
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 19 August 2020
Sec. Molecular and Structural Endocrinology
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00545
This article is part of the Research Topic GPER Control and Functions View all 11 articles
 Séverine Périan†
Séverine Périan† Jean-Marc Vanacker*
Jean-Marc Vanacker*Endocrine-disrupting chemicals (EDCs) are exogenous chemicals that interfere with endogenous hormonal systems at various levels, resulting in adverse health effects. EDCs belong to diverse chemical families and can accumulate in the environment, diet and body fluids, with different levels of persistence. Their action can be mediated by several receptors, including members of the nuclear receptor family, such as estrogen and androgen receptors. The G protein-coupled estrogen receptor (GPER), a seven-transmembrane domain receptor, has also attracted attention as a potential target of EDCs. This review summarizes our current knowledge concerning GPER as a mediator of EDCs' effects.
According to a general definition, endocrine-disrupting chemicals (EDCs) are exogenous compounds that interfere with the endogenous hormonal axes at any level (1). This includes synthesis, metabolism, transport and delivery of hormones, and also perturbation of the expression of hormone receptors as well as with the downstream signals they convey. EDCs comprise compounds that can promote or restrict a hormonal signal (acting as agonists or antagonists, respectively). Under this broad definition, EDCs include natural molecules such as the phytoestrogens (e.g., genistein, which is abundant in soy) that modulate estrogen signaling and also synthetic compounds intended for therapeutic purposes, such as the ones used as adjuvant therapy in breast cancer. Examples of the latter category include inhibitors of aromatase used to reduce the endogenous synthesis of 17β-estradiol (E2) or tamoxifen that act as an antagonist of the estrogen receptor in mammary tumors.
EDCs also comprise chemicals that are produced for various industrial purposes, being used as components of several products (plastics, paints, flame retardants, herbicides, pesticides…), that exert unintended impacts on hormonal signaling. The number and variety (in terms of chemical structure) of molecules that display suspected or validated endocrine disrupting effects increased since years (1). Furthermore, these compounds often display high levels of resistance to natural degradation leading to their accumulation in the environment as well as in body fluids [see (2–4) for examples]. Adverse effects of EDCs have been reported in domains covering all fields related to hormonal signaling, including metabolism, reproduction, induction and progression of hormone-sensitive cancers and neurodevelopment (1).
To investigate the effects of EDCs, it is essential to identify the receptors that mediate their action as well as the downstream cascades they elicit. Given that EDCs largely impact the male and female reproductive axes, it was initially suspected that their effects were largely mediated by the sex steroid receptors (5). These include the estrogen receptors (ERs) and the androgen receptor (AR), which are members of the nuclear receptor (NR) family and act as transcription factors. In line with this, several EDCs were demonstrated to modulate the activities of these NRs. However, at least in some cases, such as that of the paradigmatic EDC Bisphenol A (BPA), the affinity of these compounds for ERα appeared far lower than that of their natural ligand (6), suggesting the existence of other proteins acting as EDC receptors. Consistently, it was shown that BPA binds to ERRγ, an orphan NR which does not recognize E2, and induces its downstream activities (7, 8). As far as we are aware, the capacity of ERRγ to serve as a receptor for EDCs other than bisphenols has not been published. In contrast, an array of publications suggests that the G protein-coupled receptor (GPER) may serve as a receptor for a vast spectrum of EDCs. The purpose of this review is to (non-exhaustively) summarize what we currently know concerning the relationships between GPER and EDCs.
GPER (initially referred to as GPR30) has been identified as a membrane associated estrogen receptor 15 years ago (9, 10). This seven-transmembrane domain receptor is broadly expressed and has been detected in several sub-cellular localizations, including in internal membrane compartments, such as the endoplasmic reticulum, nucleus and even as a chromatin binding protein under certain circumstances (11). It is expected that different molecular functions could be exerted by GPER, depending on its sub-cellular localization (summarized on Figure 1). Indeed, membrane activation of GPER was shown to rapidly promote intracellular calcium mobilization, cAMP production and to induce a phosphorylation cascade in particular involving ERK1/2, PKA, and PI3K (9, 10, 12–14). On another hand, chromatin binding of GPER leads to direct transcriptional activation of target genes (11).
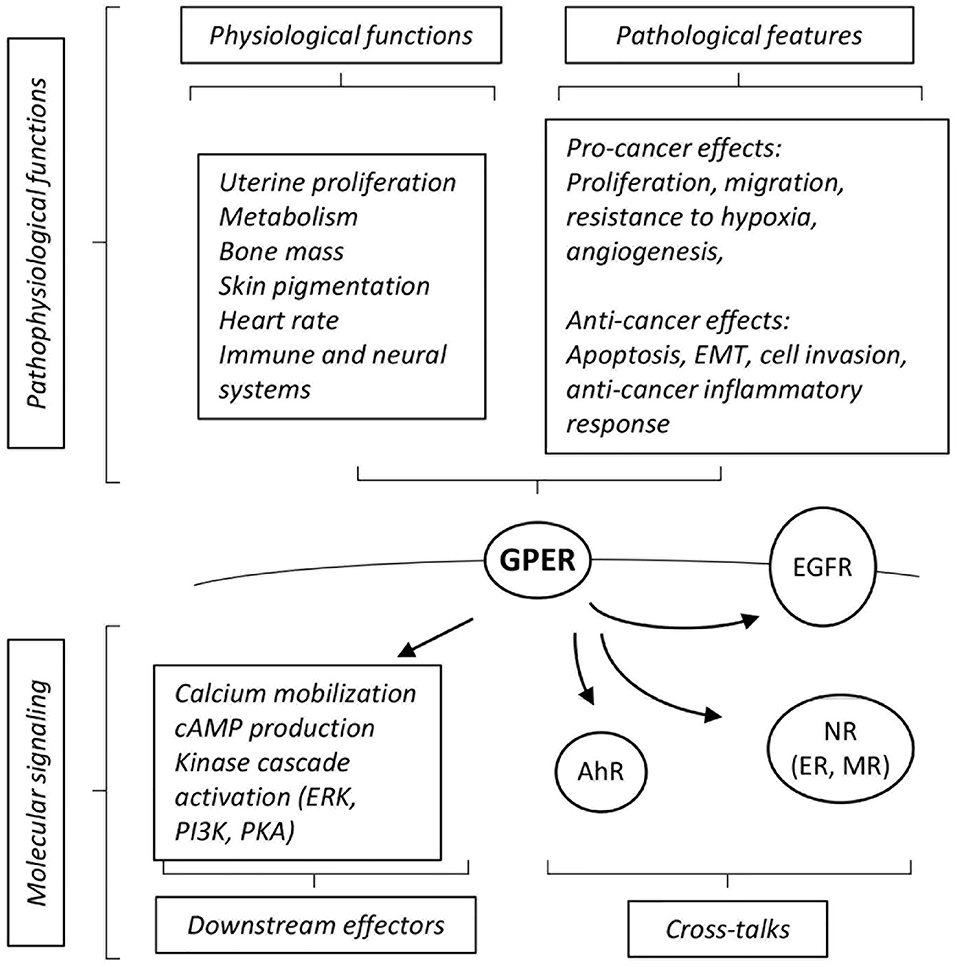
Figure 1. Summary of cross-talks, downstream effectors and pathophysiological effects elicited by GPER. See text for definition of abbreviations, details and references.
GPER cross-talks with different receptors to convey its downstream effects. For instance, functional interactions with the aryl-hydrocarbon receptor (AhR) or EGF receptors (EGFR) are instrumental for the activation of downstream MAPK activation (12, 15). GPER also functionally interacts with nuclear receptors at various levels. For instance, GPER is required for the effect of aldosterone mediated by the mineralocorticoid receptor (MR) in breast cancer cell lines (16). A more indirect level of cross-talks can be illustrated by the regulation of the circulating level of thyroid hormone which in turn modulates embryonic heart rate in a thyroid hormone receptor-dependent manner (17). Functional interactions between GPER and ER have been abundantly documented, may depend on the cell type considered and may lead to congruent or opposing effects [reviewed in (18)]. For example, in ovarian cancer cells, both GPER- and ER-mediated signals are involved in the activation of ERK1/2 leading to increased c-fos expression and induction of proliferation (19). On another hand, at least in ER-positive breast cancer cells, tamoxifen acts as an ER antagonist, but as a GPER agonist (9). Altogether, this shows that GPER displays a wide array of molecular functions and interactions with other signaling pathways. Given its broad expression spectrum and its described pathophysiological functions, GPER has emerged as a factor of clinical importance [reviewed in (20)].
The functions of GPER have been investigated using in vivo and in vitro approaches. GPER knocked-out mice [reviewed in (21)] reproduce normally, indicating that GPER is not absolutely required for reproduction. However, pharmacological studies (i.e., using treatments with agonists and antagonists) suggest that GPER intervenes in uterine epithelial proliferation, suggesting a subtle impact on reproductive function that may be compensated for in the absence of the receptor. Other in vivo studies have indicated that GPER is involved, amongst others, in glucose and lipid metabolism, bone mass, skin pigmentation, regulation of heart rate, and immune and neural systems [(17, 22–24), reviewed in (25)].
The impact of GPER, as a novel estrogen receptor, on cancers has been extensively analyzed, in particular on hormone-related cancers (e.g., breast, ovary, and endometrium). Several studies report a pro-cancer effect of GPER (26, 27). Indeed, high GPER expression correlates to a poor prognosis in breast and endometrial carcinoma (28, 29). Consistently, GPER activation promotes various traits of cancer progression including cell migration in triple negative breast cancer cells, resistance to hypoxia and proangiogenic response (30–32). GPER is also active in cancer-associated fibroblasts (CAFs) where it favors tumor-promoting activities (33, 34).
In contrast, other studies rather indicate that GPER may exert anti-cancer roles. For instance, high expression of GPER has been reported as a factor of favorable prognosis in triple-negative breast cancers (35). Similarly, low level of GPER protein expression in the cytoplasm is associated with lower levels of disease free survival in breast cancer, even when eliminating potentially confounding factors such as ER/PR/HER2 status (36). Consistently, reports indicate that GPER activation leads to cell cycle arrest, apoptosis and cell death in ER-positive and –negative cell lines (37, 38). Interestingly, an inhibitory effect of GPER has also been noted in cancers that do not depend on estrogen signaling. Indeed, GPER inhibits epithelial-to-mesenchymal transition and cell invasion in prostate and pancreatic cancer cells (39). Furthermore, tamoxifen-mediated GPER activation impairs the conversion of pancreatic stellate cells into myofibroblasts (an equivalent of CAFs in pancreatic tumors), which in turn leads to reduced cancer cell survival (40, 41). Moreover, GPER-deficient mice display increased inflammation in induced liver tumorigenesis resulting in accelerated tumor growth (42).
To date, the roles of GPER in cancer thus appear unclear. However, it is possible to propose non-mutually exclusive hypotheses to solve these apparent contradictions. (i) GPER sub-cellular localization may impact its prognosis value (and its activities). In this respect, in contrast to its detection in the cytoplasm, the low nuclear expression of GPER does not correlate to breast cancer aggressiveness (36). (ii) GPER activities may depend on the tissues in which they are studied. It may indeed be envisioned that, in pancreas and liver, the anti-inflammatory effects displayed by GPER in non-cancer cells may overcome its capacity to promote tumor growth in cancer cells. (iii) GPER may exert different activities on the various steps of cancer progression. In a mouse model of mammary cancer, GPER indeed appears dispensable for cancer initiation but contributes to the establishment of metastasis (43). (iv) GPER may play different roles depending on the expression of cross-talking factor. For example, GPER promotes the growth of ER-negative SKBr3 cells, but reduces that of ER-positive MCF7 cells (44). Furthermore, the stimulating effect of GPER on ovarian cancer cells depends on EGFR (19). More work is obviously required to refine our knowledge on the impact of GPER on cancers.
GPER was identified as a functional estrogen receptor in ER-negative cells by a combination of binding and functional studies (i.e., detection of GPER-dependent calcium mobilization or adenylyl cyclase activation) (9, 10), suggesting a shared repertoire between compounds acting on ER and on GPER (summarized on Figure 2). ER-binding ligands were thus examined and this led to the surprising finding that tamoxifen and ICI182, 780 (two ER-antagonist used in adjuvant breast cancer therapy) actually acted as GPER-agonists. Furthermore, EDCs, acting as xenoestrogens on ER, including genistein, BPA, and DDT derivatives also impacted GPER, as shown by binding assays coupled to functional signaling assays (48). Although the affinity of these compounds for GPER is less than that of E2, they broadly display similar binding constants as those displayed on ER. However, the repertoires of compounds bound by GPER and ER are not strictly similar. For instance, the potent ER-agonist DES does not bind GPER (10). Moreover, functional screening identified specific synthetic GPER ligands (i.e., not recognizing the nuclear estrogen receptors) that act as agonist (G-1) or antagonists (G-15 and G-36) for GPER [reviewed in (49)]. Altogether, this shows that GPER and ER display both overlapping and distinct repertoires of compound recruitment. Furthermore, molecular modeling and in silico docking studies indicated that GPER offers several cavities to accommodate large volume ligands and suggested a broad number of possible binding compounds (50, 51). Indeed, competition assays and measurement of cAMP accumulation revealed that organochlorides, such as polychlorinated biphenyls (PCBs) and kepone (aka chlordecone), act (amongst others) as GPER agonists (48).
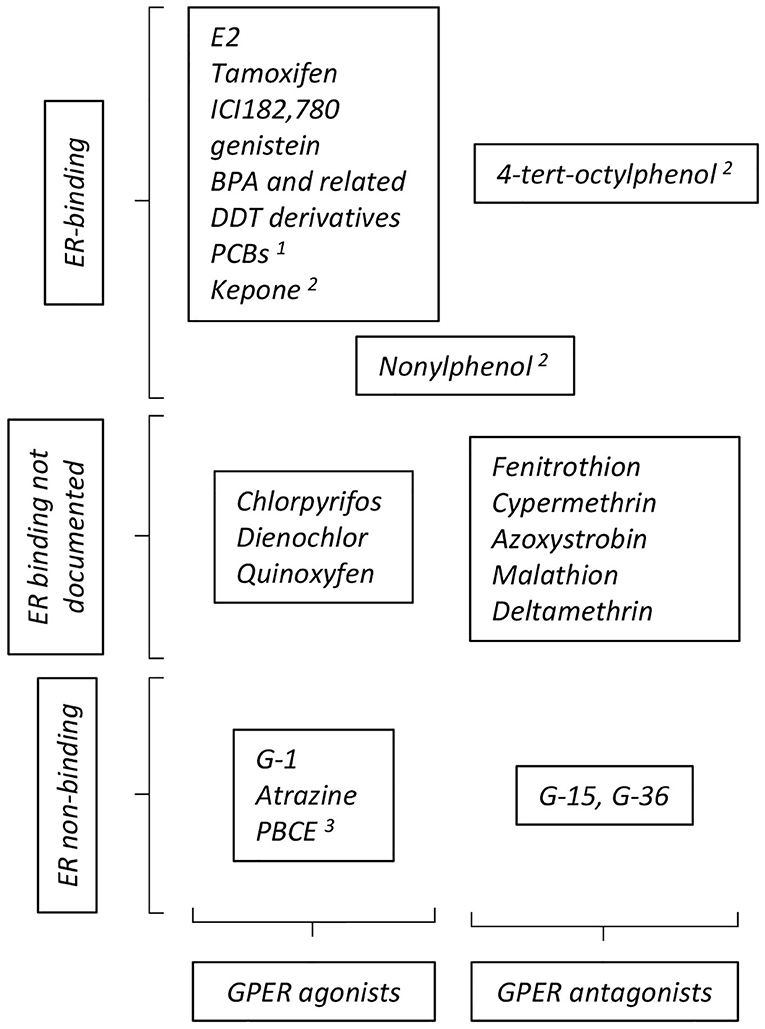
Figure 2. Summary of compounds reported to act as GPER- agonists and antagonists. The particular position occupied by nonylphenol reflects contradictory reports ascribing this compound as GPER- agonist or antagonist. See text for definition of abbreviations, details and reference. Additional references for ER binding: 1: (45); 2: (46); 3: (47).
The GPER-dependent consequences of EDC exposure in terms of molecular outcomes, as well as at the cellular, phenotypical levels have also been studied. The pesticide atrazine does not transactivate ER but induces GPER-dependent ERK activation in ovarian cancer cells and CAF, leading to increased proliferation and migration (52).
BPA induces proliferation and migration of ER-negative breast cancer cells and CAFs in a GPER-dependent manner (53, 54). Proliferation of mouse spermatogonial and Sertoli cells has also been shown as induced by BPA through GPER (55, 56). Intriguingly, analysis of the dose-response indicated a non-monotonous effect in form of an inverted U-shaped curve. Other bisphenols, used as substitutes for BPA and found in high concentrations (similar to or higher than those of BPA) in the environment and body fluids (57) have also been tested. As compared to BPA, some of these analogs, such as BPAF and BPB, display comparable binding affinities to GPER (as determined by E2 displacement), GPER activation capacities (as assessed by calcium mobilization and cAMP production) and, GPER-dependent induction of cell migration (58). Intriguingly, BPF did not display such activities although other studies indicated that its effects on hormonal axes was comparable to those of BPA [reviewed in (59)]. Other compounds such as polybrominated diphenyl ether (PBCE, used as flame retardant additives) that, as BPA, display a diphenyl core, also display GPER binding with an affinity in the micromolar range (60). These compounds induce cAMP accumulation, calcium mobilization and cell migration in ER-negative breast cancer cells.
Nonylphenol (NP) induces cardiac contractility in a non-monotonic manner (61). The effect at low doses is antagonized by G-15, suggesting that NP acts as a GPER agonist. Such an effect of NP has also been suggested on human ER-negative cells (48) as well as on zebrafish oocyte maturation, where this compound (as well as other alkylphenols, including BPA) blocks oocyte maturation, as does G-1 (62). In contrast, NP has been shown to counteract the action of G-1 as a moderator of asthma symptoms in mouse models (63), suggesting that this compounds acts as a GPER-antagonist. Whether these apparent discrepancies originate from the differences in the pathophysiological situations that are analyzed remains to be established.
GPER is a promiscuous receptor displaying a broad spectrum of compound recognition, including toward EDCs. There is however a specificity of GPER-recognition within given chemical families, as exemplified by the bisphenol derivatives. It should be noted that most if not all of the studies examining the effects of EDC on GPER have been performed using cell cultures systems and seldom in vivo. In vitro cell models provide irreplaceable tools for their capacity to be experimentally manipulated. However, comparing the effects of EDCs in wild type and GPER-inactivated animals will greatly increase our understanding of the action of these compounds.
Although several of the compounds impacting on GPER have also been demonstrated to bind ERs, there is a level of selectivity, discriminating these receptors. Various levels of cross-talks have been demonstrated between GPER and other proteins such as ERs, EGFR, or AhR. Whether or not these cross-talks are effective in a given cellular system and may influence the outcome of GPER activation is not always understood. It will thus be of interest to assess the effects of EDCs as GPER modulators under conditions where these cross-talks are controlled.
GPER exerts a large array of pathophysiological functions. A level of overlap between these functions and the perturbations induced by exposure to EDCs is worth noting. Together, this places GPER as a strong candidate to mediate, at least part, of the adverse effects displayed by EDCs. As discussed above, the exact role of GPER in cancer initiation and progression is a matter of debate and may depend on the considered tissue and/or disease stage. How the modulation of GPER activities by EDCs impact cancer features is thus unclear but should be an important field of investigations in the near future.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Work in our laboratory is funded by Ligue contre le Cancer (comité Rhône), Région Auvergne Rhône Alpes (grant SCUSI OPE2017_004), ANSES (grant EST15-076), and ENS Lyon (programme JoRISS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to apologize to colleagues whose work could not be cited for the sake of space.
1. Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, et al. EDC-2: The Endocrine Society's Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev. (2015) 36:E1–150. doi: 10.1210/er.2015-1010
2. Ritter R, Scheringer M, MacLeod M, Moeckel C, Jones KC, Hungerbühler K. Intrinsic human elimination half-lives of polychlorinated biphenyls derived from the temporal evolution of cross-sectional biomonitoring data from the United Kingdom. Environ Health Perspect. (2011) 119:225–31. doi: 10.1289/ehp.1002211
3. Saoudi A, Fréry N, Zeghnoun A, Bidondo ML, Deschamps V, Göen T, et al. Serum levels of organochlorine pesticides in the French adult population: the French National Nutrition and Health Study (ENNS), 2006-2007. Sci Total Environ. (2014) 472:1089–99. doi: 10.1016/j.scitotenv.2013.11.044
4. Gogola J, Hoffmann M, Ptak A. Persistent endocrine-disrupting chemicals found in human follicular fluid stimulate the proliferation of granulosa tumor spheroids via GPR30 and IGF1R but not via the classic estrogen receptors. Chemosphere. (2019) 217:100–10. doi: 10.1016/j.chemosphere.2018.11.018
5. Sonnenschein C, Soto AM. An updated review of environmental estrogen and androgen mimics and antagonists. J Steroid Biochem Mol Biol. (1998) 65:143–50. doi: 10.1016/S0960-0760(98)00027-2
6. Shanle EK, Xu W. Endocrine disrupting chemicals targeting estrogen receptor signaling: identification and mechanisms of action. Chem Res Toxicol. (2011) 24:6–19. doi: 10.1021/tx100231n
7. Matsushima A, Kakuta Y, Teramoto T, Koshiba T, Liu X, Okada H, et al. Structural evidence for endocrine disruptor bisphenol A binding to human nuclear receptor ERR gamma. J Biochem. (2007) 142:517–24. doi: 10.1093/jb/mvm158
8. Tohmé M, Prud'homme SM, Boulahtouf A, Samarut E, Brunet F, Bernard L, et al. Estrogen-related receptor γ is an in vivo receptor of bisphenol A. FASEB J. (2014) 28:3124–33. doi: 10.1096/fj.13-240465
9. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307:1625–30. doi: 10.1126/science.1106943
10. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. (2005) 146:624–32. doi: 10.1210/en.2004-1064
11. Madeo A, Maggiolini M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res. (2010) 70:6036–46. doi: 10.1158/0008-5472.CAN-10-0408
12. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
13. Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. (2002) 16:70–84. doi: 10.1210/mend.16.1.0758
14. Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol Cell Endocrinol. (2009) 308:32–8. doi: 10.1016/j.mce.2009.03.026
15. Cirillo F, Lappano R, Bruno L, Rizzuti B, Grande F, Guzzi R, et al. AHR and GPER mediate the stimulatory effects induced by 3-methylcholanthrene in breast cancer cells and cancer-associated fibroblasts (CAFs). J Exp Clin Cancer Res. (2019) 38:335. doi: 10.1186/s13046-019-1337-2
16. Rigiracciolo DC, Scarpelli A, Lappano R, Pisano A, Santolla MF, Avino S, et al. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget. (2016) 7:94–111. doi: 10.18632/oncotarget.6475
17. Romano SN, Edwards HE, Souder JP, Ryan KJ, Cui X, Gorelick DA. G protein-coupled estrogen receptor regulates embryonic heart rate in zebrafish. PLoS Genet. (2017) 13:e1007069. doi: 10.1371/journal.pgen.1007069
18. Romano SN, Gorelick DA. Crosstalk between nuclear and G protein-coupled estrogen receptors. Gen Comp Endocrinol. (2018) 261:190–7. doi: 10.1016/j.ygcen.2017.04.013
19. Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. (2007) 67:1859–66. doi: 10.1158/0008-5472.CAN-06-2909
20. Barton M. Not lost in translation: emerging clinical importance of the G protein-coupled estrogen receptor GPER. Steroids. (2016) 111:37–45. doi: 10.1016/j.steroids.2016.02.016
21. Prossnitz ER, Hathaway HJ. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol. (2015) 153:114–26. doi: 10.1016/j.jsbmb.2015.06.014
22. Ford J, Hajibeigi A, Long M, Hahner L, Gore C, Hsieh JT, et al. GPR30 deficiency causes increased bone mass, mineralization, and growth plate proliferative activity in male mice. J Bone Miner Res. (2011) 26:298–307. doi: 10.1002/jbmr.209
23. Natale CA, Duperret EK, Zhang J, Sadeghi R, Dahal A, O'Brien KT, et al. Sex steroids regulate skin pigmentation through nonclassical membrane-bound receptors. Elife. (2016) 5:e15104. doi: 10.7554/eLife.15104
24. Triplett KD, Pokhrel S, Castleman MJ, Daly SM, Elmore BO, Joyner JA, et al. GPER activation protects against epithelial barrier disruption by Staphylococcus aureus α-toxin. Sci Rep. (2019) 9:1343. doi: 10.1038/s41598-018-37951-3
25. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. (2011) 7:715–26. doi: 10.1038/nrendo.2011.122
26. Lappano R, Pisano A, Maggiolini M. GPER function in breast cancer: an overview. Front Endocrinol. (2014) 5:66. doi: 10.3389/fendo.2014.00066
27. Jacenik D, Cygankiewicz AI, Krajewska WM. The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol Cell Endocrinol. (2016) 429:10–8. doi: 10.1016/j.mce.2016.04.011
28. Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, et al. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res. (2006) 12:6359–66. doi: 10.1158/1078-0432.CCR-06-0860
29. Smith HO, Leslie KK, Singh M, Qualls CR, Revankar CM, Joste NE, et al. GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol. (2007) 196:386.e1-9. doi: 10.1016/j.ajog.2007.01.004
30. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. (2009) 28:523–32. doi: 10.1038/emboj.2008.304
31. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S, et al. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. (2014) 74:4053–64. doi: 10.1158/0008-5472.CAN-13-3590
32. Rigiracciolo DC, Santolla MF, Lappano R, Vivacqua A, Cirillo F, Galli GR, et al. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J Exp Clin Cancer Res. (2019) 38:58. doi: 10.1186/s13046-019-1056-8
33. Luo H, Liu M, Luo S, Yu T, Wu C, Yang G, et al. Dynamic monitoring of GPER-mediated estrogenic effects in breast cancer associated fibroblasts: an alternative role of estrogen in mammary carcinoma development. Steroids. (2016) 112:1–11. doi: 10.1016/j.steroids.2016.03.013
34. Lappano R, Maggiolini M. GPER is involved in the functional liaison between breast tumor cells and cancer-associated fibroblasts (CAFs). J Steroid Biochem Mol Biol. (2018) 176:49–56. doi: 10.1016/j.jsbmb.2017.02.019
35. Chen ZJ, Wei W, Jiang GM, Liu H, Wei WD, Yang X, et al. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol Oncol. (2016) 10:775–88. doi: 10.1016/j.molonc.2016.01.002
36. Martin SG, Lebot MN, Sukkarn B, Ball G, Green AR, Rakha EA, et al. Low expression of G protein-coupled oestrogen receptor 1 (GPER) is associated with adverse survival of breast cancer patients. Oncotarget. (2018) 9:25946–56. doi: 10.18632/oncotarget.25408
37. Wei W, Chen ZJ, Zhang KS, Yang XL, Wu YM, Chen XH, et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. (2014) 5:e1428. doi: 10.1038/cddis.2014.398
38. Vo DH, Hartig R, Weinert S, Haybaeck J, Nass N. G-protein-coupled estrogen receptor (GPER)-specific agonist G1 induces ER stress leading to cell death in MCF-7 cells. Biomolecules. (2019) 9:503. doi: 10.3390/biom9090503
39. Rice A, Cortes E, Lachowski D, Oertle P, Matellan C, Thorpe SD, et al. GPER activation inhibits cancer cell mechanotransduction and basement membrane invasion via RhoA. Cancers. (2020) 12:E289. doi: 10.3390/cancers12020289
40. Cortes E, Lachowski D, Robinson B, Sarper M, Teppo JS, Thorpe SD, et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. (2019) 20:e46557. doi: 10.15252/embr.201846557
41. Cortes E, Sarper M, Robinson B, Lachowski D, Chronopoulos A, Thorpe SD, et al. GPER is a mechanoregulator of pancreatic stellate cells and the tumor miroenvironement. EMBO Rep. (2019) 20:e46556. d doi: 10.15252/embr.201846556
42. Wei T, Chen W, Wen L, Zhang J, Zhang Q, Yang J, et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett. (2016) 382:195–202. doi: 10.1016/j.canlet.2016.08.012
43. Marjon NA, Hu C, Hathaway HJ, Prossnitz ER. G protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol Cancer Res. (2014) 12:1644–54. doi: 10.1158/1541-7786.MCR-14-0128-T
44. Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. (2010) 70:1184–94. doi: 10.1158/0008-5472.CAN-09-3068
45. Roszko M, Kaminska M, Szymczyk K, Piasecka-Józwiak K, Chabłowska B. Optimized yeast-based in vitro bioassay for determination of estrogenic and androgenic activity of hydroxylated/methoxylated metabolites of BDEs/CBs and related lipophilic organic pollutants. J Environ Sci Health B. (2018) 53:692–706. doi: 10.1080/03601234.2018.1474564
46. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. (1998) 139:4252–63. doi: 10.1210/endo.139.10.6216
47. Li X, Gao Y, Guo LH, Jiang G. Structure-dependent activities of hydroxylated polybrominated diphenyl ethers on human estrogen receptor. Toxicology. (2013) 309:15–22 doi: 10.1016/j.tox.2013.04.001
48. Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. (2006) 102:175–9. doi: 10.1016/j.jsbmb.2006.09.017
49. Prossnitz ER, Arterburn JB. International Union of Basic and Clinical Pharmacology. XCVII G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol Rev. (2015) 67:505–40. doi: 10.1124/pr.114.009712
50. Méndez-Luna D, Martínez-Archundia M, Maroun RC, Ceballos-Reyes G, Fragoso-Vázquez MJ, González-Juárez DE, et al. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J Biomol Struct Dyn. (2015) 33:2161–72. doi: 10.1080/07391102.2014.994102
51. Rosano C, Ponassi M, Santolla MF, Pisano A, Felli L, Vivacqua A, et al. Macromolecular modelling and docking simulations for the discovery of selective GPER ligands. AAPS J. (2016) 18:41–6. doi: 10.1208/s12248-015-9844-3
52. Albanito L, Lappano R, Madeo A, Chimento A, Prossnitz ER, Cappello AR, et al. Effects of atrazine on estrogen receptor α- and G protein-coupled receptor 30-mediated signaling and proliferation in cancer cells and cancer-associated fibroblasts. Environ Health Perspect. (2015) 123:493–9. doi: 10.1289/ehp.1408586
53. Pupo M, Pisano A, Lappano R, Santolla MF, De Francesco EM, Abonante S, et al. Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ Health Perspect. (2012) 120:1177–82. doi: 10.1289/ehp.1104526
54. Castillo Sanchez R, Gomez R, Perez Salazar E. Bisphenol a induces migration through a GPER-, FAK-, Src-, and ERK2-dependent pathway in MDA-MB-231 breast cancer cells. Chem Res Toxicol. (2016) 29:285–95. doi: 10.1021/acs.chemrestox.5b00457
55. Sheng ZG, Zhu BZ. Low concentrations of bisphenol A induce mouse spermatogonial cell proliferation by G protein-coupled receptor 30 and estrogen receptor-α. Environ Health Perspect. (2011) 119:1775–80. doi: 10.1289/ehp.1103781
56. Ge LC, Chen ZJ, Liu HY, Zhang KS, Liu H, Huang HB, et al. Involvement of activating ERK1/2 through G protein coupled receptor 30 and estrogen receptor α/β in low doses of bisphenol A promoting growth of Sertoli TM4 cells. Toxicol Lett. (2014) 226:81–9. doi: 10.1016/j.toxlet.2014.01.035
57. Chen D, Kannan K, Tan H, Zheng Z, Feng YL, Wu Y, et al. Bisphenol analogues other than BPA: environmental occurrence, human exposure, and toxicity-A review. Environ Sci Technol. (2016) 50:5438–53. doi: 10.1021/acs.est.5b05387
58. Cao LY, Ren XM, Li CH, Zhang J, Qin WP, Yang Y, et al. Bisphenol AF and bisphenol B exert higher estrogenic effects than bisphenol A via G protein-coupled estrogen receptor pathway. Environ Sci Technol. (2017) 51:11423–30. doi: 10.1021/acs.est.7b03336
59. Rochester JR, Bolden AL. Bisphenol S and F: a systematic review and comparison of the hormonal activity of bisphenol a substitutes. Environ Health Perspect. (2015) 123:643–50. doi: 10.1289/ehp.1408989
60. Cao LY, Ren XM, Yang Y, Wan B, Guo LH, Chen D, et al. Hydroxylated polybrominated diphenyl ethers exert estrogenic effects via non-genomic G protein-coupled estrogen receptor mediated pathways. Environ Health Perspect. (2018) 126:057005. doi: 10.1289/EHP2387
61. Gao Q, Liu S, Guo F, Liu S, Yu X, Hu H, et al. Nonylphenol affects myocardial contractility and L-type Ca(2+) channel currents in a non-monotonic manner via G protein-coupled receptor 30. Toxicology. (2015) 334:122–9. doi: 10.1016/j.tox.2015.06.004
62. Fitzgerald AC, Peyton C, Dong J, Thomas P. Bisphenol A and related alkylphenols exert nongenomic estrogenic actions through a G protein-coupled estrogen receptor 1 (Gper)/epidermal growth factor receptor (Egfr) pathway to inhibit meiotic maturation of zebrafish oocytes. Biol Reprod. (2015) 93:135. doi: 10.1095/biolreprod.115.132316
Keywords: GPER, hormone, estrogen, pathophysiology, endocrine-disrupting chemicals (EDCs)
Citation: Périan S and Vanacker J-M (2020) GPER as a Receptor for Endocrine-Disrupting Chemicals (EDCs). Front. Endocrinol. 11:545. doi: 10.3389/fendo.2020.00545
Received: 05 May 2020; Accepted: 06 July 2020;
Published: 19 August 2020.
Edited by:
Sarah H. Lindsey, Tulane University, United StatesReviewed by:
Peter Thomas, University of Texas at Austin, United StatesCopyright © 2020 Périan and Vanacker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Marc Vanacker, amVhbi1tYXJjLnZhbmFja2VyQGVucy1seW9uLmZy
†Present address: Séverine Périan, Centre International de Recherche en Infectiologie, Université de Lyon, Université Lyon1, Inserm U1111, CNRS UMR5308, Ecole Normale Supérieure de Lyon, Lyon, France
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.