 Miguel Martin-Aragon Baudel
Miguel Martin-Aragon Baudel Ricardo Espinosa-Tanguma
Ricardo Espinosa-Tanguma Madeline Nieves-Cintron
Madeline Nieves-Cintron Manuel F. Navedo
Manuel F. Navedo- 1Department of Pharmacology, University of California, Davis, Davis, CA, United States
- 2Departamento de Fisiologia y Biofisca, Universidad Autónoma San Luis Potosí, San Luis Potosí, Mexico
The activation of purinergic receptors by nucleotides and/or nucleosides plays an important role in the control of vascular function, including modulation of vascular smooth muscle excitability, and vascular reactivity. Accordingly, purinergic receptor actions, acting as either ion channels (P2X) or G protein-coupled receptors (GCPRs) (P1, P2Y), target diverse downstream effectors, and substrates to regulate vascular smooth muscle function and vascular reactivity. Both vasorelaxant and vasoconstrictive effects have been shown to be mediated by different purinergic receptors in a vascular bed- and species-specific manner. Purinergic signaling has been shown to play a key role in altering vascular smooth muscle excitability and vascular reactivity following acute and short-term elevations in extracellular glucose (e.g., hyperglycemia). Moreover, there is evidence that vascular smooth muscle excitability and vascular reactivity is severely impaired during diabetes and that this is mediated, at least in part, by activation of purinergic receptors. Thus, purinergic receptors present themselves as important candidates mediating vascular reactivity in hyperglycemia, with potentially important clinical and therapeutic potential. In this review, we provide a narrative summarizing our current understanding of the expression, function, and signaling of purinergic receptors specifically in vascular smooth muscle cells and discuss their role in vascular complications following hyperglycemia and diabetes.
Introduction
Nucleotides can act as extracellular signaling molecules engaging plasma membrane bound purinergic receptors in different tissues (1). In endothelial cells, these receptors mediate nitric oxide production and relaxation (2). In vascular smooth muscle cells, purinergic receptors primarily mediate vasoconstrictive actions (3–7), although there are some examples in which they can also mediate vasorelaxation (8–10). This dual short-term control by purinergic receptors contributes to the regulation of vascular reactivity and myogenic tone. Purinergic receptors are also involved in the long-term development of trophic events by participating in cell proliferation, differentiation, migration, and death, all of which are associated with development of vascular diseases (11). Nucleotides are released from endothelial cells due to shear stress, hypoxia, and low pH, which together with neural release of adenosine triphosphate (ATP) and uridine triphosphate (UTP) acting on smooth muscle cells, contribute to the regulation of blood flow in the vascular system. Besides the granular ATP secretion by platelets and nerve terminals through vesicular exocytosis, non-vesicular release of nucleotides occurs in virtually all cells (12). Such release occurs upon agonist, chemical, or mechanical stimulation, appearing to involve a variety of anionic pore-forming membrane proteins, such as pannexins, connexins, P2X7 receptors, or ATP-binding cassette transporters (13). Extracellular breakdown of ATP and UTP is also necessary for purinergic signaling in the vasculature and is mediated by ectonucleotidases, including ectonucleoside triphosphate diphosphohydrolase (E-NTPDases), ectonucleotide pyrophosphatase/phosphodiesterase (E-NPPS), alkaline phosphatase, and ecto-5′-nucleotidose (14). The combined action of purinergic receptors together with purinergic transporters and converting enzymes create a complex signaling network that has been argued to play a central role in different pathological conditions, including diabetic hyperglycemia (15).
Vascular function gradually decline with age manifesting as biochemical and structural changes in blood vessel function that compromises vascular health (16). Diabetes is a complex chronic metabolic/cardiovascular disorder with multiple pathophysiological abnormalities and a recognized cause of accelerated vascular aging (17). Elevated blood glucose levels (e.g., hyperglycemia) is a defining characteristic of diabetes whether produced by insulin-deficiency (type 1 diabetes) or insulin-resistance (type 2 diabetes) (18, 19). Both hyperglycemia and diabetes promote vascular complications that increase the risk of suffering from hypertension, stroke, coronary disease, and organ failure (20, 21). Hyperglycemia and diabetes are also associated with decreased cognitive function, retinopathy, and nephropathy (22). Hyperglycemia-induced vascular complications are due in part to altered vascular reactivity (23). In addition to endothelial dysfunction, which results in impaired vascular regulation, endothelium-independent mechanisms, including altered vascular smooth muscle cell excitability, are emerging as critical in the development, and progression of vascular complications in diabetic hyperglycemia (24–27). It has to be noted that both reduced and enhanced vasoconstriction have been described in both human and animal models of diabetes (28–32). Intriguingly, purinergic signaling has been observed to be altered in endothelial and vascular smooth muscle from both experimental animal models and humans with type-2 diabetes (33–35). Due to the involvement of purinergic receptors in regulating vascular tone, they may represent potential targets for the treatment of vascular complications during diabetic hyperglycemia. The contributions of purinergic signaling to endothelial cell function in health and disease has been extensively examined in recent studies (1, 2, 36–38). Here, we focus on how purinergic receptors regulate vascular smooth muscle function in health, in response to hyperglycemia, and during diabetes.
Functional expression of ion channels, which may be modulated by purinergic signaling (39–41), regulate vascular smooth muscle excitability and therefore vascular reactivity and myogenic tone (42). Vascular reactivity is the response of blood vessels to constrict or dilate in response to a given stimulus while myogenic tone refers to a sustained state of smooth muscle contraction. The expression and function of different types of K+ channels as well as the L-type Ca2+ channel CaV1.2 are essential for modulation of vascular reactivity and myogenic tone (42), and their functional expression is altered in response to elevated glucose and diabetes (25, 27, 42, 43). Changes in vascular smooth muscle ion channels' functional expression during hyperglycemia and diabetes can be underlined by activation of purinergic signaling. Accordingly, recent exciting findings have revealed a novel mechanism involving purinergic signaling in the regulation of L-type Ca2+ channels in vascular smooth muscle upon acute hyperglycemia exposure and during diabetes. This mechanism may contribute to modulate vascular smooth muscle excitation-contraction and excitation-transcription coupling. The findings revealed an elegant signaling complex that is engaged in response to hyperglycemia and diabetes and may have important clinical and therapeutic implications. In this review, we summarize current knowledge about the expression and function of purinergic receptors in vascular smooth muscle. Our main goals are to discuss prior literature and exciting findings describing the effect of hyperglycemia and diabetes on purinergic signaling and how it alters vascular smooth muscle function and vascular reactivity during this pathological condition/stimulus.
Expression and Function of Purinergic Receptors in Vascular Smooth Muscle
It is now well-established that purinergic signaling plays a pivotal role in the control of vascular smooth muscle function and corresponding regulation of myogenic tone and vascular reactivity (44, 45). The release of ATP, UTP, and/or its breakdown products from either endothelial cells, epithelial cells, platelets or sympathetic, and sensory-motor nerves, can induce either vasoconstrictor and vasodilatory effects through the activation of purinergic receptors in vascular smooth muscle cell (1, 46). Indeed, the endothelium itself could release as much as 300 nM ATP to the extracellular milieu (47), which may activate purinergic signaling in adjacent vascular smooth muscle cells. In addition, autocrine nucleotide release from vascular smooth muscle cells during hyperglycemia has been associated with changes in Ca2+ signaling, activation of transcription factors, and modulation of cell excitability, which seems to be mediated via engagement of one or more purinergic receptors (48, 49). Furthermore, in a rat model of streptozotocin (STZ)-induced diabetes (50), zebrafish (51), and retinal cultures (52) exposed to elevated extracellular glucose, and both type 1 (53) and type 2 diabetic patients (53–55), purine blood and/or extracellular levels have been shown to be elevated, which could be responsible for increased purinergic receptor signaling.
The first evidence for purines having a physiological effects in the cardiovascular system was reported more than 90 years ago (56). Two different families of purinergic receptors were subsequently identified and classified according to their primary agonist: (1) adenosine conforming the P1 family and (2) ATP/UTP corresponding to the P2 family (57). Four P1 GPCR subtypes (A1, A2A, A2B, and A3), 7 P2X ion channel receptor subtypes (P2X1−7), and 8 P2Y GPCR subtypes (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14) are recognized (58, 59). Different purinergic receptors have been identified in vascular smooth muscle cells of different vascular beds and species, including P2X1, P2X2, P2X4, P2X5, P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P1A1, P1A2, and P1A3 (1). These purinergic receptors have been shown to mediate both vasoconstrictor and vasodilatory effect in a species- and vascular bed-dependent manner, as well as having a role in vascular smooth muscle cells differentiation and proliferation (4, 48, 49, 60). In the following section, we will describe each family and receptor subtype and their involvement in the regulation of the vascular system highlighting their role in hyperglycemia and diabetes. Table 1 summarizes the expression, physiological agonist, physiological effect and involvement in pathophysiology of different purinergic receptors in smooth muscle cells from different vascular beds.
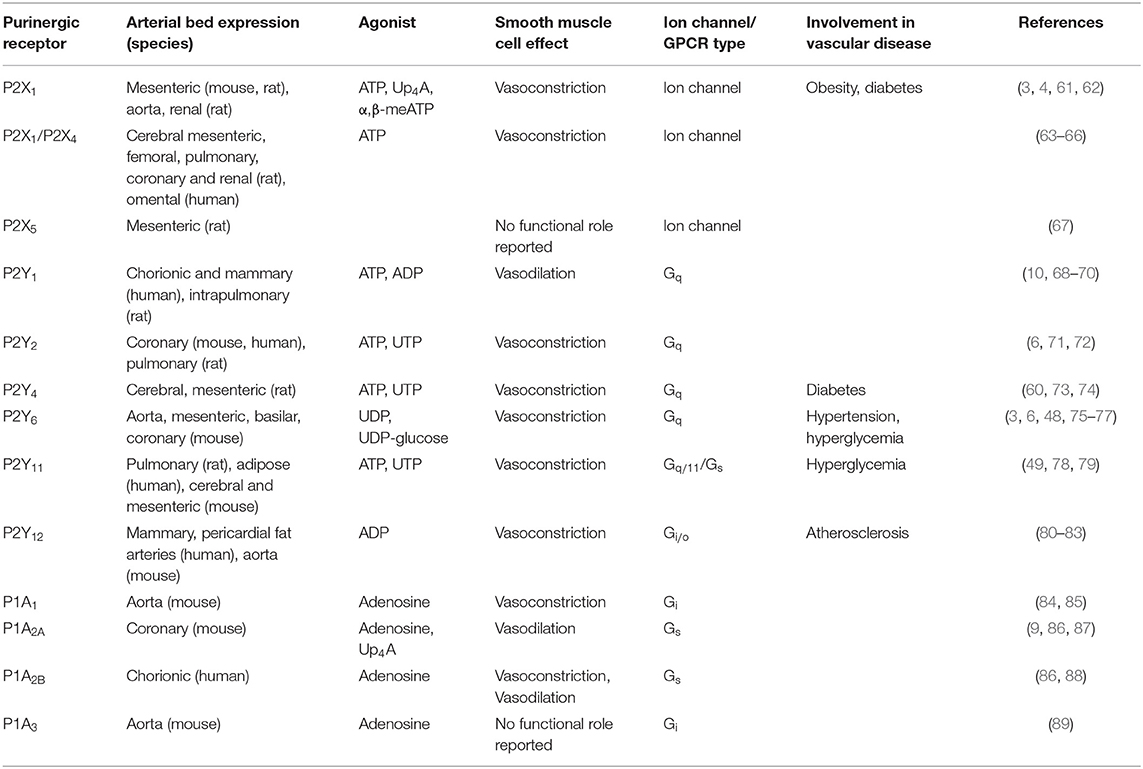
Table 1. Summary of the different purinergic receptors expressed in smooth muscle cells discussed in this review.
P2X Receptors in Vascular Smooth Muscle Cells
P2X receptors are cation permeable ligand-gated ion channels (90). Their activation by ATP leads to a rapid response involving Ca2+ and Na2+ entry directly through the P2X channel pore (91). The subsequent membrane depolarization of vascular smooth muscle due to activation of P2X channels contribute to calcium influx via voltage-gated L-type Ca2+ channels. The source of ATP for activation of P2X channels may come from sympathetic nerves in the adventitia as a co-transmitter with noradrenaline and neuropeptide Y, but also from contracting smooth muscle or damaged cells to induce vasoconstriction (1, 13, 92, 93)
The contractile actions of ATP released from perivascular sympathetic nerves in smooth muscle cells was confirmed to involve principally homomeric P2X1 receptors (3). In this study, the involvement of P2X1 receptors was corroborated using P2X1 knockout mice. P2X1 receptors appear in close proximity to sympathetic nerve varicosities where they form clusters that seem to be associated with lipid rafts (94, 95). Depolarizations mediated by neural release of ATP engaging P2X1 receptors are known as excitatory junction potentials, which can summate to produce vasoconstriction involving L-type Ca2+ channel activation (96–98). This process is vessel-dependent, as rat mesenteric artery vasoconstriction is entirely mediated via the receptor pore (97). Noradrenaline, which is co-released with ATP by sympathetic nerves, induces longer depolarizations, and contractions as it involves α-adrenoceptor coupling to Gq proteins. This elicits Ca2+ release from internal stores via inositol 1,4,5-trisphosphate (IP3), and this combined rise in cytoplasmic Ca2+, together with Ca2+ sensitization induces contraction (99). The vasoconstrictive contribution of ATP and noradrenaline is also vessel-dependent, with ATP mediating 10% of the peak response in rat tail artery (100), 20–60% in rabbit central ear artery (101) and 100% in rabbit mesenteric artery (102). Thus, sympathetic nerve-mediated vasoconstriction and the relative contribution of noradrenaline and ATP is influenced by many factors. In rat intrapulmonary arteries, ATP released by non-adrenergic non-cholinergic nerves (NANC) can also elicit excitatory junction potential (103). This is in contrast to most blood vessels in which ATP released from NANC nerves induces endothelium-dependent vasorelaxation (104, 105).
Uridine adenosine tetraphosphate (Up4A) is an endothelium-derived vasoconstricting factor that when released by different stimuli engages P2X1 receptors to promote contraction of rat aortic vascular smooth muscle cells (4), and perhaps cells in other vascular beds. This contraction was attenuated by L-type Ca2+ channel antagonists and Rho-kinase inhibitors (4), suggesting a crosstalk between P2X1 receptor, L-type Ca2+ channels, and Rho-kinase signaling that regulates vascular reactivity.
Other P2X receptors have been recently identified in vascular smooth muscle cells. For example, heteromeric P2X1/P2X4 receptors have been shown to mediate vasoconstriction of rat cerebral arteries (63). Heteromeric P2X1/P2X4 receptors are also expressed in human omental arteries (64) and rat mesenteric, femoral, pulmonary, coronary, and renal arteries (65, 66, 106). The functional role of these purinergic receptors in these vascular beds as well as their role in health and disease, however, remains to be elucidated. P2X5 receptors are also expressed in rat small mesenteric small arteries with no functional role reported to date (67).
P2Y Receptors in Vascular Smooth Muscle Cells
P2Y receptors are GPCRs with ligand selectivity (107). ATP is the primary physiological P2Y11 agonist. P2Y2 and P2Y4 are activated by ATP but also by UTP. Adenosine diphosphate (ADP) activates P2Y1, P2Y12, and P2Y13. Finally, P2Y6 and P2Y14 are activated by uridine diphosphate (UDP) and UDP-glucose, respectively. In addition, the G protein subtype of each receptor defines the specificity of the intracellular signal elicited (107). For example, P2Y1, P2Y2, P2Y4, and P2Y6 couple primarily with Gq, P2Y12, P2Y13, and P2Y14 couple with Gi/o and P2Y11 couple to both Gs and Gq/11 in vascular smooth muscle cells (59, 78, 108). Given the number of different P2Y receptor subtypes, the variety of functional signaling cascades engaged, the biased agonism of GPCR activation and the relative lack of subtype-specific agonists and inhibitors, research into the (patho)physiological role of these receptors has been challenging and remains poorly understood.
P2Y1 receptors are mainly expressed in endothelial cells mediating vasodilation (109). Although P2Y1 expression has been detected in vascular smooth muscle cells from human mammary arteries (68) and in the rat intrapulmonary artery at low levels, they seem to play no role in the response to ATP in this tissues (10). In endothelium-denuded human chorionic arteries, P2Y1 expression has been shown to be higher (69) and to mediate vasoconstriction (70). Interestingly, this study demonstrates a micro-regionalized distribution of P2Y1 receptors into lipid rafts, which when disrupted, abrogated P2Y1 receptor-mediated vasoconstriction. Interestingly, differential expression of P2Y1 was observed along the human placental vascular tree, with a decline in receptor expression in the vascular smooth muscle layer as the tree approaches the capillary network (69). A concomitant reduction in agonist-mediated vasoconstriction and a shift in vascular response to vasodilation was observed as the size of the vessels decreased. Robust P2Y1 expression was also found in canine coronary vascular smooth muscle cells (109). In this study, P2Y1 activity was found to promote agonist-induced vasodilation of coronary arteries via an endothelium-dependent mechanism in in vitro and in vivo settings. However, these receptors did not seem to play a role in pressure-flow autoregulation, thus revealing distinct mechanisms by which P2Y1 can control vascular reactivity.
P2Y2 receptors are expressed in smooth muscle cells of coronary arteries in different species and their activation by ATP or UTP has been shown to promote vasoconstriction (6, 71). In small pulmonary veins, ATP-induced vasoconstriction was associated with the stimulation of P2Y2 receptors in vascular smooth muscle cells (72). This effect was linked to the activation of PLC-β and the generation of intracellular Ca2+ oscillations mediated by cyclic Ca2+ release events via IP3 receptor activation. In vascular smooth muscle cells, P2Y2 receptors also have trophic roles, stimulating DNA synthesis, proliferation, and migration of human and rat aortic vascular smooth muscle cells, which are key events in vascular remodeling (1).
P2Y4, which are selectively activated by pyrimidines, are present in smooth muscle cells of cerebral arteries where their activation leads to vasoconstriction (73). The mechanism by which P2Y4 are activated and mediate vasoconstriction in intraparenchymal cerebral arterioles is proposed to be mechanically linked instead of through the release of endogenous nucleotides, and likely involves activation of TRP channels, inhibition of K+ channels, or direct activation of L-type Ca2+ channels (60). P2Y4 receptors have also been shown to mediate proliferation of rat aortic smooth muscle cells (110).
Different studies in rodents have highlighted the role of P2Y6 receptor in vascular smooth muscle cells as mediator of contraction in aorta, mesenteric, and basilar arteries (3, 75, 76). In mouse large diameter segments of the coronary artery, P2Y6 activation promotes contraction of vascular smooth muscle in response to UDP, whereas in smaller diameter segments, its activation causes vasodilation via an endothelium-dependent mechanism (6). P2Y6 is the most expressed P2Y receptor in resistance arteries and contributes to the maintenance of the myogenic tone through an autocrine/paracrine activation loop (7). Interestingly this action is independent of intracellular Ca2+ increase through the Gq/PLCβ/IP3 pathway and is proposed to involve phosphorylation of mitogen-activated protein kinases P38/P42–44/c-Jun N-terminal and the Rho-kinase Ca2+ sensitizing pathway (111). These mitogen-activated protein kinases are activated by different external stressors such as heat, UV irradiation, osmotic shock, or cell stretch (112–114). Therefore, P2Y6-mediated phosphorylation and activation of these kinases could represent a novel stress response mechanism. Kauffenstein et al., argued that cell stretching caused by a rise in intraluminal pressure induces the release of nucleotides that stimulate P2Y6 and promote smooth muscle cell contraction (autocrine/paracrine activation loop). However, other studies have suggested that the P2Y6 regulation of myogenic tone, at least in parenchymal arterioles, is not mediated by extracellular nucleotides, but rather by direct stretch-induced activation of the receptor (60). Yet, the mechanisms by which P2Y6 “sense” mechanical stretch remain to be elucidated. The importance of P2Y6 in regulating blood pressure has been recently highlighted by P2Y6 knockout mice that displayed attenuated angiotensin II (AngII) induced hypertension and vascular remodeling in mice (77). Intriguingly, this study also revealed heterodimer formation between angiotensin type 1 receptors (AT1R) and P2Y6 and an age-related increase of this heterodimerization (77), which could contribute to age-associated high blood pressure (115). P2Y6 activation by UDP has also been shown to act as a growth factor stimulating mitogenesis in vascular smooth muscle cells (116).
P2Y11 is primarily stimulated by ATP, although it can also be stimulated by ADP, but not pyrimidines (59, 78, 117). This receptor has the unique property of coupling to Gq/11 and Gs proteins (78). Consequently, activation of P2Y11 can stimulate PLCβ/protein kinase C (PKC) and adenylyl cyclase (AC)/protein kinase A (PKA) signaling (78). However, its role in the vascular system has so far remained unclear. In cardiomyocytes, P2Y11 has been shown to mediate positive ionotropic effects via activation of PLC and cyclic adenosine monophosphate (cAMP) signaling, and its function seems to be impaired in a desmin-deficient mouse model of cardiomyopathy that produces congestive heart failure (118). Furthermore, a polymorphism (Ala-87-Thr) in P2Y11 has been associated to increased risk of acute myocardial infarction and C-reactive protein blood levels (119). In vascular smooth muscle cells from rat pulmonary arteries, a Ca2+-dependent chloride current was activated by ATP through a P2Y receptor that was suggested to resemble a P2Y11 receptor (79). In colonic smooth muscle, P2Y11 receptors are involved in both fast and slow relaxations through KCa channels conforming parasympathetic inhibition of the gut (120). However, its role in the vasculature has remained unclear. A recent study linked P2Y11 to glucose-mediated regulation of vascular smooth muscle excitability (further discussed below) (49).
The role of ADP-selective P2Y12 in platelet aggregation is well-recognized, being the target of the antithrombotic drug clopidogrel (121). Clopidogrel has also been shown to improve endothelial dysfunction in AngII-induced hypertensive rats by improving endothelial-mediated relaxation (122). However, this effect does not seem to be directly mediated by P2Y12 receptors expressed in the endothelium. P2Y12 has also been shown to be expressed in vascular smooth muscle cells and mediate contraction in human internal mammary arteries (80). In these experiments, clopidogrel did not reduced ADP-induced contraction, which was deemed to be due to the low penetration and high instability of this drug. A modified version of clopidogrel with increased half-life (AZD6140) has been shown to block P2Y12-mediated contractions in mice aorta and human internal mammary arteries and pericardial fat arteries (81). Recent evidence also suggests an increase in the expression of this receptor in vascular smooth muscle cells in atherosclerosis, and in playing a role in migration and potentiating atherogenesis (82, 83).
P1A Receptors in Vascular Smooth Muscle Cells
P1 receptors are GPCRs that respond to adenosine upon hydrolysis of ATP (123). All P1 receptor subtypes expression (P1A1, P1A2A, P1A2B, and P1A3) have been described in smooth muscle cells where they mediate either vasoconstriction or vasorelaxation in a species- and vascular bed-specific form (45). P1 receptors A2A and A2B are coupled to AC through the activation of Gs proteins where they mediate relaxation, whereas A1 and A3 are coupled to Gi proteins and mediate vasoconstriction (124).
P1A1 receptors mediate aortic vascular smooth muscle cell contraction through the PLC pathway and its activation also reduces vascular smooth muscle relaxation mediated by P1A2B and P1A2A receptors (84). Activation of adenosine receptors leads to the metabolism of arachidonic acid via the PLC second messenger diacylglycerol (DAG). Studies utilizing P1A1 knockout () mice demonstrated that arachidonic acid metabolites can activate PKCα and the ERK1/2 pathway in aortic vascular smooth muscle leading to vasoconstriction (5). Furthermore, PKCα activation via P1A1 receptors can lead to the inhibition of large-conductance Ca2+-activated K+ (BKCa) channel activity (125), which can further contribute to contraction of vascular smooth muscle cells. P1A1-deficient mice have also been shown to present impaired autoregulation of the renal vascular resistance by removal of the P1A1-dependent vasoconstrictor tone (126), suggesting the involvement of the purinergic system in pressure-induced resistance changes. P1A3 receptor has been shown to mediate aortic vasoconstriction through an endothelium-dependent mechanism that is associated with reactive oxygen species (ROS) generation via Nox2 (89). However, the role that vascular smooth muscle P1A3 play in the control of the myogenic tone has not yet been fully determined.
The activation of P1A2A/B receptors is generally thought to mediate vasodilation in the coronary circulation in an endothelium-dependent and -independent manner via activation of the Gs/AC/cAMP/PKA signaling pathway (86). In the coronary circulation, P1A2A-mediated vasorelaxation is achieved via modulation of both endothelial and smooth muscle cells (9, 86) and involves ATP-sensitive K+ (KATP) channels (127). Up4A has been shown to mediate coronary smooth muscle relaxation through P1A2A-induced H2O2 production and subsequent activation of BKCa and voltage-gated K+ (KV) channels leading to vasodilation (87). A contractile role of P1A2B in chorionic vascular smooth muscle cells has also been demonstrated (88). Here, activation of the P1A2B is coupled to the synthesis of an arachidonate metabolite, likely thromboxane A2, which might activate a thromboxane receptor as the final effector of the adenosine contractile response.
Vascular Smooth Muscle Purinergic Signaling in Hyperglycemia and Diabetes
The vascular system is severely impaired in response to hyperglycemia, affecting both endothelial, and vascular smooth muscle cells. Mechanisms mediating endothelial dysfunction during hyperglycemia have been extensively examined (19, 25, 28, 128–130), and therefore will not be further considered here. In diabetes, the distribution and location of purinergic receptors is altered and can also be shifted between endothelial and smooth muscle cells, which can affect vascular reactivity (69). In addition, hyperglycemia can alter vascular smooth muscle function and vascular reactivity, at least in part, through engagement of purinergic signaling (33, 49, 131). Indeed, elevations in extracellular glucose have been shown to induce a paracrine and/or autocrine release of nucleotides that could engage purinergic receptors to modulate vascular function (48, 49, 52). Purinergic receptors, but also nucleotide and nucleoside converting enzymes and transporters, are affected in the hyperglycemic vascular system (33). Surprisingly, the role of purinergic receptors in modulating vascular smooth muscle excitability in response to hyperglycemia and during diabetes has not been extensively examined. This open new opportunities for future research on how changes in the functional expression of purinergic receptors in vascular smooth muscle could impact myogenic tone and vascular reactivity during hyperglycemic and diabetic states.
The sympathetic system, through the release of ATP, is responsible for the activation of P2X1 receptors to induce vasoconstriction. Interestingly, accumulating data from animal and human studies suggest an overactivity of the sympathetic system as a defining factor in the development and maintenance of diabetes (132–134). In a type 2 animal model of diabetes [Goto-Kakizaki (GK) rats], Up4A-induced contraction of renal artery rings was shown to be increased due to the activation of the cyclooxygenase (COX)/thromboxane (Tx) receptor pathway (61). Similar observations were found in type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats (135). In these OLETF rats, enhanced contraction was further increased with aging and suppressed by COX inhibition (135). Interestingly, increased Up4A concentration was detected in circulating plasma levels of human juvenile hypertensives (136). Furthermore, in mesenteric resistance arteries of diet-induced obesity rats, sympathetic nerve-mediated vasoconstriction is augmented, and involves upregulation of purinergic P2X1 signaling (62). Taken together, these studies suggest a potential role for P2X1 in vascular complications during hyperglycemia, obesity, and diabetes.
The role of P2Y1 in modulating vascular smooth muscle function in response to hyperglycemia and diabetes is unclear. Interestingly, however, a study employing P2Y1 knockout () mice, which present increased blood glucose levels (10 mM compared to 8 mM in wild type animals), showed that this receptor plays a physiological role in the maintenance of glucose homeostasis by regulating insulin secretion (137, 138). Although this mechanism is not directly involved in smooth muscle cell regulation of contractility, it highlights the need of subtype-specific purinergic receptor modulators for therapeutic use in order to avoid side-effects.
In mesenteric arteries from GK rats, the expression of P2Y4 was found to be decreased compared with control groups, and P2Y2/4-mediated contractions were shown to be increased (74). The diabetes-related enhancement of ATP-mediated vasoconstriction was due to P2Y receptor-dependent activation of the cPLA(2)/COX pathway. However, the cellular type (endothelial cell vs. smooth muscle cell) responsible for these effects was not identified in this study.
A role for P2Y6 receptors in modulating Ca2+ signaling and activation of the transcription factor NFATc3 during chronic elevations in extracellular glucose has been described (48). NFAT has been linked to vascular development during embryogenesis (139) and to cause enhanced vascular excitability in hypertension (140). Nilsson et al., demonstrated that high glucose promoted both nuclear translocation of NFATc3 and decreased its export from the nucleus via a P2Y6-dependent increase in intracellular Ca2+concentration (48). In this study, elevations in glucose (11.5–20 mmol/L) led to a global rise in intracellular calcium concentration through an autocrine/paracrine activation of P2Y6 receptors and subsequent activation of calcineurin, combined with inhibition of glycogen synthase kinase 3 (GSK3-b), and c-Jun N-terminal kinase (JNK). This combined action leads to an increase in NFAT nuclear accumulation and transcriptional activity, presenting this transcription factor as a P2Y6-dependent metabolic sensor in the vascular wall with relevance for vascular dysfunction in diabetes. Interestingly, NFAT3c activation has been shown to downregulate Kv channels (140) and BKCa channels (141), which indirectly increases L-type Ca2+ channel function leading to enhanced excitability of smooth muscle cells. Therefore, the activation of P2Y6 receptors following exposure to elevated levels of glucose could contribute to the activation of a molecular cascade leading to enhanced vascular contractility in diabetes.
In aorta of a mouse model of streptozotocin (STZ)-induced diabetes, P1A1 receptor-mediated signaling is modified without changes in overall protein expression (85). This study shows that P1A1 receptor-mediated vasoconstriction was decreased, and P1A2A receptor-mediated vasodilation impaired in diabetic mice compared to control mice. The differences were attributed to changes in receptor sensitivity. The authors showed no differences in vascular reactivity in mesenteric arteries of diabetic mice, revealing the differences in tissue specific signaling. In addition, adenosine and adenosine receptors have important non-vascular regulatory roles on glucose homeostasis and lipid metabolism and therefore in the development of diabetes that can contribute to vascular complications observed during this pathological condition (142).
Recently, new data examining how acute hyperglycemia alters vascular smooth muscle excitability have revealed an unexpected role for a Gs-coupled purinergic receptor that fits the profile of P2Y11 (49). Activation of this purinergic receptor in response to hyperglycemic conditions has been shown to modulate intracellular Ca2+ signaling and vascular reactivity (49). Given the unforeseen discovery of this P2Y11 in mediating the glucose effects in vascular smooth muscle as well as the novel signaling pathway described, we provide an extended discussion on this subject. Indeed, this study has great significance as it completely dissects the molecular events that link high glucose with changes in vascular reactivity in ex vivo and in vivo experiments utilizing both animal and human tissue.
P2Y11 and cAMP/AC Signaling in Hyperglycemia
P2Y11 receptors were first cloned from human samples and thereafter in other species (59, 78). In a recent study, immunoreactive bands of expected and similar molecular weight for P2Y11 were detected in side-by-side samples of human adipose artery and mouse mesenteric artery lysates (49). Because the ubiquitous expression P2Y11 in many cell types, including endothelial cells, expression of P2Y11 was confirmed in freshly isolated human adipose, and mouse mesenteric vascular smooth muscle cells using immunofluorescence imaging. Although the gene for P2Y11 has not been found within the expected region of the mouse genome, a number of recent rodent annotations has been made (e.g., XM_008766009.2 and XM_0130655917.2) and a growing number of functional studies based on pharmacological data suggest at least the presence of a P2Y11-like receptor in rodents (78). Nevertheless, while further studies should be undertaken to confirm the P2Y11-like receptor in rodent tissue, data suggested the presence of a P2Y11/P2Y11-like receptors in human and murine vascular smooth muscle, respectively.
Interest for a role for P2Y11 in glucose-induced changes in vascular smooth muscle Ca2+ homeostasis was fostered after the unexpected finding that elevated glucose could stimulate L-type Ca2+ channel activity leading to increased intracellular Ca2+ and vasoconstriction of mouse cerebral arteries via a mechanism requiring PKA (143, 144). P2Y11 is the only P2Y receptor subtype that couples to Gs proteins (145), which can stimulate AC activation to promote cAMP synthesis and PKA activity [Figure 1, (78, 146)]. Indeed, using innovative Förster resonance energy transfer (FRET) based cAMP biosensors expressed in human-derived tsA-201 cells, it was found that application of the highly selective P2Y11 agonist NF546 (147) increased cAMP synthesis (49). This NF546-induced cAMP response was blocked in cells treated with the selective P2Y11 inhibitor NF340 (147), but not with selective P2Y1 and P2Y6 inhibitors (49). In unpassaged human and mouse vascular smooth muscle cells expressing the same biosensor as above, similar NF546-induced cAMP responses were observed (49). Moreover, stimulating cells with an elevated glucose concentration (e.g., 15–20 mM D-glucose) that is comparable to that observed in diabetic patients and animal models (28, 143, 144, 148–150), cAMP synthesis of about the same magnitude as that observed with application of NF546 was observed. This response was not further amplified by the simultaneous application of both stimuli (49), suggesting that they may be acting via activation of the same signaling pathway. In addition, glucose-induced cAMP synthesis required glucose transport and metabolization as experiments in the presence of the membrane impermeable mannitol or non-metabolizable L-glucose failed to promote cAMP production (49). Importantly, the glucose and NF546 induced cAMP synthesis (as an independent stimulus or in combination) in human and mouse vascular smooth muscle cells was blocked if cells were first pre-treated with NF340. These findings provided robust data for the involvement of human P2Y11 and mouse P2Y11-like receptors in elevated glucose-induced cAMP synthesis.
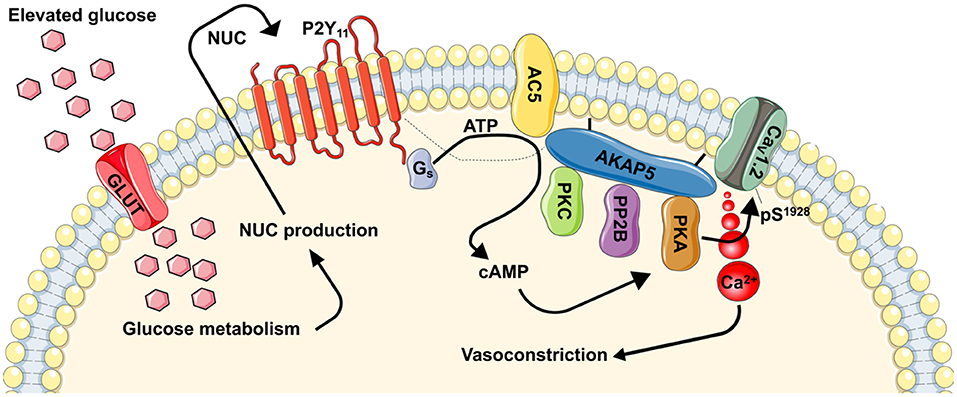
Figure 1. Proposed model for regulation of vascular contractility by P2Y11-dependent regulation of L-type Ca2+ channels during hyperglycemia and diabetes. During hyperglycemic conditions, glucose is transported into the cells via a glucose transporter (GLUT). Inside the cell, glucose is metabolized resulting in the production of nucleotides (NUC), such as ATP and UTP. These NUC are released to the extracellular space where they activate purinergic receptors coupled to Gs proteins (i.e., P2Y11). Activation of P2Y11 promotes AC5 activity and localized cAMP production. This cAMP microdomain can enable a pool of PKA that is intimately associated with L-type Ca2+ channels to increase CaV1.2 phosphorylation at S1928, which will potentiate channel activity. Hyperactive L-type Ca2+ channels result in increased global [Ca2+]i and contraction of vascular smooth muscle. Dotted line is to reflect potential close association between proteins. This figure was created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
The classic model for the production of cAMP by GsPCRs, including P2Y11/P2Y11-like receptors, suggest the involvement of intermediate regulatory enzymes such as AC [Figure 1, (151, 152)]. Nine membrane-bound AC isoforms have been described (153). Of these isoforms, AC3, AC5, and AC6 are the most abundantly expressed in vascular smooth muscle (154–156), and changes in their expression and/or function have been linked to vascular complications during diabetic hyperglycemia (157–159). AC3 and AC6 have been associated with vasodilatory pathways via regulation of K+ channels (155, 156). Moreover, a reduction in AC6 expression has been correlated with decreased vascular smooth muscle relaxation during diabetic hyperglycemia (158). Intriguingly a recent study revealed that AC5 was necessary for glucose-induced cAMP synthesis (160). Indeed, cAMP production in response to elevated glucose was completely prevented in vascular smooth muscle from AC5 knockout (AC5−/−), but not wild type or heterozygous (AC5−/+) mice. The lack of glucose-induced cAMP synthesis in AC5−/− cells was comparable to results observed in wild type cells treated with the P2Y11 inhibitor NF340 (160). Thus, it is tempting to speculate that glucose-induced activation of P2Y11/P2Y11-like receptor leading to cAMP synthesis proceeds via engagement of AC5. This exciting possibility should be examined in future investigations.
P2Y11, L-type Ca2+ Channel CaV1.2, and Myogenic Tone in Hyperglycemia
Evidence linking hyperglycemia to changes in vascular smooth muscle intracellular Ca2+ dates back decades (161–163). Yet, the association between hyperglycemia, Ca2+ homeostasis and purinergic signaling was only reported about 13 years ago (48), and a more comprehensive mechanism is just surfacing [Figure 1, (49, 143, 144, 160, 164)]. In this emerging mechanism (and as stated above), glucose is transported into the cell likely via one or more glucose transporters. Indeed, vascular smooth muscle cells express insulin-independent (e.g., Glut1) and insulin-dependent (e.g., Glut4) glucose transporters (165–167). While there is evidence for a role for both pathways, pe-treatment with indinavir, which is consider a selective Glut4 inhibitor (168, 169), prevented glucose-mediated potentiation of L-type Ca2+ channels in isolated cerebral vascular smooth muscle cells (49). Once inside the cell, glucose is metabolized (49, 149). This promotes the release of nucleotides (48, 49, 52, 144, 170, 171), perhaps via pannexins, connexins, P2X7 receptors, or ATP-binding cassette transporters (13). These nucleotides activate P2Y11 receptors to stimulate the activity of coupled Gs proteins and subsequently AC to generate cAMP (Figure 1). Data suggest that the AC5 isoform underlies the glucose-induced cAMP synthesis (160), although more studies are needed to conclusively link P2Y11 with AC5. Glucose-induced production of cAMP then activates one of its effector proteins, in this case PKA, to stimulate L-type Ca2+ channel activity (Figure 1) (143, 144, 164). The increase in L-type Ca2+ channel activity is mediated by an elevation in the phosphorylation state of residue serine 1928 (S1928) in the pore-forming CaV1.2 α1c subunit (49, 144). This results in an increase in global intracellular Ca2+ in vascular smooth muscle cells to promote contraction and vasoconstriction (Figure 1). Glucose-induced vasoconstriction is observed in both in ex vivo and in vivo preparations (49, 144, 160), thus underscoring the significance of the activation of this signaling pathway.
Support for this mechanism is robust. First, glucose-induced L-type Ca2+ channel potentiation was prevented if glucose transport into vascular smooth muscle cells was blocked with the glucose transporter inhibitor indinavir (144). Second, the role of extracellular nucleotides was confirmed in experiments in which the ectonucleotidase apyrase prevented glucose-induced S1928 phosphorylation, L-type Ca2+ channel stimulation, and vasoconstriction (49). Moreover, experiments under continuous perfusion or static bath conditions corroborated the importance of extracellular nucleotides in potentiating L-type Ca2+ channel activity upon elevated glucose. Third, genetically depleting AC5 (e.g. AC5−/−) or pre-treating cells/tissue with the P2Y11 receptor inhibitor NF340 blocked glucose-induced L-type Ca2+ channel stimulation, global increases in intracellular Ca2+, and vasoconstriction (49, 160). Fourth, cell/arteries pre-treated with a PKA inhibitor (144) or from a S1928A mouse in which the phosphorylation of this amino acid residue is prevented (144, 172) failed to show S1928 phosphorylation, L-type Ca2+ channel potentiation, and vasoconstriction in response to elevated glucose or the P2Y11 agonist NF546. Based on these data, it is tempting to speculate that P2Y11, AC5, PKA, and CaV1.2 are part of a signaling complex that facilitate its activation in response to elevations in extracellular glucose. Consistent with this possibility, super-resolution microscopy, and proximity ligation assay experiments have confirmed close association between subpopulations of these proteins, including CaV1.2 and PKA (49, 144), CaV1.2 and AC5 (160), and CaV1.2 and P2Y11 (49). Further studies will be required to assess the interaction between all members of the signaling complex and not just their link to CaV1.2.
A key question raised by the prior observations is what is the mechanism(s) for assembly of a potential P2Y11/AC5/PKA/CaV1.2 signaling complex? It is well-known that compartmentalization of proteins is facilitated scaffold proteins such as AKAPs (151). The AKAP 5 isoform (AKAP5; murine AKAP150 and human AKAP79) is known to interact with AC5, PKA, and CaV1.2 (173–178). Thus, AKAP5 could mediate AC5-mediated localized cAMP signaling and PKA compartmentalization to specifically stimulate vascular L-type Ca2+ channels upon elevated glucose. Evidence in support of this notion is provided by data indicating that genetic depletion of AKAP5 increases the distance between pools of CaV1.2 and PKA and blocked S1928 phosphorylation, L-type Ca2+ channel potentiation, and vasoconstriction upon elevated glucose (144). Whether AKAP5 interacts with AC5 and P2Y11 in vascular smooth muscle, however, is unknown and should be investigated in future studies. These studies may confirm the formation of a unique signaling complex with broad implications in health and disease not only in vascular smooth muscle, but other excitable and non-excitable cells.
Vascular Purinergic Signaling and Therapeutic
Drugs affecting purinoreceptor signaling have been tested in clinical trials for treating diabetic vascular complications. BVT-115959 (Cambridge Biotechnology), a selective P1A2A agonist reached Phase II clinical trial for the treatment of neuropathic pain in diabetic patients with positive results (179). Unfortunately, the drug was discontinued as the company terminated its small molecule research program. Sonedenoson, a P1A2A agonist, later found to be tissue plasminogen activator-dependent, entered a phase II clinical trial as a topical gel for the treatment of diabetic ulcers (180). Again, this trial was terminated due to poor enrollment. Polydeoxyribonucleotide (PDRN) has also been used for treating poorly vascularized foot ulcers by increasing neovascularization and angiogenesis and thought to be mediated through the activation of P1A2A receptors (181). Although the effect of these drugs seems to be endothelium-dependent they present purinergic receptor modulation as a valid clinical strategy that could be extrapolated to vascular smooth muscle cells.
It is necessary to highlight current limitations for the use of purinergic modulators as therapeutic options and further research directions required. There is a lack of specific agonists/antagonists able to correctly distinguish between different purinergic subtypes. Investigations into basic mechanisms can overcome this issue by employing more generic modulators combined with the use of genetically manipulated systems or animal models, but in order to translate basic findings into therapeutic approaches there is an urgent need for better, more specific drugs. Furthermore, given the heterogeneity in tissue expression of purinergic receptors and the different functions carried in different cellular types (e.g., vascular smooth muscle cells vs. endothelial cells), basic research unraveling molecular cascades elicited by purinergic receptors will facilitate the development of more targeted and cell-specific therapeutic approaches.
Conclusions
Purinergic signaling is a key modulator of vascular function and reactivity. In this review we have examined the expression, physiological role and their involvement in vascular physiology, and pathology. We focused on how purinergic receptors and downstream mediators are responsible for regulating vascular smooth muscle cell excitability. Furthermore, we have provided different examples in which this finely tuned purinergic system can be modified following hyperglycemia and diabetes. The pathophysiological roles of purinergic signaling in blood vessels are therefore clear. Given the availability of purinergic antagonists in clinical trials for different disorders, they represent promising therapeutic targets. However, it should be noted that given the ubiquity in expression of purinergic receptors, the selectivity of therapeutic strategies will be challenging. A major effort should be put into further understanding the interactions of purinergic signaling with other established signaling systems as well as in the development of inhibitors of extracellular ATP breakdown and transport in combination with more specific purinergic receptor agonists and antagonists.
Substantial efforts are being directed into understanding the mechanisms underlying enhanced vascular smooth muscle excitability during diabetes. In particular, the role of hyperglycemia in modifying the smooth muscle contractile state is the subject of intense investigation. Recent studies provided a direct link between high glucose and activation of P2Y11 leading to changes in vascular smooth muscle excitability via engagement of L-type Ca2+ channels (Figure 1). These studies have uncovered a detailed model of the molecular events that lead to altered vascular smooth muscle excitability and how this signaling response to elevations in extracellular glucose is compartmentalized. The clinical implications of this signaling complex are significant as they shed light on a mechanism underlying altered vascular reactivity during hyperglycemia and perhaps diabetes, providing novel targets that could be exploited for improving treatment of diabetic vasculopathy.
Author Contributions
Each author wrote a section of the manuscript. M-MA integrated all the parts and generated figures and tables. MN provided overall supervision and direction to the project. All authors revised the manuscript and approved the final version.
Funding
This work was supported by NIH grants R01HL098200, R01HL121059, and R01HL149127 (to MN), UC MEXUS-CONACYT CN-19-147 (to MN and RE-T) and a UC Davis Academic Federation Innovative Development Award (to MN-C).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Navedo/Nieves-Cintron Lab for critically reading early versions of the manuscript.
References
1. Burnstock G. Purinergic signaling in the cardiovascular system. Circ Res. (2017) 120:207–28. doi: 10.1161/CIRCRESAHA.116.309726
2. Ralevic V, Dunn WR. Purinergic transmission in blood vessels. Auton Neurosci Basic Clin. (2015) 191:48–66. doi: 10.1016/j.autneu.2015.04.007
3. Vial C, Evans RJ. P2X(1) receptor-deficient mice establish the native P2X receptor and a P2Y6-like receptor in arteries. Mol Pharmacol. (2002) 62:1438–45. doi: 10.1124/mol.62.6.1438
4. Linder AE, Tumbri M, Linder FFP, Webb RC, Leite R. Uridine adenosine tetraphosphate induces contraction and relaxation in rat aorta. Vascul Pharmacol. (2008) 48:202–7. doi: 10.1016/j.vph.2008.03.003
5. Kunduri SS, Mustafa SJ, Ponnoth DS, Dick GM, Nayeem MA. Adenosine A1 receptors link to smooth muscle contraction via CYP4a, protein kinase C-α, and ERK1/2. J Cardiovasc Pharmacol. (2013) 62:78–83. doi: 10.1097/FJC.0b013e3182919591
6. Haanes KA, Spray S, Syberg S, Jørgensen NR, Robaye B, Boeynaems JM, et al. New insights on pyrimidine signalling within the arterial vasculature - different roles for P2Y2 and P2Y6 receptors in large and small coronary arteries of the mouse. J Mol Cell Cardiol. (2016) 93:1–11. doi: 10.1016/j.yjmcc.2016.01.025
7. Kauffenstein G, Tamareille S, Prunier F, Roy C, Ayer A, Toutain B, et al. Central role of P2Y 6 UDP receptor in arteriolar myogenic tone. Arterioscler Thromb Vasc Biol. (2016) 36:1598–606. doi: 10.1161/ATVBAHA.116.307739
8. Wang R, Wu Y, Tang G, Wu L, Hanna ST. Altered L-type Ca(2+) channel currents in vascular smooth muscle cells from experimental diabetic rats. Am J Physiol Heart Circ Physiol. (2000) 278:H714–22. doi: 10.1152/ajpheart.2000.278.3.H714
9. Ponnoth DS, Sanjani MS, Ledent C, Roush K, Krahn T, Mustafa SJ. Absence of adenosine-mediated aortic relaxation in A2A adenosine receptor knockout mice. Am J Physiol Hear Circ Physiol. (2009) 297:H1655–60. doi: 10.1152/ajpheart.00192.2009
10. Mitchell C, Syed NIH, Tengah A, Gurney AM, Kennedy C. Identification of contractile P2Y1, P2Y6, and P2Y12 receptors in rat intrapulmonary artery using selective ligands. J Pharmacol Exp Ther. (2012) 343:755–62. doi: 10.1124/jpet.112.198051
11. Erlinge D, Burnstock G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. (2008) 4:1–20. doi: 10.1007/s11302-007-9078-7
12. Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. (2012) 8:359–73. doi: 10.1007/s11302-012-9304-9
13. Lohman AW, Billaud M, Isakson BE. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res. (2012) 95:269–80. doi: 10.1093/cvr/cvs187
14. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta - Mol Cell Res. (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
15. Fotino C, Dal Ben D, Adinolfi E. Emerging roles of purinergic signaling in diabetes. Med Chem. (2018) 14:428–38. doi: 10.2174/1573406414666180226165204
16. Nilsson PM. Early vascular aging (EVA): consequences and prevention. Vasc Health Risk Manag. (2008) 4:547–52. doi: 10.2147/VHRM.S1094
17. Morley JE. Diabetes and aging: epidemiologic overview. Clin Geriatr Med. (2008) 24:395–405. doi: 10.1016/j.cger.2008.03.005
18. Brown A, Reynolds LR, Bruemmer D. Intensive glycemic control and cardiovascular disease: an update. Nat Rev Cardiol. (2010) 7:369–75. doi: 10.1038/nrcardio.2010.35
19. Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, et al. Diabetes mellitus-associated vascular impairment. J Am Coll Cardiol. (2013) 62:667–76. doi: 10.1016/j.jacc.2013.03.089
20. Loader J, Montero D, Lorenzen C, Watts R, Méziat C, Reboul C, et al. Acute hyperglycemia impairs vascular function in healthy and cardiometabolic diseased subjects: systematic review and meta-analysis. Arterioscler Thromb Vasc Biol. (2015) 35:2060–72. doi: 10.1161/ATVBAHA.115.305530
21. Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Hypertension. (2018) 71:1269–324. doi: 10.1161/HYP.0000000000000066
22. Stehouwer CDA. Microvascular dysfunction and hyperglycemia: a vicious cycle with widespread consequences. Diabetes. (2018) 67:1729–41. doi: 10.2337/dbi17-0044
23. Funk SD, Yurdagul A, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. Int J Vasc Med. (2012) 2012:569654. doi: 10.1155/2012/569654
24. Aronson D. Hyperglycemia and the Pathobiology of Diabetic Complications. In: Cardiovascular Diabetology: Clinical, Metabolic and Inflammatory Facets. Basel: KARGER (2008). p. 1–16. doi: 10.1159/000115118
25. Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease. Pathophysiology, clinical consequences, and medical therapy: part I. Circulation. (2003) 108:1527–32. doi: 10.1161/01.CIR.0000091257.27563.32
26. Montero D, Walther G, Pérez-Martin A, Vicente-Salar N, Roche E, Vinet A. Vascular smooth muscle function in type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia. (2013) 56:2122–33. doi: 10.1007/s00125-013-2974-1
27. Nieves-Cintrón M, Syed AU, Nystoriak MA, Navedo MF. Regulation of voltage-gated potassium channels in vascular smooth muscle during hypertension and metabolic disorders. Microcirculation. (2018) 25:e12423. doi: 10.1111/micc.12423
28. Lagaud GJL, Masih-Khan E, Kai S, Van Breemen C, Dubé GP. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res. (2001) 38, 578–89. doi: 10.1159/000051094
29. Schofield I, Malik R, Izzard A, Austin C, Heagerty A. Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation. (2002) 106:3037–43. doi: 10.1161/01.CIR.0000041432.80615.A5
30. Jarajapu YPR, Guberski DL, Grant MB, Knot HJ. Myogenic tone and reactivity of cerebral arteries in type II diabetic BBZDR/Wor rat. Eur J Pharmacol. (2008) 579:298–307. doi: 10.1016/j.ejphar.2007.10.028
31. Kold-Petersen H, Brøndum E, Nilsson H, Flyvbjerg A, Aalkjaer C. Impaired myogenic tone in isolated cerebral and coronary resistance arteries from the goto-kakizaki rat model of type 2 diabetes. J Vasc Res. (2012) 49:267–78. doi: 10.1159/000335487
32. Sauvé M, Hui SK, Dinh DD, Foltz WD, Momen A, Nedospasov SA, et al. Tumor necrosis factor / sphingosine-1-phosphate signaling augments resistance artery myogenic tone in diabetes. Diabetes. (2016) 65:1916–28. doi: 10.2337/db15-1450
33. Burnstock G, Novak I. Purinergic signalling and diabetes. Purinergic Signal. (2013) 9:307–24. doi: 10.1007/s11302-013-9359-2
34. Matsumoto T, Kobayashi S, Ando M, Iguchi M, Takayanagi K, Kojima M, et al. Alteration of vascular responsiveness to uridine adenosine tetraphosphate in aortas isolated from male diabetic otsuka long-evans tokushima fatty rats: the involvement of prostanoids. Int J Mol Sci. (2017) 18:2378. doi: 10.3390/ijms18112378
35. Zhou Z, Sorop O, de Beer VJ, Heinonen I, Cheng C, Jan Danser AH, et al. Altered purinergic signaling in uridine adenosine tetraphosphate-induced coronary relaxation in swine with metabolic derangement. Purinergic Signal. (2017) 13:319–29. doi: 10.1007/s11302-017-9563-6
36. Ray FR, Huang W, Slater M, Barden JA. Purinergic receptor distribution in endothelial cells in blood vessels: a basis for selection of coronary artery grafts. Atherosclerosis. (2002) 162:55–61. doi: 10.1016/S0021-9150(01)00681-5
37. Guns PJDF, Korda A, Crauwels HM, Van Assche T, Robaye B, Boeynaems JM, et al. Pharmacological characterization of nucleotide P2Y receptors on endothelial cells of the mouse aorta. Br J Pharmacol. (2005) 146:288–95. doi: 10.1038/sj.bjp.0706326
38. Surprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol. (2009) 71:333–59. doi: 10.1146/annurev.physiol.70.113006.100630
39. So YL, O'Grady SM. Modulation of ion channel function by P2Y receptors. Cell Biochem Biophys. (2003) 39:75–88. doi: 10.1385/CBB:39:1:75
40. Lechner SG, Boehm S. Regulation of neuronal ion channels via P2Y receptors. Purinergic Signal. (2004) 1:31–41. doi: 10.1007/s11302-004-4746-3
41. Rajasekhar P, Poole DP, Liedtke W, Bunnett NW, Veldhuis NA. P2Y1 receptor activation of the TRPV4 ion channel enhances purinergic signaling in satellite glial cells. J Biol Chem. (2015) 290:29051–62. doi: 10.1074/jbc.M115.689729
42. Tykocki NR, Boerman EM, Jackson WF. Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr Physiol. (2017) 7:485–581. doi: 10.1002/cphy.c160011
43. Fleischhacker E, Esenabhalu VE, Spitaler M, Holzmann S, Skrabal F, Koidl B, et al. Human diabetes is associated with hyperreactivity of vascular smooth muscle cells due to altered subcellular Ca2+ distribution. Diabetes. (2000) 48:1323–30. doi: 10.2337/diabetes.48.6.1323
44. Burnstock G. Purinergic regulation of vascular tone and remodelling. Auton Autacoid Pharmacol. (2009) 29:63–72. doi: 10.1111/j.1474-8673.2009.00435.x
45. Burnstock G, Ralevic V. Purinergic signaling and blood vessels in health and disease. Pharmacol Rev. (2014) 66:102–92. doi: 10.1124/pr.113.008029
46. Bell PD, Lapointe JY, Sabirov R, Hayashi S, Peti-Peterdi J, Manabe K, et al. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci USA. (2003) 100:4322–7. doi: 10.1073/pnas.0736323100
47. Kwon YM, Shinozuka K, Kagota S, Yamaguchi Y, Nakamura K, Kunitomo M. Both extracellular ATP and shear stress regulate the release of nitric oxide in rat caudal artery. Clin Exp Pharmacol Physiol. (1999) 26:465–9. doi: 10.1046/j.1440-1681.1999.03062.x
48. Nilsson J, Nilsson LM, Chen YW, Molkentin JD, Erlinge D, Gomez MF. High glucose activates nuclear factor of activated T cells in native vascular smooth muscle. Arterioscler Thromb Vasc Biol. (2006) 26:794–800. doi: 10.1161/01.ATV.0000209513.00765.13
49. Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, Ghosh D, et al. A Gs-coupled purinergic receptor boosts ca2+ influx and vascular contractility during diabetic hyperglycemia. Elife. (2019) 8:e42214. doi: 10.7554/eLife.42214.074
50. Donoso MV, Mascayano MJ, Poblete IM, Huidobro-Toro JP. Increased ATP and ADO overflow from sympathetic nerve endings and mesentery endothelial cells plus reduced nitric oxide are involved in diabetic neurovascular dysfunction. Front Pharmacol. (2018) 9:546. doi: 10.3389/fphar.2018.00546
51. Capiotti KM, Siebel AM, Kist LW, Bogo MR, Bonan CD, Da Silva RS. Hyperglycemia alters E-NTPDases, ecto-5′-nucleotidase, and ectosolic and cytosolic adenosine deaminase activities and expression from encephala of adult zebrafish (Danio Rerio). Purinergic Signal. (2016) 12:211–20. doi: 10.1007/s11302-015-9494-z
52. Costa G, Pereira T, Neto AM, Cristóvão AJ, Ambrósio AF, Santos PF. High glucose changes extracellular adenosine triphosphate levels in rat retinal cultures. J Neurosci Res. (2009) 87:1375–80. doi: 10.1002/jnr.21956
53. Michno A, Bielarczyk H, Pawełczyk T, Jankowska-Kulawy A, Klimaszewska J, Szutowicz A. Alterations of adenine nucleotide metabolism and function of blood platelets in patients with diabetes. Diabetes. (2007) 56:462–7. doi: 10.2337/db06-0390
54. Xia JF, Liang QL, Hu P, Wang YM, Li P, Luo GA. Correlations of six related purine metabolites and diabetic nephropathy in Chinese type 2 diabetic patients. Clin Biochem. (2009) 42:215–20. doi: 10.1016/j.clinbiochem.2008.10.009
55. Xia J, Wang Z, Zhang F. Association between related purine metabolites and diabetic retinopathy in type 2 diabetic patients. Int J Endocrinol. (2014) 2014:651050. doi: 10.1155/2014/651050
56. Drury AN, Szent-Györgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol. (1929) 68:213–37. doi: 10.1113/jphysiol.1929.sp002608
57. Burnstock G. A basis for distinguishing two types of purinergic receptor. Cell Membr Recept Drugs Horm. A Multidiscip Approach. (1978) 107–118. Available Online at: http://ci.nii.ac.jp/naid/10007119388/en/%5Cnpapers3://publication/uuid/D4FE293F-C87C-49CE-8C60-CD636A3A066A (accessed October 8, 2019).
58. Burnstock G. Purine and pyrimidine receptors. Cell Mol Life Sci. (2007) 64:1471–83. doi: 10.1007/s00018-007-6497-0
59. von Kugelgen I, Harden TK. Molecular pharmacology, physiology, and structure of the P2Y receptors. In: Advances in Pharmacology. Academic Press Inc (2011). p. 373–415. doi: 10.1016/B978-0-12-385526-8.00012-6
60. Brayden JE, Li Y, Tavares MJ. Purinergic receptors regulate myogenic tone in cerebral parenchymal arterioles. J Cereb Blood Flow Metab. (2013) 33:293–9. doi: 10.1038/jcbfm.2012.169
61. Matsumoto T, Watanabe S, Kawamura R, Taguchi K, Kobayashi T. Enhanced uridine adenosine tetraphosphate-induced contraction in renal artery from type 2 diabetic Goto-Kakizaki rats due to activated cyclooxygenase/thromboxane receptor axis. Pflugers Arch Eur J Physiol. (2014) 466:331–42. doi: 10.1007/s00424-013-1330-0
62. Haddock RE, Hill CE. Sympathetic overdrive in obesity involves purinergic hyperactivity in the resistance vasculature. J Physiol. (2011) 589:3289–307. doi: 10.1113/jphysiol.2011.207944
63. Harhun MI, Povstyan OV, Albert AP, Nichols CM. ATP-evoked sustained vasoconstrictions mediated by Heteromeric P2X1/4 receptors in cerebral arteries. Stroke. (2014) 2444–50. doi: 10.1161/STROKEAHA.114.005544
64. Nichols CM, Povstyan OV, Albert AP, Gordienko DV, Khan O, Vasilikostas G, et al. Vascular smooth muscle cells from small human omental arteries express P2X1 and P2X4 receptor subunits. Purinergic Signal. (2014) 10:565–72. doi: 10.1007/s11302-014-9415-6
65. Lewis CJ, Evans RJ. P2X receptor immunoreactivity in different arteries from the femoral, pulmonary, cerebral, coronary and renal circulations. J Vasc Res. (2001) 38:332–40. doi: 10.1159/000051064
66. Harhun MI, Sukhanova K, Gordienko D, Dyskina Y. Molecular identification of P2X receptors in vascular smooth muscle cells from rat anterior, posterior, and basilar arteries. Pharmacol Rep. (2015) 67:1055–60. doi: 10.1016/j.pharep.2015.03.014
67. Reho JJ, Shetty A, Dippold RP, Mahurkar A, Fisher SA. Unique gene program of rat small resistance mesenteric arteries as revealed by deep RNA sequencing. Physiol Rep. (2015) 3:e12450. doi: 10.14814/phy2.12450
68. Wang L, Karlsson L, Moses S, Hultgårdh-Nilsson A, Andersson M, Borna C, et al. P2 receptor expression profiles in human vascular smooth muscle and endothelial cells. J Cardiovasc Pharmacol. (2002) 40:841–53. doi: 10.1097/00005344-200212000-00005
69. Buvinic S, Poblete MI, Donoso MV, Delpiano AM, Briones R, Miranda R, et al. P2Y1 and P2Y2 receptor distribution varies along the human placental vascular tree: role of nucleotides in vascular tone regulation. J Physiol. (2006) 573:427–43. doi: 10.1113/jphysiol.2006.105882
70. Norambuena A, Poblete MI, Donoso MV, Espinoza CS, González A, Huidobro-Toro JP. P2Y1 receptor activation elicits its partition out of membrane rafts and its rapid internalization from human blood vessels: Implications for receptor signaling. Mol Pharmacol. (2008) 74:1666–77. doi: 10.1124/mol.108.048496
71. Malmsjö M, Hou M, Harden TK, Pendergast W, Pantev E, Edvinsson L, et al. Characterization of contractile P2 receptors in human coronary arteries by use of the stable pyrimidines uridine 5'-O-thiodiphosphate and uridine 5'- O-3-thiotriphosphate. J Pharmacol Exp Ther. (2000) 293:755–60.
72. Henriquez M, Fonseca M, Perez-Zoghbi JF. Purinergic receptor stimulation induces calcium oscillations and smooth muscle contraction in small pulmonary veins. J Physiol. (2018) 596:2491–506. doi: 10.1113/JP274731
73. Malmsjö M, Hou M, Pendergast W, Erlinge D, Edvinsson L. The stable pyrimidines UDPβS and UTPγS discriminate between contractile cerebrovascular P2 receptors. Eur J Pharmacol. (2003) 458:305–11. doi: 10.1016/S0014-2999(02)02787-5
74. Ishida K, Matsumoto T, Taguchi K, Kamata K, Kobayashi T. Mechanisms underlying altered extracellular nucleotide-induced contractions in mesenteric arteries from rats in later-stage type 2 diabetes: Effect of ANG II type 1 receptor antagonism. Am J Physiol Hear Circ Physiol. (2011) 301:H1850–61. doi: 10.1152/ajpheart.00502.2011
75. Bar I, Guns PJ, Metallo J, Cammarata D, Wilkin F, Boeynams JM, et al. Knockout mice reveal a role for P2Y6 receptor in macrophages, endothelial cells, and vascular smooth muscle cells. Mol Pharmacol. (2008) 74:777–84. doi: 10.1124/mol.108.046904
76. Kauffenstein G, Drouin A, Thorin-Trescases N, Bachelard H, Robaye B, D'Orléans-Juste P, et al. NTPDase1 (CD39) controls nucleotide-dependent vasoconstriction in mouse. Cardiovasc Res. (2010) 85:204–13. doi: 10.1093/cvr/cvp265
77. Nishimura A, Sunggip C, Tozaki-Saitoh H, Shimauchi T, Numaga-Tomita T, Hirano K, et al. Purinergic P2Y6 receptors heterodimerize with angiotensin AT1 receptors to promote angiotensin II-induced hypertension. Sci Signal. (2016) 9:ra7. doi: 10.1126/scisignal.aac9187
78. Kennedy C. P2Y11 receptors: Properties, distribution and functions. In: Advances in Experimental Medicine and Biology. San Diego, CA: Springer New York LLC (2017). p. 107–22. doi: 10.1007/5584_2017_89
79. Chootip K, Gurney AM, Kennedy C. Multiple P2Y receptors couple to calcium-dependent, Chloride channels in smooth muscle cells of the rat pulmonary artery. Respir Res. (2005) 6:124. doi: 10.1186/1465-9921-6-124
80. Wihlborg AK, Wang L, Braun OÖ, Eyjolfsson A, Gustafsson R, Gudbjartsson T, et al. ADP receptor P2Y12 is expressed in vascular smooth muscle cells and stimulates contraction in human blood vessels. Arterioscler Thromb Vasc Biol. (2004) 24:1810–5. doi: 10.1161/01.ATV.0000142376.30582.ed
81. Högberg C, Svensson H, Gustafsson R, Eyjolfsson A, Erlinge D. The reversible oral P2Y12 antagonist AZD6140 inhibits ADP-induced contractions in murine and human vasculature. Int J Cardiol. (2010) 142:187–92. doi: 10.1016/j.ijcard.2008.12.091
82. West LE, Steiner T, Judge HM, Francis SE, Storey RF. Vessel wall, not platelet, P2Y12 potentiates early atherogenesis. Cardiovasc Res. (2014) 102:429–35. doi: 10.1093/cvr/cvu028
83. Niu X, Pi S-L, Baral S, Xia Y-P, He Q-W, Li Y-N, et al. P2Y12 promotes migration of vascular smooth muscle cells through cofilin dephosphorylation during atherogenesis. Arterioscler Thromb Vasc Biol. (2017) 37:515–24. doi: 10.1161/ATVBAHA.116.308725
84. Tawfik HE, Schnermann J, Oldenburg PJ, Mustafa SJ. Role of A1 adenosine receptors in regulation of vascular tone. Am J Physiol Heart Circ Physiol. (2005) 288:H1411–6. doi: 10.1152/ajpheart.00684.2004
85. Labazi H, Teng B, Mustafa SJ. Functional changes in vascular reactivity to adenosine receptor activation in type I diabetic mice. Eur J Pharmacol. (2018) 820:191–7. doi: 10.1016/j.ejphar.2017.12.034
86. Headrick JP, Ashton KJ, Rose'Meyer RB, Peart JN. Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol Ther. (2013) 140:92–111. doi: 10.1016/j.pharmthera.2013.06.002
87. Sun C, Jiao T, Merkus D, Duncker DJ, Mustafa SJ, Zhou Z. Activation of adenosine A2A but not A2B receptors is involved in uridine adenosine tetraphosphate-induced porcine coronary smooth muscle relaxation. J Pharmacol Sci. (2019) 141:64–9. doi: 10.1016/j.jphs.2019.09.006
88. Donoso MV, López R, Miranda R, Briones R, Huidobro-Toro JP. A2B adenosine receptor mediates human chorionic vasoconstriction and signals through arachidonic acid cascade. Am J Physiol - Hear Circ Physiol. (2005) 288:H2439–49. doi: 10.1152/ajpheart.00548.2004
89. El-Awady MS, Ansari HR, Fil D, Tilley SL, Mustafa SJ. NADPH oxidase pathway is involved in aortic contraction induced by A 3 adenosine receptor in mice. J Pharmacol Exp Ther. (2011) 338:711–7. doi: 10.1124/jpet.111.180828
90. Ralevic V. P2X receptors in the cardiovascular system P2X RECEPTORS. WIREs Membr Transp Signal. (2012) 1:663–74. doi: 10.1002/wmts.58
91. Evans RJ, Lewis C, Virginio C, Lundstrom K, Buell G, Surprenant A, et al. Ionic permeability of, and divalent cation effects on, two ATP-gated cation channels (P2X receptors) expressed in mammalian cells. J Physiol. (1996) 497:413–22. doi: 10.1113/jphysiol.1996.sp021777
92. Lew MJ, White TD. Release of endogenous ATP during sympathetic nerve stimulation. Br J Pharmacol. (1987) 92:349–55. doi: 10.1111/j.1476-5381.1987.tb11330.x
93. Billaud M, Lohman AW, Straub AC, Parpaite T, Johnstone SR, Isakson BE. Characterization of the thoracodorsal artery: morphology and reactivity. Microcirculation. (2012) 19:360–72. doi: 10.1111/j.1549-8719.2012.00172.x
94. Hansen MA, Dutton JL, Balcar VJ, Barden JA, Bennett MR. P2X (purinergic) receptor distributions in rat blood vessels. J Auton Nerv Syst. (1999) 75:147–55. doi: 10.1016/S0165-1838(98)00189-1
95. Vial C, Evans RJ. Disruption of lipid rafts inhibits P2X1 receptor-mediated currents and arterial vasoconstriction. J Biol Chem. (2005) 280:30705–11. doi: 10.1074/jbc.M504256200
96. Omote S, Kigoshi S, Muramatsu I. Selective inhibition by nifedipine of the purinergic component of neurogenic vasoconstriction in the dog mesenteric artery. Eur J Pharmacol. (1989) 160:239–45. doi: 10.1016/0014-2999(89)90496-2
97. Gitterman DP, Evans RJ. Nerve evoked P2X receptor contractions of rat mesenteric arteries; dependence on vessel size and lack of role of L-type calcium channels and calcium induced calcium release. Br J Pharmacol. (2001) 132:1201–8. doi: 10.1038/sj.bjp.0703925
98. Rummery NM, Brock JA, Pakdeechote P, Ralevic V, Dunn WR. ATP is the predominant sympathetic neurotransmitter in rat mesenteric arteries at high pressure. J Physiol. (2007) 582:745–54. doi: 10.1113/jphysiol.2007.134825
99. Kennedy C. ATP as a cotransmitter in the autonomic nervous system. Auton Neurosci. (2015) 191:2–15. doi: 10.1016/j.autneu.2015.04.004
100. Bao JX. Sympathetic neuromuscular transmission in rat tail artery: A study based on electrochemical, electrophysiological and mechanical recording. Acta Physiol Scand Suppl. (1993) 610:1–58.
101. Kennedy C, Saville VL, Burnstock G. The contributions of noradrenaline and ATP to the responses of the rabbit central ear artery to sympathetic nerve stimulation depend on the parameters of stimulation. Eur J Pharmacol. (1986) 122:291–300. doi: 10.1016/0014-2999(86)90409-7
102. Ramme D, Regenold JT, Starke K, Busse R, Illes P. Identification of the neuroeffector transmitter in jejunal branches of the rabbit mesenteric artery. Naunyn Schmiedebergs Arch Pharmacol. (1987) 336:267–73. doi: 10.1007/BF00172677
103. Inoue T, Kannan MS. Noradrenergic and noncholinergic excitatory neurotransmission in rat intrapulmonary artery. Am J Physiol - Hear Circ Physiol. (1988) 254:H1142–8. doi: 10.1152/ajpheart.1988.254.6.H1142
104. Simonsen U, García-Sacristán A, Prieto D. Involvement of ATP in the non-adrenergic non-cholinergic inhibitory neurotransmission of lamb isolated coronary small arteries. Br J Pharmacol. (1997) 120:411–20. doi: 10.1038/sj.bjp.0700918
105. Draid M, Shiina T, El-Mahmoudy AB, Boudaka A, Shimizu Y, Takewaki T. Neurally released ATP mediates endothelium-dependent hyperpolarization in the circular smooth muscle cells of chicken anterior mesenteric artery. Br J Pharmacol. (2005) 146:983–9. doi: 10.1038/sj.bjp.0706413
106. Inscho EW, Cook AK, Imig JD, Vial C, Evans RJ. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. J Clin Invest. (2003) 112:1895–905. doi: 10.1172/JCI18499
107. Nishimura A, Sunggip C, Oda S, Numaga-Tomita T, Tsuda M, Nishida M. Purinergic P2Y receptors: molecular diversity and implications for treatment of cardiovascular diseases. Pharmacol Ther. (2017) 180:113–28. doi: 10.1016/j.pharmthera.2017.06.010
108. Erb L, Weisman GA. Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip Rev Membr Transp Signal. (2012) 1:789–803. doi: 10.1002/wmts.62
109. Bender SB, Berwick ZC, Laughlin MH, Tune JD. Functional contribution of P2Y1 receptors to the control of coronary blood flow. J Appl Physiol. (2011) 111:1744–50. doi: 10.1152/japplphysiol.00946.2011
110. Harper S, Webb TE, Charlton SJ, Ng LL, Boarder MR. Evidence that P2Y4 nucleotide receptors are involved in the regulation of rat aortic smooth muscle cells by UTP and ATP. Br J Pharmacol. (1998) 124:703–10. doi: 10.1038/sj.bjp.0701895
111. Dubroca C, Loyer X, Retailleau K, Loirand G, Pacaud P, Feron O, et al. RhoA activation and interaction with Caveolin-1 are critical for pressure-induced myogenic tone in rat mesenteric resistance arteries. Cardiovasc Res. (2007) 73:190–7. doi: 10.1016/j.cardiores.2006.10.020
112. Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. (1996) 274:1194–7. doi: 10.1126/science.274.5290.1194
113. Loufrani L, Lehoux S, Tedgui A, Lévy BI, Henrion D. Stretch induces mitogen-activated protein kinase activation and myogenic tone through 2 distinct pathways. Arterioscler Thromb Vasc Biol. (1999) 19:2878–83. doi: 10.1161/01.ATV.19.12.2878
114. Massett MP, Ungvari Z, Csiszar A, Kaley G, Koller A. Different roles of PKC and MAP kinases in arteriolar constrictions to pressure and agonists. Am J Physiol Hear Circ Physiol. (2002) 283:H2282–7. doi: 10.1152/ajpheart.00544.2002
115. O'Rourke MF, Nichols WW. Aortic diameter, aortic stiffness, and wave reflection increase with age and isolated systolic hypertension. Hypertension. (2005) 652–8. doi: 10.1161/01.HYP.0000153793.84859.b8
116. Hou M, Harden TK, Kuhn CM, Baldetorp B, Lazarowski E, Pendergast W, et al. UDP acts as a growth factor for vascular smooth muscle cells by activation of P2Y 6 receptors. Am J Physiol Circ Physiol. (2002) 282:H784–92. doi: 10.1152/ajpheart.00997.2000
117. Schuchardt M, Tolle M, van der Giet M. P2Y purinoceptors as potential emerging therapeutical target in vascular disease. Curr Pharm Des. (2012) 18:6169–80. doi: 10.2174/138161212803582504
118. Balogh J, Wihlborg AK, Isackson H, Joshi BV, Jacobson KA, Arner A, et al. Phospholipase C and cAMP-dependent positive inotropic effects of ATP in mouse cardiomyocytes via P2Y11-like receptors. J Mol Cell Cardiol. (2005) 39:223–30. doi: 10.1016/j.yjmcc.2005.03.007
119. Amisten S, Melander O, Wihlborg AK, Berglund G, Erlinge D. Increased risk of acute myocardial infarction and elevated levels of C-reactive protein in carriers of the Thr-87 variant of the ATP receptor P2Y11. Eur Heart J. (2007) 28:13–8. doi: 10.1093/eurheartj/ehl410
120. King BF, Townsend-Nicholson A. Involvement of P2Y1 and P2Y11 purinoceptors in parasympathetic inhibition of colonic smooth muscle. J Pharmacol Exp Ther. (2008) 324:1055–63. doi: 10.1124/jpet.107.131169
121. Cattaneo M. P2Y12 receptors: structure and function. J Thromb Haemost. (2015) 13:S10–6. doi: 10.1111/jth.12952
122. Giachini FR, Leite R, Osmond DA, Lima VV, Inscho EW, Webb RC, et al. Anti-platelet therapy with clopidogrel prevents endothelial dysfunction and vascular remodeling in aortas from hypertensive rats. PLoS ONE. (2014) 9:e91890. doi: 10.1371/journal.pone.0091890
123. Jacobson KA, Balasubramanian R, Deflorian F, Gao ZG. G protein-coupled adenosine (P1) and P2Y receptors: Ligand design and receptor interactions. Purinergic Signal. (2012) 8:419–36. doi: 10.1007/s11302-012-9294-7
124. Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. (2006) 5:247–64. doi: 10.1038/nrd1983
125. Kunduri SS, Dick GM, Nayeem MA, Mustafa SJ. Adenosine A 1 receptor signaling inhibits BK channels through a PKCα-dependent mechanism in mouse aortic smooth muscle. Physiol Rep. (2013) 1:e00037. doi: 10.1002/phy2.37
126. Hashimoto S, Huang Y, Briggs J, Schnermann J. Reduced autoregulatory effectiveness in adenosine 1 receptor-deficient mice. Am J Physiol Physiol. (2006) 290:F888–91. doi: 10.1152/ajprenal.00381.2005
127. Sanjani MS, Teng B, Krahn T, Tilley S, Ledent C, Mustafa J. Contributions of A2A and A2B adenosine receptors in coronary flow responses in relation to the KATP channel using A2B and A2A/2B double-knockout mice. Am J Physiol - Hear Circ Physiol. (2011) 301:2322–33. doi: 10.1152/ajpheart.00052.2011
128. Sena CM, Pereira AM, Seiça R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta Mol Basis Dis. (2013) 1832:2216–31. doi: 10.1016/j.bbadis.2013.08.006
129. Zhang HN., Xu QQ, Thakur A, Alfred MO, Chakraborty M, Ghosh A, et al. Endothelial dysfunction in diabetes and hypertension: role of microRNAs and long non-coding RNAs. Life Sci. (2018) 213:258–68. doi: 10.1016/j.lfs.2018.10.028
130. Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int J Mol Sci. (2019) 20:3775. doi: 10.3390/ijms20153775
131. Jackson R, Brennan S, Fielding P, Sims MW, Challiss RJ, Adlam D, et al. Distinct and complementary roles for α and β isoenzymes of PKC in mediating vasoconstrictor responses to acutely elevated glucose. Br J Pharmacol. (2016) 173:870–87. doi: 10.1111/bph.13399
132. Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler M. Regional sympathetic nervous activity and oxygen consumption in obese normotensive human subjects. Circulation. (1997) 96:3423–9. doi: 10.1161/01.CIR.96.10.3423
133. Lee ZSK, Critchley JAJH, Tomlinson B, Young RP, Thomas GN, Cockram CS, et al. Urinary epinephrine and norepinephrine interrelations with obesity, insulin, and the metabolic syndrome in Hong Kong Chinese. Metab Clin Exp. (2001) 50:135–43. doi: 10.1053/meta.2001.19502
134. Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res. (2015) 2015:341583. doi: 10.1155/2015/341583
135. Matsumoto T, Watanabe S, Ando M, Yamada K, Iguchi M, Taguchi K, et al. Diabetes and age-related differences in vascular function of renal artery: possible involvement of endoplasmic reticulum stress. Rejuvenation Res. (2016) 19:41–52. doi: 10.1089/rej.2015.1662
136. Jankowski V, Meyer AA, Schlattmann P, Gui Y, Zheng XL, Stamcou I, et al. Increased uridine adenosine tetraphosphate concentrations in plasma of juvenile hypertensives. Arterioscler Thromb Vasc Biol. (2007) 27:1776–81. doi: 10.1161/ATVBAHA.107.143958
137. Léon C, Freund M, Latchoumanin O, Farret A, Petit P, Cazenave JP, et al. The P2Y1 receptor is involved in the maintenance of glucose homeostasis and in insulin secretion in mice. Purinergic Signal. (2005) 1:145–51. doi: 10.1007/s11302-005-6209-x
138. Khan S, Ferdaoussi M, Bautista A, Bergeron V, Smith N, Poitout V, et al. A role for PKD1 in insulin secretion downstream of P2Y 1 receptor activation in mouse and human islets. Physiol Rep. (2019) 7:e14250. doi: 10.14814/phy2.14250
139. Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. (2001) 105:863–75. doi: 10.1016/S0092-8674(01)00396-8
140. Amberg GC, Rossow CF, Navedo MF, Santana LF. NFATc3 regulates Kv2.1 expression in arterial smooth muscle. J Biol Chem. (2004) 279:47326–34. doi: 10.1074/jbc.M408789200
141. Nieves-Cintrón M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci USA. (2008) 105:15623–8. doi: 10.1073/pnas.0808759105
142. Koupenova M, Ravid K. Adenosine, adenosine receptors and their role in glucose homeostasis and lipid metabolism. J Cell Physiol. (2013) 228:1703–12. doi: 10.1002/jcp.24352
143. Navedo MF, Takeda Y, Nieves-Cintrón M, Molkentin JD, Santana LF. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am J Physiol Cell Physiol. (2010) 298:211–20. doi: 10.1152/ajpcell.00267.2009
144. Nystoriak MA, Nieves-Cintrón M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, et al. Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel Cav1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal. (2017) 10:eaaf9647. doi: 10.1126/scisignal.aaf9647
145. Communi D, Robaye B, Boeynaems JM. Pharmacological characterization of the human P2Y11 receptor. Br J Pharmacol. (1999) 128:1199–206. doi: 10.1038/sj.bjp.0702909
146. Torres B, Zambon AC, Insel PA. P2Y11 receptors activate adenylyl cyclase and contribute to nucleotide-promoted cAMP formation in MDCK-D1 cells: a mechanism for nucleotide-mediated autocrine-paracrine regulation. J Biol Chem. (2002) 277:7761–5. doi: 10.1074/jbc.M110352200
147. Meis S, Hamacher A, Hongwiset D, Marzian C, Wiese M, Eckstein N, et al. NF546 [4,4′-(carbonylbis(imino-3,1-phenylene-carbonylimino-3,1-(4- methyl-phenylene)-carbonylimino))-bis(1,3-xylene-α,α′- diphosphonic acid) tetrasodium salt] is a non-nucleotide P2Y11 agonist and stimulates release of interleukin-8 from human monocyte-derived dendritic cells. J Pharmacol Exp Ther. (2010) 332:238–47. doi: 10.1124/jpet.109.157750
148. Moien-Afshari F, Ghosh S, Elmi S, Khazaei M, Rahman MM, Sallam N, et al. Exercise restores coronary vascular function independent of myogenic tone or hyperglycemic status in db/db mice. Am J Physiol - Hear Circ Physiol. (2008) 295:H1470–80. doi: 10.1152/ajpheart.00016.2008
149. Nystoriak MA, Nieves-Cintrón M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, et al. AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res. (2014) 114:607–15. doi: 10.1161/CIRCRESAHA.114.302168
150. Nieves-Cintrón M, Nystoriak MA, Prada MP, Johnson K, Fayer W, Dell'Acqua ML, et al. Selective down-regulation of KV2.1 function contributes to enhanced arterial tone during diabetes. J Biol Chem. (2015) 290:7918–29. doi: 10.1074/jbc.M114.622811
151. Langeberg LK, Scott JD. Signalling scaffolds and local organization of cellular behaviour. Nat Rev Mol Cell Biol. (2015) 16:232–44. doi: 10.1038/nrm3966
152. Johnstone TB, Agarwal SR, Harvey RD, Ostrom RS. CAMP signaling compartmentation: Adenylyl cyclases as anchors of dynamic signaling complexes. Mol Pharmacol. (2018) 93:270–6. doi: 10.1124/mol.117.110825
153. Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, Seifert R. International Union of Basic and Clinical Pharmacology. CI structures and small molecule modulators of mammalian adenylyl cyclases. Pharmacol Rev. (2017) 69:93–139. doi: 10.1124/pr.116.013078
154. Webb JG, Yates PW, Yang Q, Mukhin YV, Lanier SM. Adenylyl cyclase isoforms and signal integration in models of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. (2001) 281:H1545–52. doi: 10.1152/ajpheart.2001.281.4.H1545
155. Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, Insel PA. Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells: Expression in caveolin-rich and noncaveolin domains. Mol Pharmacol. (2002) 62:983–92. doi: 10.1124/mol.62.5.983
156. Nelson CP, Rainbow RD, Brignell JL, Perry MD, Willets JM, Davies NW, et al. Principal role of adenylyl cyclase 6 in K+ channel regulation and vasodilator signalling in vascular smooth muscle cells. Cardiovasc Res. (2011) 91:694–702. doi: 10.1093/cvr/cvr137
157. Hashim S, Li Y, Nagakura A, Takeo S, Anand-Srivastava MB. Modulation of G-protein expression and adenylyl cyclase signaling by high glucose in vascular smooth muscle. Cardiovasc Res. (2004) 63:709–18. doi: 10.1016/j.cardiores.2004.04.021
158. Matsumoto T, Wakabayashi K, Kobayashi T, Kamata K. Functional changes in adenylyl cyclases and associated decreases in relaxation responses in mesenteric arteries from diabetic rats. Am J Physiol Hear Circ Physiol. (2005) 289:H2234–43. doi: 10.1152/ajpheart.00971.2004
159. Li Y, Descorbeth M, Anand-Srivastava MB. Role of oxidative stress in high glucose-induced decreased expression of Giα proteins and adenylyl cyclase signaling in vascular smooth muscle cells. Am J Physiol Hear Circ Physiol. (2008) 294:H2845–54. doi: 10.1152/ajpheart.91422.2007
160. Syed AU, Reddy GR, Ghosh D, Prada MP, Nystoriak MA, Morotti S, et al. Adenylyl cyclase 5-generated cAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J Clin Invest. (2019) 129: 3140–52. doi: 10.1172/JCI124705
161. Resnick LM, Barbagallo M, Gupta RK, Laragh JH. Ionic basis of hypertension in diabetes mellitus role of hyperglycemia. Am J Hypertens. (1993) 6:397–406. doi: 10.1093/ajh/6.5.413
162. Barbagallo M, Shan J, Pang PKT, Resnick LM. Glucose-induced alterations of cytosolic free calcium in cultured rat tail artery vascular smooth muscle cells. J Clin Invest. (1995) 95:763–7. doi: 10.1172/JCI117724
163. Barbagallo M, Gupta RK, Resnick LM. Cellular ions in NIDDM: relation of calcium to hyperglycemia and cardiac mass. Diabetes Care. (1996) 19:1393–8. doi: 10.2337/diacare.19.12.1393
164. Morotti S, Nieves-Cintrón M, Nystoriak MA, Navedo MF, Grandi E. Predominant contribution of L-type Cav1.2 channel stimulation to impaired intracellular calcium and cerebral artery vasoconstriction in diabetic hyperglycemia. Channels. (2017) 11:1–7. doi: 10.1080/19336950.2017.1293220
165. Howard RL. Down-regulation of glucose transport by elevated extracellular glucose concentrations in cultured rat aortic smooth muscle cells does not normalize intracellular glucose concentrations. J Lab Clin Med. (1996) 127:504–15. doi: 10.1016/S0022-2143(96)90068-2
166. Park JL, Loberg RD, Duquaine D, Zhang H, Deo BK, Ardanaz N, et al. GLUT4 facilitative glucose transporter specifically and differentially contributes to agonist-induced vascular reactivity in mouse aorta. Arterioscler Thromb Vasc Biol. (2005) 25:1596–602. doi: 10.1161/01.ATV.0000170137.41079.ab
167. Pyla R, Poulose N, Jun JY, Segar L. Expression of conventional and novel glucose transporters, GLUT1,−9,−10, and-12, in vascular smooth muscle cells. Am J Physiol Cell Physiol. (2013) 304:C574–89. doi: 10.1152/ajpcell.00275.2012
168. Murata H, Hruz PW, Mueckler M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J Biol Chem. (2000) 275:20251–4. doi: 10.1074/jbc.C000228200
169. Murata H, Hruz PW, Mueckler M. Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. AIDS. (2002) 16:859–63. doi: 10.1097/00002030-200204120-00005
170. Hazama A, Hayashi S, Okada Y. Cell surface measurements of ATP release from single pancreatic β cells using a novel biosensor technique. Pflugers Arch Eur J Physiol. (1998) 437:31–5. doi: 10.1007/s004240050742
171. Parodi J, Flores C, Aguayo C, Rudolph MI, Casanello P, Sobrevia L. Inhibition of nitrobenzylthioinosine-sensitive adenosine transport by elevated D-glucose involves activation of P2Y2 purinoceptors in human umbilical vein endothelial cells. Circ Res. (2002) 90:570–7. doi: 10.1161/01.RES.0000012582.11979.8B
172. Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, et al. Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J Biol Chem. (2008) 283:34738–44. doi: 10.1074/jbc.M804981200
173. Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, et al. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. (1997) 19:185–96. doi: 10.1016/S0896-6273(00)80358-X
174. Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, et al. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. (2006) 23:925–31. doi: 10.1016/j.molcel.2006.07.025
175. Navedo MF, Nieves-Cintrón M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, et al. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res. (2008) 102:e1–11. doi: 10.1161/CIRCRESAHA.107.167809
176. Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, et al. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and−6 to α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem. (2010) 285:14450–8. doi: 10.1074/jbc.M110.109769
177. Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, et al. Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic β-adrenergic signaling. J Biol Chem. (2013) 288:17918–31. doi: 10.1074/jbc.M112.449462
178. Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, et al. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep. (2014) 7:1577–88. doi: 10.1016/j.celrep.2014.04.027
179. Knezevic NN, Cicmil N, Knezevic I, Candido KD. Discontinued neuropathic pain therapy between 2009–2015. Expert Opin Investig Drugs. (2015) 24:1631–46. doi: 10.1517/13543784.2015.1099627
180. Jacobson KA, Tosh DK, Jain S, Gao ZG. Historical and current adenosine receptor agonists in preclinical and clinical development. Front Cell Neurosci. (2019) 13. doi: 10.3389/fncel.2019.00124
Keywords: purinergic receptors, diabetes, ion channels, myogenic tone, vascular reactivity, P2Y11
Citation: Martin-Aragon Baudel M, Espinosa-Tanguma R, Nieves-Cintron M and Navedo MF (2020) Purinergic Signaling During Hyperglycemia in Vascular Smooth Muscle Cells. Front. Endocrinol. 11:329. doi: 10.3389/fendo.2020.00329
Received: 13 December 2019; Accepted: 28 April 2020;
Published: 22 May 2020.
Edited by:
Janos Peti-Peterdi, University of Southern California, United StatesReviewed by:
Natassia Rodrigo, University of Sydney, AustraliaTsuneo Takenaka, International University of Health and Welfare (IUHW), Japan
Copyright © 2020 Martin-Aragon Baudel, Espinosa-Tanguma, Nieves-Cintron and Navedo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Martin-Aragon Baudel, bWFydGluYXJhZ29uQHVjZGF2aXMuZWR1; Manuel F. Navedo, bWZuYXZlZG9AdWNkYXZpcy5lZHU=