 David Perna-Barrull
David Perna-Barrull Anna Gieras
Anna Gieras Silvia Rodriguez-Fernandez
Silvia Rodriguez-Fernandez Eva Tolosa
Eva Tolosa Marta Vives-Pi
Marta Vives-Pi- 1Immunology Section, Germans Trias i Pujol Research Institute, Autonomous University of Barcelona, Badalona, Spain
- 2Department of Immunology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Type 1 diabetes (T1D) is a multifactorial disease of unknown aetiology. Studies focusing on environment-related prenatal changes, which might have an influence on the development of T1D, are still missing. Drugs, such as betamethasone, are used during this critical period without exploring possible effects later in life. Betamethasone can interact with the development and function of the two main players in T1D, the immune system and the pancreatic β-cells. Short-term or persistent changes in any of these two players may influence the initiation of the autoimmune reaction against β-cells. In this review, we focus on the ability of betamethasone to induce alterations in the immune system, impairing the recognition of autoantigens. At the same time, betamethasone affects β-cell gene expression and apoptosis rate, reducing the danger signals that will attract unwanted attention from the immune system. These effects may synergise to hinder the autoimmune attack. In this review, we compile scattered evidence to provide a better understanding of the basic relationship between betamethasone and T1D, laying the foundation for future studies on human cohorts that will help to fully grasp the role of betamethasone in the development of T1D.
Betamethasone as an Emerging Environmental Factor in T1D
Type 1 diabetes (T1D) is an autoimmune disease caused by the selective destruction of insulin-producing β-cells. The trigger, however, remains unknown. Postnatal environmental determinants have been thoroughly studied as risk factors (1, 2) but a crucial phase for the immune system development, the late prenatal stage, has been poorly investigated. Specifically, the interaction of drugs commonly used during late pregnancy with T1D and the pancreatic β-cells remains unexplored. Nonetheless, some studies reveal the importance of the prenatal stage and the prematurity of the newborn in the development of T1D (3–5). An indirect demonstration of how critical the in utero environment is in T1D development arises from the studies in twins: heterozygotic twins have an increased concordance of T1D when compared to non-twin siblings (6, 7), underlining the potential relevance of prenatal factors and their influence in the development of autoimmunity.
Synthetic glucocorticoids, most often betamethasone, are routinely given to mothers at risk of preterm birth between 24 and 34 weeks of gestation. A single course of prenatal betamethasone reduces the occurrence and severity of respiratory distress syndrome and improves the survival chances in premature infants (8, 9). Another glucocorticoid used for lung maturation is dexamethasone and produces similar results on the newborn survivability (10). These synthetic glucocorticoids cross the placenta and accelerate foetal lung maturation, achieving maximum benefit between 24 h and 7 days after administration (11). Betamethasone is a poor substrate for the glucocorticoid inactivating enzyme 11beta-hydroxysteroid-dehydrogenase 2 (11βHSD2), therefore, its bioactivity in the foetus lasts for several days (12) and it is known to exert long-lasting effects on the hypothalamic-pituitary-adrenal (HPA) axis and cognition in children (13, 14).
Glucocorticoids exert their effects by binding nuclear receptors that are ligand-dependent transcription factors. They can regulate gene transcription, either by direct binding to DNA or by interacting with other transcription factors (15). Glucocorticoid receptors (GR) are ubiquitously expressed; however, due to the variation in the genomic location of GR binding, the transcriptional responses to glucocorticoids are cell type-specific (16). Moreover, polymorphisms of the GR result in alterations in their responsiveness to glucocorticoids and in gene expression (17, 18). In addition, human GR receptor can be a target of endoncrine disruptors such as pesticides (19) that, in combination with antenatal glucocorticoids, could increase developmental neurotoxicity (20).
The general effects of glucocorticoids administered during pregnancy have been thoroughly reviewed (21). Considering the overwhelming use of betamethasone as the treatment of choice for respiratory distress syndrome in premature infants and the cell-specific response to glucocorticoids, in this review we will dissect the specific effects of betamethasone on the main cellular players in the context of T1D, namely immune cells and their targets, the β-cells of the pancreas.
Direct Effects of Betamethasone on the Immune System
Several cell types of the immune system are involved in the development of T1D, and disturbances in the activity of these cells, such as enhanced proinflammatory activity, can increase the risk to develop T1D (22). Below, the effect of betamethasone on different cell types of the immune system is detailed.
Innate Immune Cells
Prenatal administration of betamethasone can induce an anti-inflammatory status in the newborn during the first days after delivery (23), and this fact could be due to the immunomodulatory effects of betamethasone on innate immune cells.
Neutrophils
Neutrophils have gained interest in T1D aetiology due to their participation in the initial steps of autoimmunity against β-cells (24). Moreover, neutrophils are part of the islet leukocytic infiltrates of patients with T1D, and are accordingly reduced in peripheral blood at disease onset (25, 26).
A described effect of betamethasone is the increase in leukocyte counts in peripheral blood after treatment (27), similarly to the effects of natural glucocorticoids during stress (28). Accordingly, neutrophil number and percentage were increased in human blood after betamethasone treatment (29), correlating with the described neutrophil demargination into the blood vessels (30–32). Moreover, in humans, betamethasone reduces neutrophil motility and chemotaxis (33), and can affect metabolism and cytokine production, i.e., reducing interleukin (IL)-8 and macrophage inflammatory protein alpha (MIP-1α) release (34). The inflammatory capacity of neutrophils is therefore reduced, as demonstrated in a lamb model of lung inflammation after betamethasone treatment, where gene expression of IL-1, IL-6, IL-8, and CCL2 was suppressed (35).
Monocytes
Monocytes are circulating innate immune cells that can become antigen-presenting cells (APCs), either macrophages, or dendritic cells (DCs). Thus, reprogramming monocytes may lead to changes in both differentiated cells. Betamethasone has an acute effect on the metabolism of monocytes, transiently reducing the production, and the secretion of IL-6 and reactive oxygen species. By contrast, the phagocytic activity of monocyte-derived APCs was not altered by betamethasone (36). In newborn children with low weight at birth, prenatal betamethasone administration induced a transient immunomodulatory effect in monocytes, causing diminished IL-6 and IL-10 release and downregulation of human leukocyte antigen DR (HLA-DR) expression (37). Moreover, the total number of monocytes was reduced by betamethasone (38). This effect was also assessed in vitro, demonstrating that glucocorticoids induce apoptosis in human monocytes (39). Nevertheless, these results are controversial, and other authors reported that betamethasone does not affect monocytes' IL-6 production (40).
Macrophages
Macrophages are crucial in the initial damage to β-cells in T1D. These tissue-resident APCs contribute to initiate specific immune responses (41). In macrophages, betamethasone diminishes cytokine secretion (IL-8 and TNFα) (42) and impairs their ability for antigen presentation to T cells (43). These effects point to the induction of a regulatory profile in macrophages, similar as described in M2 macrophages (44). Indeed, dexamethasone induces the polarization of the M2 phenotype (45). Moreover, it was recently reported that dexamethasone increases the migration of macrophages by CD26 overexpression, a membrane glycoprotein with enzymatic capabilities involved in inflammation, and this could contribute to the egress of macrophages from inflamed tissue (46).
Dendritic Cells
DCs are professional APCs with the ability to stimulate naïve T cells. In T1D, DCs are responsible for the presentation of β-cell autoantigens to T lymphocytes, initiating the adaptive autoimmune response against the insulin-producing cells. Similarly to the observed effect of dexamethasone on this cell type, DCs differentiated in vitro in the presence of betamethasone failed to achieve a fully mature status, showing a reduced capacity to stimulate the production of IL-17, a cytokine involved in autoimmune responses, by T lymphocytes (32). Furthermore, the release of proinflammatory cytokines was reduced by this drug in DCs. Betamethasone has been reported to induce tolerogenic Langerhans DCs (LDCs) in the skin of patients with psoriasis (47) and atopic dermatitis, which in turn arrest T helper (Th) 1 and Th2 responses (48). Similarly to the effects found in monocytes, human DCs differentiated with betamethasone showed a reduction of membrane expression of costimulatory molecules, such as CD40 and CD86, accompanied by a decrease in IL-12 secretion, an important cytokine for Th1 responses. These effects resulted in tolerogenic function in DCs and impaired ability to induce T lymphocyte proliferation (39).
Natural Killer Cells
Natural Killer cells (NKc) are effector lymphocytes of the innate immune system. Their role in T1D is not completely understood, but abnormalities in this cell type may contribute to trigger autoimmune reactions against β-cells (49). Little is known about the effects of betamethasone on NKc. Betamethasone tends to increase NKc activity in very preterm newborn babies (<32 weeks of gestation), supporting the maturation of this cell type (40). However, other studies have reported that betamethasone reduces the number of NKc in newborn infants (50). In adults, NKc showed a reduced cytolytic activity after topical betamethasone administration (51).
Adaptative Immune Cells
Innate immune cells are crucial in the first phases of autoimmune diseases, but the final effector cells are the adaptative immune system cells, T and B lymphocytes. Modulation of these cells can dampen or exacerbate an autoimmune reaction.
T Lymphocytes
Betamethasone treatment results in T cell precursor apoptosis and, to a lesser extent, of mature T cells. Transient reduction in thymus weight and thymocyte numbers have been described after prenatal betamethasone administration in mice (32, 52). In humans, the thymus of the foetus of mothers that were prenatally treated with steroids showed delayed growth (53). Moreover, a reduction of 20–30% of peripheral lymphocyte counts was observed in pregnant women after treatment with betamethasone, although this effect only lasted for 3 days (27, 38, 54). Other glucocorticoids, like dexamethasone, have comparable effects on lymphocytes after prenatal treatment (55). In newborn children, a similar effect on lymphocyte counts has been described, mainly affecting CD4+ T lymphocytes (56). Data on lymphocyte counts are still controversial, since a different study reported an increase in CD3+ T lymphocytes in very preterm newborn babies (<32 gestational weeks) after betamethasone treatment (50). Prenatal betamethasone administered to the experimental model of T1D, the non-obese diabetic (NOD) mouse, resulted in long-lasting changes in the T Cell Receptor (TCR) Vβ repertoire that persisted into adulthood (32). Importantly, the TCR Vβ families that diminished in frequency after prenatal steroid treatment included pathogenic Vβ domains (57, 58), so it is reasonable to speculate that betamethasone will protect against T1D. In humans, betamethasone reduces T lymphocyte proliferation capacity (40), thus reducing clonal expansion. Overall, T cells will have an impaired capacity of interacting with β-cell antigens, thus contributing to prevent the autoimmune response.
B Lymphocytes
The role of B lymphocytes in the development of T1D is not completely understood. B cells produce autoantibodies to islet antigens that, even if extremely useful as predictive biomarkers for disease, do not appear pathogenic. Also, B cells are critical as APCs during the first stages of autoimmunity in T1D (59). Betamethasone has a deleterious effect on mature B cells of NOD mouse (32) and reduces their ability to produce antibodies, specifically IgE and IgG (60, 61). Other glucocorticoids, such as dexamethasone, show a similar deleterious effect (62), affecting early precursor B cells, whereas mature B cells –IgD positive– are resistant to glucocorticoid-induced apoptosis (63). The reduction observed in antibody production could be the result of impaired B cell receptor and Toll-like receptor 7 signalling, since without these signals B cells cannot switch their Ig isotype, thus reducing their functionality. At the same time, B cells have increased transcriptional activity of IL-10, amplifying the immunomodulatory capacity of these cells induced by glucocorticoids (16).
Effects of Betamethasone on the Target Cells of Autoimmune Diabetes, the Islet β-Cells
β-cells are the insulin-producing cells of the islets of Langerhans. The autoimmune destruction of these cells is the ultimate cause of T1D. β-cells also have an active role in their own destruction, facilitating the interaction with the immune system, and contributing to their own demise (64). Thus, the identification of changes induced by betamethasone may help to understand the possible outcome of this drug in the context of T1D. Studies performed in subjects with long term glucocorticoid treatment indicate that glucocorticoids can induce dysglycaemia, leading to diabetes (65, 66). Glucocorticoids increase insulin resistance (67) without affecting β-cell mass (68). On the other hand, physiological endogenous levels of glucocorticoids are necessary for maintaining the regulation of insulin secretion by β-cells (69). Moreover, prenatal glucocorticoids support the maturation of β-cells by enhancing their glucose sensitivity due to increased expression of Glut2 and Gck genes and by reducing apoptosis, similarly as the overexpression of surfactant proteins induced by glucocorticoids helps with the maturation of the foetal lungs (70). In a similar way, prenatal glucocorticoids enhance insulin secretion in rats due to the overexpression of Gck, Slca2, and Ins2 genes in β-cells, despite β-cell mass is smaller than in non-treated animals (71). Experimental data demonstrate that prenatal betamethasone reduces the risk of developing T1D in the NOD mice (57), correlating to altered expression of genes related to metabolism and autoimmunity in β-cells. In a previous study we reported a reduction of Ccl2 gene expression in β-cells, which may lead to a reduced recruitment of macrophages and monocytes. Moreover, an increase in Gad1 gene expression, could promote tolerance to β-cells in NOD mice (32). Recent studies in other T1D experimental models indicate that short-term treatment with betamethasone during late pregnancy does not affect β-cell metabolism in later life (72, 73). Glucocorticoid signalling can also cause epigenetic modifications in these cells. In fact, glucocorticoids impair the methylation of the DNA by altering the enzymes responsible for this process. Moreover, the prenatal period is a very sensitive phase during which the epigenome shows heightened plasticity to methylation modifications and these changes can be accumulated throughout life (74). Important β-cell functions, such as insulin secretion and islet cell mass homeostasis, are controlled by epigenetic mechanisms (75), and glucocorticoids can modify the epigenome of β-cells inducing changes that can affect their function in adults (76), altering the efficiency of glucose metabolism (77, 78).
How can Betamethasone Affect the Interplay Between the Immune System and β-Cells?
T1D is a multifactorial disease with complex interactions between the immune system and the pancreatic β-cells. Glucocorticoids are potent immune suppressors and are commonly used in patients with autoimmune diseases such as psoriasis or rheumatoid arthritis (79, 80). Betamethasone, like other synthetic glucocorticoids, can reduce cytokine production and release, thereby inhibiting specific immune responses and blocking the initiation of an autoimmune attack to β-cells (2). Dampening the autoimmune reaction can be the most efficient form of preventing T1D, and it might be a consequence of the impaired functionality of innate and adaptative immune cells (Figure 1). On the one side, betamethasone diminishes the proinflammatory action of innate immune cells (neutrophils, macrophages, and NK cells). On the other side, this drug induces a tolerogenic antigen presentation in macrophages and DCs, limiting the possibilities to activate autoreactive T lymphocytes in the lymph nodes. In turn, lymphocytes are also affected by betamethasone, as aforementioned. T lymphocytes reduce their proliferation capacity, decreasing the number of cells that can kill the β-cells. At the same time, prenatal treatment critically reduces the number of developing thymocytes and induces a skewed TCR repertoire towards T cells with less affinity to β-cell autoantigens (57). This fact will impair autoreactivity when APCs expose β-cell autoantigens in Major Histocompatibility Complex (MHC) molecules. Moreover, in the presence of betamethasone, T lymphocytes tend to differentiate to Th2 rather than autoimmunity-prone Th1 or Th17 cells (81), and it has been demonstrated that glucocorticoids exposure during foetal development can alter the HPA axis, impairing CD8+ T lymphocytes function later in life, making them less responsive against viral antigens (82), or blunting cortisol response against rhinovirus (83). In this sense, concerns have been raised about multiple doses of prenatal betamethasone, including an increased susceptibility to infections in children (84, 85). B cell precursors are also affected by betamethasone, showing reduced antibody production (63). However, how these effects altogether might influence the development of T1D is not yet known. Taken together, these alterations suggest a rather positive effect leading to T1D protection, but this effect could depend, among other factors, on the concentration, and duration of the prenatal treatment. In addition, β-cell changes induced by betamethasone may enhance this protective effect. Especially, β-cell maturation and the acquisition of an apoptosis-resistant phenotype may be key factors in thwarting undesired autoimmune reactions (70). Moreover, alterations found in the expression of genes related to interactions between the immune system and β-cell reduce β-cell immunogenicity, hindering their direct interaction with immune system cells (32). This could help to avoid the activation of stray cytotoxic T cells with affinity to β-cell autoantigens. How long this effect is maintained is unknown, but it is reasonable to speculate that prenatal betamethasone could reprogramme some aspects of β-cell function until adult life, without affecting their intrinsic capacities as described for insulin secretion (86). Furthermore, glucocorticoid stimulation also induces epigenetic changes in the precursor β-cells (76), and this could be the main actor behind the long-term effects observed after prenatal administration of betamethasone. We have summarised those studies that used betamethasone in their research (Table 1), focusing on their findings in the context of the immune system and β-cells, and the expected effect these changes would have on T1D. Further studies are needed to reveal the long-term effects of prenatal betamethasone treatment in the immune system and T1D (87).
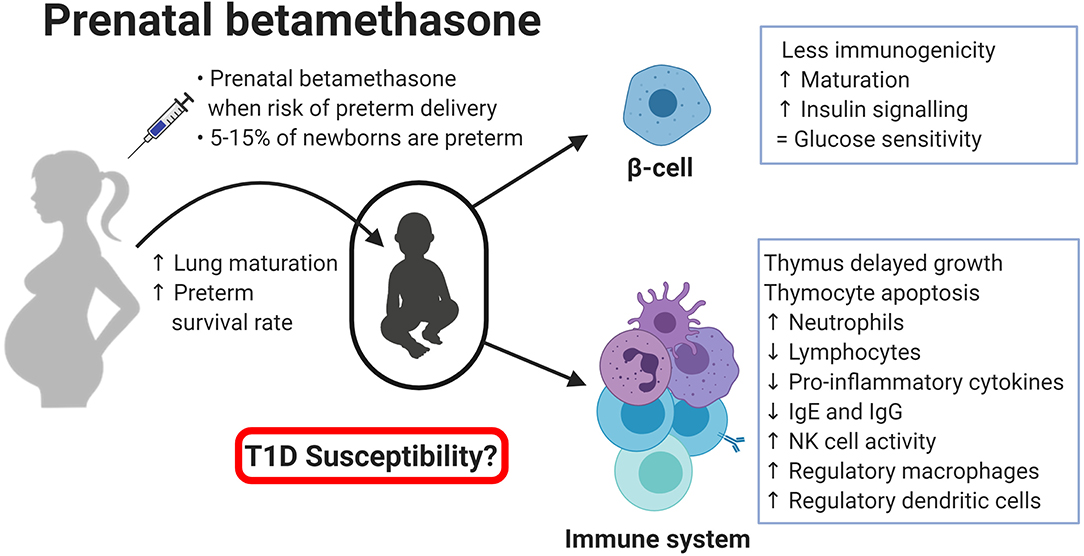
Figure 1. Effects of prental betamethasone on β-cells and the immune system of the newborn. Created with BioRender.com.
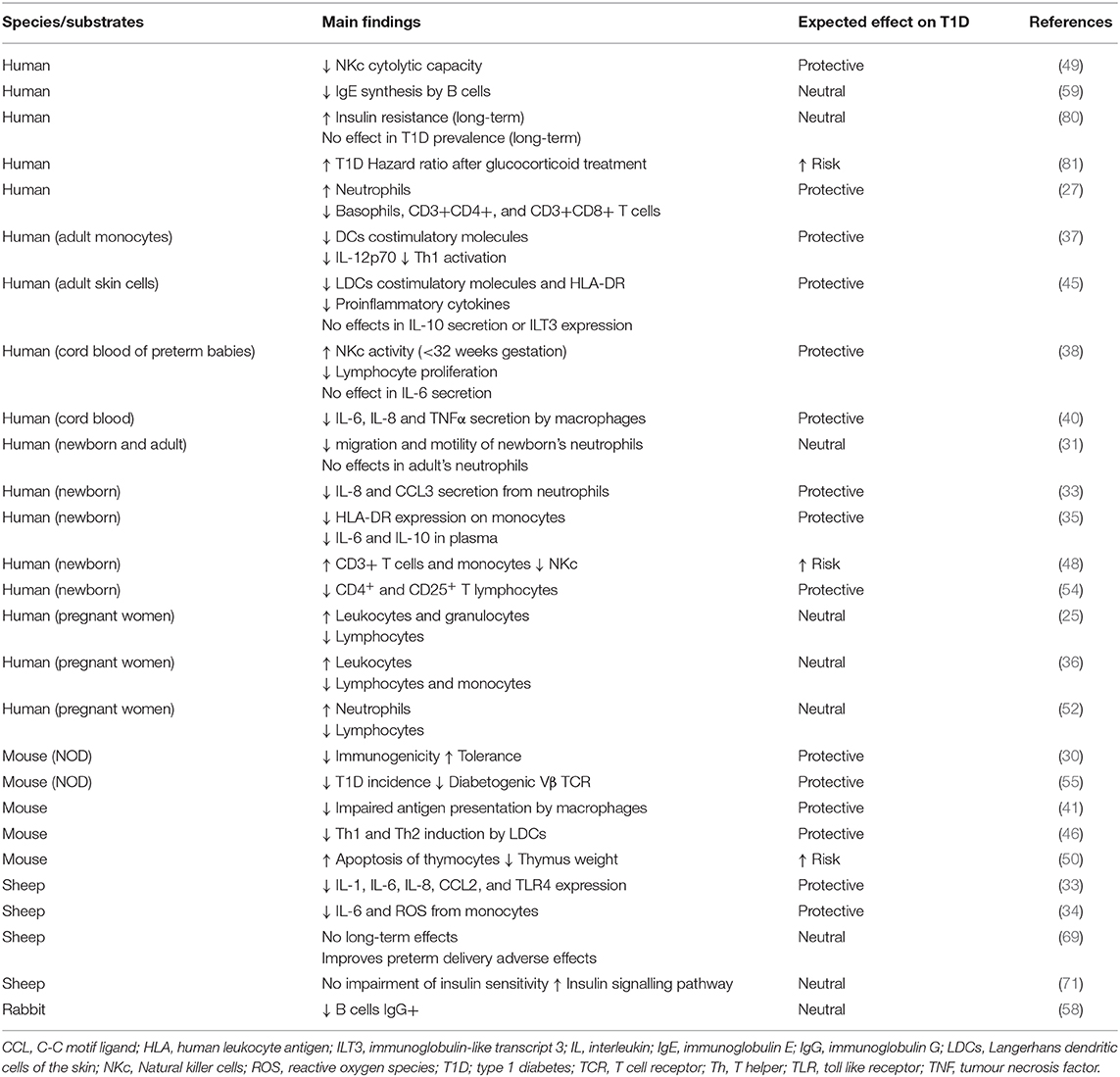
Table 1. Studies on the effect of betamethasone in the immune system and impact on T1D.
Future Perspectives
The effectiveness of glucocorticoids has been demonstrated for a wide range of immunologically related diseases. However, their effects during the prenatal period, both in the immune system and the target tissue of T1D, are not fully characterised. Expanding the understanding of how they can affect self-tolerance and T1D could contribute to reduce the increasing incidence of this and other autoimmune diseases. Another key point is to determine whether the effects of glucocorticoid treatment found in immune system cells could result from changes induced in hematopoietic stem cells, thus explaining the alterations found in many immune cell types. Further studies are required to dissect the exact mechanism and the magnitude of these changes in the immune system and β-cells, because other factors, such as maternal nutrition or stress during pregnancy, could also veil betamethasone effects (88). Finally, epidemiological studies are needed to explore the effect of prenatal betamethasone on T1D. Finding pieces of evidence of the precise effects of betamethasone in T1D development could lead to improved neonatal care, with special focus on children with higher genetic risk to suffer from T1D.
Conclusion
Knowledge about betamethasone action on the immune system is currently increasing, but it is very limited in the prenatal stage as well as in its consequences in the adulthood phase. Glucocorticoids have shown a plethora of effects, which depend on the duration of the treatment, the route of administration, the target tissue, etc. The key message of this review is that prenatal betamethasone affects the immune system cells, and these alterations may have long-term consequences. Simultaneously, betamethasone also alters β-cells towards a less immunogenic phenotype and could induce epigenetic modifications in immune cells and β-cell precursors. Considering these effects, it is tempting to speculate that betamethasone may act as a protective agent against human T1D when administered shortly before birth or in the perinatal period. Future immunological, metabolic, and epidemiological studies, together with the extrapolation of data from other glucocorticoids like dexamethasone, will shed light on the unanswered questions related to this prenatal treatment.
Author Contributions
DP-B and MV-P wrote the manuscript. AG, SR-F, and ET participated in the literature review, and edited the manuscript while adding additional insights. All authors read and approved the final version of the manuscript.
Funding
This work has been funded by the German Research Council (KFO296). DP-B was granted by DiabetesCero Foundation.
Conflict of Interest
MV-P holds a patent that relate to immunotherapy for T1D and is co-founder of Ahead Therapeutics S.L. SR-F is part-time employed at Ahead Therapeutics S.L. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Marta Murillo, Dr. Joan Bel, Dr. Federico Vázquez, and Prof. Manel Puig-Domingo, from University Hospital Germans Trias i Pujol, for fruitful discussions. Special thanks to DiabetesCero Foundation for their support.
References
1. Esposito S, Toni G, Tascini G, Santi E, Berioli MG, Principi N. Environmental factors associated with type 1 diabetes. Front Endocrinol. (2019) 10:1–12. doi: 10.3389/fendo.2019.00592
2. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. (2018) 391:2449–62. doi: 10.1016/S0140-6736(18)31320-5
3. Li S, Zhang M, Tian H, Liu Z, Yin X, Xi B. Preterm birth and risk of type 1 and type 2 diabetes: systematic review and meta-analysis. Obes Rev. (2014) 15:804–11. doi: 10.1111/obr.12214
4. Størdal K, McArdle HJ, Hayes H, Tapia G, Viken MK, Lund-Blix NA, et al. Prenatal iron exposure and childhood type 1 diabetes. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-018-27391-4
5. Samuelsson U, Oikarinen S, Hyöty H, Ludvigsson J. Low zinc in drinking water is associated with the risk of type 1 diabetes in children. Pediatr Diabetes. (2011) 12:156–64. doi: 10.1111/j.1399-5448.2010.00678.x
6. Nisticò L, Iafusco D, Galderisi A, Fagnani C, Cotichini R, Toccaceli V, et al. Emerging effects of early environmental factors over genetic background for type 1 diabetes susceptibility: evidence from a nationwide Italian twin study. J Clin Endocrinol Metab. (2012) 97:E1483–91. doi: 10.1210/jc.2011-3457
7. Kumar D, Gemayel NS, Deapen D, Kapadia D, Yamashita PH, Lee M, et al. North-American twins with IDDM: genetic, etiological, and clinical significance of disease concordance according to age, zygosity, and the interval after diagnosis in first twin. Diabetes. (1993) 42:1351–63. doi: 10.2337/diabetes.42.9.1351
8. Liggins GC, Howie RN. A controlled trial of antepartum glucoccorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. (1972) 50:515–25.
9. Roberts D, Brown J, Medley N, Dalziel SR. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. (2017) 3:CD004454. doi: 10.1002/14651858.CD004454.pub3
10. Schmidt AF, Kemp MW, Kannan PS, Kramer BW, Newnham JP, Kallapur SG, et al. Antenatal dexamethasone vs. betamethasone dosing for lung maturation in fetal sheep. Pediatr Res. (2017) 81:496–503. doi: 10.1038/pr.2016.249
11. Vidaeff AC, Belfort MA, Steer PJ. Antenatal corticosteroids: a time for more careful scrutiny of the indications? BJOG. (2016) 123:1067–9. doi: 10.1111/1471-0528.13853
12. Kajantie E, Raivio T, Jänne OA, Hovi P, Dunkel L, Andersson S. Circulating glucocorticoid bioactivity in the preterm newborn after antenatal betamethasone treatment. J Clin Endocrinol Metab. (2004) 89:3999–4003. doi: 10.1210/jc.2004-0013
13. Alexander N, Rosenlöcher F, Stalder T, Linke J, Distler W, Morgner J, et al. Impact of antenatal synthetic glucocorticoid exposure on endocrine stress reactivity in term-born children. J Clin Endocrinol Metab. (2012) 97:3538–44. doi: 10.1210/jc.2012-1970
14. Alexander N, Rosenlöcher F, Dettenborn L, Stalder T, Linke J, DIstler W, et al. Impact of antenatal glucocorticoid therapy and risk of preterm delivery on intelligence in term-born children. J Clin Endocrinol Metab. (2016) 101:581–9. doi: 10.1210/jc.2015-2453
15. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. (2017) 17:233–47. doi: 10.1038/nri.2017.1
16. Kardava L, Germain RN, Kumar P, Tsang JS, Gadkari M, Franco LM, et al. Immune regulation by glucocorticoids can be linked to cell type–dependent transcriptional responses. J Exp Med. (2019) 216:384–406. doi: 10.1084/jem.20180595
17. Jewell CM, Cidlowski JA. Molecular evidence for a link between the N363S glucocorticoid receptor polymorphism and altered gene expression. J Clin Endocrinol Metab. (2007) 92:3268–77. doi: 10.1210/jc.2007-0642
18. Moraitis AG, Block T, Nguyen D, Belanoff JK. The role of glucocorticoid receptors in metabolic syndrome and psychiatric illness. J Steroid Biochem Mol Biol. (2017) 165:114–20. doi: 10.1016/j.jsbmb.2016.03.023
19. Zhang J, Zhang J, Liu R, Gan J, Liu J, Liu W. Endocrine-disrupting effects of pesticides through interference with human glucocorticoid receptor. Environ Sci Technol. (2016) 50:435–43. doi: 10.1021/acs.est.5b03731
20. Slotkin TA, Card J, Seidler FJ. Chlorpyrifos developmental neurotoxicity: interaction with glucocorticoids in PC12 cells. Neurotoxicol Teratol. (2012) 34:505–12. doi: 10.1016/j.ntt.2012.07.002
21. Solano ME, Holmes MC, Mittelstadt PR, Chapman KE, Tolosa E. Antenatal endogenous and exogenous glucocorticoids and their impact on immune ontogeny and long-term immunity. Semin Immunopathol. (2016) 38:739–63. doi: 10.1007/s00281-016-0575-z
22. Drexhage HA, Dik WA, Leenen PJM, Versnel MA. The immune pathogenesis of type 1 diabetes: not only thinking outside the cell but also outside the islet and out of the box. Diabetes. (2016) 65:2130–3. doi: 10.2337/dbi16-0030
23. Faden M, Holm M, Allred E, Fichorova R, Dammann O, Leviton A. Antenatal glucocorticoids and neonatal inflammation-associated proteins. Cytokine. (2016) 88:199–208. doi: 10.1016/j.cyto.2016.09.015
24. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. (2013) 19:65–73. doi: 10.1038/nm.3042
25. Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes. (2013) 62:2072–7. doi: 10.2337/db12-1345
26. Battaglia M, Petrelli A, Vecchio F. Neutrophils and type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. (2019) 26:201–6. doi: 10.1097/MED.0000000000000485
27. Kadanali S, Ingeç M, Küçüközkan T, Börekçi B, Kumtepe Y. Changes in leukocyte, granulocyte and lymphocyte counts following antenatal betamethasone administration to pregnant women. Int J Gynecol Obstet. (1997) 58:269–74. doi: 10.1016/S0020-7292(97)00085-4
28. Cavalcanti DMH, Lotufo CMC, Borelli P, Ferreira ZS, Markus RP, Farsky SHP. Endogenous glucocorticoids control neutrophil mobilization from bone marrow to blood and tissues in non-inflammatory conditions. Br J Pharmacol. (2007) 152:1291–300. doi: 10.1038/sj.bjp.0707512
29. Jobe AH, Milad MA, Peppard T, Jusko WJ. Pharmacokinetics and pharmacodynamics of intramuscular and oral betamethasone and dexamethasone in reproductive age women in india. Clin Transl Sci. (2019) 13:391–9. doi: 10.1111/cts.12724
30. Fay ME, Myers DR, Kumar A, Turbyfield CT, Byler R, Crawford K, et al. Cellular softening mediates leukocyte demargination and trafficking, thereby increasing clinical blood counts. Proc Natl Acad Sci USA. (2016) 113:1987–92. doi: 10.1073/pnas.1508920113
31. Weber PSD. Mechanisms of glucocorticoid-induced down-regulation of neutrophil L-selectin in cattle: evidence for effects at the gene-expression level and primarily on blood neutrophils. J Leukoc Biol. (2003) 75:815–27. doi: 10.1189/jlb.1003505
32. Perna-Barrull D, Rodriguez-Fernandez S, Pujol-Autonell I, Gieras A, Ampudia-Carrasco RM, Villalba A, et al. Prenatal betamethasone interferes with immune system development and alters target cells in autoimmune diabetes. Sci Rep. (2019) 9:1235. doi: 10.1038/s41598-018-37878-9
33. Fuenfer MM, Herson VC, Raye JR, Woronick CL, Eisenfeld L, Ingardia CJ, et al. The effect of betamethasone on neonatal neutrophil chemotaxis. Pediatr Res. (1987) 22:150–2. doi: 10.1203/00006450-198708000-00009
34. Irakam A, Miskolci V, Vancurova I, Davidson D. Dose-related inhibition of proinflammatory cytokine release from neutrophils of the newborn by dexamethasone, betamethasone, and hydrocortisone. Neonatology. (2002) 82:89–95. doi: 10.1159/000063094
35. Kuypers E, Collins JJP, Kramer BW, Ofman G, Nitsos I, Jane Pillow J, et al. Intra-amniotic LPS and antenatal betamethasone: inflammation and maturation in preterm lamb lungs. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L380. doi: 10.1152/ajplung.00338.2011
36. Kramer BW, Ikegami M, Moss TJM, Nitsos I, Newnham JP, Jobe AH. Antenatal betamethasone changes cord blood monocyte responses to endotoxin in preterm lambs. Pediatr Res. (2004) 55:764–8. doi: 10.1203/01.PDR.0000120678.72485.19
37. Palojärvi A, Andersson S, Turpeinen U, Janér C, Petäjä J. Antenatal betamethasone associates with transient immunodepression in very low birth weight infants. Neonatology. (2013) 104:275–82. doi: 10.1159/000353964
38. Diebel ND, Parsons MT, Spellacy WN. The effects of betamethasone on white blood cells during pregnancy with PPROM. J Perinat Med. (1998) 26:204–7. doi: 10.1515/jpme.1998.26.3.204
39. Kalthoff FS, Chung J, Musser P, Stuetz A. Pimecrolimus does not affect the differentiation, maturation and function of human monocyte-derived dendritic cells, in contrast to corticosteroids. Clin Exp Immunol. (2003) 133:350–9. doi: 10.1046/j.1365-2249.2003.02225.x
40. Kavelaars A, Van Der Pompe G, Bakker JM, Van Hasselt PM, Cats B, Visser GHA, et al. Altered immune function in human newborns after prenatal administration of betamethasone: enhanced natural killer cell activity and decreased T cell proliferation in cord blood. Pediatr Res. (1999) 45:306–12. doi: 10.1203/00006450-199903000-00003
41. Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci USA. (2017) 114:E10418–27. doi: 10.1073/pnas.1713543114
42. Kasat K, Patel H, Predtechenska O, Vancurova I, Davidson D. Anti-inflammatory actions of endogenous and exogenous interleukin-10 versus glucocorticoids on macrophage functions of the newly born. J Perinatol. (2014) 34:380–5. doi: 10.1038/jp.2014.16
43. Ponzin D, Bellini F, Chizzolini C. Antigen presentation is inhibited in vivo by betamethasone. Life Sci. (1995) 56:1595–9. doi: 10.1016/0024-3205(95)00126-Q
44. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
45. Teixeira CJ, Santos-Silva JC, de Souza DN, Rafacho A, Anhe GF, Bordin S. Dexamethasone during pregnancy impairs maternal pancreatic β-cell renewal during lactation. Endocr Connect. (2019) 8:120–31. doi: 10.1530/EC-18-0505
46. Diaz-Jimenez D, Petrillo MG, Busada JT, Hermoso MA, Cidlowski JA. Glucocorticoids mobilize macrophages by transcriptionally up-regulating the exopeptidase DPP4. J Biol Chem. (2020) 295:3213–27. doi: 10.1074/jbc.RA119.010894
47. Alhadj Ali M, Thrower SL, Hanna SJ, Coulman SA, Birchall JC, Wong FS, et al. Topical steroid therapy induces pro-tolerogenic changes in langerhans cells in human skin. Immunology. (2015) 146:411–22. doi: 10.1111/imm.12518
48. Matsui K, Tamai S, Ikeda R. Betamethasone, but not tacrolimus, suppresses the development of Th2 cells mediated by langerhans cell-like dendritic cells. Biol Pharm Bull. (1980) 39:1220–3. doi: 10.1248/bpb.b16-00075
49. La Marca V, Gianchecchi E, Fierabracci A. Type 1 diabetes and its multi-factorial pathogenesis: the putative role of NK cells. Int J Mol Sci. (2018) 19:794. doi: 10.3390/ijms19030794
50. Kotiranta-Ainamo A, Apajasalo M, Pohjavuori M, Rautonen N, Rautonen J. Mononuclear cell subpopulations in preterm and full-term neonates: independent effects of gestational age, neonatal infection, maternal pre- eclampsia, maternal betamethason therapy, and mode of delivery. Clin Exp Immunol. (1999) 115:309–14. doi: 10.1046/j.1365-2249.1999.00795.x
51. Lesko MJ, Lever RS, Mackie RM, Parrott DMV. The effect of topical steroid application on natural killer cell activity. Clin Exp Allergy. (1989) 19:633–6. doi: 10.1111/j.1365-2222.1989.tb02759.x
52. Diepenbruck I, Much CC, Krumbholz A, Kolster M, Thieme R, Thieme D, et al. Effect of prenatal steroid treatment on the developing immune system. J Mol Med. (2013) 91:1293–302. doi: 10.1007/s00109-013-1069-2
53. Jones CA, Nisenbaum R, De Souza LR, Berger H. Antenatal corticosteroid administration is associated with decreased growth of the fetal thymus: a prospective cohort study. J Perinatol. (2020) 40:30–8. doi: 10.1038/s41372-019-0554-z
54. Wallace EM, Ekkel K, Cotter T, Tippetr C, Catalano J. Haematological effects of betamethasone treatment in late pregnancy. Aust N Zeal J Obstet Gynaecol. (1998) 38:396–8. doi: 10.1111/j.1479-828X.1998.tb03095.x
55. Denison FC, Elliott CL, Wallace EM. Dexamethasone-induced leucocytosis in pregnancy. Br J Obstet Gynaecol. (1997) 104:851–3. doi: 10.1111/j.1471-0528.1997.tb12035.x
56. Chabra S, Cottrill C, Rayens MK, Cross R, Lipke D, Bruce M. Lymphocyte subsets in cord blood of preterm infants: effect of antenatal steroids. Biol Neonate. (1998) 74:200–7. doi: 10.1159/000014025
57. Gieras A, Gehbauer C, Perna-Barrull D, Engler JB, Diepenbruck I, Glau L, et al. Prenatal administration of betamethasone causes changes in the T cell receptor repertoire influencing development of autoimmunity. Front Immunol. (2017) 8:1505. doi: 10.3389/fimmu.2017.01505
58. Quinn A, McInerney M, Huffman D, McInerney B, Mayo S, Haskins K, et al. T cells to a dominant epitope of GAD65 express a public CDR3 motif. Int Immunol. (2006) 18:967–79. doi: 10.1093/intimm/dxl033
59. Wong FS, Wen L, Tang M, Ramanathan M, Visintin I, Daugherty J, et al. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. (2004) 53:2581–7. doi: 10.2337/diabetes.53.10.2581
60. Oehling A, Crisci CD, Sanz ML, Subirá ML. Immunosuppressive effect of corticosteroids on rabbit's humoral and cellular response. Allergol Immunopathol. (1976) 4:255–68.
61. Franco Del Prete G, Vercelli D, Tiri A, Maggi E, Rossi O, Romagnani S, et al. Effect of in vitro irradiation and cell cycle-inhibitory drugs on the spontaneous human IgE synthesis in vitro. J Allergy Clin Immunol. (1987) 79:69–77. doi: 10.1016/S0091-6749(87)80019-2
62. Gruver-Yates AL, Quinn MA, Cidlowski JA. Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine B cells during development. Endocrinology. (2014) 155:463–74. doi: 10.1210/en.2013-1473
63. Lill-Elghanian D, Schwartz K, King L, Fraker P. Glucocorticoid-induced apoptosis in early B cells from human bone marrow. Exp Biol Med. (2002) 227:763–70. doi: 10.1177/153537020222700907
64. Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, et al. How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited. Diabetes. (2011) 60:1370–9. doi: 10.2337/db10-1797
65. Fichna M, Fichna P. Glucocorticoids and beta-cell function. Endokrynol Pol. (2017) 68:568–78. doi: 10.5603/EP.2017.0060
66. Rose AJ, Herzig S. Metabolic control through glucocorticoid hormones: an update. Mol Cell Endocrinol. (2013) 380:65–78. doi: 10.1016/j.mce.2013.03.007
67. Sheen JM, Hsieh CS, Tain YL, Li SW, Yu HR, Chen CC, et al. Programming effects of prenatal glucocorticoid exposure with a postnatal high-fat diet in diabetes mellitus. Int J Mol Sci. (2016) 17:533. doi: 10.3390/ijms17040533
68. Sato S, Saisho Y, Inaishi J, Kou K, Murakami R, Yamada T, et al. Effects of glucocorticoid treatment on β- and α-cell mass in Japanese adults with and without diabetes. Diabetes. (2015) 64:2915–27. doi: 10.2337/db15-0151
69. Fine NHF, Doig CL, Elhassan YS, Vierra NC, Marchetti P, Bugliani M, et al. Glucocorticoids reprogram b-cell signaling to preserve insulin secretion. Diabetes. (2018) 67:278–90. doi: 10.2337/db16-1356
70. Ghazalli N, Wu X, Walker S, Trieu N, Hsin LY, Choe J, et al. Glucocorticoid signaling enhances expression of glucose-sensing molecules in immature pancreatic beta-like cells derived from murine embryonic stem cells in vitro. Stem Cells Dev. (2018) 27:898–909. doi: 10.1089/scd.2017.0160
71. Santos-Silva JC, da Silva PMR, de Souza DN, Teixeira CJ, Bordin S, Anhê GF. In utero exposure to dexamethasone programs the development of the pancreatic β- and α-cells during early postnatal life. Life Sci. (2020) 255:117810. doi: 10.1016/j.lfs.2020.117810
72. Bansal A, Alsweiler JM, Oliver MH, Jaquiery A, Phua HH, Dragunow M, et al. Effects of preterm birth induced with or without exogenous glucocorticoids on the ovine glucose-insulin axis. Dev Orig Health Dis. (2020) 1–13. doi: 10.1017/S2040174419000916
73. De Matteo R, Hodgson DJ, Bianco-Miotto T, Nguyen V, Owens JA, Harding R, et al. Betamethasone-exposed preterm birth does not impair insulin action in adult sheep. J Endocrinol. (2017) 232:175–87. doi: 10.1530/JOE-16-0300
74. Zannas AS, Chrousos GP. Epigenetic programming by stress and glucocorticoids along the human lifespan. Mol Psychiatry. (2017) 22:640–6. doi: 10.1038/mp.2017.35
75. Gilbert ER, Liu D. Epigenetics the missing link to understanding β-cell dysfunction in the pathogenesis of type 2 diabetes. Epigenetics. (2012) 7:841–52. doi: 10.4161/epi.21238
76. Seckl JR. Prenatal glucocorticoids and long-term programming. Eur J Endocrinol. (2004) 151(Suppl. 3):U49–62. doi: 10.1530/eje.0.151u049
77. Kelly BA, Lewandowski AJ, Worton SA, Davis EF, Lazdam M, Francis J, et al. Antenatal glucocorticoid exposure and long-term alterations in aortic function and glucose metabolism. Pediatrics. (2012) 129:e1282–90. doi: 10.1542/peds.2011-3175
78. Norberg H, Stålnacke J, Nordenström A, Norman M. Repeat antenatal steroid exposure and later blood pressure, arterial stiffness, and metabolic profile. J Pediatr. (2013) 163:711–6. doi: 10.1016/j.jpeds.2013.03.074
79. Federman DG, Froelich CW, Kirsner RS. Topical psoriasis therapy. Am Fam Physician. (1999) 59:957–62.
80. Hench PS, Kendall EC. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone; compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proc Staff Meet Mayo Clin. (1949) 24:181–97.
81. Walker LSK, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol. (2016) 183:16–29. doi: 10.1111/cei.12672
82. Hong JY, Lim J, Carvalho F, Cho JY, Vaidyanathan B, Yu S, et al. Long-term programming of CD8 T cell immunity by perinatal exposure to glucocorticoids. Cell. (2020) 180:847–61.e15. doi: 10.1016/j.cell.2020.02.018
83. Korhonen LS, Kortesluoma S, Lukkarinen M, Peltola V, Pesonen H, Pelto J, et al. Prenatal maternal distress associates with a blunted cortisol response in rhinovirus-positive infants. Psychoneuroendocrinology. (2019) 107:187–90. doi: 10.1016/j.psyneuen.2019.05.023
84. Vermillion ST, Soper DE, Newman RB. Neonatal sepsis and death after multiple courses of antenatal betamethasone therapy. Am J Obstetr Gynecol. (2000) 183:810–4. doi: 10.1067/mob.2000.108838
85. Wang YC, Tseng HI, Yang SN, Lu CC, Wu JR, Dai ZK, et al. Effects of antenatal corticosteroids on neonatal outcomes in very-low-birth-weight preterm newborns: a 10-year retrospective study in a medical center. Pediatr Neonatol. (2012) 53:178–83. doi: 10.1016/j.pedneo.2012.04.004
86. Dalziel SR, Walker NK, Parag V, Mantell C, Rea HH, Rodgers A, et al. Cardiovascular risk factors after antenatal exposure to betamethasone: 30-Year follow-up of a randomised controlled trial. Lancet. (2005) 365:1856–62. doi: 10.1016/S0140-6736(05)66617-2
87. Greene NH, Pedersen LH, Liu S, Olsen J. Cohort profile: prenatal prescription corticosteroids and offspring diabetes: a national cohort study. Int J Epidemiol. (2013) 42:186–93. doi: 10.1093/ije/dys228
Keywords: prenatal betamethasone, Type 1 diabetes, immune system, β cell, glucocorticoid
Citation: Perna-Barrull D, Gieras A, Rodriguez-Fernandez S, Tolosa E and Vives-Pi M (2020) Immune System Remodelling by Prenatal Betamethasone: Effects on β-Cells and Type 1 Diabetes. Front. Endocrinol. 11:540. doi: 10.3389/fendo.2020.00540
Received: 25 May 2020; Accepted: 03 July 2020;
Published: 11 August 2020.
Edited by:
Teresa Rodriguez-Calvo, Helmholtz Zentrum München, GermanyReviewed by:
Guoqiang Gu, Vanderbilt University, United StatesBarak Blum, University of Wisconsin-Madison, United States
Copyright © 2020 Perna-Barrull, Gieras, Rodriguez-Fernandez, Tolosa and Vives-Pi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Vives-Pi, bXZpdmVzQGlndHAuY2F0