 Andrea Bast-Habersbrunner
Andrea Bast-Habersbrunner Tobias Fromme
Tobias Fromme- 1Chair of Molecular Nutritional Medicine, TUM School of Life Sciences, Technical University of Munich, Munich, Germany
- 2EKFZ - Else Kröner-Fresenius Center for Nutritional Medicine, Technical University of Munich, Munich, Germany
Non-shivering thermogenesis in mammalian brown adipose tissue is a powerful mechanism to defend normothermia in cold climates. To minimize the loss of chemical energy, the central functional component, mitochondrial uncoupling protein 1, UCP1, must be tightly regulated. The canonical pathway of UCP1 activation includes lipolytic release of free fatty acids in response to an adrenergic signal. Activating fatty acids overcome constitutive inhibition of UCP1 by the di- and triphosphate forms of purine nucleotides, i.e., ATP, ADP, GTP, and GDP. Cellular concentrations of inhibitory, free nucleotides are subject to significant, adrenergically induced alterations. The regulatory components involved may constitute novel drug targets to manipulate brown fat thermogenesis and thereby organismic energy balance. We here review evidence for and against a dominant role of nucleotides in thermogenic control, describe conceptual routes to endogenously and pharmacologically alter free nucleotide pool size, speculate on a signaling role of degradation products released from active brown fat, and highlight gaps in our understanding of signaling and metabolic pathways involved.
Introduction
Brown adipose tissue (BAT) provides a mechanism for adaptive, non-shivering thermogenesis to endotherm mammals [reviewed in (1)]. Isolated mitochondria from this tissue display intense oxygen consumption in the absence of ATP production, i.e., uncoupled respiration. The di- and triphosphate forms of purine nucleotides (ATP, ADP, GTP, and GDP) have long been known to restore respiratory control to such mitochondrial preparations (2–4). The nucleotide binding site was found to reside within the central thermogenic protein of the mitochondrial inner membrane later named uncoupling protein 1 (UCP1) (5–7). Nucleotides proved a constitutive inhibitor of UCP1 mediated proton conductance of the mitochondrial inner membrane and thus constitute the default shut-off mechanism in the absence of thermogenic demand. Upon BAT activation by the sympathetic nervous system, lipolytically liberated free fatty acids are thought to displace nucleotides from UCP1 in a competitive manner to act as thermogenic activators (8–10). Challenging this concept of mere competitive relief of UCP1 inhibition, several lines of evidence support an additional, active regulation of purine nucleotides in response to adrenergic stimulation.
Nucleotides Are Actively Controlled Regulators of Ucp1 Mediated Thermogenesis
For decades, the divergent roles of actively controlled free fatty acid levels as UCP1 activators and passively acting nucleotides as constitutively present inhibitors remained the widely accepted model of thermogenic regulation in BAT. The arguments seemed obvious enough: millimolar cytosolic concentration of ATP alone seemed sufficient to fully and efficiently shut down UCP1 function at all times, exceeding the nanomolar or low micromolar dissociation constant by several orders of magnitude (11–13). To actively release the inhibitory effect of purine nucleotides on UCP1, their concentrations would need to be tremendously reduced, which seemed prohibitively wasteful, as resynthesis of one nucleotide would demand an energy investment equivalent to 50 ATP (14). However, considerable degradation of nucleotides also occurs routinely in contracting muscle, where ADP is degraded in order to preserve the ATP/ADP ratio in conditions of high ATP hydrolysis (15, 16). Furthermore, in active brown adipocytes, where large amounts of chemical energy are released as heat instead of being converted into ATP, energy efficiency does not appear a priority.
Remaining doubts were convincingly countered and brought to the point in a publication by Klingenberg, in which he finds that “the conclusion that nucleotides do not play a role in intracellular regulation of UCP1 because of a too high affinity, seems to be unfounded” (17). Arguments put forward were, first, the phospholipid composition of the mitochondrial inner membrane is very different from the phosphatidylcholine proteoliposomes in which UCP1 was initially characterized (11). Adding phosphatidylserine and especially cardiolipin not only changed absolute UCP1 activity but in particular attenuated the inhibitory potential of purine nucleotides in a dose dependent manner (17). At the 12% molar amount cardiolipin typically found in the inner membrane of BAT mitochondria (18, 19), the dissociation constant must be expected to be increased by more than an order of magnitude. Second and even more important, UCP1 is only inhibited by free, non-complexed nucleotides (20). The preferred form to be used as substrate by ATPases, however, is a complex with one of the divalent cations magnesium and calcium and accordingly, the vast majority of cytosolic purine nucleotides is cation complexed and irrelevant for the inhibition of UCP1 (21, 22). Furthermore, total purine nucleotide concentration is relatively low in brown adipocytes, e.g., by a factor of two to six compared to white adipocytes or skeletal muscle cells, respectively (23). Taking into account both a higher than anticipated dissociation constant and a lower than anticipated free nucleotide concentration, further modulation of nucleotide levels appears a plausible regulator of UCP1 activity.
If active regulation of free nucleotides is assumed to be implicated in the activation of UCP1 mediated non-shivering thermogenesis, brown adipocytes require a mechanism to reduce their concentration in response to an adrenergic stimulus, either by a shift in the balance between free and complexed nucleotides or by an altered overall amount. There is evidence for both.
Complexation of purine nucleotides with magnesium increases with increasing cytosolic pH, which is indeed elevated upon α-adrenergic stimulation of brown adipocytes (21, 24–26). In addition, the inhibitory potency of free purine nucleotides on UCP1 is also a function of pH, with decreased purine affinity in response to pH elevation (20, 24). Furthermore, after thermogenic activation of brown adipocytes, total calcium content of these cells increases 15-fold by uptake from the external medium (23). Cytosolic concentrations of both calcium and magnesium sharply increase, both by uptake from the extracellular medium and release from intracellular stores (27–30). The concentration of remaining free, non-complexed purine nucleotides will strongly decrease accordingly (Figure 1).
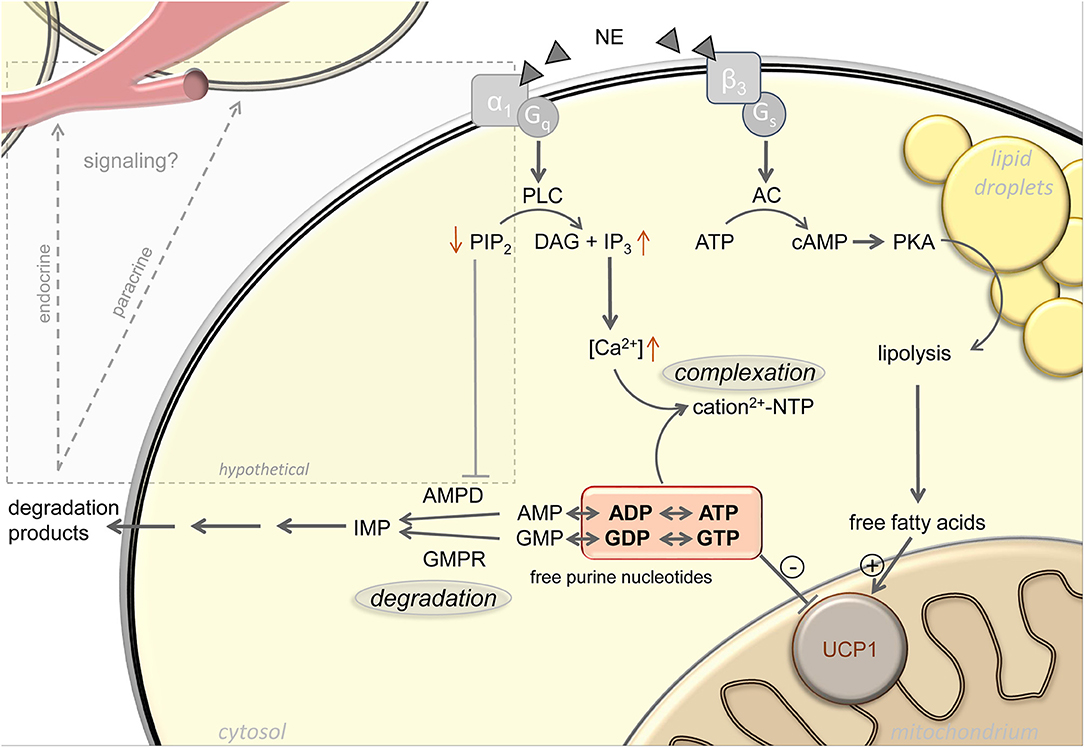
Figure 1. Active regulation of free purine nucleotides upon adrenergic stimulation of brown adipocytes. Norepinephrine (NE) stimulation of brown adipocytes mediates activation of uncoupling protein 1 (UCP1) via increasing free fatty acid and decreasing free purine nucleotide levels. Lipolytic release of free fatty acids occurs in response to Gs-coupled β3-adrenergic receptor activation. The pool of free purine nucleotides is regulated via complexation and degradation mediated by Gq-coupled α1-adrenergic receptor signaling. Phospholipase C (PLC), phosphatidylinositol 4,5-bisphosphate (PIP2), diacylglycerol (DAG), inositol 1,4,5-trisphosphate (IP3), adenylyl cyclase (AC), protein kinase A (PKA), AMP deaminase (AMPD), GMP reductase (GMPR), inosine monophosphate (IMP), adenosine mono-/di-/triphosphate (AMP/ADP/ATP), guanosine mono-/di-/triphosphate (GMP/GDP/GTP). Black arrows represent established signaling pathways, gray box and arrows indicate hypothetical pathways.
A first indication for cold induced changes in BAT purine nucleotide concentrations themselves came from the observation of drastic changes in the transcript abundance of purine metabolism gene products. For instance, GMP reductase (Gmpr) expression is strongly and very quickly induced in BAT by cold exposure, both on the transcript and the protein level (31, 32). At peak expression after ~24 h of cold exposure, Gmpr is among the 50 most abundant transcripts in murine BAT, together with core functional components including Ucp1, citrate synthase and subunits of respiratory chain complexes [GEO accession GSE119452 (23)]. These changes in gene expression are far from incidental. Total nucleotide pool size is essentially a function of nucleotide monophosphate degradation, because mono-, di- and triphosphate forms are mutually interconvertible not only by classical ATPases, but dominantly by multiple, widely expressed adenylate- and guanylate kinases. These enzymes bi-directionally convert two diphosphates into one tri- and one monophosphate, thereby constantly rearranging the ratios between mono-, di- and triphosphate nucleotides (33). Indeed, an adrenergic stimulus leads to a loss of purine nucleotides specifically in brown adipocytes, especially ATP, ADP, and GTP. The sum of UCP1-inhibiting di- and triphosphate purine nucleotides drops to nearly one half of control levels. In accordance with gene expression data, this loss is not merely a transition toward GMP and AMP, but an actual decrease in total purine nucleotide pool size associated with a release of the respective breakdown products [(23); Figure 1].
In summary, even under control conditions, inhibitory purine nucleotide concentrations in the cytosol of brown adipocytes are in the range of their UCP1 dissociation constant. Adrenergic stimulation of non-shivering thermogenesis decreases both total pool size by enzymatic degradation and the fraction of inhibitory free nucleotides by changes in calcium concentration and pH. Activation of UCP1 includes the concerted action of both free fatty acid liberating and nucleotide degrading processes.
Control of Nucleotide Metabolism in Brown Adipocytes
Adrenergic stimulation leads to degradation of purine nucleotides in brown adipocytes contributing to the activation of UCP1 mediated thermogenesis. Enzymes involved in purine metabolism are targets of a large number of approved and experimental drugs for indications as diverse as gout [e.g., allopurinol (34)], viral infection [e.g., ribavirin (35)], post-transplantive immunosuppression [e.g., mycophenolate (36)], and Alzheimer's disease [e.g., lumacaftor (37)]. Some of these are widely used, well-studied and feature favorable safety profiles. At least for mycophenolate, we demonstrated interference with thermogenic activation in brown adipocytes (23). Whether this phenomenon is an unexpected side effect of this and other such drugs in vivo remains to be studied. If so, inhibitors of human brown fat thermogenesis may be considered for the treatment of cachexia, a progressive body mass loss in cancer patients recently linked to brown fat energy wasting (38–40).
Therapeutic targeting of human brown fat thermogenesis, however, is more often discussed in the context of metabolic disease and envisions activation, not inhibition, of the immense oxidative capacity of this tissue. At least young adults feature an amount of brown adipose tissue that, fully activated, would cause a sizeable negative shift in energy balance (41), potentially exploitable to combat obesity (42), diabetes (10, 43, 44), dyslipidemia (45), and hyperphagia (46). Neither thermogenic activation, nor brown adipose tissue specific action is plausibly achieved with inhibitors of purine nucleotide metabolism. More promising targets may be found in the intracellular signaling components connecting adrenergic receptors with purine degrading enzymes, a pathway completely unresolved and of immense interest for future study.
Adrenergic stimulation leads to purine nucleotide loss in brown adipocytes. Mere changes in phosphorylation state between ADP/ATP and GDP/GTP can bi-directionally be caused by a large variety of enzymes and pathways. Similarly, the monophosphate forms AMP and GMP are readily formed or converted back to diphosphates by adenylate and guanylate kinases. Immediately upon thermogenic activation, formation of the second messengers cAMP and cGMP may, to some extent, contribute to a reduction in ATP and GTP concentration before being eliminated to AMP and GMP (47). Since cytosolic ATP/GTP and ADP/GDP together are orders of magnitude more abundant than AMP, any meaningful change in these UCP1-inhibting nucleotides by dephosphorylation alone would cause a most dramatic AMP accumulation impossible to overlook but not observed (23). Thus, total pool size of all adenine and guanine nucleotides must decrease. In order to leave the total pool of adenine and guanine nucleotides entirely, metabolites have to pass one of the two gatekeeper enzymes GMPR or AMP deaminase (AMPD), respectively (Figure 1). These two enzymes must be considered central players in Ucp1 regulation, and expression of both is indeed cold induced (31, 32).
Since full activation of UCP1 mediated thermogenesis and changes in nucleotide concentration occur rapidly upon adrenergic stimulation (23, 48, 49), the activity of gatekeepers GMPR and AMPD must be expected to be regulated on the post-translational level. To our knowledge, the regulation of mammalian GMPR activity is poorly understood, at least by anything beyond its substrate, product and highly related metabolites, in particular not by any known protein modifications or interactors (50). With its prominent role in preserving ATP/ADP ratio in contracting skeletal muscle, AMPD regulation is far better studied and understood (15, 16, 51, 52). Isoforms of AMPD are expressed from three genes, all of which well-detectable in brown adipose tissue on the transcript level [GEO accession GSE119452 (53)]. Similar to GMPR, AMPD activity is a function of nucleotide concentrations, i.e., it is activated by ADP, inhibited by ATP and therefore effectively regulated by changes in ATP/ADP ratio. Adrenergically stimulated uncoupling of respiration from ATP synthesis in brown adipocytes drastically reduces mitochondrial ATP synthesis, reducing the ATP/ADP ratio and increasing GMPR and AMPD activities, thereby enhancing nucleotide degradation (54). In striated muscle, multiple protein-protein interactions have been reported, mostly with dominant functional component of muscle contraction, e.g., myosin and troponin, some of which appear to modify kinetic properties [reviewed in (55)]. While these interactions are a plausible mechanism to couple AMPD activity to contraction, and thus rapid ATP loss, in muscle where they are exceedingly abundant, they seem unlikely to mediate adrenergically induced activity changes in brown fat.
More promisingly, facultative binding of AMPD to the plasma membrane strongly impedes catalytic activity (56), a phenomenon mediated by the inhibitory binding to phosphoinositides, especially phosphatidylinositol 4,5-bisphosphate (PIP2) [(57); Figure 1]. Intriguingly, membrane sequestered AMPD thereby forms a pool of inactive enzyme rapidly mobilizable by the action of phospholipases. In parallel to the well-known Gs-coupled β3-adrenoreceptor cascade, brown adipocytes strongly express Gq-coupled α1-adrenoreceptors that activate phospholipase C to hydrolyze PIP2 into inositol 1,4,5-trisphosphate and diacylglycerol (58). The second messenger inositol 1,4,5-trisphosphate is already responsible for the resulting increase in cytosolic calcium and thus for the sequestration of free UCP1-inhibitory nucleotides into non-inhibitory complexes (59). It is conceivable that in parallel, phospholipase C hydrolysis of PIP2 rapidly converts membrane-bound, inactive AMPD into soluble, active enzyme to decrease adenine nucleotide pool size in response to an adrenergic signal, as observed (23). If corroborated, pharmacological targeting of Gq-coupled α1-adrenoreceptors and their downstream signal transduction cascade may prove a valid alternative to the classical β-adrenergic pathway in the endeavor to identify safe activators of brown fat thermogenesis. For decades, it has been attempted to pharmacologically target the β3-receptor to treat aspects of human metabolic syndrome, to date without clinical breakthrough (60–62). The inherent challenge, i.e., unintended effects from receptor expression outside the target tissue, must be expected to be similar for α-agonists. Considered together with a completely different and partly unresolved intracellular second messenger cascade, α-adrenergic recruitment of nucleotide metabolism enzymes may prove a complementary approach worthwhile to follow.
Summary and Outlook
The significance of purine nucleotides in the control of non-shivering thermogenesis was one of the first milestones in the mechanistic understanding of brown adipose tissue mitochondria, predating the identification of their target, uncoupling protein 1. As a means of acute thermogenic activation, nucleotides have long been neglected as passive inhibitors being displaced by actively regulated free fatty acid levels. In fact, cytosolic free purine nucleotide concentrations are adrenergically modified by several routes acting in concert, including calcium complex formation and enzymatic nucleotide degradation. The signaling cascade connecting norepinephrine and purine metabolic enzymes remains unresolved, and at least in the case of adenine nucleotides, plausibly relies on α1-adrenoreceptors and phospholipase C. Beyond the diligence work of elaborating these unknown pathways, the release of degradation products by active brown adipocytes presents the fascinating opportunity to discover novel markers and endogenous indicators of non-shivering thermogenesis (Figure 1). While the well-established endocrine/paracrine agents adenosine and ATP (63) are not detectably released from brown adipocytes, abundant guanosine, inosine, and AMP have all been reported to act as non-canonical transcellular messengers through known purinergic or novel receptors (64–69). Taken together, novel targets to manipulate or detect brown fat thermogenesis may be found both up- and downstream of the thermogenic modulation conferred by purine nucleotide metabolism.
Author Contributions
AB-H and TF wrote this review article in concert. All authors read and approved the final manuscript.
Funding
AB-H received support from a grant by the German Research Foundation (DFG KL973/12). TF received support from a grant by the German Research Foundation (DFG FR3628/2-1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Klingenspor M, Andrea B, Florian B, Yongguo L, Stefanie M, Sabine S, et al. Brown adipose tissue. In: Michael Symonds E, editor. Adipose Tissue Biology. Cham: Springer International Publishing (2017). doi: 10.1007/978-3-319-52031-5_4
2. Hohorst HJ, Rafael J. [Oxidative phosphorylation by mitochondria from brown adipose tissue]. Hoppe Seylers Z Physiol Chem. (1968) 349:268–70.
3. Rafael J, Ludolph HJ, Hohorst HJ. [Mitochondria from brown adipose tissue: uncoupling of respiratory chain phosphorylation by long fatty acids and recoupling by guanosine triphosphate]. Hoppe Seylers Z Physiol Chem. (1969) 350:1121–31. doi: 10.1515/bchm2.1969.350.2.1121
4. Pedersen JI. Coupled endogenous respiration in brown adipose tissue mitochondria. Eur J Biochem. (1970) 16:12–18. doi: 10.1111/j.1432-1033.1970.tb01047.x
5. Heaton GM, Wagenvoord RJ, Kemp AJr, Nicholls DG. Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur J Biochem. (1978) 82:515–21. doi: 10.1111/j.1432-1033.1978.tb12045.x
6. Ricquier D, Gervais C, Kader JC, Hemon P. Partial purification by guanosine-5-diphosphate–agarose affinity chromatography of the 32,000 molecular weight polypeptide from mitochondria of brown adipose tissue. FEBS Lett. (1979) 101:35–8. doi: 10.1016/0014-5793(79)81289-2
7. Lin CS, Klingenberg M. Isolation of the uncoupling protein from brown adipose tissue mitochondria. FEBS Lett. (1980) 113:299–303. doi: 10.1016/0014-5793(80)80613-2
8. Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem. (2004) 279:38236–48. doi: 10.1074/jbc.M402375200
9. Bertholet AM, Kirichok Y. The mechanism FA-dependent H(+) transport by UCP1. Handb Exp Pharmacol. (2019) 251:143–59. doi: 10.1007/164_2018_138
10. Schweizer S, Oeckl J, Klingenspor M, Fromme T. Substrate fluxes in brown adipocytes upon adrenergic stimulation and uncoupling protein 1 ablation. Life Sci Alliance. (2018) 1:e201800136. doi: 10.26508/lsa.201800136
11. Klingenberg M, Winkler E. The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J. (1985) 4:3087–92. doi: 10.1002/j.1460-2075.1985.tb04049.x
12. Rial E, Poustie A, Nicholls DG. Brown-adipose-tissue mitochondria: the regulation of the 32000-Mr uncoupling protein by fatty acids and purine nucleotides. Eur J Biochem. (1983) 137:197–203. doi: 10.1111/j.1432-1033.1983.tb07815.x
13. Cannon B, Sundin U, Romert L. Palmitoyl coenzyme A: a possible physiological regulator of nucleotide binding to brown adipose tissue mitochondria. FEBS Lett. (1977) 74:43–6. doi: 10.1016/0014-5793(77)80748-5
14. Lynch M, Marinov GK. The bioenergetic costs of a gene. Proc Natl Acad Sci U S A. (2015) 112:15690–5. doi: 10.1073/pnas.1514974112
15. Parnas JK. Über die Ammoniakbildung im Muskel und ihren Zusammenhang mit Funktion und Zustandsänderung. VI. Mitteilung: der Zusammenhang der Ammoniakbildung mit der Umwandlung des Adeninnucleotids zu Inosinsäure. Biochem Zschr. (1929) 206:16–38.
16. Hancock CR, Brault JJ, Terjung RL. Protecting the cellular energy state during contractions: role of AMP deaminase. J Physiol Pharmacol. (2006) 57(Suppl. 10):17–29.
17. Klingenberg M. Cardiolipin and mitochondrial carriers. Biochim Biophys Acta. (2009) 1788:2048–58. doi: 10.1016/j.bbamem.2009.06.007
18. Goubern M, Chapey MF, Senault C, Laury MC, Yazbeck J, Miroux B, et al. Effect of sympathetic de-activation on thermogenic function and membrane lipid composition in mitochondria of brown adipose tissue. Biochim Biophys Acta. (1992) 1107:159–64. doi: 10.1016/0005-2736(92)90342-J
19. Senault C, Yazbeck J, Goubern M, Portet R, Vincent M, Gallay J. Relation between membrane phospholipid composition, fluidity and function in mitochondria of rat brown adipose tissue. Effect of thermal adaptation and essential fatty acid deficiency. Biochim Biophys Acta. (1990) 1023:283–9. doi: 10.1016/0005-2736(90)90424-M
20. Klingenberg M. Nucleotide binding to uncoupling protein. Mechanism of control by protonation. Biochemistry. (1988) 27:781–91. doi: 10.1021/bi00402a044
21. Luthi D, Gunzel D, McGuigan JA. Mg-ATP binding: its modification by spermine, the relevance to cytosolic Mg2+ buffering, changes in the intracellular ionized Mg2+ concentration and the estimation of Mg2+ by 31P-NMR. Exp Physiol. (1999) 84:231–52. doi: 10.1017/S0958067099017996
22. Corkey BE, Duszynski J, Rich TL, Matschinsky B, Williamson JR. Regulation of free and bound magnesium in rat hepatocytes and isolated mitochondria. J Biol Chem. (1986) 261:2567–74.
23. Fromme T, Kleigrewe K, Dunkel A, Retzler A, Li Y, Maurer S, et al. Degradation of brown adipocyte purine nucleotides regulates uncoupling protein 1 activity. Mol Metab. (2018) 8:77–85. doi: 10.1016/j.molmet.2017.12.010
24. Chinet A, Friedli C, Seydoux J, Girardier L. Does cytoplasmic alkalinization trigger mitochondrial energy dissipation in the brown adipocyte? Experientia Suppl. (1978) 32:25–32. doi: 10.1007/978-3-0348-5559-4_2
25. Lee SC, Hamilton JS, Trammell T, Horwitz BA, Pappone PA. Adrenergic modulation of intracellular pH in isolated brown fat cells from hamster and rat. Am J Physiol. (1994) 267:C349–C356. doi: 10.1152/ajpcell.1994.267.2.C349
26. Giovannini P, Seydoux J, Girardier L. Evidence for a modulating effect of Na+/H+ exchange on the metabolic response of rat brown adipose tissue. Pflugers Arch. (1988) 411:273–7. doi: 10.1007/BF00585114
27. Hayato R, Higure Y, Kuba M, Nagai H, Yamashita H, Kuba K. beta(3)-Adrenergic activation of sequential Ca(2+) release from mitochondria and the endoplasmic reticulum and the subsequent Ca(2+) entry in rodent brown adipocytes. Cell Calcium. (2011) 49:400–14. doi: 10.1016/j.ceca.2011.02.011
28. Wilcke M, Nedergaard J. Alpha 1- and beta-adrenergic regulation of intracellular Ca2+ levels in brown adipocytes. Biochem Biophys Res Commun. (1989) 163:292–300. doi: 10.1016/0006-291X(89)92134-7
29. Nedergaard J. Effects of cations on brown adipose tissue in relation to possible metabolic consequences of membrane depolarisation. Eur J Biochem. (1981) 114:159–67. doi: 10.1111/j.1432-1033.1981.tb06187.x
30. Connolly E, Nanberg E, Nedergaard J. Na+-dependent, alpha-adrenergic mobilization of intracellular (mitochondrial) Ca2+ in brown adipocytes. Eur J Biochem. (1984) 141:187–93. doi: 10.1111/j.1432-1033.1984.tb08173.x
31. Salvatore D, Bartha T, Larsen PR. The guanosine monophosphate reductase gene is conserved in rats and its expression increases rapidly in brown adipose tissue during cold exposure. J Biol Chem. (1998) 273:31092–6. doi: 10.1074/jbc.273.47.31092
32. Watanabe M, Yamamoto T, Kakuhata R, Okada N, Kajimoto K, Yamazaki N, et al. Synchronized changes in transcript levels of genes activating cold exposure-induced thermogenesis in brown adipose tissue of experimental animals. Biochim Biophys Acta. (2008) 1777:104–12. doi: 10.1016/j.bbabio.2007.10.014
33. Panayiotou C, Solaroli N, Karlsson A. The many isoforms of human adenylate kinases. Int J Biochem Cell Biol. (2014) 49:75–83. doi: 10.1016/j.biocel.2014.01.014
34. Yue TF, Gutman AB. Effect of allopurinol (4-hydroxypyrazolo-(3,4-D)pyrimidine) on serum and urinary uric acid in primary and secondary gout. Am J Med. (1964) 37:885–98. doi: 10.1016/0002-9343(64)90131-7
35. Hedstrom L. IMP dehydrogenase: structure, mechanism, and inhibition. Chem Rev. (2009) 109:2903–28. doi: 10.1021/cr900021w
36. Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. (2000) 47:85–118. doi: 10.1016/S0162-3109(00)00188-0
37. Liu HD, Luo K, Luo DH. Guanosine monophosphate reductase 1 is a potential therapeutic target for Alzheimers disease. Scientific Rep. (2018) 8:2759. doi: 10.1038/s41598-018-21256-6
38. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. (2014) 513:100–4. doi: 10.1038/nature13528
39. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. (2014) 20:433–47. doi: 10.1016/j.cmet.2014.06.011
40. Tsoli M, Moore M, Burg D, Painter A, Taylor R, Lockie SH, et al. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res. (2012) 72:4372–82. doi: 10.1158/0008-5472.CAN-11-3536
41. Gerngross C, Schretter J, Klingenspor M, Schwaiger M, Fromme T. Active brown fat during 18FDG-PET/CT imaging defines a patient group with characteristic traits and an increased probability of brown fat redetection. J Nucl Med. (2017) 58:1104–10. doi: 10.2967/jnumed.116.183988
42. Poher AL, Altirriba J, Veyrat-Durebex C, Rohner-Jeanrenaud F. Brown adipose tissue activity as a target for the treatment of obesity/insulin resistance. Front Physiol. (2015) 6:4. doi: 10.3389/fphys.2015.00004
43. Maurer SF, Fromme T, Mocek S, Zimmermann A, Klingenspor M. Uncoupling protein 1 and the capacity for non-shivering thermogenesis are components of the glucose homeostatic system. Am J Physiol Endocrinol Metab. (2019) 318:E198–215. doi: 10.1152/ajpendo.00121.2019
44. Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. (2013) 123:215–23. doi: 10.1172/JCI62308
45. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. (2011) 17:200–5. doi: 10.1038/nm.2297
46. Li Y, Schnabl K, Gabler SM, Willershauser M, Reber J, Karlas A, et al. Secretin-activated brown fat mediates prandial thermogenesis to induce satiation. Cell. (2018) 175:1561–74.e12. doi: 10.1016/j.cell.2018.10.016
47. Jennissen K, Haas B, Kunz WS, Pfeifer A. cGMP and cAMP differentially regulate differentiation and function of brown adipocytes. BMC Pharmacol. (2011) 11:P37. doi: 10.1186/1471-2210-11-S1-P37
48. Li Y, Fromme T, Klingenspor M. Meaningful respirometric measurements of UCP1-mediated thermogenesis. Biochimie. (2017) 134:56–61. doi: 10.1016/j.biochi.2016.12.005
49. Li Y, Fromme T, Schweizer S, Schottl T, Klingenspor M. Taking control over intracellular fatty acid levels is essential for the analysis of thermogenic function in cultured primary brown and brite/beige adipocytes. EMBO Rep. (2014) 15:1069–76. doi: 10.15252/embr.201438775
50. Spector T, Jones TE, Miller RL. Reaction mechanism and specificity of human GMP reductase. Substrates, inhibitors, activators, and inactivators. J Biol Chem. (1979) 254:2308–15.
51. Chapman AG, Atkinson DE. Stabilization of adenylate energy charge by the adenylate deaminase reaction. J Biol Chem. (1973) 248:8309–12.
52. Sahlin K, Broberg S. Adenine nucleotide depletion in human muscle during exercise: causality and significance of AMP deamination. Int J Sports Med. (1990) 11(Suppl. 2):S62–S67. doi: 10.1055/s-2007-1024856
53. Morisaki T, Sabina RL, Holmes EW. Adenylate deaminase. A multigene family in humans and rats. J Biol Chem. (1990) 265:11482–6.
54. Ronca-Testoni S, Raggi A, Ronca G. Muscle AMP aminohydrolase. 3. A comparative study on the regulatory properties of skeletal muscle enzyme from various species. Biochim Biophys Acta. (1970) 198:101–12. doi: 10.1016/0005-2744(70)90038-0
55. Sabina RL, Mahnke-Zizelman DK. Towards an understanding of the functional significance of N-terminal domain divergence in human AMP deaminase isoforms. Pharmacol Ther. (2000) 87:279–83. doi: 10.1016/S0163-7258(00)00040-1
56. Pipoly GM, Nathans GR, Chang D, Deuel TF. Regulation of the interaction of purified human erythrocyte AMP deaminase and the human erythrocyte membrane. J Clin Invest. (1979) 63:1066–76. doi: 10.1172/JCI109376
57. Sims B, Mahnke-Zizelman DK, Profit AA, Prestwich GD, Sabina RL, Theibert AB. Regulation of AMP deaminase by phosphoinositides. J Biol Chem. (1999) 274:25701–7. doi: 10.1074/jbc.274.36.25701
58. Bronnikov GE, Zhang SJ, Cannon B, Nedergaard J. A dual component analysis explains the distinctive kinetics of cAMP accumulation in brown adipocytes. J Biol Chem. (1999) 274:37770–80. doi: 10.1074/jbc.274.53.37770
59. Leaver EV, Pappone PA. Beta-adrenergic potentiation of endoplasmic reticulum Ca(2+) release in brown fat cells. Am J Physiol Cell Physiol. (2002) 282:C1016–C1024. doi: 10.1152/ajpcell.00204.2001
60. Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. (2015) 21:33–8. doi: 10.1016/j.cmet.2014.12.009
61. Lowell BB, Flier JS. Brown adipose tissue, beta 3-adrenergic receptors, and obesity. Annu Rev Med. (1997) 48:307–16. doi: 10.1146/annurev.med.48.1.307
62. Himms-Hagen J, Danforth E. The potential role of beta3 adrenoceptor agonists in the treatment of obesity and diabetes. Curr Opin Endocrinol Diab. (1996) 3:59–65. doi: 10.1097/00060793-199602000-00010
63. Tozzi M, Novak I. Purinergic receptors in adipose tissue as potential targets in metabolic disorders. Front Pharmacol. (2017) 8:878. doi: 10.3389/fphar.2017.00878
64. Tilley SL, Wagoner VA, Salvatore CA, Jacobson MA, Koller BH. Adenosine and inosine increase cutaneous vasopermeability by activating A(3) receptors on mast cells. J Clin Invest. (2000) 105:361–7. doi: 10.1172/JCI8253
65. Guinzberg R, Cortes D, Diaz-Cruz A, Riveros-Rosas H, Villalobos-Molina R, Pina E. Inosine released after hypoxia activates hepatic glucose liberation through A3 adenosine receptors. Am J Physiol Endocrinol Metab. (2006) 290:E940–51. doi: 10.1152/ajpendo.00173.2005
66. Volpini R, Marucci G, Buccioni M, Dal Ben D, Lambertucci C, Lammi C, et al. Evidence for the existence of a specific g protein-coupled receptor activated by guanosine. ChemMedChem. (2011) 6:1074–80. doi: 10.1002/cmdc.201100100
67. Rittiner JE, Korboukh I, Hull-Ryde EA, Jin J, Janzen WP, Frye SV, et al. AMP is an adenosine A1 receptor agonist. J Biol Chem. (2012) 287:5301–9. doi: 10.1074/jbc.M111.291666
68. Lanznaster D, Dal-Cim T, Piermartiri TC, Tasca CI. Guanosine: a neuromodulator with therapeutic potential in brain disorders. Aging Dis. (2016) 7:657–79. doi: 10.14336/AD.2016.0208
Keywords: brown adipose tissue, uncoupling protein 1, nucleotides, non-shivering thermogenesis, mitochondria, GMP reductase, AMP deaminase
Citation: Bast-Habersbrunner A and Fromme T (2020) Purine Nucleotides in the Regulation of Brown Adipose Tissue Activity. Front. Endocrinol. 11:118. doi: 10.3389/fendo.2020.00118
Received: 27 November 2019; Accepted: 24 February 2020;
Published: 10 March 2020.
Edited by:
Takeshi Yoneshiro, University of California, San Francisco, United StatesReviewed by:
Andrea Armani, San Raffaele Pisana (IRCCS), ItalyLei Zhang, Garvan Institute of Medical Research, Australia
Copyright © 2020 Bast-Habersbrunner and Fromme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tobias Fromme, ZnJvbW1lQHR1bS5kZQ==