 Rong Yu
Rong Yu Urban Lendahl2
Urban Lendahl2 Monica Nistér
Monica Nistér Jian Zhao
Jian Zhao- 1Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital Solna, Stockholm, Sweden
- 2Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden
Mitochondria are highly dynamic organelles and important for a variety of cellular functions. They constantly undergo fission and fusion events, referred to as mitochondrial dynamics, which affects the shape, size, and number of mitochondria in the cell, as well as mitochondrial subcellular transport, mitochondrial quality control (mitophagy), and programmed cell death (apoptosis). Dysfunctional mitochondrial dynamics is associated with various human diseases. Mitochondrial dynamics is mediated by a set of mitochondria-shaping proteins in both yeast and mammals. In this review, we describe recent insights into the potential molecular mechanisms underlying mitochondrial fusion and fission, particularly highlighting the coordinating roles of different mitochondria-shaping proteins in the processes, as well as the roles of the endoplasmic reticulum (ER), the actin cytoskeleton and membrane phospholipids in the regulation of mitochondrial dynamics. We particularly focus on emerging roles for the mammalian mitochondrial proteins Fis1, Mff, and MIEFs (MIEF1 and MIEF2) in regulating the recruitment of the cytosolic Drp1 to the surface of mitochondria and how these proteins, especially Fis1, mediate crosstalk between the mitochondrial fission and fusion machineries. In summary, this review provides novel insights into the molecular mechanisms of mammalian mitochondrial dynamics and the involvement of these mechanisms in apoptosis and autophagy.
Introduction
Mitochondria are double membrane-bound organelles present in most eukaryotic cells. They are traditionally considered to function as the powerhouses of the cell, generating adenosine triphosphate (ATP) through oxidative phosphorylation, used for cellular chemical energy. Mitochondria have their own genome, known as mitochondrial DNA (mtDNA), which in mammalian cells contains 37 genes that are essential for normal mitochondrial function. The mitochondrial genome encodes 2 rRNAs, 22 tRNAs, and 13 protein subunits that are essential for the oxidative phosphorylation process. A reason why these particular genes are found in the mitochondrial genome is that the encoded proteins are highly hydrophobic and thus would have a problem entering the mitochondria (1–3). Besides these proteins, however, most (99%) of mitochondrial proteins in mammalian cells are encoded by nuclear genes, synthesized in the cytoplasm, and imported into mitochondria (4–7). In addition to producing ATP, mitochondria are involved in regulating numerous cellular activities, including cell-cycle progression and cell proliferation, the maintenance and differentiation of cells, mitophagy, and autophagy, the mitochondria-mediated intrinsic cell-death pathway and transduction of calcium signaling. Additionally, mitochondria are the major organelles generating and detoxifying reactive oxygen species (ROS) to adjust cellular redox homeostasis (8).
Mitochondria are highly dynamic and constantly alter their shape through fusion and fission events. The balance of fission and fusion events is termed mitochondrial dynamics, which is controlled by a number of mitochondria-shaping proteins encoded by genes in the nuclear genome. In addition to mitochondrial morphology, mitochondrial dynamics regulates the size, number, distribution, quality control, and transport of mitochondria in cells. Activated fission and/or inhibited fusion can lead to fragmented mitochondria, whereas active fusion and/or inhibited fission conversely can cause mitochondrial elongation (9–11). Over the last decades, emerging evidence indicates that deregulation of mitochondrial dynamics causes mitochondrial dysfunction, impacting on a broad range of cellular functions, and associated with a number of human diseases (11–17).
In this review, we discuss how mitochondrial fusion and fission is regulated by the mitochondria-shaping proteins, and by other co-factors such as the endoplasmic reticulum (ER) and the actin cytoskeleton, as well as how mitochondrial dynamics influences a variety of cellular biological processes such as apoptosis and mitophagy.
The Mitochondrial Fusion Machinery
The Yeast Mitochondrial Fusion Machinery
Mitochondrial dynamics has been studied in yeast for many years. The mitochondrial fusion machinery characterized in yeast is composed of three key proteins, Fzo1p and Ugo1p, which are anchored in the mitochondrial outer membrane (MOM), and Mgm1p, localized in the mitochondrial inner membrane (MIM) (Figure 1). Depleting any of these proteins results in mitochondrial fragmentation (18–20).
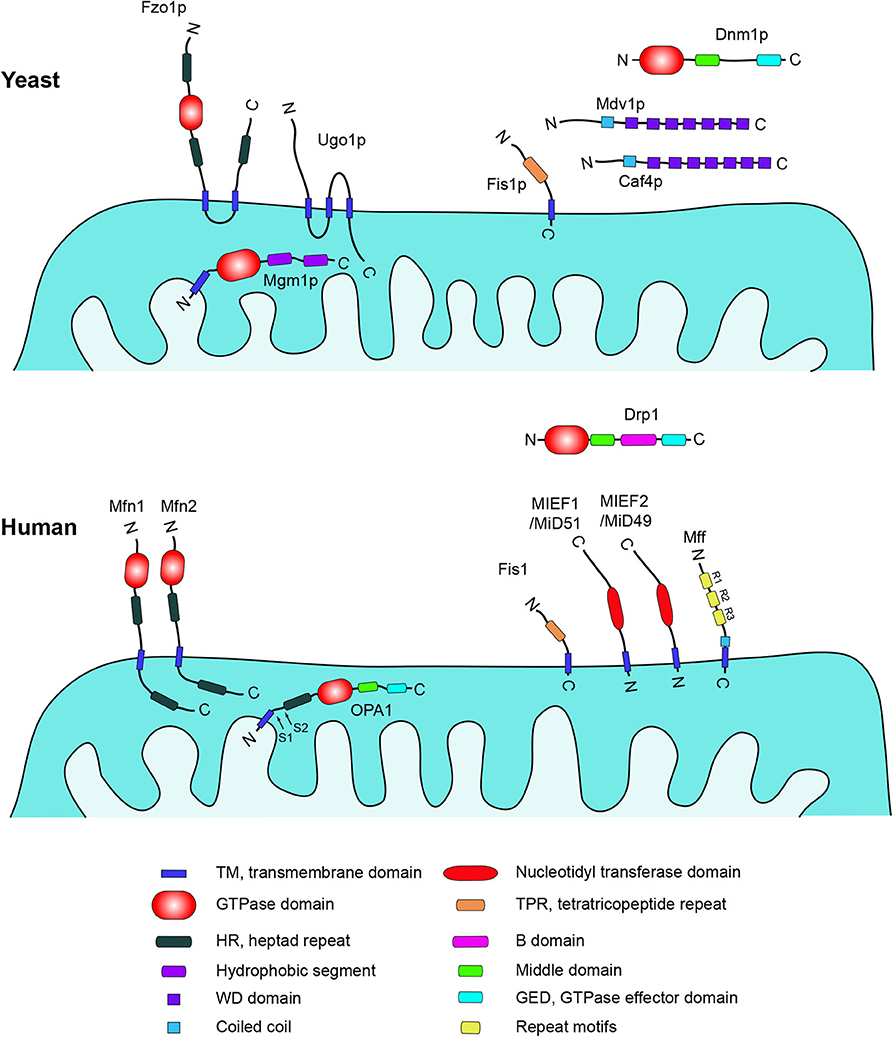
Figure 1. Schematic representation of structures, domains, and locations of key mitochondria-shaping proteins in yeast and human cells. Data were compiled from UniprotKB (http://www.uniprot.org). Yeast: Fzo1p (855aa, P38297), Ugo1p (502aa, Q03327), Mgm1p (881aa, P32266), Dnm1p (757aa, P54861), Mdv1p (714aa, P47025), Caf4p (643aa, P36130), Fis1p (155aa, P40515). Homo sapiens: Mfn1 (741aa, Q8IWA4), Mfn2 (757aa, O95140), OPA1 (960aa, O60313), Drp1 (736aa, O00429), Fis1 (152aa, Q9Y3D6), MIEF1/MiD51 (463aa, Q9NQG6), MIEF2/MiD49 (454aa, Q96C03), Mff (342aa, Q9GZY8). Literature references are provided in the main text.
Fzo1p, a large dynamin-related GTPase, is integrated in the MOM through two adjacent transmembrane (TM) domains near its C-terminus, and with a highly conserved N-terminal GTPase domain facing the cytoplasm. A functional Fzo1p is a prerequisite for fusion of the MOM (21, 22). Mgm1p is also a dynamin-related GTPase, anchored in the MIM via an N-terminal TM domain and a GTPase domain and two hydrophobic segments close to the C terminus, which are exposed in the intermembrane space. Mgm1p is required for both mitochondrial outer and inner membrane fusion, and disruption of the MGM1 gene can result in mitochondrial fragmentation and loss of mtDNA in a Dnm1p-dependent manner (23–25). Both Fzo1p and Mgm1p are evolutionally conserved from yeast to human. There are two homologs of Fzo1p in mammalian cells, termed mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2), and the mammalian homolog of Mgm1p is known as OPA1 (26, 27).
Ugo1p was initially identified through the screening for yeast mutants that lost mtDNA in Dnm1p-dependent mitochondrial division (28). Ugo1p is a mitochondrial outer membrane-anchored protein and contains three TM domains in the middle region. The N-terminus faces the cytosol interacting with Fzo1p directly and the C-terminal domain is exposed to the intermembrane space for Mgm1p binding; thus Ugo1p acts as a molecular bridge between Fzo1p and Mgm1p in mitochondrial fusion (29, 30). Ugo1p is required for both outer and inner mitochondrial membrane fusion (31), and loss of Ugo1p causes mitochondrial fragmentation in yeast (28). Two recent studies identified the nuclear-encoded mitochondrial outer membrane protein SLC25A46 as the mammalian protein most similar to the yeast Ugo1p, but knockdown of SLC25A46 results in mitochondrial hyperfusion, indicating an opposite effect in human mitochondrial dynamics compared to the role of Ugo1p in yeast (32, 33). Yet other proteins are involved in the mitochondrial fusion process in yeast. For example, the F-box protein Mdm30p is essential for Fzo1p ubiquitylation and degradation after GTP hydrolysis in the late stage of outer membrane fusion to maintain fusion-competent mitochondria in yeast, and this step requires Ugo1p (34–36). Pcp1p and Ups1p are required for the processing of Mgm1p to control mitochondrial morphology (37, 38).
The Mammalian Mitochondrial Fusion Machinery
In mammals, there are three key dynamin-related proteins controlling mitochondrial fusion, mitofusin 1 (Mfn1), mitofusin 2 (Mfn2) and optic atrophy 1 (OPA1) (Figure 1). All of them are large dynamin-related GTPases and their activation proceeds via a three-step mitochondrial fusion process: Two mitochondria are tethered and form a docking ring structure around the contact point between the outer membranes. Then, the two outer membranes fuse together triggered by GTP hydrolysis, followed by a final fusion of the two inner membranes [(39, 40); Figure 2A].
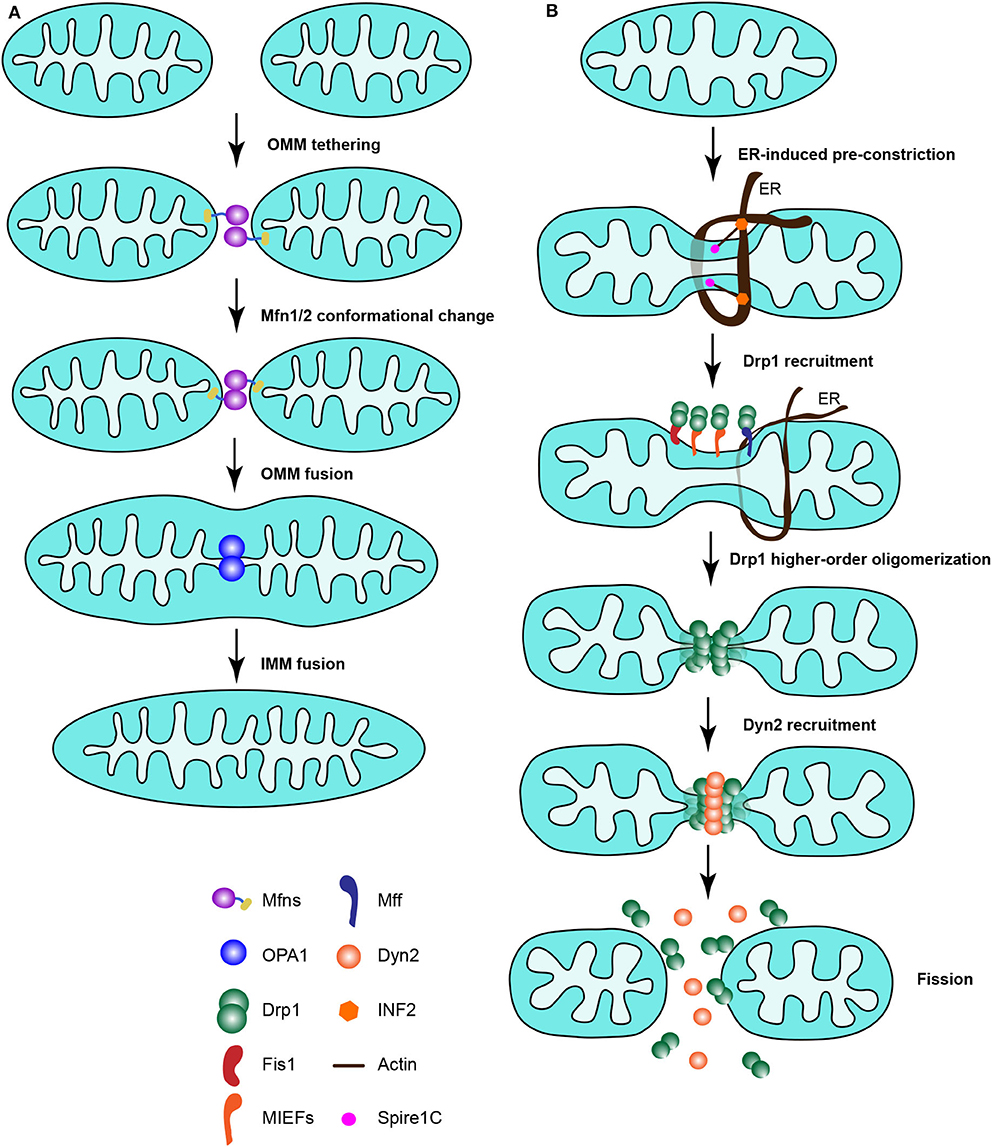
Figure 2. The mitochondrial fusion machinery and canonical Drp1-dependent mitochondrial fission machinery in mammals. (A) The mitochondrial fusion machinery. Two mitochondria are tethered and the MOM fused together by Mfn1/Mfn2, followed by fusion of the IMM by OPA1. (B) The canonical Drp1-dependent mitochondrial fission machinery in mammalian cells. Mitochondria are pre-constricted by the ER, together with actin filaments associated with mitochondria and the ER via Spire1C and INF2, respectively. At the ER constriction site, Drp1 is recruited by its receptors to mitochondria and assembled to form higher-order oligomers around the mitochondrial surface. Then mitochondria are further constricted by Drp1 oligomerization, and Dyn2 is instantly recruited to the constricted site to finalize the scission of mitochondria through GTP hydrolysis.
Both mammalian Mfn1 and its paralog Mfn2 reside in the outer mitochondrial membrane. They are homologs of Drosophila fzo, and similar to the fusion protein Fzo1p in yeast. Based on the topology from the ortholog Fzo1p in yeast, it is generally accepted that both Mfns have a bipartite TM domain, with the N-terminal and C-terminal domains of the protein exposed toward the cytoplasm and with a GTPase domain close to the N-terminus (26). However, it has been demonstrated recently that Mfns in mammals only have a single TM domain, which makes the N-terminal GTPase and HR1 domains face the cytoplasm and the C-terminus with HR2 domain reside in the mitochondrial intermembrane space [(41); Figure 1]. Crystal structure studies prove that both Mfn1 and Mfn2 can form stable homodimers through their GTPase domains, which are critical for outer membrane fusion (42–44). Mfn1 and Mfn2 can also form heterodimers via GTPase domains in a nucleotide-dependent manner (42). Overexpression of either Mfn1 or Mfn2 can induce perinuclear mitochondrial clustering, whereas Mfn1/2-deficient cells contain severely fragmented mitochondria. Furthermore, Mfn1 and Mfn2 can form homo- and hetero-oligomers, and these three types of complexes can work in concert to tether the outer mitochondrial membranes of adjacent mitochondria (45). In spite of that both proteins are essential for the maintenance of mitochondrial morphology, Mfn1 has considerably higher GTPase activity than Mfn2 (46). In addition to controlling mitochondrial morphology, Mfn2 has other specific functions not observed for Mfn1. For instance, Mfn2 is involved in ER-mitochondria connections (47), energy metabolism and insulin signaling (48), mitophagy (49), and apoptosis (50). Strikingly, mutations in MFN2 but not MFN1 can cause Charcot-Marie-Tooth disease type 2A (CMT2A), a peripheral neuropathy characterized by axonal degeneration (51). However, Mfn1 (but not wild-type Mfn2) has been shown to compensate for mitochondrial fusion deficiency caused by MFN2 mutations through the formation of Mfn1-Mfn2 heterooligomeric complexes (52) and rescues axonal degeneration induced by Mfn2 mutations (53). Along these lines, it is further shown that increased levels of Mfn1 expression in the nervous system prevents axonal degeneration in a CMT2A mouse model, suggesting a potential therapeutic strategy for this disease (54). Together, these data suggest that the two mammalian mitofusin proteins, Mfn1 and Mfn2, are functionally correlated but non-redundant.
OPA1 is the mammalian homolog of yeast Mgm1p. It is also a dynamin-related GTPase localized in the MIM and essential for fusion of the MIM. OPA1 was originally discovered by gene mutation screening of autosomal dominant optic atrophy (27). There are at least eight mRNA variants identified from the OPA1 gene through alternative splicing, generating long and short isoforms (55). Membrane–anchored long forms of OPA1 (L-OPA1) undergo proteolytic cleavage at the S1 or S2 sites by a group of proteases residing in the mitochondrial intermembrane space (e.g., PARL, PRELI, Yme1L, and OMA1), resulting in soluble short forms of OPA1 (S-OPA1) (13, 56–58). Recently, crystal structures of short Mgm1p (S-Mgm1) from C. thermophilum (59) and S. cerevisiae (60), orthologs of S-OPA1, have been determined, providing a mechanistic platform for understanding the functions of Mgm1/OPA1 during mitochondrial inner membrane fusion. It has been suggested that both long and short forms of OPA1 are required for inner membrane fusion (57). In contrast, there are reports suggesting that long forms of OPA1 are sufficient for mediating mitochondrial fusion when expressed in Yme1l−/−and Oma1−/−cells. This may indicate that Opa1 processing is not necessary for fusion, and that overexpression of short OPA1 forms trigger mitochondrial fragmentation in Yme1l−/− cells (61). However, recent studies show that S-OPA1 play a role in MIM fusion, and cryo-electron microscopy (cryo-EM) studies reveal that S-OPA1 has a dynamin-like structure and employs dynamin-like power stroke membrane remodeling for MIM fusion (62). Moreover, moderate levels of S-OPA1 together with L-OPA1 are required for efficient and fast membrane pore opening during the fusion process, but excess levels of S-OPA1 inhibit fusion activity (63). OPA1 processing can also be affected by mitochondrial membrane potential and proapoptotic stimuli. For example, Caspase-3 can lead to N-terminal cleavage of OPA1 resulting in mitochondrial fragmentation (64), and CCCP treatment is also known to induce cleavage of long OPA1 forms by OMA1 (65, 66). Knockdown of OPA1 causes mitochondrial fragmentation (67), and overexpression of OPA1 induces mitochondrial elongation (68). Although Mfn1, Mfn2, and OPA1 are all essential for controlling mitochondrial fusion, overexpression of OPA1 can counteract the effect of Mfn2 knockout but not the effect of Mfn1 loss on mitochondrial morphology (68). This confirms that there is a functional difference between Mfn1 and Mfn2.
The Mitochondrial Fission Machinery
The Yeast Mitochondrial Fission Machinery
In yeast, four key proteins are involved in the mitochondrial division process: Dnm1p, Fis1p, Mdv1p, and Caf4p (Figure 1). The dynamin-related GTPase Dnm1p is a central component in the mitochondrial fission machinery, and was initially discovered by screening yeast mutants with defective mitochondrial morphology (69, 70). The re-localization of Dnm1p from the cytosol to mitochondria is a key step in mitochondrial fission. Fis1p is the mitochondrial receptor recruiting Dnm1p to the surface of mitochondria via one of the adaptors Mdv1p or Caf4p, which act as protein bridges between Fis1p and Dnm1p. Then Dnm1p self-assembles around the mitochondrial surface forming spiral-like structures at constriction sites leading to mitochondrial scission (71–74).
Fis1p is anchored in the MOM via the C-terminal tail and the N-terminal domain is facing the cytosol (75). The cytoplasmic domain of Fis1p contains a tetratricopeptide repeat (TPR) domain forming a concave surface, and a short N-terminal helix, which is required for binding and recruiting Mdv1p to the concave surface (76). Both Mdv1p and its paralog Caf4p are soluble cytosolic proteins containing an N-terminal extension, a middle coiled-coil domain, and a C-terminal WD repeat domain. Acting as molecular adaptors and bridges between Dnm1p and Fis1p, these proteins can bind to Dnm1p through the WD repeat domain and then associate with Fis1p through the N-terminal extension (74, 77).
In Dnm1p-null cells, when mitochondrial fission is blocked, mitochondria form long tubular networks and mitochondrial membranes collapse to one side of the cell (69). Fis1p-null mutation also causes mitochondrial reticular formation (75), indicating that both Dnm1p and Fis1p are critical for mitochondrial division in yeast. In yeast cells where only one of the Mdv1p or Caf4p genes has been deleted, mitochondrial morphology is similar to wild type, but in cells with both genes deleted, like in Fis1p-null mutant cells, mitochondria show an elongated and net-like morphology, and most of Dnm1p stays in the cytosol. Interestingly, overexpression of Mdv1p or Caf4p can also inhibit mitochondrial fission, possibly because overexpressed Mdv1p or Caf4p blocks the recruitment of Dnm1p to mitochondria (74, 78). Mdv1p and Caf4p are also endowed with unique functions. For instance, Mdv1p is more active than Caf4p in promoting fission, whereas Caf4p but not Mdv1p in association with Fis1p can determine the polarized localization of Dnm1p clusters on the mitochondrial surface after Dnm1p recruitment (79). Furthermore, a truncated form of Mdv1p lacking the N-terminal extension (that binds with Fis1p), fused with the TM domain of Tom20, can be tethered to the outer mitochondrial membrane, and this is sufficient to recruit Dnm1p to mitochondria and trigger fission in the absence of Fis1p (80).
The Canonical Mitochondrial Fission Machinery in Mammals
Drp1—A Central Regulator of the Canonical Mitochondrial Fission Machinery
In mammals, the regulation of mitochondrial fission is considerably more complicated than in yeast. The dynamin-related protein 1 (Drp1) is the ortholog of yeast Dnm1p and shares 42% homology with Dnm1p (81). Structural analysis demonstrates that Drp1 exists in a dynamic equilibrium between multiple oligomeric states in cells, and the minimal functional assembly subunit is a dimer (82). Like its yeast ortholog, Drp1 acts as the master regulator and plays a central role in mammalian mitochondrial fission. In mammalian cells, Drp1 is primarily present in the cytosol, but can be recruited via any of its mitochondrial receptors (such as Fis1, Mff, MIEF1/MiD51, and MIEF2/MiD49, see Figure 1) to the mitochondrial surface, where it is assembled into higher-order complexes that wrap around the mitochondrial surface triggering mitochondrial fission through its GTPase activity (83). Thus, Drp1 and its four mitochondrial receptors Fis1, Mff, and MIEFs (MIEF1 and MIEF2) constitute the core components of the canonical mitochondrial fission machinery in mammals. In contrast to the strong evolutionary conservation between Drp1 and Dnm1p, their interacting factors, i.e., Mdv1p and Caf4p in yeast vs. Mff and MIEFs in mammals, are quite evolutionarily diverged: The yeast Mdv1p and Caf4p proteins have no mammalian homologs, whereas counterparts of mammalian Mff and MIEFs have not yet been identified in yeast (13). Even though mammalian Fis1 has been identified as the homolog of yeast Fis1p (84, 85), a growing body of evidence suggests that Fis1p and Fis1 have become functionally diverged in yeast and mammals (80, 86–88). Mammalian Fis1 is no longer an essential mitochondrial receptor responsible for recruitment of the cytosolic Drp1 to the mitochondrial surface. Instead, three additional mitochondrial proteins, Mff, MIEF1, and MIEF2, have been identified as major receptors for translocation of Drp1 to mitochondria in mammals (89–92). Another member of the dynamin-related family, Dynamin 2 (Dyn2) promotes membrane remodeling of multiple organelles (93). Recently, it was reported that Dyn2 is recruited to mitochondrial constriction sites transiently after Drp1 puncta accumulation and acts in the final step of Drp1-mediated mitochondrial division (94). However, a recent study suggests that although Dyn2 plays a role in mitochondrial fission, it is not essential for Drp1-mediated fission (95). Further supporting this notion, Drp1 is shown to be essential for mitochondrial and peroxisomal fission, whereas Dnm1, Dnm2 (Dyn2), and Dnm3 are dispensable (96).
Overall, during the mitochondrial fission process, Drp1 recruitment to mitochondria is a critical step, but the mechanisms underlying this process are not fully understood. A number of issues remain to be elucidated, for instance, why multiple mitochondrial receptors of Drp1 simultaneously exist in the cell; whether these receptors work independently of each other or in a coordinating way; and how the post-translational modifications and the oligomeric state of Drp1 impact on the process of mitochondrial division. Below, the different Drp1 receptors in mammals will be discussed in further detail.
The Mitochondrial Receptors for Drp1
In mammalian cells, the recruitment of Drp1 to mitochondria is mediated by four MOM-anchored proteins: fission protein 1 (Fis1), mitochondrial fission factor (Mff), mitochondrial elongation factor 1 (MIEF1/MiD51), and mitochondrial elongation factor 2 (MIEF2/MiD49).
Fis1
Mammalian Fis1 is the ortholog of yeast Fis1p and was initially identified as the receptor for the recruitment of Drp1 to mitochondria in mammals based on the studies of Fis1p. Like yeast Fis1p, mammalian Fis1 is localized to the MOM via its C-terminal TM domain, and with the N-terminal region of Fis1 facing the cytosol (84, 85). Structural analysis shows that the N-terminal region contains a TPR-like core domain, which is different from the typical TPR motif, but still important for binding to other proteins (97). In yeast, Fis1p acts as the receptor for the recruitment of Dnm1p (Drp1 in mammals) to mitochondria through the adaptors Mdv1p or Caf4p, thereby regulating mitochondrial morphology (98, 99). Since Fis1 is evolutionarily conserved from yeast to human, mammalian Fis1 was initially believed to have similar functions to its ortholog Fis1p in yeast, i.e., serving as the receptor for the recruitment of Drp1 to mitochondria, promoting fission. In keeping with a pro-fission role, several early studies showed that overexpression of human Fis1 (hFis1) results in extensive mitochondrial fragmentation, whereas knockdown of hFis1 causes mitochondrial elongation (84, 85, 100–102). However, due to the absence of yeast adaptors Mdv1p and Caf4p in mammals, whether hFis1 promotes mitochondrial fragmentation through a similar mechanism as in yeast has remained quite controversial. For example, it was reported that depletion of hFis1 induces mitochondrial elongation in HeLa cells and in Fis1-null mouse embryonic fibroblasts (MEFs) (86, 103, 104), but does not affect mitochondrial morphology in HCT116 cells (90). Furthermore, increased or decreased levels of Fis1 do not seem to affect the subcellular distribution of Drp1 between the cytosol and mitochondria in mammalian cells. For instance, elevated levels of hFis1 in 293T cells do not affect the subcellular distribution of Drp1, but promote mitochondrial fragmentation (91). Likewise, knockdown of hFis1 in HeLa and HCT116 cells does not reduce levels of Drp1 on mitochondria (90, 104). However, in Fis1-null MEFs, Drp1 puncta on mitochondria are reduced to some extent (86). Taken together, these studies suggest that mammalian Fis1 is not essential for Drp1 recruitment and Drp1-mediated mitochondrial fission, but might play specific roles in some physiological processes or certain types of mammalian cells.
In spite of the facts presented above, however, it is found that overexpression of Fis1 can induce extensive mitochondrial fragmentation in all mammalian cells analyzed. Given the absence of Mdv1p and Caf4p in mammals as well as a minor role of Fis1 in Drp1 recruitment and Drp1-dependent fission, the molecular mechansms underlying mammalian Fis1-induced mitochondrial fragmentation have remained enigmatic. Moreover, human Fis1 cannot rescue the mutant phenotype in yeast cells lacking Fis1p (100), while yeast Fis1p, when expressed in human HeLa cells, is targeted to mitochondria, but does not affect mitochondrial morphology (101). These data support the notion that despite similarities in protein structure, membrane topology and mitochondrial localization, Fis1p in yeast and hFis1 in humans have functionally diverged during evolution. Thus, the precise roles of hFis1 in regulating mitochondrial dynamics remain to be clarified.
However, despite the fact that hFis1 is dispensable for Drp1 recruitment to mitochondria in many conditions, it should be emphasized that hFis1 does play a role in regulating mitochondrial fission by recruiting Drp1 to mitochondria, especially in response to cell stress-induced mitochondrial fragmentation, such as mitophagy/apoptosis-related fission, or pathophysiology-associated fission (105–109). In normal conditions, the interaction between Fis1 and Drp1 is weak in mammalian cells, but the interaction can be enhanced after treatment with different apoptotic or autophagic stimuli, and is accompanied by mitochondrial fragmentation (91, 106, 110–112).
Mff
Mff was originally identified through a small interfering RNA (siRNA) screen in Drosophila melanogaster cells, and it exists in metazoans but not in yeast. The Mff gene generates at least nine different isoforms by alternative splicing (89). Similar to Fis1, Mff has a C-terminal TM domain, by which it is anchored in the MOM, while its N-terminal region facing the cytosol contains three short amino acid repeats (R1-R3 motifs) and a coiled-coil domain. The first 50 N-terminal residues containing R1 and R2 motifs are essential for Drp1 recruitment, and this is also the minimal region required for Drp1-Mff interaction (89, 90, 113). Depletion of Mff in HeLa cells (89, 90) or MEFs (86) severely inhibits mitochondrial fission and largely abrogates Drp1 recruitment to mitochondria, whereas overexpression of Mff in HeLa cells recruits most of Drp1 from the cytosol to mitochondria and induces extensive mitochondrial fission (90). It has become increasingly clear that Mff is the major receptor for Drp1 recruitment to mitochondria and thus actively promotes mitochondrial fission in mammals.
MIEF1 and MIEF2
MIEF1 and its paralog MIEF2 (also known as MiD51 and MiD49) were initially characterized by us and others as novel mitochondrial receptors for Drp1 recruitment to mitochondria in mammals (91, 92, 114). MIEF1 and MIEF2 are highly conserved vertebrate-specific mitochondrial proteins, not yet identified in invertebrates and plants (91, 114). Similar to the mitochondrial receptor Mff (89, 90), overexpression of either MIEF leads to extensive recruitment of the cytosolic Drp1 to mitochondria in a hFis1- and Mff-independent manner (91, 115). In contrast, MIEF overexpression induces mitochondrial elongation rather than fission in most of cells. Most likely, MIEFs act as Drp1 inhibitors sequestering it on the surface of mitochondria and inhibiting its GTPase activity (91, 92, 114, 115).
MIEF1 and MIEF2 are highly similar with respect to protein sequence, sharing 45% amino acid identity in human. Unlike hFis1 and Mff, both MIEF1 and MIEF2 have an N-terminal TM domain to anchor them in the MOM (91, 92, 114). Additionally, MIEF1 and MIEF2 can form homodimers and heterodimers (114). MIEF1 and MIEF2 however also differ in some aspects. For example, biochemical analysis shows that in addition to the monomeric form, MIEF1 appears predominantly as dimers, whereas MIEF2 appears as oligomers. Importantly, the first 1–49 residues including the TM domain are required for oligomerization of MIEF2, whereas the region between residues 109–154, but not the TM domain, is crucial for dimerization of MIEF1 (114). Moreover, their different crystal structures also indicate distinct functions. Both proteins have a nucleotidyl transferase domain, but MIEF1 can bind nucleotide diphosphates (ADP and GDP), while MIEF2 does not, and MIEF1 binding to ADP can stimulate Drp1 oligomerization, self-assembly and its GTPase activity (116–118). Interestingly, treatment of MIEF1- or MIEF2-overexpressing cells with antimycin A, an inhibitor of complex III of the electron transport chain, leads to mitochondrial fragmentation in cells overexpressing MIEF1 but not in cells overexpressing MIEF2. Moreover, MIEF1-induced fission requires ADP binding to MIEF1 in this process (86, 117). A recent cryo-EM structural analysis has revealed four interfaces of Drp1 that mediate the interaction with MIEFs, providing a structural basis for Drp1 recruitment to mitochondria (119).
Interplay Between Different Drp1 Receptors in Mitochondrial Dynamics
An increasing number of studies have suggested that Mff and MIEFs play a significant role in recruitment of the cytosolic Drp1 to mitochondria in mammals, but the action of MIEFs seems to be more complicated than that of Mff in regulating mitochondrial fission and fusion. In general, it is observed that Mff overexpression leads to the extensive recruitment of Drp1 on mitochondria promoting mitochondrial fission, whereas depletion of Mff reduces Drp1 on mitochondria resulting in mitochondrial elongation. In contrast, the mechanisms by which MIEFs can regulate mitochondrial morphology remain puzzling with discordant results. For example, in 293T and HeLa cells, expression of exogenous MIEF1 or MIEF2 induces mitochondrial elongation. Both MIEF1 and MIEF2 can interact with and recruit Drp1 to mitochondria and distribute in a punctate manner on the mitochondrial surface (91, 114). In Cos-7 monkey cells, expression of exogenous MIEF1 or MIEF2 also leads to mitochondrial elongation and accumulation of Drp1 on mitochondria, however, at low levels of the MIEF protein, mitochondrial morphology is relatively normal (92). Further studies show that long-term expression of MIEF1 results in a variety of morphological changes of mitochondria from fragmentation to a network state, suggesting that low levels of exogenous MiD51/MIEF1 can increase fission events, whereas at high expression levels, fission events are inhibited, resulting in mitochondrial fusion (120). On the other hand, knockdown of MIEF1 and MIEF2 by siRNA leads to inconsistent effects on mitochondrial morphology. It was reported that in Cos-7 cells, knockdown of both MIEF genes is required to cause a mitochondrial fusion phenotype (92). In line with this, knockdown of both MIEFs or either of the two genes similarly induces mitochondrial elongation in MEFs (86). However, it was also reported that in HeLa cells, down-regulation of MIEF1 by siRNA resulted in mitochondrial fission (91), while complete knockout of either MIEF alone, or double-knockout of both MIEF1/2 in HeLa cells caused mitochondrial elongation (121). The reasons for these inconsistent results can be that the different cell types used for experiments might have different endogenous levels of the fission/fusion proteins. Furthermore, it has been experimentally shown that introduction of low/intermediate or high levels of MIEF receptor proteins in cells can lead to opposite effects as described in detail below, possibly due to competitive Drp1 binding to the different receptor populations available (122).
It should be stressed, however, that the four Drp1 receptors Mff, MIEF1, MIEF2, and hFis1 are usually expressed simultaneously in mammalian cells. Moreover, each receptor is able to independently interact with and recruit Drp1 to mitochondria. This seems to imply a redundant mode of action for the Drp1 receptors in mammals, but these receptors appear to have an additive effect on Drp1 accumulation on mitochondria. However, whether these receptors work coordinately in the process of Drp1 recruitment is largely unclear. A recent study by Yu et al. reports that MIEFs can interact with both Drp1 and Mff and act as adaptors linking Drp1 and Mff in a trimeric protein complex, thereby facilitating a direct interaction between Mff and Drp1 (122). Ablation of both MIEFs largely reduces the association of endogenous Mff with Drp1, and also greatly impairs the Drp1 recruitment and accumulation on mitochondria mediated by Mff overexpression (122). This indicates that endogenous MIEFs regulate Mff-mediated mitochondrial recruitment of Drp1, although Mff also can serve as an independent receptor for Drp1. In contrast, depletion of endogenous Mff does not affect the association of MIEFs with Drp1 and neither has any evident impact on MIEF overexpression-induced recruitment and accumulation of Drp1 on mitochondria, indicating that Drp1 recruitment by MIEFs occurs independently of Mff (122). However, increased levels of MIEFs can decrease the association between Drp1 and Mff, and conversely, elevated expression of Mff reduces the interaction of MIEFs with Drp1. These results suggest that Mff and MIEFs compete for the binding to Drp1. Based on this, we have proposed a working model to illustrate how MIEFs and Mff coordinately balance mitochondrial dynamics (Figure 3). When higher levels of MIEFs exist in cells, Drp1 is sequestered in Drp1-MIEF-Mff and/or Drp1-MIEF complexes, inhibiting a direct Drp1-Mff binding and resulting in mitochondrial fusion (Figure 3A). When MIEFs are present at moderate levels in cells, Mff can receive sufficient Drp1 via MIEFs acting as a molecular bridge facilitating a direct interaction between Mff and Drp1, thereby maintaining normal and balanced mitochondrial morphology (Figure 3B). However, in the absence of MIEFs, Mff cannot directly capture sufficient Drp1 from the cytosol by itself, thus the balance shifts to mitochondrial fusion, resulting in mitochondrial elongation (Figure 3C).
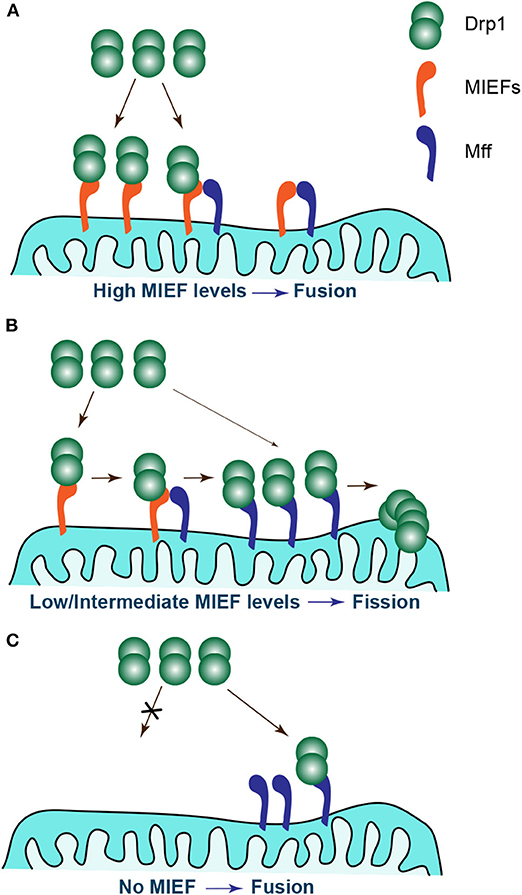
Figure 3. Different levels of MIEFs coordinate with Mff to regulate the balance of mitochondrial dynamics. (A) At high levels of MIEFs, Drp1 is sequestered in Drp1-MIEF-Mff and/or Drp1-MIEF complexes on mitochondria, inhibiting a direct Drp1-Mff interaction and resulting in mitochondrial fusion. (B) At moderate levels of MIEFs, Mff receives sufficient Drp1 facilitating a direct Drp1-Mff interaction via MIEFs acting as a molecular bridge, and a normal balance of mitochondrial dynamics is maintained. (C) In the absence of MIEFs, Mff cannot directly capture sufficient Drp1 from the cytosol by itself, thus the balance shifts to mitochondrial fusion, resulting in mitochondrial elongation.
In addition to the interplay between MIEFs and Mff regulating Drp1 recruitment as mentioned above, accumulating evidence suggests that MIEFs play a role in regulating hFis1-induced mitochondrial fragmentation (91, 114). It is reported that MIEFs exhibit a robust interaction with hFis1, in a manner independent of the interaction between MIEFs and Drp1. Thus, elevated levels of MIEFs partially reverse hFis1-induced mitochondrial fragmentation, whereas increased levels of hFis1 reduce the interaction between Drp1 and MIEF1 or MIEF2, respectively, and also partially reverse MIEF overexpression-induced mitochondrial elongation (91, 114). However, it is still largely unclear how MIEFs and hFis1 can regulate mitochondrial morphology via their mutual interactions.
Overall, the recruitment of cytosolic Drp1 to mitochondria is a key step in Drp1-mediated fission. Due to the simultaneous existence of four Drp1 receptors on mitochondria, the regulation of Drp1 recruitment and subsequent fission induction appears to be subject to a much more complex regulation in mammals than in yeast. Therefore, it will be an important topic for future research to further explore the molecular mechanisms mediating the interplay between different Drp1 receptors, in order to better understand their functions in the regulation of mitochondrial dynamics in mammals.
Crosstalk Between the Mitochondrial Fission and Fusion Machineries
Mitochondrial morphology is dynamically regulated through the two counteracting apparatuses on mitochondria, i.e., the fission and fusion machineries. It is generally believed that the pro-fission proteins (such as Drp1, Fis1, Mff, and MIEFs) and the pro-fusion proteins (such as Mfn1, Mfn2, and OPA1) separately work in the fission and fusion machineries. However, emerging evidence suggests a crosstalk between the fission and fusion machineries. For example, it was reported that Drp1 colocalizes with Mfn2 and Bax at mitochondrial fission sites during apoptosis (123). Interestingly, another study suggests that Drp1 can interact with Mfn2 on the mitochondrial surface and overexpression of Drp1 facilitates mitochondrial tethering and fusion in Mfn2- or Mfn1-deficient cells, indicating that Drp1 participates in mitochondrial fusion in addition to acting as a pro-fission protein (124). Anand et al. provide additional evidence for a crosstalk between the fission and fusion machineries through studying the actions of the long and short forms of OPA1 in mitochondrial dynamics. The short forms (produced by processing) of OPA1 are found to trigger mitochondrial fragmentation and colocalize with the fission machinery, suggesting that the processing of OPA1 can regulate the balance of mitochondrial fission and fusion (61).
It was recently observed that hFis1 overexpression can induce extensive mitochondrial fragmentation even in the absence of Drp1 or/and Dyn2 (88). This indicates that Drp1/Dyn2 is largely dispensable for hFis1-induced mitochondrial fragmentation, i.e., the existence of Fis1-dependent but Drp1/Dyn2-independent mitochondrial fission pathways in mammals. hFis1 was found to robustly interact with the pro-fusion GTPases Mfn1, Mfn2, and OPA1 at endogenous levels, which supports the idea that hFis1 regulates mitochondrial dynamics via affecting the fusion machinery. In line with this notion, hFis1 reduces the GTPase activity of Mfn1, Mfn2, and OPA1 in vitro. Overexpression of hFis1 reduces mitochondrial fusion, whereas knockdown of hFis1 enhances exchange of the matrix content between mitochondria regardless of whether Drp1 is present or absent in cells. Disruption of the fusion machinery by CRISPR-Cas9 genome editing-based knockout technology phenocopied the hFis1 overexpression-induced mitochondrial fragmentation phenotype. Overall, several lines of evidence suggested that hFis1 can induce mitochondrial fragmentation by inhibiting the activity of the mitochondrial fusion machinery (88). This work emphasizes the importance of hFis1 in bidirectional regulation of the mitochondrial fission and fusion machineries and highlights that hFis1, known as a pro-fission factor, is not limited to promoting mitochondrial fission; it can also actively inhibit mitochondrial fusion (Figure 4).
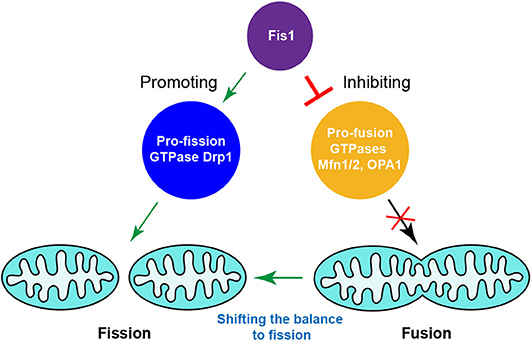
Figure 4. Fis1-mediated crosstalk between the mitochondrial fission and fusion machineries in mammals. Fis1, as a pro-fission factor, is not limited to promoting mitochondrial fission, simultaneously inhibiting the activities of pro-fusion GTPases to shift the balance to fission.
Collectively, all these data provide evidence for a functional crosstalk between the mitochondrial fission and fusion machineries, and suggest that proteins known as components of the fission machinery may act concurrently in the fusion machinery, and likewise, fusion proteins may also affect the fission machinery. While many details remain largely unknown, the bidirectional regulation of mitochondrial morphology through functional interactions between fission and fusion proteins represents an emerging field of research in mammalian mitochondrial dynamics. Supporting this notion, a recent study reveals that the fission and fusion machineries are present at the same ER-mitochondria membrane contact sites to regulate mitochondrial morphology (125).
Roles of the Endoplasmic Reticulum in Mitochondrial Dynamics
The endoplasmic reticulum (ER) communicates with mitochondria through mitochondria-associated ER membranes (MAMs) in yeast and mammals, and these connections have been shown to participate in different physiological functions, such as phospholipid synthesis, Ca2+-mediated signal transduction, protein import, mitochondrial distribution and mitophagy (126–128). It is reported that some of the mitochondria-shaping proteins are involved in this ER-MAM intercommunication. The mitochondrial profusion protein Mfn2 is localized on both mitochondria and the ER, and Mfn2 on the ER associates with Mfn1/2 on mitochondria and tethers the ER and mitochondria together to maintain efficient mitochondrial Ca2+ uptake, which is essential for ATP production (47).
The ER also plays an active role in mitochondrial division. ER tubules were observed to wrap around mitochondria, mark the prospective sites of mitochondrial division and reduce the mitochondrial diameter by about 30% before Drp1 recruitment. The ER tubules mainly occur at positions of Drp1 and Mff foci on mitochondria. In fact, ER tubules also mark positions of mitochondrial constriction in the absence of Mff or Drp1 (129). In addition to Drp1 and Mff, two other receptors of Drp1, i.e., MIEF1 and MIEF2, are also observed at mitochondria-ER contact sites, and co-localized with other fission proteins, such as Drp1 and Mff. However, <40% of observed mitochondria-ER contacts at MIEF foci are constriction sites, implicating that MIEFs are not the essential factors to determine ER-mitochondria constriction sites (120). Furthermore, MIEFs require the presence of Drp1 to form foci, whereas Mff can form foci in cells lacking Drp1 (118, 129). Another known Drp1 receptor, Fis1, has been shown in C. elegans to enter into a complex containing Drp1, Mff, and ER proteins at the ER-mitochondrial interface during stimulation of mitophagy (112). Strikingly, it is recently reported that endoplasmic reticulum–associated degradation (ERAD), an ER quality control mechanism, regulates mitochondrial dynamics (130). Additionally, in human cells, a subset of ER-mitochondria contacts spatially couple mtDNA replication with downstream mitochondrial division (131). However, it is still unclear to what extent ER tubules are essential for mitochondrial fission and fusion.
Roles of the Actin Cytoskeleton in Mitochondrial Dynamics
Besides the ER, the actin cytoskeleton is also involved in regulating mitochondrial fission (40, 132). The cycling of actin assembly and disassembly around mitochondrial subpopulations efficiently promotes local Drp1-dependent mitochondrial fission. However, inhibiting Drp1 activity by transfection of Drp1-K38A does not affect actin cycling onto mitochondrial subpopulations as observed through live-cell imaging (133). Furthermore, some actin-related proteins play important roles in mitochondrial fission. An ER-localized actin regulator, INF2 (inverted formin 2) induces actin filaments to drive the initial mitochondrial constriction and then promotes Drp1 recruitment to ER-mitochondria constriction sites (134). An isoform of the actin-nucleating protein Spire1C, localized to the MOM, interacts with INF2, promoting actin assembly at the mitochondrial surface. Disturbing Spire1C or its formin-binding activities can affect mitochondrial constriction and further mitochondrial fission (135). Myosin II plays similar roles in mitochondrial fission, it is enriched at mitochondrial constriction sites, and deletion of Myosin II reduces Drp1 accumulation on mitochondria (136). Interestingly, silencing of the cytoskeletal component septin 2 (Sept2) also decreases the association of Drp1 with mitochondria and increases the average distance between Drp1 clusters (137). In addition, knockdown of the actin-regulating factors cortactin, cofilin, or the Arp2/3 complexes results in elongated mitochondria and disassembles mitochondrial F-actin around mitochondrial subpopulations (133, 138).
The proposed models of mitochondrial fission in mammalian cells are summarized in Figure 2B. Mitochondria are pre-constricted by the ER, together with actin filaments that associate with mitochondria by the action of Spire1C and with the ER by INF2. At the ER constriction site, Drp1 is recruited by its receptors to mitochondria and assembled to form higher-order oligomers around the mitochondrial surface. Then mitochondria are further constricted by Drp1 oligomers, and Dyn2 is recruited to the constricted site instantly to finalize the scission of mitochondria through GTP hydrolysis. In summary, it is a great challenge to understand how the ER, actin cytoskeleton and Drp1 receptors interplay and coordinate their actions to mediate mitochondrial division.
Roles of Membrane Phospholipids in Mitochondrial Dynamics
In addition to the roles of the ER and actin cytoskeleton in regulating mitochondrial dynamics, mitochondrial membrane phospholipids also communicate with the fusion/fission machinery. Phospholipids are the fundamental building blocks of mitochondrial membrane bilayers. Emerging evidence indicates that at least two types of phospholipids, phosphatidic acid (PA) and cardiolipin (CL), play important roles in mitochondrial dynamics. PA is transported mainly from the ER to the mitochondrial inner membrane, where PA can be converted to CL via multiple enzymatic reactions (139). Some of the CL synthesized in the inner membrane can also be transported to the outer membrane. It has been reported that the pro-fission GTPase Drp1 on the outer membrane directly interacts with CL, and the B insert (also called the variable domain) of Drp1 is responsible for its interaction with CL (140, 141). This interaction between Drp1 and CL stimulates both oligomerization and GTPase activity of Drp1, enhancing mitochondrial division (82, 142, 143). In the outer membrane, CL can be converted back to PA by the MOM-localized phospholipase D (MitoPLD) (144). Drp1 also interacts with PA in the outer membrane, which restrains Drp1 activity, leading to mitochondrial fusion (142). Along these lines, MitoPLD interacts with Drp1, Mfn1, and OPA1, inhibiting Drp1-mediated fission and promoting mitofusin-dependent fusion (142, 144). There is evidence showing that CL located in the inner membrane is directly involved in OPA1-mediated fusion (145), while PA that is generated through the cleavage of CL by MitoPLD in the outer membrane participates in Mfn-mediated fusion (144). Moreover, overexpression of MitoPLD induces mitochondrial fusion, while its knockdown inhibits fusion resulting in mitochondrial fragmentation (144). In line with this, overexpression of lipin 1b [a phosphatase that dephosphorylates PA to form diacylglycerol (DAG)] results in mitochondrial fragmentation (142, 146). Similarly, overexpression of PA-PLA1 (PA-preferring phospholipase A1), which hydrolyzes PA to produce lysoPA, triggers mitochondrial fragmentation, whereas knockdown of PA-PLA1 induces elongation (147). CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation (148).
Overall, it has become increasingly clear that the interactions between the fusion/fission proteins and the phospholipids in mitochondrial membranes add another layer of complexity to the regulation of mitochondrial fusion-fission dynamics and contributes significantly to our understanding of this process (149–151).
DRP1-Independent Mitochondrial Fission Pathways in Mammals
Accumulating data suggest that apart from the canonical Drp1-dependent mitochondrial fission, there may be additional Drp1-independent mechanisms that contribute to mitochondrial division in mammalian cells, as partly described above (Figure 5). Several previous studies show that Fis1 is involved in such a Drp1-independent mitochondrial fission pathway. Onoue et al. reports that Fis1 interacts with TBC1D15 (a GTPase activating protein for the small GTPases Rab7 and Rab11) and recruits the cytosolic TBC1D15 to mitochondria. Fis1, together with TBC1D15, regulates mitochondrial morphology independently of Drp1 (152). This finding is further supported by a recent study showing that Fis1 mediates crosstalk between mitochondria and lysosomes via recruiting TBC1D15 to the mitochondrial surface, and in turn TBC1D15 interacts with lysosomal GTP-bound Rab7 at mitochondria-lysosome contact sites to drive hydrolysis of Rab7-GTP, thereby regulating both mitochondrial and lysosomal dynamics independently of Drp1 (87, 153). Although these findings that the Fis1-TBC1D15-Rab7 interaction involves mitochondria-lysosome contact sites can largely promote our understanding of how Fis1 functions in regulating mitochondrial dynamics, it still seems difficult to fully understand the mechanisms by which Fis1 overexpression induces extensive mitochondrial fragmentation.
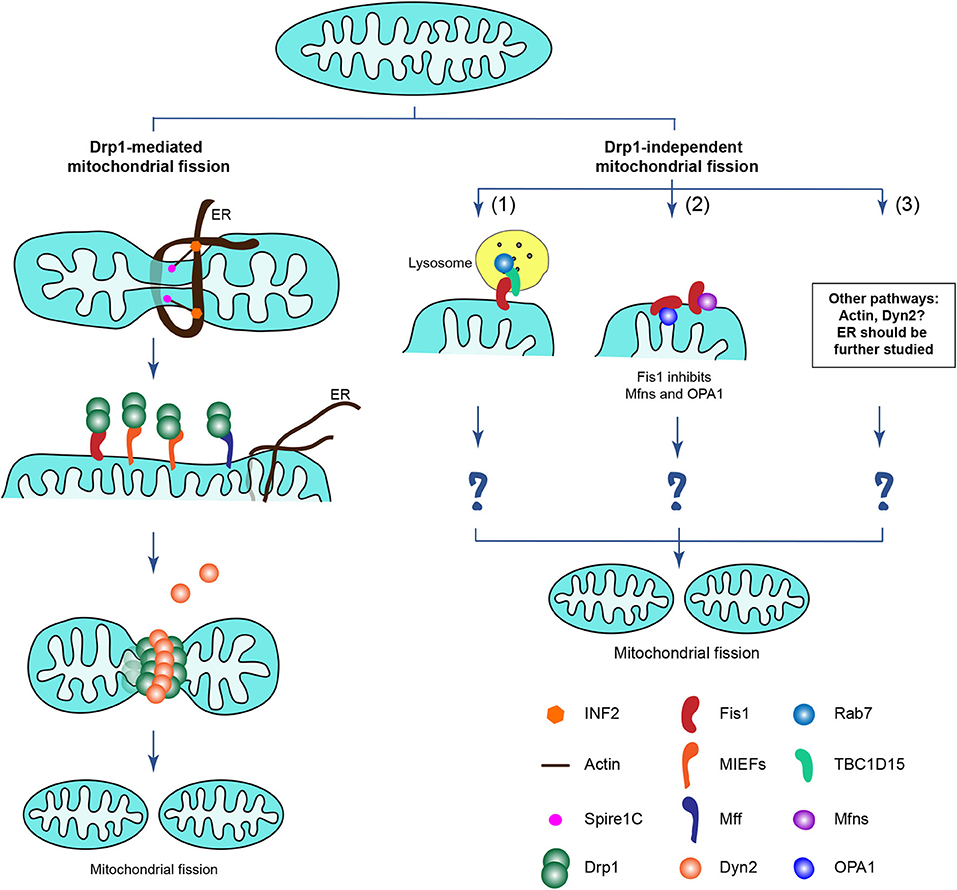
Figure 5. Proposed potential mechanisms driving mammalian mitochondrial fission, including Drp1-dependent and Drp1-independent mitochondrial fission. (Left) The Drp1-dependent mitochondrial fission as described in Figure 2B, including Drp1 and its four mitochondrial receptors Fis1, Mff, MIEF1, and MIEF2. (Right) Apart from the canonical Drp1-dependent mitochondrial fission, there may be multiple Drp1-independent mechanisms contributing to mitochondrial division in mammals although little is known about their details in the regulation of mitochondrial division. Emerging evidence implies that the Drp1-independent mitochondrial fission may involve: (1) Fis1 together with TBC1D15 and Rab7 regulate both mitochondrial and lysosomal dynamics, which likely is important in mitophagy; (2) Fis1 binds to the pro-fusion GTPases Mfn1/2 and OPA1 and inhibits the activity of the fusion machinery, thereby shifting mitochondrial morphology to a fission phenotype, probably via Drp1-dependent and -independent mechanisms (the details in this process still remain poorly understood); (3) The actin cytoskeleton and Dyn2, as well as the ER may have Drp1-independent functions in regulating mitochondrial division (the details are unclear).
In a more recent study mentioned above, hFis1 overexpression was reported to induce extensive mitochondrial fragmentation regardless of the presence or absence of Drp1 (88), further suggesting that Drp1 is dispensable for hFis1-mediated mitochondrial fragmentation. Further analysis showed that hFis1 binds to and inhibits the pro-fusion GTPases Mfn1/2 and OPA1, thereby reducing the activity of the fusion machinery in Drp1-deficient cells, shifting mitochondrial morphology to a fission phenotype (88). Given the importance of the Fis1-TBC1D15-Rab7 involvement in a Drp1-independent mitochondrial fission pathway, ablation of TBC1D15 would be expected to rescue the hFis1 overexpression-induced fragmentation phenotype, but this still remains to be explored. Thus, the underlying mechanisms by which hFis1 can induce mitochondrial fragmentation in the absence of endogenous Drp1 remains to be further explored.
The ER and the actin cytoskeleton have been reported to be involved in mitochondrial dynamics in cooperation with Drp1 (129, 132, 134), but whether the ER and actin cytoskeleton play a role in Drp1-independent fission is unknown. Emerging evidence, however, suggests that F-actin depolymerization significantly prevents hFis1 overexpression-induced mitochondrial fragmentation in a Drp1-independent manner, implying that the actin cytoskeleton plays a Drp1-independent role in regulating mitochondrial fission (88). The ER is generally considered to be the first step of Drp1-mediated mitochondrial fission, promoting mitochondrial constriction before Drp1 recruitment (129), but whether the ER induces mitochondrial scission directly, independently of Drp1 needs to be further explored.
Dyn2 has been reported to work at the final step of Drp1-dependent mitochondrial scission (94), but another study suggests that Drp1-mediated mitochondrial division still occurs in Dyn2-deficient cells (95), indicating Dyn2 is not essential for Drp1-dependent fission. However, a recent report presents that Dyn2 knockdown partially reversed the mitochondrial fission phenotype that was induced when both fission and fusion machineries had simultaneously been destroyed by double knockout of Drp1 and OPA1 through CRISPR/Cas9 technology (88), indicating that Dyn2 may act via a separate mechanism to promote mitochondrial fission when Drp1-mediated fission is blocked.
Collectively, it is becoming increasingly clear that there may be multiple Drp1-independent mechanisms regulating mitochondrial division in mammalian cells. Although substantial progress has been achieved through studies of Drp1-mediated mitochondrial fission, little is known about the regulation of mitochondrial division in mammalian cells lacking Drp1. Undoubtedly, elucidating the underlying mechanisms that drive Drp1-independent mitochondrial division is of fundamental importance to fully understand mammalian mitochondrial dynamics.
Crosstalk Between Mitochondrial Dynamics and Other Cellular Processes
Accumulating evidence indicates that the carefully orchestrated balance of mitochondrial fusion and fission is critical for maintaining a healthy population of mitochondria. Fission allows the exclusion of damaged mitochondria via mitophagy and mitochondrial biogenesis, whereas fusion allows mitochondria to exchange their substance including proteins and mtDNA among individual mitochondria (154). Abnormal mitochondrial dynamics is directly associated with mitochondrial dysfunction, which impacts on a wide range of cellular processes, such as mitochondrial transport and biogenesis, cell-cycle regulation, cell proliferation and differentiation, energy metabolism, ROS production, Ca2+ signaling, mtDNA maintenance, mitochondrial quality control via autophagy (mitophagy), and ultimately programmed cell death (155–160). Moreover, some mitochondrial fission and fusion proteins are known to be critical for embryonic development in mice (45, 161–163), further emphasizing that functions of mitochondrial dynamics go beyond the appearance of mitochondria and have important physiological consequences. In this review, we will focus on the potential roles of mitochondrial dynamics in apoptosis and mitophagy.
Mitochondrial Dynamics and Apoptosis
Apoptosis, a programmed cell death, occurs in physiological and pathological conditions, and can be activated through intrinsic or extrinsic mechanisms. It is well-established that the Bcl-2 protein family plays crucial roles in the regulation of apoptosis. The Bcl-2 family includes both pro-apoptotic proteins (such as Bax, Bak, Bid, Bad, and Bik) and the anti-apoptotic proteins (such as Bcl-2, Bcl-XL, Bcl-W, and MCL1), which promote and inhibit programmed cell death, respectively (164–167).
Mitochondria have been shown to be involved in the intrinsic apoptotic pathway (168). Emerging data indicate that mitochondrial dynamics participates in the regulation of cell death pathways and remodeling of the mitochondrial network takes place in response to cellular stress, such as hypoxia, drug treatments, and in various pathological conditions. During apoptosis, mitochondria undergo extensive fragmentation and some proteins of the mitochondrial fusion/fission machinery are directly involved in the regulation of apoptosis (169, 170). In general, the prevalent idea is that elongated mitochondria confer cellular resistance to apoptosis, whereas fragmented mitochondria make cells sensitive to apoptotic stimuli (169, 171). For example, knockdown of the pro-fission proteins hFis1 or Drp1 leads to mitochondrial elongation and resistance to different apoptotic stimuli, whereas depletion of the pro-fusion proteins Mfn1, Mfn2, or OPA1 leads to mitochondrial division and makes cells more sensitive to apoptosis (170, 172). Conversely, Mfn2 overexpression reduces sensitivity of cells to radical-induced apoptosis (173). Upon apoptotic stimulation, Drp1 is recruited to the mitochondrial outer membrane, where it co-localizes with Bax and Mfn2 at fission sites, and Drp1 is required for apoptotic mitochondrial fission (104, 123, 174). In agreement with the notion that mitochondrial dynamics regulates the sensitivity of cells to apoptotic stimuli, it was reported that overexpression of MTGM/ROMO1 (a MIM anchored fission protein) results in fragmented mitochondria and triggers a significant release of the death factor Smac/Diablo from mitochondria to the cytosol, but not of other death factors (for instance cytochrome c, AIF, or Omi/HtrA2), and does not lead to spontaneous apoptosis. However, down-regulation of MTGM results in elongated mitochondria and increases cell proliferation as well as resistance of cells to apoptotic stimuli (175). Furthermore, it was reported that MIEFs are also involved in the regulation of apoptosis and autophagy. Expression of exogenous MIEF1 induces elevated autophagic activity and decreases the sensitivity of cells to apoptotic stimuli. Conversely, human cells depleted of MIEF1 are more sensitive to apoptotic stimuli (91, 176). However, loss of both MIEF1 and MIEF2 together with Mff in MEFs confers resistance to apoptosis (115). Additionally, Drp1-mediated mitochondrial division controls cristae remodeling through MIEF1/2 during intrinsic apoptosis (121). Moreover, the OMM-associated E3 ubiquitin ligase MARCH5 coupled with MIEF2 controls Drp1-dependent mitochondrial fission and cell sensitivity to stress-induced apoptosis (177).
In healthy cells, Bax is normally localized predominantly in the cytosol, but during apoptosis, Bax translocates from the cytosol to the surface of mitochondria (178), where Bax, in coordination with Bak, results in mitochondrial outer membrane permeabilization (MOMP) (179). Several lines of evidence show that some mitochondria-shaping proteins participate in the Bax translocation. For example, hFis1 is required for translocation of Bax from the cytosol to the surface of mitochondria (104), Drp1 is required for activation and oligomerization of Bax (180, 181), and OPA1 regulates cristae remodeling and triggers the release of cytochrome c to the cytosol (182, 183). More recently, MIEF1 was reported to regulate the mitochondrial translocation and oligomerization of Bax via BCL2L1 (also a member of Bcl-2 family), but independently of Drp1 (176).
On the other hand, extensive data suggest that the Bcl-2 family also modulates mitochondrial fusion/fission dynamics (184, 185). Bax/Bak are not just involved in Drp1-mediated mitochondrial fission during apoptosis (186–188), but also regulate normal mitochondrial fusion by affecting Mfn2 in healthy cells (189). Intriguingly, it is reported that Bak can induce mitochondrial fragmentation during apoptosis by differentially interacting with Mfn1 and Mfn2 (190). Bcl-XL interacts with Drp1 to promote Drp1-mediated fission and increases the rates between fission and fusion in normal neurons (191, 192). MCL1 is known to have two splice isoforms, and the long isoform (MCL1L) is anchored to the MOM as an anti-apoptotic protein binding to Drp1, while the short isoform (MCL1S) acts as a pro-apoptotic factor. A lower MCL1L/S ratio inhibits Drp1 translocation from the cytosol to mitochondria, leading to mitochondrial elongation (193).
In addition, mitochondrial membrane phospholipids have been suggested to play a role in the regulation of apoptosis (194), and accumulating evidence suggests that CL acts as an interaction platform to recruit apoptotic factors such as tBid and Bax (195–198). CL is also essential for promoting the association between BAX dimers, hence CL plays an important role in controlling the formation of active BAX oligomers (199). Ceramide and sphingolipid are also reported to be critical for the formation of Bax oligomers during apoptosis (200, 201). A recent study further shows that ceramides interact directly with the voltage-dependent anion channel VDAC2, a mitochondrial platform for Bax/Bak translocation, and trigger mitochondrial apoptosis (202).
Furthermore, emerging evidence implicates that mitochondrial dynamics influences appearance, distribution and stability of mtDNA nucleoids in mitochondrial networks (203). For instance, loss of Drp1 function results in severe mtDNA nucleoid clustering and loss of mtDNA (204–206), and disruption of mitochondrial fusion leads to mtDNA instability (207–209). Interestingly, a recent study reported that increased mitochondrial fission by Drp1 overexpression triggers the release of mtDNA into the cytosol, resulting in cytosolic mtDNA stress in hepatocellular carcinoma (HCC) cells (210). There is also evidence that Bak/Bax macropores facilitate mtDNA release from mitochondria during apoptosis regardless of mitochondrial fusion/fission dynamics (211). Obviously, more work is needed to understand the role of mitochondrial dynamics in these processes.
In summary, mitochondrial fragmentation occurs in most forms of apoptosis through activation of the mitochondrial fission machinery and/or inhibition of the fusion machinery in various physiological and pathophysiological conditions. However, how this process is regulated and the precise mechanisms by which the mitochondria-shaping proteins take part in apoptotic progression is largely unclear. Furthermore, whether Drp1-mediated fission is necessary during apoptosis is still controversial. There are reports suggesting that Drp1-mediated fission is an early step during apoptosis, since Drp1 depletion delays the cytochrome c release and subsequent apoptosis (212–214). Conversely, it is suggested that mitochondrial fission and apoptosis are two separate processes, because mitochondrial hyper-fusion caused by down-regulation of Drp1/Fis1 does not prevent cell death after apoptotic stimuli (215, 216). Moreover, it is reported that the mitochondrial network in DRP1−/− MEFs undergoes extensive fragmentation during apoptosis (162). Consistent with these data, apoptotic mitochondrial fragmentation still occurs during Bax/Bak-dependent apoptosis in Drp1-deficient cells albeit loss of Drp1 results in mitochondrial network super-fusion (211). Mechanistically, these data suggest that Drp1-independent fission might be involved in mitochondrial fragmentation during apoptosis in addition to canonical Drp1-dependent fission.
Mitochondrial Dynamics and Mitophagy
The mitochondrial quality control is executed through mitophagy, which selectively removes senescent or damaged mitochondria and balances the overall mitochondrial mass between biogenesis and degradation. Mitophagy is regulated mainly by Parkin and PINK1 in mammals, and mutations in the genes encoding these proteins are associated with Parkinson's disease (217, 218). While Parkin normally exists in the cytosol, when cells lose the mitochondrial membrane potential, such as by carbonyl cyanide m-chlorophenylhydrazone (CCCP) treatment, PINK1 accumulates at the surface of impaired mitochondria and recruits Parkin from the cytosol to mitochondria. Parkin, recruited by PINK1 to the surface of damaged mitochondria, ubiquitylates certain mitochondrial proteins (e.g., Mfn1/2) and promotes engulfment of damaged mitochondria by lysosomes (218, 219). There are a number of Parkin ubiquitylation substrates on the MOM and in the cytosol (220, 221). The mitochondrial outer membrane protein Miro1 (Mitochondrial Rho GTPase 1), essential for mitochondrial transport (222), interacts with Parkin and functions as a Ca2+-sensitive docking site for Parkin during mitochondrial damage. Knockdown of Miro1 reduces Parkin translocation to mitochondria and suppresses mitophagy (223).
In addition to the canonical Parkin-dependent mitophagy pathway mentioned above, increasing evidence shows that mitophagy can occur through Parkin-independent pathways, such as other ubiquitin ligase- and receptor-mediated mitophagy processes as well as mitochondrial lipid-mediated mitophagy (224). In addition to Parkin, several other E3 ligases have been shown to be involved in mitophagy, e.g., MUL1, ARIH1, and MARCH5 (225–227). Several mitophagy-specific receptors, such as FUDNC1, NIX, BNIP3, PHB2, and BCL2L13, have been reported to recognize damaged mitochondria, interact with LC3 and recruit autophagosomes decorated by LC3 (228–230). NLRX1 has recently been identified as a new mitophagy receptor in L. monocytogenes (231).
Additionally, mitochondrial membrane lipids also play important roles in regulating mitophagy. For instance, ceramide (a bioactive sphingolipid) in the MOM is found to interact with LC3B-II and trigger lethal mitophagy through the selective targeting of LC3B-II-containing autophagosomes to mitochondria (232). It has also been shown that CL externalization (i.e., CL is translocated from the MIM to the MOM) plays an important role in modulating mitophagy. During mitophagic stimulation, the externalized CL can form an essential platform on the surface of mitochondria where CL binds to LC3, and initiates autophagosome formation, ultimately resulting in mitophagy (233). The role of CL in mitophagy is supported by the findings that Beclin 1, a central regulator of mitophagy, directly interacts with CL on the MOM, regulating mitophagy (234). Another study suggests that LC3B, a structural protein of autophagosomal membranes, is translocated to the MOM by interaction with externalized CL (235).
Hypoxia-induced/FUNDC1-mediated mitophagy and mitochondrial dynamics are likewise important. It has been reported that the MOM-anchored protein FUNDC1 drives hypoxia-induced mitophagy through interacting with the key autophagy factor LC3 in mammals (236). Upon mitophagy induced by hypoxia or treatment with FCCP, FUNDC1 binds and recruits ULK1 (a Ser/Thr kinase required for early autophagosome formation) to damaged mitochondria, where FUNDC1 is phosphorylated by ULK1, inducing mitophagy (237). FUNDC1 also interacts with Drp1 and OPA1, and increased association of FUNDC1 with Drp1 and less interaction with OPA1 promote mitochondrial fission and mitophagy (238). FUNDC1 is also found to accumulate at the mitochondria-associated ER membranes (MAMs) and interact with the ER membrane protein calnexin. Under hypoxic conditions, FUNDC1 associated more with Drp1 accompanied with less interaction with calnexin, triggering mitochondrial fission and mitophagy (239). Additionally, Bcl-2 family protein Bcl-xL also participates in FUNDC1-mediated mitophagy. Bcl-xL but not Bcl-2, can inhibit the dephosphorylation of FUNDC1 to block further mitophagy (240).
Growing evidence links mitochondrial dynamics with mitochondrial quality control via selective removal of damaged mitochondria (i.e., mitophagy). The mitochondrial fusion factors OPA1, Mfn1, and Mfn2 are involved in the regulation of Parkin/PINK1-mediated mitophagy. OPA1 mutations or impaired cleavage of OPA1 affect mitophagy and Mfn1 and Mfn2 are ubiquitinated in a PINK1/Parkin-dependent manner upon mitophagic stimulation (241–243). Mfn2, but not Mfn1, was reported to mediate Parkin recruitment to damaged mitochondria. PINK1 can phosphorylate Mfn2, thereby promoting the interaction of Mfn2 with Parkin, the translocation of Parkin to mitochondria and Parkin-mediated ubiquitylation of mitochondrial proteins. Loss of Mfn2 prevents the translocation of Parkin to mitochondria (49). Emerging data suggest that Drp1 and its four receptors also participate in mitophagy. For example, overexpressing exogenous hFis1 in MEF cells triggers mitophagy (244), whereas cells lacking one or more Drp1 receptors (Fis1, Mff, and MIEFs) are resistant to CCCP-induced mitochondrial fragmentation to different degrees (86, 115). However, the resistance to apoptosis resulting from Fis1/Mff depletion can be reversed through overexpression of MIEF1 or MIEF2 (86). Of note, knockdown of MIEF1 by RNAi triggers and overexpression of MIEF1 blocks Parkin recruitment to mitochondria at the early stages of CCCP treatment (176). Interestingly, transient transfection of exogenous MIEF1 without CCCP treatment still leads to elevated expression of LC3B-II, implicating increased mitophagic activity (91). Additionally, some apoptotic factors in the Bcl-2 family are reported to be involved in the Parkin/PINK1-mediated mitophagic process. Bcl-xL, MCL1, and Bcl-W, but not Bcl-2, Bax, and Bak can inhibit this type of mitophagy (167).
Additionally, the SNARE protein syntaxin 17 (STX17), which is involved in the process of autophagy, is shown to cooperate with mitochondria-shaping proteins in mitophagy (245, 246). Intriguingly, it is reported that loss of Fis1 leads to aberrant accumulation of STX17 on mitochondria and induces mitophagy in Fis1-deficient cells in a way independent of Parkin/PINK1 (247). Furthermore, STX17 also localizes at the ER-mitochondria contact sites and regulates the localization and GTPase activity of Drp1 (248, 249). These data support the notion of an intricate crosstalk between autophagy factors and mitochondria-shaping proteins during mitophagy. Overall, although a number of mitochondria-shaping proteins are implicated in mitophagy, the detailed roles of these proteins in the process remain poorly understood. A deeper understanding of these mechanisms will be of major importance especially in the field of neurodegeneration.
In Summary
In the past 20 years, considerable progress has been made in terms of identifying core components and regulatory factors of the mitochondrial fission/fusion machineries in mammals, and huge efforts have been made to elucidate the potential molecular mechanisms that control and regulate mitochondrial fission and fusion events. One outcome of these efforts is a shift from the view that the mitochondrial fission and fusion machineries are evolutionarily conserved from yeast to human, to an understanding that these processes are much more complicated in mammals than in single-cell yeast. So far, at least four Drp1 receptors (Fis1, Mff, MIEF1, and MIEF2) in mammalian cells are responsible for recruitment of Drp1 to mitochondria with an additive effect, although they are not functionally redundant. Increasing evidence indicates that these receptors also coordinately work in Drp1-mediated mitochondrial fission. Thus, further research is warranted to elucidate the interplay between these Drp1 receptors in health and disease. Although the pro-fission and pro-fusion proteins are thought to regulate mitochondrial fission and fusion events separately, emerging data suggest that there is also extensive crosstalk between the fission and fusion machineries. It is also becoming increasingly clear that there are additional Drp1-independent mechanisms that drive mitochondrial division in mammalian cells. Finally, research into the mechanisms of apoptosis and mitophagy has excitingly placed the mitochondrial fission and fusion machineries at the center of these physiological processes. Elucidation of the underlying molecular mechanisms involved in these processes will be critical for better understanding of mammalian mitochondrial dynamics.
Author Contributions
RY and JZ conceptualized and wrote the review with contributions from MN and UL. RY and JZ made the figures. All authors discussed and approved the final manuscript.
Funding
This work was supported by grants from the Swedish Research Council (VR-NT and VR-MH, No. K2014-67X-15399-10-4, ÄR-MH2018-02452, ÄR-NT2015-05542, ÄR-NT2012-5140), the Swedish Cancer Society (No. CAN2014/836, can2017/737, 2019/19 0316 Pj01H), the Cancer Society in Stockholm (No. 2015-151213, 2018-181223), Karolinska Institutet and the Stockholm County Council (No. 2016fobi47658).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to apologize to those colleagues whose work could not be cited or discussed in sufficient detail due to space limitations.
References
1. Neupert W. Mitochondrial gene expression: a playground of evolutionary tinkering. Annu Rev Biochem. (2016) 85:65–76. doi: 10.1146/annurev-biochem-011116-110824
2. von Heijne G. Why mitochondria need a genome. FEBS Lett. (1986) 198:1–4. doi: 10.1016/0014-5793(86)81172-3
3. Bjorkholm P, Ernst AM, Hagstrom E, Andersson SG. Why mitochondria need a genome revisited. FEBS Lett. (2017) 591:65–75. doi: 10.1002/1873-3468.12510
4. Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. (1999) 1410:103–23. doi: 10.1016/S0005-2728(98)00161-3
5. Sun X, St John JC. The role of the mtDNA set point in differentiation, development and tumorigenesis. Biochem J. (2016) 473:2955–71. doi: 10.1042/BCJ20160008
6. El-Hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1539–55. doi: 10.1016/j.bbadis.2017.02.017
7. Calvo SE, Clauser KR, Mootha VK. MitoCarta 2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. (2016) 44:D1251–7. doi: 10.1093/nar/gkv1003
8. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. (2006) 16:R551–60. doi: 10.1016/j.cub.2006.06.054
9. Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. (2009) 18:R169–76. doi: 10.1093/hmg/ddp326
10. Senft D, Ronai ZA. Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. (2016) 39:43–52. doi: 10.1016/j.ceb.2016.02.001
11. Altieri DC. Mitochondrial dynamics and metastasis. Cell Mol Life Sci. (2018) 76:827–35. doi: 10.1007/s00018-018-2961-2
12. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. (2012) 46:265–87. doi: 10.1146/annurev-genet-110410-132529
13. Zhao J, Lendahl U, Nister M. Regulation of mitochondrial dynamics: convergences and divergences between yeast and vertebrates. Cell Mol Life Sci. (2013) 70:951–76. doi: 10.1007/s00018-012-1066-6
14. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. (2014) 15:634–46. doi: 10.1038/nrm3877
15. Roy M, Reddy PH, Iijima M, Sesaki H. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol. (2015) 33:111–8. doi: 10.1016/j.ceb.2015.02.001
16. Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. (2017) 26:39–48. doi: 10.1016/j.cmet.2017.05.016
17. Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci. (2017) 74:1999–2017. doi: 10.1007/s00018-016-2451-3
18. Braun RJ, Westermann B. Mitochondrial dynamics in yeast cell death and aging. Biochem Soc Trans. (2011) 39:1520–6. doi: 10.1042/BST0391520
19. Merz S, Hammermeister M, Altmann K, Durr M, Westermann B. Molecular machinery of mitochondrial dynamics in yeast. Biol Chem. (2007) 388:917–26. doi: 10.1515/BC.2007.110
20. Shaw JM, Nunnari J. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. (2002) 12:178–84. doi: 10.1016/S0962-8924(01)02246-2
21. Rapaport D, Brunner M, Neupert W, Westermann B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J Biol Chem. (1998) 273:20150–5. doi: 10.1074/jbc.273.32.20150
22. Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, et al. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. (1998) 143:359–73. doi: 10.1083/jcb.143.2.359
23. Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J. The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol. (2000) 151:341–52. doi: 10.1083/jcb.151.2.341
24. Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, et al. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol. (2003) 160:303–11. doi: 10.1083/jcb.200209015
25. Sesaki H, Southard SM, Yaffe MP, Jensen RE. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell. (2003) 14:2342–56. doi: 10.1091/mbc.e02-12-0788
26. Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. (2001) 114:867–74.
27. Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. (2000) 26:211–5. doi: 10.1038/79944
28. Sesaki H, Jensen RE. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol. (2001) 152:1123–34. doi: 10.1083/jcb.152.6.1123
29. Sesaki H, Jensen RE. Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J Biol Chem. (2004) 279:28298–303. doi: 10.1074/jbc.M401363200
30. Coonrod EM, Karren MA, Shaw JM. Ugo1p is a multipass transmembrane protein with a single carrier domain required for mitochondrial fusion. Traffic. (2007) 8:500–11. doi: 10.1111/j.1600-0854.2007.00550.x
31. Hoppins S, Horner J, Song C, McCaffery JM, Nunnari J. Mitochondrial outer and inner membrane fusion requires a modified carrier protein. J Cell Biol. (2009) 184:569–81. doi: 10.1083/jcb.200809099
32. Janer A, Prudent J, Paupe V, Fahiminiya S, Majewski J, Sgarioto N, et al. SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for leigh syndrome. EMBO Mol Med. (2016) 8:1019–38. doi: 10.15252/emmm.201506159
33. Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet. (2015) 47:926–32. doi: 10.1038/ng.3354
34. Fritz S, Weinbach N, Westermann B. Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol Biol Cell. (2003) 14:2303–13. doi: 10.1091/mbc.e02-12-0831
35. Anton F, Fres JM, Schauss A, Pinson B, Praefcke GJ, Langer T, et al. Ugo1 and Mdm30 act sequentially during Fzo1-mediated mitochondrial outer membrane fusion. J Cell Sci. (2011) 124:1126–35. doi: 10.1242/jcs.073080
36. Cohen MM, Amiott EA, Day AR, Leboucher GP, Pryce EN, Glickman MH, et al. Sequential requirements for the GTPase domain of the mitofusin Fzo1 and the ubiquitin ligase SCFMdm30 in mitochondrial outer membrane fusion. J Cell Sci. (2011) 124:1403–10. doi: 10.1242/jcs.079293
37. Sesaki H, Dunn CD, Iijima M, Shepard KA, Yaffe MP, Machamer CE, et al. Ups1p, a conserved intermembrane space protein, regulates mitochondrial shape and alternative topogenesis of Mgm1p. J Cell Biol. (2006) 173:651–8. doi: 10.1083/jcb.200603092
38. Sesaki H, Southard SM, Hobbs AE, Jensen RE. Cells lacking Pcp1p/Ugo2p, a rhomboid-like protease required for Mgm1p processing, lose mtDNA and mitochondrial structure in a Dnm1p-dependent manner, but remain competent for mitochondrial fusion. Biochem Biophys Res Commun. (2003) 308:276–83. doi: 10.1016/S0006-291X(03)01348-2
39. Brandt T, Cavellini L, Kuhlbrandt W, Cohen MM. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. Elife. (2016) 5:e14618. doi: 10.7554/eLife.14618
40. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. (2018) 62:341–60. doi: 10.1042/EBC20170104
41. Mattie S, Riemer J, Wideman JG, McBride HM. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J Cell Biol. (2018) 217:507–15. doi: 10.1083/jcb.201611194
42. Li YJ, Cao YL, Feng JX, Qi Y, Meng S, Yang JF, et al. Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat Commun. (2019) 10:4914. doi: 10.1038/s41467-019-12912-0
43. Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature. (2017) 542:372–6. doi: 10.1038/nature21077
44. Qi Y, Yan L, Yu C, Guo X, Zhou X, Hu X, et al. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J Cell Biol. (2016) 215:621–9. doi: 10.1083/jcb.201609019
45. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
46. Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. (2004) 117:6535–46. doi: 10.1242/jcs.01565
47. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008) 456:605–10. doi: 10.1038/nature07534
48. Zorzano A, Hernandez-Alvarez MI, Sebastian D, Munoz JP. Mitofusin 2 as a driver that controls energy metabolism and insulin signaling. Antioxid Redox Signal. (2015) 22:1020–31. doi: 10.1089/ars.2014.6208
49. Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science. (2013) 340:471–5. doi: 10.1126/science.1231031
50. Perumalsamy LR, Nagala M, Sarin A. Notch-activated signaling cascade interacts with mitochondrial remodeling proteins to regulate cell survival. Proc Natl Acad Sci USA. (2010) 107:6882–7. doi: 10.1073/pnas.0910060107
51. Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause charcot-marie-tooth neuropathy type 2A. Nat Genet. (2004) 36:449–51. doi: 10.1038/ng1341
52. Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. (2007) 176:405–14. doi: 10.1083/jcb.200611080
53. Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci. (2012) 32:4145–55. doi: 10.1523/JNEUROSCI.6338-11.2012
54. Zhou Y, Carmona S, Muhammad A, Bell S, Landeros J, Vazquez M, et al. Restoring mitofusin balance prevents axonal degeneration in a charcot-marie-tooth type 2A model. J Clin Invest. (2019) 130:1756–71. doi: 10.1172/JCI124194
55. Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet. (2001) 109:584–91. doi: 10.1007/s00439-001-0633-y
56. Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. (2006) 25:2966–77. doi: 10.1038/sj.emboj.7601184
57. Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. (2007) 178:749–55. doi: 10.1083/jcb.200704110
58. Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. (2014) 19:630–41. doi: 10.1016/j.cmet.2014.03.011
59. Faelber K, Dietrich L, Noel JK, Wollweber F, Pfitzner AK, Muhleip A, et al. Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature. (2019) 571:429–33. doi: 10.1038/s41586-019-1372-3
60. Yan L, Qi Y, Ricketson D, Li L, Subramanian K, Zhao J, et al. Structural analysis of a trimeric assembly of the mitochondrial dynamin-like GTPase Mgm1. Proc Natl Acad Sci USA. (2020) 117:4061–70. doi: 10.1073/pnas.1919116117
61. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. (2014) 204:919–29. doi: 10.1083/jcb.201308006
62. Zhang D, Zhang Y, Ma J, Zhu C, Niu T, Chen W, et al. Cryo-EM structures of S-OPA1 reveal its interactions with membrane and changes upon nucleotide binding. Elife. (2020) 9:e50294. doi: 10.7554/eLife.50294
63. Ge Y, Shi X, Boopathy S, McDonald J, Smith AW, Chao LH. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. Elife. (2020) 9:e50973. doi: 10.7554/eLife.50973
64. Loucks FA, Schroeder EK, Zommer AE, Hilger S, Kelsey NA, Bouchard RJ, et al. Caspases indirectly regulate cleavage of the mitochondrial fusion GTPase OPA1 in neurons undergoing apoptosis. Brain Res. (2009) 1250:63–74. doi: 10.1016/j.brainres.2008.10.081
65. Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. (2009) 187:1023–36. doi: 10.1083/jcb.200906084
66. Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. (2009) 187:959–66. doi: 10.1083/jcb.200906083
67. Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. (2004) 279:18792–8. doi: 10.1074/jbc.M400920200
68. Cipolat S, de Brito MO, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. (2004) 101:15927–32. doi: 10.1073/pnas.0407043101
69. Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, et al. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J Cell Biol. (1998) 143:333–49. doi: 10.1083/jcb.143.2.333
70. Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, et al. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. (1999) 1:298–304. doi: 10.1038/13014
71. Legesse-Miller A, Massol RH, Kirchhausen T. Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol Biol Cell. (2003) 14:1953–63. doi: 10.1091/mbc.e02-10-0657
72. Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. (2005) 170:1021–7. doi: 10.1083/jcb.200506078
73. Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. (2000) 151:353–66. doi: 10.1083/jcb.151.2.353
74. Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. (2005) 170:237–48. doi: 10.1083/jcb.200503148
75. Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. (2000) 151:367–80. doi: 10.1083/jcb.151.2.367
76. Suzuki M, Neutzner A, Tjandra N, Youle RJ. Novel structure of the N terminus in yeast Fis1 correlates with a specialized function in mitochondrial fission. J Biol Chem. (2005) 280:21444–52. doi: 10.1074/jbc.M414092200
77. Tieu Q, Okreglak V, Naylor K, Nunnari J. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J Cell Biol. (2002) 158:445–52. doi: 10.1083/jcb.200205031
78. Cerveny KL, Jensen RE. The WD-repeats of Net2p interact with Dnm1p and Fis1p to regulate division of mitochondria. Mol Biol Cell. (2003) 14:4126–39. doi: 10.1091/mbc.e03-02-0092
79. Schauss AC, Bewersdorf J, Jakobs S. Fis1p and Caf4p, but not Mdv1p, determine the polar localization of Dnm1p clusters on the mitochondrial surface. J Cell Sci. (2006) 119:3098–106. doi: 10.1242/jcs.03026
80. Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, et al. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci USA. (2013) 110:E1342–51. doi: 10.1073/pnas.1300855110
81. Pitts KR, Yoon Y, Krueger EW, McNiven MA. The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol Biol Cell. (1999) 10:4403–17. doi: 10.1091/mbc.10.12.4403
82. Macdonald PJ, Stepanyants N, Mehrotra N, Mears JA, Qi X, Sesaki H, et al. A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol Biol Cell. (2014) 25:1905–15. doi: 10.1091/mbc.e14-02-0728
83. Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. (2013) 1833:1256–68. doi: 10.1016/j.bbamcr.2013.02.002
84. James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. (2003) 278:36373–9. doi: 10.1074/jbc.M303758200
85. Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. (2003) 23:5409–20. doi: 10.1128/MCB.23.15.5409-5420.2003
86. Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. (2013) 24:659–67. doi: 10.1091/mbc.e12-10-0721
87. Wong YC, Ysselstein D, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. (2018) 554:382–6. doi: 10.1038/nature25486
88. Yu R, Jin SB, Lendahl U, Nister M, Zhao J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. (2019) 38:e99748. doi: 10.15252/embj.201899748
89. Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. (2008) 19:2402–12. doi: 10.1091/mbc.e07-12-1287
90. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. (2010) 191:1141–58. doi: 10.1083/jcb.201007152
91. Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, et al. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. (2011) 30:2762–78. doi: 10.1038/emboj.2011.198
92. Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. (2011) 12:565–73. doi: 10.1038/embor.2011.54
93. Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Bio. (2012) 13:75–88. doi: 10.1038/nrm3266
94. Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. (2016) 540:139–43. doi: 10.1038/nature20555
95. Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ, Ryan MT. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat Commun. (2018) 9:5239. doi: 10.1038/s41467-018-07543-w
96. Fonseca TB, Sanchez-Guerrero A, Milosevic I, Raimundo N. Mitochondrial fission requires DRP1 but not dynamins. Nature. (2019) 570:E34–42. doi: 10.1038/s41586-019-1296-y
97. Suzuki M, Jeong SY, Karbowski M, Youle RJ, Tjandra N. The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J Mol Biol. (2003) 334:445–58. doi: 10.1016/j.jmb.2003.09.064
98. Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. (2005) 39:503–36. doi: 10.1146/annurev.genet.38.072902.093019
99. Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. (2007) 76:751–80. doi: 10.1146/annurev.biochem.76.071905.090048
100. Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. (2004) 117:1201–10. doi: 10.1242/jcs.01058
101. Jofuku A, Ishihara N, Mihara K. Analysis of functional domains of rat mitochondrial Fis1, the mitochondrial fission-stimulating protein. Biochem Biophys Res Commun. (2005) 333:650–9. doi: 10.1016/j.bbrc.2005.05.154
102. Yu T, Fox RJ, Burwell LS, Yoon Y. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J Cell Sci. (2005) 118:4141–51. doi: 10.1242/jcs.02537
103. Arai R, Ito K, Wakiyama M, Matsumoto E, Sakamoto A, Etou Y, et al. Establishment of stable hFis1 knockdown cells with an siRNA expression vector. J Biochem. (2004) 136:421–5. doi: 10.1093/jb/mvh139
104. Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. (2004) 15:5001–11. doi: 10.1091/mbc.e04-04-0294
105. Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. (2013) 126:789–802. doi: 10.1242/jcs.114439
106. Kaddour-Djebbar I, Choudhary V, Brooks C, Ghazaly T, Lakshmikanthan V, Dong Z, et al. Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells. Int J Oncol. (2010) 36:1437–44. doi: 10.3892/ijo_00000629
107. Joshi AU, Saw NL, Shamloo M, Mochly-Rosen D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer's disease. Oncotarget. (2018) 9:6128–43. doi: 10.18632/oncotarget.23640
108. Joshi AU, Saw NL, Vogel H, Cunnigham AD, Shamloo M, Mochly-Rosen D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol Med. (2018) 10:e8166. doi: 10.15252/emmm.201708166
109. Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, et al. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. (2011) 44:532–44. doi: 10.1016/j.molcel.2011.08.045
110. Ciarlo L, Manganelli V, Garofalo T, Matarrese P, Tinari A, Misasi R, et al. Association of fission proteins with mitochondrial raft-like domains. Cell Death Differ. (2010) 17:1047–58. doi: 10.1038/cdd.2009.208
111. De Palma C, Falcone S, Pisoni S, Cipolat S, Panzeri C, Pambianco S, et al. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. (2010) 17:1684–96. doi: 10.1038/cdd.2010.48
112. Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JT, Wang C, et al. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell. (2014) 25:145–59. doi: 10.1091/mbc.e13-09-0525
113. Liu R, Chan DC. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol Biol Cell. (2015) 26:4466–77. doi: 10.1091/mbc.E15-08-0591
114. Liu T, Yu R, Jin SB, Han L, Lendahl U, Zhao J, et al. The mitochondrial elongation factors MIEF1 and MIEF2 exert partially distinct functions in mitochondrial dynamics. Exp Cell Res. (2013) 319:2893–904. doi: 10.1016/j.yexcr.2013.07.010
115. Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, et al. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci. (2016) 129:2170–81. doi: 10.1242/jcs.185165
116. Loson OC, Meng S, Ngo H, Liu R, Kaiser JT, Chan DC. Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein Sci. (2015) 24:386–94. doi: 10.1002/pro.2629
117. Loson OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, et al. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure. (2014) 22:367–77. doi: 10.1016/j.str.2014.01.001
118. Richter V, Palmer CS, Osellame LD, Singh AP, Elgass K, Stroud DA, et al. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J Cell Biol. (2014) 204:477–86. doi: 10.1083/jcb.201311014
119. Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature. (2018) 558:401–5. doi: 10.1038/s41586-018-0211-2
120. Elgass KD, Smith EA, LeGros MA, Larabell CA, Ryan MT. Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J Cell Sci. (2015) 128:2795–804. doi: 10.1242/jcs.169136
121. Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. (2016) 212:531–44. doi: 10.1083/jcb.201508099
122. Yu R, Liu T, Jin SB, Ning C, Lendahl U, Nister M, et al. MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci Rep. (2017) 7:880. doi: 10.1038/s41598-017-00853-x
123. Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. (2002) 159:931–8. doi: 10.1083/jcb.200209124
124. Huang P, Galloway CA, Yoon Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS ONE. (2011) 6:e20655. doi: 10.1371/journal.pone.0020655
125. Abrisch RG, Gumbin SC, Wisniewski BT, Lackner LL, Voeltz GK. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J Cell Biol. (2020) 219:e201911122. doi: 10.1083/jcb.201911122
126. de Brito OM, Scorrano L. An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. (2010) 29:2715–23. doi: 10.1038/emboj.2010.177
127. Lang A, John Peter AT, Kornmann B. ER-mitochondria contact sites in yeast: beyond the myths of ERMES. Curr Opin Cell Biol. (2015) 35:7–12. doi: 10.1016/j.ceb.2015.03.002
128. Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, et al. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife. (2013) 2:e00422. doi: 10.7554/eLife.00422
129. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. (2011) 334:358–62. doi: 10.1126/science.1207385
130. Zhou Z, Torres M, Sha H, Halbrook CJ, Van den Bergh F, Reinert RB, et al. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science. (2020) 368:54–60. doi: 10.1126/science.aay2494
131. Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science. (2016) 353:aaf5549. doi: 10.1126/science.aaf5549
132. Moore AS, Holzbaur ELF. Mitochondrial-cytoskeletal interactions: dynamic associations that facilitate network function and remodeling. Curr Opin Physiol. (2018) 3:94–100. doi: 10.1016/j.cophys.2018.03.003
133. Moore AS, Wong YC, Simpson CL, Holzbaur EL. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat Commun. (2016) 7:12886–98. doi: 10.1038/ncomms12886
134. Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. (2013) 339:464–7. doi: 10.1126/science.1228360
135. Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, et al. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife. (2015) 4:e08828. doi: 10.7554/eLife.08828
136. Korobova F, Gauvin TJ, Higgs HN. A role for myosin II in mammalian mitochondrial fission. Curr Biol. (2014) 24:409–14. doi: 10.1016/j.cub.2013.12.032
137. Pagliuso A, Tham TN, Stevens JK, Lagache T, Persson R, Salles A, et al. A role for septin 2 in Drp1-mediated mitochondrial fission. EMBO Rep. (2016) 17:858–73. doi: 10.15252/embr.201541612
138. Li S, Xu S, Roelofs BA, Boyman L, Lederer WJ, Sesaki H, et al. Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J Cell Biol. (2015) 208:109–23. doi: 10.1083/jcb.201404050
139. Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic. (2015) 16:1–18. doi: 10.1111/tra.12230
140. Stepanyants N, Macdonald PJ, Francy CA, Mears JA, Qi X, Ramachandran R. Cardiolipin's propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell. (2015) 26:3104–16. doi: 10.1091/mbc.E15-06-0330
141. Bustillo-Zabalbeitia I, Montessuit S, Raemy E, Basanez G, Terrones O, Martinou JC. Specific interaction with cardiolipin triggers functional activation of dynamin-related protein 1. PLoS ONE. (2014) 9:e102738. doi: 10.1371/journal.pone.0102738
142. Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, et al. Coincident phosphatidic acid interaction restrains Drp1 in mitochondrial division. Mol Cell. (2016) 63:1034–43. doi: 10.1016/j.molcel.2016.08.013
143. Ugarte-Uribe B, Muller HM, Otsuki M, Nickel W, Garcia-Saez AJ. Dynamin-related protein 1 (Drp1) promotes structural intermediates of membrane division. J Biol Chem. (2014) 289:30645–56. doi: 10.1074/jbc.M114.575779
144. Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. (2006) 8:1255–62. doi: 10.1038/ncb1487
145. Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. (2017) 19:856–63. doi: 10.1038/ncb3560
146. Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell. (2011) 20:376–87. doi: 10.1016/j.devcel.2011.01.004
147. Baba T, Kashiwagi Y, Arimitsu N, Kogure T, Edo A, Maruyama T, et al. Phosphatidic acid (PA)-preferring phospholipase A1 regulates mitochondrial dynamics. J Biol Chem. (2014) 289:11497–511. doi: 10.1074/jbc.M113.531921
148. Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, et al. CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell. (2019) 177:1536–52.e23. doi: 10.1016/j.cell.2019.05.008
149. Kameoka S, Adachi Y, Okamoto K, Iijima M, Sesaki H. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. (2018) 28:67–76. doi: 10.1016/j.tcb.2017.08.011
150. Basu Ball W, Neff JK, Gohil VM. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. (2018) 592:1273–90. doi: 10.1002/1873-3468.12887
151. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: molecular and pharmacological aspects. Cells. (2019) 8:728. doi: 10.3390/cells8070728
152. Onoue K, Jofuku A, Ban-Ishihara R, Ishihara T, Maeda M, Koshiba T, et al. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J Cell Sci. (2013) 126:176–85. doi: 10.1242/jcs.111211
153. Wong YC, Kim S, Peng W, Krainc D. Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. (2019) 29:500–13. doi: 10.1016/j.tcb.2019.02.004
154. Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. (2008) 9:505–18. doi: 10.1038/nrn2417
155. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. (2019) 21:85–100. doi: 10.1038/s41580-019-0173-8
156. Fu W, Liu Y, Yin H. Mitochondrial dynamics: biogenesis, fission, fusion, and mitophagy in the regulation of stem cell behaviors. Stem Cells Int. (2019) 2019:9757201. doi: 10.1155/2019/9757201
157. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. (2016) 212:379–87. doi: 10.1083/jcb.201511036
158. Seo BJ, Yoon SH, Do JT. Mitochondrial dynamics in stem cells and differentiation. Int J Mol Sci. (2018) 19:3893. doi: 10.20944/preprints201811.0024.v1
159. Yu SB, Pekkurnaz G. Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J Mol Biol. (2018) 430:3922–41. doi: 10.1016/j.jmb.2018.07.027
160. Horbay R, Bilyy R. Mitochondrial dynamics during cell cycling. Apoptosis. (2016) 21:1327–35. doi: 10.1007/s10495-016-1295-5
161. Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. (2007) 16:1307–18. doi: 10.1093/hmg/ddm079
162. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. (2009) 11:958–66. doi: 10.1038/ncb1907
163. Chen H, Ren S, Clish C, Jain M, Mootha V, McCaffery JM, et al. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol. (2015) 211:795–805. doi: 10.1083/jcb.201507035
165. Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. (1998) 1366:127–37. doi: 10.1016/S0005-2728(98)00108-X
166. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. (2008) 9:47–59. doi: 10.1038/nrm2308
167. Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell. (2014) 55:451–66. doi: 10.1016/j.molcel.2014.06.001
168. Jeong SY, Seol DW. The role of mitochondria in apoptosis. BMB Rep. (2008) 41:11–22. doi: 10.5483/BMBRep.2008.41.1.011
169. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. (2008) 22:1577–90. doi: 10.1101/gad.1658508
170. Otera H, Mihara K. Mitochondrial dynamics: functional link with apoptosis. Int J Cell Biol. (2012) 2012:821676. doi: 10.1155/2012/821676
171. Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. (2005) 2:R283–9. doi: 10.1093/hmg/ddi270
172. Ugarte-Uribe B, Garcia-Saez AJ. Membranes in motion: mitochondrial dynamics and their role in apoptosis. Biol Chem. (2014) 395:297–311. doi: 10.1515/hsz-2013-0234
173. Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. (2005) 280:25060–70. doi: 10.1074/jbc.M501599200
174. Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. (2001) 1:515–25. doi: 10.1016/S1534-5807(01)00055-7
175. Zhao J, Liu T, Jin SB, Tomilin N, Castro J, Shupliakov O, et al. The novel conserved mitochondrial inner-membrane protein MTGM regulates mitochondrial morphology and cell proliferation. J Cell Sci. (2009) 122:2252–62. doi: 10.1242/jcs.038513
176. Xian H, Liou YC. Loss of MIEF1/MiD51 confers susceptibility to BAX-mediated cell death and PINK1-PRKN-dependent mitophagy. Autophagy. (2019) 15:2107–25. doi: 10.1080/15548627.2019.1596494
177. Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, et al. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell. (2016) 27:349–59. doi: 10.1091/mbc.e15-09-0678
178. Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. (1997) 139:1281–92. doi: 10.1083/jcb.139.5.1281
179. Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. (2001) 153:1265–76. doi: 10.1083/jcb.153.6.1265
180. Wang P, Liu B, Zhao J, Pang Q, Agrawal SG, Jia L, et al. Dynamin-related protein Drp1 is required for Bax translocation to mitochondria in response to irradiation-induced apoptosis. Oncotarget. (2015) 6:22598–612. doi: 10.18632/oncotarget.4200
181. Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. (2010) 142:889–901. doi: 10.1016/j.cell.2010.08.017
182. Saita S, Ishihara T, Maeda M, Iemura SI, Natsume T, Mihara K, et al. Distinct types of protease systems are involved in homeostasis regulation of mitochondrial morphology via balanced fusion and fission. Genes Cells. (2016) 21:408–24. doi: 10.1111/gtc.12351
183. Mopert K, Hajek P, Frank S, Chen C, Kaufmann J, Santel A. Loss of Drp1 function alters OPA1 processing and changes mitochondrial membrane organization. Exp Cell Res. (2009) 315:2165–80. doi: 10.1016/j.yexcr.2009.04.016
184. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. (2010) 37:299–310. doi: 10.1016/j.molcel.2010.01.025
185. Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. (2008) 31:570–85. doi: 10.1016/j.molcel.2008.08.002
186. Wu S, Zhou F, Zhang Z, Xing D. Bax is essential for Drp1-mediated mitochondrial fission but not for mitochondrial outer membrane permeabilization caused by photodynamic therapy. J Cell Physiol. (2011) 226:530–41. doi: 10.1002/jcp.22362
187. Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, et al. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. (2005) 15:2112–8. doi: 10.1016/j.cub.2005.10.041
188. Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. (2007) 177:439–50. doi: 10.1083/jcb.200610042
189. Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. (2006) 443:658–62. doi: 10.1038/nature05111
190. Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA. (2007) 104:11649–54. doi: 10.1073/pnas.0703976104
191. Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB 3rd, Goebbels S, et al. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. (2009) 184:707–19. doi: 10.1083/jcb.200809060
192. Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA. (2008) 105:2169–74. doi: 10.1073/pnas.0711647105
193. Morciano G, Giorgi C, Balestra D, Marchi S, Perrone D, Pinotti M, et al. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol Biol Cell. (2016) 27:20–34. doi: 10.1091/mbc.E15-01-0028
194. Praharaj PP, Naik PP, Panigrahi DP, Bhol CS, Mahapatra KK, Patra S, et al. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: its implication in cancer therapeutics. Cell Mol Life Sci. (2019) 76:1641–52. doi: 10.1007/s00018-018-2990-x
195. Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. (2000) 2:754–61. doi: 10.1038/35036395
196. Lucken-Ardjomande S, Montessuit S, Martinou JC. Contributions to Bax insertion and oligomerization of lipids of the mitochondrial outer membrane. Cell Death Differ. (2008) 15:929–37. doi: 10.1038/cdd.2008.9
197. Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. (2002) 111:331–42. doi: 10.1016/S0092-8674(02)01036-X
198. Raemy E, Montessuit S, Pierredon S, van Kampen AH, Vaz FM, Martinou JC. Cardiolipin or MTCH2 can serve as tBID receptors during apoptosis. Cell Death Differ. (2016) 23:1165–74. doi: 10.1038/cdd.2015.166
199. Lai YC, Li CC, Sung TC, Chang CW, Lan YJ, Chiang YW. The role of cardiolipin in promoting the membrane pore-forming activity of BAX oligomers. Biochim Biophys Acta Biomembr. (2019) 1861:268–80. doi: 10.1016/j.bbamem.2018.06.014
200. Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. (2012) 148:988–1000. doi: 10.1016/j.cell.2012.01.038
201. Lee H, Rotolo JA, Mesicek J, Penate-Medina T, Rimner A, Liao WC, et al. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS ONE. (2011) 6:e19783. doi: 10.1371/journal.pone.0019783
202. Dadsena S, Bockelmann S, Mina JGM, Hassan DG, Korneev S, Razzera G, et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat Commun. (2019) 10:1832. doi: 10.1038/s41467-019-09654-4
203. Jayashankar V, Rafelski SM. Integrating mitochondrial organization and dynamics with cellular architecture. Curr Opin Cell Biol. (2014) 26:34–40. doi: 10.1016/j.ceb.2013.09.002
204. Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE. (2008) 3:e3257. doi: 10.1371/journal.pone.0003257
205. Ishihara T, Ban-Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol. (2015) 35:211–23. doi: 10.1128/MCB.01054-14
206. Ota A, Ishihara T, Ishihara N. Mitochondrial nucleoid morphology and respiratory function are altered in Drp1-deficient HeLa cells. J Biochem. (2020) 167:287–94. doi: 10.1093/jb/mvz112
207. Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. (2010) 141:280–9. doi: 10.1016/j.cell.2010.02.026
208. Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes. Brain. (2008) 131:338–51. doi: 10.1093/brain/awm298
209. Elachouri G, Vidoni S, Zanna C, Pattyn A, Boukhaddaoui H, Gaget K, et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. (2011) 21:12–20. doi: 10.1101/gr.108696.110
210. Bao D, Zhao J, Zhou X, Yang Q, Chen Y, Zhu J, et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene. (2019) 38:5007–20. doi: 10.1038/s41388-019-0772-z
211. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. (2018) 359:eaao6047. doi: 10.1126/science.aao6047
212. Yuan H, Gerencser AA, Liot G, Lipton SA, Ellisman M, Perkins GA, et al. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. (2007) 14:462–71. doi: 10.1038/sj.cdd.4402046
213. Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. (2005) 6:657–63. doi: 10.1038/nrm1697
214. Oettinghaus B, D'Alonzo D, Barbieri E, Restelli LM, Savoia C, Licci M, et al. DRP1-dependent apoptotic mitochondrial fission occurs independently of BAX, BAK and APAF1 to amplify cell death by BID and oxidative stress. Biochim Biophys Acta. (2016) 1857:1267–76. doi: 10.1016/j.bbabio.2016.03.016
215. Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, et al. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. (2006) 26:7397–408. doi: 10.1128/MCB.02282-05
216. Parone PA, Martinou JC. Mitochondrial fission and apoptosis: an ongoing trial. Biochim Biophys Acta. (2006) 1763:522–30. doi: 10.1016/j.bbamcr.2006.04.005
217. Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta. (2012) 1833:205–12. doi: 10.1016/j.bbamcr.2012.02.012
218. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) 12:9–14. doi: 10.1038/nrm3028
219. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. (2014) 33:2142–56. doi: 10.15252/embj.201488104
220. Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. (2013) 496:372–6. doi: 10.1038/nature12043
221. Martinez A, Lectez B, Ramirez J, Popp O, Sutherland JD, Urbe S, et al. Quantitative proteomic analysis of parkin substrates in Drosophila neurons. Mol Neurodegener. (2017) 12:29–47. doi: 10.1186/s13024-017-0170-3
222. Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. (2013) 5:a011304. doi: 10.1101/cshperspect.a011304
223. Safiulina D, Kuum M, Choubey V, Gogichaishvili N, Liiv J, Hickey MA, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. (2018) 38:e99384. doi: 10.15252/embj.201899384
224. Cai Q, Jeong YY. Mitophagy in Alzheimer's disease and other age-related neurodegenerative diseases. Cells. (2020) 9:150. doi: 10.3390/cells9010150
225. Yun J, Puri R, Yang H, Lizzio MA, Wu C, Sheng ZH, et al. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. (2014) 3:e01958. doi: 10.7554/eLife.01958
226. Villa E, Proics E, Rubio-Patino C, Obba S, Zunino B, Bossowski JP, et al. Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. (2017) 20:2846–59. doi: 10.1016/j.celrep.2017.08.087
227. Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. (2017) 18:495–509. doi: 10.15252/embr.201643309
228. Chen G, Kroemer G, Kepp O. Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol. (2020) 8:200. doi: 10.3389/fcell.2020.00200
229. Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells. (2018) 41:18–26. doi: 10.14348/molcells.2018.2277
230. Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. (2018) 28:R170–85. doi: 10.1016/j.cub.2018.01.004
231. Zhang Y, Yao Y, Qiu X, Wang G, Hu Z, Chen S, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. (2019) 20:433–46. doi: 10.1038/s41590-019-0324-2
232. Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. (2012) 8:831–8. doi: 10.1038/nchembio.1059
233. Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. (2013) 15:1197–205. doi: 10.1038/ncb2837
234. Huang W, Choi W, Hu W, Mi N, Guo Q, Ma M, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. (2012) 22:473–89. doi: 10.1038/cr.2012.24
235. Anton Z, Landajuela A, Hervas JH, Montes LR, Hernandez-Tiedra S, Velasco G, et al. Human Atg8-cardiolipin interactions in mitophagy: specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy. (2016) 12:2386–403. doi: 10.1080/15548627.2016.1240856
236. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. (2012) 14:177–85. doi: 10.1038/ncb2422
237. Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. (2014) 15:566–75. doi: 10.1002/embr.201438501
238. Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. (2016) 12:689–702. doi: 10.1080/15548627.2016.1151580
239. Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. (2016) 35:1368–84. doi: 10.15252/embj.201593102
240. Wu H, Xue D, Chen G, Han Z, Huang L, Zhu C, et al. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy. (2014) 10:1712–25. doi: 10.4161/auto.29568
241. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. (2010) 19:4861–70. doi: 10.1093/hmg/ddq419
242. Liao C, Ashley N, Diot A, Morten K, Phadwal K, Williams A, et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology. (2017) 88:131–42. doi: 10.1212/WNL.0000000000003491
243. MacVicar TD, Lane JD. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci. (2014) 127:2313–25. doi: 10.1242/jcs.144337
244. Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. (2008) 1777:860–6. doi: 10.1016/j.bbabio.2008.05.442
245. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. (2013) 495:389–93. doi: 10.1038/nature11910
246. Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. (2012) 151:1256–69. doi: 10.1016/j.cell.2012.11.001
247. Xian H, Yang Q, Xiao L, Shen HM, Liou YC. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat Commun. (2019) 10:2059. doi: 10.1038/s41467-019-10096-1
248. Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell. (2015) 32:304–17. doi: 10.1016/j.devcel.2014.12.011
Keywords: mitochondrial dynamics, fission, fusion, apoptosis, mitophagy
Citation: Yu R, Lendahl U, Nistér M and Zhao J (2020) Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 11:374. doi: 10.3389/fendo.2020.00374
Received: 11 February 2020; Accepted: 12 May 2020;
Published: 11 June 2020.
Edited by:
Cecilia Poderoso, University of Buenos Aires, ArgentinaReviewed by:
Rajesh Ramachandran, Case Western Reserve University, United StatesYushan Zhu, Nankai University, China
Kasturi Mitra, University of Alabama at Birmingham, United States
Copyright © 2020 Yu, Lendahl, Nistér and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica Nistér, bW9uaWNhLm5pc3RlckBraS5zZQ==; Jian Zhao, amlhbi56aGFvQGtpLnNl