 Diana Cruz-Topete
Diana Cruz-Topete Robert H. Oakley
Robert H. Oakley John A. Cidlowski
John A. Cidlowski- 1Department of Molecular and Cellular Physiology, Center for Cardiovascular Diseases and Sciences, LSU Health Sciences Center, Shreveport, LA, United States
- 2Signal Transduction Laboratory, Department of Health and Human Services, National Institute of Environmental Health Sciences, National Institutes of Health, Durham, NC, United States
A decline in normal physiological functions characterizes the aging process. While some of these changes are benign, the decrease in the function of the cardiovascular system that occurs during aging leads to the activation of pathological processes associated with an increased risk for heart disease and its complications. Imbalances in endocrine function are also common occurrences during the aging process. Glucocorticoids are primary stress hormones and are critical regulators of energy metabolism, inflammation, and cardiac function. Glucocorticoids exert their actions by binding the glucocorticoid receptor (GR) and, in some instances, to the mineralocorticoid receptor (MR). GR and MR are members of the nuclear receptor family of ligand-activated transcription factors. There is strong evidence that imbalances in GR and MR signaling in the heart have a causal role in cardiac disease. The extent to which glucocorticoids play a role in the aging heart, however, remains unclear. This review will summarize the positive and negative direct and indirect effects of glucocorticoids on the heart and the latest molecular and physiological evidence on how alterations in glucocorticoid signaling lead to changes in cardiac structure and function. We also briefly discuss the effects of other hormones systems such as estrogens and GH/IGF-1 on different cardiovascular cells during aging. We will also review the link between imbalances in glucocorticoid levels and the molecular processes responsible for promoting cardiomyocyte dysfunction in aging. Finally, we will discuss the potential for selectively manipulating glucocorticoid signaling in cardiomyocytes, which may represent an improved therapeutic approach for preventing and treating age-related heart disease.
Introduction
Aging is characterized by a gradual decline in all physiological functions, a decrease in repair mechanisms, senescence, and eventually death. The endocrine system is a complex network system that regulates virtually all of an organism's biological processes, including development, growth, reproduction, metabolism, blood pressure, and responses to stressors (1, 2). Aging leads to significant alterations in the endocrine system, but imbalances in the endocrine system also affect the aging process (1–3). For example, the secretory patterns of hormones produced by the hypothalamic–pituitary axis are altered in the elderly population as well as the sensitivity to hormones by their target organs. Conversely, imbalances in the production of hormones or alterations in their negative feedback loops have been shown to accelerate the aging process by leading to disturbances in metabolism, cardiovascular function, and cognition (1, 2). Therefore, there is increasing interest in understanding the association between aging and endocrine function so that novel therapies can be developed to prevent/treat age-related endocrine disorders and the deleterious effects of endocrine imbalances during the aging process.
Aging is closely associated with alterations in cardiovascular function. Structural and functional changes in the heart and vasculature during aging are characterized by vascular stiffening, increased left ventricular wall thickness, hypertrophy, fibrosis, changes in maximal heart rate, and alterations in cardiac diastolic function (4–6). Age-related cardiac changes result from molecular alterations in the expression and function of proteins involved in maintaining cardiomyocyte structure, survival, calcium handling, redox balance, and metabolism (7). Additionally, changes in the expression profile of cardiac interstitial cells, including endothelial cells and fibroblasts, contribute to phenotypic changes in the aging heart (8). Although these alterations are not necessarily pathological in nature by themselves, they do increase the risk for cardiac damage and heart failure in the elderly population. There are several factors that further exacerbate the aging effect and make the “old heart” more likely to fail. For example, aging-associated alterations in inflammatory and fibrogenic pathways are exacerbated by metabolic imbalances, which are intertwined with a decline in endocrine function (9).
The relationship between the endocrine system, aging, and the cardiovascular system has been illustrated by the increased incidence of cardiovascular complications associated with the decline in gonadal hormone production. The risk of heart disease in women undergoing menopause is significantly higher than that in younger women (6, 10). Studies have suggested that the increased risk of heart disease in post-menopausal women is associated with a decline in ovarian hormone (estrogen and progesterone) production (11). Decreased ovarian function correlates with an elevation in the serum concentrations of atherogenic lipids (low-density lipoproteins and total cholesterol) and a decrease in the levels of cardioprotective lipids (high-density proteins) (12, 13). A decline in androgen production in males during aging has also been associated with higher cardiovascular risk due to increases in fat mass and the development of insulin resistance (12–14). The age-related decline in sex hormones has been considered one of the most clinically significant associations between the endocrine and cardiovascular systems. However, aging clearly influences other endocrine systems, and conversely, these endocrine alterations further influence the aging process. For example, with aging, glucose homeostasis is altered, which leads to an increased risk for metabolic complications such as type 2 diabetes that significantly elevate the risk for vascular and heart disease in the elderly population (2). Additionally, aging leads to changes in growth hormone (GH) and insulin growth factor 1 (IGF-1) levels. Alterations in GH and IGF-1 have profound effects on body composition and muscle strength, bone density, metabolism and the lipid profile, which in turn contribute to the deterioration of cardiac function (15–17). Therefore, a deeper understanding of the interactions between the endocrine system, aging, and the cardiovascular system is of great clinical interest, since it is estimated that more than 70% of individuals over 60 years of age suffer from cardiovascular diseases (3), and by 2050, ~17% of the population worldwide will be over 65 years of age. In the present review, we will discuss the effects of aging on the production and biological function of the primary stress hormones glucocorticoids and how alterations in glucocorticoid signaling affect the aging heart. We will also review the potential contribution of GR signaling to the vasculature morphological and functional changes that occur during the aging process. In addition, we will briefly review the cardiac effects of glucocorticoids signaling via MR in the heart. Finally, we will discuss whether the manipulation of glucocorticoid signaling in cardiomyocytes could prevent/revert aged-related heart disease.
Mechanisms Regulating Glucocorticoid Secretion and Physiological Effects
Regulation of Glucocorticoid Secretion
Glucocorticoids (cortisol in humans; corticosterone in rodents) are steroid hormones produced by the zona fasciculata of the adrenal cortex in a circadian manner and in response to stress (Figure 1). Glucocorticoid secretion is regulated by the hypothalamic-pituitary axis (18, 19). Exposure to physical, psychological, and environmental stressors stimulates parvocellular neurons within the paraventricular nucleus of the hypothalamus to release corticotropin releasing hormone (CRH) at the median eminence into the capillary plexus of the hypothalamo-hypophyseal portal system (Figure 1). CRH is then carried to the anterior lobe of the pituitary gland, where it stimulates the production and secretion of adrenocorticotropic hormone (ACTH) by corticotroph cells (Figure 1). Once in circulation, ACTH binds G protein-coupled receptors located on the extracellular membranes of the zona fasciculata and zona reticularis of the adrenal cortex (20). ACTH binding to its receptors leads to the activation of adenylyl cyclase and the production of intracellular cyclic adenosine monophosphate (cAMP). Increased formation of cAMP triggers the activation of protein kinase A (PKA), which then phosphorylates and induces hormone-sensitive lipase to hydrolyze cholesteryl esters into cholesterol (21, 22). PKA also leads to the activation of the steroidogenic acute regulatory protein (StAR) (23), which then transports cholesterol into the mitochondria, where glucocorticoids are synthesized (steroidogenesis) by the action of mitochondrial and smooth endoplasmic reticulum enzymes (Figure 1).
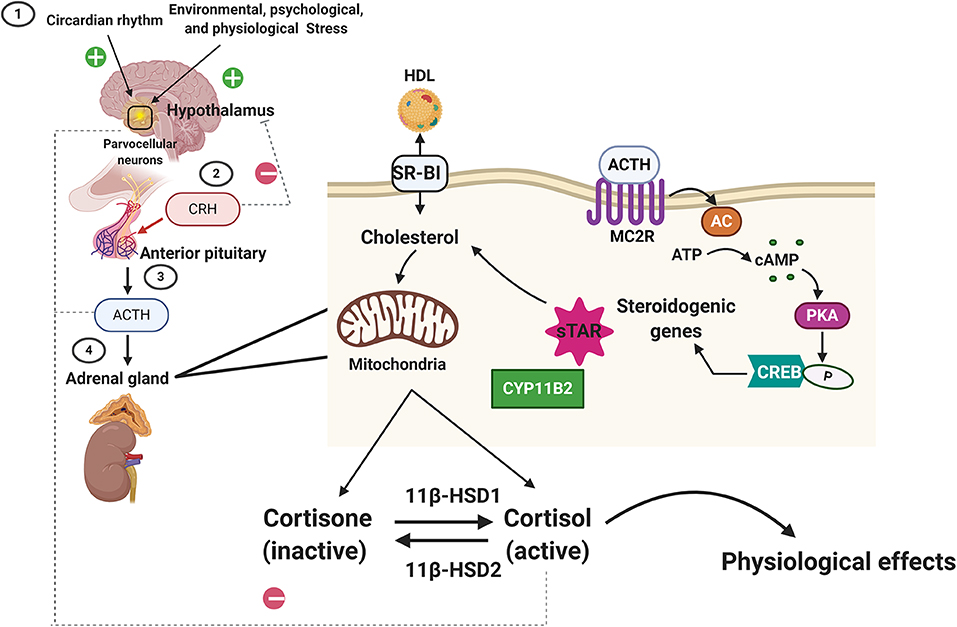
Figure 1. Regulation of glucocorticoid synthesis and secretion by the hypothalamic-pituitary-adrenal axis. Exposure to stressors and changes in our daily cycle (circadian rhythms) stimulate the parvocellular neurons within the paraventricular nucleus of the hypothalamus to release corticotropin releasing hormone (CRH). CRH then triggers the secretion of the adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. ACTH then binds to its receptors (melanocortin receptor 2, MC2R) located on the cortex of the adrenal gland. ACTH binding to MC2R leads to the activation of adenylyl cyclase (AC) and the production of intracellular cyclic adenosine monophosphate (cAMP). cAMP then activates protein kinase A (PKA), which then phosphorylates cAMP response element-binding protein (CREB), which then promotes steroidogenic gene expression (Cytochrome P450 Family 11 Subfamily B Member, CYPB2, and the steroidogenic acute regulatory protein, StAR) that leads to the transport of cholesterol (imported from the blood into the cortical cells via the scavenger receptor type B class 1, SARB1) into the mitochondria, where glucocorticoids are synthesized (steroidogenesis) by the action of mitochondrial and smooth endoplasmic reticulum enzymes. The biologically active form of the glucocorticoid is the unbound cortisol that can be converted to the inactive form, cortisone by type 2 11β-hydroxysteroid dehydrogenase. Type 1 11β-hydroxysteroid dehydrogenase converts the cortisone to cortisol. Homeostasis in glucocorticoids synthesis and secretion is maintained by the negative feedback loop suppressing ACTH levels in the anterior pituitary and CRH levels in the hypothalamus.
Following synthesis, glucocorticoids are released from the adrenal glands and are bound to plasma proteins, in particular to corticosteroid-binding globulins (CBGs). Approximately 80% of circulating glucocorticoids are bound to CBG (24, 25). Glucocorticoids are released from their binding proteins by the action of neutrophil elastases at sites of inflammation (25, 26). Free glucocorticoids then diffuse through cell membranes. The cellular levels of glucocorticoids are controlled by 2 enzymes working in an opposing manner: 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) oxidizes cortisol into the inactive metabolite cortisone whereas 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) converts cortisone to cortisol (27). Once inside the cell, glucocorticoids bind their receptor, the glucocorticoid receptor (GR, NR3C1). GR is a member of the nuclear receptor family of ligand-activated transcription factors and is ubiquitously expressed in all nucleated cells throughout the body. Glucocorticoids can also bind the closely related mineralocorticoid receptor (MR, NR3C2) which has a more restricted tissue distribution than GR (28). In most cell types, MR appears to be principally bound by aldosterone due to the action of 11βHSD2. However, glucocorticoid occupancy of MR can occur in certain tissues, such as the heart, that are deficient in this enzyme. In the face of an acute stressor, the increase in glucocorticoid levels and their signaling via GR is beneficial and aids the body in restoring homeostasis by modulating the immune response, metabolism, and cardiovascular function (Figure 1). However, exposure to chronic stress or imbalances in glucocorticoid synthesis and secretion (e.g., Cushing's disease or Addison's disease) leads to an array of pathologies, ranging from immune disorders to metabolic and cardiovascular complications (29). Therefore, in normal physiology, glucocorticoid levels are tightly regulated by a negative feedback loop at the level of the hypothalamus and pituitary gland, the availability of CBG in circulation and at target tissues by the action of 11βHSD1 and 11βHSD2 (26).
As elegantly discussed by McEwen BS (30), depending on the context, glucocorticoids can exert both protective and deleterious effects on the body. An acute increase in glucocorticoids in response to stress is critical to maintaining homeostasis and allostasis (survival) (30). However, imbalances in glucocorticoid secretion patterns and levels due to exogenous administration (increased levels) or pathological states (overproduction or deficiency) can lead to or accelerate disease processes, including metabolic and cardiovascular complications (30), which are commonly found in the elderly. During the aging process, glucocorticoid secretion patterns undergo several modifications characterized by impairments in their circadian profile. While cortisol increases early during the day in the young, a flattening in glucocorticoid rhythm is seen in a subpopulation of the elderly in particular on those suffering from chronic disease, including cognitive impairments such Alzheimer's disease (2, 31, 32). This dysregulation in natural glucocorticoid rhythm seems attributable to a reduction in sensitivity to hypothalamic-pituitary-adrenal (HPA) axis negative feedback control due to an increased glucocorticoid level (32). However, there is no cause and effect relationship established between HPA axis dysregulation and cognitive impairments, dementia, depression, anxiety, as well as an increased risk of Alzheimer's disease, diabetes, and hypertension in the elderly (33–36). Studies are needed to fully establish if there is a physiological effect between the HPA axis dysregulation and increased glucocorticoid levels and the risk for cognitive impairments, anxiety, depression, and chronic inflammatory disorders in the elderly.
Glucocorticoid Receptor Signaling and Physiology
The GR is encoded by the NR3C1 gene located on chromosome 5 (5q31) in humans and chromosome 18 in mice. The GR gene is composed of 9 exons, and three domains comprise the GR protein: (1) an amino-terminal transactivation domain (NTD), encoded by exon 2, which mediates some interactions with co-regulators and the transcriptional machinery; (2) a DNA binding domain (DBD), encoded by exons 3 and 4, which contains two zinc-finger motifs involved in genomic interactions; and (3) a ligand-binding domain (LBD), encoded by exons 5–9, that contains a hydrophobic pocket for glucocorticoid binding and an activation function (AF2) that interacts with transcriptional coregulators. The DBD and LBD are separated by a region known as the hinge region that is involved in receptor dimerization. Additionally, there are two nuclear localization signals, NL1 and NL2, located in the DBD/hinge region junction and within the LBD, respectively. Although only one gene encodes GR, alternative splicing in exon 9 results in two receptor isoforms, GRα and GRβ. GRα is thought to mediate the majority of physiological actions of glucocorticoids; however, recent studies suggest that GRβ can also regulate the expression of numerous genes involved in inflammation (37) at the level of transcription. In addition to these most abundant isoforms, the GR gene can also give rise to three additional splice isoforms known as GRγ, GR-A, and GR-P. Furthermore, GRα can also undergo alternative translation in exon 2, leading to the formation of eight additional GR isoforms, GRα: GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2, and GRα-D3. These isoforms exhibit unique expression patterns and gene regulatory profiles and are thought to play an important role in the tissue-specific actions of glucocorticoids (38).
An inactive GR is located in the cytoplasm in complex with chaperone proteins (hsp90, hsp70, and p23) and immunophilins of the FK506 family (FKBP51 and FKBP52) (38–40). Upon hormone binding, GR undergoes a conformational rearrangement that results in the dissociation of the associated proteins and the exposure of two nuclear localization signals that trigger GR rapid translocation into the nucleus (38). Once in the nucleus, GR exerts effects on the expression of target genes by directly binding to DNA or by interacting with other transcription factors (genomic mechanisms) (29, 38, 41, 42). Additionally, GR can act via protein-protein interactions in the cytoplasm (non-genomic mechanisms) that lead to rapid cellular responses that trigger the activation of various kinases, including PI3K, AKT, and MAPKs (43–45).
In addition to the different GR isoforms and genomic and non-genomic mechanisms of action, post-translational modifications (PTMs) affect the transcriptional and physiological activity of GR. The effects of phosphorylation have been extensively characterized and have been found to be critical for GR genomic and non-genomic interactions, including cellular localization, half-life, and interaction with DNA and coregulators. Additional PTMs, including ubiquitination, acetylation, and sumoylation, have been shown to influence GR activity. The GR gene and protein structure, isoforms, PTMs, and mechanisms of signaling are described in detail in recent reviews by Oakley et al. (38) and Scheschowitsch et al. (46).
Further complexity and diversity in the physiological responses of glucocorticoids are achieved by these hormones acting in a sex- and tissue-specific manner (47–50), and by glucocorticoid activation of MR in cells deficient in 11βHSD2 Like GR, MR is comprised of an NTD that regulates transcriptional activity, a DBD for interacting with specific genomic sequences, and a LBD that binds glucocorticoids (or aldosterone) (51). The DBD of MR is 94% identical to the DBD of GR. Therefore, in response to glucocorticoids, GR and MR can bind the same DNA response element but these receptors elicit distinct transcriptional effects on target genes (52, 53). Figure 2 summarizes some of the effects of glucocorticoids and GR in different organ systems.
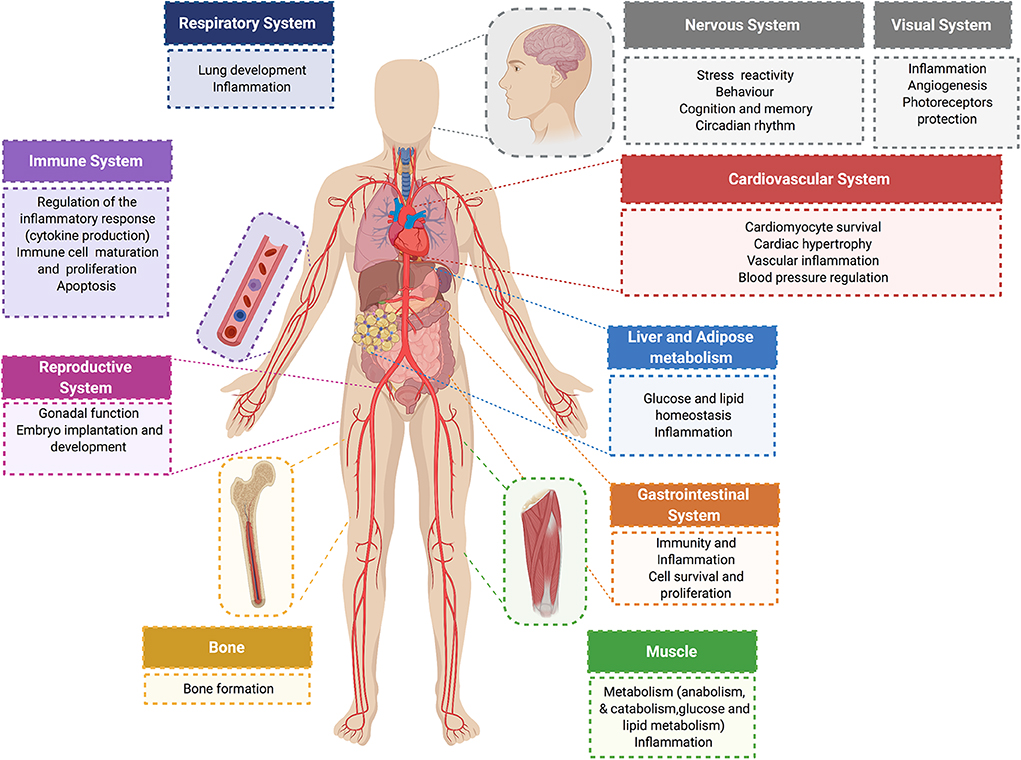
Figure 2. Physiological glucocorticoid effects. Glucocorticoids exert direct actions in major organ systems of the human body. Their effects range from modulation of the inflammatory and immune response to the regulation of glucose and lipid metabolism in different tissues. Glucocorticoid signaling plays an essential role on the cardiovascular system. In normal physiology, glucocorticoids exert cardioprotective and anti-inflammatory effects, and have an essential role in controlling blood pressure and cardiac function.
How aging alters the molecular mechanisms of glucocorticoid signaling remains largely unknown. A study by Murphy et al. (54) found that intracellular GR trafficking is impaired in the aging hippocampus due to a deficit in chaperone proteins, which diminish GR signaling within this area of the brain. A later study showed that GR mRNA levels in the cortex of the brain rise between infancy and adolescence and decline between adulthood and advanced age (55). The same study found that GR mRNA levels remain stable across the life span in the hippocampus (55). However, the mechanism that causes this aged-related change in GR expression in the cortex is unknown.
Little is also known about the effects of aging on GR signaling in the cardiovascular system. For example, no studies have been performed to test whether the expression of GR isoforms changes with age or whether there are alterations in GR phosphorylation or in other PTMs that affect GR cellular signaling. Additionally, no data exist on how activation of GR signaling by exposure to acute or chronic stressors alters the gene expression profile of the aging heart. In the next section, we will discuss the latest research on glucocorticoids and GR signaling in the heart and the potential interplay between age and stress in cardiac health.
Glucocorticoid Signaling and the Aging Cardiovascular System
Glucocorticoids and the Vasculature in Aging
Several mechanisms have been shown to contribute to vascular pathology in aging. However, oxidative stress [increased reactive oxygen species (ROS) production] is a significant contributor to coronary artery disease, myocardial ischemia, and stroke in the elderly (56). There is strong evidence that endothelial dysfunction caused by ROS leads to both impaired dilation of coronary arteries and a pro-atherogenic vascular phenotype in aging (57–61) by altering endothelium nitric oxide (NO) production (62), which is a major endogenous vasodilator, anti-inflammatory, and anti-thrombotic molecule (59–61). The decreased in NO synthesis has been proposed to be responsible for many of the age-associated vasoconstriction and reduced tissue perfusion (63–66). NO bioavailability during aging is also affected by alterations in endothelial nitric oxide synthase (eNOS) activation status, concentrations of L-arginine (NO substrate) and decreasing expression of guanosine triphosphate cyclohydrolase 1 (GTPCH1) mRNA, which is the rate-limiting enzyme in the production of the cofactor tetrahydrobiopterin (BH4) (59–61, 67). Glucocorticoids exert effects on the vasculature by GR regulation of a vast array of signaling pathways that are involved in development, angiogenesis, oxidative stress, and inflammation in vascular smooth and endothelial cells (59, 68). Among the most well-characterized effects of glucocorticoids on the vasculature are those mediated by GR modulation of NO biosynthesis (68, 69). NO production is elevated in the early response to glucocorticoids, but its levels are significantly repressed at later phases of the stress response, or when exposed to sustained increases in systemic glucocorticoids (70). Studies using mouse models have shown that systemic glucocorticoid administration leads to hypertension by a mechanism involving inhibition of NO metabolites, NO2–, and NO3– (indicators of total NO levels), and by downregulating the gene expression of NO synthase III in endothelial cells (71). More recent studies on transgenic mouse models showed that these effects are mediated directly by vascular smooth muscle and endothelial cells GR expression (72, 73). Although mice lacking GR in vascular smooth muscle cells displayed a normal phenotype under basal physiology, when glucocorticoids were acutely administered, the smooth cell-specific knockout mice were protected from glucocorticoid-induced hypertension as compared to their littermate controls (72). These results suggest a role for smooth cells-GR in regulating the hypertensive response in vivo (72). An intact GR signaling seems to play a more critical role in vascular endothelial cells. Mice lacking GR in endothelial cells were relatively resistant to dexamethasone-induced hypertension (74) and displayed increased expression of eNOS and inducible nitric oxide synthase (iNOS), which are critical enzymes in NO synthesis (73, 74). Endothelial cell GR knockout mice also have an exacerbated hypotensive response after lipopolysaccharide (LPS) administration due to increase NO production by endothelial cells (74).
In addition to the effects of glucocorticoids on NO biosynthesis, glucocorticoids can increase the expression of angiotensin II type I receptors in smooth muscle cells affecting blood pressure (75). Moreover, glucocorticoids can influence the influx of Na+ and Ca2+ into vascular smooth muscle affecting contractility and therefore leading to alterations in blood pressure. However, this effect could be mediated by the closely related mineralocorticoid receptor (MR). In addition, glucocorticoids exert effects on the vascular tone regulation by inhibiting angiogenesis, cell proliferation, viability, and cell migration via GR regulation of the anti-angiogenic thrombospondin-1 (74, 76). Moreover, glucocorticoids have indirect effects on the vasculature throughout their actions on inflammatory cells within the vasculature that contribute to endothelial cell responses and function. In the context of aging, the role of GR signaling in the vasculature has not been studied or characterized. However, based on the data discussed above, glucocorticoid signaling is most likely influencing the aged vasculature, and depending on the context, the effects may be beneficial or detrimental. Future studies are needed to fully elucidate the role of GR signaling in the vasculature in aging, and whether pharmacological regulation of this signaling pathway can be potentially used to control hypertension in the elderly.
Glucocorticoids and the Heart
In the last decade, studies have shown that depending on the physiological context (e.g., sex, disease state, etc.), type and duration of the stress (e.g., environmental, psychological, acute or chronic), and mechanisms of signaling (via GR or the closely related MR) glucocorticoids have many effects on the heart. Some of these effects are positive and essential for life, whereas other effects can be detrimental for cardiac health. Clinical data suggest that decreased systemic GR signaling is associated with a reduction in cardiac contractile force, systolic dysfunction, coronary artery disease, dilated cardiomyopathy, and progression to heart failure (77–81). Similarly, overactivated glucocorticoid signaling has been shown to lead to negative cardiac outcomes. For example, prenatal exposure to glucocorticoids due to increased stress levels during pregnancy increases the risk for developing cardiovascular disease in adulthood (82). Additionally, excessive glucocorticoid levels due to endocrine disorders or pharmacological treatment are linked to major risk factors for cardiovascular disease, including metabolic syndrome and hypertension, and to pathological cardiac hypertrophy and failure (83–86). A limitation of many these studies is that they do not distinguish between systemic actions of glucocorticoids and direct local actions of glucocorticoids on the heart and the vasculature.
Both in vitro and in vivo models have provided significant insights into the direct role played by cardiomyocyte GR. Since global deletion of GR leads to death soon after birth (87), Rog-Zielinska et al. performed prenatal studies on mice with global knockout of GR and on mice with conditional knockout of GR in cardiomyocytes and vascular smooth muscle cells to investigate the role of glucocorticoid signaling in fetal heart maturation (88). Their studies showed that prenatal inactivation of global GR decreases the size of the heart, impairs diastolic function, and alters cardiomyocyte structure and fibril organization. Hearts from these mice also presented with defects in the levels of alpha myosin heavy chain (Myh6), the major contractile protein in adult hearts, and in calcium handling proteins, including the cardiac ryanodine receptor (Ryr2), sodium calcium exchanger (NCX1), and the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA2a) (88). These findings highlight the role of glucocorticoid signaling in the development of the heart, particularly in regulating the maturation and expression of genes critical for cardiomyocyte architecture and function (88).
The role of GR signaling in the postnatal heart has also been demonstrated with transgenic mouse models. An elegant study by Sainte-Marie et al. (89) demonstrated that cardiomyocyte overexpression of human GR in the mouse heart leads to an abnormal heart rhythm (bradycardia) and electrical deficits in the heart, including an extended PQ interval, long QRS duration, and increased QTc dispersion (89). In vitro studies using cardiomyocytes isolated from the heart of these transgenic mice showed that the observed phenotype resulted from defects in sodium and potassium currents and increases in L-type calcium currents, calcium transient amplitudes, and the sarcoplasmic reticulum (SR) calcium content (89). These results suggested that overactivation of GR signaling in cardiomyocytes leads to abnormalities in the cardiac conduction system, but no structural abnormalities were found in this mouse model.
Studies by Oakley et al. (90) on mice lacking GR only in cardiomyocytes showed that inactivation of cardiomyocyte GR leads to premature death (median survival age is ~7 months) due to the development of systolic dysfunction and dilated cardiomyopathy. In agreement with the prenatal and overexpression findings, this study also found that cardiomyocyte GR plays a role in the regulation of genes involved in cardiac structure and calcium handling (90). Interesting the deleterious effects of GR inactivation occur earlier and exacerbated in males compared to females (91).
Recent studies by the same group using a transgenic mouse model lacking both GR and MR in cardiomyocytes showed that inactivation of MR rescued the left ventricular dysfunction and premature mortality observed in the absence of GR (92). Despite exhibiting a reduction in several Ca2+ handling genes as well as increased expression of pathological cardiac hypertrophy markers, the double knockout mice have normal heart morphology and function compared to the single GR knockout mouse model (92). These data suggest that under conditions of myocardial stress, such as that triggered by inactivation of cardiomyocyte GR, MR signaling becomes deleterious and promotes cardiac disease (93–96). Cardiac MR signaling has also been shown to be harmful in other models of heart dysfunction. Evidence suggests that pathological conditions resulting in the accumulation of reactive oxygen species may be responsible for the inappropriate gene regulatory activity of glucocorticoid occupied MR in the heart (97–99). Currently, there is much interest in cardiac MR signaling since treatment with the MR antagonists eplerenone or spironolactone leads to a reduction in morbidity and mortality in heart failure patients (100). The role of MR signaling in the heart is described in detail in recent reviews by Oakley et al. (78) and Young et al. (101). Figure 3 summarizes some of the effects of glucocorticoids on the heart.
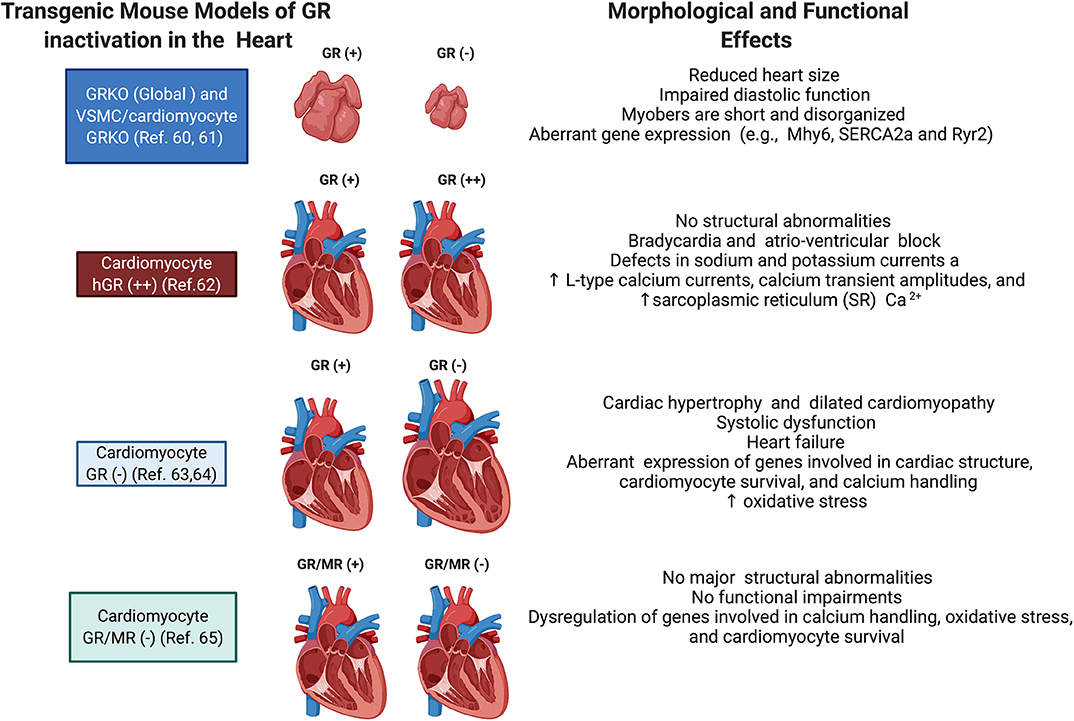
Figure 3. Schematic representation of the phenotypes of transgenic mice targeting GR in cardiomyocytes. Shown are the major morphological and functional phenotypes associated with GR overexpression (GR+), GR inactivation (GR–), and GR and MR inactivation (GR/MR–) in the whole heart, vascular smooth muscle cells (VSMC), and cardiomyocytes.
Regarding the role of glucocorticoids in the aging process, some studies show that an increase in glucocorticoid levels accelerates the aging process and increases the risk of premature mortality due to negative effects on vasculature, adipose tissue, and lipid and carbohydrate metabolism (102–104). There are no studies, however, on the direct effects of glucocorticoid signaling on aging cardiomyocytes and the heart. Therefore, it is still unclear whether increased glucocorticoid levels directly contribute to cardiovascular complications during aging or whether the interplay of glucocorticoid signaling with additional risk factors, such as obesity, hypertension, and diabetes, drives the negative systemic actions of these hormones on the heart. In the next section, we will discuss the potential role of glucocorticoids and GR signaling in cardiomyocyte dysfunction in aging and whether manipulation of this system could be potentially used to maintain a healthy heart.
Glucocorticoids and Cardiomyocyte Dysfunction in Aging
Cardiac hypertrophy is a hallmark of aging and a major cause of heart failure (3, 4, 105, 106). Increased glucocorticoid levels have been associated with pathological cardiac hypertrophy (107). In vitro and in vivo studies show that glucocorticoid administration under non-stress conditions leads to cardiomyocyte hypertrophy characterized by cellular structural and morphological changes and an increase in the levels of the cardiac hypertrophy markers atrial natriuretic factor (ANF), β-myosin heavy chain (MHC), and skeletal actin (SKA). Additionally, changes in the expression of the pro-hypertrophic genes α1b-adrenergic receptor (ADRα1b), insulin growth factor (IGF)-IR), and interleukin (IL)-6R were found in response to glucocorticoid treatment (108–110). IGF-I signaling is one of the best characterized pathways that is known to be involved in regulating lifespan in animal models (111). Decreased activation of this signaling has been associated with improved cardiomyocyte performance and an attenuation of age-related structural changes in cardiomyocytes (112, 113); however, studies in humans have shown a correlation between low IGF-I levels and an increased risk for heart failure in elderly patients (114). Additionally, an increase in circulating levels of IGF-I resulting from growth hormone (GH) therapy has been shown to be beneficial for the treatment of heart failure patients (115–117). IGF-I signaling has been proposed to exert this cardioprotective effect on aging by decreasing reactive oxygen species production by the mitochondria and thereby reducing cellular oxidative stress (118, 119). Therefore, some of the effects of glucocorticoids on the aging heart may be mediated by GR-dependent regulation of this pathway. Future studies are needed to further elucidate the relevance of the crosstalk between GR and IGF-I signaling in the context of heart aging and heart failure.
Under stressful conditions (e.g., starvation), studies show that glucocorticoids protect cardiomyocytes from cell death through GR-dependent upregulation of anti-apoptotic proteins such as Bcl-xL and repression of pro-apoptotic proteins, such as Gas2 (108). These results suggest that the elevation in glucocorticoid levels during aging may have a direct protective role in the heart by preventing cardiomyocyte death triggered by age-related changes in the levels of proteins associated with cardiomyocyte architecture and mechanical properties (e.g., myosin heavy chain and sarcomeric actin isoforms), calcium handling [e.g., SERCA2 [SR (sarcoplasmic reticulum)/ER (endoplasmic reticulum) Ca2+-ATPase 2]], and DNA repair mechanisms (120).
Collectively, these findings suggest that glucocorticoids can both positively and negatively influence the function of the heart through direct effects on cardiomyocytes. Future research needs to be focused on elucidating the mechanisms by which glucocorticoids exert these positive/negative actions on cardiomyocytes, in which context glucocorticoids signal through GR and/or MR, and whether the majority of their beneficial effects on the heart are mediated by cardiomyocyte GR signaling or MR signaling.
Conclusions and Future Perspectives
Aging has a significant impact on the endocrine system, affecting hormonal secretion patterns and hormone sensitivity, but the endocrine system also modulates aging by triggering changes in gene expression and the cellular structure of different organ systems. There is a clear connection between the endocrine system and cardiac health in aging. Whether this connection leads to positive or negative effects depends on the influence of intercurrent chronic diseases, nutritional status, and other age-related changes. In the context of the heart, the aging process is associated with significant morphological and functional alterations, including cardiac hypertrophy and fibrosis, and changes in end-systolic and diastolic volume, and cardiac filling pressure and diastolic relaxation, among other parameters. The aging heart also displayed marked abnormalities in the cardiac conduction system that impacts the heart electrical properties (6, 121). Given the role of GR signaling in regulating the expression of key genes involved in Ca2+ handling, cell death pathways, and cardiac hypertrophy and fibrosis, future studies are needed to test if targeting cardiomyocyte activation of GR with high affinity agonists holds promise for the development of new therapies for combatting cardiac disease in the elderly.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This research was supported by the Molecular and Cellular Physiology Department and the Center for Cardiovascular Diseases and Sciences (DC-T), NIH: P20GM121307 and K01 HL144882-01A1 (DC-T), and the Intramural Research Program of the NIH (RO and JC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to extend our apologies to colleagues whose work we were unable to cite owing to space limitations. We would like to thank and acknowledge these individuals for their contributions to our current understanding of aging, the endocrine system, and glucocorticoid signaling in the heart.
References
1. Cai H, McNeilly AS, Luttrell LM, Martin B. Endocrine function in aging. Int J Endocrinol. (2012) 2012:872478. doi: 10.1155/2012/872478
2. van den Beld AW, Kaufman JM, Zillikens MC, S.Lamberts WJ, Egan JM, van der Lely AJ. The physiology of endocrine systems with ageing. Lancet Diabetes Endocrinol. (2018) 6:647–58. doi: 10.1016/S2213-8587(18)30026-3
3. Chiao YA. Rabinovitch PS. The aging heart. Cold Spring Harb Perspect Med. (2015) 5:a025148. doi: 10.1101/cshperspect.a025148
4. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. (2003) 107:346–54. doi: 10.1161/01.CIR.0000048893.62841.F7
5. Scholz DG, Kitzman DW, Hagen PT, Ilstrup DM, Edwards WD. Age-related changes in normal human hearts during the first 10 decades of life. Part I (Growth): a quantitative anatomic study of 200 specimens from subjects from birth to 19 years old. Mayo Clin Proc. (1988) 63:126–36. doi: 10.1016/S0025-619664945-3
6. Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin. (2012) 8:143–64. doi: 10.1016/j.hfc.2011.08.011
7. Volkova M, Garg R, Dick S, Boheler KR. Aging-associated changes in cardiac gene expression. Cardiovasc Res. (2005) 66:194–204. doi: 10.1016/j.cardiores.2004.11.016
8. Burlew BS. Diastolic dysfunction in the elderly–the interstitial issue. Am J Geriatr Cardiol. (2004) 13:29–38. doi: 10.1111/j.1076-7460.2004.00059.x
9. Rhee SS, Pearce EN. Update: systemic diseases and the cardiovascular system (II). the endocrine system and the heart: a review. Rev Esp Cardiol. (2011) 64:220–31. doi: 10.1016/j.rec.2010.10.016
10. Jones CM, Boelaert K. The endocrinology of ageing: a mini-review. Gerontology. (2015) 61:291–300. doi: 10.1159/000367692
11. Marjoribanks J, Farquhar CM, Roberts H, Lethaby A. Cochrane corner: long-term hormone therapy for perimenopausal and postmenopausal women. Heart. (2018) 104:93–95. doi: 10.1136/heartjnl-2017-311583
12. Chahal HS, Drake WM. The endocrine system and ageing. J Pathol. (2007) 211:173–80. doi: 10.1002/path.2110
13. Abate N, Haffner SM, Garg A, Peshock RM, Grundy SM. Sex steroid hormones, upper body obesity, and insulin resistance. J Clin Endocrinol Metab. (2002) 87:4522–7. doi: 10.1210/jc.2002-020567
14. Hak AE. Witteman JC, de Jong FH, Geerlings MI, Hofman A, Pols HA. Low levels of endogenous androgens increase the risk of atherosclerosis in elderly men: the Rotterdam study. J Clin Endocrinol Metab. (2002) 87:3632–9. doi: 10.1210/jcem.87.8.8762
15. Corpas E. Harman SM, Blackman MR. Human growth hormone and human aging. Endocr Rev. (1993) 14:20–39. doi: 10.1210/edrv-14-1-20
16. Bartke A, List EO, Kopchick JJ. The somatotropic axis and aging: Benefits of endocrine defects. Growth Horm IGF Res. (2016) 27:41–5. doi: 10.1016/j.ghir.2016.02.002
18. Burford NG, Webster NA, Cruz-Topete D. Hypothalamic-pituitary-adrenal axis modulation of glucocorticoids in the cardiovascular system. Int J Mol Sci. (2017) 18:2150. doi: 10.3390/ijms18102150
19. Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. (2015) 22:20–32. doi: 10.1159/000362724
20. Allen MJ, Sharma S. Physiology, Adrenocorticotropic Hormone (ACTH). Treasure Island, FL: StatPearls (2020).
21. Kraemer FB, Shen WJ, Natu V, Patel S, Osuga J, Ishibashi S, et al. Adrenal neutral cholesteryl ester hydrolase: identification, subcellular distribution, and sex differences. Endocrinology. (2002) 143:801–6. doi: 10.1210/endo.143.3.8693
22. Kraemer FB, Shen WJ. Hormone-sensitive lipase: control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J Lipid Res. (2002) 43:1585–94. doi: 10.1194/jlr.R200009-JLR200
23. Miller WL. Steroidogenic acute regulatory protein (StAR) a novel mitochondrial cholesterol transporter. Biochim Biophys Acta. (2007) 1771:663–76. doi: 10.1016/j.bbalip.2007.02.012
24. Hammond GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. J Endocrinol. (2016) 230: R13–25. doi: 10.1530/JOE-16-0070
25. Hammond GL, Smith CL, Paterson NA, Sibbald WJ. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab. (1990) 71:34–9. doi: 10.1210/jcem-71-1-34
26. Timmermans S, Souffriau J, Libert C, Sibbald WJ. A general introduction to glucocorticoid biology. Front Immunol. (2019) 10:1545. doi: 10.3389/fimmu.2019.01545
27. Chapman K, Holmes M, Seckl J. 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. (2013) 93:1139–206. doi: 10.1152/physrev.00020.2012
28. Young MJ. Targeting the mineralocorticoid receptor in cardiovascular disease. Expert Opin Ther Targets. (2013) 17:321–31. doi: 10.1517/14728222.2013.748750
29. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. (2013) 34:518–30. doi: 10.1016/j.tips.2013.07.003
30. McEwen BS. Allostasis allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology. (2000) 22:108–24. doi: 10.1016/S0893-133X(99)00129-3
31. Ferrari E, Cravello L, Falvo F, Barili L, Solerte SB, Fioravanti M, et al. Neuroendocrine features in extreme longevity. Exp Gerontol. (2008) 43:88–94. doi: 10.1016/j.exger.2007.06.010
32. Piazza JR, Almeida DM, Dmitrieva NO, Klein LC. Frontiers in the use of biomarkers of health in research on stress and aging. J Gerontol B Psychol Sci Soc Sci. (2010) 65:513–25. doi: 10.1093/geronb/gbq049
33. Hallstrom T, Samuelsson S, Balldin J, Walinder J, Bengtsson C, Nystrom E, et al. Abnormal dexamethasone suppression test in normal females. Br J Psychiatry. (1983) 142:489–97. doi: 10.1192/bjp.142.5.489
34. Balldin J, Gottfries CG, Karlsson I, Lindstedt G, Langstrom G, Walinder J. Dexamethasone suppression test and serum prolactin in dementia disorders. Br J Psychiatry. (1983) 143:277–81. doi: 10.1192/bjp.143.3.277
35. Ennis GE, An Y, Resnick SM, Ferrucci L, O'Brien RJ, Moffat SD. Long-term cortisol measures predict Alzheimer disease risk. Neurology. (2017) 88:371–8. doi: 10.1212/WNL.0000000000003537
36. Schoorlemmer RM, Peeters GM, van Schoor NM, Lips P. Relationships between cortisol level, mortality and chronic diseases in older persons. Clin Endocrinol. (2009) 71:779–86. doi: 10.1111/j.1365-2265.2009.03552.x
37. He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA. Human glucocorticoid receptor beta regulates gluconeogenesis and inflammation in mouse liver. Mol Cell Biol. (2015) 36:714–30. doi: 10.1128/MCB.00908-15
38. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. (2013) 132:1033–44. doi: 10.1016/j.jaci.2013.09.007
39. Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. (2007) 275:2–12. doi: 10.1016/j.mce.2007.05.018
40. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. (1997) 18:306–60. doi: 10.1210/edrv.18.3.0303
41. Beato M, Chalepakis G, Schauer M, Slater EP. DNA regulatory elements for steroid hormones. J Steroid Biochem. (1989) 32:737–47. doi: 10.1016/0022-4731(89)90521-9
42. John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. (2011) 43:264–8. doi: 10.1038/ng.759
43. Groeneweg FL, Karst H, de Kloet ER, Joels M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol. (2012) 350:299–309. doi: 10.1016/j.mce.2011.06.020
44. Samarasinghe RA, Witchell SF, DeFranco DB. Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle. (2012) 11:2819–27. doi: 10.4161/cc.21018
45. Croxtall JD, Choudhury Q, Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br J Pharmacol. (2000) 130:289–98. doi: 10.1038/sj.bjp.0703272
46. Scheschowitsch K, Leite JA, Assreuy J. New insights in glucocorticoid receptor signaling-more than just a ligand-binding receptor. Front Endocrinol. (2017) 8:16. doi: 10.3389/fendo.2017.00016
47. Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. (2009) 1179:167–78. doi: 10.1111/j.1749-6632.2009.04986.x
48. Duma D, Collins JB, Chou JW, Cidlowski JA. Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci Signal. (2010) 3:ra74. doi: 10.1126/scisignal.2001077
49. Quinn MA, Cidlowski JA. Endogenous hepatic glucocorticoid receptor signaling coordinates sex-biased inflammatory gene expression. FASEB J. (2016) 30:971–82. doi: 10.1096/fj.15-278309
50. Quinn M, Ramamoorthy S, Cidlowski JA. Sexually dimorphic actions of glucocorticoids: beyond chromosomes and sex hormones. Ann N Y Acad Sci. (2014) 1317:1–6. doi: 10.1111/nyas.12425
51. Fuller PJ, Yang J, Young MJ. Mechanisms of mineralocorticoid receptor signaling. Vitam Horm. (2019) 109:37–68. doi: 10.1016/bs.vh.2018.09.004
52. Rivers CA, Rogers MF, Stubbs FE, Conway-Campbell BL, Lightman SL, Pooley JR. Glucocorticoid receptor-tethered mineralocorticoid receptors increase glucocorticoid-Induced Transcriptional Responses. Endocrinology. (2019) 160:1044–56. doi: 10.1210/en.2018-00819
53. van Weert L, Buurstede JC, Mahfouz A, Braakhuis SM, Polman AE, Sips CM, et al. NeuroD factors discriminate mineralocorticoid from glucocorticoid receptor DNA binding in the male rat brain. Endocrinology. (2017) 158:1511–22. doi: 10.1210/en.2016-1422
54. Murphy EK, Spencer RL, Sipe KJ, Herman JP. Decrements in nuclear glucocorticoid receptor (GR) protein levels and DNA binding in aged rat hippocampus. Endocrinology. (2002) 143:1362–70. doi: 10.1210/endo.143.4.8740
55. Perlman WR, Webster MJ, Herman MM, Kleinman JE, Weickert CS. Age-related differences in glucocorticoid receptor mRNA levels in the human brain. Neurobiol Aging. (2007) 28:447–58. doi: 10.1016/j.neurobiolaging.2006.01.010
56. Madhavan MV, Gersh BJ, Alexander KP, Granger CB, Stone GW. Coronary artery disease in patients >/=80 years of age. J Am Coll Cardiol. (2018) 71:2015–40. doi: 10.1016/j.jacc.2017.12.068
57. Ganz P, Vita JA. Testing endothelial vasomotor function: nitric oxide, a multipotent molecule. Circulation. (2003) 108:2049–53. doi: 10.1161/01.CIR.0000089507.19675.F9
58. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. (1991) 43:109–42.
59. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. (2018) 123:849–67. doi: 10.1161/CIRCRESAHA.118.311378
60. Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. (2018) 15:555–65. doi: 10.1038/s41569-018-0030-z
61. Ungvari Z, Yabluchanskiy A, Hasko G, Pacher P. Age-dependent cardiovascular effects of sepsis in a murine model of cecal ligation and puncture: implications for the design of interventional studies. Am J Physiol Heart Circ Physiol. (2018) 315:H1356–7. doi: 10.1152/ajpheart.00528.2018
62. Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. (2007) 27:1908–18. doi: 10.1038/sj.jcbfm.9600491
63. Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. (2002) 90:1159–66. doi: 10.1161/01.RES.0000020401.61826.EA
64. van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. (2000) 192:1731–44. doi: 10.1084/jem.192.12.1731
65. Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. (2009) 297: H1829–36. doi: 10.1152/ajpheart.00230.2009
66. Sun D, Huang A, Yan EH, Wu Z, Yan C, Kaminski PM, et al. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol. (2004) 286:H2249–56. doi: 10.1152/ajpheart.00854.2003
67. Dai DF, Chiao YA, Marcinek DJ, Szeto HH, Rabinovitch PS. Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan. (2014) 3:6. doi: 10.1186/2046-2395-3-6
68. Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. (2002) 8:473–9. doi: 10.1038/nm0502-473
69. Ong SL, Whitworth JA. Glucocorticoid-induced hypertension and the nitric oxide system. Expert Rev Endocrinol Metab. (2012) 7:273–80. doi: 10.1586/eem.12.19
70. Puzserova A, Bernatova I. Blood pressure regulation in stress: focus on nitric oxide-dependent mechanisms. Physiol Res. (2016) 65:S309–42. doi: 10.33549/physiolres.933442
71. Baum M, Moe OW. Glucocorticoid-mediated hypertension: does the vascular smooth muscle hold all the answers? J Am Soc Nephrol. (2008) 19:1251–3. doi: 10.1681/ASN.2008040410
72. Goodwin JE, Zhang J, Geller DS. A critical role for vascular smooth muscle in acute glucocorticoid-induced hypertension. J Am Soc Nephrol. (2008) 19:1291–9. doi: 10.1681/ASN.2007080911
73. Goodwin JE, Geller DS. Glucocorticoid-induced hypertension. Pediatr Nephrol. (2012) 27:1059–66. doi: 10.1007/s00467-011-1928-4
74. Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci USA. (2013) 110:306–11. doi: 10.1073/pnas.1210200110
75. Sato A, Suzuki H, Murakami M, Nakazato Y, Iwaita Y, Saruta T. Glucocorticoid increases angiotensin II type 1 receptor and its gene expression. Hypertension. (1994) 23:25–30. doi: 10.1161/01.HYP.23.1.25
76. Logie JJ, Ali S, Marshall KM, Heck MM, Walker BR, Hadoke PW. Glucocorticoid-mediated inhibition of angiogenic changes in human endothelial cells is not caused by reductions in cell proliferation or migration. PLoS ONE. (2010) 5:e14476. doi: 10.1371/journal.pone.0014476
77. Krug JJ. Cardiac arrest secondary to Addison's disease. Ann Emerg Med. (1986) 15:735–7. doi: 10.1016/S0196-0644(86)80438-3
78. Oakley RH, Cidlowski JA. Glucocorticoid signaling in the heart: a cardiomyocyte perspective. J Steroid Biochem Mol Biol. (2015) 153:27–34. doi: 10.1016/j.jsbmb.2015.03.009
79. Geelhoed JJ, van Duijn C, van Osch-Gevers L, Steegers EA, Hofman A, Helbing WA, et al. Glucocorticoid receptor-9beta polymorphism is associated with systolic blood pressure and heart growth during early childhood. The Generation R Study. Early Hum Dev. (2011) 87:97–102. doi: 10.1016/j.earlhumdev.2010.11.006
80. Otte C. Wust S, Zhao S, Pawlikowska L, Kwok PY, Whooley MA. Glucocorticoid receptor gene, low-grade inflammation, and heart failure: the heart and soul study. J Clin Endocrinol Metab. (2010) 95:2885–91. doi: 10.1210/jc.2009-2251
81. van den Akker EL, Koper JW, van Rossum EF, Dekker MJ, Russcher H, de Jong FH, et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. (2008) 168:33–9. doi: 10.1001/archinternmed.2007.41
82. Plana-Ripoll O, Liu X, Momen NC, Parner E, Olsen J, Li J. Prenatal exposure to maternal stress following bereavement and cardiovascular disease: a nationwide population-based and sibling-matched cohort study. Eur J Prev Cardiol. (2016) 23:1018–28. doi: 10.1177/2047487315585294
83. Brotman DJ, Girod JP, Posch A, Jani JT, Patel JV, Gupta M, et al. Effects of short-term glucocorticoids on hemostatic factors in healthy volunteers. Thromb Res. (2006) 118:247–52. doi: 10.1016/j.thromres.2005.06.006
84. Souverein PC, Berard A, Van Staa TP, Cooper C, Egberts AC, Leufkens HG, et al. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart. (2004) 90:859–65. doi: 10.1136/hrt.2003.020180
85. Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med. (2004) 141:764–70. doi: 10.7326/0003-4819-141-10-200411160-00007
86. De Leo M, Pivonello R, Auriemma RS, Cozzolino A, Vitale P, Simeoli C, et al. Cardiovascular disease in Cushing's syndrome: heart versus vasculature. Neuroendocrinology. (2010) 92(Suppl. 1):50–4. doi: 10.1159/000318566
87. Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. (1995) 9:1608–21. doi: 10.1101/gad.9.13.1608
88. Rog-Zielinska EA, Thomson A, Kenyon CJ, Brownstein DG, Moran CM, Szumska D, et al. Glucocorticoid receptor is required for foetal heart maturation. Hum Mol Genet. (2013) 22:3269–82. doi: 10.1093/hmg/ddt182
89. Sainte-Marie Y, Nguyen Dinh Cat A, Perrier R, Mangin L, Soukaseum C, Peuchmaur M, et al. Conditional glucocorticoid receptor expression in the heart induces atrio-ventricular block. FASEB J. (2007) 21:3133–41. doi: 10.1096/fj.07-8357com
90. Oakley RH, Ren R, Cruz-Topete D, Bird GS, Myers PH, Boyle MC, et al. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc Natl Acad Sci USA. (2013) 110:17035–40. doi: 10.1073/pnas.1302546110
91. Cruz-Topete D, Oakley RH, Carroll NG, He B, Myers PH, Xu X, et al. Deletion of the cardiomyocyte glucocorticoid receptor leads to sexually dimorphic changes in cardiac gene expression and progression to heart failure. J Am Heart Assoc. (2019) 8:e011012. doi: 10.1161/JAHA.118.011012
92. Oakley RH, Cruz-Topete D, He B, Foley JF, Myers PH, Xu X, et al. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci Signal. (2019) 12:eaau9685. doi: 10.1126/scisignal.aau9685
93. Fraccarollo D, Berger S, Galuppo P, Kneitz S, Hein L, Schutz G, et al. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation. (2011) 123:400–8. doi: 10.1161/CIRCULATIONAHA.110.983023
94. Fraccarollo D, Bauersachs J. Cardiomyocyte mineralocorticoid receptor function post myocardial infarction. Trends Cardiovasc Med. (2011) 21:42–7. doi: 10.1016/j.tcm.2012.02.003
95. Lother A, Berger S, Gilsbach R, Rosner S, Ecke A, Barreto F, et al. Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension. (2011) 57:746–54. doi: 10.1161/HYPERTENSIONAHA.110.163287
96. Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston GA, Shen JZ, et al. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension. (2012) 60:1443–50. doi: 10.1161/HYPERTENSIONAHA.112.203158
97. Funder JW. Mineralocorticoid receptor antagonists: emerging roles in cardiovascular medicine. Integr Blood Press Control. (2013) 6:129–38. doi: 10.2147/IBPC.S13783
98. Funder JW. Mineralocorticoid receptor activation oxidative stress. Hypertension. (2007) 50:840–1. doi: 10.1161/HYPERTENSIONAHA.107.098012
99. Rossier MF, Lenglet S, Vetterli L, Python M, Maturana A. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertension. (2008) 52:721–8. doi: 10.1161/HYPERTENSIONAHA.108.114223
100. Greenberg B, Zannad F, Pitt B. Role of aldosterone blockade for treatment of heart failure and post-acute myocardial infarction. Am J Cardiol. (2006) 97:34F−40F. doi: 10.1016/j.amjcard.2006.03.009
101. Young MJ, Adler GK. Aldosterone, the mineralocorticoid receptor and mechanisms of cardiovascular disease. Vitam Horm. (2019) 109:361–85. doi: 10.1016/bs.vh.2018.10.003
102. Anderson RM, Birnie AK, Koblesky NK, Romig-Martin SA, Radley JJ. Adrenocortical status predicts the degree of age-related deficits in prefrontal structural plasticity and working memory. J Neurosci. (2014) 34:8387–97. doi: 10.1523/JNEUROSCI.1385-14.2014
103. Zambrano E, Reyes-Castro LA, Nathanielsz PW. Aging, glucocorticoids and developmental programming. Age. (2015) 37:9774. doi: 10.1007/s11357-015-9774-0
104. Girod JP, Brotman DJ. Does altered glucocorticoid homeostasis increase cardiovascular risk? Cardiovasc Res. (2004) 64:217–26. doi: 10.1016/j.cardiores.2004.07.006
105. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. (2003) 107:139–46. doi: 10.1161/01.CIR.0000048892.83521.58
106. Lakatta EG. Arterial cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular molecular clues to heart arterial aging. Circulation. (2003) 107:490–7. doi: 10.1161/01.CIR.0000048894.99865.02
107. Ohtani T, Mano T, Hikoso S, Sakata Y, Nishio M, Takeda Y, et al. Cardiac steroidogenesis and glucocorticoid in the development of cardiac hypertrophy during the progression to heart failure. J Hypertens. (2009) 27:1074–83. doi: 10.1097/HJH.0b013e328326cb04
108. Ren R, Oakley RH, Cruz-Topete D, Cidlowski JA. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology. (2012) 153:5346–60. doi: 10.1210/en.2012-1563
109. de Vries WB, van der Leij FR, Bakker JM, Kamphuis PJ, van Oosterhout MF, Schipper ME, et al. Alterations in adult rat heart after neonatal dexamethasone therapy. Pediatr Res. (2002) 52:900–6. doi: 10.1203/00006450-200212000-00015
110. La Mear NS, MacGilvray SS, Myers TF. Dexamethasone-induced myocardial hypertrophy in neonatal rats. Biol Neonate. (1997) 72:175–80. doi: 10.1159/000244481
111. Vitale G, Pellegrino G, Vollery M, Hofland LJ. ROLE of IGF-1 system in the modulation of longevity: controversies and new insights from a centenarians' perspective. Front Endocrinol. (2019) 10:27. doi: 10.3389/fendo.2019.00027
112. Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet. (2004) 36:1275–81. doi: 10.1038/ng1476
113. Li Q, Ceylan-Isik AF, Li J, Ren J. Deficiency of insulin-like growth factor 1 reduces sensitivity to aging-associated cardiomyocyte dysfunction. Rejuvenation Res. (2008) 11:725–33. doi: 10.1089/rej.2008.0717
114. Vasan RS, Sullivan LM, D'Agostino RB, Roubenoff R, Harris T, Sawyer DB, et al. Serum insulin-like growth factor I and risk for heart failure in elderly individuals without a previous myocardial infarction: the framingham heart study. Ann Intern Med. (2003) 139:642–8. doi: 10.7326/0003-4819-139-8-200310210-00007
115. Broglio F, Fubini A, Morello M, Arvat E, Aimaretti G, Gianotti L, et al. Activity of GH/IGF-I axis in patients with dilated cardiomyopathy. Clin Endocrinol. (1999) 50:417–30. doi: 10.1046/j.1365-2265.1999.00696.x
116. Khan AS, Sane DC, Wannenburg T, Sonntag WE. Growth hormone, insulin-like growth factor-1 and the aging cardiovascular system. Cardiovasc Res. (2002) 54:25–35. doi: 10.1016/S0008-6363(01)00533-8
117. Isgaard J, Arcopinto M, Karason K, Cittadini A. GH and the cardiovascular system: an update on a topic at heart. Endocrine. (2015) 48:25–35. doi: 10.1007/s12020-014-0327-6
118. Groban L, Pailes NA, Bennett CD, Carter CS, Chappell MC, Kitzman DW, et al. Growth hormone replacement attenuates diastolic dysfunction and cardiac angiotensin II expression in senescent rats. J Gerontol A Biol Sci Med Sci. (2006) 61:28–35. doi: 10.1093/gerona/61.1.28
119. Perez R, Garcia-Fernandez M, Diaz-Sanchez M, Puche JE, Delgado G, Conchillo M, et al. Mitochondrial protection by low doses of insulin-like growth factor- I in experimental cirrhosis. World J Gastroenterol. (2008) 14:2731–9. doi: 10.3748/wjg.14.2731
120. Shimizu I, Minamino T. Cellular senescence in cardiac diseases. J Cardiol. (2019) 74:313–9. doi: 10.1016/j.jjcc.2019.05.002
Keywords: glucocorticoids, aging, heart, cardiomyocytes, myocardium
Citation: Cruz-Topete D, Oakley RH and Cidlowski JA (2020) Glucocorticoid Signaling and the Aging Heart. Front. Endocrinol. 11:347. doi: 10.3389/fendo.2020.00347
Received: 21 February 2020; Accepted: 04 May 2020;
Published: 27 May 2020.
Edited by:
Yang Yang, Northwest University, ChinaReviewed by:
Claude Libert, Flanders Institute for Biotechnology, BelgiumKaren Chapman, University of Edinburgh, United Kingdom
Onno Meijer, Leiden University, Netherlands
Copyright © 2020 Cruz-Topete, Oakley and Cidlowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Cruz-Topete, ZGNydXoyQGxzdWhzYy5lZHU=; John A. Cidlowski, Y2lkbG93czFAbmllaHMubmloLmdvdg==