 Shuang Zhou
Shuang Zhou Yi-Min Dai
Yi-Min Dai Xiao-Feng Zeng
Xiao-Feng Zeng Hou-Zao Chen
Hou-Zao Chen- 1Department of Rheumatology, National Clinical Research Center for Dermatologic and Immunologic Diseases (NCRC-DID), Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2State Key Laboratory of Medical Molecular Biology, Department of Biochemistry and Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Diabetes-induced tissue injuries in target organs such as the kidney, heart, eye, liver, skin, and nervous system contribute significantly to the morbidity and mortality of diabetes. However, whether the lung should be considered a diabetic target organ has been discussed for decades. Accumulating evidence shows that both pulmonary histological changes and functional abnormalities have been observed in diabetic patients, suggesting that the lung is a diabetic target organ. Mechanisms underlying diabetic lung are unclear, however, oxidative stress, systemic inflammation, and premature aging convincingly contribute to them. Circadian system and Sirtuins have been well-documented to play important roles in above mechanisms. Circadian rhythms are intrinsic mammalian biological oscillations with a period of near 24 h driven by the circadian clock system. This system plays an important role in the regulation of energy metabolism, oxidative stress, inflammation, cellular proliferation and senescence, thus impacting metabolism-related diseases, chronic airway diseases and cancers. Sirtuins, a family of adenine dinucleotide (NAD+)-dependent histone deacetylases, have been demonstrated to regulate a series of physiological processes and affect diseases such as obesity, insulin resistance, type 2 diabetes (T2DM), heart disease, cancer, and aging. In this review, we summarize recent advances in the understanding of the roles of the circadian clock and Sirtuins in regulating cellular processes and highlight the potential interactions of the circadian clock and Sirtuins in the context of diabetic lung.
Introduction
Diabetes mellitus appears to be one of the most common chronic diseases worldwide and leads to high premature mortality in human. The complications of diabetes can be separated into two main subgroups, microvascular complications, and macrovascular complications. Vascular damage plays a central role in diabetic complications. Despite a large capillary network, the lung is frequently overlooked because of its subclinical characteristic in diabetic patients. Recently, accumulating evidence has indicated a correlation between diabetes and impaired pulmonary structures and functions. In addition, diabetes increases the risk of some chronic pulmonary diseases. For instance, T2DM can deteriorate the progression and prognosis of COPD (1–4). Moreover, diabetes increases the severity of pulmonary hypertension secondary to COPD (5). A multicenter investigation proved that diabetes increases the odds of mortality for COPD patients (3). Therefore, the lung is certainly a diabetic target organ.
Mechanisms of lung damage caused by diabetes are still unclear, but some are convincing. For instance, glycosylation of proteins in the lung and chest wall promotes collagen accumulation in lung connective tissue and ultimately leads to a reduction in lung compliance (6–8). Hyperglycemia triggers vascular oxidative damage resulting in a loss of microvascular reserve in the lung. Systemic inflammation exaggerates vascular damage through endothelial dysfunction (9–11). Insulin resistance has been shown to disturb lung volume through leptin (12, 13). Interestingly, the anatomical and biological changes in the diabetic lung are similar to those described in the aging lung (14, 15), suggesting that mechanisms associated with premature aging may contribute to diabetic pulmonary injuries.
Mammalian Sirtuins, a family of adenine dinucleotide (NAD+)-dependent histone deacetylases, play important roles in age-related diseases including T2DM (16, 17). According to previous studies, Sirtuins are speculated to act on all of the known mechanisms underlying diabetic pulmonary injuries. However, few articles refer to the roles of Sirtuins in pulmonary pathophysiology, let alone in diabetic pulmonary injuries.
The circadian clock system drives mammalian intrinsic biological oscillations with a period of near 24 h (18). Current studies highlight the critical role of the circadian clock system in regulating cellular processes such as metabolism, oxidative stress, inflammation, cellular proliferation, and senescence (19, 20). Disrupted circadian rhythms are common in patients with chronic airway diseases and may trigger cellular senescence, especially among tobacco smokers, and disturb inflammatory responses in the lungs of COPD patients (21). Sirtuins regulate both the circadian clock in the brain as well as in peripheral tissues, including the lungs. Thus, it is rational to hypothesize that Sirtuins affect diabetic pulmonary injuries by interacting with the circadian clock system.
In this review, we provide a general view of the regulatory effects of Sirtuins and the circadian clock system in pulmonary pathophysiology and diabetic pulmonary injuries. Moreover, we focus on the interactions of Sirtuins and the circadian clock system to provide new ideas for viewing diabetes complications in the lung and to provide novel targets for therapies.
The Lung Is a Diabetic Target Organ
Histological Changes in the Diabetic Lung
Microangiopathy is a well-known diabetic complication involving the retina, kidney and peripheral or autonomic nervous system. A study comparing the histological changes in the lungs of diabetic patients showed significantly increased thickness of alveolar epithelial basal lamina (BL), endothelial capillary BL, and both fused BL (22). Moreover, researchers found the same thickening magnitude of BL in the lung and kidney in this study. The clinical findings of diabetic pulmonary microangiopathy have also been demonstrated in a streptozotocin (STZ)-induced diabetic rat model (23). In addition to microangiopathy, the increased glycosylation of insoluble collagen in human lung parenchyma found in young diabetic patients is similar to that in non-diabetic aged individuals (24). This phenomenon has also been reported in STZ-induced diabetic rats (25). These findings show accelerated aging in the diabetic lung. Data from a retrospective longitudinal cohort study shows that lung fibrosis is significantly enhanced in diabetic subjects (26). Lung fibrosis is representative of an important cause of premature mortality in patients.
Function Abnormalities in the Diabetic Lung
Lung function mainly consists of pulmonary diffusion function and pulmonary mechanical function. Pulmonary diffusing capacity for carbon monoxide (DLCO) was reported to be reduced among diabetes patients compared with healthy subjects (27). The reduction in DLCO in diabetic patients is parallel to the severity of retinopathy and nephropathy (15). Moreover, glycemic control increases DLCO (28). Pulmonary mechanical function is reflected by several parameters, including forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC), and total lung capacity (TLC). An increasing number of recent studies support the notion that there is a correlation between diabetes and decreased pulmonary mechanical function (28–30). The meta-analysis carried by Klein et al., showed a decline in lung function, including FEV1, FVC, and DLCO, in diabetic patients compared with healthy subjects (31). The reduction in lung function is negatively correlated with blood glucose level and the duration and severity of diabetes (31). In four longitudinal studies, two studies demonstrated a significant decline in lung function in diabetic patients compared with healthy individuals, and two other studies showed lower FEV1 and FVC in patients before diabetes onset than in subjects who did not develop diabetes (31).
The bronchomotor tone (15, 32–34), the chemosensibility to hypoxia (35, 36) and the respiratory muscle strength (37–40) are damaged in diabetic patients. On the basis of histological and functional changes in the lungs of diabetic patients and animals, it can be concluded that the lung is a definite diabetic target organ (Figure 1).
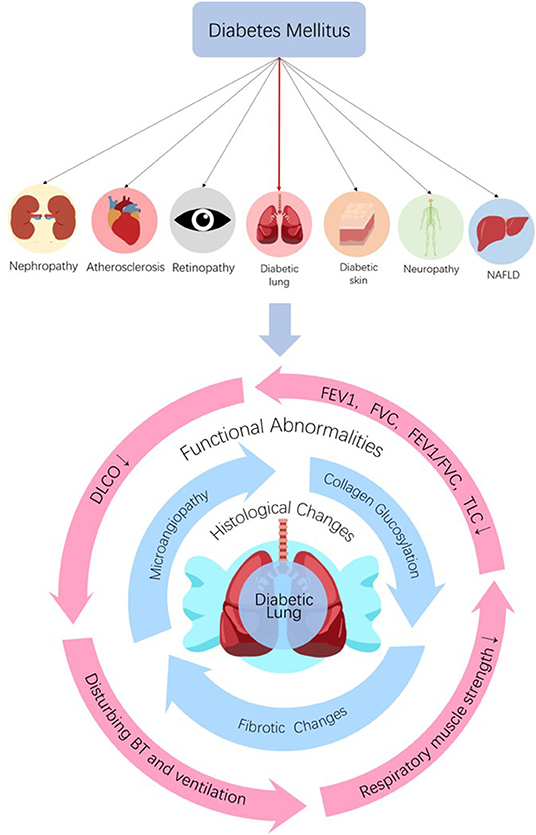
Figure 1. The lung is a diabetic target organ. Diabetes can induce tissue injuries in target organs including nephropathy, retinopathy, neuropathy, NAFLD, dermopathy, and atherosclerosis in coronary arteries, cerebrovascular and peripheral vascular systems. Despite the subclinical characteristic of the lung, diabetes induces pulmonary structural and functional abnormities, so the lung is a diabetic target organ. Diabetes can induce histological and functional changes to the lung. Histological changes, such as microangiopathy, fibrotic changes, and accelerated collagen aging, can be seen in diabetic lungs. Impaired pulmonary functions such as reductions in FEV1, FVC, FEV1/FVC, TLC, and DLCO, abnormal bronchomotor tone, decreased ventilatory response to hypercapnia and reduction in the strength of respiratory muscle have been implicated in the diabetic lung. NAFLD, non-alcoholic fatty liver disease; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; TLC, total lung capacity; DLCO, diffusing capacity for carbon monoxide; BT, bronchomotor tone.
Molecular Mechanisms Underlying Diabetic Pulmonary Injuries
Mechanisms underlying diabetic pulmonary injuries remain unclear; however, advanced glycosylation end-products (AGEs), oxidative stress and inflammation, endothelial dysfunction, and hypercoagulation convincingly contribute to pulmonary injuries, which have well-summarized in other reviews (15, 41–43). All of the above-mentioned pathogenic factors are regulated in a circadian manner. Patients with chronic airway diseases, including COPD and asthma, develop more frequently, and worsen mostly in the evening or early morning (44–46). Accumulating evidence shows that circadian rhythm regulation is upstream of known mechanisms. Next, we will discuss the mechanisms underlying diabetic pulmonary injuries in the context of circadian regulation (Figure 2).
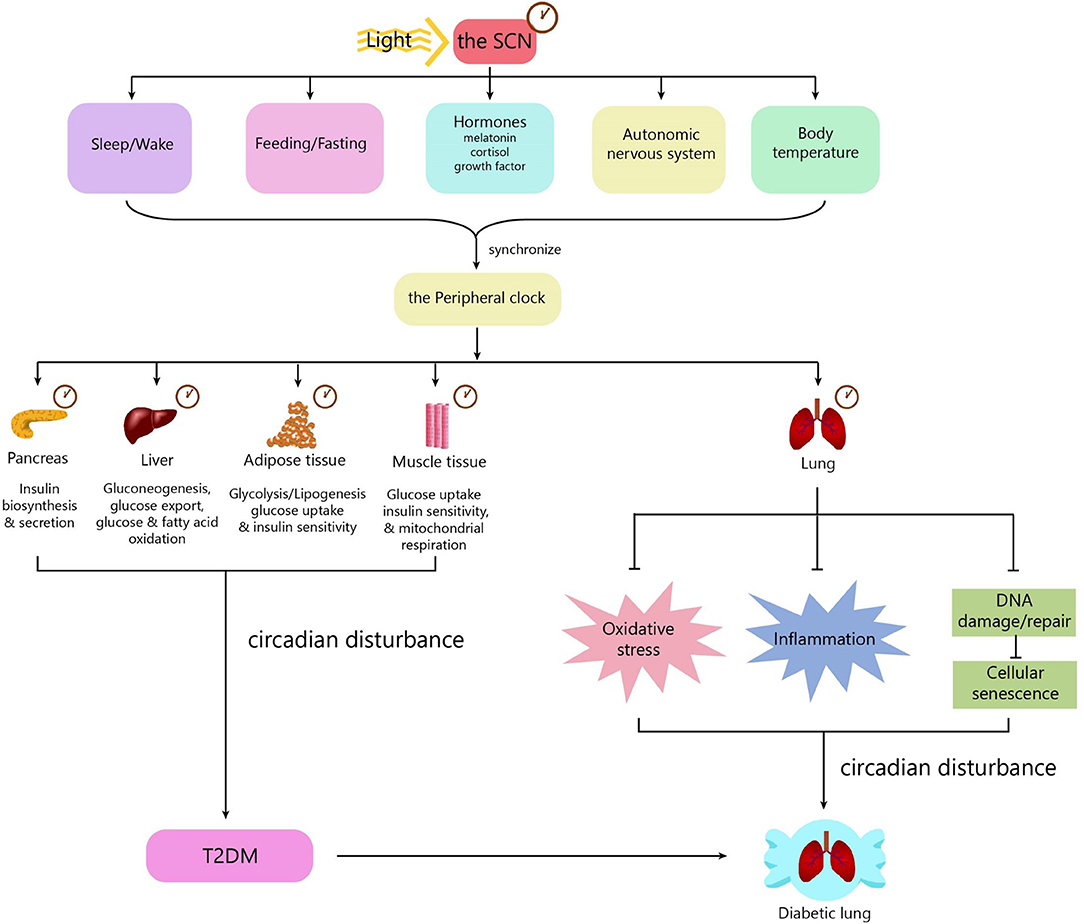
Figure 2. The role of the circadian clock in the diabetic lung. The circadian clock system consists of a central clock located in the SCN and peripheral clocks located in the pancreas, muscle, liver, adipose tissue, gut, kidney, and lung. Light is the most important zeitgeber for the SCN. The SCN synchronizes the peripheral clocks through signals including biological behaviors, hormones, the autonomic nervous system, and body temperature. The central clock and peripheral clocks regulate insulin secretion, insulin sensitivity, lipid, and glucose metabolism. Circadian disturbance contributes to the development of T2DM. The peripheral clock in the lung can influence some pathophysiological processes, including oxidative stress, inflammation, and cellular senescence. Disturbance of the circadian clock in the lung promotes the development of diabetic lung. SCN, suprachiasmatic nucleus; T2DM, type 2 diabetes.
Circadian Regulation in the Diabetic Lung
The Circadian Clock System
In mammals, the circadian clock system consists of a central clock located in the hypothalamic suprachiasmatic nucleus (SCN) and peripheral clocks, which drive 24 h rhythms of physiology and behavior. The SCN clock is set mainly by environmental light and then sends the entrained timing signal to peripheral clocks via neural signals, hormonal signals, and body temperature. At the cellular level, the circadian rhythms are generated by clock genes. The core clock genes include Bmal1 and Clock encoding activators, period genes (Per1-3) and cryptochrome genes (Cry1-2) encoding repressors and genes encoding the nuclear receptors REV-ERB (NR1D1 and NR1D2) and ROR (Rorα, Rorβ, and Rorγ).
Clock genes forms a transcriptional autoregulatory feedback loop. The BMAL1/CLOCK heterodimer translocate to the nucleus and transcriptionally activate expression of the core clock genes including Per1-3, Cry1-2, and nuclear receptors Rev-erbα and Rorα. Conversely, once PER and CRY accumulate to a certain level, they form heterodimer and translocate back to the nucleus to block transcriptional activity of the BMAL1/CLOCK complex and ultimately repress their own transcription. REV-ERB and ROR drive the rhythmic expression of BMAL1 and CLOCK via competitively binding to the REV-ERB/ROR binding site, thus repressing or activating transcription of Bmal1 and Clock, respectively (47, 48). In addition, posttranslational modifications have been established to regulate clock gene expression. For instance, SIRT1 binds BMAL1/CLOCK and promotes deacetylation and degradation of PER2 (49). Phosphorylation of PER2, mediated by casein kinase Iε, recruits the ubiquitin ligase adaptor protein β-TrCP and leads to polyubiquitination and proteasome-mediated degradation of PER2 (50). CRY binds PER2 and prevents its nuclear export, thus preventing the ubiquitylation and subsequent degradation of PER2 (51). Likewise, PER2 can prevent the ubiquitylation and subsequent degradation of CRY (51). CLOCK can induce sumoylation of BMAL1 at Lys259 and control BMAL1 stability (52). Both BMAL1 and CLOCK undergo phosphorylation during the circadian cycle (53), which is coupled to nuclear translocation and the subsequent degradation of CLOCK (54). CLOCK has intrinsic histone acetyltransferase activity and can acetylate BMAL1 on the Lys537 residue, which facilitates the recruitment of CRY1 to the BMAL1/CLOCK complex, resulting in transcription repression (55).
These core clock genes function not only as active or repressive components of a cell-autonomous clock but also as regulators of clock-controlled genes (CCGs). Mechanistically, the core clock genes interact with chromatin-modifying complexes, co-activators and co-repressors to regulate CCG expression. The BMAL1/CLOCK complex drives the expression of numerous CCGs, thus regulating a series of biological processes, including metabolism. At the beginning of transcription, the BMAL1/CLOCK complex interacts with chromatin and recruits chromatin-modifying complexes such as histone acetyltransferases P300 and CBP (56), histone deacetylases SIRT1 (57, 58) and SIRT6 (59), methyltransferases MLL1 (60) and MLL3 (61), and histone lysine demethylases JARID1a and LSD1 (62) to promote chromatin accessibility and activate CCG transcription. The PER-CRY repressor complex translocate to the nucleus and recruits a series of co-repressors to block BMAL1/CLOCK complex activity (48). In general, in the circadian cycle, transcriptional activation and repression of rhythmic genes involve dynamic chromatin epigenetic transition. In addition, transcription factors, including NF-κB, nuclear receptor hepatocyte nuclear factor 4A (HNF4A) and USF1, can compete with the BMAL1/CLOCK complex for binding to target genes and repress the transcriptional activity of the BMAL1/CLOCK complex (63–65). MYC can inhibit the expression and oscillation of BMAL1 by inducing REV-ERBα expression (66). Furthermore, REV-ERBα can recruit the N-CoR/HDAC3 co-repressor to regulate the expression of some metabolic genes (67). Conversely, transcription factors, including PDX1 and HIF1α, act synergistically with BMAL1 to activate target gene expression (68, 69).
The Role of the Circadian System in Diabetes
As described in a series of reviews, the circadian clock system plays a pivotal role in regulating energy metabolism and maintaining energy homeostasis. The SCN clock drives sleep-wake and feeding-fasting cycles and functions as the basic biological clock of metabolism. Except for tuning by the SCN, peripheral clocks can be set by feeding as well, having autonomic circadian oscillators in their respective tissues and contributing to metabolic processes. Insulin resistance and pancreatic β cell dysfunction are critical pathophysiological processes in the development of T2DM. Glucose metabolism and insulin secretion occur in a circadian manner. Internal circadian system dysfunction induces insulin resistance and glucose intolerance. The roles of the SCN clock and peripheral clocks located in different tissues in insulin secretion, insulin sensitivity and glucose metabolism regulation will be discussed individually.
First, the SCN clock controls the sleep-wake cycle as well as rhythmic feeding behavior, which is critical in determining organism nutritional status and in the development of diabetes. The SCN dives the rhythmic release of several hormones that affect the secretion and/or action of insulin. For instance, melatonin, a hormone synthesized by the pineal gland at night, is orchestrated by output from the SCN and coordinates circadian activity in turn by regulating clock gene expression (70). Furthermore, melatonin exerts its function through two specific receptors, MT1 and MT2, in different peripheral tissues. Both of these receptors are present in human islets. The protective roles of melatonin in maintaining glucose homeostasis and suppressing insulin resistance and T2DM have been described in substantial human studies. For instance, lower nocturnal melatonin secretion is linked with increased insulin resistance in non-diabetic individuals and is an independent risk factor for developing T2DM (71). Notably, diabetic patients mostly lack circadian melatonin rhythm (72). Specific single nucleotide polymorphisms of MT2 are related to higher fasting glucose levels and HbA1c (73, 74). Further, loss-of-function mutations of MT2 are associated with the highest incidence of T2DM (75). In addition to regulating insulin secretion, melatonin has other functions, such as stimulating antioxidant enzymes (76) and attenuating the production of proinflammatory cytokines in high-fat diet (HFD)-induced insulin-resistant rats (77), suppressing mitochondrial dysfunction in diabetic rats (78), reducing cortisol secretion (75), and regulating glucose metabolism in adipocytes (79), skeletal muscle cells (80), and hepatocytes (81). In addition, the SCN affects the production and release of cortisol via regulating the activity of the hypothalamic-pituitary-adrenal axis (HPA) (82). Endogenous hypercortisolism can cause pancreatic β cell dysfunction and induce insulin resistance in the liver, adipose tissue and skeletal muscle (83). Disorder of circadian rhythm caused by obstructive sleep apnea causes HPA hyperactivity, contributing to insulin resistance (84). The SCN regulates the diurnal rhythm of growth hormone, which exerts anabolic effects and favors body composition and physical fitness (85). Moreover, the SCN is responsible for circadian regulation of energy expenditure for thermogenesis (82).
Convincingly, pancreatic β cell dysfunction contributes to T2DM. As mentioned above, insulin from rodents (86) and human (87, 88) islet cells is secreted in a circadian manner. Insulin secretion lacking a rhythmic release pattern has been observed in T2DM patients (89). The pancreatic clock is synchronized to the light-dark cycle by the SCN via signals such as melatonin, cortisol and body temperature (86–88). Pancreatic islet cells in mice have self-sustained clock genes and protein oscillations of BMAL1 and CLOCK, which act with co-activator PDX1 to activate the transcription of genes involved in insulin biosynthesis, transport and secretion (68). Moreover, specific ablation of these clock components disrupted insulin secretion leading to diabetes in mice (90). Saini et al. reported that circadian clock disruption via small interfering RNA perturbed insulin secretion in human pancreatic islet cells (88).
Adipose tissue, liver and skeletal muscle are important insulin target organs responsible for energy metabolism, and insulin resistance in these organs contributes to the development of T2DM. These organs have autonomous circadian clocks that are synchronized by the SCN (91) and signals from food intake (92–96). Misalignment of the peripheral clocks in these organs by disruption of the normal fasting-feeding cycle contributes to the development of diabetes in HFD-fed mice (97). Furthermore, germ-line Bmal1 disruption mice exhibit increased total fat content, glucose intolerance comparable to mice lacking protein kinase Akt2 (98) as well as reduced insulin production after refeeding following overnight fasting (99). Adipocytes from humans present rhythmic glucose uptake due to an intrinsic diurnal rhythm in insulin sensitivity (96), which has been mechanistically demonstrated to be associated with circadian regulation of retinol-binding protein receptor STRA6 (100). The circadian clock regulates the expression of key enzymes involved in lipolysis (101) and lipogenesis (102), and disruption of the clock promotes triglyceride accumulation in white adipose tissue (101). As the liver plays a pivotal role in maintaining blood glucose homeostasis by regulating glycogenolysis, glycogenesis, and gluconeogenesis, it is strongly affected by the fasting-feeding cycle. Abundant genes in the liver responsible for glucose metabolism exhibit circadian regulation (103). Liver-specific Bmal1 knockout (KO) mice showed abnormalities in both glucose storage and production resulting from disturbed expression of CCGs, including Glut2, GCK, Pepck2, and L-PK, which confirms the essential role of the liver clock in maintaining euglycemia (99). Human muscle exhibits diurnal rhythms in mitochondrial oxidative capacity and insulin sensitivity (104, 105). Muscle-specific Bmal1 KO mice showed insulin resistance and impaired glucose metabolism in skeletal muscles (106). The BMAL1/CLOCK complex regulates the expression and membrane translocation of the insulin-sensitive glucose transporter GLUT4 and affects pyruvate dehydrogenase (PDH) activity by regulating the expression of PDH regulators, including Pdp1 and Pdk4, ultimately impacting glucose oxidation (106). In addition, the BMAL1/CLOCK complex improves insulin sensitivity through the upregulation of SIRT1 expression in cultured C2C12 myotubes and mouse skeletal muscle (107). Recently, the muscle clock was reported to regulate insulin sensitivity and glucose utilization by affecting genomic recruitment of HDAC3 and subsequently disturbing the expression of metabolic genes (108).
The Role of the Circadian System in the Lung
As early as two decades ago, researchers observed clock gene expression in the lungs of rats (109). Later, the link between circadian rhythm and lung pathophysiology was well-documented. For example, patients with asthma show a circadian rhythm in the bronchial response to challenges such as cold dry air, dust mite, histamine, etc. (110). Nocturnal worsening in lung function in asthma has been linked to diurnal alterations of inflammation and airway narrowing (111). Core clock genes are expressed strongly in Clara cells lining the bronchioles, and these cells are critical for maintaining circadian oscillations in both mouse and human lung tissue (112). Subsequent studies declared that environmental factors such as air pollutants, cigarette smoke (CS), allergens, pathogens, jet lag, and shift work can disturb molecular clock function in the lung and lead to exacerbations of chronic lung diseases, including COPD, lung fibrosis and asthma (113–120).
The Role of the Circadian Clock in Pulmonary Redox Regulation
A growing body of evidence indicates that the molecular clock regulates redox in multiple tissues. For instance, global Bmal1 KO mice showed significant ROS accumulation in the kidney, heart, brain, and spleen compared to wild-type mice, indicating that BMAL1 controls ROS homeostasis (121). These Bmal1−/− mice showed reduced expression of redox genes, including Aldh2 encoding ALDH2, which scavenges reactive aldehydes generated during mitochondrial respiration, and Nqo1 encodes NADPH dehydrogenase, which decreases toxic quinones and ultimately increases lipid peroxidation in the brain, promoting neurodegeneration (122). Bmal1 depletion in macrophages reduced the NRF2 response to LPS challenge, resulting in ROS accumulation and production of the proinflammatory factors IL-1β and IL-6 (123). Furthermore, Bmal1 depletion predisposes pancreatic β cells to oxidative-induced β cell dysfunction, generating a diabetic phenotype in mice (124). In humans, impaired redox balance has been associated with several chronic pulmonary diseases, including COPD, lung fibrosis, asthma, and lung cancer (125–128), which contributes to diabetic pulmonary dysfunction, as previously mentioned. The critical role of NRF2 in cellular antioxidant defense has been well-documented in a substantial number of studies. In response to oxidative stress, NRF2 translocates to the nucleus and induces the expression of a series of antioxidant genes, including glutathione cysteine ligase (GCL), glutathione S-transferase (GST), and haeme oxygenase 1 (HMOX1) (129, 130). In ClockΔ19 mice expressing a dominant negative mutation of the CLOCK protein, NRF2 expression in the lung is constitutively low and arrhythmic and is accompanied by reduced glutathione levels, increased markers of oxidative damage and fibrotic phenotype (131). Several antioxidant enzymes, including SODs and GPxs, are under transcriptional control of PPARs (132–137). Interestingly, PPARα is involved in a positive regulatory feedback loop with BMAL1 in rodent liver circadian clock (138). PER2 interacts with PPARγ directly and represses its activity (139). These findings suggest that the circadian clock may exert an antioxidant role by regulating PPARs. In addition, the circadian clock has been shown to drive NAD+ oscillations and control mitochondrial oxidative metabolism (140). REV-ERBα is reported to improve skeletal muscle oxidative capacity by reducing mitochondrial autophagy and biogenesis (141). As mitochondrial dysfunction is the major cause of ROS production, the circadian clock is supposed to protect against oxidative damage to the diabetic lung via regulating mitochondrial function.
The Role of the Circadian Clock in the Pulmonary Inflammatory Response
The molecular clock powerfully regulates the inflammatory response (142, 143). Global Bmal1 KO mice display significantly increased expression of proinflammatory cytokines, including TNF-α, COX2 and prostaglandin synthase gene (ptgs2), in the brain, suggesting that molecular clock disruption is directly related to the inflammatory response (122). Myeloid cell-specific Bmal1 depletion disrupts rhythmic mobilization of Ly6Chi monocytes and fortifies inflammatory responses, potentiating metabolic inflammation, and predisposing experimental animals to insulin resistance and metabolic dysfunction (144). Mechanistically, the BMAL1/CLOCK complex recruits polycomb repressive complex 2 (PRC2) to chemokine gene promoters, such as Ccl2, Ccl8, and S100a8, and silences expression of these CCGs in monocytes and macrophages (144). The BMAL1/CLOCK complex induces NRF2 expression via binding the E-box sites in the Nrf2 promoter, which contributes to suppressing the inflammatory response of macrophages (123). NRF2 further inhibits proinflammatory cytokine IL-6 and IL-1β expression by reducing ROS levels (123) as well as by inhibiting the recruitment of RNA polymerase II to the transcription start sites (TSSs) of IL-6 and IL-1β (145). BMAL1 can exert an anti-inflammatory effect by recruiting glucocorticoid receptors to promoters of proinflammatory cytokines such as CXCL5 (116). Bronchiole-specific Bmal1 depletion enhances CXCL5 expression, driving pulmonary neutrophil recruitment and augmenting pulmonary inflammation and responses to pathogen (116). Deletion of Bmal1 in pulmonary airway epithelial cells increases neutrophil infiltration in mouse lungs, alters lung mechanic functions and impairs influenza defense (146). An HFD has been shown to induce insulin resistance partly via activation of the NF-κB signaling pathway (147). CLOCK interacts with the p65 subunit of NF-κB and enhances NF-κB-dependent transcription; however, BMAL1 counteracts NF-κB activation by sequestering CLOCK (148). Positively regulated by BMAL1, the nuclear receptor REV-ERBα has been demonstrated to be an important intermediary molecule linking the core clock and inflammatory pathways in macrophages. As a transcriptional repressor, REV-ERBα inhibits IL-6 and Ccl2 production by direct DNA binding to their promoters (149, 150). REV-ERBs repress macrophage genes, including Mmp9 and Cx3cr1 expression, by inhibiting the function of distal enhancers selected by macrophage lineage-determining factors (151). Deletion of Rev-erbα in bronchial epithelia exaggerates pulmonary inflammation (152). Inflammatory stimuli can induce REV-ERBα protein degradation, which can be blocked by its inverse agonist GSK1362 (152). Deletion of Cry releases its inhibition of cAMP production and leads to constitutive activation of PKA, which results in activation of NF-κB via phosphorylation of the p65 subunit and subsequent induction of proinflammatory cytokines (153).
In recent years, Although the role of the circadian clock has been well-documented in the development of insulin resistance and T2DM via the regulation of oxidative stress, inflammation and energy metabolism, far less is known about circadian regulation in the diabetic lung. SIRT1, a well-known member of Sirtuins, has been well-demonstrated to bridge metabolism and circadian rhythms, and together with other family members, SIRT1 plays a powerful role in metabolic homeostasis, oxidative stress, inflammation, and aging. Next, we will retrospectively examine the roles of Sirtuins in diabetic pulmonary injuries and discuss the interactions of Sirtuins with circadian clocks in this context.
The Circadian Roles of Sirtuins in the Lung
The mammalian Sirtuins are a family of NAD+-dependent deacetylases consisting of seven members (SIRT1-SIRT7) (154). These seven members display distinct subcellular localization and biological functions (17). Sirtuin family members regulate a wide variety of cellular processes and affect a series of diseases such as obesity, insulin resistance, T2DM, cardiovascular disease, cancer, and aging (155, 156). Sirtuins are supposed to prevent diabetic lung through attenuating diabetes. Roles of Sirtuins in insulin resistance, NAFLD and T2DM etc. have been summarized in our and other reviews (17, 155). Here, we will focus on the interaction of Sirtuins and the circadian clock (Figure 3) and discuss their possible role in diabetic pulmonary injuries (Figure 4).
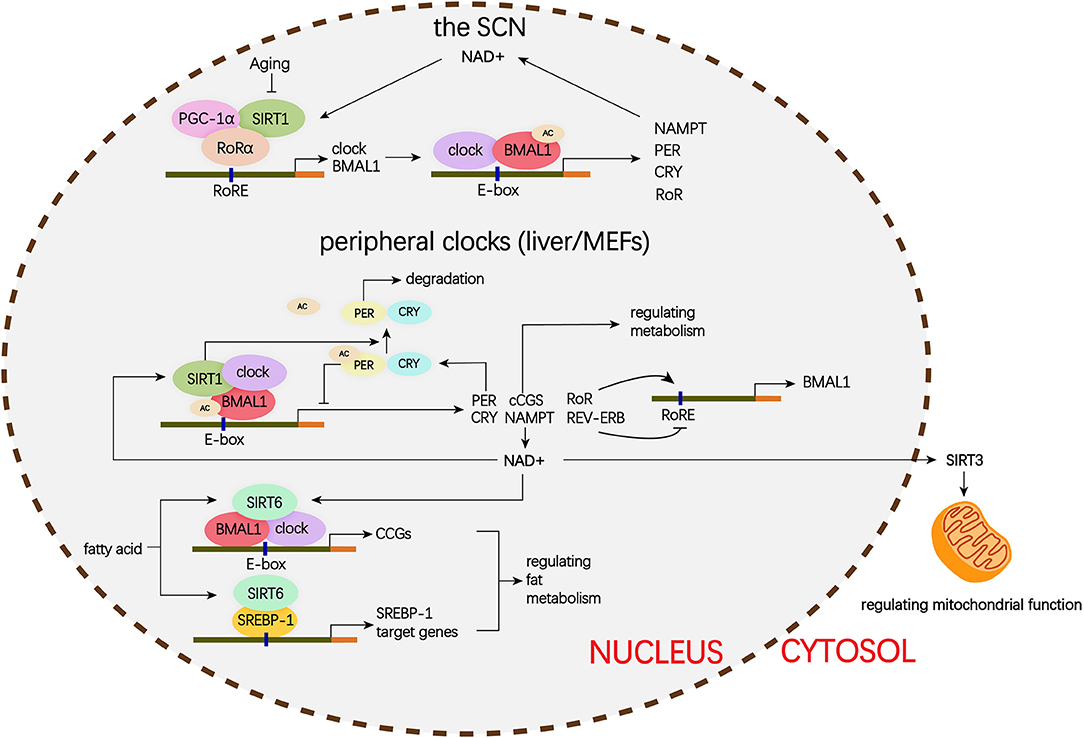
Figure 3. Schematic representation of the role of Siruins in clock regulation. Clock genes forms a transcriptional autoregulatory feedback loop. The BMAL1/CLOCK heterodimer transcriptionally activate expression of the core clock genes including Per1-3, Cry1-2, and nuclear receptors Rev-erbα and Rorα. Conversely, once PER and CRY accumulate to a certain level, they form complex with BMAL1/CLOCK and block transcriptional activity of the BMAL1/CLOCK complex and ultimately repress their own transcription. In addition, REV-ERB represses and ROR activates BMAL1 transcription. (1) In the SCN, the binding of SIRT1 and PGC-1α to the Bmal1 promoter containing RORE through a synergistic action with RORα amplifies circadian gene expression, thereby, brain SIRT1 prevents circadian function decline with aging. (2) In the peripheral clock located in the liver and MEFs, there is a new negative feedback loop. BMAL1/CLOCK positively regulates the transcription of NAMPT, the rate-limiting enzyme in mammalian NAD+ biosynthesis. NAD+ is a classic co-enzyme for Sirtuins. Conversely, SIRT1 deacetylates BMAL1 and represses BMAL1/CLOCK activity. In addition, SIRT1 binds to BMAL1/CLOCK complex and promotes the deacetylation and degradation of PER2. SIRT6 controls different circadian genome subdomains from SIRT1. SIRT6 interacts with BMAL1/CLOCK complex and controls their chromatin recruitment to CCGS promoter. Meanwhile, SIRT6 governs circadian chromatin recruitment of SREBP-1. The core clock regulates circadian oscillation of SIRT3 activity by driving changes in NAD+ levels. MEFs, mouse embryonic fibroblasts; PGC-1α, peroxisome proliferator-activated receptor gamma co-activator 1-alpha; BMAL1, brain, and muscle aryl hydrocarbon receptor nuclear translocator-like 1; CLOCK, circadian locomotor output cycles protein kaput; PER2, period 2; SREBP-1, sterol regulatory element-binding protein 1; CCGs, clock-controlled genes; mTOR, mechanistic target of rapamycin; IGF, insulin-like growth factor; NF-κB, nuclear factor κB; mtDNA, mitochondrial DNA; TGFβ1, transforming growth factor β1; NAD, nicotinamide adenine dinucleotide; NAMPT, nicotinamide phosphoribosyl transferase; FOXO3, forkhead protein box 3; E-box, enhancer box; RORα, retinoic acid receptor– related orphan receptor a; RORE, retinoic acid receptor–related orphan receptor response element.
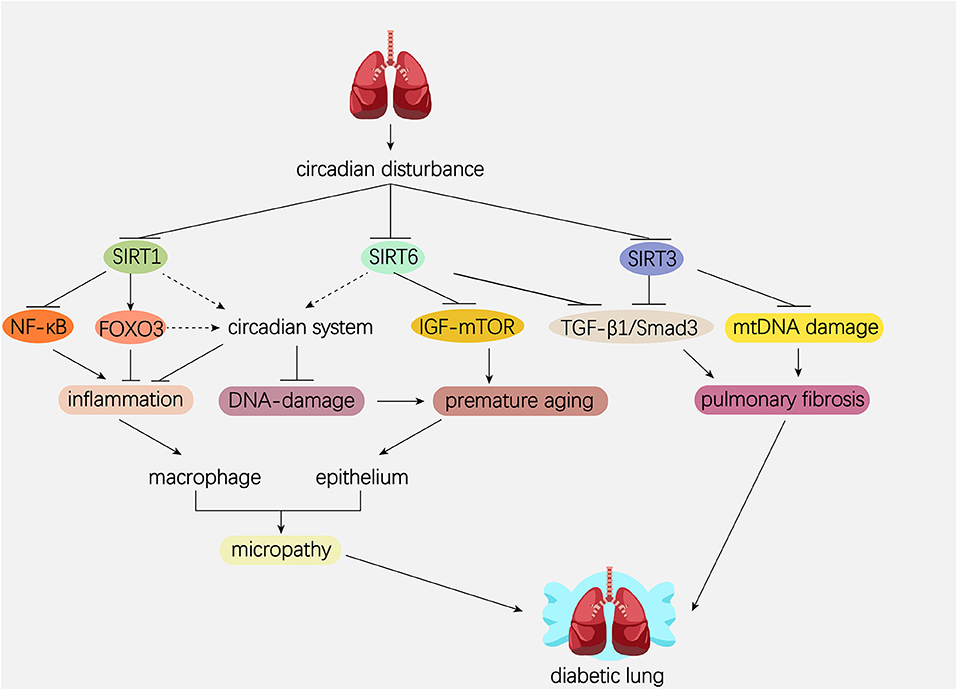
Figure 4. Hypothetical model showing the possible role of Sirtuins and circadian system in the pathogenesis of diabetic lung. Dotted line represents the pathway needed to be elucidated. Solid line represents the pathway which has been proved in former studies. Circadian disturbance decreases deacetylase activity of Sirtuins via impairing NAD+ oscillation. SIRT1 inhibits pulmonary inflammation through regulating circadian clock or through regulating transcriptional factors such as NF-κB and FOXO3. SIRT1 and SIRT6 rescue pulmonary cells from premature aging via regulating the DNA damage response synergistically with circadian clock. In addition, SIRT6 prevents premature aging by attenuating the IGF-mTOR signaling pathway. Both SIRT3 and SIRT6 inhibit pulmonary fibrosis by inactivating the TGFβ1/Smad3 signaling pathway. SIRT3 inhibits pulmonary fibrosis by maintaining mtDNA stability. NAD, nicotinamide adenine dinucleotide; NF-κB, nuclear factor κB; FOXO3, forkhead protein box 3; mTOR, mechanistic target of rapamycin; IGF, insulin-like growth factor; TGFβ1, transforming growth factor β1; mtDNA, mitochondrial DNA.
The Interaction of SIRT1 and the Circadian Clock in Diabetes and Pathogenesis of the Diabetic Lung
The Role of SIRT1 in the Central Clock
Sirt1 mRNA is highly expressed in the hypothalamus (157), which is an important metabolism-relevant region as well as the location of the central clock. Loss of brain SIRT1 activity in mice results in an abnormal extension of the intrinsic period and an inability to reset a new light-dark regimen (158). Expression of core clock genes, including BMAL1, CLOCK, and PER2, in the SCN significantly decreases in brain-specific Sirt1 knockout (BSKO) mice and increases in brain Sirt1 transgenic mice (BSTG) (158). Mechanistically, in N2a neuroblastoma cells, SIRT1 and PGC-1α bind cooperatively to the Bmal1 promoter, driving its expression and enhancing the amplitude of circadian machinery (158). In addition, SIRT1 in the ventromedial hypothalamus (VMH) has been shown to function as a metabolic sensor by sending nutritional information to the SCN via efferent signals and synchronizing the central clock to feeding conditions (159). Sirt1 gene ablation in the Sf1 neurons of the VMH perturbs the activity and circadian gene expression in the SCN and subsequently disrupts the connection of food intake and circadian behavior (159). These studies support the crucial role of SIRT1 in linking metabolism with the central pacemaker.
The Role of SIRT1 in Peripheral Clocks
Studies on the interactions of SIRT1 and peripheral clocks have mainly been carried out in mouse liver cells and embryo fibroblasts (MEFs). First, SIRT1 is expressed in a circadian manner in mouse liver cells, MEFs, and NIH 3T3 cells and is essential for augmenting the expression of several core clock genes, such as Bmal1, Rorγ, Per2, and Cry1 (49). This study also reported that SIRT1 binding to the BMAL1/CLOCK complex rhythmically drove the deacetylation and degradation of the PER2 protein (49). A secondary study revealed that SIRT1 deacetylase activity rather than transcript and protein levels was regulated in a circadian manner in mouse liver cells and MEFs (57). SIRT1 associates with CLOCK and is then recruited to the BMAL1/CLOCK complex at the CCG promoters, where it is responsible for the rhythmic deacetylation of H3 Lys9/Lys14 and BMAL1 at Lys537, counteracting the acetyltransferase activity of CLOCK (160), and prevents the transcriptional activating effect of the BMAL1/CLOCK complex (57). This study revealed the crucial role of SIRT1 in acting as the molecular rheostat of CLOCK. Subsequent studies declared that the SIRT1 activity rhythm was generated from the oscillation of intracellular NAD+ levels, which are regulated by NAMPT, the rate-limiting enzyme in the NAD+ salvage pathway (58, 161). NAMPT synthesis is positively regulated by the BMAL1/CLOCK complex, and SIRT1 activation due to NAMPT-mediated NAD+ biosynthesis, in turn, suppresses BMAL1/CLOCK complex activity, forming a circadian clock feedback loop (58, 161). In addition to histone acetylation, circadian transcription is associated with MLL1-mediated H3K4 trimethylation. MLL1-dependent H3K4me3 on the CCG promoter favors the recruitment of the BMAL1/CLOCK complex and activates CCG transcription (60). MLL1 exhibits rhythmic acetylation of two conserved lysine residues, K1130 and K1133, and SIRT1 deacetylates these two lysine residues of MLL1, inhibiting its methyltransferase activity (162). Some studies demonstrated other interactions of SIRT1 and clock genes. For instance, BMAL1/CLOCK regulates mouse hepatocyte insulin sensitivity via circadian regulation of the expression of SIRT1 (163). Similarly, BMAL1/CLOCK regulates muscle insulin sensitivity through circadian regulation of SIRT1 expression (107). PER2 negatively regulates mouse hepatocyte SIRT1 expression by binding to the BMAL1/CLOCK binding E-box sites in the Sirt1 promoter in vivo and in vitro. In turn, Sirt1 deficiency leads to increased acetylation of H4K16 in the Per2 promoter and subsequent transcriptional activation of Per2, which results in misalignment of the circadian rhythm in the liver (164).
SIRT1 Regulates the Circadian Clock in Response to Inflammation in the Lung
SIRT1 protein levels and activity decrease in macrophages, lung epithelium and peripheral lung tissues of smokers and COPD patients, leading to increased acetylation of RelA/p65 and subsequent activation of the NF-κB pathway (165). SIRT1 activity in peripheral blood mononuclear cells is positively associated to lung function of COPD patients (166). Similar results were observed in the lungs of rats and mice exposed to CS (167, 168). BMAL1 levels also decrease in the lungs of COPD patients compared with non-smoking individuals (169). Acute and chronic CS exposure reduces the amplitude of core clock genes, especially BMAL1 (169). Moreover, lung epithelial-specific Bmal1 deletion mice show enhanced pulmonary inflammation in response to CS (169). However, SRT1720, a pharmacological activator of SIRT1, failed to inhibit CS-induced pulmonary inflammation in lung epithelial-specific Bmal1 deletion mice (169). These findings show a pivotal role of the SIRT1-BMAL1 pathway in regulating pulmonary inflammation in response to environmental stress (119, 169). Although SIRT1 may play an important role in regulating several pulmonary pathophysiological processes, including oxidative stress, inflammation, and endothelial dysfunction, which are involved in the development of the diabetic lung, studies about the direct effects of SIRT1 on the diabetic lung are absent.
Roles of SIRT1 and the Circadian Clock in Pulmonary Premature Aging
Interestingly, the anatomical and biological changes in the diabetic lung are similar to those described in the aging lung, which suggests that we can discuss the roles of SIRT1 and the circadian clock in the diabetic lung from the perspective of cellular senescence-induced organ dysfunction and aging.
CS is a well-known factor leading to rapid decline in lung function and increases cellular senescence in the lungs of COPD patients (167). Mechanistically, CS induces DNA damage and impairs double-strand break (DSB) repair (47, 170, 171). Persistent DNA damage in the lung causes stress-induced senescence (SIPS) and a senescence-associated secretory phenotype (SASP), as characterized in COPD (172, 173). As mentioned above, CS also induces molecular clock dysfunction in the lung. The molecular clock is established to play a critical role in regulating the cellular response to DNA damage (174, 175). Bmal1-deficient mice show age-related pathologies and increased levels of ROS in a series of organs (121). Circadian clock protein also mediates cellular DNA damage/repair responses by interacting with factors such as Ku70, Ku80 and ataxia telangiectasia mutated (ATM) (175–177). p21 is induced by p53 following DNA damage and is known as a prosenescence gene. The p21 gene is negatively regulated by REV-ERBα, which in turn is positively regulated by the CLOCK/BMAL1 complex (178). Therefore, Bmal1 deficiency results in aberrant p21 expression and decreased hepatocyte proliferation (178). In addition, PER1 inhibits p21 expression and interacts with the checkpoint proteins ATM and Chk2, leading to significant growth reduction and sensitizing human cancer cells to DNA damage-induced apoptosis (179). These studies suggest that the circadian clock can influence cellular senescence by regulating the DNA damage response pathway.
In aged mice, levels of SIRT1 and core clock genes, including BMAL1, CLOCK, CYR1, and PER2, in the SCN decrease, leading to a longer intrinsic period and the inability to reset to a new light-dark regimen (158). Young BSKO mice have the same circadian changes as aged mice (158). SIRT1 and PGC-1α synergistically activate expression of the circadian activator Bmal1 in the SCN (158). This study suggests that SIRT1 links the clock with aging in the mammalian brain. In response to DNA damage, SIRT1 is recruited to DSBs and is required for efficient DSB repair and genomic stability; however, this process results in deregulation of genes causing aging (167, 180, 181). SIRT1 overexpression promotes survival in a mouse model of genomic instability and inhibits age-related transcriptional changes (167, 180, 181). Furthermore, SIRT1 activation by both overexpression and pharmacological activator SRT1720 can reduce cellular senescence via the SIRT1-FOXO3 axis and then attenuate emphysema (167). This protective role of SIRT1 in emphysema is not attributed to its effect on NF-κB-mediated inflammation (167). CLOCK is reported to be a transcriptional target of FOXO3, and FOXO3 knockdown dampens circadian amplitude in the mouse liver (182). Hence, it is rational that SIRT1 affects SIPS and SASP by regulating molecular clock directly or indirectly in the lung. In addition, SIRT6 deletion also decreases genomic stability via reducing base excision DNA repair and causes accelerating aging in mice (183). Accumulating evidence indicates that SIRT1 and SIRT6 play an important role in regulating the DNA damage response, maintaining genomic stability and defending against aging. However, the roles of SIRT1 and SIRT6 in circadian function related to DNA damage response and to SIPS and SASP in the lung need to be clarified.
The Role of SIRT3 in Diabetic Lung
In the STZ-induced diabetic rat model, decreased NADH/NAD+ redox imbalance, mitochondrial abnormalities, and decreased SIRT3 expression were present in the diabetic lung (184). Lungs from idiopathic pulmonary fibrosis patients show decreased SIRT3 activity, as indicated by acetylated mitochondrial SOD (MnSOD) levels, particularly in the lung epithelium. Sirt3 deletion promotes lung fibrosis by augmenting mitochondrial DNA (mtDNA) damage and apoptosis in mouse alveolar epithelial cells and myofibroblasts (185, 186). SIRT3 can prevent the fibrosis phenotype via inhibition of the TGFβ1/Smad3 signaling pathway (187, 188).
The Role of SIRT3 in Circadian Mitochondrial Functions
Mitochondria are the factory for metabolism and energy generation in the body. SIRT3 is localized in the mitochondria and plays important roles in regulating metabolism and ROS production, maintaining mtDNA integrity and preventing aging. Analysis of the liver acetylome from the Clock-deficient mouse revealed that a large number of mitochondrial proteins influenced by circadian acetylation are involved in amino acid and fatty acid metabolism, glycolysis and gluconeogenesis, and the citric acid cycle (189). The core clock regulates circadian oscillation of SIRT3 activity together with oxidative enzyme activity by driving changes in NAD+ levels. In ClockΔ19 mice, the rhythms of SIRT3 activity are disrupted in young mice (190). MEFs and livers from Bmal1-deficient mice show impaired mitochondrial function due to decreased fatty acid oxidation (FAO), glucose oxidation and NAD+ concentrations, whereas Cry1- and Cry2-deficient mice show the opposite trend (140). The low NAD+ concentrations in the livers of Bmal1 KO mice are correlated with impaired SIRT3 activity, resulting in enhanced protein acetylation and decreased enzymatic activity of SIRT3 targets, including OTC, MCAD, LCAD, MnSOD, and IDH2 (140). NAD+ supplementation with nicotinamide mononucleotide (NAN) restores SIRT3 activity and thereby mitochondrial oxidative capacity (140). A later study showed that both circadian and feeding rhythms coordinated the liver acetylome, including mitochondrial protein rhythmic acetylation, by impacting NAD+-dependent SIRT3 deacetylase activity (191). SIRT3 expression decreases in aged rats (192). As ROS are the main cause of aging and increased SIRT3 expression has been considered to contribute to human longevity, the circadian clock may influence cellular senescence and organic aging through SIRT3. In addition, SIRT3 interacts with Ku70 and deacetylates it, thus protecting cardiomyocytes from aging and stress-induced death (193). As mentioned above, the circadian clock protein also interacts with Ku70; hence, SIRT3 may be involved in circadian oscillator-mediated DNA damage/repair responses. Further studies on the circadian role of SIRT3 in the lung are needed.
SIRT6 and Partitioning Circadian Transcription
SIRT6 is uniquely located in the nucleus and constitutively binds to the chromatin (183, 194). The genome-wide occupancy of SIRT6 is mainly at TSSs of active genomic loci, which are also binding sites for serine 5 phosphorylated RNA polymerase II (195). The chromatin binding of SIRT6 is reported to be dynamic in response to stimuli (196, 197). SIRT6 deacetylates H3K9 and H3K56 in a NAD+-dependent manner, regulating gene expression, genome stability and telomere maintenance, thereby impacting metabolic diseases, heart disease and cancer (198, 199). DNA microarray analysis of liver-specific Sirt1 KO mice and liver-specific Sirt6 KO mice revealed that SIRT6 significantly regulates hepatic CCG expression, which is exclusive to CCGs regulated by SIRT1 (59). SIRT6 interacts with the BMAL1/CLOCK complex and is responsible for chromatin recruitment of the BMAL1/CLOCK complex to the promoter regions of CCGs (59). Furthermore, SIRT6 governs circadian SREBP-1 chromatin recruitment, leading to circadian regulation of genes such as Fasn that are implicated in fatty acid and lipid metabolism (59). SIRT6 inhibits pulmonary fibrosis by inactivating the TGFβ1/Smad3 signaling pathway (200). Lung-targeted Sirt6 delivery via injection of adeno-associated virus-Sirt6 attenuates bleomycin-induced pulmonary fibrosis (200). Moreover, SIRT6 can inhibit human bronchial cell (HBEC) senescence by inactivating the TGFβ1/Smad3 signaling pathway (201). SIRT6 induces apoptosis of HBECs by attenuating the IGF-Akt-mTOR signaling pathway, which contributes to the prevention of CSE-induced HBEC senescence (202). As CSE-induced HBEC senescence has been implicated in the pathogenesis of COPD, SIRT6 is supposed to be a protective factor in chronic airway diseases (202). Consistent with this hypothesis, SIRT6 levels are positively correlated with FEV1/FVC, and its expression in the lungs of COPD patients is decreased (202). SIRT6 is speculated to be a protective factor in the diabetic lung.
Of the seven members of the Sirtuin family, only SIRT1, SIRT3, and SIRT6 have been implicated directly in circadian clock regulation, but their roles in pulmonary pathophysiology and the interactions of these Sirtuins with circadian clock are largely unknown. The majority of Sirtuins are implicated in metabolic regulation, oxidative stress, inflammation, DNA damage/repair response, and telomere length regulation, which are mainly related to aging processes as well as lung disease. Circadian rhythms are intimately related to pulmonary pathophysiology. There should be an interaction of Sirtuins and the circadian clock in lung disease, especially in metabolism-related lung disease.
Conclusions and Future Directions
In summary, we gave a mechanistic perspective about the role of circadian clock and Sirtuins in diabetic lung based on the strengths of their roles in metabolic disturbance, oxidative stress, inflammation, and cellular DNA damage/repair responses. However, our understanding of the diabetic lung is still poor. Further studies are still needed to elucidate the following the questions. (1) Although we have summarized the potential interactions of the circadian clock and Sirtuins, the exact roles of these two systems underlying diabetic lung remain unknown. Clock genes or Sirtuins tissue-specific knock out or transgenic animals need to be used to evaluate the definite role of these molecules in diabetic lung. (2) With the development of high-throughput and epigenetic methodologies, more clearly molecular regulatory network will be identified. Therefore, more cell omic sequencing methods, which include single cell sequencing (scRNA-seq) can be used to analysis expression differences in different types of pulmonary cells and further elucidate roles of these cells in the development of diabetic lung. Moreover, assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) can be used to analyze epigenetic mechanisms of clock genes or Sirtuins. (3) Given the apparent roles of the circadian clock and Sirtuins in regulating a series of pathophysiologic processes and the subsequent demonstration of therapeutic value in animal models, the utility of natural or synthetic small molecules that can activate or inhibit one or more clock genes and Sirtuins would grow increasingly broader. Sirtuin-activating compounds (STACs), such as resveratrol, SRT1720 and SRT2183 (203), and synthetic REV-ERB and ROR ligands, such as GSK4112, SR9009, and SR9011 (204), have been well-documented in vivo. These small molecules provide potential treatment strategies for diabetic lung.
Author Contributions
SZ and H-ZC concepted the review. SZ is responsible for literature collection and article draft. Y-MD designed the figures. X-FZ and H-ZC revised the manuscript. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81600038, 31571193), National Key Research and Development Project of China (2019YFA0801500), and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS2017-I2M-1-008, 2019-RC-HL-006). H-ZC is also supported by the National Ten Thousand Talent Program and the Youth Yangtze River Scholar Program in China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the De-Pei Liu laboratory for helpful discussions.
References
1. Parappil A, Depczynski B, Collett P, Marks GB. Effect of comorbid diabetes on length of stay and risk of death in patients admitted with acute exacerbations of COPD. Respirology. (2010) 15:918–22. doi: 10.1111/j.1440-1843.2010.01781.x
2. Baker EH, Janaway CH, Philips BJ, Brennan AL, Baines DL, Wood DM, et al. Hyperglycaemia is associated with poor outcomes in patients admitted to hospital with acute exacerbations of chronic obstructive pulmonary disease. Thorax. (2006) 61:284–9. doi: 10.1136/thx.2005.051029
3. Miller J, Edwards LD, Agusti A, Bakke P, Calverley PM, Celli B, et al. Comorbidity, systemic inflammation and outcomes in the ECLIPSE cohort. Respir Med. (2013) 107:1376–84. doi: 10.1016/j.rmed.2013.05.001
4. Ho TW, Huang CT, Ruan SY, Tsai YJ, Lai F, Yu CJ. Diabetes mellitus in patients with chronic obstructive pulmonary disease-The impact on mortality. PLoS ONE. (2017) 12:e0175794. doi: 10.1371/journal.pone.0175794
5. Makarevich AE, Valevich VE, Pochtavtsev AU. Evaluation of pulmonary hypertension in COPD patients with diabetes. Adv Med Sci. (2007) 52:265–72.
6. Ofulue AF, Thurlbeck WM. Experimental diabetes and the lung. II. In vivo connective tissue metabolism. Am Rev Respir Dis. (1988) 138:284–9. doi: 10.1164/ajrccm/138.2.284
7. Cavan DA, Parkes A, O'Donnell MJ, Freeman W, Cayton RM. Lung function and diabetes. Respir Med. (1991) 85:257–8. doi: 10.1016/S0954-6111(06)80092-2
8. Wang CM, Hsu CT, Niu HS, Chang CH, Cheng JT, Shieh JM. Lung damage induced by hyperglycemia in diabetic rats: the role of signal transducer and activator of transcription 3 (STAT3). J Diab Compl. (2016) 30:1426–33. doi: 10.1016/j.jdiacomp.2016.07.005
9. Ford ES. Body mass index, diabetes, and C-reactive protein among U.S. adults. Diabetes Care. (1999) 22:1971–7. doi: 10.2337/diacare.22.12.1971
10. Rodriguez-Moran M, Guerrero-Romero F. Increased levels of C-reactive protein in noncontrolled type II diabetic subjects. J Diab Compl. (1999) 13:211–5. doi: 10.1016/S1056-8727(99)00047-1
11. Tan KC, Chow WS, Tam SC, Ai VH, Lam CH, Lam KS. Atorvastatin lowers C-reactive protein and improves endothelium-dependent vasodilation in type 2 diabetes mellitus. J Clin Endocrinol Metab. (2002) 87:563–8. doi: 10.1210/jcem.87.2.8249
12. O'Donnell CP, Tankersley CG, Polotsky VP, Schwartz AR, Smith PL. Leptin, obesity, and respiratory function. Res Physiol. (2000) 119:163–70. doi: 10.1016/S0034-5687(99)00111-5
13. Sin DD, Man SF. Impaired lung function and serum leptin in men and women with normal body weight: a population based study. Thorax. (2003) 58:695–8. doi: 10.1136/thorax.58.8.695
14. Hamlin CR, Kohn RR, Luschin JH. Apparent accelerated aging of human collagen in diabetes mellitus. Diabetes. (1975) 24:902–4. doi: 10.2337/diabetes.24.10.902
15. Pitocco D, Fuso L, Conte EG, Zaccardi F, Condoluci C, Scavone G, et al. The diabetic lung–a new target organ? Rev Diab Stud. (2012) 9:23–35. doi: 10.1900/RDS.2012.9.23
16. Masri S. Sirtuin-dependent clock control: new advances in metabolism, aging and cancer. Curr Opin Clin Nutr Metab Care. (2015) 18:521–7. doi: 10.1097/MCO.0000000000000219
17. Zhou S, Tang X, Chen H-Z. Sirtuins and insulin resistance. Front Endocrinol. (2018) 9:748. doi: 10.3389/fendo.2018.00748
18. Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. (2012) 74:246–60. doi: 10.1016/j.neuron.2012.04.006
19. Orozco-Solis R, Sassone-Corsi P. Circadian clock: linking epigenetics to aging. Curr Opin Genet Dev. (2014) 26:66–72. doi: 10.1016/j.gde.2014.06.003
20. Panda S. Circadian physiology of metabolism. Science. (2016) 354:1008–15. doi: 10.1126/science.aah4967
21. Sundar IK, Yao H, Sellix MT, Rahman I. Circadian molecular clock in lung pathophysiology. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L1056–L75. doi: 10.1152/ajplung.00152.2015
22. Weynand B, Jonckheere A, Frans A, Rahier J. Diabetes mellitus induces a thickening of the pulmonary basal lamina. Respiration. (1999) 66:14–9. doi: 10.1159/000029331
23. Popov D, Simionescu M. Alterations of lung structure in experimental diabetes, and diabetes associated with hyperlipidaemia in hamsters. Eur Respir J. (1997) 10:1850–8. doi: 10.1183/09031936.97.10081850
24. Schnider SL, Kohn RR. Glucosylation of human collagen in aging and diabetes mellitus. J Clin Invest. (1980) 66:1179–81. doi: 10.1172/JCI109950
25. Kida K, Utsuyama M, Takizawa T, Thurlbeck WM. Changes in lung morphologic features and elasticity caused by streptozotocin-induced diabetes mellitus in growing rats. Am Rev Resp Dis. (1983) 128:125–31. doi: 10.1164/arrd.1983.128.1.125
26. Ehrlich SF, Quesenberry CP Jr, Van Den Eeden SK, Shan J, Ferrara A. Patients diagnosed with diabetes are at increased risk for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and pneumonia but not lung cancer. Diab Care. (2010) 33:55–60. doi: 10.2337/dc09-0880
27. Sandler M, Bunn AE, Stewart RI. Cross-section study of pulmonary function in patients with insulin-dependent diabetes mellitus. Am Rev Resp Dis. (1987) 135:223–9.
28. Tai H, Wang MY, Zhao YP, Li LB, Dong QY, Liu XG, et al. The effect of alogliptin on pulmonary function in obese patients with type 2 diabetes inadequately controlled by metformin monotherapy. Medicine. (2016) 95:e4541. doi: 10.1097/MD.0000000000004541
29. Lange P, Groth S, Kastrup J, Mortensen J, Appleyard M, Nyboe J, et al. Diabetes mellitus, plasma glucose and lung function in a cross-sectional population study. Eur Respir J. (1989) 2:14–9.
30. Niranjan V, McBrayer DG, Ramirez LC, Raskin P, Hsia CC. Glycemic control and cardiopulmonary function in patients with insulin-dependent diabetes mellitus. Am J Med. (1997) 103:504–13. doi: 10.1016/S0002-9343(97)00251-9
31. Klein OL, Krishnan JA, Glick S, Smith LJ. Systematic review of the association between lung function and Type 2 diabetes mellitus. Diab Med. (2010) 27:977–87. doi: 10.1111/j.1464-5491.2010.03073.x
32. Antonelli Incalzi R, Fuso L, Giordano A, Pitocco D, Maiolo C, Calcagni ML, et al. Neuroadrenergic denervation of the lung in type I diabetes mellitus complicated by autonomic neuropathy. Chest. (2002) 121:443–51. doi: 10.1378/chest.121.2.443
33. Bottini P, Scionti L, Santeusanio F, Casucci G, Tantucci C. Impairment of the respiratory system in diabetic autonomic neuropathy. Diab Nutr Metab. (2000) 13:165–72.
34. Giordano A, Calcagni ML, Rossi B, Fuso L, Accardo D, Valente S, et al. Potential use of iodine-123 metaiodobenzylguanidine radioaerosol as a marker of pulmonary neuroadrenergic function. Eur J Nucl Med. (1997) 24:52–8. doi: 10.1007/BF01728309
35. Nishimura M, Miyamoto K, Suzuki A, Yamamoto H, Tsuji M, Kishi F, et al. Ventilatory and heart rate responses to hypoxia and hypercapnia in patients with diabetes mellitus. Thorax. (1989) 44:251–7. doi: 10.1136/thx.44.4.251
36. Montserrat JM, Cochrane GM, Wolf C, Picado C, Roca J, Agusti Vidal A. Ventilatory control in diabetes mellitus. Euro J Resp Dis. (1985) 67:112–7.
37. Heimer D, Brami J, Lieberman D, Bark H. Respiratory muscle performance in patients with type 1 diabetes. Diab Med. (1990) 7:434–7. doi: 10.1111/j.1464-5491.1990.tb01419.x
38. Wanke T, Formanek D, Auinger M, Popp W, Zwick H, Irsigler K. Inspiratory muscle performance and pulmonary function changes in insulin-dependent diabetes mellitus. Am Rev Resp Dis. (1991) 143:97–100. doi: 10.1164/ajrccm/143.1.97
39. Scano G, Seghieri G, Mancini M, Filippelli M, Duranti R, Fabbri A, et al. Dyspnoea, peripheral airway involvement and respiratory muscle effort in patients with type I diabetes mellitus under good metabolic control. Clin Sci. (1999) 96:499–506. doi: 10.1042/cs0960499
40. Fuso L, Pitocco D, Longobardi A, Zaccardi F, Contu C, Pozzuto C, et al. Reduced respiratory muscle strength and endurance in type 2 diabetes mellitus. Diab Metab Res Rev. (2012) 28:370–5. doi: 10.1002/dmrr.2284
41. Domingueti CP, Dusse LMSA, Carvalho MdG, de Sousa LP, Gomes KB, Fernandes AP. Diabetes mellitus: the linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J Diab Compl. (2016) 30:738–45. doi: 10.1016/j.jdiacomp.2015.12.018
42. Kaparianos A, Argyropoulou E, Sampsonas F, Karkoulias K, Tsiamita M, Spiropoulos K. Pulmonary complications in diabetes mellitus. Chron Respir Dis. (2008) 5:101–8. doi: 10.1177/1479972307086313
43. Goldman MD. Lung dysfunction in diabetes. Diab Care. (2003) 26:1915–8. doi: 10.2337/diacare.26.6.1915
44. Tsai CL, Brenner BE, Camargo CA Jr. Circadian-rhythm differences among emergency department patients with chronic obstructive pulmonary disease exacerbation. Chronobiol Int. (2007) 24:699–713. doi: 10.1080/07420520701535753
45. Petty TL. Circadian variations in chronic asthma and chronic obstructive pulmonary disease. Am J Med. (1988) 85:21–3. doi: 10.1016/0002-9343(88)90237-9
46. Tirlapur VG. Nocturnal deaths among patients with chronic bronchitis and emphysema. Br Med J. (1984) 289:1540. doi: 10.1136/bmj.289.6457.1540
47. Sundar IK, Yao H, Sellix MT, Rahman I. Circadian clock-coupled lung cellular and molecular functions in chronic airway diseases. Am J Respir Cell Mol Biol. (2015) 53:285–90. doi: 10.1165/rcmb.2014-0476TR
48. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. (2017) 18:164–79. doi: 10.1038/nrg.2016.150
49. Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. (2008) 134:317–28. doi: 10.1016/j.cell.2008.06.050
50. Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, et al. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol. (2005) 25:2795–807. doi: 10.1128/MCB.25.7.2795-2807.2005
51. Yagita K, Tamanini F, Yasuda M, Hoeijmakers JH, van der Horst GT, Okamura H. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. (2002) 21:1301–14. doi: 10.1093/emboj/21.6.1301
52. Cardone L, Hirayama J, Giordano F, Tamaru T, Palvimo JJ, Sassone-Corsi P. Circadian clock control by SUMOylation of BMAL1. Science. (2005) 309:1390–4. doi: 10.1126/science.1110689
53. Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. (2001) 107:855–67. doi: 10.1016/S0092-8674(01)00610-9
54. Kondratov RV, Chernov MV, Kondratova AA, Gorbacheva VY, Gudkov AV, Antoch MP. BMAL1-dependent circadian oscillation of nuclear CLOCK: posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. (2003) 17:1921–32. doi: 10.1101/gad.1099503
55. Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, Nakahata Y, et al. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature. (2007) 450:1086–90. doi: 10.1038/nature06394
56. Etchegaray JP, Lee C, Wade PA, Reppert SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature. (2003) 421:177–82. doi: 10.1038/nature01314
57. Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. (2008) 134:329–40. doi: 10.1016/j.cell.2008.07.002
58. Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. (2009) 324:654–7. doi: 10.1126/science.1170803
59. Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C, et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell. (2014) 158:659–72. doi: 10.1016/j.cell.2014.06.050
60. Katada S, Sassone-Corsi P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat Struct Mol Biol. (2010) 17:1414–21. doi: 10.1038/nsmb.1961
61. Valekunja UK, Edgar RS, Oklejewicz M, van der Horst GT, O'Neill JS, Tamanini F, et al. Histone methyltransferase MLL3 contributes to genome-scale circadian transcription. Proc Natl Acad Sci USA. (2013) 110:1554–9. doi: 10.1073/pnas.1214168110
62. DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, Secombe J, et al. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. (2011) 333:1881–5. doi: 10.1126/science.1206022
63. Qu M, Duffy T, Hirota T, Kay SA. Nuclear receptor HNF4A transrepresses CLOCK:BMAL1 and modulates tissue-specific circadian networks. Proc Natl Acad Sci USA. (2018) 115:E12305–12. doi: 10.1073/pnas.1816411115
64. Hong HK, Maury E, Ramsey KM, Perelis M, Marcheva B, Omura C, et al. Requirement for NF-kappaB in maintenance of molecular and behavioral circadian rhythms in mice. Genes Dev. (2018) 32:1367–79. doi: 10.1101/gad.319228.118
65. Shimomura K, Kumar V, Koike N, Kim TK, Chong J, Buhr ED, et al. Usf1, a suppressor of the circadian Clock mutant, reveals the nature of the DNA-binding of the CLOCK:BMAL1 complex in mice. eLif. (2013) 2:e00426. doi: 10.7554/eLife.00426
66. Altman BJ, Hsieh AL, Sengupta A, Krishnanaiah SY, Stine ZE, Walton ZE, et al. MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab. (2015) 22:1009–19. doi: 10.1016/j.cmet.2015.09.003
67. Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, et al. GENE REGULATION. Discrete functions of nuclear receptor Rev-erbalpha couple metabolism to the clock. Science. (2015) 348:1488–92. doi: 10.1126/science.aab3021
68. Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, et al. Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science. (2015) 350:aac4250. doi: 10.1126/science.aac4250
69. Wu Y, Tang D, Liu N, Xiong W, Huang H, Li Y, et al. Reciprocal regulation between the circadian clock and hypoxia signaling at the genome level in mammals. Cell Metab. (2017) 25:73–85. doi: 10.1016/j.cmet.2016.09.009
70. Sharma S, Singh H, Ahmad N, Mishra P, Tiwari A. The role of melatonin in diabetes: therapeutic implications. Arch Endocrinol Metab. (2015) 59:391–9. doi: 10.1590/2359-3997000000098
71. McMullan CJ, Schernhammer ES, Rimm EB, Hu FB, Forman JP. Melatonin secretion and the incidence of type 2 diabetes. JAMA. (2013) 309:1388–96. doi: 10.1001/jama.2013.2710
72. O'Brien IA, Lewin IG, O'Hare JP, Arendt J, Corrall RJ. Abnormal circadian rhythm of melatonin in diabetic autonomic neuropathy. Clin Endocrinol. (1986) 24:359–64. doi: 10.1111/j.1365-2265.1986.tb01639.x
73. Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. (2009) 41:89–94. doi: 10.1038/ng.277
74. Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G, et al. Variants in MTNR1B influence fasting glucose levels. Nature genetics. (2009) 41:77–81. doi: 10.1038/ng.290
75. Bonnefond A, Clement N, Fawcett K, Yengo L, Vaillant E, Guillaume JL, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. (2012) 44:297–301. doi: 10.1038/ng.1053
76. Reiter RJ, Tan DX, Osuna C, Gitto E. Actions of melatonin in the reduction of oxidative stress. A review. J Biomed Sci. (2000) 7:444–58. doi: 10.1007/BF02253360
77. Cano Barquilla P, Pagano ES, Jimenez-Ortega V, Fernandez-Mateos P, Esquifino AI, Cardinali DP. Melatonin normalizes clinical and biochemical parameters of mild inflammation in diet-induced metabolic syndrome in rats. J Pine Res. (2014) 57:280–90. doi: 10.1111/jpi.12168
78. Agil A, El-Hammadi M, Jimenez-Aranda A, Tassi M, Abdo W, Fernandez-Vazquez G, et al. Melatonin reduces hepatic mitochondrial dysfunction in diabetic obese rats. J Pine Res. (2015) 59:70–9. doi: 10.1111/jpi.12241
79. Brydon L, Petit L, Delagrange P, Strosberg AD, Jockers R. Functional expression of MT2 (Mel1b) melatonin receptors in human PAZ6 adipocytes. Endocrinology. (2001) 142:4264–71. doi: 10.1210/endo.142.10.8423
80. Ha E, Yim SV, Chung JH, Yoon KS, Kang I, Cho YH, et al. Melatonin stimulates glucose transport via insulin receptor substrate-1/phosphatidylinositol 3-kinase pathway in C2C12 murine skeletal muscle cells. J Pine Res. (2006) 41:67–72. doi: 10.1111/j.1600-079X.2006.00334.x
81. Poon AM, Choy EH, Pang SF. Modulation of blood glucose by melatonin: a direct action on melatonin receptors in mouse hepatocytes. Biol Sign Recept. (2001) 10:367–79. doi: 10.1159/000046904
82. Stenvers DJ, Scheer F, Schrauwen P, la Fleur SE, Kalsbeek A. Circadian clocks and insulin resistance. Nat Rev Endocrinol. (2019) 15:75–89. doi: 10.1038/s41574-018-0122-1
83. van Raalte DH, Diamant M. Steroid diabetes: from mechanism to treatment? Netherl J Med. (2014) 72:62–72.
84. Buckley TM, Schatzberg AF. On the interactions of the hypothalamic-pituitary-adrenal (HPA) axis and sleep: normal HPA axis activity and circadian rhythm, exemplary sleep disorders. J Clin Endocrinol Metab. (2005) 90:3106–14. doi: 10.1210/jc.2004-1056
85. Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. (2009) 30:152–77. doi: 10.1210/er.2008-0027
86. Peschke E, Peschke D. Evidence for a circadian rhythm of insulin release from perifused rat pancreatic islets. Diabetologia. (1998) 41:1085–92. doi: 10.1007/s001250051034
87. Pulimeno P, Mannic T, Sage D, Giovannoni L, Salmon P, Lemeille S, et al. Autonomous and self-sustained circadian oscillators displayed in human islet cells. Diabetologia. (2013) 56:497–507. doi: 10.1007/s00125-012-2779-7
88. Saini C, Petrenko V, Pulimeno P, Giovannoni L, Berney T, Hebrok M, et al. A functional circadian clock is required for proper insulin secretion by human pancreatic islet cells. Diab Obes Metab. (2016) 18:355–65. doi: 10.1111/dom.12616
89. Polonsky KS, Given BD, Hirsch LJ, Tillil H, Shapiro ET, Beebe C, et al. Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med. (1988) 318:1231–9. doi: 10.1056/NEJM198805123181903
90. Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. (2010) 466:627–31. doi: 10.1038/nature09253
91. Guo H, Brewer JM, Champhekar A, Harris RB, Bittman EL. Differential control of peripheral circadian rhythms by suprachiasmatic-dependent neural signals. Proc Natl Acad Sci USA. (2005) 102:3111–6. doi: 10.1073/pnas.0409734102
92. Opperhuizen AL, Wang D, Foppen E, Jansen R, Boudzovitch-Surovtseva O, de Vries J, et al. Feeding during the resting phase causes profound changes in physiology and desynchronization between liver and muscle rhythms of rats. Euro J Neurosci. (2016) 44:2795–806. doi: 10.1111/ejn.13377
93. Reznick J, Preston E, Wilks DL, Beale SM, Turner N, Cooney GJ. Altered feeding differentially regulates circadian rhythms and energy metabolism in liver and muscle of rats. Biochim Biophys Acta. (2013) 1832:228–38. doi: 10.1016/j.bbadis.2012.08.010
94. Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the circadian clock in the liver by feeding. Science. (2001) 291:490–3. doi: 10.1126/science.291.5503.490
95. Wehrens SMT, Christou S, Isherwood C, Middleton B, Gibbs MA, Archer SN, et al. Meal timing regulates the human circadian system. Curr Biol. (2017) 27:1768–75e3. doi: 10.1016/j.cub.2017.04.059
96. Carrasco-Benso MP, Rivero-Gutierrez B, Lopez-Minguez J, Anzola A, Diez-Noguera A, Madrid JA, et al. Human adipose tissue expresses intrinsic circadian rhythm in insulin sensitivity. FASEB J. (2016) 30:3117–23. doi: 10.1096/fj.201600269RR
97. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. (2012) 15:848–60. doi: 10.1016/j.cmet.2012.04.019
98. Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. (2001) 292:1728–31. doi: 10.1126/science.292.5522.1728
99. Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA. (2008) 105:15172–7. doi: 10.1073/pnas.0806717105
100. Gliniak CM, Brown JM, Noy N. The retinol-binding protein receptor STRA6 regulates diurnal insulin responses. J Biol Chem. (2017) 292:15080–93. doi: 10.1074/jbc.M117.782334
101. Shostak A, Meyer-Kovac J, Oster H. Circadian regulation of lipid mobilization in white adipose tissues. Diabetes. (2013) 62:2195–203. doi: 10.2337/db12-1449
102. Delezie J, Dumont S, Dardente H, Oudart H, Grechez-Cassiau A, Klosen P, et al. The nuclear receptor REV-ERBalpha is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. (2012) 26:3321–35. doi: 10.1096/fj.12-208751
103. Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, et al. Extensive and divergent circadian gene expression in liver and heart. Nature. (2002) 417:78–83. doi: 10.1038/nature744
104. van Moorsel D, Hansen J, Havekes B, Scheer F, Jorgensen JA, Hoeks J, et al. Demonstration of a day-night rhythm in human skeletal muscle oxidative capacity. Mol Metab. (2016) 5:635–45. doi: 10.1016/j.molmet.2016.06.012
105. Verrillo A, De Teresa A, Martino C, Di Chiara G, Pinto M, Verrillo L, et al. Differential roles of splanchnic and peripheral tissues in determining diurnal fluctuation of glucose tolerance. Am J Physiol. (1989) 257:E459–65. doi: 10.1152/ajpendo.1989.257.4.E459
106. Dyar KA, Ciciliot S, Wright LE, Bienso RS, Tagliazucchi GM, Patel VR, et al. Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab. (2014) 3:29–41. doi: 10.1016/j.molmet.2013.10.005
107. Liu J, Zhou B, Yan M, Huang R, Wang Y, He Z, et al. CLOCK and BMAL1 regulate muscle insulin sensitivity via SIRT1 in male mice. Endocrinology. (2016) 157:2259–69. doi: 10.1210/en.2015-2027
108. Hong S, Zhou W, Fang B, Lu W, Loro E, Damle M, et al. Dissociation of muscle insulin sensitivity from exercise endurance in mice by HDAC3 depletion. Nat Med. (2017) 23:223–34. doi: 10.1038/nm.4245
109. Oishi K, Sakamoto K, Okada T, Nagase T, Ishida N. Antiphase circadian expression between BMAL1 and period homologue mRNA in the suprachiasmatic nucleus and peripheral tissues of rats. Biochem Biophys Res Commun. (1998) 253:199–203. doi: 10.1006/bbrc.1998.9779
110. Jarjour NN. Circadian variation in allergen and nonspecific bronchial responsiveness in asthma. Chronobiol Int. (1999) 16:631–9. doi: 10.3109/07420529908998732
111. Martin RJ. Location of airway inflammation in asthma and the relationship to circadian change in lung function. Chronobiol Int. (1999) 16:623–30. doi: 10.3109/07420529908998731
112. Gibbs JE, Beesley S, Plumb J, Singh D, Farrow S, Ray DW, et al. Circadian timing in the lung; a specific role for bronchiolar epithelial cells. Endocrinology. (2009) 150:268–76. doi: 10.1210/en.2008-0638
113. Barnes PJ. Circadian variation in airway function. Am J Med. (1985) 79:5–9. doi: 10.1016/0002-9343(85)90080-4
114. Ehlers A, Xie W, Agapov E, Brown S, Steinberg D, Tidwell R, et al. BMAL1 links the circadian clock to viral airway pathology and asthma phenotypes. Mucosal Immunol. (2018) 11:97–111. doi: 10.1038/mi.2017.24
115. Gebel S, Gerstmayer B, Kuhl P, Borlak J, Meurrens K, Muller T. The kinetics of transcriptomic changes induced by cigarette smoke in rat lungs reveals a specific program of defense, inflammation, and circadian clock gene expression. Toxicol Sci. (2006) 93:422–31. doi: 10.1093/toxsci/kfl071
116. Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat Med. (2014) 20:919–26. doi: 10.1038/nm.3599
118. Sundar IK, Ahmad T, Yao H, Hwang JW, Gerloff J, Lawrence BP, et al. Influenza A virus-dependent remodeling of pulmonary clock function in a mouse model of COPD. Sci Rep. (2015) 4:9927. doi: 10.1038/srep09927
119. Yao H, Sundar IK, Huang Y, Gerloff J, Sellix MT, Sime PJ, et al. Disruption of Sirtuin 1-mediated control of circadian molecular clock and inflammation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2015) 53:782–92. doi: 10.1165/rcmb.2014-0474OC
120. Khan NA, Yogeswaran S, Wang Q, Muthumalage T, Sundar IK, Rahman I. Waterpipe smoke and e-cigarette vapor differentially affect circadian molecular clock gene expression in mouse lungs. PLoS ONE. (2019) 14:e0211645–e. doi: 10.1371/journal.pone.0211645
121. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. (2006) 20:1868–73. doi: 10.1101/gad.1432206
122. Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Investig. (2013) 123:5389–400. doi: 10.1172/JCI70317
123. Early JO, Menon D, Wyse CA, Cervantes-Silva MP, Zaslona Z, Carroll RG, et al. Circadian clock protein BMAL1 regulates IL-1beta in macrophages via NRF2. Proc Natl Acad Sci USA. (2018) 115:E8460–e8. doi: 10.1073/pnas.1800431115
124. Lee J, Moulik M, Fang Z, Saha P, Zou F, Xu Y, et al. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol Cell Biol. (2013) 33:2327–38. doi: 10.1128/MCB.01421-12
125. Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci USA. (1999) 96:12731–6. doi: 10.1073/pnas.96.22.12731
126. Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. (2006) 8:76–87. doi: 10.1089/ars.2006.8.76
127. Cho HY, Reddy SP, Yamamoto M, Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. (2004) 18:1258–60. doi: 10.1096/fj.03-1127fje
128. Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. (2009) 34:176–88. doi: 10.1016/j.tibs.2008.12.008
129. Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. (2010) 38:5718–34. doi: 10.1093/nar/gkq212
130. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. (2009) 284:13291–5. doi: 10.1074/jbc.R900010200
131. Pekovic-Vaughan V, Gibbs J, Yoshitane H, Yang N, Pathiranage D, Guo B, et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. (2014) 28:548–60. doi: 10.1101/gad.237081.113
132. Heo KS, Kim DU, Ryoo S, Nam M, Baek ST, Kim L, et al. PPARgamma activation abolishes LDL-induced proliferation of human aortic smooth muscle cells via SOD-mediated down-regulation of superoxide. Biochem Biophys Res Commun. (2007) 359:1017–23. doi: 10.1016/j.bbrc.2007.06.006
133. Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. (2002) 16:2793–801. doi: 10.1210/me.2002-0020
134. Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res. (2007) 76:269–79. doi: 10.1016/j.cardiores.2007.06.027
135. Chung SS, Kim M, Youn BS, Lee NS, Park JW, Lee IK, et al. Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol Cell Biol. (2009) 29:20–30. doi: 10.1128/MCB.00544-08
136. Toyama T, Nakamura H, Harano Y, Yamauchi N, Morita A, Kirishima T, et al. PPARalpha ligands activate antioxidant enzymes and suppress hepatic fibrosis in rats. Biochem Biophys Res Commun. (2004) 324:697–704. doi: 10.1016/j.bbrc.2004.09.110
137. Inoue I, Noji S, Awata T, Takahashi K, Nakajima T, Sonoda M, et al. Bezafibrate has an antioxidant effect: peroxisome proliferator-activated receptor alpha is associated with Cu2+, Zn2+-superoxide dismutase in the liver. Life Sci. (1998) 63:135–44. doi: 10.1016/S0024-3205(98)00249-5
138. Canaple L, Rambaud J, Dkhissi-Benyahya O, Rayet B, Tan NS, Michalik L, et al. Reciprocal regulation of brain and muscle Arnt-like protein 1 and peroxisome proliferator-activated receptor alpha defines a novel positive feedback loop in the rodent liver circadian clock. Mol Endocrinol. (2006) 20:1715–27. doi: 10.1210/me.2006-0052
139. Grimaldi B, Bellet MM, Katada S, Astarita G, Hirayama J, Amin RH, et al. PER2 controls lipid metabolism by direct regulation of PPARgamma. Cell Metab. (2010) 12:509–20. doi: 10.1016/j.cmet.2010.10.005
140. Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. (2013) 342:1243417. doi: 10.1126/science.1243417
141. Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, et al. Rev-erb-alpha modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med. (2013) 19:1039–46. doi: 10.1038/nm.3213
142. Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol. (2013) 13:190–8. doi: 10.1038/nri3386
143. Man K, Loudon A, Chawla A. Immunity around the clock. Science. (2016) 354:999–1003. doi: 10.1126/science.aah4966
144. Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. (2013) 341:1483–8. doi: 10.1126/science.1240636
145. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. (2016) 7:11624. doi: 10.1038/ncomms11624
146. Zhang Z, Hunter L, Wu G, Maidstone R, Mizoro Y, Vonslow R, et al. Genome-wide effect of pulmonary airway epithelial cell-specific Bmal1 deletion. FASEB J. (2019) 33:6226–38. doi: 10.1096/fj.201801682R
147. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. (2005) 11:183–90. doi: 10.1038/nm1166
148. Spengler ML, Kuropatwinski KK, Comas M, Gasparian AV, Fedtsova N, Gleiberman AS, et al. Core circadian protein CLOCK is a positive regulator of NF-kappaB-mediated transcription. Proc Natl Acad Sci USA. (2012) 109:E2457–65. doi: 10.1073/pnas.1206274109
149. Sato S, Sakurai T, Ogasawara J, Takahashi M, Izawa T, Imaizumi K, et al. A circadian clock gene, Rev-erbalpha, modulates the inflammatory function of macrophages through the negative regulation of Ccl2 expression. J Immunol. (2014) 192:407–17. doi: 10.4049/jimmunol.1301982
150. Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, et al. The nuclear receptor REV-ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc Natl Acad Sci USA. (2012) 109:582–7. doi: 10.1073/pnas.1106750109
151. Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. (2013) 498:511–5. doi: 10.1038/nature12209
152. Pariollaud M, Gibbs JE, Hopwood TW, Brown S, Begley N, Vonslow R, et al. Circadian clock component REV-ERBalpha controls homeostatic regulation of pulmonary inflammation. J Clin Invest. (2018) 128:2281–96. doi: 10.1172/JCI93910
153. Narasimamurthy R, Hatori M, Nayak SK, Liu F, Panda S, Verma IM. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc Natl Acad Sci USA. (2012) 109:12662–7. doi: 10.1073/pnas.1209965109
154. Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiological reviews. (2012) 92:1479–514. doi: 10.1152/physrev.00022.2011
155. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free radical biology & medicine. (2013) 56:133–71. doi: 10.1016/j.freeradbiomed.2012.10.525
156. Guarente L. Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. (2011) 364:2235–44. doi: 10.1056/NEJMra1100831
157. Ramadori G, Lee CE, Bookout AL, Lee S, Williams KW, Anderson J, et al. Brain SIRT1: anatomical distribution and regulation by energy availability. J Neurosci. (2008) 28:9989–96. doi: 10.1523/JNEUROSCI.3257-08.2008
158. Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. (2013) 153:1448–60. doi: 10.1016/j.cell.2013.05.027
159. Orozco-Solis R, Ramadori G, Coppari R, Sassone-Corsi P. SIRT1 relays nutritional inputs to the circadian clock through the sf1 neurons of the ventromedial hypothalamus. Endocrinology. (2015) 156:2174–84. doi: 10.1210/en.2014-1805
160. Hardin PE, Yu W. Circadian transcription: passing the HAT to CLOCK. Cell. (2006) 125:424–6. doi: 10.1016/j.cell.2006.04.010
161. Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. (2009) 324:651–4. doi: 10.1126/science.1171641
162. Aguilar-Arnal L, Katada S, Orozco-Solis R, Sassone-Corsi P. NAD(+)-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat Struct Mol Biol. (2015) 22:312–8. doi: 10.1038/nsmb.2990
163. Zhou B, Zhang Y, Zhang F, Xia Y, Liu J, Huang R, et al. CLOCK/BMAL1 regulates circadian change of mouse hepatic insulin sensitivity by SIRT1. Hepatology. (2014) 59:2196–206. doi: 10.1002/hep.26992
164. Wang RH, Zhao T, Cui K, Hu G, Chen Q, Chen W, et al. Negative reciprocal regulation between Sirt1 and Per2 modulates the circadian clock and aging. Sci Rep. (2016) 6:28633. doi: 10.1038/srep28633
165. Di Vincenzo S, Heijink IH, Noordhoek JA, Cipollina C, Siena L, Bruno A, et al. SIRT1/FoxO3 axis alteration leads to aberrant immune responses in bronchial epithelial cells. J Cell Mol Med. (2018) 22:2272–82. doi: 10.1111/jcmm.13509
166. Conti V, Corbi G, Manzo V, Malangone P, Vitale C, Maglio A, et al. SIRT1 activity in peripheral blood mononuclear cells correlates with altered lung function in patients with chronic obstructive pulmonary disease. Oxid Med Cell Longev. (2018) 2018:9391261. doi: 10.1155/2018/9391261
167. Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. (2012) 122:2032–45. doi: 10.1172/JCI60132
168. Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L567–76. doi: 10.1152/ajplung.00308.2006
169. Hwang JW, Sundar IK, Yao H, Sellix MT, Rahman I. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J. (2014) 28:176–94. doi: 10.1096/fj.13-232629
170. Caramori G, Adcock IM, Casolari P, Ito K, Jazrawi E, Tsaprouni L, et al. Unbalanced oxidant-induced DNA damage and repair in COPD: a link towards lung cancer. Thorax. (2011) 66:521–7. doi: 10.1136/thx.2010.156448
171. Deslee G, Adair-Kirk TL, Betsuyaku T, Woods JC, Moore CH, Gierada DS, et al. Cigarette smoke induces nucleic-acid oxidation in lung fibroblasts. Am J Respir Cell Mol Biol. (2010) 43:576–84. doi: 10.1165/rcmb.2009-0221OC
172. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Resp Crit Care Med. (2006) 174:886–93. doi: 10.1164/rccm.200509-1374OC
173. Tuder RM, Kern JA, Miller YE. Senescence in chronic obstructive pulmonary disease. Proc Am Thoracic Soc. (2012) 9:62–3. doi: 10.1513/pats.201201-012MS
174. Gaddameedhi S, Reardon JT, Ye R, Ozturk N, Sancar A. Effect of circadian clock mutations on DNA damage response in mammalian cells. Cell Cycle. (2012) 11:3481–91. doi: 10.4161/cc.21771
175. Sancar A, Lindsey-Boltz LA, Kang TH, Reardon JT, Lee JH, Ozturk N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. (2010) 584:2618–25. doi: 10.1016/j.febslet.2010.03.017
176. Kang TH, Leem SH. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. (2014) 42:4427–34. doi: 10.1093/nar/gku094
177. Kondratov RV, Antoch MP. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. (2007) 17:311–7. doi: 10.1016/j.tcb.2007.07.001
178. Grechez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem. (2008) 283:4535–42. doi: 10.1074/jbc.M705576200
179. Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell. (2006) 22:375–82. doi: 10.1016/j.molcel.2006.03.038
180. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. (2008) 135:907–18. doi: 10.1016/j.cell.2008.10.025
181. Yao H, Rahman I. Perspectives on translational and therapeutic aspects of SIRT1 in inflammaging and senescence. Biochem Pharmacol. (2012) 84:1332–9. doi: 10.1016/j.bcp.2012.06.031
182. Chaves I, van der Horst GT, Schellevis R, Nijman RM, Koerkamp MG, Holstege FC, et al. Insulin-FOXO3 signaling modulates circadian rhythms via regulation of clock transcription. Curr Biol. (2014) 24:1248–55. doi: 10.1016/j.cub.2014.04.018
183. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. (2006) 124:315–29. doi: 10.1016/j.cell.2005.11.044
184. Wu J, Jin Z, Yan LJ. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox Biol. (2017) 11:51–9. doi: 10.1016/j.redox.2016.11.003
185. Jablonski RP, Kim SJ, Cheresh P, Williams DB, Morales-Nebreda L, Cheng Y, et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. (2017) 31:2520–32. doi: 10.1096/fj.201601077R
186. Bindu S, Pillai VB, Kanwal A, Samant S, Mutlu GM, Verdin E, et al. SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L68–l78. doi: 10.1152/ajplung.00188.2016
187. Chen T, Li J, Liu J, Li N, Wang S, Liu H, et al. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-beta/Smad3 pathway. Ame J Physiol Heart Circ Physiol. (2015) 308:H424–34. doi: 10.1152/ajpheart.00454.2014
188. Sosulski ML, Gongora R, Feghali-Bostwick C, Lasky JA, Sanchez CG. Sirtuin 3 deregulation promotes pulmonary fibrosis. J Gerontol Series A. (2017) 72:595–602. doi: 10.1093/gerona/glw151
189. Masri S, Patel VR, Eckel-Mahan KL, Peleg S, Forne I, Ladurner AG, et al. Circadian acetylome reveals regulation of mitochondrial metabolic pathways. Proc Natl Acad Sci USA. (2013) 110:3339–44. doi: 10.1073/pnas.1217632110
190. Gong C, Li C, Qi X, Song Z, Wu J, Hughes ME, et al. The daily rhythms of mitochondrial gene expression and oxidative stress regulation are altered by aging in the mouse liver. Chronobiol Int. (2015) 32:1254–63. doi: 10.3109/07420528.2015.1085388
191. Mauvoisin D, Atger F, Dayon L, Nunez Galindo A, Wang J, Martin E, et al. Circadian and feeding rhythms orchestrate the diurnal liver acetylome. Cell Rep. (2017) 20:1729–43. doi: 10.1016/j.celrep.2017.07.065
192. Kwon Y, Kim J, Lee CY, Kim H. Expression of SIRT1 and SIRT3 varies according to age in mice. Anatomy Cell Biol. (2015) 48:54–61. doi: 10.5115/acb.2015.48.1.54
193. Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. (2008) 28:6384–401. doi: 10.1128/MCB.00426-08
194. Wang Y, Zhu Y, Xing S, Ma P, Lin D. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO3. Cell Stress Chaperones. (2015) 20:805–10. doi: 10.1007/s12192-015-0599-7
195. Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. (2011) 147:1628–39. doi: 10.1016/j.cell.2011.09.057
196. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. (2009) 136:62–74. doi: 10.1016/j.cell.2008.10.052
197. Kawahara TL, Rapicavoli NA, Wu AR, Qu K, Quake SR, Chang HY. Dynamic chromatin localization of Sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet. (2011) 7:e1002153. doi: 10.1371/journal.pgen.1002153
198. Vitiello M, Zullo A, Servillo L, Mancini FP, Borriello A, Giovane A, et al. Multiple pathways of SIRT6 at the crossroads in the control of longevity, cancer, and cardiovascular diseases. Ageing Res Rev. (2017) 35:301–11. doi: 10.1016/j.arr.2016.10.008
199. Tennen RI, Chua KF. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci. (2011) 36:39–46. doi: 10.1016/j.tibs.2010.07.009
200. Tian K, Chen P, Liu Z, Si S, Zhang Q, Mou Y, et al. Sirtuin 6 inhibits epithelial to mesenchymal transition during idiopathic pulmonary fibrosis via inactivating TGF-beta1/Smad3 signaling. Oncotarget. (2017) 8:61011–24. doi: 10.18632/oncotarget.17723
201. Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2011) 300:L391–401. doi: 10.1152/ajplung.00097.2010
202. Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol. (2014) 192:958–68. doi: 10.4049/jimmunol.1302341
203. Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. (2014) 35:146–54. doi: 10.1016/j.tips.2013.12.004
Keywords: circadian clock, Sirtuins, diabetic lung, oxidative stress, inflammation, aging
Citation: Zhou S, Dai Y-M, Zeng X-F and Chen H-Z (2020) Circadian Clock and Sirtuins in Diabetic Lung: A Mechanistic Perspective. Front. Endocrinol. 11:173. doi: 10.3389/fendo.2020.00173
Received: 19 October 2019; Accepted: 10 March 2020;
Published: 03 April 2020.
Edited by:
Liang-Jun Yan, University of North Texas Health Science Center, United StatesReviewed by:
Isaac Kirubakaran Sundar, University of Rochester, United StatesGraziamaria Corbi, University of Molise, Italy
Copyright © 2020 Zhou, Dai, Zeng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuang Zhou, ZG9jdG9yY2F0MThAMTYzLmNvbQ==; Hou-Zao Chen, Y2hlbmhvdXphb0BpYm1zLmNhbXMuY24=; aG91emFvQGdtYWlsLmNvbQ==