, glutamine may fuel the TCA cycle to support oxidative phosphorylation in DCs as human pDCs increased oxidative phosphorylation following pRNA stimulation and inhibition of glutaminolysis in these cells caused a significant decrease in activation, IFNα secretion and mitochondrial respiration (21). Since activation of human CD1c+ myeloid DCs using pRNA resulted in reduced oxidative phosphorylation and as immunogenicity remained unaffected by inhibition of glutaminolysis, this effect of glutamine may be selectively associated with DC subsets that depend on mitochondrial respiration upon activation, such as pDCs (21).
Apart from glutamine, the importance of availability of different amino acids on DC biology are still poorly defined and are mainly addressed in moDCs. Possibly, due to the low proliferative capacity of differentiated DCs and therefore expected relative little dependency on anabolic metabolism, general amino acid availability may be less of critical factor for DC function than for other more proliferating cells. Nonetheless, as studies exploring the role of glutamine on DC function suggest, specific amino acids may be important in regulation of certain metabolic properties of DCs that are essential for their functional output. Hence, single amino acid depletion studies under more physiological nutrient levels may unravel novel roles of amino acids in DC function.
Lipids
In contrast to activated DCs, in which glycolysis is often the main bioenergetic pathway, immature quiescent BMDCs and Flt3L-induced cDC1s rely on FAO for energy generation, which would support a longer lifespan for these immature cells (Figure 1A) (16, 17, 40). Lipids from the local micro-environment may function as important nutrients for FAO in resting DCs. In human Lin-HLA-DR+ and murine CD11c+ hepatic DCs, high lipid content is associated with a stronger immune response (41). These lipids derived from both fatty acid (FA) uptake and synthesis and are stored in lipid bodies. Short term priming with triacyl glycerides of murine DCs containing few lipid bodies did not boost their immunogenic capacities, suggesting that pre-stored lipids rather than direct lipid availability in the micro-environment is important for hepatic DC immunogenicity (41). Mechanistically, lipid bodies in murine BMDCs and splenic CD11c+ DCs have been shown to boost CD8+ T cell priming by supporting cross-presentation, a process by which peptides from exogenous antigens are presented in MHC-I (42). It is therefore tempting to speculate that resting DCs may not only utilize FAs from the extracellular environment to fuel FAO for their bioenergetic homeostasis, but also to form lipid bodies to help prepare them for potent T cell priming after activation (Figure 1A).
In contract to conventional DCs, FAO can increase upon TLR7/8/9 stimulation of pDCs (20, 43). Interestingly, FAO in murine bone marrow-derived pDCs is fueled with FAs that are synthesized de novo (43). LPS is also known to increase FAS in BMDCs, possibly suggesting that FAO during an immune response predominantly depends on de novo synthesis and not on the FA availability in the micro-environment (Figure 1B) (18, 23). Correspondingly, in rats it was found that lipid content between different cell types in the same micro-environment was more similar than lipid content between DCs from distinct lymph nodes. Additionally, in vivo LPS stimulation diminished the differences observed between distinct DCs, supporting the notion that lipid accumulation during inflammation is independent of FA availability, while lipid storage during homeostasis does appear to be determined by the micro-environment (Figure 1A) (44).
In summary, it appears that during homeostasis lipid availability influences the types and amount of lipids stored in DCs and at least in some tissues this is important for their immunogenic potential. During an immune response, both conventional and pDCs accumulate lipids, most likely independent of the FA availability in the local micro-environment, but fueled by FAS. For cDCs the reduced ability of activated DCs to burn FAs by FAO may also contribute to lipid accumulation (16). Cultures in lipid-restricted conditions, 13C-labeled lipid metabolic flux analysis and lipid profiling of DCs during homeostasis and upon activation can further elucidate the role of extracellular lipids on DC function.
Concluding Remarks
The metabolic demands of DCs in non-pathological conditions are dependent on the subset, their location and the maturation stage, as summarized in Figure 1. Given that most of these data are obtained from in vitro studies it is important to realize that in vitro nutrient availability is often not limiting and exceed the levels that occur in vivo. Furthermore, nutrient competition with cells in the proximity of DCs and metabolites secreted by these surrounding cells are metabolic settings that are hard to mimic in vitro and difficult to measure in vivo, but likely to affect the metabolic micro-environment. Nevertheless, it is evident that nutrient availability is of great importance for the functional output by DCs.
Effects of the Metabolic Environment on DCs in Cancer
Metabolic Properties of the Tumor Micro-Environment
Disturbance of nutrient homeostasis is a cause and consequence of many pathologies. A well-studied and complex disease is cancer, which is characterized by a wide range of local metabolic alterations, including nutrient deficiency, hypoxia and oxidative stress. Cancer is a heterogenous disease that arises from cells with traits that allow uncontrolled proliferation. One of these traits, or hallmarks, is avoiding immune detection, required to prevent elimination by the immune system (45). Cells within the tumor micro-environment (TME), including tumor cells, fibroblasts, endothelial cells, and immune cells secrete immunomodulatory signals that regulate the anti-tumor immune response (46). Among these factors are cytokines, growth factors and metabolites. During the initial phases of tumor growth, tumor-associated DCs (TADCs) are able to recognize tumor antigens and initiate an anti-tumor T cell response. However, during tumor progression DCs gain tolerogenic rather than pro-inflammatory properties (47–49). A major contributor to immune suppression and another hallmark of cancer is deregulated cellular metabolism (45). The best known metabolic adaptation of cancer cells is the Warburg effect, the conversion of glucose to lactate under aerobic conditions, which allows for rapid production of ATP and biosynthetic precursors (50). In addition, tumors also use amino acids and lipids to fuel the TCA cycle, which promotes ATP generation via oxidative phosphorylation and synthesis of macromolecules to support cell growth and proliferation (51–53). Another cancer-specific metabolic feature is the accumulation of oncometabolites due to mutations in metabolic enzymes. L- or D-2-hydroxyglutarate (L- or D2-HG) is a well-known oncometabolite that promotes tumor growth by regulating DNA and histone modifying enzymes (54). Finally, malignancies are often characterized by unusually high concentrations of extracellular ATP and adenosine, hypoxia and by large quantities of ROS in poorly vascularized regions (52, 55). The above mentioned metabolic changes and stressors do not only affect tumor cells, but also reach stromal cells residing in the TME. Here we will describe how these metabolic cues affect DCs.
Nutrient Starvation
The excessive utilization of carbohydrates, amino acids and lipids by cancer cells results in a limited nutrient supply for cells residing in the TME. Although there are several seminal papers showing how nutrient limitation in the TME impairs CD8+ T cell function, there are few studies that have directly interrogated the contribution of nutrient starvation to the known suppressive effects of the TME on DCs (56, 57). Initially upon entering of DCs into the TME, one could imagine that TADCs may be able to utilize internal stores of glycogen and lipids to support the metabolic demands for their survival and immunogenic activation (19, 41). However, sustained limited access to glucose may impair metabolic rewiring and thereby DC maturation and migration to tumor draining lymph nodes (16, 24, 26). Likewise, based on in vitro studies, as discussed in section Amino Acids, it stands to reason that also insufficient access to amino acids may compromise TADC function by affecting mitochondrial respiration, redox homeostasis and antigen presenting capacity (33, 36, 39). However, to date no studies have directly addressed this. On the other hand, the effects of lipids on the function of TADCs have been studied more extensively and hence will be discussed in a separate section.
Tolerogenic properties of DCs have been linked to increased FAO and mitochondrial respiration (18, 58). These processes are both stimulated by activation of AMPK, an energy-sensing enzyme that is activated under nutrient limiting conditions (59). AMPK has been shown to be inactivated in DCs upon LPS-induced activation (16). Conversely, in TADCs of mice inoculated with MC38 colon adenocarcinoma cells, activation of LKB1, one of the main activating kinases of AMPK, was elevated (60). Hence, the nutrient-poor TME may boost AMPK signaling in DCs to induce catabolic metabolism that favors tolerogenic properties. Although no study to date has examined it directly, it is likely that limited nutrient access in the TME contributes to immune suppression of DCs (Table 1). Addressing the influence of tumors with different bioenergetic profiles on DC activation in vivo and in situ will provide more insights into the effects of nutrient deprivation on DC-driven immune suppression.
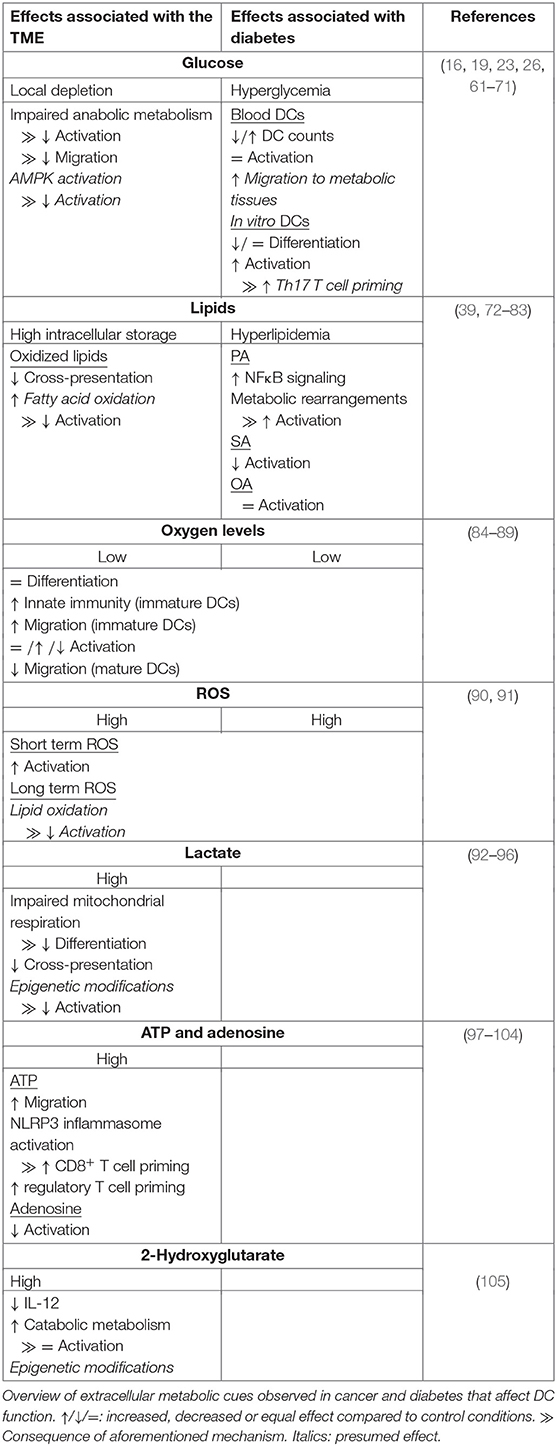
Table 1. Metabolic determinants from pathological environments that influence DC function.
Lipid Accumulation in Tumor-Associated DCs
Although tumor cells are generally characterized by high lipid uptake, TADCs can also take up large amounts of FAs from the TME (106). Acquisition of lipids by TADCs is facilitated by the upregulation of genes involved in lipid uptake, including lipoprotein lipase (LPL), fatty acid binding protein 4 (FABP4) and macrophage scavenger receptor (Msr1). The lipids are stored in large lipid droplets, which are associated with a reduced capacity to activate T cells (72–74). As discussed earlier, high lipid content in hepatic DCs is associated with higher immunogenicity and LPS stimulation of BMDCs stimulates lipid droplet formation (23, 41). However, in contrast to lipid droplets of these DCs, lipid droplets from TADCs contain high levels of oxidized polyunsaturated FAs. These oxidized FAs cause accumulation of peptide-bound MHC-I complexes in late endosomes and lysosomes via capturing of heat shock protein 70, an important mediator of cross-presentation (75, 76). This limits cross-presentation and thereby priming of cytotoxic T cell responses (Table 1). Given the importance of FAO in supporting a tolerogenic phenotype by DCs it is tempting to speculate that perhaps oxidation of these lipids in the mitochondria contributes to DC tolerogenicity within the TME (Table 1) (18, 58). What signals trigger the initial increase in lipid uptake by DCs in the TME remains to be determined.
Hypoxia
Rapid tumor growth results in poorly vascularized regions where oxygen supply is limited. The main metabolic response to hypoxia is stabilization of HIF1α and subsequent activation of glycolysis, which can also occur independently from HIF1α activation during DC activation (16, 17, 107). This may explain why some studies did not find changes in expression of maturation markers or T cell priming capacity when moDCs were stimulated with LPS after differentiation in a 1% O2 hypoxic chamber (Table 1) (84, 89). Also differentiation itself of moDCs is mostly unaffected by hypoxic conditions (84–86, 89). Nonetheless there is also some evidence that 1% oxygen can impair LPS-induced moDC maturation and T cell priming potential (Table 1) (85, 87). In addition, hypoxia impaired migration of in vitro cultured LPS-treated moDCs and human primary myeloid DCs. Mouse CD34+-derived myeloid DCs injected in the footpad of mice after they were treated with LPS and deferoxamine (DFX), a drug that mimics hypoxia, showed reduced migration to the draining lymph node compared to untreated cells, indicating that hypoxia reduces migration both in vitro and in vivo (87, 89). Interestingly, enhanced expression of migratory genes was found in immature moDCs cultured under low oxygen conditions (86). This could indicate that the hypoxic TME has immunosuppressive effects via capturing mature DCs and elimination of immature DCs. Interestingly, immature moDCs cultured under hypoxic conditions had increased expression of genes involved in sensing chemotactic signals from pro-inflammatory sites and induced secretion of chemotactic factors that attract neutrophils (84, 85, 87). Additionally, murine myeloid DCs treated with DFX increased local leukocyte infiltration in vivo (87). Altogether these data obtained from moDCs, may indicate that an oxygen-poor environment triggers DCs to boost innate rather than adaptive immune responses. However, contrary to human moDCs, murine BMDCs cultured in hypoxic conditions enhanced expression of costimulatory molecules, pro-inflammatory cytokine secretion and T cell proliferation upon LPS stimulation (Table 1) (88). Additional studies are needed to determine whether these discrepancies are due to inherent differences between human and murine DCs in their response to hypoxia or caused by differences in experimental setup. In addition, the metabolic context in which DCs are exposed to hypoxia may also affect how DCs respond. For instance, under nutrient replete conditions hypoxia may not compromise DC function, such as in lymph nodes in which hypoxic region have been described (108). In contrast, under pathological conditions, such as in the TME where hypoxia may be also accompanied by nutrient restriction, hypoxia may have anti-inflammatory effects on DCs. Studies addressing the effects of hypoxia on DC biology particularly in vivo during homeostasis as well as in pathological settings are needed to fully understand the role of oxygen availability on DC function in situ.
Oxidative Stress
The main sources of ROS in tumor cells are dysfunctional mitochondria and NADPH oxidases. This is further enhanced by intracellular ROS production of stromal cells, as a consequence of the metabolic alterations within the TME (109, 110). Intracellular ROS production in DCs during an immune response can have both pro-inflammatory and anti-inflammatory effects, via modulation of cross-presentation and of signaling pathways (39, 61, 77, 110–112). In general, extracellular ROS seems to have a pro-inflammatory effect, although data is limited (Table 1). Treatment of immature moDCs with hydrogen peroxide enhanced maturation and their capacity to induce T cell proliferation upon LPS stimulation (90). The inflammatory response of primary DCs to the malaria parasite Plasmodium falciparum also increased upon exposure to cells treated with xanthine oxidase, a malaria-induced enzyme that increases extracellular ROS levels (91). However, while transient ROS exposure following DC activation may have pro-inflammatory effects, what the functional consequences are of chronic ROS exposure, a situation DCs presumably have to deal within the TME, remains unclear. Possibly, the highly oxidized lipids that are found in TADCs, are one of the byproducts of chronic ROS exposure (113). Through this mechanism long-term ROS exposure in the TME could lead to impaired DC immunogenicity. Studies addressing the functional consequences of transient vs. chronic ROS exposure as well as of the different types of ROS on DCs, will be required to better define what role tumor-associated ROS and oxidative stress play in DC function in the TME.
Lactate
Tumor cells are known for the Warburg effect, which goes along with secretion of high levels of lactate. Lactate has a major influence on the immune-priming efficiency of DCs. Lactate derived from tumor spheroids, mesenchymal stromal cells or endogenously produced, affects the differentiation and maturation of moDCs. High concentrations of lactate reduce the differentiation capacity of moDCs, as higher numbers of monocyte like CD14+/CD1a− cells were detected at the end of the cultures (92–94). This was accompanied by a lactate-dependent reduction of oxidative phosphorylation, but enhanced respiratory capacity in immature moDCs (94). Also maturation of DCs is affected by high lactate levels, reflected by lower levels of maturation markers, an increase in immunosuppressive cytokine secretion, a decrease in pro-inflammatory cytokine secretion and reduced ability to induce T cell proliferation (92, 93, 95). The latter can be caused by detrimental effects of lactate on cross presentation. Using tumor conditioned Flt3L-DCs stimulated with CpG/PolyI:C and OVA-peptide, it was found that high lactate concentrations accelerate antigen processing via lowering the endosomal pH, resulting in impaired preservation of MHC-I epitopes. Thus, high concentrations of lactate in the local environment of differentiating or maturing DCs induces tolerance in DCs, via altering metabolism and antigen processing (Table 1). Extracellular lactate can mediate its anti-inflammatory function via binding to lactate receptor Gi-protein–coupled receptor 81 (GPR81) as was recently shown in DCs derived from murine mammary gland tumors (114). Alternatively, lactate can enter the cells via monocarboxylate transporter 1 (MCT1) as was shown in moDCs (92). Intracellular lactate may also hamper the immune response via a recently discovered epigenetic modification termed lactylation. In M1 macrophages endogenously produced lactate promoted lactylation of lysine residues, thereby promoting M2-like gene expression (115). Whether histone lactylation is another immunosuppressive feature of lactate in DCs is an interesting question that warrants further study.
ATP and Adenosine
Whereas during homeostasis extracellular ATP levels are negligible, ATP is highly abundant in the TME where it functions as a signaling molecule that provokes inflammation (55, 116). It has been proposed that diffusion of ATP out of the TME recruits DCs to the TME, given that BDMCs treated with 500 uM ATP increased migratory speed (Table 1) (97, 98). Moreover, extracellular ATP released by tumor cells after chemotherapy can promote anti-tumor immunity via signaling through ATP-receptors P2RX7 on DCs, thereby activating the NLRP3 inflammasome, enhancing IL-1β secretion and boosting CD8+ T cell priming (99, 100). In contrast, moDCs co-cultured with acute myeloid leukemia cells treated with chemotherapy drugs displayed increased potency to expand regulatory T cells in an extracellular ATP-dependent manner (Table 1) (101). Hence, there is great value in understanding what factors determine the balance between the pro- and anti-inflammatory effects by extracellular ATP after chemotherapy, as it may be an important predictor for treatment efficacy.
Paradoxically, immunosuppressive nucleoside adenosine, derived from conversion of ATP by membrane-bound ectonucleosides CD39 and CD73, is also abundantly present in the TME (117, 118). Adenosine interacts with four different receptors, of which A2AR and A2BR are most highly expressed on immune cells (119). Irradiation of mouse breast tumors caused upregulation of CD73 expression in tumor cells and increased local adenosine concentrations. Anti-CD73 treatment enhanced cDC1 tumor infiltration, increased the antitumor T cell response and reduced tumor growth (102). Additionally, in mice in which adenosine receptor A2BR was selectively knocked out in CD11c+ DCs, the growth of B16-melanoma was delayed, supporting a role for adenosine signaling in rendering DCs immunosuppressive (103). Furthermore, LPS-stimulated BMDCs treated with adenosine analog NECA increased intracellular cAMP levels, which lowered secretion of IL-12 and TNF-α secretion and enhanced IL-10 release via protein kinase A (PKA) and exchange protein directly activated by cAMP (Epac) signaling (104). Overall most studies indicate that extracellular ATP enhances immunogenicity of DCs and anti-tumor immune responses, while adenosine does the opposite (Table 1) (98, 120). Shifting the balance in favor of ATP by blocking CD73, CD39 or adenosine receptors is therefore a promising immunotherapy.
2-Hydroxyglutarate
In various tumors the oncometabolite 2-HG accumulates, which in non-malignant tissues is found at low concentrations (54). 2-HG has been shown to contribute to immune suppression in the TME via anti-inflammatory effects on T cells (121, 122). Immature moDCs cultured for 24 h with LPS and 2-HG secreted reduced levels of IL-12, enhanced mitochondrial respiration and lowered lactate secretion, indicating that accumulation of 2-HG affects moDCs via metabolic reprogramming. However, the ability of DCs to induce T cell proliferation remained the same, suggesting that the 2-HG-induced metabolic rearrangement in DCs does not affect their T cell priming capacity (Table 1) (105). However, in this context 2-HG was added simultaneously with the TLR ligand, while in the TME immature DCs may reside in a 2-HG-rich environment without immediate activation. Long-term exposure to 2-HG may have a stronger effect on the immunogenic capacities of DCs, potentially via the changes in gene expression, given that 2-HG affects activity of DNA and histone modifying enzymes, but this remains to be determined (54).
Concluding Remarks
Thus far the effects of metabolic perturbations characteristic of the TME on DC biology have been primarily studied in in vitro systems using moDCs. However, we have still limited knowledge about the real contribution of those metabolic changes on the functional properties of conventional as well as inflammatory DCs residing in the TME in situ. Likewise, if and to what extent these different metabolic perturbations interact and synergize to affect the functional properties of DCs remains to be determined. For successful activation of the immune system via DC-based therapy it is important to know how DCs deal with these metabolic rearrangements in the TME. For instance, how do DCs respond to adjuvants in the metabolic context of the TME? Is there a way to make these cells less vulnerable to potential immunosuppressive metabolic cues from the TME? And once out of the immunosuppressive metabolic TME, how quickly can DCs regain immunogenic function, if at all possible? To answer these questions and to gain better understanding of the immunosuppressive effects of the metabolic TME on DCs, in depth characterization of the metabolic TME and DC phenotype in primary tumors will be key.
Effects of the Metabolic Environment on DCs in Diabetes
Interplay Between Metabolic Disturbances and Inflammation Leading to Diabetes
Not only nutrient deprivation, but also excessive amounts of nutrients can disturb immune homeostasis and DC function. A well-known example of a disease that is characterized by elevated concentrations of glucose and lipids is diabetes. The two main types of diabetes are type I and type II Diabetes Mellitus (T1D/T2D), both characterized by dysfunctional insulin regulation and subsequent hyperglycemia. While T1D develops as a consequence of an auto-immune reaction against beta-cells, common causes for T2D are aging and obesity. Obesity causes hyperglycemia, elevated levels of free fatty acids, hypoxia, oxidative stress and an imbalance in many other metabolites, hormones and cytokines (123–126). This causes a switch in the composition and phenotype of immune cells in metabolic tissues from an anti-inflammatory to a more pro-inflammatory profile and thereby induces chronic low-grade inflammation, which ultimately drives insulin resistance (127–129). cDCs and pDCs are among the immune cells present in adipose tissue and there is a clear correlation between insulin resistance and number of CD11c+ DCs present in adipose tissue (62, 130–132). Moreover, several studies have shown that in response to high-fat diet DCs present in murine adipose tissue transition from Th1- to Th17-priming cells, an inflammatory profile linked to the pathogenesis of diabetes (62, 63, 133).
We will here describe how DCs are affected by the metabolic changes in their environment and focus on hyperglycemia and elevated levels of free fatty acids. Oxidative stress and hypoxia are also major metabolic players in diabetes, but to our knowledge there is no data available looking at the effects of these conditions on DC function in diabetic context (125, 126). Hence, we refer to the previous section for the effects of oxygen deprivation and excessive ROS levels on DC function.
Hyperglycemia
Decreased insulin secretion by beta-cells and lowered sensitivity to insulin signaling reduces the uptake of glucose by cells, which subsequently results in elevated blood glucose levels. As glucose availability plays a major role in DC activation it is conceivable that this glucose imbalance affects DC function. Several studies addressed the effects of hyperglycemia on primary dendritic cells from blood. Both a reduction and an increase in myeloid and pDC counts in blood of patients with T1D and T2D have been reported (Table 1) (64–67, 71). Reduced counts seems to be stronger in patients with poor glycemic control (66). Pro- and anti-inflammatory cytokine secretion by DCs from diabetic patients was not altered following ex vivo TLR stimulation, indicating that high blood glucose levels do not directly affect the function of circulating DCs, but primarily their numbers (65, 68, 71). It should be noted that hyperglycemia is not the only (metabolic) difference in blood from diabetic patients and other factors may also influence DC frequencies and function. Since a study in mice showed that hyperglycemia does not influence CD11c+ DC differentiation in the bone marrow, it is unlikely that the decrease in circulating DCs is a consequence of impaired DC generation (69). Instead, lower numbers of circulating DCs are possibly a reflection of enhanced migration of DCs to metabolic tissues, where DCs are known to accumulate and contribute to the low-grade inflammation observed in metabolic tissues of T2D patients. As previously mentioned DCs in obese adipose tissue drive a Th17 inflammatory response (62, 63). Interestingly, moDCs exposed to 5.5, 15, and 30 mM glucose for 24 h increase CD83 and CD86 expression and secretion of IL-6 and IL-12 in a dose-dependent manner (61). IL-6 is involved in Th17 differentiation of naïve T cells and was also found to be highly secreted by CD11c+ DCs from obese adipose tissue, suggesting that high glucose levels in adipose tissue may contribute to conditioning DCs for Th17 priming (Table 1) (63, 134). However, in vivo data connecting glucose levels to DC function in metabolic tissues is currently lacking. In vitro generation of tolerogenic moDCs was less efficient with monocytes derived from T1D patients with poor glycemic control in comparison to patients who maintained glycemic control, supporting the hypothesis that a hyperglycemic environment promotes a more pro-inflammatory profile (135). On the other hand, moDCs derived from T2D donors compared to healthy control donors expressed lower levels of maturation markers (64). Moreover, moDC differentiation in high-glucose medium (25 mM) or media supplemented with sera from hyperglycemic T2D patients reduced the number of moDCs, expression of maturation markers and the capacity to induce T cell proliferation after LPS stimulation. In addition, glucose-rich micro-environments increase ROS production and promote activation of the p38 MAPK and Wnt/b-catenin pathways, which are associated with tolerogenic properties of DCs (70, 136–138). Together these in vitro studies may indicate that over-abundance of glucose drives monocyte differentiation toward less-proinflammatory DCs, while differentiated moDCs and potentially CD11c+ DCs residing in adipose tissue may become more immunogenic in a hyperglycemic environment (Table 1).
Free Fatty Acids
A cause and consequence of obesity and insulin resistance in T2D is the release of free fatty acids by expanding fat mass (124, 139). FAs are well-known regulators of the immune response. Polyunsaturated FAs (PUFAs) often have anti-inflammatory effects while many saturated fatty acids (SFAs) serve as pro-inflammatory molecules (140, 141). Examples of the latter are palmitic acid (PA) and stearic acid (SA), which together with unsaturated oleic acid (OA) are among the most abundant dysregulated FFAs in obese and T2D patients (123, 142). Especially PA is known for its pro-inflammatory effects and detrimental role in T2D pathogenesis (143). This is partly caused via its effects on DCs. PA in combination with LPS can enhance Th1-associated inflammation, which is driven by TLR4-dependent activation of the NFκB pathway and ROS in moDCs (77, 78, 144). PA also boosts inflammatory properties of activated BMDCs in a TLR4-independent manner, via inhibition of hexokinase (HK) during the late stages of metabolic reprogramming. This inhibition of HK and thereby glycolysis resulted in enhanced mitochondrial respiration, increased mitochondrial ROS levels, elevated activation of the unfolded protein response (UPR) and subsequent induction of IL-23 expression. UPR-dependent IL-23 expression was also confirmed in mice fed a high fat diet (39). BMDCs derived from obese mice additionally increased IL-1β secretion in a NLRP3 inflammasome-dependent manner following stimulation with PA (79). IL-1β and IL-23 are key cytokines involved in promoting Th17 responses and hence PA is a potential driver of insulin resistance (133). However, a direct causal link between enhanced pro-inflammatory cytokine secretion by DCs and Th17 induction in settings of FA exposure still needs to be established as for instance DCs isolated from human blood displayed a reduced capacity to prime T cell responses upon stimulation with PA, despite having increased IL-1β and TNF secretion (80). In contrast to PA, SA does not seem to affect DC function. SA treatment of LPS-stimulated moDCs did not affect expression of maturation markers nor the capacity to induce T cell proliferation (81). Although data on the effects of SA treatment of DCs is still limited, this appears to be different from what is known for macrophages, where SA and PA have been reported to have a similar pro-inflammatory effect (145, 146). OA is a mono-unsaturated FA that has beneficial effects on insulin resistance. In macrophages, this is partly mediated by counteracting the pro-inflammatory effects of SFA (143). Thus far, there is no data available indicating an anti-inflammatory role for OA in DCs. OA treatment had either no effect, or boosted a pro-inflammatory immune response, but has never been tested in combination with SFA stimulation (78, 80, 82, 83).
The balance in dietary intake of SFAs and PUFAs can have great influence on the clinical outcome of diabetes. A comparison of 6, 12, and 24% of SFA in the diet of mice without changing the total dietary fat contribution had a profound effect on macrophage function and insulin resistance, with 12% SFA as the greatest contributor to inflammation and insulin resistance (147). Human data is however thus far inconclusive about the beneficial effects of PUFA-rich and SFA-poor diets on glycemic control of T2D patients (148). Therefore, studies in humans and mice with a focus on the composition in dietary fat and profiling of DCs in metabolic tissues may tell us whether DCs contribute to PUFA-mediated protection against diabetes and/or SFA-mediated development of diabetes, potentially via a PA-induced Th17 response.
In conclusion, compared to other immune cells, there is still little known about the effects on DCs of the FFAs that are most abundant in obesity and T2D patients. While PA stimulation of DCs seems to have the expected pro-inflammatory effects, it is remains unclear if SA and OA influence insulin resistance via DCs (Table 1).
Concluding Remarks
T1D is characterized by an active immune response against beta-cells, while T2D is associated with chronic low-grade inflammation. Hence, it is perhaps somewhat surprising that ex vivo data indicate that hyperglycemia has minimal effect on the function of DCs, that most in vitro studies describe a tolerogenic effect of excessive glucose levels on DC differentiation, and that only in vitro mature DCs are likely to become more pro-inflammatory. Although currently it cannot be excluded that the latter observations may be due the use of in vitro model systems, such as in vitro generated moDCs that possibly cannot sufficiently mimic the metabolic alterations and glucose-rich environment that cDCs and pDCs are exposed to in vivo, it may in fact indicate that hyperglycemia is not a major driver of the pro-inflammatory profiles of DCs observed in diabetes and that other metabolic and/or immunological cues are more important (62, 130–132). This idea would be consistent with the fact that hyperglycemia is generally associated with impaired immune response against for example infections and tumors (149, 150). A better understanding of how overabundance of various nutrients may act in concert to modulate DC function and to thereby contribute to local inflammation in the context of diabetes will be important for identification of the pathways that lead to inflammation-driven insulin resistance that could be targeted to control diabetes.
Perspectives and Outlook
It is becoming evident that the metabolic micro-environment has a major influence on DC function and that disturbance of metabolic homeostasis can impact immune responses. We aimed to provide an overview of key metabolites that influence DC phenotype and function. Cancer and diabetes are examples of highly prevalent disorders in which metabolic homeostasis is disturbed, but many more pathologies are associated with dysregulated metabolism. Eating disorders alter nutrient availability, organ-specific pathologies, such as hepatic steatosis impair systemic metabolism and oxidative stress is associated with many diseases including atherosclerosis, cardiovascular diseases and neurodegenerative disorders. Hence, understanding the impact of nutrient availability on the function of immune cells, including DCs, is relevant for a broad range of diseases. Reprogramming the metabolic state of DCs by intervening with nutrient availability can be an effective way to control inflammation. This could be achieved by systemic approaches, including nutritional interventions which are commonly used to control inflammation (151). For instance, lowering caloric intake, by reducing fat and glucose content, can improve glycemic control and subsequently reduce diabetes-associated low-grade inflammation (152–154). Given the pro-inflammatory effects of high glucose and SFA concentration on tissue-associated DCs, it is reasonable to assume that dietary interventions that lead to normalization of glucose and SFA concentrations in the tissue that these cells reside in, will render them less pro-inflammatory, thereby contributing to reduction of local tissue inflammation and eventually improvement of metabolic homeostasis. Alternatively, molecular approaches that directly target cellular nutrient uptake or bioenergetic pathways can make DCs potentially less vulnerable to extracellular nutritional changes and interventions that target energy-sensing enzymes like AMPK can also control inflammation (155). In addition, targeting metabolism of non-immune cells can also have a beneficial effect on the metabolic micro-environment of DCs. For example, therapies that interfere with cancer metabolism to directly impair tumor growth could also have indirect anti-tumor effects by potentially creating a TME with higher nutrient availability that would be more permissive to effective anti-tumor immune responses (156).
Current studies addressing the effects of the metabolic micro-environment on DCs are mostly performed in vitro using human moDCs or murine BMDCs. While these studies have provided us important new insights into how nutrient availability can shape DC function, in vitro culture conditions often do not fully mimic the complexity and concentrations of various nutrients and metabolites these cells are exposed to in situ. For instance, In vitro-generated DCs are commonly cultured in media supplemented with 10% fetal calf serum (FCS) and glutamine. Serum is a source for lipids, vitamins, hormones, growth factors, and other compounds, but the exact amounts of these compounds are unknown, differ per batch and may not correspond with concentrations found in tissues that DCs reside in Yao and Asayama (157). Moreover, commonly used culture media, such as RPMI 1640 and DMEM contain supraphysiological levels of glucose (11 and 25 mM, respectively, vs. ~5.5 mM in situ) and lower levels of electrolytes including calcium and magnesium (158). The effects of nutrient availability on the function of DC subsets in a more physiological environment, with other metabolic and non-metabolic immunomodulatory signals around, need to be further evaluated, to be able to better assess what the in vivo contribution of the metabolic micro-environment is on the functional properties of DCs. To tackle this issue, there have been recent efforts to develop human plasma-like, physiological medium, which contains components, such as amino acids, metabolites, salt ions, and vitamins that are absent from standard media and holds physiologically relevant concentrations of common media components. To minimize the effects of FCS-derived components, medium can be supplemented with either a low percentage (2.5%) of FCS or dialyzed FCS (159–161). Studies using these media found enhanced in vitro T cell activation and increased biological similarity between cultured breast cancer cells and primary mammary tumors, providing promising first evidence that these types of media could be used to better mimic physiological setting in vitro than classically used culture media (160, 161). These tools will likely also be key to further the field of DC metabolism and to better delineate the interplay between DC function and extra- or intracellular metabolism. In addition, various novel mass-spectrometry, high dimensional flow cytometry and transcriptomics platforms have been developed in recent years that enable one to assess metabolic profiles in tissues at high spatial resolution as well as to characterize metabolic and immunological phenotypes of immune cells present in those tissues at the single cell level. These novel techniques will no doubt greatly improve our understanding of how nutrients shape DC function in situ.
Even though many open questions remain, recent work has revealed profound effects of the metabolic micro-environment on DC function in health and disease, which may pave the way for developing DC metabolism-based approaches to treat metabolic and inflammatory disorders.
Author Contributions
EB wrote the manuscript and prepared the figure. BE supervised and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This review was supported by an LUMC fellowship awarded to BE.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
2. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol. (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
3. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. (2018) 154:3–20. doi: 10.1111/imm.12888
4. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus iuterleukin 4 and downregulated by tumor necrosis factor A. J Exp Med. (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
5. Schultze JL, Aschenbrenner AC. Systems immunology allows a new view on human dendritic cells. Semin Cell Dev Biol. (2019) 86:15–23. doi: 10.1016/j.semcdb.2018.02.017
6. Sander J, Schmidt SV, Cirovic B, McGovern N, Papantonopoulou O, Hardt AL, et al. Cellular differentiation of human monocytes is regulated by time-dependent interleukin-4 signaling and the transcriptional regulator NCOR2. Immunity. (2017) 47:1051–66 e12. doi: 10.1016/j.immuni.2017.11.024
7. Inaba BK, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
8. Guo X, Zhou Y, Wu T, Zhu X, Lai W, Wu L. Generation of mouse and human dendritic cells in vitro. J Immunol Methods. (2016) 432:24–9. doi: 10.1016/j.jim.2016.02.011
9. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. (2005) 174:6592–7. doi: 10.4049/jimmunol.174.11.6592
10. Joffre O, Nolte MA, Spörri R, Reis e Sous C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. (2009) 227:234–47. doi: 10.1111/j.1600-065X.2008.00718.x
11. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
12. Tiberio L, Del Prete A, Schioppa T, Sozio F, Bosisio D, Sozzani S. Chemokine and chemotactic signals in dendritic cell migration. Cell Mol Immunol. (2018) 15:346–52. doi: 10.1038/s41423-018-0005-3
13. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
14. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
15. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun. (2019) 10:1. doi: 10.1038/s41467-019-10015-4
16. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
17. Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. (2012) 120:1422–31. doi: 10.1182/blood-2012-03-419747
18. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol. (2015) 194:5174–86. doi: 10.4049/jimmunol.1303316
19. Thwe PM, Pelgrom L, Cooper R, Beauchamp S, Reisz JA, D'Alessandro A, et al. Cell-intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab. (2017) 26:558–67 e5. doi: 10.1016/j.cmet.2017.08.012
20. Basit F, de Vries IJM. Dendritic cells require PINK1-mediated phosphorylation of BCKDE1alpha to promote fatty acid oxidation for immune function. Front Immunol. (2019) 10:2386. doi: 10.3389/fimmu.2019.02386
21. Basit F, Mathan T, Sancho D, de Vries IJM. Human dendritic cell subsets undergo distinct metabolic reprogramming for immune response. Front Immunol. (2018) 9:2489. doi: 10.3389/fimmu.2018.02489
22. Bajwa G, DeBerardinis RJ, Shao B, Hall B, Farrar JD, Gill MA. Cutting edge: critical role of glycolysis in human plasmacytoid dendritic cell antiviral responses. J Immunol. (2016) 196:2004–9. doi: 10.4049/jimmunol.1501557
23. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
24. Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, et al. Glucose represses dendritic cell-induced T cell responses. Nat Commun. (2017) 8:15620. doi: 10.1038/ncomms15620
25. Randolph GJ, Ochando J, Partida-Sanchez S. Migration of dendritic cell subsets and their precursors. Annu Rev Immunol. (2008) 26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254
26. Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY, et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun. (2018) 9:2463. doi: 10.1038/s41467-018-04804-6
27. Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, et al. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol. (2012) 189:2151–8. doi: 10.4049/jimmunol.1103741
28. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. (2018) 28:463–75 e4. doi: 10.1016/j.cmet.2018.08.012
29. Marquez S, Fernandez JJ, Teran-Cabanillas E, Herrero C, Alonso S, Azogil A, et al. Endoplasmic reticulum stress sensor IRE1alpha enhances IL-23 expression by human dendritic cells. Front Immunol. (2017) 8:639. doi: 10.3389/fimmu.2017.00639
30. Thwe PM, Amiel E. The role of nitric oxide in metabolic regulation of dendritic cell immune function. Cancer Lett. (2018) 412:236–42. doi: 10.1016/j.canlet.2017.10.032
31. Zhang D, Chia C, Jiao X, Jin W, Kasagi S, Wu R, et al. D-mannose induces regulatory T cells and suppresses immunopathology. Nat Med. (2017) 23:1036–45. doi: 10.1038/nm.4375
32. Kakazu E, Ueno Y, Kondo Y, Fukushima K, Shiina M, Inoue J, et al. Branched chain amino acids enhance the maturation and function of myeloid dendritic cells ex vivo in patients with advanced cirrhosis. Hepatology. (2009) 50:1936–45. doi: 10.1002/hep.23248
33. Kakazu E, Kondo Y, Kogure T, Ninomiya M, Kimura O, Ueno Y, et al. Plasma amino acids imbalance in cirrhotic patients disturbs the tricarboxylic acid cycle of dendritic cell. Sci Rep. (2013) 3:3459. doi: 10.1038/srep03459
34. Kakazu E, Kanno N, Ueno Y, Shimosegawa T. Extracellular branched-chain amino acids, especially valine, regulate maturation and function of monocyte-derived dendritic cells. J Immunol. (2007) 179:7137–46. doi: 10.4049/jimmunol.179.10.7137
35. Sun X, Zemel MB. Leucine modulation of mitochondrial mass and oxygen consumption in skeletal muscle cells and adipocytes. Nutr Metab (Lond). (2009) 6:26. doi: 10.1186/1743-7075-6-26
36. D'Angelo JA, Dehlink E, Platzer B, Dwyer P, Circu ML, Garay J, et al. The cystine/glutamate antiporter regulates dendritic cell differentiation and antigen presentation. J Immunol. (2010) 185:3217–26. doi: 10.4049/jimmunol.1001199
37. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. (2007) 104:19345–50. doi: 10.1073/pnas.0709747104
38. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. (2011) 481:380–4. doi: 10.1038/nature10602
39. Mogilenko DA, Haas JT, L'Homme L, Fleury S, Quemener S, Levavasseur M, et al. Metabolic and innate immune cues merge into a specific inflammatory response via the UPR. Cell. (2019) 177:1201–16 e19. doi: 10.1016/j.cell.2019.06.017
40. Kratchmarov R, Viragova S, Kim MJ, Rothman NJ, Liu K, Reizis B, et al. Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol Cell Biol. (2018) 96:863–71. doi: 10.1111/imcb.12040
41. Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. (2012) 143:1061–72. doi: 10.1053/j.gastro.2012.06.003
42. Bougneres L, Helft J, Tiwari S, Vargas P, Chang BH, Chan L, et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity. (2009) 31:232–44. doi: 10.1016/j.immuni.2009.06.022
43. Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity. (2016) 44:1325–36. doi: 10.1016/j.immuni.2016.06.006
44. Mattacks CA, Sadler D, Pond CM. Site-specific differences in fatty acid composition of dendritic cells and associated adipose tissue in popliteal depot, mesentery, and omentum and their modulation by chronic inflammation and dietary lipids. Lymphat Res Biol. (2004) 2:107–29. doi: 10.1089/lrb.2004.2.107
45. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
46. Giovanelli P, Sandoval TA, Cubillos-Ruiz JR. Dendritic cell metabolism and function in tumors. Trends Immunol. (2019) 40:699–718. doi: 10.1016/j.it.2019.06.004
47. Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. (2012) 209:495–506. doi: 10.1084/jem.20111413
48. Monjazeb AM, Zamora AE, Grossenbacher SK, Mirsoian A, Sckisel GD, Murphy WJ. Immunoediting and antigen loss: overcoming the achilles heel of immunotherapy with antigen non-specific therapies. Front Oncol. (2013) 3:197. doi: 10.3389/fonc.2013.00197
49. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. (2002) 3:991–8. doi: 10.1038/ni1102-991
50. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
51. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (2018) 38:1. doi: 10.1186/s40880-018-0301-4
52. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
53. Vettore L, Westbrook RL, Tennant DA. New aspects of amino acid metabolism in cancer. Br J Cancer. (2020) 122:150–56. doi: 10.1038/s41416-019-0620-5
54. Losman JA, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. (2013) 27:836–52. doi: 10.1101/gad.217406.113
55. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS ONE. (2008) 3:e2599. doi: 10.1371/journal.pone.0002599
56. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
57. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
58. Zhao F, Xiao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, et al. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. (2018) 48:147–60.e7. doi: 10.1016/j.immuni.2017.12.004
59. Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. (2017) 66:789–800. doi: 10.1016/j.molcel.2017.05.032
60. Wang Y, Du X, Wei J, Long L, Tan H, Guy C, et al. LKB1 orchestrates dendritic cell metabolic quiescence and anti-tumor immunity. Cell Res. (2019) 29:391–405. doi: 10.1038/s41422-019-0157-4
61. Lu H, Yao K, Huang D, Sun A, Zou Y, Qian J, et al. High glucose induces upregulation of scavenger receptors and promotes maturation of dendritic cells. Cardiovasc Diabetol. (2013) 12:80. doi: 10.1186/1475-2840-12-80
62. Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. (2012) 61:2238–47. doi: 10.2337/db11-1274
63. Chen Y, Tian J, Tian X, Tang X, Rui K, Tong J, et al. Adipose tissue dendritic cells enhances inflammation by prompting the generation of Th17 cells. PLoS ONE. (2014) 9:e92450. doi: 10.1371/journal.pone.0092450
64. Musilli C, Paccosi S, Pala L, Gerlini G, Ledda F, Mugelli A, et al. Characterization of circulating and monocyte-derived dendritic cells in obese and diabetic patients. Mol Immunol. (2011) 49:234–8. doi: 10.1016/j.molimm.2011.08.019
65. Vuckovic S, Withers G, Harris M, Khalil D, Gardiner D, Flesch I, et al. Decreased blood dendritic cell counts in type 1 diabetic children. Clin Immunol. (2007) 123:281–8. doi: 10.1016/j.clim.2007.03.002
66. Seifarth CC, Hinkmann C, Hahn EG, Lohmann T, Harsch IA. Reduced frequency of peripheral dendritic cells in type 2 diabetes. Exp Clin Endocrinol Diabetes. (2008) 116:162–6. doi: 10.1055/s-2007-990278
67. Peng R, Li Y, Brezner K, Litherland S, Clare-Salzler MJ. Abnormal peripheral blood dendritic cell populations in type 1 diabetes. Ann N Y Acad Sci. (2003) 1005:222–25. doi: 10.1196/annals.1288.031
68. Nam HW, Cho YJ, Lim JA, Kim SJ, Kim H, Sim SY, et al. Functional status of immune cells in patients with long-lasting type 2 diabetes mellitus. Clin Exp Immunol. (2018) 194:125–36. doi: 10.1111/cei.13187
69. Loomans CJ, van Haperen R, Duijs JM, Verseyden C, de Crom R, Leenen PJ, et al. Differentiation of bone marrow-derived endothelial progenitor cells is shifted into a proinflammatory phenotype by hyperglycemia. Mol Med. (2009) 15:152–9. doi: 10.2119/molmed.2009.00032
70. Gilardini Montani MS, Granato M, Cuomo L, Valia S, Di Renzo L, D'Orazi G, et al. High glucose and hyperglycemic sera from type 2 diabetic patients impair DC differentiation by inducing ROS and activating Wnt/beta-catenin and p38 MAPK. Biochim Biophys Acta. (2016) 1862:805–13. doi: 10.1016/j.bbadis.2016.01.001
71. Blank SE, Johnson EC, Weeks DK, Wysham CH. Circulating dendritic cell number and intracellular TNF-alpha production in women with type 2 diabetes. Acta Diabetol. (2012) 49:25–32. doi: 10.1007/s00592-010-0190-8
72. Gao F, Liu C, Guo J, Sun W, Xian L, Bai D, et al. Radiation-driven lipid accumulation and dendritic cell dysfunction in cancer. Sci Rep. (2015) 5:9613. doi: 10.1038/srep09613
73. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
74. Gardner JK, Mamotte CD, Patel P, Yeoh TL, Jackaman C, Nelson DJ. Mesothelioma tumor cells modulate dendritic cell lipid content, phenotype and function. PLoS ONE. (2015) 10:e0123563. doi: 10.1371/journal.pone.0123563
75. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. (2017) 8:2122. doi: 10.1038/s41467-017-02186-9
76. Cao W, Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol. (2014) 192:2920–31. doi: 10.4049/jimmunol.1302801
77. Nicholas DA, Zhang K, Hung C, Glasgow S, Aruni AW, Unternaehrer J, et al. Palmitic acid is a toll-like receptor 4 ligand that induces human dendritic cell secretion of IL-1beta. PLoS ONE. (2017) 12:e0176793. doi: 10.1371/journal.pone.0176793
78. Stelzner K, Herbert D, Popkova Y, Lorz A, Schiller J, Gericke M, et al. Free fatty acids sensitize dendritic cells to amplify TH1/TH17-immune responses. Eur J Immunol. (2016) 46:2043–53. doi: 10.1002/eji.201546263
79. Reynolds CM, McGillicuddy FC, Harford KA, Finucane OM, Mills KH, Roche HM. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol Nutr Food Res. (2012) 56:1212–22. doi: 10.1002/mnfr.201200058
80. Miyake T, Akbar SMF, Yoshida O, Chen S, Hiasa Y, Matsuura B, et al. Impaired dendritic cell functions disrupt antigen-specific adaptive immune responses in mice with nonalcoholic fatty liver disease. J Gastroenterol. (2010) 45:859–67. doi: 10.1007/s00535-010-0218-4
81. Wang H, Hao Q, Li QR, Yan XW, Ye S, Li YS, et al. Omega-3 polyunsaturated fatty acids affect lipopolysaccharide-induced maturation of dendritic cells through mitogen-activated protein kinases p38. Nutrition. (2007) 23:474–82. doi: 10.1016/j.nut.2007.04.002
82. Carlsson JA, Wold AE, Sandberg AS, Ostman SM. The polyunsaturated fatty acids arachidonic acid and docosahexaenoic acid induce mouse dendritic cells maturation but reduce T-cell responses in vitro. PLoS ONE. (2015) 10:e0143741. doi: 10.1371/journal.pone.0143741
83. Zeyda M, Saemann MD, Stuhlmeier KM, Mascher DG, Nowotny PN, Zlabinger GJ, et al. Polyunsaturated fatty acids block dendritic cell activation and function independently of NF-kappaB activation. J Biol Chem. (2005) 280:14293–301. doi: 10.1074/jbc.M410000200
84. Elia AR, Cappello P, Puppo M, Fraone T, Vanni C, Eva A, et al. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J Leukoc Biol. (2008) 84:1472–82. doi: 10.1189/jlb.0208082
85. Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W, et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology. (2009) 128:e237–49. doi: 10.1111/j.1365-2567.2008.02954.x
86. Ricciardi A, Elia AR, Cappello P, Puppo M, Vanni C, Fardin P, et al. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res. (2008) 6:175–85. doi: 10.1158/1541-7786.MCR-07-0391
87. Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, et al. Divergent effects of hypoxia on dendritic cell functions. Blood. (2008) 112:3723–34. doi: 10.1182/blood-2008-02-142091
88. Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, et al. Hypoxia and hypoxia-inducible factor-1 modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. (2008) 180:4697–705. doi: 10.4049/jimmunol.180.7.4697
89. Zhao W, Darmanin S, Fu Q, Chen J, Cui H, Wang J, et al. Hypoxia suppresses the production of matrix metalloproteinases and the migration of humanmonocyte-derived dendritic cells. Eur J Immunol. (2005) 35:3468–77. doi: 10.1002/eji.200526262
90. Rutault K, Alderman C, Chain BM, Katz DR. Reactive oxygen species activate human peripheral blood dendritic cells. Free Radic Biol Med. (1999) 26:232–8. doi: 10.1016/S0891-5849(98)00194-4
91. Gotz A, Ty MC, Rodriguez A. Oxidative stress enhances dendritic cell responses to Plasmodium falciparum. Immunohorizons. (2019) 3:511–8. doi: 10.4049/immunohorizons.1900076
92. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. (2006) 107:2013–21. doi: 10.1182/blood-2005-05-1795
93. Nasi A, Fekete T, Krishnamurthy A, Snowden S, Rajnavolgyi E, Catrina AI, et al. Dendritic cell reprogramming by endogenously produced lactic acid. J Immunol. (2013) 191:3090–9. doi: 10.4049/jimmunol.1300772
94. Selleri S, Bifsha P, Civini S, Pacelli C, Dieng MM, Lemieux W, et al. Human mesenchymal stromal cell-secreted lactate induces M2macrophage differentiation by metabolic reprogramming. Oncotarget. (2016) 7:30193–210. doi: 10.18632/oncotarget.8623
95. Puig-Kroger A, Pello OM, Muniz-Pello O, Selgas R, Criado G, Bajo MA, et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: effect of lactate and glucose-degradation products. J Leukoc Biol. (2003) 73:482–92. doi: 10.1189/jlb.0902451
96. Caronni N, Simoncello F, Stafetta F, Guarnaccia C, Ruiz-Moreno JS, Opitz B, et al. Downregulation of membrane trafficking proteins and lactate conditioning determine loss of dendritic cell function in lung cancer. Cancer Res. (2018) 78:1685–99. doi: 10.1158/0008-5472.Can-17-1307
97. Sáez PJ, Vargas P, Shoji KF, Harcha PA, Lennon-Duménil AM, Sáez JC. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci Signal. (2017) 10:506. doi: 10.1126/scisignal.aah7107
98. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
99. Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. (2010) 70:855–8. doi: 10.1158/0008-5472.CAN-09-3566
100. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. doi: 10.1038/nm.2028
101. Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, et al. ATP release from chemotherapy-treated dying leukemia cells elicits an immune suppressive effect by increasing regulatory T cells and tolerogenic dendritic cells. Front Immunol. (2017) 8:1918. doi: 10.3389/fimmu.2017.01918
102. Wennerberg E, Spada S, Rudqvist NP, Lhuillier C, Gruber S, Chen Q, et al. CD73 blockade promotes dendritic cell infiltration of irradiated tumors and tumor rejection. Cancer Immunol Res. (2020) 8:465–78. doi: 10.1158/2326-6066.CIR-19-0449
103. Chen S, Akdemir I, Fan J, Linden J, Zhang B, Cekic C. The expression of adenosine A2B receptor on antigen presenting cells suppresses CD8+ T cell responses and promotes tumor growth. Cancer Immunol Res. (2020) 8:1064–74. doi: 10.1158/2326-6066.CIR-19-0833
104. Kayhan M, Koyas A, Akdemir I, Savas AC, Cekic C. Adenosine receptor signaling targets both PKA and Epac pathways to polarize dendritic cells to a suppressive phenotype. J Immunol. (2019) 203:3247–55. doi: 10.4049/jimmunol.1900765
105. Ugele I, Cárdenas-Conejo Z, Hammon K, Wehrstein M, Bruss C, Peter K, et al. D-2-hydroxyglutarate and L-2-hydroxyglutarate inhibit IL-12 secretion by human monocyte-derived dendritic cells. Int J Mol Sci. (2019) 20:3. doi: 10.3390/ijms20030742
106. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. (2019) 122:4–22. doi: 10.1038/s41416-019-0650-z
107. Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1alpha in cancer. Cancer Sci. (2019) 110:1510–7. doi: 10.1111/cas.13990
108. Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. (2017) 17:774–85. doi: 10.1038/nri.2017.103
109. Policastro LL, Ibañez IL, Notcovich C, Duran HA, Podhajcer OL. The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid Redox Signal. (2013) 19:854–95. doi: 10.1089/ars.2011.4367
110. Paardekooper LM, Vos W, van den Bogaart G. Oxygen in the tumor microenvironment: effects on dendritic cell function. Oncotarget. (2019) 10:883–96. doi: 10.18632/oncotarget.26608
111. Oberkampf M, Guillerey C, Mouries J, Rosenbaum P, Fayolle C, Bobard A, et al. Mitochondrial reactive oxygen species regulate the induction of CD8(+) T cells by plasmacytoid dendritic cells. Nat Commun. (2018) 9:2241. doi: 10.1038/s41467-018-04686-8
112. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol. (2015) 195:2624–32. doi: 10.4049/jimmunol.1501006
113. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
114. Brown TP, Bhattacharjee P, Ramachandran S, Sivaprakasam S, Ristic B, Sikder MOF, et al. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene. (2020) 39:3292–304. doi: 10.1038/s41388-020-1216-5
115. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
116. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
117. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. (1997) 57:2602–5.
118. Longhi MS, Robson SC, Bernstein SH, Serra S, Deaglio S. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J Mol Med (Berl). (2013) 91:165–72. doi: 10.1007/s00109-012-0991-z
119. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. (2016) 16:177–92. doi: 10.1038/nri.2016.4
120. Silva-Vilches C, Ring S, Mahnke K. ATP and its metabolite adenosine as regulators of dendritic cell activity. Front Immunol. (2018) 9:2581. doi: 10.3389/fimmu.2018.02581
121. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. (2018) 24:1192–203. doi: 10.1038/s41591-018-0095-6
122. Bottcher M, Renner K, Berger R, Mentz K, Thomas S, Cardenas-Conejo ZE, et al. D-2-hydroxyglutarate interferes with HIF-1alpha stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology. (2018) 7:e1445454. doi: 10.1080/2162402X.2018.1445454
123. Ma Q, Li Y, Wang M, Tang Z, Wang T, Liu C, et al. Progress in metabonomics of type 2 diabetes mellitus. Molecules. (2018) 23:7. doi: 10.3390/molecules23071834
124. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. (2008) 37:635–46. doi: 10.1016/j.ecl.2008.06.007
125. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. (2007) 56:901–11. doi: 10.2337/db06-0911
126. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. (2010) 107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545
127. Ferrante AW Jr. The immune cells in adipose tissue. Diabetes Obes Metab. (2013) 15:34–8. doi: 10.1111/dom.12154
128. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/JCI200319451
129. Coope A, Torsoni AS, Velloso LA. Metabolic and inflammatory pathways on the pathogenesis of type 2 diabetes. Eur J Endocrinol. (2016) 174:R175–87. doi: 10.1530/EJE-15-1065
130. Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. (2008) 8:301–9. doi: 10.1016/j.cmet.2008.08.015
131. Mraz M, Cinkajzlova A, Klouckova J, Lacinova Z, Kratochvilova H, Lips M, et al. Dendritic cells in subcutaneous and epicardial adipose tissue of subjects with type 2 diabetes, obesity, and coronary artery disease. Mediators Inflamm. (2019) 2019:5481725. doi: 10.1155/2019/5481725
132. Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes. (2012) 61:2330–9. doi: 10.2337/db11-1523
133. Abdel-Moneim A, Bakery HH, Allam G. The potential pathogenic role of IL-17/Th17 cells in both type 1 and type 2 diabetes mellitus. Biomed Pharmacother. (2018) 101:287–92. doi: 10.1016/j.biopha.2018.02.103
134. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
135. Danova K, Grohova A, Strnadova P, Funda DP, Sumnik Z, Lebl J, et al. Tolerogenic dendritic cells from poorly compensated type 1 diabetes patients have decreased ability to induce stable antigen-specific T cell hyporesponsiveness and generation of suppressive regulatory T cells. J Immunol. (2017) 198:729–40. doi: 10.4049/jimmunol.1600676
136. Macdougall CE, Wood EG, Loschko J, Scagliotti V, Cassidy FC, Robinson ME, et al. Visceral adipose tissue immune homeostasis is regulated by the crosstalk between adipocytes and dendritic cell subsets. Cell Metab. (2018) 27:588–601. doi: 10.1016/j.cmet.2018.02.007
137. Oderup C, LaJevic M, Butcher EC. Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J Immunol. (2013) 190:6126–34. doi: 10.4049/jimmunol.1203002
138. Cirone M, Di Renzo L, Trivedi P, Lucania G, Borgia G, Frati L, et al. Dendritic cell differentiation blocked by primary effusion lymphoma-released factors is partially restored by inhibition of P38 MAPK. Int J Immunopathol Pharmacol. (2010) 23:1079–86. doi: 10.1177/039463201002300412
139. Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care. (2007) 10:142–8. doi: 10.1097/MCO.0b013e328042ba90
140. Weatherill AR, Lee JY, Zhao L, Lemay DG, Youn HS, Hwang DH. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol. (2005) 174:5390–7. doi: 10.4049/jimmunol.174.9.5390
141. Fritsche KL. The science of fatty acids and inflammation. Adv Nutr. (2015) 6:293S−301S. doi: 10.3945/an.114.006940
142. Staiger H, Staiger K, Stefan N, Wahl HG, Machicao F, Kellerer M, et al. Palmitate-Induced Interleukin-6 Expression in Human Coronary Artery Endothelial Cells. Diabetes. (2004) 53:3209–16. doi: 10.2337/diabetes.53.12.3209
143. Palomer X, Pizarro-Delgado J, Barroso E, Vazquez-Carrera M. Palmitic and oleic acid: the Yin and Yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol Metab. (2018) 29:178–90. doi: 10.1016/j.tem.2017.11.009
144. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. (2018) 27:1096–110 e5. doi: 10.1016/j.cmet.2018.03.014
145. Zeng J, Zhang Y, Hao J, Sun Y, Liu S, Bernlohr DA, et al. Stearic acid induces CD11c expression in proinflammatory macrophages via epidermal fatty acid binding protein. J Immunol. (2018) 200:3407–19. doi: 10.4049/jimmunol.1701416
146. Karasawa T, Kawashima A, Usui-Kawanishi F, Watanabe S, Kimura H, Kamata R, et al. Saturated fatty acids undergo intracellular crystallization and activate the NLRP3 inflammasome in macrophages. Arterioscler Thromb Vasc Biol. (2018) 38:744–56. doi: 10.1161/ATVBAHA.117.310581
147. Enos RT, Davis JM, Velazquez KT, McClellan JL, Day SD, Carnevale KA, et al. Influence of dietary saturated fat content on adiposity, macrophage behavior, inflammation, and metabolism: composition matters. J Lipid Res. (2013) 54:152–63. doi: 10.1194/jlr.M030700
148. Telle-Hansen VH, Gaundal L, Myhrstad MCW. Polyunsaturated fatty acids and glycemic control in type 2 diabetes. Nutrients. (2019) 11:5. doi: 10.3390/nu11051067
149. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. Diabetes Care. (2010) 33:1674–85. doi: 10.2337/dc10-0666
150. Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: a review of pathogenesis. Indian J Endocrinol Metab. (2012) 16:S27–36. doi: 10.4103/2230-8210.94253
151. Norling LV, Ly L, Dalli J. Resolving inflammation by using nutrition therapy: roles for specialized proresolving mediators. Curr Opin Clin Nutr Metab Care. (2017) 20:145–52. doi: 10.1097/MCO.0000000000000353
152. Sainsbury E, Kizirian NV, Partridge SR, Gill T, Colagiuri S, Gibson AA. Effect of dietary carbohydrate restriction on glycemic control in adults with diabetes: a systematic review and meta-analysis. Diabetes Res Clin Pract. (2018) 139:239–52. doi: 10.1016/j.diabres.2018.02.026
153. Jonasson L, Guldbrand H, Lundberg AK, Nystrom FH. Advice to follow a low-carbohydrate diet has a favourable impact on low-grade inflammation in type 2 diabetes compared with advice to follow a low-fat diet. Ann Med. (2014) 46:182–7. doi: 10.3109/07853890.2014.894286
154. Vitale M, Masulli M, Rivellese AA, Babini AC, Boemi M, Bonora E, et al. Influence of dietary fat and carbohydrates proportions on plasma lipids, glucose control and low-grade inflammation in patients with type 2 diabetes–the TOSCA.IT study. Eur J Nutr. (2016) 55:1645–51. doi: 10.1007/s00394-015-0983-1
155. O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. (2013) 493:346–55. doi: 10.1038/nature11862
156. Wegiel B, Vuerich M, Daneshmandi S, Seth P. Metabolic switch in the tumor microenvironment determines immune responses to anti-cancer therapy. Front Oncol. (2018) 8:284. doi: 10.3389/fonc.2018.00284
157. Yao T, Asayama Y. Animal-cell culture media: history, characteristics, and current issues. Reprod Med Biol. (2017) 16:99–117. doi: 10.1002/rmb2.12024
158. McKee TJ, Komarova SV. Is it time to reinvent basic cell culture medium? Am J Physiol Cell Physiol. (2017) 312:C624–6. doi: 10.1152/ajpcell.00336.2016
159. Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell. (2017) 169:258–72 e17. doi: 10.1016/j.cell.2017.03.023
160. Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci Adv. (2019) 5:1. doi: 10.1126/sciadv.aau7314
Keywords: dendritic cells, metabolism, nutrient availability, tumor micro-environment, diabetes
Citation: Brombacher EC and Everts B (2020) Shaping of Dendritic Cell Function by the Metabolic Micro-Environment. Front. Endocrinol. 11:555. doi: 10.3389/fendo.2020.00555
Received: 07 February 2020; Accepted: 07 July 2020;
Published: 28 August 2020.
Edited by:
Rinke Stienstra, Radboud University Nijmegen Medical Centre, NetherlandsReviewed by:
Gosse J. Adema, Radboud University Nijmegen Medical Centre, NetherlandsR. J. Joost Van Neerven, Wageningen University and Research, Netherlands
Copyright © 2020 Brombacher and Everts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bart Everts, Yi5ldmVydHNAbHVtYy5ubA==