 Jennyfer Martínez
Jennyfer Martínez Inés Marmisolle
Inés Marmisolle Doménica Tarallo
Doménica Tarallo Celia Quijano
Celia Quijano- Departamento de Bioquímica, Centro de Investigaciones Biomédicas (CEINBIO), Facultad de Medicina, Universidad de la República, Montevideo, Uruguay
Secretion is an energy consuming process that plays a relevant role in cell communication and adaptation to the environment. Among others, endocrine cells producing hormones, immune cells producing cytokines or antibodies, neurons releasing neurotransmitters at synapsis, and more recently acknowledged, senescent cells synthesizing and secreting multiple cytokines, growth factors and proteases, require energy to successfully accomplish the different stages of the secretion process. Calcium ions (Ca2+) act as second messengers regulating secretion in many of these cases. In this setting, mitochondria appear as key players providing ATP by oxidative phosphorylation, buffering Ca2+ concentrations and acting as structural platforms. These tasks also require the concerted actions of the mitochondrial dynamics machinery. These proteins mediate mitochondrial fusion and fission, and are also required for transport and tethering of mitochondria to cellular organelles where the different steps of the secretion process take place. Herein we present a brief overview of mitochondrial energy metabolism, mitochondrial dynamics, and the different steps of the secretion processes, along with evidence of the interaction between these pathways. We also analyze the role of mitochondria in secretion by different cell types in physiological and pathological settings.
Introduction
About 20% of the proteins synthesized by eukaryotic cells are secreted to the extracellular space either as soluble or membrane bound proteins (1). The secretory pathway in eukaryotic cells is responsible for biogenesis and proper distribution of a wide range of extracellular proteins, as well as complex carbohydrates and lipids. This pathway is highly dynamic and responsive to specific cellular demands and stimuli (2). Eukaryotic cells also secrete many amino acids and amino acid derivatives, such as neurotransmitters, that play key roles in intercellular communication (3).
As many other complex cellular processes secretion of proteins consumes energy, therefore requires the support of functional mitochondria. In the conventional pathway proteins are transported into the endoplasmic reticulum (ER), and folding and quality control of proteins in the ER (4–8) consumes ATP. Likewise the assembly, transport and fusion of the vesicles carrying proteins from the ER to the Golgi and to the plasma membrane requires the hydrolysis of ATP, as well as GTP (2, 9, 10). Besides, several steps in the secretion process depend on calcium ion (Ca2+) as a cofactor or signaling molecule (11, 12). Calcium pumps are required to maintain the appropriate concentrations of Ca2+ inside the ER and near the sites of exocytosis, and mitochondria play a relevant role providing energy to transport Ca2+ against its concentration gradient (13, 14). Mitochondria also modulate Ca2+ concentrations in the cytosol, sequestering the ion in the mitochondrial matrix (14, 15). Secretion of proteins by unconventional pathways also depends on mitochondria, not only as a source of ATP, but also of activation signals such as mitochondrial reactive oxygen species (ROS) and oxidized mtDNA (16, 17), as well as a structural platform for the assembly of inflammasomes (17, 18).
Work by others and us shows that impairment of mitochondrial catabolism and dynamics affects the secretion processes (6, 7, 19–26) and can underlie pathology in endocrine and neurodegenerative diseases (27–30). Relevant roles for mitochondria in protein secretion by immune and senescent cells have been recently described as well (19–21, 24–26, 31), underscoring the relevance of these interactions. This prompted us to explore the existing literature on mitochondrial interactions with the secretion machinery.
While extensive literature can be found regarding both secretion and mitochondrial bioenergetics and dynamics, the connection between these processes and the organelles involved are still largely unexplored. In this manuscript secretion pathways, mitochondrial metabolism and dynamics per se have not been extensively reviewed. We have relied on reviews by others to present an overview, and centered our efforts in exploring the requirement for mitochondrial ATP and fusion and fission proteins to sustain secretion in metazoans, excluding the events linked to ER stress. We expand on three main roles of mitochondria: (1) providing ATP for multiple steps of the secretion process; (2) buffering Ca2+ concentrations; (3) providing signals and structural scaffold for the activation of the inflammasome (Figure 1).
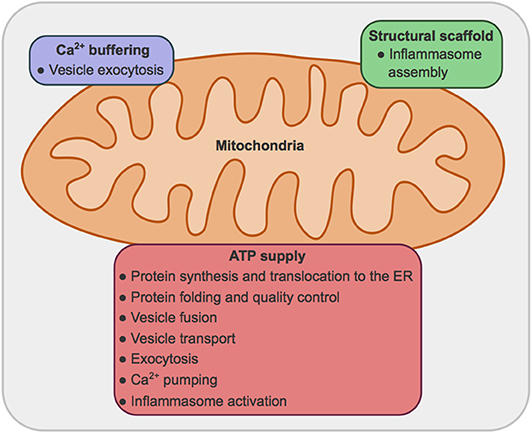
Figure 1. Main roles for mitochondria in secretory processes. (1) Mitochondria provide ATP, obtained by oxidative phosphorylation, for: protein synthesis, translocation to the ER, folding and quality control, vesicle transport, vesicle fusion and exocytosis, Ca2+ pumping across plasma and ER membranes; and inflammasome activation. (2) Mitochondria can uptake Ca2+, modulating Ca2+ concentration and therefore vesicle exocytosis. (3) Mitochondria provide a structural scaffold for the assembly of the NLRP3 inflammasome.
Overview of the Secretion Pathways and Their Energy Demands
Secreted proteins can reach the extracellular media through the conventional (classic) pathway or through unconventional pathways (32). In the conventional pathway proteins are transported into the ER, either cotranslationally or postranslationally; then travel from the ER to the Golgi complex, from which they then migrate to the trans-Golgi network and finally to the plasma membrane (33, 34). Transport from one compartment to another takes place by sequential budding and fusion of vesicles (35) and the microtubule and actin cytoskeleton plays a relevant role in vesicle transport (33, 36) (Figure 2). Unconventional secretion pathways, on the other hand, include proteins that do not present a leader sequence and proteins that by-pass the Golgi apparatus while traveling to the plasma membrane (32).
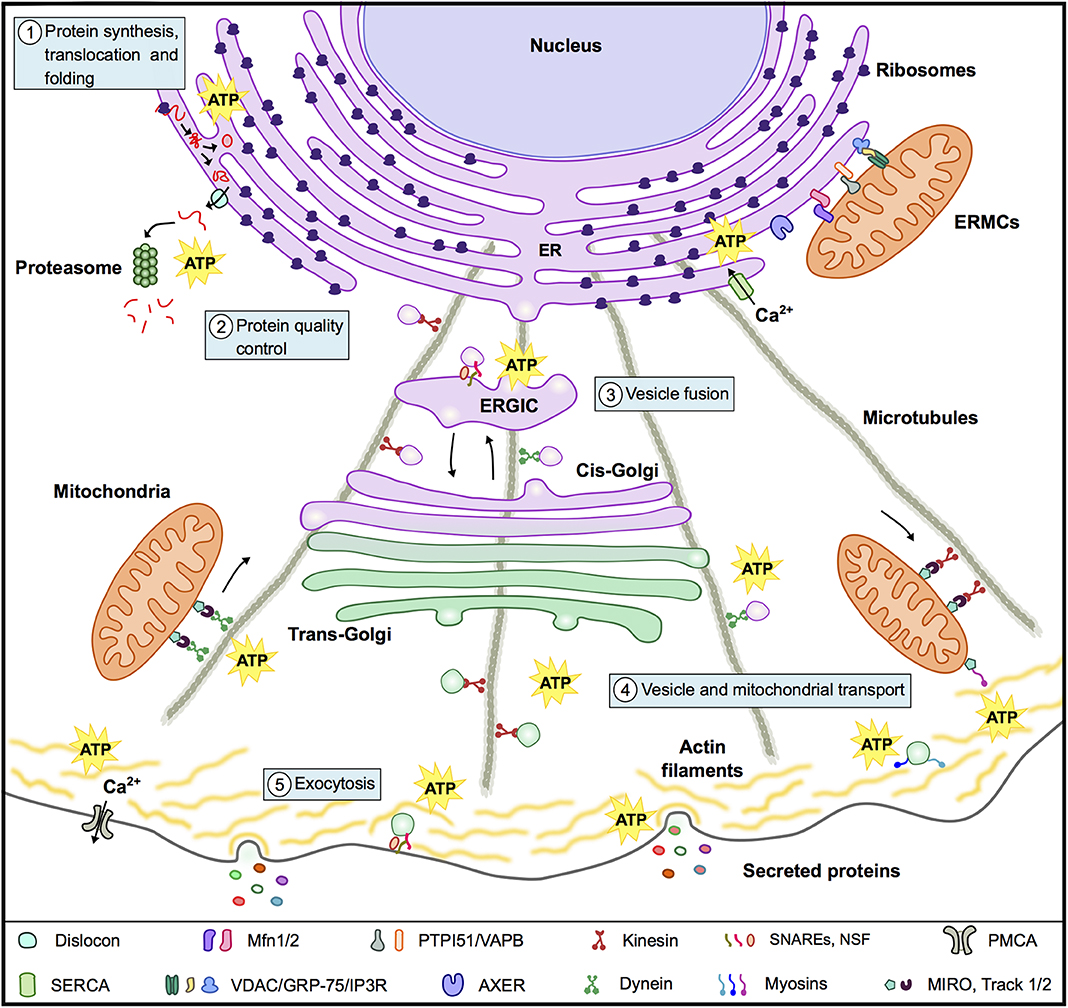
Figure 2. Mitochondria and the conventional secretion machinery. To support the energy requirements of the secretion process mitochondria interact with organelles and components of the cytoskeleton and supply ATP for: (1) Protein synthesis, translocation to the ER and folding. (2) Protein quality control in particular for the energy consuming ERAD. (3) Vesicle fusion with target membranes in the ER, ERGIC, Golgi, and plasma membrane. (4) Vesicle and mitochondrial transport along microtubules and actin filaments. (5) Exocytosis. In the panel below the figure are a series of molecules and complexes that play relevant roles in these events.
Conventional Pathway for Protein Secretion
Most of the secreted proteins in the conventional pathway are translocated to the rough ER during translation by the ribosome (37). Proteins are targeted to the ER by a hydrophobic signal peptide located in the N-terminus, which is later cleaved by a signal peptidase present in the ER lumen. The signal peptide is recognized by the signal recognition particle (SRP) that then binds to the SRP receptor (SR) (33, 38). Both SRP and SR are GTPases and the docking and release of SRP at the ER membrane requires GTP (37, 39). Docking is followed by the translocation of the nascent protein to the ER lumen through the translocon, a pore that in mammals is composed of Sec61 proteins, and associated proteins including BiP, Sec63, Sec62, translocating-chain-associated protein (TRAM), translocon-associated protein (TRAP), and ribosome-associated membrane protein (RAMP) (37, 40, 41). BiP plays a dual role during protein translocation, it seals de pore and provides the driving force to transfer of the nascent protein into the ER (4, 40). This protein is an ATP-dependent chaperone that belongs to the heat shock protein 70 (Hsp70) family. During protein translocation BiP interacts with co-chaperones Sec63 and RAMP, these ER-resident J-domain proteins (ERdjs) stimulate BiP binding to the nascent protein and ribosomes as well as ATP hydrolysis (4, 41). BiP also interacts with the nucleotide exchange factors GRP170 and Sil1 that promote the exchange of ADP for ATP (41, 42). Though ATP hydrolysis by BiP is the main driving force for translocation, GTP hydrolysis during the elongation stage of protein synthesis may also contribute to the process (4, 40) (Table 1 and Figure 2).

Table 1. The conventional pathway for protein secretion and its energy requirements.
In the ER, the oligosaccharyl transferase complex (OGT) catalyzes the transfer of the oligosaccharide (Glc3Man9GlcNAc2) from dolichol phosphate to an asparagine residue in the protein (52, 53). Exoglucosidases I and II then catalyze the removal of two terminal glucose moieties producing a monoglucosylated structure (Glc1Man9GlcNAc2) that is recognized and bound by lectin chaperones calnexin and calreticulin. These chaperones, in collaboration with BiP, GRP-94 and protein disulfide isomerase A3 (PDIA3, Erp57), help the protein acquire its native conformation (8, 38). Removal of the remaining glucose moiety by exoglucosidase II prevents further binding of the glycoprotein to calnexin and calreticulin, allowing it exit to the ER. However, if the native structure is not achieved the protein undergoes reglucosylation catalyzed by UDP-glucose:glycoprotein glucosyltransferase (GT), an enzyme that preferentially acts on incompletely folded glycoproteins. Thereby favoring a new cycle of interactions with calnexin and calreticulin and the other components of the folding machinery (8, 54, 55). Protein folding requires ATP, since chaperones BiP and GRP-94 hydrolyze ATP during their catalytic cycles (41, 44). Besides, calnexin and calreticulin bind ATP though they do not hydrolize it (56). Calnexin and calreticulin also bind Ca2+, therefore Ca2+ homeostasis must be maintained in the ER lumen (56). The sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) is responsible of coupling ATP hydrolysis to Ca2+ transport from the cytosol to the ER (4, 57) (Figure 2).
In the ER proteins undergo quality control pathways for the detection, remotion, and degradation of misfolded proteins that did not attain their native structures. Degradation by the proteasome takes place in the cytosol in a pathway known as ER-associated degradation (ERAD) (8, 45). The first step in this pathway is the recognition of the misfolded protein and several luminal and cytoplamatic factors are involved in this process (46). Among them we find ER mannosidases (ER α1,2-mannosidase and EDEMs, 1, 2, and 3) that trim the mannose residues from glycoproteins, preventing glucosylation by GT and therefore withdrawing the protein from the quality control cycle (8, 58). Mannose trimmed glycoproteins bind OS-9 and XTP-3B and associate with SEL1L that channels the misfolded protein to the dislocon where proteins are retrotranslocated to the cytosol (45). Though the dislocon is not well-characterized it is thought to include Sec61, the E3 ligase HRD1, Derlins 1-3, and ATPase VCP/p97, which converts the energy released by ATP hydrolysis into mechanical force for the extraction process (4, 45). BiP and PDIs (ERdj5 and ERp90), also play a role, at this stage, binding and oxidizing the proteins to be transported (41, 59). In the cytosol, ubiquitin activating enzymes (E1), conjugating enzymes (E2), and E3 ligases (primarly HRD1 and AMFR) transfer ubiquitin to the protein in an ATP-dependent process (46). Finally, ubiquitinated proteins are degraded by the 26S proteasome in a pathway that also consumes ATP (45, 46). Overall the ERAD is an extremely costly process that consumes ATP during the extraction, ubiquitination and degradation of misfolded proteins (4, 41, 45) (Table 1 and Figure 2).
Secretory proteins that achieve the correct folded and assembled conformation are then transported from the ER to the Golgi complex in coat protein complex II (COPII) carrier vesicles (47). Vesicles for anterograde (forward) transport are formed in ER exit sites (ERES) and key proteins involved in their assembly include SEC12, SEC16, secretion-associated Ras-related 1 (SAR1) GTPase and the two major coat subunits SEC23- SEC24 and SEC13- SEC31 (47, 60). Proteins enter the vesicles by diffusion (bulk-flow), or undergo selective uptake (cargo capture) by SEC24, and other receptors (2, 48, 61). Upon leaving the ERES COPII vesicles shed their SEC13- SEC31 coats in a process that involves the hydrolysis of GTP bound to SAR1 (62), and fuse with one another or with the ER-Golgi intermediate compartments (ERGIC) (2, 47). Rab-family small GTPases and Rab effectors are involved in vesicle tethering to the membranes. After tethering vesicles fuse with the acceptor compartment delivering the cargo to the Golgi (2, 48). Retrieval of misplaced ER luminal proteins from the ERGIC or from the Golgi is achieved by retrograde transport in coat protein complex I (COPI) carrier vesicles. Formation of COPI vesicles involves the ADP-ribosylation factor 1 (ARF) GTPase, that recruits the coatomer subunits to the membrane (48). Vesicle formation, coat shedding and tethering to target membranes are energy consuming processes that consume GTP (2). SAR1 and ARF GTPases switch between inactive GDP-bound and active GTP-bound forms. Guanine nucleotide exchange factors (GEFs) catalyze the release of GDP (allowing the union of a new GTP molecule); while GTPase-activating proteins (GAPs) promote the hydrolysis of bound GTP, regulating enzyme activity and therefore vesicle trafficking (2, 63) (Table 1).
From the cis-Golgi compartment proteins are transported via cisternal maturation to the trans-Golgi network (48), where proteins to be secreted are sorted in secretory vesicles and delivered to the plasma membrane, to the endosomal system or to immature secretory granules (33, 48). Motor proteins, kinesin, and dynein transport the cargo vesicles along microtubules. Kinesins transport secretory vesicles from the organelles where they arise to the cortical region, while dynein mediates their transport in the opposite direction (49, 64). Myosin V and II are responsible for the movement of vesicles along actin filaments in the cortical area (49, 50). The cortical actin network, at the cell borders, acts as a diffusion barrier that prevents the access of granules to secretory sites until the arrival of the triggering stimuli (50). Motor proteins, kinesin, dynein and myosin use the energy released during ATP hydrolysis to move vesicles along microtubules and actin filaments (9) (Table 1 and Figure 2).
Upon arrival to the plasma membrane secretory proteins are released to the extracellular space by exocytosis. Constitutive exocytosis occurs in all cell types to release extracellular proteins and maintain plasma membrane homeostasis and cell polarity. While regulated exocytosis occurs in specialized secretory cells and is triggered by secretion signals (13), such as increases in Ca2+ concentrations (65). Calcium ions bind sensor molecules and promote the fusion of vesicles with the plasma membrane (65). Upon the arrival of the secretion stimuli myosin II drives the movement of vesicles to the fusion sites where they dock to the plasma membrane (66). SNARE complexes (formed by syntaxin, SNP-25, and VAMP), and SM proteins (such as Munc18-1), are in charge of the fusion of vesicles with the plasma membrane (10, 13, 51), while the actin and myosin network contributes to the release of vesicle contents in an ATP-dependent manner (50, 66, 67). Fusion of vesicles with target membranes also consumes ATP. SNARE proteins localized in vesicles and target membranes interact forming a complex that pulls the two membranes together, exerting the force required for fusion. The dissociation of the SNARE complex involves soluble NSF attachment protein (SNAP) and the ATPase N-ethylmaleimide-sensitive fusion protein (NSF) that hydrolyses ATP (10, 51) (Table 1 and Figure 2).
Overall the concerted action of many cellular components is required to ensure the selective and efficient secretion of proteins. Many of these steps require energy and mitochondria play a relevant role, fulfilling the ATP requirements of the pathway (Figures 1, 2). Besides, mitochondria help to modulate the intracellular concentrations of Ca2+ (13, 14), an essential cofactor in the folding process and a potent trigger of exocytosis (65) (Figure 1).
Unconventional Pathways for Protein Secretion
Though the conventional protein secretion pathway was considered for a long time as the only mechanism for protein secretion, we now know that many proteins use alternative pathways. These include the secretion of cytosolic proteins that do not have a signal peptide (leaderless proteins) or a transmembrane domain; and proteins that contain a signal peptide or a transmembrane domain and enter the ER but are not transferred to the Golgi complex (68).
Proteins without a leader sequence are secreted along three different pathways: Type I pathways involve the translocation from the cytoplasm to the extracellular space through a pore in the plasma membrane. In Type II secretion processes the ATP-binding cassette transporter is responsible for the secretion of the protein present in the cytoplasm while in Type III secretion the proteins use autophagosomes and endosomes to reach the extracellular space. The Golgi-bypassing route is also known as Type IV secretion pathway (68). Some proteins are constitutively secreted through unconventional pathways, however these pathways are usually induced in stressful conditions, such as nutrient starvation, inflammation, and mechanical or ER stress (32, 68).
One of the most studied proteins secreted by the unconventional pathways is interleukin-1β (IL-1β) (68, 69). During inflammation IL-1β is produced as an immature and inactive form that is cleaved by caspase 1 upon recruitment and activation by the Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome (70), in a process that will be discussed below in detail. The assembly and activation of the complex is an energetically costly event that requires the hydrolysis of ATP by NLRP3 (71). The posterior secretion of mature IL-1β involves the formation of a pore that increases the permeability of the plasma membrane (69) or occurs through the autophagosome/endosmal pathway (68).
Secretion Pathway of Neurotransmitters
Many neurotransmitter molecules are amino acids or amino acid derivatives and their secretion occurs through exocytosis. The pathway starts with the synthesis of the neurotransmitters by enzymes, followed by their loading into of synaptic vesicles. The loading process involves transporters in the vesicle membrane and happens at the expense of an electrochemical proton gradient, generated by the vacuolar H+-ATPase. This pump uses the energy released by ATP hydrolysis to transport protons into the vesicle lumen (72). Loaded vesicles can then be sequestered in a reserve pool or travel to specialized release sites, known as active zones, at the presynaptic terminal where vesicles become docked at the plasma membrane. Increases in Ca2+ concentrations promote the fusion of the vesicle with the plasma membrane and discharge their content into the synaptic cleft (73). Calcium sensing, vesicle docking and exocytosis follow the same steps described for the conventional secretion pathway of proteins (51). These steps are followed by the recovery of the synaptic vesicle membranes that are recycled for refilling in preparation for the next cycle of exocytosis (74).
Overview of Mitochondrial Structure, Function and Dynamics
Mitochondrial Bioenergetics
Mitochondria are cell organelles defined by a double membrane, the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM). The OMM separates the mitochondria from the cytosol, however the voltage-dependent anion channel (VDAC or porin) allows the passage of metabolites and ions. In the OMM are also located proteins involved in apoptosis, mitochondrial dynamics, and tethering to other organelles. In the IMM, two functional and structurally different regions are described: the inner boundary membrane and the cristae, where electron transport and ATP synthesis take place in a process known as oxidative phosphorylation (75). In the cristae we find the electron transport chain (ETC) complexes: NADH-ubiquinone oxidoreductase (complex I), succinate-ubiquinone oxidoreductase (complex II), ubiquinol-cytochrome c oxidoreductase (complex III), and cytochrome c oxidase (complex IV), as well as the FoF1-ATP synthase (ATP synthase or complex V) and two mobile electron carriers, ubiquinone and cytochrome c (75).
The ETC complexes are assembled forming supercomplexes that optimize electron transport and proton shuttling through the IMM (76, 77). ATP synthase localizes on the edges of cristae forming dimers, which optimize ATP production (78). At the cristae junctions we find the mitochondrial contact site and cristae-organizing system (MICOS), a large oligomeric complex that interacts with both the IMM and OMM (79). Cristae structure influences the ETC, complex and supercomplex formation, and can present different morphologies depending on the metabolic state of the cell (80).
Mitochondria are the main source of ATP in most cells, since many catabolic pathways converge in this organelle and result in the production of ATP by oxidative phosphorylation. The catabolism of metabolites such as glucose, proteins, and fatty acids produces acetyl-CoA, which is in turn oxidized in the tricarboxylic acid cycle generating the reduced electron donors nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). Electrons are then transferred to the ETC and flow through the complexes to molecular oxygen (O2). This thermodynamically favorable electron transport releases energy, which is used to pump protons from the matrix to the intermembrane space, at complexes I, III, and IV, creating an electrochemical gradient (81). The energy released by the dissipation of the electrochemical proton gradient is then coupled to the synthesis ATP from ADP and Pi, catalyzed by ATP synthase (82, 83).
Reactive Oxygen Species in Mitochondria
Mitochondrial catabolism of nutrients and electron transport in the respiratory chain involves many redox reactions that have as by-products reactive oxygen species (ROS) (84). In particular, superoxide anion radical is formed by several flavoproteins and at complex I and III of the respiratory chain, and its dismutation gives rise to hydrogen peroxide (85). These mildly oxidant species can result in the formation of higher oxidant species such as peroxynitrite, formed in the reaction between superoxide and nitric oxide, oxo-metal complexes, nitrogen dioxide, and hydroxyl radical (86).
Under physiological conditions mitochondria produce controlled levels of oxidants, many of which participate in signaling processes. However, ROS formation can increase during cellular stress or in pathological conditions. Although mitochondrial oxidants can be detoxified by enzymatic and non-enzymatic antioxidants (87, 88), an imbalance in mitochondrial redox status may lead to mitochondrial damage (84, 89, 90). In fact, oxidants impair the activity of enzymes of the tricarboxylic acid cycle, the ETC, and ATP synthase (84, 90).
Mitochondrial Dynamics
Mitochondrial dynamics consists of fusion and fission events driven mainly by dynamin-related GTPases (91). Optic atrophy protein 1 (OPA1) and mitofusins 1 and 2 (MFN1 and MFN2) are involved in mitochondrial fusion. The long OPA1 isoform is anchored to the inner membrane, where it promotes IMM fusion. The soluble and short OPA1 isoform, found in the intermembrane space, maintains mitochondrial cristae structure (92). MFN1 and MFN2 participate in tethering and fusion of the outer membranes of two different mitochondria through the formation of homo and heteroligomeric complexes (93, 94). Phospatidic acid, a fusogenic lipid formed in a reaction catalyzed by mitochondrial phospholipase D, is also required for OMM fusion (95).
In turn, mitochondrial fission is carried out by the GTPase dynamin related protein 1 (DRP1), a cytosolic protein that is recruited to the OMM (96) by receptor proteins, such as mitochondria fission factor (97), mitochondrial dynamics protein of 49 and 51 kDa (98) and fission 1 protein (FIS1) (99). DRP1 oligomerizes around mitochondria into a ring-like structure constricting the organelle (96). ER-mitochondrial contacts play a role in mitochondrial fission, determining the position of the fission events (100) and ER tubules wrap around mitochondria and constrict them (100). Components of the cytoskeleton such as actin, myosin II and septin 2 are required for mitochondrial fission (101–104), as well as the ER protein inverted formin 2 (105) and a mitochondrial Spire1 isoform (103). The latter two cooperate to induce localized actin polymerization at the constriction sites (103). Both fusion and fission are highly regulated processes, and changes in protein levels, GTPase activity and post-translational modifications of proteins involved in mitochondrial dynamics affect not only mitochondrial morphology but also cellular bioenergetics and homeostasis (106).
Multiple studies linking mitochondrial morphology and bioenergetics can be found in the literature (107–109). At the molecular level several reports support that MFN2 is required for correct mitochondrial function (110); since its silencing results in an impairment of oxidative phosphorylation, while overexpression enhances mitochondrial metabolism (19, 111–113). Less evidence supports a role for MFN1. Although there are reports on MFN1 silencing decreasing mitochondrial respiration (114) this does not seem to be the case in all models (19). OPA1 deletion also affects mitochondrial membrane potential, respiration and cristae structure and reduces assembly, and stability of supercomplexes (80, 114–117).
In order to face increases in energy demands, such as those imposed by secretion, mitochondria also modify their cellular distribution. Motor proteins, kinesin, and dynein, transport mitochondria over long distances along microtubules toward the cell periphery or the cell center, respectively (49). While myosin 19, mediates short-range movement along the actin filaments near the cell periphery (49, 118). The mitochondrial Rho GTPase (MIRO) and the adaptor proteins trafficking kinesin-binding proteins 1 and 2 (TRAK1 and 2) support bidirectional mitochondrial movement along microtubules by binding kinesin and dynein (119–121) (Figure 2). MIRO proteins also engage myosin 19, linking mitochondria to actin filaments (122) and interact with components of the MICOS complex, linking the transport machinery to cristae organization and ensuring the appropriate provision of energy to the regions where mitochondria are delivered (123).
ER-Mitochondria Contact Sites (ERMCs)
Mitochondria can interact with several cellular compartments, including organelles involved in protein secretion such as the Golgi complex and ER (124–126). In particular, mitochondrial interactions with the ER have been extensively studied and many components and functions of the ER–mitochondrial contact sites (ERMCs)1 have been identified. ERMCs are crucial sites for the synthesis and exchange of lipids, Ca2+ transport, mitochondrial fission and inflammasome formation (4, 125, 127).
Among the proteins found in ERMCs is MFN2, and though some controversy exists (128, 129), this protein is proposed to link the ER membrane to mitochondria through interactions with MFN1 and MFN2 in the OMM (130, 131). The mitochondrial proteins MITOL and mitostatin, as well as the ER protein presenilin 2 modulate these MFN2-dependent interactions between organelles (132–134). Other relevant proteins linking mitochondria to the ER are VDAC, the OMM chaperone GRP-75 and the inositol-1,4,5-trisphosphate-sensitive channel or receptor (IP3R) in the ER. Another tethering complex is formed between the ER protein vesicle-associated membrane protein- associated protein B (VAPB) and the OMM protein tyrosine phosphatase-interacting protein 51(PTPIP51) (135–137) (Figure 2).
Physical tethering of mitochondria and the ER by mitofusins (130, 131), VDAC/IP3R/GRP-75 (14, 138), and VAPB/ PTPIP51 (135) is required for efficient transport of Ca2+ between organelles. Besides, several regulatory proteins modulate Ca2+ release from the ER and uptake by mitochondria, through interactions with IP3R or VDAC (123, 138, 139). Of particular interest is the chaperone calnexin, whose translocation to different ER domains is regulated by palmitoylation of two conserved cysteine residues (140) and interaction with regulatory proteins (138). Calnexin migration to the ERMCs is required for Ca2+ transfer from the ER to the mitochondria (141).
Calcium ion leaves the ER through the I3PR and enters the mitochondria through VDAC and the mitochondrial calcium uniporter (MCU) complex present in the OMM and IMM, respectively (14, 138). The complex is formed by the MCU, a pore that allows the passage of the ions through the lipid membrane (142, 143), essential MCU regulator (ERMES) and mitochondrial calcium uptake 1 and 2 proteins (MICU1 and 2) that act as gatekeepers regulating Ca2+ uptake (144). Calcium entry to mitochondria occurs at the expense of the H+ gradient in the IMM (144). In the matrix, Ca2+ activates several dehydrogenases (including enzymes of the tricarboxylic acid cycle) and ATP synthase enhancing oxygen consumption rate and ATP synthesis (15, 144). Thus, MERCs are extremely important for mitochondrial function. On the other hand Ca2+ uptake by mitochondria prevents or “buffers” the increase of Ca2+ concentrations near the mouth of the IP3R affecting its activity as we will describe in detail in the next section (14).
In ERMCs we can also find SAR1 and ARF. These small GTPases involved in COPII and COPI vesicle assembly are also required to maintain mitochondrial morphology and function (145, 146). SAR1 regulates the size of mitochondrial ER contact sites by affecting the curvature of the membrane (146). While ARF1 and its guanine nucleotide exchange factor (GBF1) interact with mitochondrial protein MIRO, and their depletion affects mitochondrial morphology, autophagy, positioning, and movement within the cell (145, 147). These observations point to an important role of secretory pathway components in dynamics and physiology of mitochondria.
Overall, mitochondria are dynamic organelles whose function, morphology, distribution, and contacts with other organelles (49, 109, 148) undergo changes in response to intracellular energy requirements, as well as other physiological and pathological stimuli (109).
Mitochondria in Secretion
Fulfilling ATP Requirements of the Secretion Process
As mentioned before, many of the steps in the conventional secretion pathway demand energy (Table 1 and Figures 1, 2). Chaperones involved in protein translocation to the ER and protein folding consume ATP (5–7), as does protein quality control by the ERAD (8), transport and fusion of vesicles with target membranes (2, 9, 51). Calcium signaling during regulated exocytosis also consumes ATP to maintain the levels of IP3 precursors and to pump calcium from the cytoplasm into the ER or to the extracellular space (13, 14, 149). Overall, an increase in secretion of proteins is bound to challenge the cell's bioenergetics capacity; and inhibition of ATP synthesis, using inhibitors or uncouplers of oxidative phosphorylation, prevents the secretion of many proteins (7, 150–152).
The mechanism for ATP transport into the ER in mammalian cells was elusive for many years, and different possibilities, ranging from the existence of specific transporters to non-specific transport through anion channels or leaky membranes, have been proposed (4). However, recently protein SLC35B1 was identified as an ATP/ADP exchanger present in the ER membrane, and named AXER (153). Silencing the expression of SLC35B1/AXER reduced ATP levels in the ER of different cell lines and affected BiP dependent protein import activity and Ca2+ levels inside the ER (153, 154). A decrease in ATP in the ER was observed upon inhibition of SERCA or exposure to high cytosolic Ca2+ concentrations, suggesting that a Ca2+ gradient across the ER membrane is required for ATP transport into the ER (154). Inhibitors and uncouplers of oxidative phosphorylation lead to a decrease in ATP levels inside the ER as well, suggesting mitochondria is a relevant source of ATP for the ER (154, 155). Interestingly, several components of the mitochondrial ATP synthase complex immunoprecipitate with SLC35B1/AXER; along with proteins involved in ER protein import and folding such as BiP, calnexin, and Sec61 complex (153). Transfer of ATP from mitochondria to the ER probably occurs at ERMCs, where both organelles are in close proximity, yet this is still to be established (4).
Calcium Buffering by Mitochondria in Exocytosis
As we mentioned above, Ca2+ signals for exocytosis in many secretory cells. The increase in cytosolic Ca2+ concentration triggers vesicle fusion with the plasma membrane leading to the release of the cargo to the extracellular space. Calcium enters the cytosol through channels in the plasma or the ER membranes and several processes can affect the magnitude and duration of the signal for exocytosis, among them is Ca2+ uptake by mitochondria (13). Mitochondria are located in close proximity to Ca2+ channels in the ER and plasma membrane and can take up the ion through the VDAC and MCU channels in the OMM and IMM, respectively (144). This process is known as Ca2+ buffering and is driven by the negative potential across the IMM (144). Positioning of mitochondria near the sites of Ca2+ entry to the cytosol is also a requirement for the efficient uptake of the ion and effective Ca2+ signaling for exocytosis. This depends on proteins involved in mitochondrial dynamics and movement, such as MFN2, myosin, kinesin, TRAK, and MIRO (14, 156–158). Of note, MIRO is both an adaptor protein, that links mitochondria to TRAK and kinesin, and a Ca2+ sensor which can regulate mitochondrial trafficking in response to changes in Ca2+ concentrations (157).
Ca2+ buffering has different effects on exocytosis, depending on the channel and cell type. Voltage-gated Ca2+ channels (Cav) are present in the plasma membrane. These are activated by membrane depolarization and mediate Ca2+ influx in response to action potentials and other depolarizing signals (159). The increase in cytosolic Ca2+ stimulates exocytosis by binding syaptogamins that promote SNARE mediated vesicle fusion with the membrane (65). Mitochondria can be found in the cell cortex regions, near the sites of exocytosis and in this setting Ca2+ sequestering by mitochondria reduces vesicle release (157, 160, 161).
In the plasma membrane we also find store-operated channels, known as Ca2+ release-activated (CRAC) channels. These are composed by Orai pore proteins, present in the plasma membrane, and STIM proteins from the ER and are activated by depletion of calcium stores in the ER (13, 149, 158). When Ca2+ levels fall in the ER STIM proteins migrate to ER-plasma membrane junctions where they activate Orai channels, allowing the entry of Ca2+ into the cytosol. In turn, high cytosolic Ca2+ concentrations result in the closure of the pore and both a rapid and a slow Ca2+-dependent inactivation have been described (158). Mitochondrial translocation to the plasma membrane is essential to sustain Ca2+ influx through CRAC channels (162), since it prevents the slow inactivation of the CRAC channel without affecting the fast Ca2+-dependent inactivation (158). Inhibitors and uncouplers of electron transport and oxidative phosphorylation that dissipate the proton gradient prevent Ca2+ uptake by mitochondria and promote channel inactivation (163). Inhibition of kinesin- dependent transport of mitochondria to the plasma membrane also results in channel inactivation (162).
In the ER membrane the IP3R and ryanodine-sensitive receptors are the channels responsible for Ca2+ release to the cytosol (13, 14). IP3R is activated by IP3, a second messenger formed by hydrolysis of phosphatidylinositol 2,5-biphosphate by phospholipase C. While ryanodine- sensitive receptors in secretory processes are activated by Ca2+ to release Ca2+ stored in the ER, in a process known as Ca2+ induced Ca2+ release that amplifies Ca2+ signaling (149). For both channels the response to cytosolic Ca2+ concentrations is bell shaped, since Ca2+ can act both as a positive or negative regulator of the channel depending on the concentration of the ion (14, 164). Thus, high concentrations of cytosolic Ca2+ result in a decrease in the activity of both I3PR and ryanodine sensitive receptors (14, 164). Uptake of Ca2+ by mitochondria prevents its accumulation precluding channel inhibition by high concentrations of Ca2+ (14).
From what we just described it is clear that mitochondria contribute to the regulation of intracellular Ca2+ concentrations, and therefore impact on the regulated exocytosis of many proteins and other signaling molecules such as neurotransmitters (Figure 1).
Mitochondria in the Activation of NLRP3 Inflammasome
The NLRP3 inflammasome is a multiprotein complex that participates in innate immunity. It is activated by multiple signals of infection, cellular damage, or stress, to produce inflammatory cytokines that trigger innate immune responses (71, 165). The sensor molecule NLRP3, the adaptor protein ASC and the effector caspase-1 form the complex, and recently the NIMA-related kinase 7 was identified as a fourth component. Upon stimulation, NLRP3 oligomerizes and recruits ASC that in turn recruits caspase 1. The assembly of the NLRP3 inflammasome leads to the activation of caspase 1 that then cleaves pro- IL-1β and pro-IL-18, producing the mature cytokines IL-1β and IL-18 (71). As mentioned above the assembly of the NLRP3 inflammasome requires energy that is obtained from ATP hydrolysis catalyzed by NLRP3 NACHT domain (71). Mitochondria supplies ATP for this process but contributes also in other ways to the activation of the inflammasome.
NLRP3 inflammasome activation occurs in two steps, priming and NLRP3 activation. Priming involves the recognition of pathogen-associated molecular patterns (PAMPs, such as LPS) by pattern recognition receptors or binding of cytokines to receptors. These events lead to the activation of NF-κB and consequent upregulation of NLRP3, caspase 1 and IL-1β and IL-18 gene expression (71). The second step is the activation of NLRP3 by PAMPs or damage-associated molecular patterns (DAMPs, such as ATP); which involves multiple signaling events that result in the assembly of the complex [for exhaustive reviews see (71, 165)]. Mitochondrial ROS and oxidized mtDNA released into the cytosol can trigger NLRP3 activation (16, 17).
Moreover, mitochondria are required for the assembly of the inflammasome. In absence of stimuli the NLRP3 is found in the cytoplasm associated with the ER, while its activation results in an association with mitochondria and enrichment in ERMCs (17). ASC and caspase 1 also migrate to mitochondria upon activation and are enriched at ERMCs (17, 18). In a recent report Elliot et al. suggest that priming results in NLRP3 and capase 1 linkage to the OMM, by association with the phospholipid cardiolipin, and mtROS are proposed to mediate cardiolipin transfer from the IMM to the OMM. Instead the association of ASC with mitochondria occurs in the activation step and does not require cardiolipin, but is dependent on NLRP3 and regulated by cytosolic Ca2+ (18). Interestingly in absence of VDAC, a relevant OMM channel and tether molecule in mitochondria-ER contact sites, activation is not observed (17).
Other mitochondrial proteins that are required for NLRP3 inflammasome activation are mitochondrial antiviral signaling proteins (MAVS). These proteins form aggregates in the OMM, associate with NLRP3 and promote its oligomerization, and activation of the inflammasome during RNA virus infections (166). The fusion protein MFN2 also participates in inflammasome activation and its silencing reduces IL-1β secretion in response to viral infection (167). Finally, dynein dependent transport of mitochondria along microtubules, toward the ER, is required for the activation of the inflammasome (168). Acetylation and deacetylation of tubulin by acetyltransferase MEC17 and the NAD+ dependent deacetylase sirtuin 2 regulate microtubule transport of mitochondria to the ER and inflammasome activation (168).
In sum, mitochondria provide energy, signaling molecules and a structural scaffold for the assembly and activation of the NLRP3 inflammasome (Figure 1).
Bioenergetic Failure and Secretory Defects in Disease
Considering the relevant roles of mitochondria in secretion it is not surprising that mitochondrial dysfunction underlies the pathogenesis of certain diseases. Herein we discuss the role of this organelle in some relevant physiological secretion processes, and present evidence linking bioenergetic failure to the development of disease.
Insulin Secretion by Pancreatic β-Cells in Diabetes Mellitus
Pancreatic β-cells are secretory cells located in the islets of Langerhans in the pancreas. Their main function is to synthesize and secrete insulin, a peptidic hormone responsible for regulating levels of glucose in the blood. Upon the increase in glucose concentrations in plasma, β-cells secrete insulin stored in the secretory granules and increase the synthesis of the hormone (169, 170).
Mitochondria play a key role in glucose-stimulated insulin secretion (GSIS) in pancreatic β-cells. In these cells glucose is metabolized in the glycolytic pathway and tricarboxylic acid cycle and results in ATP synthesis by oxidative phosphorylation. ATP promotes the closure of ATP-sensitive potassium channels and depolarization of the plasma membrane, leading to the opening of voltage-gated Ca2+ channels. Calcium influx then triggers the exocytosis of insulin granules (170). This process is termed KATP- dependent GSIS. A KATP- independent GSIS that depends on mitochondrial GTP and phosphoenolpyruvate metabolism has also been described (171, 172). Therefore, GSIS is extremely sensitive to genetic interventions and pharmacological agents that affect mitochondrial metabolism (22). This is clearly exemplified by the work of Kennedy et al. showing that depletion of mitochondria in the INS-1 insulin-secreting cell line precludes insulin secretion upon exposure to glucose (173). Likewise impairment of autophagy in pancreatic β-cells affects both mitochondrial respiration and GSIS, affecting glucose metabolism in vivo (174). As does inhibition of Ca2+ uptake by mitochondria after silencing of MCU or MICU1 gene expression, that also reduces ATP synthesis and insulin secretion (175).
Pancreatic β-cells present an active mitochondrial network where mitochondria constantly undergo fusion and fission events (176); and functional mitochondrial dynamics are required for GSIS in INS-1 cells and primary β-cells (23). Overexpression of FIS1 in human pancreatic β-cells resulted in fragmentation and clustering of the mitochondrial network in the perinuclear area, a decrease in mitochondrial membrane potential and ATP synthesis, along with impairment of GSIS (177). Similar results where observed upon silencing of FIS1 expression that affected respiration rates and insulin secretion (178), overexpression of a dominant negative form of fission protein DRP1 and DRP1 silencing (178, 179). Alteration in the fusion machinery also affected insulin secretion. Overexpression of MFN1 resulted in hyperfusion and aggregation of mitochondria, reduced ATP levels and GSIS (177); while OPA1 ablation impaired glucose-stimulated oxygen consumption rate, ATP production and insulin secretion (116).
It is clear that mitochondria play relevant roles in pancreatic β-cell secretion and a growing body of evidence links mitochondrial dysfunction to impaired insulin release in diabetes mellitus. Diabetes mellitus are a group of metabolic disorders characterized by hyperglycemia, resulting from defects in insulin secretion, insulin sensitivity, or both (180). In type 1 diabetes the cause is a total deficiency of insulin secretion, while in type 2 diabetes the cause is a combination of resistance to insulin action of the liver, skeletal muscle, and adipose tissue and an inadequate compensatory insulin secretory response (180, 181).
To start with, diabetes mellitus (type 1 and 2) is frequently observed in patients with inherited mitochondrial diseases, which are caused by defects in mtDNA or in nuclear genes encoding mitochondrial proteins that affect mitochondrial ATP synthesis (27). Several point mutations as well as deletions and rearrangements in mtDNA are strongly associated with the onset of diabetes (27, 182). As are mutations in nuclear genes encoding proteins involved in maintenance of the mitochondrial genome (27). The most common defect is the mutation m.3243A>G in MT-TL1, which encodes a mitochondrial tRNA for leucine (183) and can lead to maternally inherited mitochondrial diabetes and deafness (MIDD) (27). Other point mutations in mitochondrial tRNA genes MT-TL1, MT-TK, MT-TS2, MT-TE, and MT-TT as well as in MT-ND6, a core subunit of complex I, have also been associated with diabetes mellitus (27, 184). Patients, carrying the m.3243A>G mutation presented reduced pancreatic insulin secretion (185–187), yet increases in insulin resistance and alterations in glucose metabolism cannot be discarded as causes of the disease (188).
Additionally profound changes in mitochondrial metabolism have been observed in pancreatic islets of type 2 diabetes patients and animal models. These include down regulation of components of the tricarboxylic cycle, ETC, ATP synthase and proteins involved in transport across the OMM and IMM (29, 189); as well as a decrease in glucose oxidation and a loss of glucose-stimulated increases in NADH, ATP, and oxygen consumption (29, 190–192). Other authors have observed an upregulation of uncoupling protein 2 (UCP-2), an IMM protein that dissipates the proton gradient; in pancreatic islets in type 2 diabetes (190, 193, 194). Genetic deletion of UCP-2 in mice enhances islet ATP generation and insulin secretion during glucose stimulation and reduces hyperglycemia in the ob/ob mice (a well-known diabetes type 2 mouse model), providing further support for the role of UCP-2 in the pathogenesis of the disease (194). Regardless of the mechanism, the impairment in mitochondrial function in pancreatic β cells precludes the glucose-induced increase in ATP levels. Lack of ATP will prevent the closure of ATP-sensitive K+ channels (that is required to activate insulin secretion), impair the steps of the secretion pathway that require energy, resulting in lower levels of the circulating hormone and hyperglycemia.
Neurotransmitter Release in Alzheimer's Disease
Neurons are excitable cells that communicate with other cells at synapses. At the chemical synapse neurotransmitters are released by pre-synaptic neurons and bind their receptor at the post-synaptic terminal of target cells. Vesicle discharge at the synaptic cleft occurs in response to an influx of Ca2+ ions through voltage-gated Ca2+ channels, triggered by the action potential. Three different pools of synaptic vesicles can be found in neurons readily releasable pool, the recycling pool, and the reserve pool. The vesicles in the readily releasable pool are released under moderate or intense neuronal activity, while vesicles from the reserve pool are recruited only upon intense stimulation. The latter constitute the majority of vesicles in presynaptic terminals (28, 160).
Maintaining the electrochemical gradients required for action potentials, transporting, discharging, recycling, and refilling synaptic vesicles, and regulating Ca2+ concentrations are energy demanding processes, that require functional mitochondria (28, 195). Regarding secretion in particular, loading of vesicles with neurotransmitters requires ATP to fuel the vacuole type H+-ATPase (72). Besides, the mobilization of the reserve pool requires mitochondrial ATP that is used by myosins to move the vesicles along actin filaments (196). Additionally, Ca2+ signaling for vesicle exocytosis requires a constant export of the ion from the cytosol to the extracellular space, the ER and mitochondria. Plasma membrane Ca2+ ATPase (PMCA) and SERCA at the ER actively consume ATP (149). Mitochondria provide ATP for these pumps and directly remove Ca2+ from the cytosol through the MCU, as described previously (28, 196). Therefore, mitochondria are very abundant in neurons, where they form a highly dynamic network and their position and morphology change continually (28). In particular we find a high number of mitochondria at chemical synapses, where they provide ATP and buffer local rises in Ca2+ concentrations (156, 160, 197). Since mitochondrial biogenesis and elimination (by mitophagy) occurs mainly in the neuronal soma, mitochondria travel between the axon terminals and the cell body (156) via the microtubule network. Motor proteins kinesin (anterograde) and dynein (retrograde), adaptor proteins TRAK, MIRO, and syntabulin as well as fission protein DRP1 are involved in mitochondrial trafficking (28, 156, 196). Docking to microtubules, by syntaphilin, or to actin filaments limits mitochondrial mobility, retaining these organelles near to the sites where energy is required (156, 160).
Due to their high-energy demands, neurons are extremely affected in mitochondrial diseases, caused by mutations in mtDNA, and in other pathologies where mitochondrial function is compromised (30, 198). An example of the latter is Alzheimer's disease (AD), a neurodegenerative disease that involves progressive synaptic dysfunction and death of neurons in cortical and subcortical regions. AD is characterized by the accumulation of extracellular plaques of the peptide amyloid-β (Aβ) and the formation of intracellular tangles of the microtubule-associated protein tau (198, 199).
Alterations in energy metabolism can be found in patients and animal models of AD. Reduced glucose metabolism was observed in brain regions affected by AD and the reduction correlated with cognitive decline in AD patients (200, 201). Analysis of brain samples from patients with AD showed abnormalities in mitochondrial cristae of hippocampal neurons (202); and reduced activity of enzymes involved in oxidative metabolism, namely pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase and complex IV (203–205). Studies of brain mitochondrial function in a transgenic AD mouse model also revealed decreases in PDH, complex IV and respiratory control ratios (RCR) and the decay in PDH and neuronal mitochondrial function preceded plaque formation (206). Moreover, in recent reports mitochondrial dysfunction was detected in hippocampal synaptosomes from transgenic AD rat model (207, 208). Decreased complex I activity, oxygen consumption rate, and ATP synthesis, along with an increase in hydrogen peroxide formation were measured, evidencing bioenergetics defects as well as oxidative stress at the presynaptic level (208). Alterations in mitochondrial dynamics and motility have also been observed in transgenic animal models of AD, and result in lower number of mitochondria at the presynapses (28, 209, 210). Regarding the causes behind bioenergetic failure in neurons, the Aβ peptide has been found to accumulate in mitochondria (206, 211), where it directly impairs complex IV activity and ATP synthesis, and increases ROS formation (212). Aβ can also directly affect mitochondrial dynamics, promoting mitochondrial fragmentation (213, 214). Defects in Ca2+ handling by mitochondria have also been observed in AD and Aβ can alter Ca2+ homeostasis at several levels (28, 73). Hyperphosphorylated tau also affects mitochodrial respiration and ATP synthesis, through the inhibition of complex I, impairs mitochondrial trafficking and induces mitochondrial fission (215). Overall in AD patients impaired mitochondrial energy metabolism, dynamics, Ca2+ homeostasis and oxidant formation are affected in neurons, at presynaptic terminals where neurotransmitter secretion takes place (28, 198).
Synapse loss is an early event in AD animal models and patients, and exhibits a strong correlation with cognitive deficits (73). Presynaptic release of neurotransmissors is affected in AD, and Aβ can affect various steps of the secretion process including SNARE mediated fusion, recovery of vesicle membranes by endocytosis and Ca2+ homeostasis (73). The decrese in mitochondrial ATP synthesis and Ca2+ buffering, observed in AD at the presynaptic level, probably affects neurotransmitter release as well, and should be considered in the design of pharmacological strategies to target the disease (215). Supporting this concept are results obtained in a transgenic AD rat model, where treatment with a pharmacological inducer of mitochondrial biogenesis helped to recover mitochondrial function and protected against cognitive impairment (208).
Mitochondria and Secretion in Other Pathological Settings
In this section we present two examples of mitochondrial involvement in secretion processes that underlie pathology. This is not an extensive list, but rather a couple of interesting examples that support the idea that mitochondria might be interesting targets in the design of drugs to modulate secretory processes.
Mast Cells Secretion in Atopic Dermatitis
Mast cells are cells of hematopoietic origin that participate in host defense and immunity as well as tissue repair, wound healing, angiogenesis and are also responsible for the development of allergies (216). Mast cells can be activated by foreign pathogens, toxins chemical agents and particles, as well as by mediators of the immune response, such as immunoglobulin E (IgE) and complement components (216, 217). Upon activation mast cells release pre-stored mediators, and/or de novo synthesize and secrete a wide range of biologically active molecules (216). Pre-stored mediators include proteases, histamine, proteoglycans, and cytokines such as tumor necrosis factor (TNF), these are released immediately upon activation and are critical for initiating mast cell-mediated innate immune responses. Differently from pre-stored mediators, other molecules are synthesized de novo (i.e., lipids, cytokines, chemokines, antimicrobial peptides, growth and angiogenic factors) and play multiple roles in the immune response and other physiological and pathological processes (216, 217).
Mast cell degranulation and TNF secretion requires Ca2+ and mitochondrial ATP (31, 218). Interestingly stimuli that induce mast cell degranulation, promote the translocation of mitochondria to sites of exocytosis, close to the cell surface; and pharmacological or genetic inhibition of DRP1 inhibits both TNF secretion and mitochondrial translocation, in a cell line derived from human mast cell leukemia (24). In this report authors also show that calcium is required for DRP1 activation, as well as for mitochondrial translocation, and postulate that calcineurin is involved in DRP1 recruitment to mitochondrial surface (24). The relevance of this observation was further supported by studies in human mast cells from patients with atopic dermatitis where increased degranulation and mitochondrial translocation was also observed, along with an increase in DRP1 and calcineurin expression (24). Mast cell degranulation also results in the secretion of mitochondrial particles outside cells including mtDNA, that in turn promote cytokine release from other mast cells, keratinocytes, and endothelial cells promoting inflammation (219).
Senescence Associated Secretory Phenotype in Disease
Cellular senescence is triggered in response to stress stimuli. Agents that damage DNA and strong mitogenic signals, such as the expression of oncogenes or loss of tumor suppressors, are strong senescence inducers (220–223). Senescent cells are characterized by permanent proliferation arrest, activation of the DNA damage response (DDR), increase in size and β-galactosidase activity and acquisition of a secretory phenotype (224, 225). The senescence-associated secretory phenotype (SASP) includes numerous cytokines and chemokines (e.g., IL-1α, IL-1β and IL-6, IL-8), growth factors and extracellular proteases. These mediators can induce senescence in neighboring cells, alter the extracellular matrix, contribute to local inflammation, and recruit immune cells among other effects (226–228); and contribute to several physiological and pathological processes (229, 230). At the transcriptional level most of the SASP components are regulated by NF-κB, C/EBPβ, and NOTCH (231). IL-1β is a component of the SASP that is secreted through unconventional pathways (32). However, many of the SASP factors are most probably secreted through the conventional pathway, though this is practically unexplored.
Several studies support that establishment of senescence and the secretory phenotype is accompanied by metabolic reprogramming including increases in mitochondrial oxygen consumption rates, biogenesis and dynamics. In particular, important alterations have been observed in oncogene and therapy induced senescence (19–21, 232–235). Interestingly replicative senescent cells that present mitochondrial dysfunction (236, 237) secrete lower levels of cytokines and other mediators than oncogene induced senescent cells (20, 224).
Moreover, mitochondria are required to sustain the SASP and mitochondrial depletion down regulates the expression and secretion of multiple cytokines and other factors in senescent cells (21, 26). In oncogene induced senescence inhibition of carnitine palmitoyl transferase I (CPTI), a key enzyme of the mitochondrial fatty acid oxidation pathway, decreases the secretion of several cytokines and growth factors, but not of IL-1β (20). Similar results were obtained upon inhibition of nicotinamide phosphoribosyltransferase (NAMPT), and enzyme from the NAD+ synthesis pathway (25), or silencing of mitochondrial sirtuin 3, known to impair mitochondrial electron transport (26, 238). Silencing of mitochondrial fusion proteins, MFN1 and 2 also impaired the secretion of IL-6 in senescent cells (19). Since mitofusins participate in the tethering of the mitochondria to the ER, these results suggest the association between these two organelles might be relevant in secretion processes in senescence. Finally mitochondrial ROS formation by the ETC (239, 240) and NADPH oxidase 4 (241, 242) are increased in senescent cells, and contribute to the establishment of senescence and the SASP [through the activation of NF-kB (243) and the DDR (244–246)]. Taken together these results point to a link between mitochondrial oxidative metabolism and dynamics and the proinflammatory secretory phenotype.
Since the SASP has been implied in numerous diseases (229, 247), the search for senolytics (that will kill senescent cells) or senomorphics (that will modulate the SASP) has become an active area of research (248, 249). Mitochondria appear as promising targets for the modulation of the SASP in pathological settings were senescent cells are involved.
Concluding Remarks
Herein we present evidence that mitochondria are required for the successful export of proteins to the extracellular space. Mitochondrial dynamics, bioenergetics and distribution as well as their interactions with other organelles, in particular with the ER, can undergo profound changes in response to secretion stimuli. Mitochondria contribute and support the secretion of proteins providing ATP for energy requiring processes, buffering Ca2+ concentrations and offering structural support and signals for NLRP3 inflammasome activation. A better understanding, at the molecular level, of the role of mitochondria in secretion processes is required and will help in the development of new genetic and pharmacological strategies to modulate protein secretion in pathological contexts.
Author Contributions
JM, IM, DT, and CQ contributed to the writing of the manuscript, CQ supervised all the activities. JM made the figures. All authors reviewed contents and approved the final version of the manuscript.
Funding
Agencia Nacional de Investigación e Innovación (ANII) FCE_1_2017_1_136021 to CQ and DM; Comisión Académica de Posgrados (CAP) of the UdelaR to JM and IM, Espacio Interdisciplinario Centros 2015, UdelaR; Programa de Desarrollo de las Ciencias Básicas (PEDECIBA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Adriana Cassina for reading the manuscript and providing insightful comments.
Abbreviations
ARF, ADP-ribosylation factor; BiP, ER chaperone BiP; Ca2+, calcium; COPI, coat protein complex I; COPII, coat protein complex II; DRP1, GTPase dynamin related protein 1; EDEM, ER degradation-enhancing alpha-mannosidase-like proteins; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum- associated degradation; ERES, ER exit sites; ERGIC, ER-Golgi intermediate compartment; ERMCs, ER–mitochondrial contact sites; ETC, electron transport chain; FIS1, fission 1 protein; GRP, glucose regulated protein; GSIS, glucose-stimulated insulin secretion; GT, UDP-glucose:glycoprotein glucosyltransferase; IL-1β, interleukin-1β; IMM, inner mitochondrial membrane; IP3, inositol-1, 4, 5-trisphosphate; IP3R, inositol-1, 4, 5-trisphosphate- sensitive channel or receptor; MCU, mitochondrial calcium uniporter; MICOS, mitochondrial contact site and cristae-organizing system; MICU, MCU regulator and mitochondrial calcium uptake; MFN1, mitofusin 1; MFN2, mitofusin 2; MIRO, mitochondrial Rho GTPase; NLRP3, Nod-like receptor family, pyrin domain containing 3; OMM, outer mitochondrial membrane; OPA1, optic atrophy protein 1; ROS, reactive oxygen species; SAR1, secretion associated Ras related 1 GTPAse; SASP, senescence-associated secretory phenotype; SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase; SRP, signal recognition particle; TNF, tumor necrosis factor, VDAC, voltage-dependent anion channel.
Footnotes
1. ^ERMCs are also known as MERCs or mitochondria-associated ER membranes (MAMs).
References
1. Kanapin A, Batalov S, Davis MJ, Gough J, Grimmond S, Kawaji H, et al. Mouse proteome analysis. Genome Res. (2003) 13(6B):1335–44. doi: 10.1101/gr.978703
2. Brandizzi F, Barlowe C. Organization of the ER-Golgi interface for membrane traffic control. Nat Rev Mol Cell Biol. (2013) 14:382–92. doi: 10.1038/nrm3588
3. Bajjalieh SM, Scheller RH. The biochemistry of neurotransmitter secretion. J Biol Chem. (1995) 270:1971–4. doi: 10.1074/jbc.270.5.1971
4. Depaoli MR, Hay JC, Graier WF, Malli R. The enigmatic ATP supply of the endoplasmic reticulum. Biol Rev Cambridge Philos Soc. (2019) 94:610–28. doi: 10.1111/brv.12469
5. Braakman I, Helenius J, Helenius A. Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature. (1992) 356:260–2. doi: 10.1038/356260a0
6. Dorner AJ, Kaufman RJ. The levels of endoplasmic reticulum proteins and ATP affect folding and secretion of selective proteins. Biologicals. (1994) 22:103–12. doi: 10.1006/biol.1994.1016
7. Dorner AJ, Wasley LC, Kaufman RJ. Protein dissociation from GRP78 and secretion are blocked by depletion of cellular ATP levels. Proc Natl Acad Sci USA. (1990) 87:7429–32. doi: 10.1073/pnas.87.19.7429
8. Caramelo JJ, Parodi AJ. A sweet code for glycoprotein folding. FEBS Lett. (2015) 589:3379–87. doi: 10.1016/j.febslet.2015.07.021
9. Sweeney HL, Holzbaur ELF. Motor proteins. Cold Spring Harbor Perspect Biol. (2018) 10:a021931. doi: 10.1101/cshperspect.a021931
10. Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. (2009) 323:474–7. doi: 10.1126/science.1161748
11. Anantharam A, Kreutzberger AJB. Unraveling the mechanisms of calcium-dependent secretion. J General Physiol. (2019) 151:417–34. doi: 10.1085/jgp.201812298
12. Carreras-Sureda A, Pihan P, Hetz C. Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium. (2018) 70:24–31. doi: 10.1016/j.ceca.2017.08.004
13. Burgoyne RD, Morgan A. Secretory granule exocytosis. Physiol Rev. (2003) 83:581–632. doi: 10.1152/physrev.00031.2002
14. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. (2012) 13:566–78. doi: 10.1038/nrm3412
15. Finkel T, Menazza S, Holmstrom KM, Parks RJ, Liu J, Sun J, et al. The ins and outs of mitochondrial calcium. Circul Res. (2015) 116:1810–9. doi: 10.1161/CIRCRESAHA.116.305484
16. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
17. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
18. Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, et al. Cutting edge: mitochondrial assembly of the NLRP3 Inflammasome complex is initiated at priming. J Immunol. (2018) 200:3047–52. doi: 10.4049/jimmunol.1701723
19. Martinez J, Tarallo D, Martinez-Palma L, Victoria S, Bresque M, Rodriguez-Bottero S, et al. Mitofusins modulate the increase in mitochondrial length, bioenergetics and secretory phenotype in therapy-induced senescent melanoma cells. Biochem J. (2019) 476:2463–86. doi: 10.1042/BCJ20190405
20. Quijano C, Cao L, Fergusson MM, Romero H, Liu J, Gutkind S, et al. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle. (2012) 11:1383–92. doi: 10.4161/cc.19800
21. Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. (2016) 35:724–42. doi: 10.15252/embj.201592862
22. Jhun BS, Lee H, Jin ZG, Yoon Y. Glucose stimulation induces dynamic change of mitochondrial morphology to promote insulin secretion in the insulinoma cell line INS-1E. PLoS ONE. (2013) 8:e60810. doi: 10.1371/journal.pone.0060810
23. Stiles L, Shirihai OS. Mitochondrial dynamics and morphology in beta-cells. Best Pract Res Clin Endocrinol Metab. (2012) 26:725–38. doi: 10.1016/j.beem.2012.05.004
24. Zhang B, Alysandratos KD, Angelidou A, Asadi S, Sismanopoulos N, Delivanis DA, et al. Human mast cell degranulation and preformed TNF secretion require mitochondrial translocation to exocytosis sites: relevance to atopic dermatitis. J Allergy Clin Immunol. (2011) 127:1522–31 e8. doi: 10.1016/j.jaci.2011.02.005
25. Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. (2019) 21:397–407. doi: 10.1038/s41556-019-0287-4
26. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. (2016) 23:303–14. doi: 10.1016/j.cmet.2015.11.011
27. Chow J, Rahman J, Achermann JC, Dattani MT, Rahman S. Mitochondrial disease and endocrine dysfunction. Nat Rev Endocrinol. (2017) 13:92–104. doi: 10.1038/nrendo.2016.151
28. Devine MJ, Kittler JT. Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci. (2018) 19:63–80. doi: 10.1038/nrn.2017.170
29. Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells. Nat Commun. (2019) 10:2474. doi: 10.1038/s41467-019-10189-x
30. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. (2005) 39:359–407. doi: 10.1146/annurev.genet.39.110304.095751
31. Weatherly LM, Shim J, Hashmi HN, Kennedy RH, Hess ST, Gosse JA. Antimicrobial agent triclosan is a proton ionophore uncoupler of mitochondria in living rat and human mast cells and in primary human keratinocytes. J Appl Toxicol. (2016) 36:777–89. doi: 10.1002/jat.3209
32. Kim J, Gee HY, Lee MG. Unconventional protein secretion - new insights into the pathogenesis and therapeutic targets of human diseases. J Cell Sci. (2018) 131:jcs213686. doi: 10.1242/jcs.213686
33. Viotti C. ER to Golgi-dependent protein secretion: the conventional pathway. Methods Mol Biol. (2016) 1459:3–29. doi: 10.1007/978-1-4939-3804-9_1
34. Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. (2004) 20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307
35. Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. (2004) 116:153–66. doi: 10.1016/S0092-8674(03)01079-1
36. Hehnly H, Stamnes M. Regulating cytoskeleton-based vesicle motility. FEBS Lett. (2007) 581:2112–8. doi: 10.1016/j.febslet.2007.01.094
37. Mandon EC, Trueman SF, Gilmore R. Protein translocation across the rough endoplasmic reticulum. Cold Spring Harbor Perspect Biol. (2013) 5:a013342. doi: 10.1101/cshperspect.a013342
38. Benham AM. Protein secretion and the endoplasmic reticulum. Cold Spring Harbor Perspect Biol. (2012) 4:a012872. doi: 10.1101/cshperspect.a012872
39. Connolly T, Gilmore R. The signal recognition particle receptor mediates the GTP-dependent displacement of SRP from the signal sequence of the nascent polypeptide. Cell. (1989) 57:599–610. doi: 10.1016/0092-8674(89)90129-3
40. Zimmermann R, Eyrisch S, Ahmad M, Helms V. Protein translocation across the ER membrane. Biochim Biophys Acta. (2011) 1808:912–24. doi: 10.1016/j.bbamem.2010.06.015
41. Pobre KFR, Poet GJ, Hendershot LM. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J Biol Chem. (2019) 294:2098–108. doi: 10.1074/jbc.REV118.002804
42. Melnyk A, Rieger H, Zimmermann R. Co-chaperones of the mammalian endoplasmic reticulum. Sub Cell Biochem. (2015) 78:179–200. doi: 10.1007/978-3-319-11731-7_9
43. Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 6th ed. New York, NY: W. H. Freeman and Company (2008).
44. Frey S, Leskovar A, Reinstein J, Buchner J. The ATPase cycle of the endoplasmic chaperone Grp94. J Biol Chem. (2007) 282:35612–20. doi: 10.1074/jbc.M704647200
45. Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harbor Perspect Biol. (2013) 5:a013185. doi: 10.1101/cshperspect.a013185
46. Ruggiano A, Foresti O, Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol. (2014) 204:869–79. doi: 10.1083/jcb.201312042
47. Barlowe C, Helenius A. Cargo capture and bulk flow in the early secretory pathway. Annu Rev Cell Dev Biol. (2016) 32:197–222. doi: 10.1146/annurev-cellbio-111315-125016
48. Szul T, Sztul E. COPII and COPI traffic at the ER-Golgi interface. Physiology. (2011) 26:348–64. doi: 10.1152/physiol.00017.2011
49. Barlan K, Gelfand VI. Microtubule-based transport and the distribution, tethering, and organization of organelles. Cold Spring Harbor Perspect Biol. (2017) 9:a025817. doi: 10.1101/cshperspect.a025817
50. Gutierrez LM. New insights into the role of the cortical cytoskeleton in exocytosis from neuroendocrine cells. Int Rev Cell Mol Biol. (2012) 295:109–37. doi: 10.1016/B978-0-12-394306-4.00009-5
51. Rizo J, Xu J. The synaptic vesicle release machinery. Annu Rev Biophys. (2015) 44:339–67. doi: 10.1146/annurev-biophys-060414-034057
52. Dempski RE Jr, Imperiali B. Oligosaccharyl transferase: gatekeeper to the secretory pathway. Curr Opin Chem Biol. (2002) 6:844–50. doi: 10.1016/S1367-5931(02)00390-3
53. Mohorko E, Glockshuber R, Aebi M. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J Inherited Metab Dis. (2011) 34:869–78. doi: 10.1007/s10545-011-9337-1
54. Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. (2000) 69:69–93. doi: 10.1146/annurev.biochem.69.1.69
55. Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem. (2008) 283:10221–5. doi: 10.1074/jbc.R700048200
56. Williams DB. Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J Cell Sci. (2006) 119(Pt 4):615–23. doi: 10.1242/jcs.02856
57. Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. (2007) 35:430–42. doi: 10.1002/mus.20745
58. Svedine S, Wang T, Halaban R, Hebert DN. Carbohydrates act as sorting determinants in ER-associated degradation of tyrosinase. J Cell Sci. (2004) 117(Pt 14):2937–49. doi: 10.1242/jcs.01154
59. Anelli T, Sitia R. Protein quality control in the early secretory pathway. EMBO J. (2008) 27:315–27. doi: 10.1038/sj.emboj.7601974
60. Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, et al. COPII: a membrane coat formed by sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. (1994) 77:895–907. doi: 10.1016/0092-8674(94)90138-4
61. Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, et al. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. (2003) 114:497–509. doi: 10.1016/S0092-8674(03)00609-3
62. Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev Cell. (2007) 13:635–45. doi: 10.1016/j.devcel.2007.10.006
63. Jackson CL, Bouvet S. Arfs at a glance. J Cell Sci. (2014) 127(Pt 19):4103–9. doi: 10.1242/jcs.144899
64. Ross JL, Ali MY, Warshaw DM. Cargo transport: molecular motors navigate a complex cytoskeleton. Curr Opin Cell Biol. (2008) 20:41–7. doi: 10.1016/j.ceb.2007.11.006
65. Pang ZP, Sudhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. (2010) 22:496–505. doi: 10.1016/j.ceb.2010.05.001
66. Li P, Bademosi AT, Luo J, Meunier FA. Actin remodeling in regulated exocytosis: toward a mesoscopic view. Trends Cell Biol. (2018) 28:685–97. doi: 10.1016/j.tcb.2018.04.004
67. Wen PJ, Grenklo S, Arpino G, Tan X, Liao HS, Heureaux J, et al. Actin dynamics provides membrane tension to merge fusing vesicles into the plasma membrane. Nat Commun. (2016) 7:12604. doi: 10.1038/ncomms12604
68. Rabouille C. Pathways of unconventional protein secretion. Trends Cell Biol. (2017) 27:230–40. doi: 10.1016/j.tcb.2016.11.007
69. Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Different. (2016) 23:1219–31. doi: 10.1038/cdd.2015.176
70. Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol. (2015) 33:49–77. doi: 10.1146/annurev-immunol-032414-112306
71. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
72. Blakely RD, Edwards RH. Vesicular and plasma membrane transporters for neurotransmitters. Cold Spring Harbor Perspect Biol. (2012) 4:a025817. doi: 10.1101/cshperspect.a005595
73. Ovsepian SV, O'Leary VB, Zaborszky L, Ntziachristos V, Dolly JO. Synaptic vesicle cycle and amyloid beta: biting the hand that feeds. Alzheimer's Dement. (2018) 14:502–13. doi: 10.1016/j.jalz.2018.01.011
74. Chanaday NL, Cousin MA, Milosevic I, Watanabe S, Morgan JR. The synaptic vesicle cycle revisited: new insights into the modes and mechanisms. J Neurosci. (2019) 39:8209–16. doi: 10.1523/JNEUROSCI.1158-19.2019
75. Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. (2016) 41:261–73. doi: 10.1016/j.tibs.2016.01.001
76. Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. (2008) 32:529–39. doi: 10.1016/j.molcel.2008.10.021
77. Letts JA, Sazanov LA. Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat Struct Mol Biol. (2017) 24:800–8. doi: 10.1038/nsmb.3460
78. Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, et al. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA. (2011) 108:14121–6. doi: 10.1073/pnas.1103621108
79. Harner M, Korner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U, et al. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. (2011) 30:4356–70. doi: 10.1038/emboj.2011.379
80. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. (2013) 155:160–71. doi: 10.1016/j.cell.2013.08.032
81. Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. (2018) 20:745–54. doi: 10.1038/s41556-018-0124-1
82. Neupane P, Bhuju S, Thapa N, Bhattarai HK. ATP synthase: structure, function and inhibition. Biomol Concepts. (2019) 10:1–10. doi: 10.1515/bmc-2019-0001
83. Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA. (2010) 107:16823–7. doi: 10.1073/pnas.1011099107
84. Quijano C, Trujillo M, Castro L, Trostchansky A. Interplay between oxidant species and energy metabolism. Redox Biol. (2016) 8:28–42. doi: 10.1016/j.redox.2015.11.010
85. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. (2003) 552(Pt 2):335–44. doi: 10.1113/jphysiol.2003.049478
86. Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc Natl Acad Sci USA. (2018) 115:5839–48. doi: 10.1073/pnas.1804932115
87. Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem J. (2010) 425:313–25. doi: 10.1042/BJ20091541
88. Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. (1995) 64:97–112. doi: 10.1146/annurev.bi.64.070195.000525
89. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. (2000) 408:239–47. doi: 10.1038/35041687
90. Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. (2002) 33:1451–64. doi: 10.1016/S0891-5849(02)01111-5
91. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. (2012) 337:1062–5. doi: 10.1126/science.1219855
92. Lee H, Smith SB, Yoon Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem. (2017) 292:7115–30. doi: 10.1074/jbc.M116.762567
93. Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. (2004) 117(Pt 26):6535–46. doi: 10.1242/jcs.01565
94. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. (2004) 305:858–62. doi: 10.1126/science.1099793
95. Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. (2006) 8:1255–62. doi: 10.1038/ncb1487
96. Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. (2001) 12:2245–56. doi: 10.1091/mbc.12.8.2245
97. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. (2010) 191:1141–58. doi: 10.1083/jcb.201007152
98. Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. (2013) 24:659–67. doi: 10.1091/mbc.e12-10-0721
99. Zhang Y, Chan DC. Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc Natl Acad Sci USA. (2007) 104:18526–30. doi: 10.1073/pnas.0706441104
100. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. (2011) 334:358–62. doi: 10.1126/science.1207385
101. De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr Biol. (2005) 15:678–83. doi: 10.1016/j.cub.2005.02.064
102. Korobova F, Gauvin TJ, Higgs HN. A role for myosin II in mammalian mitochondrial fission. Curr Biol. (2014) 24:409–14. doi: 10.1016/j.cub.2013.12.032
103. Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, et al. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife. (2015) 4:e08828. doi: 10.7554/eLife.08828
104. Pagliuso A, Tham TN, Stevens JK, Lagache T, Persson R, Salles A, et al. A role for septin 2 in Drp1-mediated mitochondrial fission. EMBO Rep. (2016) 17:858–73. doi: 10.15252/embr.201541612
105. Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. (2013) 339:464–7. doi: 10.1126/science.1228360
106. Kar UP, Dey H, Rahaman A. Regulation of dynamin family proteins by post-translational modifications. J Biosci. (2017) 42:333–44. doi: 10.1007/s12038-017-9680-y
107. Bulthuis EP, Adjobo-Hermans MJW, Willems P, Koopman WJH. Mitochondrial morphofunction in mammalian cells. Antioxidants Redox Signal. (2019) 30:2066–109. doi: 10.1089/ars.2018.7534
108. Sauvanet C, Duvezin-Caubet S, di Rago JP, Rojo M. Energetic requirements and bioenergetic modulation of mitochondrial morphology and dynamics. Semin Cell Dev Biol. (2010) 21:558–65. doi: 10.1016/j.semcdb.2009.12.006
109. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. (2016) 212:379–87. doi: 10.1083/jcb.201511036
110. Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol cell. (2016) 61:683–94. doi: 10.1016/j.molcel.2016.02.022
111. Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. (2003) 278:17190–7. doi: 10.1074/jbc.M212754200
112. Mourier A, Motori E, Brandt T, Lagouge M, Atanassov I, Galinier A, et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol. (2015) 208:429–42. doi: 10.1083/jcb.201411100
113. Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, et al. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. (2005) 14:1405–15. doi: 10.1093/hmg/ddi149
114. Son JM, Sarsour EH, Kakkerla Balaraju A, Fussell J, Kalen AL, Wagner BA, et al. Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell. (2017) 16:1136–45. doi: 10.1111/acel.12649
115. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. (2005) 280:26185–92. doi: 10.1074/jbc.M503062200
116. Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S, et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell. (2011) 22:2235–45. doi: 10.1091/mbc.e10-12-0933
117. Patten DA, Wong J, Khacho M, Soubannier V, Mailloux RJ, Pilon-Larose K, et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. (2014) 33:2676–91. doi: 10.15252/embj.201488349
118. Quintero OA, DiVito MM, Adikes RC, Kortan MB, Case LB, Lier AJ, et al. Human Myo19 is a novel myosin that associates with mitochondria. Curr Biol. (2009) 19:2008–13. doi: 10.1016/j.cub.2009.10.026
119. Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. (2006) 173:545–57. doi: 10.1083/jcb.200601067
120. Morlino G, Barreiro O, Baixauli F, Robles-Valero J, Gonzalez-Granado JM, Villa-Bellosta R, et al. Miro-1 links mitochondria and microtubule Dynein motors to control lymphocyte migration and polarity. Mol Cell Biol. (2014) 34:1412–26. doi: 10.1128/MCB.01177-13
121. van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. (2013) 77:485–502. doi: 10.1016/j.neuron.2012.11.027
122. Lopez-Domenech G, Covill-Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, et al. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. (2018) 37:321–36. doi: 10.15252/embj.201696380
123. Modi S, Lopez-Domenech G, Halff EF, Covill-Cooke C, Ivankovic D, Melandri D, et al. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat Commun. (2019) 10:4399. doi: 10.1038/s41467-019-12382-4
124. Dolman NJ, Gerasimenko JV, Gerasimenko OV, Voronina SG, Petersen OH, Tepikin AV. Stable Golgi-mitochondria complexes and formation of Golgi Ca(2+) gradients in pancreatic acinar cells. J Biol Chem. (2005) 280:15794–9. doi: 10.1074/jbc.M412694200
125. Moltedo O, Remondelli P, Amodio G. The mitochondria-endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Front Cell Dev Biol. (2019) 7:172. doi: 10.3389/fcell.2019.00172
126. Xia M, Zhang Y, Jin K, Lu Z, Zeng Z, Xiong W. Communication between mitochondria and other organelles: a brand-new perspective on mitochondria in cancer. Cell Biosci. (2019) 9:27. doi: 10.1186/s13578-019-0289-8
127. Elbaz Y, Schuldiner M. Staying in touch: the molecular era of organelle contact sites. Trends Biochem Sci. (2011) 36:616–23. doi: 10.1016/j.tibs.2011.08.004
128. Cosson P, Marchetti A, Ravazzola M, Orci L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS ONE. (2012) 7:e46293. doi: 10.1371/journal.pone.0046293
129. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci USA. (2015) 112:E2174-81. doi: 10.1073/pnas.1504880112
130. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008) 456:605–10. doi: 10.1038/nature07534
131. Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, et al. Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA. (2016) 113:11249–54. doi: 10.1073/pnas.1606786113
132. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. (2016) 15:2226–38. doi: 10.1016/j.celrep.2016.05.013
133. Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, et al. MITOL regulates endoplasmic reticulum-mitochondria contacts via mitofusin2. Mol Cell. (2013) 51:20–34. doi: 10.1016/j.molcel.2013.04.023
134. Cerqua C, Anesti V, Pyakurel A, Liu D, Naon D, Wiche G, et al. Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep. (2010) 11:854–60. doi: 10.1038/embor.2010.151
135. De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. (2012) 21:1299–311. doi: 10.1093/hmg/ddr559
136. Gomez-Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr Biol. (2017) 27:371–85. doi: 10.1016/j.cub.2016.12.038
137. Gomez-Suaga P, Perez-Nievas BG, Glennon EB, Lau DHW, Paillusson S, Morotz GM, et al. The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol Commun. (2019) 7:35. doi: 10.1186/s40478-019-0688-4
138. Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta. (2013) 1833:213–24. doi: 10.1016/j.bbamcr.2012.04.013
139. Lee S, Lee KS, Huh S, Liu S, Lee DY, Hong SH, et al. Polo kinase phosphorylates miro to control ER-mitochondria contact sites and mitochondrial Ca(2+) homeostasis in neural stem cell development. Dev Cell. (2016) 37:174–89. doi: 10.1016/j.devcel.2016.03.023
140. Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, et al. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. (2012) 31:457–70. doi: 10.1038/emboj.2011.384
141. Lynes EM, Raturi A, Shenkman M, Ortiz Sandoval C, Yap MC, Wu J, et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J Cell Sci. (2013) 126(Pt 17):3893–903. doi: 10.1242/jcs.125856
142. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. (2011) 476:341–5. doi: 10.1038/nature10234
143. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. (2011) 476:336–40. doi: 10.1038/nature10230
144. De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem. (2016) 85:161–92. doi: 10.1146/annurev-biochem-060614-034216
145. Ackema KB, Hench J, Bockler S, Wang SC, Sauder U, Mergentaler H, et al. The small GTPase Arf1 modulates mitochondrial morphology and function. EMBO J. (2014) 33:2659–75. doi: 10.15252/embj.201489039
146. Ackema KB, Prescianotto-Baschong C, Hench J, Wang SC, Chia ZH, Mergentaler H, et al. Sar1, a novel regulator of ER-mitochondrial contact sites. PLoS ONE. (2016) 11:e0154280. doi: 10.1371/journal.pone.0154280
147. Walch L, Pellier E, Leng W, Lakisic G, Gautreau A, Contremoulins V, et al. GBF1 and Arf1 interact with Miro and regulate mitochondrial positioning within cells. Sci Rep. (2018) 8:17121. doi: 10.1038/s41598-018-35190-0
148. Sack MN. Mitochondrial fidelity and metabolic agility control immune cell fate and function. J Clin Investig. (2018) 128:3651–61. doi: 10.1172/JCI120845
149. Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. (2006) 86:369–408. doi: 10.1152/physrev.00004.2005
150. Antoine JC, Jouanne C. Multiple effects of the phenylhydrazone derivative FCCP on the secretory pathway in rat plasma cells. Eur J Biol. (1986) 42:68–73.
151. Argon Y, Burkhardt JK, Leeds JM, Milstein C. Two steps in the intracellular transport of IgD are sensitive to energy depletion. J Immunol. (1989) 142:554–61.
152. Burkhardt JK, Argon Y. Intracellular transport of the glycoprotein of VSV is inhibited by CCCP at a late stage of post-translational processing. J Cell Sci. (1989) 92(Pt 4):633–42.
153. Klein MC, Zimmermann K, Schorr S, Landini M, Klemens PAW, Altensell J, et al. AXER is an ATP/ADP exchanger in the membrane of the endoplasmic reticulum. Nat Commun. (2018) 9:3489. doi: 10.1038/s41467-018-06003-9
154. Yong J, Bischof H, Burgstaller S, Siirin M, Murphy A, Malli R, et al. Mitochondria supply ATP to the ER through a mechanism antagonized by cytosolic Ca2+. eLife. (2019) 8:e49682. doi: 10.7554/eLife.49682
155. Vishnu N, Jadoon Khan M, Karsten F, Groschner LN, Waldeck-Weiermair M, Rost R, et al. ATP increases within the lumen of the endoplasmic reticulum upon intracellular Ca2+ release. Mol Biol Cell. (2014) 25:368–79. doi: 10.1091/mbc.e13-07-0433
156. Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. (2012) 13:77–93. doi: 10.1038/nrn3156
157. Vaccaro V, Devine MJ, Higgs NF, Kittler JT. Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Rep. (2017) 18:231–40. doi: 10.15252/embr.201642710
158. Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev. (2015) 95:1383–436. doi: 10.1152/physrev.00020.2014
159. Catterall WA. Voltage-gated calcium channels. Cold Spring Harbor Perspect Biol. (2011) 3:a003947. doi: 10.1101/cshperspect.a003947
160. Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. (2010) 2:139. doi: 10.3389/fnsyn.2010.00139
161. Griesche N, Sanchez G, Hermans C, Idevall-Hagren O. Cortical mitochondria regulate insulin secretion by local Ca(2+) buffering in rodent beta cells. J Cell Sci. (2019) 132:jcs228544. doi: 10.1242/jcs.228544
162. Quintana A, Schwarz EC, Schwindling C, Lipp P, Kaestner L, Hoth M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem. (2006) 281:40302–9. doi: 10.1074/jbc.M607896200
163. Hoth M, Button DC, Lewis RS. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci USA. (2000) 97:10607–12. doi: 10.1073/pnas.180143997
164. Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. (1991) 351:751–4. doi: 10.1038/351751a0
165. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. (2019) 10:128. doi: 10.1038/s41419-019-1413-8
166. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. (2013) 191:4358–66. doi: 10.4049/jimmunol.1301170
167. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci USA. (2013) 110:17963–8. doi: 10.1073/pnas.1312571110
168. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. (2013) 14:454–60. doi: 10.1038/ni.2550
169. Boland BB, Rhodes CJ, Grimsby JS. The dynamic plasticity of insulin production in beta-cells. Mol Metab. (2017) 6:958–73. doi: 10.1016/j.molmet.2017.04.010
170. Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia. (2010) 53:1019–32. doi: 10.1007/s00125-010-1685-0
171. Jesinkey SR, Madiraju AK, Alves TC, Yarborough OH, Cardone RL, Zhao X, et al. Mitochondrial GTP links nutrient sensing to beta cell health, mitochondrial morphology, and insulin secretion independent of oxphos. Cell Rep. (2019) 28:759–72 e10. doi: 10.1016/j.celrep.2019.06.058
172. Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, et al. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J Biol Chem. (2009) 284:26578–90. doi: 10.1074/jbc.M109.011775
173. Kennedy ED, Maechler P, Wollheim CB. Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS-1 cells. Diabetes. (1998) 47:374–80. doi: 10.2337/diabetes.47.3.374
174. Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. (2009) 1:425–37. doi: 10.18632/aging.100038
175. Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, et al. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J Biol Chem. (2012) 287:34445–54. doi: 10.1074/jbc.M112.392084
176. Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. (2009) 58:2303–15. doi: 10.2337/db07-1781
177. Park KS, Wiederkehr A, Kirkpatrick C, Mattenberger Y, Martinou JC, Marchetti P, et al. Selective actions of mitochondrial fission/fusion genes on metabolism-secretion coupling in insulin-releasing cells. J Biol Chem. (2008) 283:33347–56. doi: 10.1074/jbc.M806251200
178. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. (2008) 27:433–46. doi: 10.1038/sj.emboj.7601963
179. Kabra UD, Pfuhlmann K, Migliorini A, Keipert S, Lamp D, Korsgren O, et al. Direct substrate delivery into mitochondrial fission-deficient pancreatic islets rescues insulin secretion. Diabetes. (2017) 66:1247–57. doi: 10.2337/db16-1088
180. American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care. (2008) 31(Suppl 1):S55–60. doi: 10.2337/dc08-S055
181. DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers. (2015) 1:15019. doi: 10.1038/nrdp.2015.19
182. Ballinger SW, Shoffner JM, Hedaya EV, Trounce I, Polak MA, Koontz DA, et al. Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat Genet. (1992) 1:11–5. doi: 10.1038/ng0492-11
183. van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, et al. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. (1992) 1:368–71. doi: 10.1038/ng0892-368
184. Li K, Wu L, Liu J, Lin W, Qi Q, Zhao T. Maternally inherited diabetes mellitus associated with a novel m.15897G>A mutation in mitochondrial tRNA(Thr) gene. J Diabetes Res. (2020) 2020:2057187. doi: 10.1155/2020/2057187
185. Maassen JA, 'T Hart LM, Van Essen E, Heine RJ, Nijpels G, Jahangir Tafrechi RS, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. (2004) 53(Suppl 1):S103–9. doi: 10.2337/diabetes.53.2007.S103
186. Suzuki Y, Iizuka T, Kobayashi T, Nishikawa T, Atsumi Y, Kadowaki T, et al. Diabetes mellitus associated with the 3243 mitochondrial tRNA(Leu)(UUR) mutation: insulin secretion and sensitivity. Metabolism. (1997) 46:1019–23. doi: 10.1016/S0026-0495(97)90272-9
187. Brandle M, Lehmann R, Maly FE, Schmid C, Spinas GA. Diminished insulin secretory response to glucose but normal insulin and glucagon secretory responses to arginine in a family with maternally inherited diabetes and deafness caused by mitochondrial tRNA(LEU(UUR)) gene mutation. Diabetes Care. (2001) 24:1253–8. doi: 10.2337/diacare.24.7.1253
188. Gebhart SS, Shoffner JM, Koontz D, Kaufman A, Wallace D. Insulin resistance associated with maternally inherited diabetes and deafness. Metabolism. (1996) 45:526–31. doi: 10.1016/S0026-0495(96)90231-0
189. Hou J, Li Z, Zhong W, Hao Q, Lei L, Wang L, et al. Temporal transcriptomic and proteomic landscapes of deteriorating pancreatic islets in type 2 diabetic rats. Diabetes. (2017) 66:2188–200. doi: 10.2337/db16-1305
190. Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. (2005) 48:282–9. doi: 10.1007/s00125-004-1627-9
191. Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. (2005) 54:727–35. doi: 10.2337/diabetes.54.3.727
192. Fernandez-Alvarez J, Conget I, Rasschaert J, Sener A, Gomis R, Malaisse WJ. Enzymatic, metabolic and secretory patterns in human islets of type 2 (non-insulin-dependent) diabetic patients. Diabetologia. (1994) 37:177–81. doi: 10.1007/s001250050090
193. Chan CB, De Leo D, Joseph JW, McQuaid TS, Ha XF, Xu F, et al. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes. (2001) 50:1302–10. doi: 10.2337/diabetes.50.6.1302
194. Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. (2001) 105:745–55. doi: 10.1016/S0092-8674(01)00378-6
195. Liotta A, Rosner J, Huchzermeyer C, Wojtowicz A, Kann O, Schmitz D, et al. Energy demand of synaptic transmission at the hippocampal Schaffer-collateral synapse. J Cereb Blood Flow Metab. (2012) 32:2076–83. doi: 10.1038/jcbfm.2012.116
196. Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. (2005) 47:365–78. doi: 10.1016/j.neuron.2005.06.018
197. Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. (2012) 75:762–77. doi: 10.1016/j.neuron.2012.08.019
198. Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. (2008) 60:748–66. doi: 10.1016/j.neuron.2008.10.010
199. Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. (2006) 314:777–81. doi: 10.1126/science.1132814
200. Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging. (2005) 32:486–510. doi: 10.1007/s00259-005-1762-7
201. Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, et al. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging. (2008) 29:676–92. doi: 10.1016/j.neurobiolaging.2006.12.008
202. Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. (2001) 21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001
203. Silva DF, Selfridge JE, Lu J, E L, Cardoso SM, Swerdlow RH. Mitochondrial abnormalities in Alzheimer's disease: possible targets for therapeutic intervention. Adv Pharmacol. (2012) 64:83–126. doi: 10.1016/B978-0-12-394816-8.00003-9
204. Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Trans. (1998) 105:855–70. doi: 10.1007/s007020050099
205. Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. (2000) 21:455–62. doi: 10.1016/S0197-4580(00)00112-3
206. Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. (2009) 106:14670–5. doi: 10.1073/pnas.0903563106
207. Martino Adami PV, Galeano P, Wallinger ML, Quijano C, Rabossi A, Pagano ES, et al. Worsening of memory deficit induced by energy-dense diet in a rat model of early-Alzheimer's disease is associated to neurotoxic Abeta species and independent of neuroinflammation. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:731–43. doi: 10.1016/j.bbadis.2016.12.014
208. Martino Adami PV, Quijano C, Magnani N, Galeano P, Evelson P, Cassina A, et al. Synaptosomal bioenergetic defects are associated with cognitive impairment in a transgenic rat model of early Alzheimer's disease. J Cereb Blood Flow Metab. (2017) 37:69–84. doi: 10.1177/0271678X15615132
209. Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. (2011) 20:4515–29. doi: 10.1093/hmg/ddr381
210. Zhao XL, Wang WA, Tan JX, Huang JK, Zhang X, Zhang BZ, et al. Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J Neurosci. (2010) 30:1512–22. doi: 10.1523/JNEUROSCI.3699-09.2010
211. Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. (2006) 15:1437–49. doi: 10.1093/hmg/ddl066
212. Veereshwarayya V, Kumar P, Rosen KM, Mestril R, Querfurth HW. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular beta-amyloid-induced inhibition of complex IV and limit apoptosis. J Biol Chem. (2006) 281:29468–78. doi: 10.1074/jbc.M602533200
213. Joshi AU, Saw NL, Shamloo M, Mochly-Rosen D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer's disease. Oncotarget. (2018) 9:6128–43. doi: 10.18632/oncotarget.23640
214. Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA. (2008) 105:19318–23. doi: 10.1073/pnas.0804871105
215. Cai Q, Tammineni P. Mitochondrial aspects of synaptic dysfunction in Alzheimer's disease. J Alzheimer's Dis. (2017) 57:1087–103. doi: 10.3233/JAD-160726
216. Rao KN, Brown MA. Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann NY Acad Sci. (2008) 1143:83–104. doi: 10.1196/annals.1443.023
217. Metz M, Maurer M. Mast cells–key effector cells in immune responses. Trends Immunol. (2007) 28:234–41. doi: 10.1016/j.it.2007.03.003
218. Douglas WW, Kagayama M. Calcium and stimulus-secretion coupling in the mast cell: stimulant and inhibitory effects of calcium-rich media on exocytosis. J Physiol. (1977) 270:691–703. doi: 10.1113/jphysiol.1977.sp011976
219. Zhang B, Asadi S, Weng Z, Sismanopoulos N, Theoharides TC. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS ONE. (2012) 7:e49767. doi: 10.1371/journal.pone.0049767
220. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. (2013) 75:685–705. doi: 10.1146/annurev-physiol-030212-183653
221. Chen QM, Tu VC, Liu J. Measurements of hydrogen peroxide induced premature senescence: senescence-associated beta-galactosidase and DNA synthesis index in human diploid fibroblasts with down-regulated p53 or Rb. Biogerontology. (2000) 1:335–9. doi: 10.1023/a:1026590501344
222. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
223. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. (1997) 88:593–602. doi: 10.1016/S0092-8674(00)81902-9
224. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
225. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. (2011) 192:547–56. doi: 10.1083/jcb.201009094
226. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. (2008) 133:1006–18. doi: 10.1016/j.cell.2008.03.038
227. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
228. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. (2009) 9:81–94. doi: 10.1038/nrc2560
229. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. doi: 10.1038/nrm3823
230. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. (2015) 15:397–408. doi: 10.1038/nrc3960
231. Ito Y, Hoare M, Narita M. Spatial and temporal control of senescence. Trends Cell Biol. (2017) 27:820–32. doi: 10.1016/j.tcb.2017.07.004
232. Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. (2013) 501:421–5. doi: 10.1038/nature12437
233. Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. (2014) 14:547–58. doi: 10.1038/nrc3773
234. Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. (2009) 29:4495–507. doi: 10.1128/MCB.01868-08
235. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. (2013) 498:109–12. doi: 10.1038/nature12154
236. Hutter E, Renner K, Pfister G, Stockl P, Jansen-Durr P, Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. (2004) 380(Pt 3):919–28. doi: 10.1042/bj20040095
237. Zwerschke W, Mazurek S, Stockl P, Hutter E, Eigenbrodt E, Jansen-Durr P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J. (2003) 376(Pt 2):403–11. doi: 10.1042/bj20030816
238. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. (2008) 105:14447–52. doi: 10.1073/pnas.0803790105
239. Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. (1999) 274:7936–40. doi: 10.1074/jbc.274.12.7936
240. Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. (2007) 5:e110. doi: 10.1371/journal.pbio.0050110
241. Sakai Y, Yamamori T, Yoshikawa Y, Bo T, Suzuki M, Yamamoto K, et al. NADPH oxidase 4 mediates ROS production in radiation-induced senescent cells and promotes migration of inflammatory cells. Free Radical Res. (2018) 52:92–102. doi: 10.1080/10715762.2017.1416112
242. Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene. (2012) 31:1117–29. doi: 10.1038/onc.2011.327
243. Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev. (2018) 170:30–6. doi: 10.1016/j.mad.2017.08.005
244. Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. (2010) 6:347. doi: 10.1038/msb.2010.5
245. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
246. Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. (2011) 124(Pt 1):68–81. doi: 10.1242/jcs.071340
247. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: aging, cancer, and injury. Physiol Rev. (2019) 99:1047–78. doi: 10.1152/physrev.00020.2018
248. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. (2017) 21:21–8. doi: 10.1016/j.ebiom.2017.04.013
Keywords: mitochondria, secretion, bioenergetics, ATP, calcium, dynamics, endoplasmic reticulum, exocytosis
Citation: Martínez J, Marmisolle I, Tarallo D and Quijano C (2020) Mitochondrial Bioenergetics and Dynamics in Secretion Processes. Front. Endocrinol. 11:319. doi: 10.3389/fendo.2020.00319
Received: 17 February 2020; Accepted: 27 April 2020;
Published: 22 May 2020.
Edited by:
Cecilia Poderoso, University of Buenos Aires, ArgentinaReviewed by:
Amandine Grimm, University of Basel, SwitzerlandTito Cali, University of Padova, Italy
Charles Affourtit, University of Plymouth, United Kingdom
Copyright © 2020 Martínez, Marmisolle, Tarallo and Quijano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celia Quijano, Y2VsaXFAZm1lZC5lZHUudXk=; Y2VsaWEucXVpamFub0BnbWFpbC5jb20=