 Yentl Huybrechts
Yentl Huybrechts Geert Mortier
Geert Mortier Eveline Boudin
Eveline Boudin Wim Van Hul
Wim Van Hul
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 09 April 2020
Sec. Bone Research
Volume 11 - 2020 | https://doi.org/10.3389/fendo.2020.00165
This article is part of the Research Topic Innovative Models in Bone Biology: What can be Learned from Rare Bone Diseases? View all 11 articles
Skeletal dysplasias are a diverse group of heritable diseases affecting bone and cartilage growth. Throughout the years, the molecular defect underlying many of the diseases has been identified. These identifications led to novel insights in the mechanisms regulating bone and cartilage growth and homeostasis. One of the pathways that is clearly important during skeletal development and bone homeostasis is the Wingless and int-1 (WNT) signaling pathway. So far, three different WNT signaling pathways have been described, which are all activated by binding of the WNT ligands to the Frizzled (FZD) receptors. In this review, we discuss the skeletal disorders that are included in the latest nosology of skeletal disorders and that are caused by genetic defects involving the WNT signaling pathway. The number of skeletal disorders caused by defects in WNT signaling genes and the clinical phenotype associated with these disorders illustrate the importance of the WNT signaling pathway during skeletal development as well as later on in life to maintain bone mass. The knowledge gained through the identification of the genes underlying these monogenic conditions is used for the identification of novel therapeutic targets. For example, the genes underlying disorders with altered bone mass are all involved in the canonical WNT signaling pathway. Consequently, targeting this pathway is one of the major strategies to increase bone mass in patients with osteoporosis. In addition to increasing the insights in the pathways regulating skeletal development and bone homeostasis, knowledge of rare skeletal dysplasias can also be used to predict possible adverse effects of these novel drug targets. Therefore, this review gives an overview of the skeletal and extra-skeletal phenotype of the different skeletal disorders linked to the WNT signaling pathway.
In the last decades, plenty of examples have been generated where the identification of causal genes for rare, monogenic diseases resulted in novel insights into the role and functioning of already known pathways. A textbook example of this is Wingless and int-1 (WNT; pronounced “wint”) signaling and its role in bone formation and homeostasis. First indications of this involvement were generated by studying rare monogenic conditions with abnormal bone mass, delivering novel targets for drug development for osteoporosis, a common disorder characterized by decreased bone mass in late life. Recently, an antibody against a WNT signaling inhibitor was approved as an anabolic agent for the treatment of osteoporosis.
The name WNT was introduced almost 30 years ago referring to the wingless-type mouse mammary tumor virus integration site (int-1) which was the first member from this gene family being described (1, 2). Meanwhile, three different pathways have been described, one canonical pathway also known as the WNT/β-catenin pathway and two non-canonical pathways, the WNT/Ca2+-dependent pathway and the WNT/planar cell polarity (PCP) pathway (Figure 1). All three are initiated by the binding of an extracellular WNT ligand to the N-terminal domain of one of the seven transmembrane-span FZD receptors. Depending on the pathway, also co-receptors are involved with the low-density lipoprotein (LDL)-related receptor (LRP)-5 and−6 being specific for canonical WNT signaling.
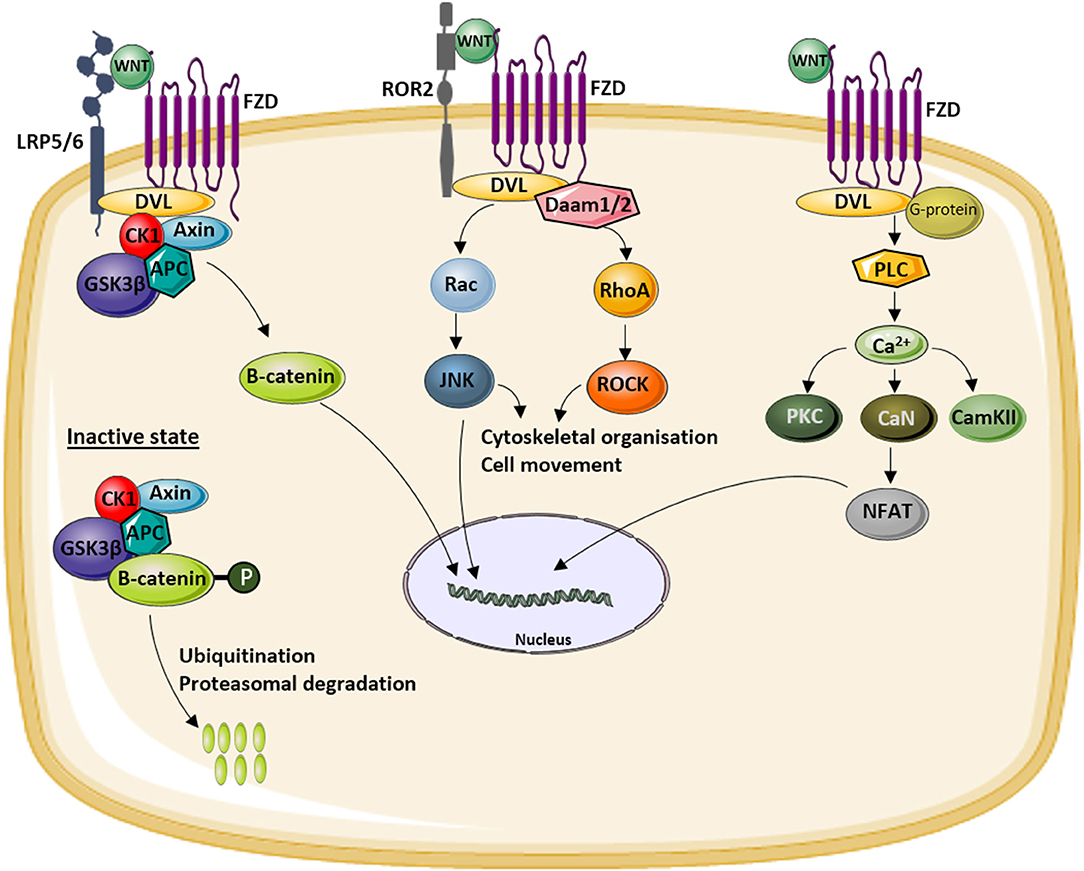
Figure 1. Overview of the different WNT signaling pathways. WNT ligands can activate the canonical or WNT/β-catenin signaling pathway (left) by binding to a receptor complex formed by low-density lipoprotein (LDL)-related receptor (LRP)5/6 and Frizzled (FZD). This activation results in the translocation of hypophosphorylated β-catenin to the nucleus. In the inactive state, β-catenin is degraded by the proteasome after phosphorylation by a glycogen synthase kinase 3β (GSK3β)-Axin-casein kinase 1 (CK1)–adenomatous polyposis coli (APC) complex and subsequent ubiquitination. The activation of the non-canonical WNT/planar cell polarity (PCP) pathway (middle) results in the activation of c-jun NH2-terminal kinase (JNK) via disheveled (DVL) and the Rac and Rho small GTPases. Activation of the non-canonical WNT/Ca2+ pathway (right) by binding of WNT to an FZD receptor results in intracellular Ca2+ release which activates a number of calcium-sensitive enzymes [protein kinase C (PKC), calcineurin (CaN), calmodulin-dependent protein kinase II (CamKII)]. More downstream nuclear factor of activated T cells (NF-AT) is activated and translocates to the nucleus to induce the expression of target genes.
In mammalians, 19 different WNT ligands have been identified and 10 FZD receptors (3), already illustrating the involvement of this pathway in a broad range of cellular processes. All three pathways have a number of functions both during embryonic development and in adult life. These include cell fate specification, cell proliferation and migration, as well as body axis patterning. Furthermore, they are also important for cell functioning as well as processes of cell death. For some processes, only one of the three pathways is involved, but for others, evidence was generated indicating convergence of some of them (4).
At the beginning of this century, an additional role of WNT signaling was discovered following new gene identifications in some rare monogenic skeletal dysplasias. As explained in detail below, the study of conditions with either decreased or increased bone mass resulted in the identification of mutations in several genes involved in especially canonical WNT signaling (5). The most recent revision of the nosology and classification of genetic skeletal disorders (6) includes 461 different diseases. These disorders are interesting experiments of nature to gain insights into the regulatory mechanisms of bone formation, resorption, and homeostasis both during development and during adult life. In this review, we aim to discuss those skeletal disorders in which abnormal WNT signaling contributes to their pathogenesis. Furthermore, the implications of the novel insights toward more common bone disorders such as osteoporosis are highlighted.
As previously mentioned, the WNT signaling pathway is activated by the binding of WNT ligands. Because of the broad functions of this pathway, additional regulation mechanisms are required to ensure proper timely and spatially functioning of the pathway. Extracellular modulators, including WNT inhibitors and activators, contribute to this complex regulation. Not surprisingly, mutations in various components of this pathway have been described in skeletal dysplasias.
WNT ligands are secreted glycoproteins with a length of 350–400 amino acids. In humans, 19 different ligands have been identified, all containing 23–24 conserved cysteine residues (7, 8). A distinction between canonical (e.g., WNT1 and WNT3) and non-canonical (e.g., WNT5A) WNTs can be made, although overlap between the different pathways has been suggested. Various WNT ligands are associated with skeletal disorders, as described below.
WNT1 is of major importance for the regulation of bone homeostasis, through binding with the co-receptor LRP5. Mutations in the WNT1 gene are found in families with osteogenesis imperfecta (OI) type XV and early-onset osteoporosis (9–11).
OI is a hereditary connective tissue disorder, characterized by bone fragility, hearing loss, and dentinogenesis imperfecta. In the majority of patients, the disease is caused by heterozygous mutations in COL1A1 and COL1A2, which encode the type I collagen pro α-chains. However, also recessive forms of OI do exist. One example is OI type XV caused by bi-allelic loss-of-function mutations in the WNT1 gene (11–15). Moreover, the importance of the WNT signaling pathway in OI is emphasized by the observation of increased serum levels of Dickkopf1 (DKK1), an antagonist of the canonical WNT pathway, in OI patients. However, so far, no disease-causing OI mutations have been described in DKK1 (16).
Osteoporosis is a common skeletal disorder characterized by low bone mass, impaired bone quality, and increased fracture risk (14, 17). Whereas, most patients show symptoms later in life, early-onset osteoporosis can already present in adolescence. Some of this early-onset forms of osteoporosis are caused by heterozygous WNT1 mutations (10, 12–15). These mutations result in disturbed bone remodeling and subsequent imbalance in bone homeostasis (13).
WNT3 is critical for axis formation and limb growth in vertebrates (18, 19), and a homozygous nonsense mutation (p.Glu83*) in the WNT3 gene has been reported in tetra-amelia syndrome type 1 (19). Tetra-amelia is an autosomal recessive disorder, characterized by the absence of all four limbs. It is extremely rare, with an incidence of 1.5–4 per 100,000 births (19–21).
Unlike the other WNT ligands discussed in this review, WNT5A belongs to the non-canonical WNT/PCP pathway. In order to exert its functions, such as promoting osteoblast differentiation, WNT5A binds to the cysteine-rich domain of receptor tyrosine kinase-like orphan receptor (ROR) 1/2 (22–24). Heterozygous loss-of-function mutations in the WNT5A gene are associated with autosomal dominant Robinow syndrome (RS) type 1 (ADRS1) (24–27). RS is characterized by skeletal abnormalities (short stature, brachydactyly, and mesomelic limb shortening predominantly of the upper limbs), genital hypoplasia, and typical facial dysmorphisms. The disorder is genetically and clinically heterogeneous, showing both dominant and recessive inheritance patterns. Additional phenotypic features of RS include hypertelorism, dental problems, and kidney abnormalities.
Acro-pectoro-vertebral dysplasia (F-syndrome) is a rare skeletal disorder with a dominant inheritance pattern (28, 29). Symptoms include carpal and tarsal synostosis, malformations of the first and second fingers with frequent syndactyly, and spina bifida occulta (28–30). Although the molecular basis remains unknown, Thiele et al. (29) mapped the disease to a 6.5 cM region on chromosome 2q36, where the WNT6 gene is located. As this gene is expressed during limb development, it is a promising candidate gene for harboring disease-causing mutations for F-syndrome (6, 29).
WNT7A forms a receptor complex with LRP6 and FZD5 in order to activate the canonical WNT signaling pathway (31, 32). It is expressed in the dorsal ectoderm and involved in the formation of the dorsoventral axis in limb development (32–34). Moreover, it also contributes to anteroposterior patterning by maintaining sonic hedgehog expression (32–34). Consistently, mutations in WNT7A cause limb malformations in Al-Awadi–Raas–Rothschild syndrome (AARRS) and Fuhrmann syndrome (FS) (31–37). Patients with AARRS display short upper limbs, severe malformations of the lower limbs, hypoplastic pelvis, anteriorly displaced genitalia, underdeveloped nails, and absence of uterus. The phenotype observed in FS is milder (e.g., solely ulnar deficiency and oligodactyly) since this is due to only a reduced function of the protein compared to complete loss-of-function mutations as seen in AARRS (32, 34–37). Although both are rare autosomal recessive disorders, different mutations contribute to the highly variable disease phenotypes (31, 34–36, 38, 39).
The WNT10B ligand is a key regulator for osteogenesis and is involved in all stages of limb development (24, 40–42). WNT10B plays an important role in the development of hands and feet, and bi-allelic mutations can cause split-hand/foot malformation type 6 (SHFM6). Generally, SHFMs are complex limb malformations that affect the central rays of hands and feet (42–44). From a clinical and genetic point of view, SHFM is highly heterogeneous. Both isolated and syndromic forms can be distinguished, as well as reduced penetrance has been reported (42–44). While most types of SHFM are autosomal dominantly inherited, SHFM6 follows an autosomal recessive pattern of inheritance (43, 44).
Sclerostin, encoded by the SOST gene, is an osteocyte-derived secreted glycoprotein with a cysteine-knot motif (45). It acts as an extracellular antagonist of the canonical WNT signaling pathway by binding to the first β-propeller domain of LRP5/6 (46–49). Hence, sclerostin functions as a key regulator in bone metabolism and, particularly, bone formation (46, 50). Moreover, an interaction with the extracellular domain of LRP4 has been described, and studies demonstrated that this interaction is essential for normal sclerostin functioning (51, 52).
SOST was identified as the disease-causing gene for several high bone mass disorders, including sclerosteosis type 1. This rare skeletal dysplasia, caused by bi-allelic loss-of-function mutations in the SOST gene, is characterized by hyperostosis, tall stature, and syndactyly. The bone overgrowth affects predominantly the skull, mandible, and tubular bones in the limbs. Involvement of the skull base may cause compression of the cranial nerves and subsequent facial palsy, hearing loss, and blindness (50, 53–55). In 1998, Van Buchem disease, another autosomal recessive high-bone mass disorder, was mapped to the same chromosomal region as sclerosteosis (56). The molecular cause was a bi-allelic 52-kb deletion 35-kb downstream of SOST (23, 57). Since the deleted region contains a myocyte-enhancer factor 2C (Mef2c) binding site, imperative for SOST expression, the deletion results in reduced transcription of SOST (23). Although there is phenotypic overlap between sclerosteosis and Van Buchem disease, the phenotype of the latter is usually milder, as the patients have normal stature and do not exhibit syndactyly (54, 55). Another very rare sclerotic bone disorder is craniodiaphyseal dysplasia (CDD), inherited in an autosomal dominant manner. Clinical manifestations include sclerosis and hyperostosis, especially of the skull and facial bones, resulting in facial dysmorphism or “leontiasis ossea” (49, 58). Mutations causing CDD are located in the signal peptide of sclerostin and affect the secretion of sclerostin in a dominant negative manner.
Gremlin has been characterized as a bone morphogenetic protein (BMP) inhibitor that regulates limb and kidney formation during development (59). However, WNT antagonistic activity has also been described since luciferase assays showed reduced stimulatory effects of WNT3A in the presence of gremlin (60, 61). In 2010, Dimitrov et al. (62) described a monoallelic 1.7 Mb duplication containing both the GREM1 and the FMN1 locus in a patient with Cenani–Lenz-like non-syndromic bilateral oligosyndactyly. FMN1 contains a cis-regulatory region that is essential for the activation of gremlin in the limb bud. Hence, genomic rearrangements can lead to an altered function of gremlin and subsequent phenotypic abnormalities (39, 62–64). Interestingly, the phenotype of the patient is similar to that of the transgenic chick with overexpression of Grem1 (65, 66). Furthermore, GREM1 has been suggested to decrease the metastatic potential of osteosarcoma. Besides reduced gremlin levels in osteosarcoma cells, downregulation of GREM1 was also associated with an increased degree of proliferation and angiogenesis (67).
Secreted FZD-related protein (sFRP) 4 is one of the five members of the sFRP family, known for antagonizing the WNT signaling pathway (68–70). sFRPs contain a C-terminal netrin-like domain (NTR) and an N-terminal cysteine-rich domain, sharing up to 50% sequence homology with the cysteine-rich domain of FZD receptors (24, 68, 70, 71). Consequently, sFRPs serve as decoy receptors for WNT ligands, thereby preventing activation of both canonical and non-canonical WNT signaling (24, 68–70). sFRP4 is a 346-amino acid protein, expressed in the majority of tissues (70). Regarding skeletal tissue, it contributes to maintaining normal osteoblast and osteoclast function during bone development and in the adult skeleton (68–70). A disrupted function of sFRP4 was discovered in Pyle disease, a recessive disorder affecting long bones with metaphyseal widening, cortex thinning, increased trabecular bone, decreased bone mineral density, and bone fragility (24, 72–74). Since 2016, various bi-allelic truncating mutations in sFRP4 have been confirmed to cause Pyle disease (70, 72, 73). These nonsense mutations prohibit appropriate WNT and BMP signaling interaction, which is crucial to maintain appropriate stability of cortical bone (70, 71, 73).
Glypicans (GPCs), a subfamily of heparin sulfate proteoglycans, are linked to the cell surface via a glycosyl-phosphatidylinositol (GPI) anchor (75–77). The main function of the GPCs is regulating the activity of growth factors, such as hedgehogs and (non-)canonical WNTs (77–80). Six highly conserved GPCs have been identified in mammals, and pathogenic variants in GPC4 and GPC6 have been found in patients clinically diagnosed with skeletal dysplasia.
GPC4 is located on chromosome Xq26 and is a regulator of the WNT/PCP pathway (79, 81). Only recently, hemizygous truncating and frameshift mutations were found in families with Keipert syndrome (82). These mutations are recognized as loss of function due to the subsequent loss of essential domains, such as the GPI anchor. Keipert syndrome is characterized by craniofacial and digital abnormalities, accompanied by learning difficulties and deafness (82, 83). Furthermore, there are indications that hemizygous pathological variants could result in an X-linked form of RS. White et al. (84) described a non-synonymous missense variant (p.Arg412Lys) in a male patient with an RS-like phenotype, including brachydactyly, mesomelia, and facial dysmorphisms.
GPC6 is the smallest protein of the GPC family and is widely expressed in the growth plate during skeletal development (76, 77, 85). Genetic null mutations in GPC6 prevent anchoring to the membrane, thereby disrupting the action of GPC6 in developmental processes (75–77). Such mutations were found in omodysplasia type 1 (OMOD1), an autosomal recessive skeletal dysplasia. Both nonsense mutations and genomic rearrangements have been described to contribute to the development of OMOD1 (76). This rare syndrome is characterized by cranial dysmorphisms, short stature of prenatal onset with severe shortening of the humeri (“omo” is the Greek term for humerus) and femora, and restricted mobility in elbows, hips, and knees. In addition, extra-skeletal manifestations, including congenital heart defects and cognitive delay, can also be observed (76, 77). There is also an autosomal dominant form of OMOD (OMOD2), which is milder with normal stature and is caused by heterozygous mutations in the FZD2 gene (see below).
R-spondins (RSPO) are secreted proteins and activators of the canonical WNT signaling pathway (86–88). The WNT agonistic activity is a direct result of the formation of a ternary complex with leucine-rich repeat-containing G protein-coupled receptor 4, 5, or 6 (LGR4/5/6) and Zinc and ring finger 3 (Znrf3) or Ring finger protein 43 (Rnf43), as this complex prevents ubiquitination of the WNT receptors (86, 89–91). RSPOs are key regulators in bone development and bone homeostasis, partly mediated by RSPO2, which has high expression levels in the apical ectodermal ridge of limb buds, but also in the postnatal skeleton, lungs, and brain tissue (86). Similar to WNT3 alterations (see above), an impaired RSPO2 function contributes to the development of tetra-amelia. However, patients affected with RSPO2 mutations exhibit additional hypoplasia or aplasia of the lungs (91). Bi-allelic nonsense mutations, deletions, and frameshift mutations were described in families with tetra-amelia type 2 (91). Furthermore, a homozygous missense mutation (p.Arg69Cys) was reported in humerofemoral hypoplasia with radiotibial ray deficiency, albeit in only one consanguineous family. The affected family members showed severe dysostosis with malformation of all four limbs, absence of tibiae, and hypoplasia of the pelvis (91).
Both the canonical and non-canonical WNT signaling pathways are activated by the binding of WNT ligands to the FZD receptors alone or in combination with specific co-receptors. Identification of single disease mutations in LRP5, LRP6, and ROR2 highlights the importance of these co-receptors in the regulation of canonical and non-canonical WNT signaling during skeletal development.
FZD2 is a member of the FZD receptor family that includes nine G protein-coupled receptors. These FZD receptors interact with disheveled (DVL) upon activation by one of the WNT ligands which results in the initiation of one of the different WNT pathways. In 2015, Saal et al. (92) demonstrated that mutations in FZD2 can cause autosomal dominant OMOD, also referred to as OMOD2. OMOD2 is a rare skeletal dysplasia which is marked by severe rhizomelic shortening of the upper limbs, mild facial dysmorphism (frontal bossing, depressed nasal bridge, and short nose), and genital hypoplasia (92). Clinically, OMOD2 can be distinguished from OMOD1 (recessive form—discussed previously) by the presence of short first metacarpals and normal stature (93).
As mentioned, heterozygous mutations in FZD2 can cause OMOD2. To date, one missense mutation (p.Gly434Val) and two nonsense mutations involving adjacent amino acids (p.Ser547* and p.Trp548*) have been reported (92–95). The nonsense mutations are located in the intracellular domain of FZD2, more precisely in the conserved DVL binding motif (KTxxW). Subsequent functional studies have demonstrated that in the presence of p.Trp548* mutant FZD2, the localization of DVL at the membrane and the activation of the canonical WNT signaling pathway upon WNT treatment are lost (92). The effect of the identified mutations on the non-canonical WNT signaling pathways was not investigated. Several of the clinical features that are reported in patients with OMOD2 are also present in patients with RS (described previously). Therefore, it is likely that the causal genes for these disorders are involved in the same pathway(s). As it is shown that WNT5A/ROR2 interaction can induce WNT/PCP signaling (see below), it is likely that FZD2 is also involved in this pathway.
ROR2 is a member of the ROR kinase family of orphan receptors with tyrosine kinase activity which is involved in the WNT signaling pathway as co-receptor of the non-canonical PCP pathway. The extracellular domain of ROR2 which is important for protein–protein interactions contains several conserved domains such as an immunoglobin-like domain, an FZD-like cysteine-rich domain (CRD), and a Kringle domain (KD). The intracellular region contains a tyrosine kinase (TK) domain followed by serine/threonine-rich and proline-rich structures. Mutations in ROR2 can cause either autosomal dominant brachydactyly type B1 (BDB1) or autosomal recessive RS type 1 (ARRS1) (96, 97).
ARRS1 is characterized by short stature, mesomelic limb shortening, genital hypoplasia, and typical facial dysmorphisms (including hypertelorism). In addition, patients with ARRS1 may also have vertebral segmentation defects such as hemivertebrae and rib fusions (98). Since the identification of ROR2 as a disease-causing gene for ARRS1, more than 25 different mutations have been identified (99). These mutations include missense, nonsense, and frameshift mutations located in both the intra- and extracellular regions of the protein (96, 98–101). Based on the phenotypic overlap between ARRS1 and Ror2 knockout mouse, it was suggested that the mutations result in loss of function of ROR2 due to decreased protein stability and intracellular entrapment of the mutant protein (98, 101). ROR2 serves as a co-receptor for WNT5A-induced non-canonical WNT signaling of which WNT/PCP signaling is shown to be most important for limb development. Loss of function of ROR2 will most likely result in decreased WNT/PCP signaling and limb deformities (102).
BDB1 is the most severe form of brachydactyly and is one of the first described types of heritable brachydactyly. It is characterized by hypoplasia and/or aplasia of the distal phalanges and nails in hand and feet resulting in an amputation-like phenotype (101, 102). The BDB1 causing mutations in ROR2 are all nonsense or frameshift mutations that are located in the intracellular region of the protein and co-localize in two domains, either immediately N-terminal or C-terminal of the TK domain (101). Genotype–phenotype correlation demonstrated that the latter group of mutations results in a more severe phenotype (103). Since parents of ARRS1 patients do not show brachydactyly, it is suggested that BDB1 causing mutations are gain-of-function mutations (96).
The LRPs are a group of evolutionary conserved receptors regulating a wide range of cellular processes. In order to regulate these processes, LRPs can modulate a variety of pathways, including canonical WNT signaling activity (104). The extracellular region of the majority of the LRP receptors contains at least one ligand-binding domain composed of cysteine-rich ligand-binding-type repeats and one epidermal growth factor (EGF)-precursor homology domain composed of EGF repeats and a YWTD/β-propeller domain. Most LRP receptors have in the intracellular region at least one NPxY-(endocytosis) motif. However, LRP5/6 are lacking this motif (105). Mutations in three members of the LRP family, namely, LRP4, LRP5, and LRP6, are reported to result in rare skeletal disorders (51, 106, 107).
Almost 20 years ago, genetic research highlighted the importance of LRP5 in the regulation of bone formation with the identification of disease-causing mutations in patients with abnormal low or high bone mass. Subsequent studies demonstrated that LRP5, as a co-receptor of the canonical WNT signaling pathway, regulates osteoblast proliferation and differentiation as well as osteocyte apoptosis (108). More specifically, homozygous loss-of-function mutations in LRP5 can cause the osteoporosis-pseudoglioma (OPPG) syndrome which is characterized by reduced bone mass, increased bone fragility, and severely reduced visual acuity (109). So far, over 70 different mutations in LRP5 have been reported to cause OPPG, and these mutations are spread throughout the gene. Mutations affecting splicing or introducing premature stop codons result in the most severe phenotypes (110). Furthermore, reduced bone mass is also reported in heterozygous mutation carriers (111). Besides the severe OPPG phenotype, it is reported that heterozygous OPPG loss-of-function mutations in LRP5 can cause juvenile-onset osteoporosis without an ocular phenotype in children (112).
As mentioned, mutations in LRP5 can also result in a skeletal phenotype with increased bone mass (106, 113, 114). When consulting OMIM or the nosology of skeletal disorders (6), mutations in LRP5 are identified in patients diagnosed with high bone mass (HBM) phenotype, autosomal dominant osteopetrosis type 1, autosomal dominant Van Buchem disease, Worth disease, endosteal hyperostosis, or osteosclerosis. Although these rare disorders have a different name, they represent the same disorder which is characterized by increased bone mass especially affecting the skull and tubular bones and a reduced fracture risk (106). Due to the increased bone mass of the skull, headaches and cranial nerve entrapment are commonly reported in these patients (115). Furthermore, these disorders are all caused by heterozygous gain-of-function mutations in LRP5. All mutations are located in the first β-propeller domain of the protein, and functional studies demonstrated that they disrupt the binding of canonical WNT signaling inhibitors sclerostin and DKK1 with the co-receptor. Although mutations in DKK1 are not reported in patients with monogenic skeletal disorders, different studies have shown that DKK1 is an important regulator of the WNT signaling pathway and bone mass via its interaction with LRP5 (116). As a result, mutations disrupting the binding of LRP5 with sclerostin and DKK1 result in increased canonical WNT signaling activity which consequently leads to increased bone formation (116–118).
LRP6 is the closest homolog of LRP5, and functional studies demonstrated that LRP6, similar to LRP5, is a co-receptor of the canonical WNT signaling pathway (104). However, mutations in LRP6 are less common compared to LRP5, most likely due to a more important role for LRP6 during embryonic development. However, recently, heterozygous mutations in the first β-propeller domain of LRP6 were identified in two families with HBM phenotype. The amino acids mutated in LRP6 are homologs to known LRP5 mutations (107). Although no functional studies were performed, it is highly likely that the mutations, similar to those reported for LRP5, disturb the binding of sclerostin and DKK1 and consequently result in increased WNT signaling activity and increased bone mass. The skeletal phenotype of LRP6 mutant patients was similar to the phenotype reported in patients with LRP5 mutations. However, in the small group of patients with HBM mutations in LRP6, no cranial nerve entrapment was reported which is commonly found in LRP5 HBM mutant patients. In addition, in patients with HBM mutations in LRP6, absence of the adult maxillary lateral incisors was reported. Since the latter is never reported in LRP5 HBM mutant patients, this finding can maybe be used to distinguish between both HBM types (107). Besides the high bone mass phenotype, heterozygous loss-of-function mutations in LRP6 are previously shown to cause coronary artery disease and tooth agenesis (119, 120). Remarkably, in the family with coronary artery disease due to a mutation in LRP6 described by Mani et al. (119), osteoporosis was also reported. Based on the role of LRP6 in WNT signaling, it was suggested that the observed osteoporosis phenotype was also caused by the mutation in LRP6.
Another LRP receptor which is implicated in the regulation of bone formation is LRP4. In contrast to LRP5/6, LRP4 is involved in the inhibition of the canonical WNT signaling pathway through its interaction with sclerostin. The identification of disease-causing LRP4 mutations in syndromic disorders demonstrated that LRP4 is not only important in the regulation of bone mass but also regulates limb and kidney development among others (51, 121, 122). Mutations in LRP4 can cause sclerosteosis type 2, Cenani–Lenz syndrome (CLS), isolated syndactyly, and congenital myasthenia gravis depending on type and location of the mutations (51, 122–124). As myasthenia gravis patients have no skeletal phenotype and the mutations do not affect WNT signaling activity (125), it is not further discussed in this review.
The clinical phenotype of sclerosteosis type 2 is highly similar to the sclerosteosis type 1 phenotype previously described. Sclerosteosis type 2 causing mutations in LRP4 are located in the third β-propeller domain and result in decreased binding of sclerostin (51, 124). Due to the impaired LRP4–sclerostin binding, serum levels of sclerostin are elevated in these patients and the activity of the canonical WNT signaling pathway is increased in osteoblasts, leading to increased bone formation (51, 124).
As mentioned, bi-allelic mutations in LRP4 can also cause CLS. CLS is marked by syndactyly, synostosis, and renal abnormalities, similar to what has been discussed above for patients with a mutation in GREM1 that interacts with LRP4. CLS-causing mutations are spread throughout LRP4, except for the third β-propeller domain (122, 123). Functional studies indicated CLS-causing mutations result in a decreased expression of the receptor at the cell membrane. This indicates that CLS-causing mutations have an effect not only on the inhibition of the canonical WNT signaling pathway by sclerostin but also on the regulation of other pathways such as MUSK-agrin signaling. In addition, in an individual with isolated syndactyly, compound heterozygous missense mutations within the fourth β-propeller domain have been described (126). In vitro studies demonstrated that the presence of the mutations results in decreased inhibition of the WNT signaling activity by LRP4 similar as reported for CLS mutations. Therefore, it is suggested that these patients might suffer from a mild type of CLS.
Activation of the different FZD receptors and co-receptors results intracellularly in a complex signaling cascade which ultimately leads to transcription of target genes and regulation of a wide range of cellular processes. There are many proteins involved in this intracellular signaling; however, only a few underlie the development of rare inherited skeletal dysplasias.
The disheveled protein family consists of three members (DVL1-3) which all act downstream of FZD receptors in both the canonical and non-canonical WNT signaling pathway. The DVL proteins share highly conserved regions, namely, the amino-terminal DIX (N-terminus) domain, a PDZ (central) domain, a carboxyl-terminal DEP (C-terminus) domain, and two regions with positively charged amino acid residues (127). These conserved regions are important to mediate protein–protein interactions and define the further propagation of the signal through either the canonical or non-canonical pathway. Furthermore, DVL can move from the cytoplasm to the nucleus, and this is also required for proper propagation of the WNT signaling. To ensure the movement of DVL to the nucleus, DVL proteins contain a nuclear export sequence (NES) and nuclear localization sequence (NLS) (128).
DVL proteins are highly important for normal functioning of the different WNT signaling pathways. Genetic defects can consequently affect the skeleton. Heterozygous mutations in both DVL1 and DVL3 have been identified in individuals with autosomal dominant RS (ADRS) types 2 and 3, respectively (129–131). As mentioned, ADRS is marked by skeletal abnormalities, genital abnormalities, and dysmorphic facial features (132).
In 2015, two independent research groups reported the identification of de novo frameshift mutations in exon 14 of DVL1 in patients with ADRS without mutations in WNT5A (129, 130). In total, nine different frameshift mutations, all located in the penultimate exon of DVL1, have been reported so far. All mutations result in the same premature stop codon in the last exon and escape nonsense-mediated mRNA decay. The mutations are located downstream of the abovementioned conserved domains, and all mutations result in a novel shared, highly basic C-terminal tail sequence that most likely exerts novel signaling functions (129–131). More in detail, in vitro luciferase reporter assays demonstrated that co-transfection of wild-type and mutant DVL1, representing the heterozygous mutations, results in significantly increased canonical WNT signaling activity compared to the effect of wild-type DVL1 alone. More interestingly, several patients with mutations in DVL1 show increased bone mass which is not seen in the other autosomal dominant or recessive forms of RS (129). Most likely, the increased bone mass in ADRS2 patients is caused by the effect of DVL1 mutations on canonical WNT signaling. In addition, ADRS2 patients often have macrocephaly and normal stature (>10% percentile) (129, 130).
In a subsequent study, it was shown that not only mutations in DVL1 but also DVL3 can cause ADRS (131). Similar as for DVL1, the mutations result in a frameshift to the−1-reading frame and a shared premature stop codon in the last exon. The DVL3 mutants are shown to escape nonsense-mediated mRNA decay. All identified mutations have 83 C-terminal amino acids in common (131, 133). The functional effect of this C-terminal tail on regulation of canonical and/or non-canonical WNT signaling pathway is still unknown. Patients with mutations in DVL3 (ADRS3) can be distinguished from ADRS2 patients based on the presence of short stature and macrocephaly. Furthermore, congenital heart defects and cleft lip and/or cleft palate were reported in the majority of the patients (131).
Nucleoredoxin (NXN) is an oxidative stress response protein which is highly expressed during murine limb bud development and can directly bind DVL via the PDZ domain. By binding to DVL, NXN can inhibit DVL signaling activity and consequently different WNT signaling pathways (134–136). It is suggested that the interaction of NXN with DVL is a key regulatory mechanism to maintain spatial and temporal balance between canonical and non-canonical WNT signaling during development (84). Recently, NXN was identified as the causative gene for autosomal recessive RS type 2 (ARRS2). In two families without mutations in the known causative gene ROR2, White et al. (84) identified bi-allelic variants which segregated with the ARRS2 phenotype consisting of short stature, mesomelic shortening, genital hypoplasia, and typical facial dysmorphisms. Mice lacking Nxn also show craniofacial defects, suggesting that the mutations reported in the patients are loss-of-function mutations (136, 137).
RAC3 is a member of the Rac subfamily of the Rho family of GTPases. Upon activation by WNT ligands, RAC3 can interact with DVL to activate downstream signaling including c-jun NH2-terminal kinase (JNK)/c-jun phosphorylation which is necessary for cytoskeletal organization. White et al. (84) reported in 2018 the identification of a rare variant in RAC3 in a patient with a Robinow-like phenotype including facial dysmorphism and developmental delay, seizures, an abnormal electroencephalogram, and a thin corpus callosum. Based on the role of RAC3 in the WNT/PCP pathway (Figure 3), they suggest that the identified de novo variant can cause the Robinow-like phenotype in the patient (84). However, additional functional studies or identification of additional affected individuals with possible pathogenic variants is necessary. In a more recent study, Costain et al. (138) also identified heterozygous missense variants in RAC3 in patients with neurodevelopmental disorder with structural brain anomalies and dysmorphic facial features. Since there is some overlap between the clinical phenotype of the patients described in both reports, most likely the patients described by both research groups suffer from the same disease.
Adenomatous polyposis coli (APC) is a tumor suppressor gene that can regulate canonical WNT signaling as a part of the multiprotein destruction complex that targets β-catenin for phosphorylation and degradation (139). APC2 or APC-like protein is a homolog of APC which is mostly expressed in the postmitotic neurons during development (140). Both proteins share a 20-amino acid repeat motif that is capable of binding β-catenin. However, in addition to this shared motif, APC also contains a second motif which is lacking in APC2 and can bind β-catenin with a higher affinity. Nevertheless, APC2 is still capable of depleting intracellular β-catenin, although less efficient than APC (141).
Homozygous loss-of-function mutations in APC2 have recently been described in patients with a Sotos-like phenotype (142). Sotos syndrome is an overgrowth syndrome, also known as cerebral gigantism. Individuals with APC2 mutations resemble patients with Sotos syndrome because they show intellectual disability and relative macrocephaly with a long face and prominent chin (142). The role of the canonical WNT signaling pathway in the development of this disorder is not yet investigated.
APC Membrane Recruitment Protein 1 (AMER1), also known as FAM123B or Wilms tumor on the X chromosome (WTX), is an intracellular inhibitor of the canonical WNT signaling pathway. AMER1 can interact with the AXIN/APC/GSK3β/β-catenin multiprotein complex and enhances the ubiquitination and degradation of β-catenin (143, 144). AMER1 is located on the X-chromosome, and loss-of-function mutations or gene deletions can cause the X-linked dominant disorder osteopathia striata with cranial sclerosis (OSCS) (145). Radiographs of patients with OSCS show cranial sclerosis and dense linear striations in the submetaphyseal regions of the long bones and pelvis. Besides these radiographic signs, patients with OSCS often also present with macrocephaly, broad nasal bridge, frontal bossing, ocular hypertelorism, hearing loss, and palate abnormalities. Less common symptoms of OSCS are cardiac malformations and cognitive defects. In males, OSCS can be lethal due to severe heart defects and/or gastrointestinal malformations (143, 145, 146).
Porcupine O-acyltransferase (PORCN) is a member of the evolutionarily conserved porcupine (PORC) gene family of endoplasmic reticulum transmembrane proteins that are involved in the processing and secretion of WNT ligands. PORCN is located on the X-chromosome, and heterozygous loss-of-function mutations can cause focal dermal hypoplasia (FDH) also known as Goltz syndrome in female patients. Goltz syndrome caused by mutations in PORCN is rarely reported in males which leads to the suspicion that non-mosaic Goltz syndrome is embryonically lethal for males (147, 148). Goltz syndrome is a multisystem syndrome that is characterized by dermal abnormalities including focal dermal hypoplasia among others, ophthalmologic features, facial anomalies, and skeletal abnormalities. The latter are reported in ~80% of the patients and include syndactyly or polydactyly, osteopathic striae, hypoplasia or absence of digits, scoliosis, and/or facial asymmetry (148, 149).
The identification of novel disease-causing genes for rare skeletal dysplasias accelerated significantly in the last decades, initially by positional cloning efforts and more recently by the availability of next-generation sequencing technology. This resulted in the identification of the disease-causing gene for 92% of the skeletal disorders (6). The increased knowledge on monogenic diseases resulted in a better understanding of the pathological mechanisms and highlighted which pathways regulate specific cellular processes. This information is also relevant for understanding more common multifactorial diseases. Furthermore, it has been shown that therapeutic targets which are based on genetic evidence from Mendelian traits as well as genome-wide association studies (GWASs) are more likely to be successful in clinical studies for multifactorial diseases (150). Here, we focused on skeletal dysplasias caused by mutations in genes that encode proteins that are directly involved in one of the WNT signaling pathways. As shown in Table 1, mutations in these genes can result in a variety of skeletal dysplasias, each with specific clinical features. The broad spectrum of clinical observations reflect the cellular and spatial functions of WNT signaling, some of them associated with embryonal development, others with bone mass and homeostasis in adult life. For example, the clinical features of RS and OMOD are similar which led to the hypothesis that all causative genes are involved in the WNT/PCP pathway which is previously shown to be important during limb development (Figure 2) (102). On the other hand, the influence of canonical WNT signaling on bone mass was highlighted by unraveling the underlying pathogenic mechanisms of disorders with a progressively increasing bone mass such as sclerosteosis, Van Buchem disease, and high bone mass phenotypes (osteosclerosis) (51, 53, 57, 107, 113). The genes causing these disorders, SOST, LRP4, LRP5, and LRP6, are all involved in the canonical WNT signaling pathway (Figure 3), and all mutations reported result in an increased canonical WNT signaling (Table 1). In addition to the conditions with increased bone mass mentioned, mutations in WNT1, a ligand inducing canonical WNT signaling, and LRP5 can also result in decreased canonical WNT signaling activity and consequently decreased bone mass (Table 1; Figure 3) (12–15, 109). All these data show that the canonical WNT signaling pathway is an important regulator of bone mass as is also corroborated by the observation in GWASs that many of these genes harbor genetic polymorphisms associated with bone mineral density or fractures (151, 152, 154) (Table 1). This is in line with a general observation that there is an enrichment in genes for Mendelian disorders near regions associated in GWASs with a similar phenotype (157).
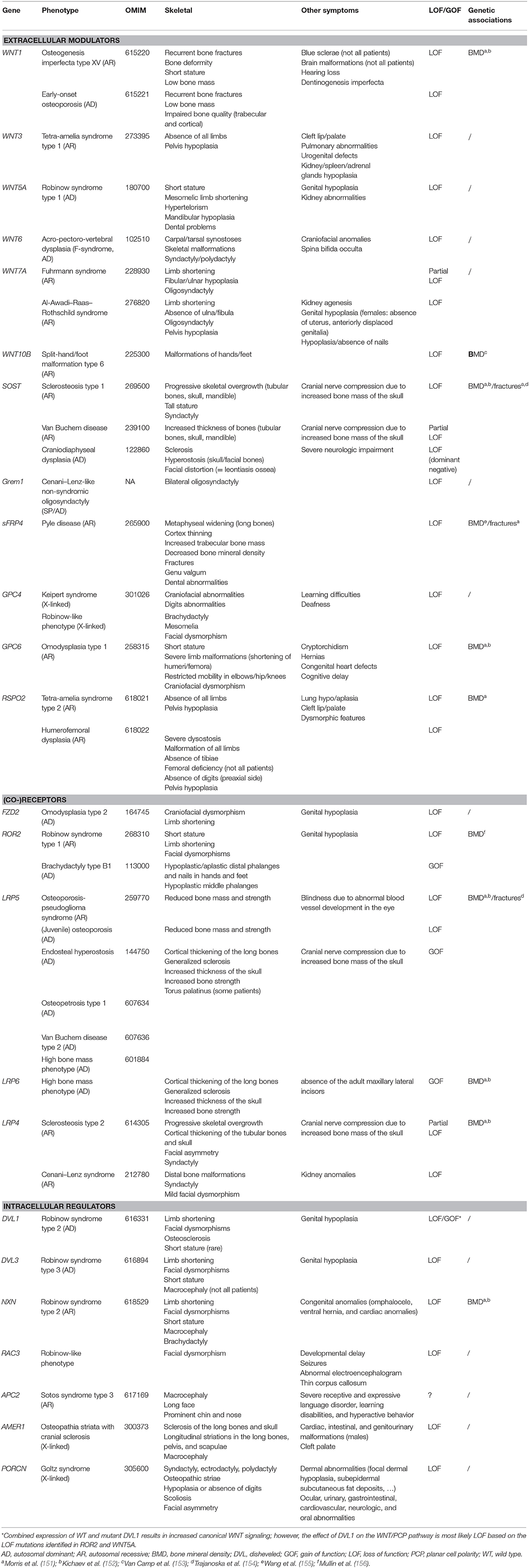
Table 1. Overview of the main skeletal dysplasias caused by mutations in genes involved in WNT signaling.
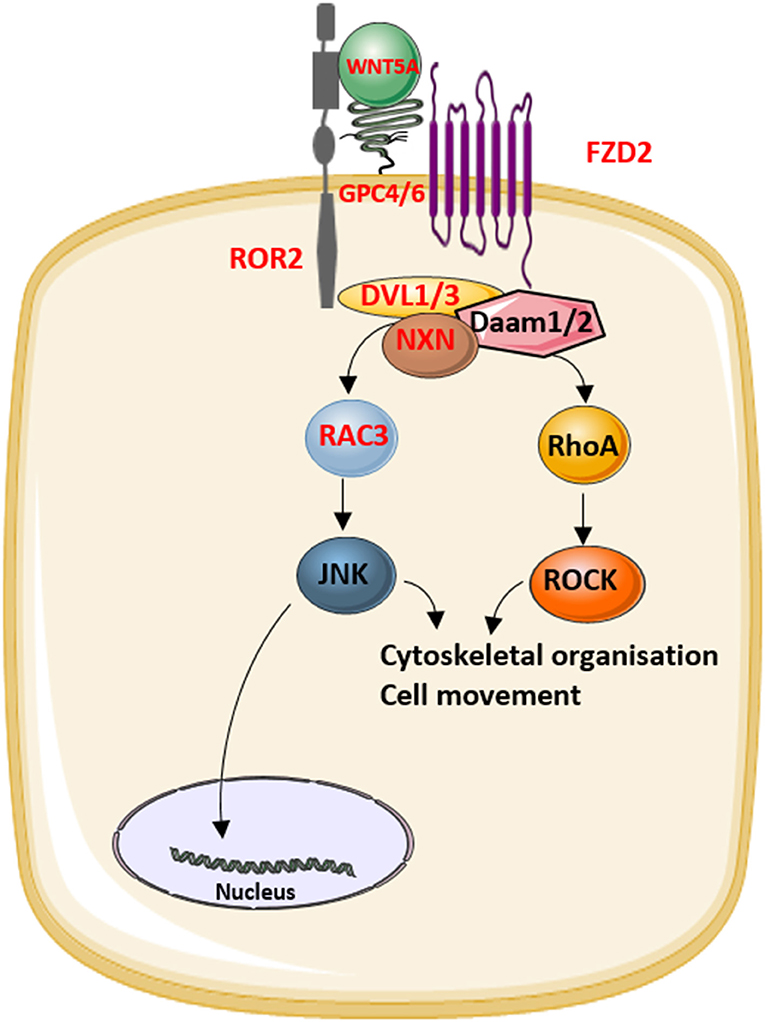
Figure 2. The role of the proteins that underlie Robinow syndrome or omodysplasia (indicated in red) in the WNT/planar cell polarity (PCP) pathway.
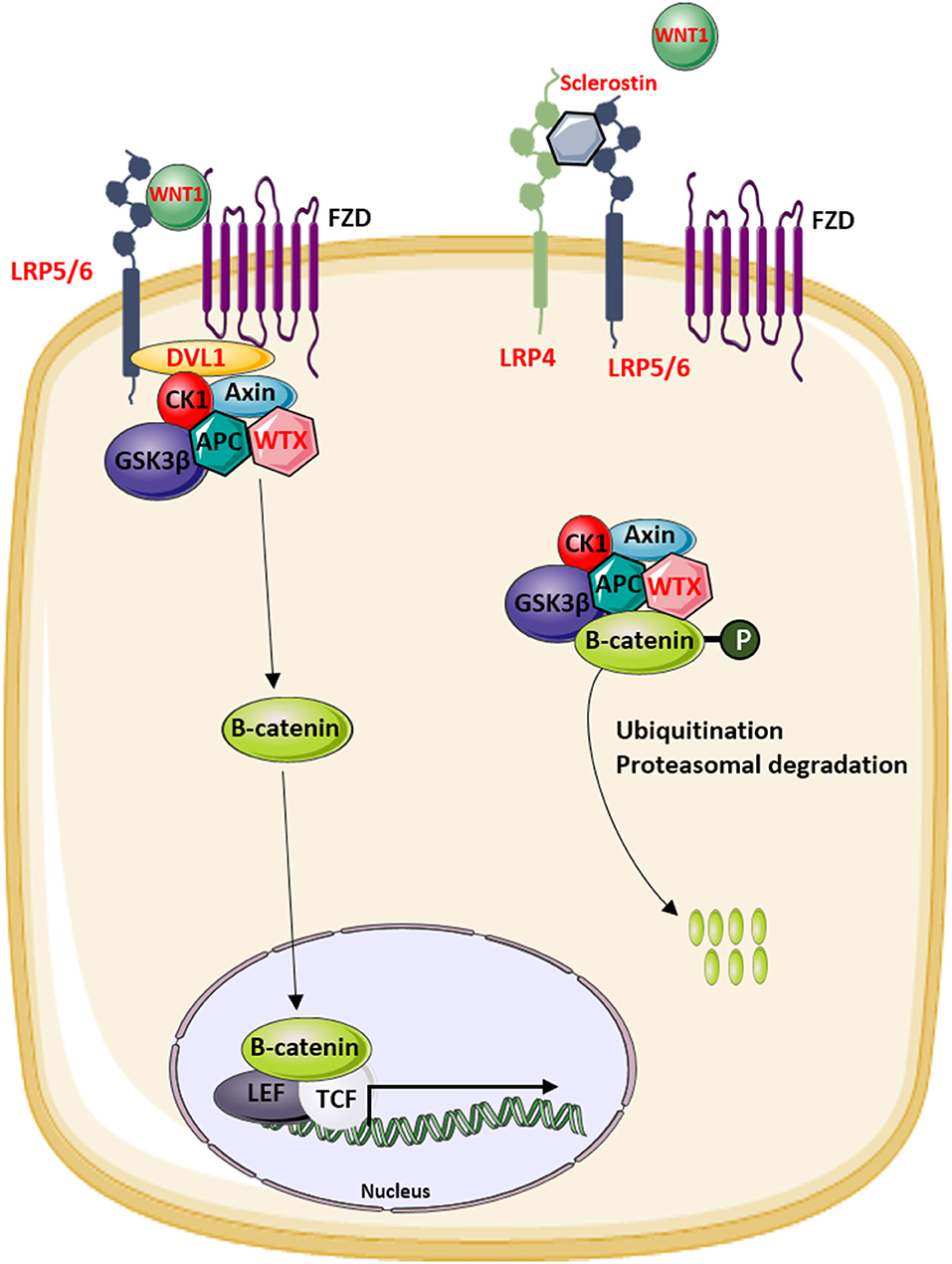
Figure 3. Overview of the modulators of the canonical WNT signaling pathway that are involved in the regulation of bone mass in human disease. Proteins indicated in red are mutated in patients with increased or decreased bone mass.
The evidence for an important role for WNT signaling in regulating bone mass makes it an obvious candidate for drug development for the treatment of osteoporosis. The incidental finding that lithium chloride (LiCl), a first-line treatment for bipolar disorders, increases canonical WNT signaling activity and increases bone formation already confirms the therapeutic potential of the pathway (158, 159). But at the same time, phenotypic features of single mutation diseases can give insights into possible adverse effects of interfering with the pathway. For example, a rare nonsense mutation (c.376C>T) was identified in the LGR4 gene, which encodes a receptor for RSPO1–4, activators of WNT signaling. The mutation is specific for the Icelandic population, as described by Styrkarsdottir et al. (160), and shows strong association with osteoporotic fractures and low bone mineral density (BMD). But at the same time, the variant is associated with an increased risk of squamous cell carcinoma of the skin and biliary tract cancer (160). Also for other types of cancer, abnormal WNT signaling has been reported. These cancers can be caused by single mutations (germline or mosaic) in, for example, APC or WTX, but also common genetic variation in WNT3, DVL1, and NXN is previously associated with increased cancer risk (161–166). However, increased cancer risk is not reported for any of the rare skeletal dysplasias described in this review.
Currently, the treatment of osteoporosis is mainly based on the use of antiresorptive agents, such as bisphosphonates, without the capacity to rebuild the lost bone. Consequently, there is an urgent need for more specific, anabolic therapies for osteoporosis, making canonical WNT signaling of major interest. The pharmaceutical industry focused on sclerostin as a therapeutic target based on the expression of sclerostin being almost exclusive in bone tissue, the progressive increase in bone mass in adults with sclerosteosis and Van Buchem disease and on the absence of non-skeletal clinical features in these patients (167). Clinical trials demonstrated that treatment with romosozumab (Evenity™, Amgen/UCB), a monoclonal antibody for sclerostin, results in increased bone mass and decreased fracture risk (168, 169). However, a phase III clinical trial demonstrated that although romosozumab is clearly beneficial for bone health, treatment was also associated with slightly increased incidence of cardiovascular events (169). Increased incidence of cardiovascular events was observed in men treated with romosozumab compared to placebo (170) and in postmenopausal women treated with romosozumab compared to women treated with alendronate (169). When compared to postmenopausal women treated with placebo, romosozumab-treated individuals have a similar incidence of cardiovascular events (168). In addition, in patients pretreated with alendronate, the incidence of cardiovascular events was lower (171). Furthermore, in postmenopausal women who were treated with alendronate after romosozumab treatment, the difference in cardiovascular events remained stable compared to patients treated with alendronate alone (169). Therefore, additional studies are needed to investigate whether combined treatment of alendronate and romosozumab can reduce the cardiovascular events. Clinical data from sclerosteosis and Van Buchem patients who lack sclerostin do not report an increased incidence of cardiovascular events in these patients (172) which suggests that targeting sclerostin does not cause cardiovascular disease. However, for now, the use of romosozumab is only approved for the treatment of severe osteoporosis in postmenopausal women but is contraindicated in patients with an increased cardiovascular risk. Therefore, the identification of new therapeutic targets potentially interfering with WNT signaling remains imperative.
In conclusion, mutations in the WNT signaling pathway can lead to a broad range of skeletal dysplasias. Especially those characterized by an increased bone mass have been instrumental in highlighting the role of WNT signaling in bone formation and homeostasis with therapeutic applications for the treatment of osteoporosis.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
EB holds a postdoctoral Grant (12A3814N) with the research foundation Flanders (FWO Vlaanderen). This work was supported by a Methusalem grant to the Genomed consortium (FFB190208).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. (1982) 31:99–109. doi: 10.1016/0092-8674(82)90409-3
2. Nusse R, Brown A, Papkoff J, Scambler P, Shackleford G, Mcmahon A, et al. A new nomenclature for int-1 and related genes: the Wnt gene family. Cell. (1991) 64:231. doi: 10.1016/0092-8674(91)90633-A
3. Katoh M. WNT and FGF gene clusters (review). Int J Oncol. (2002) 21:1269–73. doi: 10.3892/ijo.21.6.1269
4. Van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. (2009) 136:3205–14. doi: 10.1242/dev.033910
5. Boudin E, Van Hul W. Mechanisms in endocrinology: genetics of human bone formation. Eur J Endocrinol. (2017) 177:R69–83. doi: 10.1530/EJE-16-0990
6. Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. (2019) 179:2393–419. doi: 10.1002/ajmg.a.61366
7. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. (1997) 11:3286–305. doi: 10.1101/gad.11.24.3286
9. Joeng KS, Lee YC, Jiang MM, Bertin TK, Chen Y, Abraham AM, et al. The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet. (2014) 23:4035–42. doi: 10.1093/hmg/ddu117
10. Joeng KS, Lee YC, Lim J, Chen Y, Jiang MM, Munivez E, et al. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest. (2017) 127:2678–88. doi: 10.1172/JCI92617
11. Cao YJ, Zhang H, Zhang ZL. Novel Mutations In The Wnt1, Tmem38b, P4hb, And Pls3 Genes in four unrelated Chinese families with osteogenesis imperfecta. Endocr Pract. (2019) 25:230–41. doi: 10.4158/EP-2018-0443
12. Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. (2013) 50:345–8. doi: 10.1136/jmedgenet-2013-101567
13. Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. (2013) 92:565–74. doi: 10.1016/j.ajhg.2013.02.010
14. Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. (2013) 368:1809–16. doi: 10.1056/NEJMoa1215458
15. Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. (2013) 92:590–7. doi: 10.1016/j.ajhg.2013.02.009
16. Brunetti G, Papadia F, Tummolo A, Fischetto R, Nicastro F, Piacente L, et al. Impaired bone remodeling in children with osteogenesis imperfecta treated and untreated with bisphosphonates: the role of DKK1, RANKL, and TNF-alpha. Osteoporos Int. (2016) 27:2355–65. doi: 10.1007/s00198-016-3501-2
17. Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. (2012) 44:491–501. doi: 10.1038/ng.2249
18. Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. (1999) 22:361–5. doi: 10.1038/11932
19. Niemann S, Zhao C, Pascu F, Stahl U, Aulepp U, Niswander L, et al. Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am J Hum Genet. (2004) 74:558–63. doi: 10.1086/382196
20. Gupta AM, Jaipal NU. A very rare case of isolated tetra-amelia. J Evol Med Dent Sci. (2017) 6:337–9. doi: 10.14260/Jemds/2017/75
21. Kaur L, Singh D, Kaur M. P. Diagnosis of tetra-amelia in the first trimester. J Fetal Med. (2017) 4:37–9. doi: 10.1007/s40556-016-0111-1
22. Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science. (2012) 337:59–64. doi: 10.1126/science.1222879
23. Kobayashi Y, Uehara S, Udagawa N, Takahashi N. Regulation of bone metabolism by Wnt signals. J Biochem. (2016) 159:387–92. doi: 10.1093/jb/mvv124
24. Maeda K, Kobayashi Y, Koide M, Uehara S, Okamoto M, Ishihara A, et al. The regulation of bone metabolism and disorders by Wnt signaling. Int J Mol Sci. (2019) 20:5525. doi: 10.3390/ijms20225525
25. Robinow M, Silverman FN, Smith HD. A newly recognized dwarfing syndrome. Am J Dis Child. (1969) 117:645–51. doi: 10.1001/archpedi.1969.02100030647005
26. Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME, et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dyn. (2010) 239:327–37. doi: 10.1002/dvdy.22156
27. Roifman M, Marcelis CL, Paton T, Marshall C, Silver R, Lohr JL, et al. De novo WNT5A-associated autosomal dominant Robinow syndrome suggests specificity of genotype and phenotype. Clin Genet. (2015) 87:34–41. doi: 10.1111/cge.12401
28. Camera G, Camera A, Pozzolo S, Costa M, Mantero R. F-syndrome (F-form of acro-pectoro-vertebral dysplasia): report on a second family. Am J Med Genet. (1995) 57:472–5. doi: 10.1002/ajmg.1320570322
29. Thiele H, Mccann C, Van't Padje S, Schwabe GC, Hennies HC, Camera G, et al. (2004). Acropectorovertebral dysgenesis (F syndrome) maps to chromosome 2q36. J. Med. Genet. 41, 213–218. doi: 10.1136/jmg.2003.014894
30. Grosse FH, Opitz JJM. The F-form of acropectorovertebral dysplasia: the F-syndrome. Birth Defects Orig Artic Ser. (1969) 3:48–63.
31. Kantaputra PN, Mundlos S, Sripathomsawat W. A novel homozygous Arg222Trp missense mutation in WNT7A in two sisters with severe Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Med Genet A. (2010) 152a:2832–7. doi: 10.1002/ajmg.a.33673
32. Lan L, Wang W, Huang Y, Bu X, Zhao C. Roles of Wnt7a in embryo development, tissue homeostasis, and human diseases. J Cell Biochem. (2019) 120:18588–98. doi: 10.1002/jcb.29217
33. Woods CG, Stricker S, Seemann P, Stern R, Cox J, Sherridan E, et al. Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet. (2006) 79:402–8. doi: 10.1086/506332
34. Eyaid W, Al-Qattan MM, Al Abdulkareem I, Fetaini N, Al Balwi M. A novel homozygous missense mutation (c.610G>A, p.Gly204Ser) in the WNT7A gene causes tetra-amelia in two Saudi families. Am J Med Genet A. (2011) 155a:599–604. doi: 10.1002/ajmg.a.33717
35. Garavelli L, Wischmeijer A, Rosato S, Gelmini C, Reverberi S, Sassi S, et al. Al-Awadi-Raas-Rothschild (limb/pelvis/uterus-hypoplasia/aplasia) syndrome and WNT7A mutations: genetic homogeneity and nosological delineation. Am J Med Genet A. (2011) 155a:332–6. doi: 10.1002/ajmg.a.33793
36. Mutlu MB, Cetinkaya A, Koc N, Ceylaner G, Erguner B, Aydin H, et al. A novel missense mutation, p. (R102W) in WNT7A causes Al-Awadi Raas-Rothschild syndrome in a fetus. Eur J Med Genet. (2016) 59:604–6. doi: 10.1016/j.ejmg.2016.09.009
37. Sun L, Huang Y, Zhao S, Zhong W, Lin M, Guo Y, et al. Advances in understanding the genetics of syndromes involving congenital upper limb anomalies. (2019) 4:3. doi: 10.21037/aoj.2019.06.03
38. Al-Qattan MM, Al-Balwi M, Eyaid W, Al-Abdulkarim I, Al-Turki S. Congenital duplication of the palm syndrome: gene analysis and the molecular basis of its clinical features. J Hand Surg Eur. (2009) 34:247–51. doi: 10.1177/1753193408099828
39. Al-Qattan MM, Alkuraya FS. Cenani-Lenz syndrome and other related syndactyly disorders due to variants in LRP4, GREM1/FMN1, and APC: Insight into the pathogenesis and the relationship to polyposis through the WNT and BMP antagonistic pathways. Am J Med Genet A. (2019) 179:266–79. doi: 10.1002/ajmg.a.60694
40. Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA. (2005) 102:3324–9. doi: 10.1073/pnas.0408742102
41. Glass DA II, Karsenty G. Molecular bases of the regulation of bone remodeling by the canonical Wnt signaling pathway. Curr Top Dev Biol. (2006) 73:43–84. doi: 10.1016/S0070-2153(05)73002-7
42. Kantaputra PN, Kapoor S, Verma P, Intachai W, Ketudat Cairns JR. Split hand-foot malformation and a novel WNT10B mutation. Eur J Med Genet. (2018) 61:372–5. doi: 10.1016/j.ejmg.2018.02.001
43. Ugur SA, Tolun A. Homozygous WNT10b mutation and complex inheritance in Split-Hand/Foot Malformation. Hum Mol Genet. (2008) 17:2644–53. doi: 10.1093/hmg/ddn164
44. Blattner A, Huber AR, Rothlisberger B. Homozygous nonsense mutation in WNT10B and sporadic split-hand/foot malformation (SHFM) with autosomal recessive inheritance. Am J Med Genet A. (2010) 152a:2053–6. doi: 10.1002/ajmg.a.33504
45. Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, et al. Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. (2009) 284:10890–900. doi: 10.1074/jbc.M807994200
46. Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. (2003) 22:6267–76. doi: 10.1093/emboj/cdg599
47. Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. (2005) 280:19883–7. doi: 10.1074/jbc.M413274200
48. Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. (2005) 280:26770–5. doi: 10.1074/jbc.M504308200
49. Kim SJ, Bieganski T, Sohn YB, Kozlowski K, Semenov M, Okamoto N, et al. Identification of signal peptide domain SOST mutations in autosomal dominant craniodiaphyseal dysplasia. Hum Genet. (2011) 129:497–502. doi: 10.1007/s00439-011-0947-3
50. Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. (2001) 68:577–89. doi: 10.1086/318811
51. Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. (2011) 286:19489–500. doi: 10.1074/jbc.M110.190330
52. Shen C, Xiong WC, Mei L. LRP4 in neuromuscular junction and bone development and diseases. Bone. (2015) 80:101–8. doi: 10.1016/j.bone.2015.05.012
53. Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. (2001) 10:537–43. doi: 10.1093/hmg/10.5.537
54. Appelman-Dijkstra NM, Papapoulos SE. From disease to treatment: from rare skeletal disorders to treatments for osteoporosis. Endocrine. (2016) 52:414–26. doi: 10.1007/s12020-016-0888-7
55. Sebastian A, Loots GG. Genetics of Sost/SOST in sclerosteosis and van Buchem disease animal models. Metab Clin Exp. (2018) 80:38–47. doi: 10.1016/j.metabol.2017.10.005
56. Van Hul W, Balemans W, Van Hul E, Dikkers FG, Obee H, Stokroos RJ, et al. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet. (1998) 62:391–9. doi: 10.1086/301721
57. Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. (2002) 39:91–7. doi: 10.1136/jmg.39.2.91
58. Bieganski T, Baranska D, Miastkowska I, Kobielski A, Gorska-Chrzastek M, Kozlowski K. A boy with severe craniodiaphyseal dysplasia and apparently normal mother. Am J Med Genet A. (2007) 143A:2435–43. doi: 10.1002/ajmg.a.31938
59. Brazil DP, Church RH, Surae S, Godson C, Martin F. BMP signalling: agony and antagony in the family. Trends Cell Biol. (2015) 25:249–64. doi: 10.1016/j.tcb.2014.12.004
60. Gazzerro E, Pereira RC, Jorgetti V, Olson S, Economides AN, Canalis E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. (2005) 146:655–65. doi: 10.1210/en.2004-0766
61. Gazzerro E, Smerdel-Ramoya A, Zanotti S, Stadmeyer L, Durant D, Economides AN, et al. Conditional deletion of gremlin causes a transient increase in bone formation and bone mass. J Biol Chem. (2007) 282:31549–57. doi: 10.1074/jbc.M701317200
62. Dimitrov BI, Voet T, De Smet L, Vermeesch JR, Devriendt K, Fryns JP, et al. Genomic rearrangements of the GREM1-FMN1 locus cause oligosyndactyly, radio-ulnar synostosis, hearing loss, renal defects syndrome and Cenani–Lenz-like non-syndromic oligosyndactyly. J Med Genet. (2010) 47:569–74. doi: 10.1136/jmg.2009.073833
63. Zuniga A, Michos O, Spitz F, Haramis AP, Panman L, Galli A, et al. Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes Dev. (2004) 18:1553–64. doi: 10.1101/gad.299904
64. Umair MAF, Bilal M, Abbas S. Syndactyly genes and classification: a mini-review. JBC Genetics. (2018) 1:34–47. doi: 10.24911/JBCGenetics/183-1532177257
65. Capdevila J, Tsukui T, Rodriquez Esteban C, Zappavigna V, Izpisua Belmonte JC. Control of vertebrate limb outgrowth by the proximal factor Meis2 and distal antagonism of BMPs by Gremlin. Mol Cell. (1999) 4:839–49. doi: 10.1016/S1097-2765(00)80393-7
66. Merino R, Rodriguez-Leon J, Macias D, Ganan Y, Economides AN, Hurle JM. The BMP antagonist Gremlin regulates outgrowth, chondrogenesis and programmed cell death in the developing limb. Development. (1999) 126:5515–22.
67. Gu Q, Luo Y, Chen C, Jiang D, Huang Q, Wang X. GREM1 overexpression inhibits proliferation, migration and angiogenesis of osteosarcoma. Exp Cell Res. (2019) 384:111619. doi: 10.1016/j.yexcr.2019.111619
68. Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. (2003) 116:2627–34. doi: 10.1242/jcs.00623
69. Haraguchi R, Kitazawa R, Mori K, Tachibana R, Kiyonari H, Imai Y, et al. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci Rep. (2016) 6:25198. doi: 10.1038/srep25198
70. Pawar NM, Rao P. Secreted frizzled related protein 4 (sFRP4) update: A brief review. Cell Signal. (2018) 45:63–70. doi: 10.1016/j.cellsig.2018.01.019
71. Claudel M, Jouzeau JY, Cailotto F. Secreted Frizzled-related proteins (sFRPs) in osteo-articular diseases: much more than simple antagonists of Wnt signaling? FEBS J. (2019) 286:4832–51. doi: 10.1111/febs.15119
72. Kiper POS, Saito H, Gori F, Unger S, Hesse E, Yamana K, et al. Cortical-Bone Fragility–Insights from sFRP4 Deficiency in Pyle's Disease. N Engl J Med. (2016) 374:2553–62. doi: 10.1056/NEJMoa1509342
73. Galada C, Shah H, Shukla A, Girisha KM. A novel sequence variant in SFRP4 causing Pyle disease. J Hum Genet. (2017) 62:575–6. doi: 10.1038/jhg.2016.166
74. Arboleya L, Queiro R, Alperi M, Lorenzo JA, Ballina J. Pyle's disease: a human model of differentiated cortical and trabecular homeostasis. Reumatol Clin. (2018) 16:56–8.
75. Veugelers M, De Cat B, Ceulemans H, Bruystens AM, Coomans C, Durr J, et al. Glypican-6, a new member of the glypican family of cell surface heparan sulfate proteoglycans. J Biol Chem. (1999) 274:26968–77. doi: 10.1074/jbc.274.38.26968
76. Campos-Xavier AB, Martinet D, Bateman J, Belluoccio D, Rowley L, Tan TY, et al. Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia. Am J Hum Genet. (2009) 84:760–70. doi: 10.1016/j.ajhg.2009.05.002
77. Capurro M, Izumikawa T, Suarez P, Shi W, Cydzik M, Kaneiwa T, et al. Glypican-6 promotes the growth of developing long bones by stimulating Hedgehog signaling. J Cell Biol. (2017) 216:2911–26. doi: 10.1083/jcb.201605119
78. Reichsman F, Smith L, Cumberledge S. Glycosaminoglycans can modulate extracellular localization of the wingless protein and promote signal transduction. J Cell Biol. (1996) 135:819–27. doi: 10.1083/jcb.135.3.819
79. Ohkawara B, Yamamoto TS, Tada M, Ueno N. Role of glypican 4 in the regulation of convergent extension movements during gastrulation in Xenopus laevis. Development. (2003) 130:2129–38. doi: 10.1242/dev.00435
80. Song HH, Shi W, Xiang YY, Filmus J. The loss of glypican-3 induces alterations in Wnt signaling. J Biol Chem. (2005) 280:2116–25. doi: 10.1074/jbc.M410090200
82. Amor DJ, Stephenson SEM, Mustapha M, Mensah MA, Ockeloen CW, Lee WS, et al. Pathogenic variants in GPC4 cause Keipert syndrome. Am J Hum Genet. (2019) 104:914–24. doi: 10.1016/j.ajhg.2019.02.026
83. Keipert JA, Fitzgerald MG, Danks DM. A new syndrome of broad terminal phalanges and facial abnormalities. Aust Paediatr J. (1973) 9:10–3. doi: 10.1111/j.1440-1754.1973.tb02215.x
84. White JJ, Mazzeu JF, Coban-Akdemir Z, Bayram Y, Bahrambeigi V, Hoischen A, et al. WNT signaling perturbations underlie the genetic heterogeneity of robinow syndrome. Am J Hum Genet. (2018) 102:27–43. doi: 10.1016/j.ajhg.2017.10.002
85. Paine-Saunders S, Viviano BL, Saunders S. GPC6, a novel member of the glypican gene family, encodes a product structurally related to GPC4 and is colocalized with GPC5 on human chromosome 13. Genomics. (1999) 57:455–8. doi: 10.1006/geno.1999.5793
86. Shi GX, Mao WW, Zheng XF, Jiang LS. The role of R-spondins and their receptors in bone metabolism. Prog Biophys Mol Biol. (2016) 122:93–100. doi: 10.1016/j.pbiomolbio.2016.05.012
87. Kim KA, Zhao J, Andarmani S, Kakitani M, Oshima T, Binnerts ME, et al. R-Spondin proteins: a novel link to beta-catenin activation. Cell Cycle. (2006) 5:23–6. doi: 10.4161/cc.5.1.2305
88. Jin YR, Turcotte TJ, Crocker AL, Han XH, Yoon JK. The canonical Wnt signaling activator, R-spondin2, regulates craniofacial patterning and morphogenesis within the branchial arch through ectodermal-mesenchymal interaction. Dev Biol. (2011) 352:1–13. doi: 10.1016/j.ydbio.2011.01.004
89. Lebensohn AM, Rohatgi R. R-spondins can potentiate WNT signaling without LGRs. Elife. (2018) 7:e33126. doi: 10.7554/eLife.33126
90. Park S, Cui J, Yu W, Wu L, Carmon KS, Liu QJ. Differential activities and mechanisms of the four R-spondins in potentiating Wnt/beta-catenin signaling. J Biol Chem. (2018) 293:9759–69. doi: 10.1074/jbc.RA118.002743
91. Szenker-Ravi E, Altunoglu U, Leushacke M, Bosso-Lefevre C, Khatoo M, Thi Tran H, et al. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature. (2018) 557:564–9. doi: 10.1038/s41586-018-0118-y
92. Saal HM, Prows CA, Guerreiro I, Donlin M, Knudson L, Sund KL, et al. A mutation in FRIZZLED2 impairs Wnt signaling and causes autosomal dominant omodysplasia. Hum Mol Genet. (2015) 24:3399–409. doi: 10.1093/hmg/ddv088
93. Nagasaki K, Nishimura G, Kikuchi T, Nyuzuki H, Sasaki S, Ogawa Y, et al. Nonsense mutations in FZD2 cause autosomal-dominant omodysplasia: Robinow syndrome-like phenotypes. Am J Med Genet A. (2018) 176:739–42. doi: 10.1002/ajmg.a.38623
94. Turkmen S, Spielmann M, Gunes N, Knaus A, Flottmann R, Mundlos S, et al. A Novel de novo FZD2 mutation in a patient with autosomal dominant omodysplasia. Mol Syndromol. (2017) 8:318–24. doi: 10.1159/000479721
95. Warren HE, Louie RJ, Friez MJ, Frias JL, Leroy JG, Spranger JW, et al. Two unrelated patients with autosomal dominant omodysplasia and FRIZZLED2 mutations. Clin Case Rep. (2018) 6:2252–5. doi: 10.1002/ccr3.1818
96. Afzal AR, Jeffery S. One gene, two phenotypes: ROR2 mutations in autosomal recessive Robinow syndrome and autosomal dominant brachydactyly type B. Hum Mutat. (2003) 22:1–11. doi: 10.1002/humu.10233
97. Habib R, Amin-Ud-Din M, Ahmad W. A nonsense mutation in the gene ROR2 underlying autosomal dominant brachydactyly type B. Clin Dysmorphol. (2013) 22:47–50. doi: 10.1097/MCD.0b013e32835c6c8c
98. Van Bokhoven H, Celli J, Kayserili H, Van Beusekom E, Balci S, Brussel W, et al. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat Genet. (2000) 25:423–6. doi: 10.1038/78113
99. Yang K, Zhu J, Tan Y, Sun X, Zhao H, Tang G, et al. Whole-exome sequencing identified compound heterozygous variants in ROR2 gene in a fetus with Robinow syndrome. J Clin Lab Anal. (2019) 34:e23074. doi: 10.1002/jcla.23074
100. Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat Genet. (2000) 25:419–22. doi: 10.1038/78107
101. Schwarzer W, Witte F, Rajab A, Mundlos S, Stricker S. A gradient of ROR2 protein stability and membrane localization confers brachydactyly type B or Robinow syndrome phenotypes. Hum Mol Genet. (2009) 18:4013–21. doi: 10.1093/hmg/ddp345
102. Wang B, Sinha T, Jiao K, Serra R, Wang J. Disruption of PCP signaling causes limb morphogenesis and skeletal defects and may underlie Robinow syndrome and brachydactyly type B. Hum Mol Genet. (2011) 20:271–85. doi: 10.1093/hmg/ddq462
103. Schwabe GC, Tinschert S, Buschow C, Meinecke P, Wolff G, Gillessen-Kaesbach G, et al. Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am J Hum Genet. (2000) 67:822–31. doi: 10.1086/303084
104. Boudin E, Fijalkowski I, Piters E, Van Hul W. The role of extracellular modulators of canonical Wnt signaling in bone metabolism and diseases. Semin Arthritis Rheum. (2013) 43:220–40. doi: 10.1016/j.semarthrit.2013.01.004
105. Pohlkamp T, Wasser CR, Herz J. Functional roles of the interaction of APP and lipoprotein receptors. Front Mol Neurosci. (2017) 10:54. doi: 10.3389/fnmol.2017.00054
106. Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D, et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. (2003) 72:763–71. doi: 10.1086/368277
107. Whyte MP, Mcalister WH, Zhang F, Bijanki VN, Nenninger A, Gottesman GS, et al. New explanation for autosomal dominant high bone mass: Mutation of low-density lipoprotein receptor-related protein 6. Bone. (2019) 127:228–43. doi: 10.1016/j.bone.2019.05.003
108. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. (2013) 19:179–92. doi: 10.1038/nm.3074
109. Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. (2001) 107:513–23. doi: 10.1016/S0092-8674(01)00571-2
110. Astiazaran MC, Cervantes-Sodi M, Rebolledo-Enriquez E, Chacon-Camacho O, Villegas V, Zenteno JC. Novel homozygous LRP5 mutations in Mexican patients with osteoporosis-pseudoglioma syndrome. Genet Test Mol Biomarkers. (2017) 21:742–6. doi: 10.1089/gtmb.2017.0118
111. Laine CM, Chung BD, Susic M, Prescott T, Semler O, Fiskerstrand T, et al. Novel mutations affecting LRP5 splicing in patients with osteoporosis-pseudoglioma syndrome (OPPG). Eur J Hum Genet. (2011) 19:875–81. doi: 10.1038/ejhg.2011.42
112. Korvala J, Juppner H, Makitie O, Sochett E, Schnabel D, Mora S, et al. Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med Genet. (2012) 13:26. doi: 10.1186/1471-2350-13-26
113. Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. (2002) 346:1513–21. doi: 10.1056/NEJMoa013444
114. Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. (2002) 70:11–9. doi: 10.1086/338450
115. Kwee ML, Balemans W, Cleiren E, Gille JJ, Van Der Blij F, Sepers JM, et al. An autosomal dominant high bone mass phenotype in association with craniosynostosis in an extended family is caused by an LRP5 missense mutation. J Bone Miner Res. (2005) 20:1254–60. doi: 10.1359/JBMR.050303
116. Balemans W, Piters E, Cleiren E, Ai M, Van Wesenbeeck L, Warman ML, et al. The binding between sclerostin and LRP5 is altered by DKK1 and by high-bone mass LRP5 mutations. Calcif Tissue Int. (2008) 82:445–53. doi: 10.1007/s00223-008-9130-9
117. Semenov MV, He X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. (2006) 281:38276–84. doi: 10.1074/jbc.M609509200
118. Balemans W, Devogelaer JP, Cleiren E, Piters E, Caussin E, Van Hul W. Novel LRP5 missense mutation in a patient with a high bone mass phenotype results in decreased DKK1-mediated inhibition of Wnt signaling. J Bone Miner Res. (2007) 22:708–16. doi: 10.1359/jbmr.070211
119. Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. (2007) 315:1278–82. doi: 10.1126/science.1136370
120. Ockeloen CW, Khandelwal KD, Dreesen K, Ludwig KU, Sullivan R, Van Rooij I, et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet Med. (2016) 18:1158–62. doi: 10.1038/gim.2016.10
121. Karner CM, Dietrich MF, Johnson EB, Kappesser N, Tennert C, Percin F, et al. Lrp4 regulates initiation of ureteric budding and is crucial for kidney formation–a mouse model for Cenani-Lenz syndrome. PLoS ONE. (2010) 5:e10418. doi: 10.1371/journal.pone.0010418
122. Kariminejad A, Stollfuss B, Li Y, Bogershausen N, Boss K, Hennekam RC, et al. (2013). Severe Cenani-Lenz syndrome caused by loss of LRP4 function. Am. J. Med. Genet. A 161A, 1475–1479. doi: 10.1002/ajmg.a.35920
123. Li Y, Pawlik B, Elcioglu N, Aglan M, Kayserili H, Yigit G, et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am J Hum Genet. (2010) 86:696–706. doi: 10.1016/j.ajhg.2010.03.004
124. Fijalkowski I, Geets E, Steenackers E, Van Hoof V, Ramos FJ, Mortier G, et al. A Novel Domain-Specific Mutation in a Sclerosteosis Patient Suggests a Role of LRP4 as an Anchor for Sclerostin in Human Bone. J Bone Miner Res. (2016) 31:874–81. doi: 10.1002/jbmr.2782
125. Ohkawara B, Cabrera-Serrano M, Nakata T, Milone M, Asai N, Ito K, et al. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet. (2014) 23:1856–68. doi: 10.1093/hmg/ddt578
126. Sukenik Halevy R, Chien HC, Heinz B, Bamshad MJ, Nickerson DA University of Washington Center for Mendelian G. Mutations in the fourth beta-propeller domain of LRP4 are associated with isolated syndactyly with fusion of the third and fourth fingers. Hum Mutat. (2018) 39:811–15. doi: 10.1002/humu.23417
127. Sharma M, Castro-Piedras I, Simmons GE Jr, Pruitt K. Dishevelled: A masterful conductor of complex Wnt signals. Cell Signal. (2018) 47:52–64. doi: 10.1016/j.cellsig.2018.03.004
128. Itoh K, Brott BK, Bae GU, Ratcliffe MJ, Sokol SY. Nuclear localization is required for Dishevelled function in Wnt/beta-catenin signaling. J Biol. (2005) 4:3. doi: 10.1186/jbiol20
129. Bunn KJ, Daniel P, Rosken HS, O'neill AC, Cameron-Christie SR, Morgan T, et al. Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome. Am J Hum Genet. (2015) 96:623–30. doi: 10.1016/j.ajhg.2015.02.010
130. White J, Mazzeu JF, Hoischen A, Jhangiani SN, Gambin T, Alcino MC, et al. DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. Am J Hum Genet. (2015) 96:612–22. doi: 10.1016/j.ajhg.2015.02.015
131. White JJ, Mazzeu JF, Hoischen A, Bayram Y, Withers M, Gezdirici A, et al. DVL3 Alleles resulting in a−1 frameshift of the last exon mediate autosomal-dominant robinow syndrome. Am J Hum Genet. (2016) 98:553–61. doi: 10.1016/j.ajhg.2016.01.005
132. Roifman M, Brunner H, Lohr J, Mazzeu J, Chitayat D. Autosomal dominant robinow syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle, WA: University of Washington (1993) 1993–2020.
133. Danyel M, Kortum F, Dathe K, Kutsche K, Horn D. Autosomal dominant Robinow syndrome associated with a novel DVL3 splice mutation. Am J Med Genet A. (2018) 176:992–6. doi: 10.1002/ajmg.a.38635
134. Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. (2006) 8:501–8. doi: 10.1038/ncb1405
135. Funato Y, Michiue T, Terabayashi T, Yukita A, Danno H, Asashima M, et al. Nucleoredoxin regulates the Wnt/planar cell polarity pathway in Xenopus. Genes Cells. (2008) 13:965–75. doi: 10.1111/j.1365-2443.2008.01220.x
136. Funato Y, Terabayashi T, Sakamoto R, Okuzaki D, Ichise H, Nojima H, et al. Nucleoredoxin sustains Wnt/beta-catenin signaling by retaining a pool of inactive dishevelled protein. Curr Biol. (2010) 20:1945–52. doi: 10.1016/j.cub.2010.09.065
137. Boles MK, Wilkinson BM, Wilming LG, Liu B, Probst FJ, Harrow J, et al. Discovery of candidate disease genes in ENU-induced mouse mutants by large-scale sequencing, including a splice-site mutation in nucleoredoxin. PLoS Genet. (2009) 5:e1000759. doi: 10.1371/journal.pgen.1000759
138. Costain G, Callewaert B, Gabriel H, Tan TY, Walker S, Christodoulou J, et al. De novo missense variants in RAC3 cause a novel neurodevelopmental syndrome. Genet Med. (2019) 21:1021–6. doi: 10.1038/s41436-018-0323-y
139. Xing Y, Clements WK, Le Trong I, Hinds TR, Stenkamp R, Kimelman D, et al. Crystal structure of a beta-catenin/APC complex reveals a critical role for APC phosphorylation in APC function. Mol Cell. (2004) 15:523–33. doi: 10.1016/j.molcel.2004.08.001
140. Shintani T, Ihara M, Tani S, Sakuraba J, Sakuta H, Noda M. APC2 plays an essential role in axonal projections through the regulation of microtubule stability. J Neurosci. (2009) 29:11628–40. doi: 10.1523/JNEUROSCI.2394-09.2009
141. Schneikert J, Vijaya Chandra SH, Ruppert JG, Ray S, Wenzel EM, Behrens J. Functional comparison of human adenomatous polyposis coli (APC) and APC-like in targeting beta-catenin for degradation. PLoS ONE. (2013) 8:e68072. doi: 10.1371/journal.pone.0068072
142. Almuriekhi M, Shintani T, Fahiminiya S, Fujikawa A, Kuboyama K, Takeuchi Y, et al. Loss-of-function mutation in APC2 causes Sotos syndrome features. Cell Rep. (2015) 10:1585–98. doi: 10.1016/j.celrep.2015.02.011
143. Perdu B, De Freitas F, Frints SG, Schouten M, Schrander-Stumpel C, Barbosa M, et al. Osteopathia striata with cranial sclerosis owing to WTX gene defect. J Bone Miner Res. (2010) 25:82–90. doi: 10.1359/jbmr.090707
144. Tanneberger K, Pfister AS, Kriz V, Bryja V, Schambony A, Behrens J. Structural and functional characterization of the Wnt inhibitor APC membrane recruitment 1 (Amer1). J Biol Chem. (2011) 286:19204–14. doi: 10.1074/jbc.M111.224881
145. Jenkins ZA, Van Kogelenberg M, Morgan T, Jeffs A, Fukuzawa R, Pearl E, et al. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet. (2009) 41:95–100. doi: 10.1038/ng.270
146. Perdu B, Lakeman P, Mortier G, Koenig R, Lachmeijer AM, Van Hul W. Two novel WTX mutations underscore the unpredictability of male survival in osteopathia striata with cranial sclerosis. Clin Genet. (2011) 80:383–8. doi: 10.1111/j.1399-0004.2010.01553.x
147. Grzeschik KH, Bornholdt D, Oeffner F, Konig A, Del Carmen Boente M, Enders H, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. (2007) 39:833–5. doi: 10.1038/ng2052
148. Stevenson DA, Chirpich M, Contreras Y, Hanson H, Dent K. Goltz syndrome and PORCN mosaicism. Int J Dermatol. (2014) 53:1481–4. doi: 10.1111/ijd.12605
149. Bree AF, Grange DK, Hicks MJ, Goltz RW. Dermatologic findings of focal dermal hypoplasia (Goltz syndrome). Am J Med Genet C Semin Med Genet. (2016) 172C:44–51. doi: 10.1002/ajmg.c.31472
150. King EA, Davis JW, Degner JF. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. (2019) 15:e1008489. doi: 10.1371/journal.pgen.1008489
151. Morris JA, Kemp JP, Youlten SE, Laurent L, Logan JG, Chai RC, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet. (2019) 51:258–66. doi: 10.1038/s41588-018-0302-x
152. Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, et al. Leveraging Polygenic Functional enrichment to improve GWAS power. Am J Hum Genet. (2019) 104:65–75. doi: 10.1016/j.ajhg.2018.11.008
153. Van Camp JK, Beckers S, Zegers D, Boudin E, Nielsen TL, Andersen M, et al. Genetic association study of WNT10B polymorphisms with BMD and adiposity parameters in Danish and Belgian males. Endocrine. (2013) 44:247–54. doi: 10.1007/s12020-012-9869-7
154. Trajanoska K, Morris JA, Oei L, Zheng HF, Evans DM, Kiel DP, et al. Assessment of the genetic and clinical determinants of fracture risk: genome wide association and mendelian randomisation study. BMJ. (2018) 362:k3225. doi: 10.1136/bmj.k3225
155. Wang H, Zhang F, Zeng J, Wu Y, Kemper KE, Xue A, et al. Genotype-by-environment interactions inferred from genetic effects on phenotypic variability in the UK Biobank. Sci Adv. (2019) 5:eaaw3538. doi: 10.1126/sciadv.aaw3538
156. Mullin BH, Walsh JP, Zheng HF, Brown SJ, Surdulescu GL, Curtis C, et al. Genome-wide association study using family-based cohorts identifies the WLS and CCDC170/ESR1 loci as associated with bone mineral density. BMC Genomics. (2016) 17:136. doi: 10.1186/s12864-016-2481-0
157. Freund MK, Burch KS, Shi H, Mancuso N, Kichaev G, Garske KM, et al. Phenotype-Specific enrichment of mendelian disorder genes near GWAS regions across 62 complex traits. Am J Hum Genet. (2018) 103:535–52. doi: 10.1016/j.ajhg.2018.08.017
158. Clement-Lacroix P, Ai M, Morvan F, Roman-Roman S, Vayssiere B, Belleville C, et al. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci USA. (2005) 102:17406–11. doi: 10.1073/pnas.0505259102
159. Vestergaard P. Skeletal effects of central nervous system active drugs: anxiolytics, sedatives, antidepressants, lithium and neuroleptics. Curr Drug Saf. (2008) 3:185–9. doi: 10.2174/157488608785699432
160. Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature. (2013) 497:517–20. doi: 10.1038/nature12124
161. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature. (1992) 359:235–7. doi: 10.1038/359235a0
162. Rivera MN, Kim WJ, Wells J, Driscoll DR, Brannigan BW, Han M, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. (2007) 315:642–5. doi: 10.1126/science.1137509
163. Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. (2008) 47:461–70. doi: 10.1002/gcc.20553
164. Fehringer G, Kraft P, Pharoah PD, Eeles RA, Chatterjee N, Schumacher FR, et al. Cross-Cancer genome-wide analysis of lung, ovary, breast, prostate, and colorectal cancer reveals novel pleiotropic associations. Cancer Res. (2016) 76:5103–14. doi: 10.1158/0008-5472.CAN-15-2980
165. Wang M, Gu D, Du M, Xu Z, Zhang S, Zhu L, et al. Common genetic variation in ETV6 is associated with colorectal cancer susceptibility. Nat Commun. (2016) 7:11478. doi: 10.1038/ncomms11478
166. Michailidou K, Lindstrom S, Dennis J, Beesley J, Hui S, Kar S, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. (2017) 551:92–4. doi: 10.1038/nature24284
167. Appelman-Dijkstra NM, Papapoulos SE. Sclerostin inhibition in the management of osteoporosis. Calcif Tissue Int. (2016) 98:370–80. doi: 10.1007/s00223-016-0126-6
168. Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. (2016) 375:1532–43. doi: 10.1056/NEJMoa1607948
169. Saag KG, Petersen J, Brandi ML, Karaplis AC, Lorentzon M, Thomas T, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. (2017) 377:1417–27. doi: 10.1056/NEJMoa1708322
170. Lewiecki EM, Blicharski T, Goemaere S, Lippuner K, Meisner PD, Miller PD, et al. A phase III randomized placebo-controlled trial to evaluate efficacy and safety of romosozumab in men with osteoporosis. J Clin Endocrinol Metab. (2018) 103:3183–93. doi: 10.1210/jc.2017-02163
171. Langdahl BL, Libanati C, Crittenden DB, Bolognese MA, Brown JP, Daizadeh NS, et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, phase 3 trial. Lancet. (2017) 390:1585–94. doi: 10.1016/S0140-6736(17)31613-6
Keywords: skeletal dysplasias, Wingless and Int-1 (WNT)/planar cell polarity (PCP) pathway, Wingless and int-1 (WNT)/β-catenin pathway, Wingless and int-1 (WNT)/Ca2+ pathway, osteoporosis
Citation: Huybrechts Y, Mortier G, Boudin E and Van Hul W (2020) WNT Signaling and Bone: Lessons From Skeletal Dysplasias and Disorders. Front. Endocrinol. 11:165. doi: 10.3389/fendo.2020.00165
Received: 20 December 2019; Accepted: 09 March 2020;
Published: 09 April 2020.
Edited by:
Katherine A. Staines, University of Brighton, United KingdomReviewed by:
Thomas Funck-Brentano, Université de Paris, FranceCopyright © 2020 Huybrechts, Mortier, Boudin and Van Hul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wim Van Hul, d2ltLnZhbmh1bEB1YW50d2VycGVuLmJl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.