 Rebecca Berdeaux
Rebecca Berdeaux Chase Hutchins
Chase Hutchins- 1Department of Integrative Biology and Pharmacology, Center for Metabolic and Degenerative Diseases, The Brown Foundation Institute of Molecular Medicine, McGovern Medical School, University of Texas Health Science Center Houston, Houston, TX, United States
- 2Graduate Program in Biochemistry and Cell Biology, The MD Anderson-UTHealth Graduate School of Biomedical Sciences, Houston, TX, United States
cAMP is one of the earliest described mediators of hormone action in response to physiologic stress that allows acute stress responses and adaptation in every tissue. The classic role of cAMP signaling in metabolic tissues is to regulate nutrient partitioning. In response to acute stress, such as epinephrine released during strenuous exercise or fasting, intramuscular cAMP liberates glucose from glycogen and fatty acids from triglycerides. In the long-term, activation of Gs-coupled GPCRs stimulates muscle growth (hypertrophy) and metabolic adaptation through multiple pathways that culminate in a net increase of protein synthesis, mitochondrial biogenesis, and improved metabolic efficiency. This review focuses on regulation, function, and transcriptional targets of CREB (cAMP response element binding protein) and CRTCs (CREB regulated transcriptional coactivators) in skeletal muscle and the potential for targeting this pathway to sustain muscle mass and metabolic function in type 2 diabetes and cancer. Although the muscle-autonomous roles of these proteins might render them excellent targets for both conditions, pharmacologic targeting must be approached with caution. Gain of CREB-CRTC function is associated with excess liver glucose output in type 2 diabetes, and growing evidence implicates CREB-CRTC activation in proliferation and invasion of different types of cancer cells. We conclude that deeper investigation to identify skeletal muscle specific regulatory mechanisms that govern CREB-CRTC transcriptional activity is needed to safely take advantage of their potent effects to invigorate skeletal muscle to potentially improve health in people with type 2 diabetes and cancer.
Introduction
Many chronic human diseases are either caused or accompanied by skeletal muscle dysfunction and loss of muscle mass (1). In obesity and type 2 diabetes, reduced skeletal muscle glucose uptake and inefficient use of glucose and lipid-derived metabolites contributes to whole body metabolic dysfunction (2, 3). Reduced skeletal muscle mass also underlies impaired metabolism in type 2 diabetes (4). Muscle wasting and metabolic dysfunction are even more severe in cancer cachexia (5, 6). Although pharmacologic strategies to reverse these effects through activating key pro-hypertrophic and inhibiting inflammatory signaling pathways are being actively investigated (1), GPCR-activated cAMP signaling is an additional pathway with salutary effects on skeletal muscle metabolism and growth (7, 8). Chronic activation of intracellular cAMP signaling, either through exercise training or chronic administration of agonists to Gs-coupled GPCRs, results in transcriptional activation of genes that reduce muscle protein breakdown and increase mitochondrial biogenesis and metabolism, ultimately promoting muscle growth, and metabolic function [reviewed in (8)]. Conversely, muscle-specific deletion of Gnas (which encodes Gαs) in mice causes loss of muscle mass and poor glucose tolerance in concert with reduced mitochondrial abundance and function (9).
As in other cell types, the cAMP response element binding protein (CREB) is activated in response to cAMP signaling and other intracellular signaling pathways in myocytes (Figure 1). CREB becomes activated in skeletal muscle after intense exercise (10–12), fasting (13), nerve activity or muscle depolarization (10, 14, 15), and necrotizing injury (16). cAMP signaling simultaneously activates CRTCs (CREB regulated transcriptional coactivators), which interact with CREB to drive transcription of cAMP-regulated genes (17). Because of their implicated functions in muscle hypertrophy and metabolic adaptions to exercise, CREB, CRTCs, and their target genes represent potential therapeutic targets to sustain skeletal muscle mass and function in diabetes and cancer.
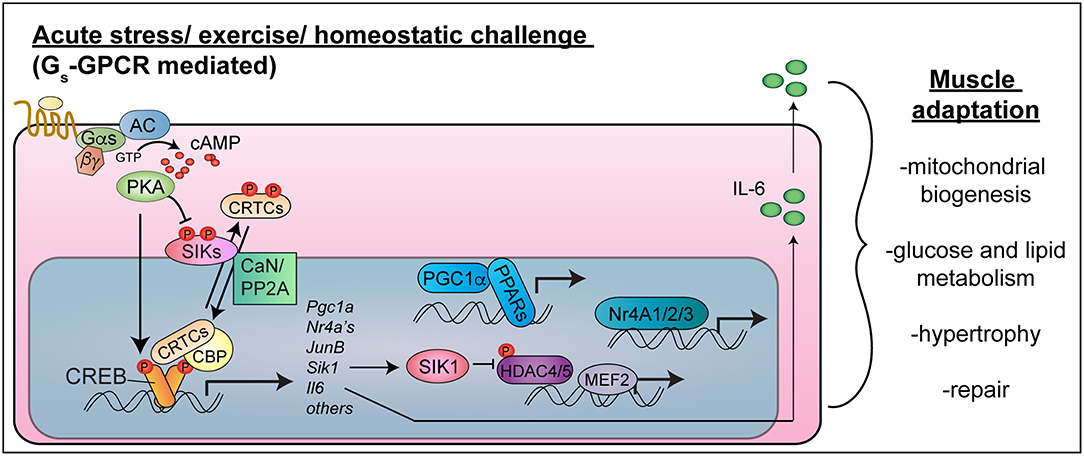
Figure 1. CREB-CRTC function in skeletal muscle. Exercise or acute stress activates Gs-coupled GPCRs to stimulate cAMP-PKA signaling. PKA phosphorylates CREB in the nucleus, leading to CBP recruitment, and phosphorylates and inhibits SIKs, leading to CRTC de-phosphorylation in concert with phosphatase activation (CaN/PP2A). CREB-CRTC-CBP complexes activate target genes encoding transcription factors (PGC1-alpha, Nr4A family nuclear hormone receptors, JunB), signaling mediators (SIK1) and myokines (IL-6), which collectively stimulate mitochondrial biogenesis, improve nutrient uptake and metabolism, stimulate hypertrophy and allow muscle repair. Targeting these pathways in a muscle cell autonomous manner would be expected to have salutary effects on skeletal muscle health and function in type 2 diabetes and cancer cachexia (Note: the nucleus is not drawn to scale).
This mini-review focuses on the known roles and transcriptional targets of CREB and CRTCs in regulation of skeletal muscle function and adaptation. We highlight how the transcriptional activity of CREB-CRTC regulates muscle function and explore dysregulation of the cAMP-CREB-CRTC pathway in skeletal muscles in the context of type 2 diabetes and cancer cachexia. Finally, we discuss literature showing that although there is potential to harness these endogenous pathways to maintain muscle mass and improve function in both disease states, there are also risks due to diabetogenic and tumor-promoting roles of CREB-CRTCs in non-skeletal muscle organs and tumor cells. Identification of skeletal muscle-specific mechanisms of CREB-CRTC regulation could allow development of novel therapeutic strategies to target this pathway to improve muscle health and avoid off-target effects in individuals with metabolic disease or cancer.
Literature Search Strategy
The authors conducted independent, unbiased reviews of published literature using numerous combinations of search strategies (e.g., CREB or CRTC and cancer, cAMP cachexia, CRTC metabolism, Gs skeletal muscle, GPCR skeletal muscle) in PubMed and GoogleScholar. We examined citations within those articles and subsequent articles that cited them. We sought association of muscle-expressed Gs-coupled GPCRs with cancer. Articles with contradictory information were not excluded. We focused discussion on articles with mechanistic findings related to skeletal muscle autonomous and non-autonomous functions of CREB-CRTCs, their target genes and upstream activating Gs-coupled GPCRs in skeletal muscle hypertrophy, metabolism, and cancer.
Regulation of CREB-CRTC Transcription Factors in Skeletal Muscle
cAMP activates CREB through direct PKA-mediated phosphorylation on serine 133, which can also be phosphorylated by other kinases [Figure 1; reviewed in (18)]. Phospho-CREB then recruits the histone acetyltransferase CREB binding protein (CBP). In addition, cAMP signaling leads to activation of CRTC co-activators, also known as TORCs (19, 20). CRTCs are held in latent cytosolic complexes with 14-3-3 proteins due to phosphorylation (21, 22) by AMPK-related kinases: salt inducible kinases (SIK1, SIK2, and SIK3) (21, 23), AMPKα1, AMPKα2 (24), and MARK2 (25). cAMP-PKA signaling inhibits these kinases, and calcium or growth-factor signaling activates calcineurin or protein phosphatase 2A (PP2A) (21, 22, 26). The convergence of these signals results in CRTC de-phosphorylation and nuclear entry. CRTC2 interacts in transcriptional complexes with CREB (19), CBP (27, 28), NONO (29), and KAT2B (30) to stimulate expression of genes containing CREB binding sites (17, 31).
In pancreatic beta cells, CRTC2 functions as a coincidence detector of both depolarization (Ca2+ dependent activation of calcineurin phosphatase) and incretin hormones (cAMP-PKA inhibition of SIK kinases), culminating in CRTC2 nuclear entry (21). In cultured skeletal myocytes, CRTCs appear to be regulated in a similar manner: catecholamines and calcium signaling synergize to induce CRTC2 and CRTC3 dephosphorylation and activation (32). The CRTC phosphatase in skeletal muscle has not been determined. All of the known CRTC kinases are expressed in skeletal muscle, either at baseline (33, 34) or in response to stimulus (35), so it will be interesting to examine CRTC activity in muscles of mice lacking these kinases. Based on similarity to hepatocytes, in which CRTC phosphorylation remained largely intact with single genetic knockout of Sik1 (36) or Sik2 (37) or with triple knockout of Sik2/Ampk-a1/ Ampk-a2 (37), there might be functional redundancy among CRTC kinases in skeletal muscle.
CREB-CRTC Functions and Target Genes in Skeletal Muscle Adaptation and Dysfunction
Skeletal Muscle Phenotypes in Mice With Altered CREB-CRTC Function
In adult skeletal muscle, exercise and other catabolic conditions activate CREB, which is associated with expression of genes involved in metabolism [(10–12); and Section CREB-CRTC Target Genes in Skeletal Muscle]. Creb knockout in mice causes perinatal lethality (38) accompanied by defective muscle progenitor cell proliferation (39) or no phenotype due to developmental compensation by related proteins (40). Transgenic over-expression of dominant-negative CREB (A-CREB) in postnatal skeletal muscles of mice caused muscle degeneration that was associated with reduced expression of MEF2 target genes and was rescued by reintroduction of SIK1 (34). Unfortunately, the developmental and degenerative phenotypes preclude the use of these animal models to study CREB function in muscle hypertrophy or metabolism in adult animals. Surprisingly, no studies to date have utilized muscle-specific Cre recombinase drivers with available conditional knock-out or knock-in alleles to achieve muscle-specific Creb knockout (41) or inducible A-CREB expression in postnatal muscles (42). Knock-in of a mild CREB gain-of-function mutant [CREB(Y134F)] did not cause hypertrophy or patent metabolic alterations, but did increase myoblast proliferation (16).
All three CRTC proteins are expressed in cultured skeletal myocytes (32, 43). CRTC2 and CRTC3 proteins are also expressed in mouse skeletal muscle, where they are activated (dephosphorylated) in response to intense exercise (32). Unlike Creb knockout mice, Crtc1, Crtc2, and Crtc3 single knockout mice develop normally without reported muscle defects (44–47). Mice lacking Crtc3 in all tissues (47) or selectively in brown fat and skeletal muscle (48) have reduced adiposity and increased brown adipose thermogenesis (47, 48). Activation of CRTC3 in adipose would therefore be expected to worsen metabolism in people with type 2 diabetes. Indeed, activating CRTC3 mutations are associated with increased adiposity in humans (47).
In skeletal muscle, CRTCs also regulate metabolism. Forced expression of CRTCs in primary myotubes promoted mitochondrial biogenesis and improved mitochondrial oxidative capacity via transactivation of Ppargc1a (encoding PGC-1α, the master regulator of mitochondrial biogenesis) (49). Bruno et al., established in vivo roles for CRTCs in skeletal muscle metabolism by demonstrating that transgenic mice with doxycycline inducible expression of CRTC2 exhibit myofiber hypertrophy, increased intramuscular glycogen and triglycerides, and enhanced exercise performance (32). Muscle over-expression of CRTC2 or CRTC3 also prevented protein degradation, which was associated with activation of the SIK1-class II HDAC pathway by transcriptional induction of Sik1 (34, 50) as well as with increased IGF1-AKT signaling due to transcriptional activation of Ppargc1a4, encoding PGC-1α4 (51). The authors suggest that CRTCs mediate transcriptional reprogramming in skeletal muscle after strenuous exercise by activation of transcriptional targets (section CREB-CRTC Target Genes in Skeletal Muscle) known to drive mitochondrial biogenesis, myofiber hypertrophy, and energy storage (32). It would be important to test whether over-expression of CRTC2 or CRTC3 in skeletal muscle in vivo is capable of stimulating mitochondrial gene expression and oxidative capacity, as in primary skeletal muscle cells in vitro (49).
CREB-CRTC Target Genes in Skeletal Muscle
PGC-1alpha
CREB-CRTC complexes bind directly to the gene encoding PGC-1α (32, 52), which is well-known to drive mitochondrial biogenesis in skeletal muscle (53). Forced expression of CRTCs stimulated Pgc-1alpha mRNA levels in skeletal myocytes (32, 49). Interestingly, CRTCs and CREB preferentially associated with the distal region of the Pgc-1alpha gene in myocytes (32), which activates expression of the pro-hypertrophic Pgc1-alpha4 isoform (51). Isoproterenol had a much stronger effect on CRTC3 occupancy in the same genomic region than on CRTC2, indicating that the signaling controlling these two isoforms may diverge in skeletal muscle cells. Notably, CRTC3, but not CRTC2, is dephosphorylated by PP2A after MAP kinase dependent priming in a non-muscle cell line (26). It is therefore possible that CRTC3 is more sensitive to isoproterenol than CRTC2 due to simultaneous activation of Gαs-activated cAMP signaling (inhibit SIKs) and Gβγ-activated MAP kinase signaling (54) (enhance PP2A recruitment). Increased expression of both Pgc-1alpha isoforms in skeletal muscle would be expected to improve both muscle metabolism and strength in type 2 diabetes and cancer cachexia.
NR4A Nuclear Hormone Receptors
Nr4a family nuclear hormone receptors are direct CREB target genes (55–58) that are strongly induced in response to exercise (in mice, rats, and humans), β-adrenergic receptor activation, cAMP signaling, or overexpression of CRTCs in muscle cells (16, 32, 34, 59–63). Transgenic mice with overexpression or knockout of Nr4a1, Nr4a2, or Nr4a3 have revealed that NR4A receptors have numerous beneficial effects on skeletal muscle metabolism and growth (59). For example, NR4A1/Nurr77 supports myogenesis (64), muscle fiber hypertrophy (65), insulin sensitivity, glucose uptake (66, 67), and lipid metabolism (68). In mice fed high fat diet, MED13 represses Nr4a2/Nurr1 transcription, which normally directly induces transcription of Glut4 and additional genes to promote glucose uptake and utilization (69). NR4A3/NOR-1 stimulates oxidative metabolism in skeletal muscle. siRNA-mediated knockdown of Nor1 (also called Nr4a3) in skeletal myocytes reduced palmitate oxidation (70). Conversely, transgenic expression of NOR-1 in mouse muscles caused a lean phenotype and increased exercise endurance, with increased muscle mitochondrial density and autophagy. At the molecular level, these phenotypes were associated with increased expression of myoglobin and genes involved in glycogen storage and aerobic metabolism (60, 71). Muscle phenotypes have not been reported in viable NOR-1 knockout mice (72).
IL-6
Interleukin-6 (IL-6) is a pro-inflammatory cytokine and myokine secreted from many cell types including skeletal muscle, in which IL-6 has pleiotropic functions (73). Basal or transient IL-6 signaling promotes muscle regeneration and growth in response to muscle damage, but chronically high IL-6 signaling in hyper-inflammatory states such as type 2 diabetes and cancer promotes muscle atrophy through activation of the JAK-STAT pathway in conjunction with elevated TNF-alpha signaling (74, 75). The Il-6 gene has a full CREB binding site (58). In C2C12 myotubes, CREB sustains basal Il6 expression (76) and cooperates with NF-κB in transcriptional activation of the Il6 promoter in myocytes treated simultaneously with a beta-adrenergic agonist and TNF-alpha (77). Muscle Il6 expression also increases after exercise (62, 76), but it has not been determined whether CREB-CRTCs regulate Il6 in this context.
JunB
A member of the AP-1 family of transcription factors, JunB is an immediate early gene induced by CREB (78). In human skeletal muscle, JUNB expression is induced by exercise (79, 80) or physiologic hyperinsulinemia (81). In mice, JunB is pro-hypertrophic: dexamethasone-induced atrophy in C2C12 myotubes dramatically reduces JunB expression, and JunB knock-down reduces muscle mass in vivo, whereas over-expression stimulates muscle hypertrophy by counteracting FoxO3 binding to atrogene promoters (82). Although JunB expression in skeletal muscle has not been directly linked to CREB activity, it is possible that JunB is part of an anabolic program induced by CREB-CRTC2 during exercise.
SIK1
Salt inducible kinase 1 (Sik1) is a direct cAMP-regulated CREB target gene in myocytes and most other cells types (34, 83–85). SIK1 phosphorylates not only CRTCs but also class II histone deacetylases (HDACs), thereby indirectly activating MEF2 (34, 86–88). In transgenic mice expressing dominant-negative A-CREB in skeletal muscle, SIK1 re-expression rescues muscle degeneration and restores class II HDAC phosphorylation (34). In cultured myocytes, knock-down of Sik1 mRNA impairs differentiation in vitro (35). Muscle deletion of the Sik1 catalytic domain in mice leads to improved muscle glucose uptake after high fat diet feeding without developmental defects, indicating that SIK1 regulates muscle metabolism (36). It will be interesting in future studies to determine how SIK1 regulates muscle metabolism and whether it is a key CREB-CRTC effector in metabolic adaptation to exercise.
Potential Therapeutic Value and Complications of CREB-CRTC Activation in Type 2 Diabetes and Cancer Cachexia
From the above discussion, it is clear that cAMP signaling to CREB-CRTCs in skeletal muscle drives gene expression programs capable of increasing muscle growth, metabolic efficiency, and exercise performance. Each of these functions would be beneficial to improve metabolism and overall health in patients with type 2 diabetes and cachexia. This might imply that pharmacologic activation of Gs-coupled GPCRs in skeletal muscle or pharmacologic blockade of CRTC inhibitory kinases would have salutary effects on skeletal muscle in people with these diseases. However, recent reports established that activation of Gs-signaling as well as CREB-CRTCs in other organs and in tumor cells would be expected to worsen metabolic disease and cancer. Therefore, to harness this pathway for therapeutic benefit, it will be important to identify CREB-CRTC regulatory mechanisms specific to skeletal muscle.
GPCR Signaling
Several Gs-coupled GPCRs expressed in skeletal muscle have been documented to promote hypertrophy and to improve muscle metabolism (Table 1). β2-adrenergic agonists, such as clenbuterol and formoterol, have shown great promise in maintaining skeletal muscle mass in animal models, but have undesirable cardiovascular side effects [reviewed in (7, 108, 109)] that might be avoided by newer-generation agonists with functional selectivity for skeletal muscle (110). Similarly, stimulation of CRFR2 with chronic administration of urocortin 2 (94–97) or transgenic expression of urocortin 3 (111) promotes muscle hypertrophy. However, CRFR2 signaling can either blunt (93) or augment (111) insulin action in skeletal muscle, depending on the ligand. Thus, stimulating cAMP signaling selectively in skeletal muscle, but not liver or heart, through β2-adrenergic receptors, Frizzled-7 (101), TGR-5 (103) or urocortin3-CRFR2 signaling (111), might be useful for treatment of type 2 diabetes.
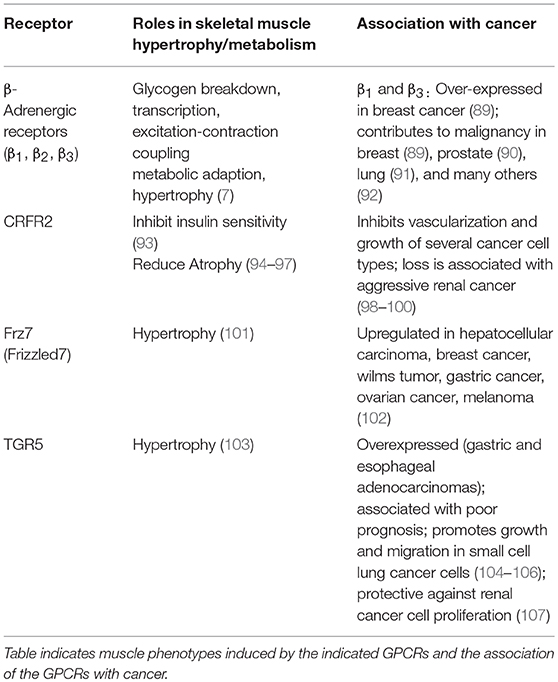
Table 1. Gs-coupled GPCRs in skeletal muscle and tumors.
In sharp contrast, stimulating Gs-coupled GPCRs in cases of cachexia would be expected to have deleterious effects on malignancy. Activating mutations of the cAMP-producing G protein Gαs (Gnas) are observed in ~5% of all human tumors (112) and 40–75% of pancreatic cancers (113). The literature points to a trend of cancer-promoting effects of many of the GS-coupled GPCRs that drive hypertrophy in skeletal muscle (Table 1). For example, β1 and β3 adrenoreceptors are overexpressed in breast tumors, when compared to normal breast tissue, and non-selective beta-blockers reduced proliferation indices in early stage breast tumors (89). Similar observations were made in many other cancers, either through in vitro studies or analysis of human specimens and correlation of clinical outcome with administration of beta-blockers (Table 1). The same is true of Fzd7 and TGR5, which are generally associated with worse cancer phenotype (proliferation, invasion, and metastasis) [(102, 104) and Table 1]. Thus, pharmacological activation of several of the Gs-coupled GPCRs that are expressed in skeletal muscle would be a poor choice for therapeutic targeting of muscle loss in cancer cachexia. The notable exception is CRFR2, which has pro-hypertrophic effects in skeletal muscle but tumor suppressive effects via inhibition of vascularization and tumor cell proliferation (98–100).
CREB-CRTCs and SIKs
Activation of CRTC2 or CRTC3 in muscle or myotubes by over-expression has beneficial effects on myocyte size (32) and metabolism (49), rendering these proteins potentially promising candidates for sustenance of muscle mass and function. It would be important to test whether transgenic expression of CRTCs in skeletal muscle could overcome atrophy or improve muscle metabolism in mouse models of cancer cachexia or type 2 diabetes. One novel therapeutic approach could be the use of bioavailable SIK inhibitors to potentiate nuclear CRTC entry (85). Currently available SIK inhibitors target all SIKs, and the most bioavailable inhibitor YKL-05-093 activates CRTC2 and class II HDACs in mice and stimulates bone formation (114). YKL-05-093 had no reported effects in lean animals, but SIK inhibitors might be expected to cause complex metabolic changes in obese animals due to functions of SIKs in liver, adipose tissue, and skeletal muscle (36, 37, 83, 115–118). More research is needed on regulation and functions of CRTCs and SIKs in skeletal muscle to predict the therapeutic value of targeting this pathway for type 2 diabetes.
Similar to GPCRs, strategies to pharmacologically activate CREB-CRTCs for cancer cachexia is questionable because gain of CREB-CRTC function in tumor cells is strongly associated with proliferation, survival, metabolic adaptation, and invasion [comprehensively reviewed in (119)]. The SIKs have emerged as tumor regulators as well. SIK1 is downregulated in several cancer types (120, 121) and has been shown to be a tumor suppressor by activating anoikis (anchorage-dependent cell death) to inhibit metastasis (120) by suppressing metabolic reprogramming (122) and epithelial to mesenchymal transitions (123). On the other hand, SIK2 activity at the centrosome is required for mitotic spindle assembly and survival of ovarian cancer cells (124, 125). A SIK2 inhibitor (ARN-3236) synergizes with paclitaxel to improve outcome in preclinical ovarian cancer models (126). The effects of SIK inhibition on muscle mass have not been evaluated, but due to the pro-oncogenic functions of CRTCs in tumor cells, strategies to activate CRTCs to increase muscle mass in cancer cachexia would require identification of skeletal muscle-specific mechanisms of CRTC regulation.
Concluding Remarks
Long known for its role as a second messenger coordinating skeletal muscle nutrient utilization with functional demands, cAMP has also been appreciated for its ability to stimulate hypertrophic growth and long-term metabolic adaptation. CREB-CRTCs are key cAMP effectors that contribute to muscle adaptation through activation of transcriptional programs that increase hypertrophic growth, mitochondrial biogenesis, metabolic efficiency, and muscle performance. Therefore, targeting CREB-CRTCs could be highly advantageous to sustain muscle mass and improve muscle function in people with type 2 diabetes and cancer cachexia. However, functions of CREB-CRTCs in other metabolic tissues as well as in tumor cells present significant obstacles to developing therapeutic approaches to safely target this pathway. Targeting CRTCs has the most potential for improving metabolism in type 2 diabetes, but should be approached with caution in cancer cachexia due to oncogenic effects of these proteins. Identification of specific signaling mechanisms that regulate CREB-CRTCs in muscle cells could enable the field to harness the potential of these proteins to improve skeletal muscle health.
Author Contributions
RB developed the thesis, designed the manuscript organization, and prepared the figure. RB and CH prepared the table, and wrote and edited the manuscript. Both authors approved the final version.
Funding
Research for this publication was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases and National Institute of Arthritis and Musculoskeletal Diseases of the National Institutes of Health under award numbers DK092590 and AR072368 as well as the American Diabetes Association under award number IBS-18-050. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Dmitry Akhmedov for critical comments on the manuscript.
Abbreviations
A-CREB, acidic-CREB (dominant-negative CREB); AKT, Protein Kinase B; AMPK, adenosine monophosphate activated protein kinase; cAMP, cyclic adenosine monophosphate; CBP, CREB binding protein; CREB, cAMP response element binding protein; CRFR, corticotrophin-releasing hormone receptor; CRTC, CREB regulated transcriptional coactivator; FoxO3, forkhead box protein; GPCR, G Protein-coupled receptor; HDAC, histone deacetylate; IGF-1, insulin like growth factor 1; IL-6, interleukin 6; JAK, Janus Kinase; MAPK, mitogen activated protein kinase; MED13, mediator complex subunit 13; MEF2, myocyte enhancer factor-2; NF-κB, nuclear factor kappa B; NR4A, Nuclear receptor family 4A; PGC-1α, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PKA, Protein Kinase A; PP2A, protein phosphatase 2A; SIK, salt inducible kinase; STAT, Signal transducer and activator of transcription proteins; TNF-alpha, tumor necrosis factor alpha; TORC, transducer of regulated CREB activity.
References
1. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. (2015) 14:58–74. doi: 10.1038/nrd4467
2. Hesselink MK, Schrauwen-Hinderling V, Schrauwen P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol. (2016) 12:633–45. doi: 10.1038/nrendo.2016.104
3. Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. (2006) 75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512
4. Cleasby ME, Jamieson PM, Atherton PJ. Insulin resistance and sarcopenia: mechanistic links between common co-morbidities. J Endocrinol. (2016) 229:R67–81. doi: 10.1530/JOE-15-0533
5. Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol. (2015) 22:100–6. doi: 10.1016/j.coph.2015.04.003
6. Penna F, Ballaro R, Beltra M, De Lucia S, Garcia Castillo L, Costelli P. The skeletal muscle as an active player against cancer cachexia. Front Physiol. (2019) 10:41. doi: 10.3389/fphys.2019.00041
7. Lynch GS, Ryall JG. Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev. (2008) 88:729–67. doi: 10.1152/physrev.00028.2007
8. Berdeaux R, Stewart R. cAMP signaling in skeletal muscle adaptation: hypertrophy, metabolism, and regeneration. Am J Physiol Endocrinol Metab. (2012) 303:E1–17. doi: 10.1152/ajpendo.00555.2011
9. Chen M, Feng HZ, Gupta D, Kelleher J, Dickerson KE, Wang J, et al. G(s)alpha deficiency in skeletal muscle leads to reduced muscle mass, fiber-type switching, and glucose intolerance without insulin resistance or deficiency. Am J Physiol Cell Physiol. (2009) 296:C930–40. doi: 10.1152/ajpcell.00443.2008
10. Popov DV, Makhnovskii PA, Shagimardanova EI, Gazizova GR, Lysenko EA, Gusev OA, et al. Contractile activity-specific transcriptome response to acute endurance exercise and training in human skeletal muscle. Am J Physiol Endocrinol Metab. (2019) 316:E605–E14. doi: 10.1152/ajpendo.00449.2018
11. Egan B, Carson BP, Garcia-Roves PM, Chibalin AV, Sarsfield FM, Barron N, et al. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol. (2010) 588(Pt 10):1779–90. doi: 10.1113/jphysiol.2010.188011
12. Widegren U, Jiang XJ, Krook A, Chibalin AV, Bjornholm M, Tally M, et al. Divergent effects of exercise on metabolic and mitogenic signaling pathways in human skeletal muscle. FASEB J. (1998) 12:1379–89. doi: 10.1096/fasebj.12.13.1379
13. Kinouchi K, Magnan C, Ceglia N, Liu Y, Cervantes M, Pastore N, et al. Fasting imparts a switch to alternative daily pathways in liver and muscle. Cell Rep. (2018) 25:3299–314 e6. doi: 10.1016/j.celrep.2018.11.077
14. Akimoto T, Sorg BS, Yan Z. Real-time imaging of peroxisome proliferator-activated receptor-gamma coactivator-1alpha promoter activity in skeletal muscles of living mice. Am J Physiol Cell Physiol. (2004) 287:C790–6. doi: 10.1152/ajpcell.00425.2003
15. Espinosa A, Leiva A, Pena M, Muller M, Debandi A, Hidalgo C, et al. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J Cell Physiol. (2006) 209:379–88. doi: 10.1002/jcp.20745
16. Stewart R, Flechner L, Montminy M, Berdeaux R. CREB is activated by muscle injury and promotes muscle regeneration. PLoS ONE. (2011) 6:e24714. doi: 10.1371/journal.pone.0024714
17. Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. (2011) 12:141–51. doi: 10.1038/nrm3072
18. Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. (2001) 2:599–609. doi: 10.1038/35085068
19. Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, et al. TORCs: transducers of regulated CREB activity. Mol Cell. (2003) 12:413–23. doi: 10.1016/j.molcel.2003.08.013
20. Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA. (2003) 100:12147–52. doi: 10.1073/pnas.1932773100
21. Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. (2004) 119:61–74. doi: 10.1016/j.cell.2004.09.015
22. Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, et al. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. (2004) 14:2156–61. doi: 10.1016/j.cub.2004.11.002
23. Katoh Y, Takemori H, Min L, Muraoka M, Doi J, Horike N, et al. Salt-inducible kinase-1 represses cAMP response element-binding protein activity both in the nucleus and in the cytoplasm. Eur J Biochem. (2004) 271:4307–19. doi: 10.1111/j.1432-1033.2004.04372.x
24. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. (2005) 310:1642–6. doi: 10.1126/science.1120781
25. Fu A, Screaton RA. Using kinomics to delineate signaling pathways: control of CRTC2/TORC2 by the AMPK family. Cell Cycle. (2008) 7:3823–8. doi: 10.4161/cc.7.24.7241
26. Sonntag T, Ostojic J, Vaughan JM, Moresco JJ, Yoon YS, Yates JR III, et al. Mitogenic signals stimulate the CREB coactivator CRTC3 through PP2A recruitment. iScience. (2019) 11:134–45. doi: 10.1016/j.isci.2018.12.012
27. Ravnskjaer K, Kester H, Liu Y, Zhang X, Lee D, Yates JR III, et al. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. (2007) 26:2880–9. doi: 10.1038/sj.emboj.7601715
28. He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. (2009) 137:635–46. doi: 10.1016/j.cell.2009.03.016
29. Amelio AL, Miraglia LJ, Conkright JJ, Mercer BA, Batalov S, Cavett V, et al. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc Natl Acad Sci U.S.A. (2007) 104:20314–9. doi: 10.1073/pnas.0707999105
30. Ravnskjaer K, Hogan MF, Lackey D, Tora L, Dent SY, Olefsky J, et al. Glucagon regulates gluconeogenesis through KAT2B- and WDR5-mediated epigenetic effects. J Clin Invest. (2013) 123:4318–28. doi: 10.1172/JCI69035
31. Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell. (2003) 11:1101–8. doi: 10.1016/S1097-2765(03)00134-5
32. Bruno NE, Kelly KA, Hawkins R, Bramah-Lawani M, Amelio AL, Nwachukwu JC, et al. Creb coactivators direct anabolic responses and enhance performance of skeletal muscle. EMBO J. (2014) 33:1027–43. doi: 10.1002/embj.201386145
33. Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab. (2004) 287:E310–7. doi: 10.1152/ajpendo.00074.2004
34. Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T, Shelton GD, et al. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med. (2007) 13:597–603. doi: 10.1038/nm1573
35. Stewart R, Akhmedov D, Robb C, Leiter C, Berdeaux R. Regulation of SIK1 abundance and stability is critical for myogenesis. Proc Natl Acad Sci USA. (2013) 110:117–22. doi: 10.1073/pnas.1212676110
36. Nixon M, Stewart-Fitzgibbon R, Fu J, Akhmedov D, Rajendran K, Mendoza-Rodriguez MG, et al. Skeletal muscle salt inducible kinase 1 promotes insulin resistance in obesity. Mol Metab. (2016) 5:34–46. doi: 10.1016/j.molmet.2015.10.004
37. Patel K, Foretz M, Marion A, Campbell DG, Gourlay R, Boudaba N, et al. The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat Commun. (2014) 5:4535. doi: 10.1038/ncomms5535
38. Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B, Schutz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci USA. (1998) 95:4481–6. doi: 10.1073/pnas.95.8.4481
39. Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature. (2005) 433:317–22. doi: 10.1038/nature03126
40. Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, et al. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci USA. (1994) 91:5647–51. doi: 10.1073/pnas.91.12.5647
41. Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. (2002) 31:47–54. doi: 10.1038/ng882
42. Li L, Fan CM. A CREB-MPP7-AMOT regulatory axis controls muscle stem cell expansion and self-renewal competence. Cell Rep. (2017) 21:1253–66. doi: 10.1016/j.celrep.2017.10.031
43. Rahnert JA, Zheng B, Hudson MB, Woodworth-Hobbs ME, Price SR. Glucocorticoids alter CRTC-CREB signaling in muscle cells: impact on PGC-1alpha expression and atrophy markers. PLoS ONE. (2016) 11:e0159181. doi: 10.1371/journal.pone.0159181
44. Altarejos JY, Goebel N, Conkright MD, Inoue H, Xie J, Arias CM, et al. The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat Med. (2008) 14:1112–7. doi: 10.1038/nm.1866
45. Wang Y, Inoue H, Ravnskjaer K, Viste K, Miller N, Liu Y, et al. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci USA. (2010) 107:3087–92. doi: 10.1073/pnas.0914897107
46. Le Lay J, Tuteja G, White P, Dhir R, Ahima R, Kaestner KH. CRTC2 (TORC2) contributes to the transcriptional response to fasting in the liver but is not required for the maintenance of glucose homeostasis. Cell Metab. (2009) 10:55–62. doi: 10.1016/j.cmet.2009.06.006
47. Song Y, Altarejos J, Goodarzi MO, Inoue H, Guo X, Berdeaux R, et al. CRTC3 links catecholamine signalling to energy balance. Nature. (2010) 468:933–9. doi: 10.1038/nature09564
48. Yoon YS, Tsai WW, Van de Velde S, Chen Z, Lee KF, Morgan DA, et al. cAMP-inducible coactivator CRTC3 attenuates brown adipose tissue thermogenesis. Proc Natl Acad Sci USA. (2018) 115:E5289–97. doi: 10.1073/pnas.1805257115
49. Wu Z, Huang X, Feng Y, Handschin C, Gullicksen PS, Bare O, et al. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci USA. (2006) 103:14379–84. doi: 10.1073/pnas.0606714103
50. Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell. (2010) 143:35–45. doi: 10.1016/j.cell.2010.09.004
51. Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, et al. A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell. (2012) 151:1319–31. doi: 10.1016/j.cell.2012.10.050
52. Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. (2001) 413:179–83. doi: 10.1038/35093131
53. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. (1999) 98:115–24. doi: 10.1016/S0092-8674(00)80611-X
54. Crespo P, Cachero TG, Xu N, Gutkind JS. Dual effect of beta-adrenergic receptors on mitogen-activated protein kinase. Evidence for a beta gamma-dependent activation and a G alpha s-cAMP-mediated inhibition. J Biol Chem. (1995) 270:25259–65. doi: 10.1074/jbc.270.42.25259
55. Saucedo-Cardenas O, Kardon R, Ediger TR, Lydon JP, Conneely OM. Cloning and structural organization of the gene encoding the murine nuclear receptor transcription factor, NURR1. Gene. (1997) 187:135–9. doi: 10.1016/S0378-1119(96)00736-6
56. Volakakis N, Kadkhodaei B, Joodmardi E, Wallis K, Panman L, Silvaggi J, et al. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc Natl Acad Sci USA. (2010) 107:12317–22. doi: 10.1073/pnas.1007088107
57. Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, et al. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. (2004) 119:1041–54. doi: 10.1016/S0092-8674(04)01159-6
58. Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA. (2005) 102:4459–64. doi: 10.1073/pnas.0501076102
59. Pearen MA, Muscat GE. Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. (2010) 24:1891–903. doi: 10.1210/me.2010-0015
60. Goode JM, Pearen MA, Tuong ZK, Wang SC, Oh TG, Shao EX, et al. The nuclear receptor, Nor-1, induces the physiological responses associated with exercise. Mol Endocrinol. (2016) 30:660–76. doi: 10.1210/me.2015-1300
61. Kawasaki E, Hokari F, Sasaki M, Sakai A, Koshinaka K, Kawanaka K. The effects of beta-adrenergic stimulation and exercise on NR4A3 protein expression in rat skeletal muscle. J Physiol Sci. (2011) 61:1–11. doi: 10.1007/s12576-010-0114-y
62. Mahoney DJ, Parise G, Melov S, Safdar A, Tarnopolsky MA. Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. FASEB J. (2005) 19:1498–500. doi: 10.1096/fj.04-3149fje
63. Fass DM, Butler JE, Goodman RH. Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J Biol Chem. (2003) 278:43014–9. doi: 10.1074/jbc.M305905200
64. Cortez-Toledo O, Schnair C, Sangngern P, Metzger D, Chao LC. Nur77 deletion impairs muscle growth during developmental myogenesis and muscle regeneration in mice. PLoS ONE. (2017) 12:e0171268. doi: 10.1371/journal.pone.0171268
65. Tontonoz P, Cortez-Toledo O, Wroblewski K, Hong C, Lim L, Carranza R, et al. The orphan nuclear receptor Nur77 is a determinant of myofiber size and muscle mass in mice. Mol Cell Biol. (2015) 35:1125–38. doi: 10.1128/MCB.00715-14
66. Chao LC, Wroblewski K, Zhang Z, Pei L, Vergnes L, Ilkayeva OR, et al. Insulin resistance and altered systemic glucose metabolism in mice lacking Nur77. Diabetes. (2009) 58:2788–96. doi: 10.2337/db09-0763
67. Chao LC, Zhang Z, Pei L, Saito T, Tontonoz P, Pilch PF. Nur77 coordinately regulates expression of genes linked to glucose metabolism in skeletal muscle. Mol Endocrinol. (2007) 21:2152–63. doi: 10.1210/me.2007-0169
68. Maxwell MA, Cleasby ME, Harding A, Stark A, Cooney GJ, Muscat GE. Nur77 regulates lipolysis in skeletal muscle cells. Evidence for cross-talk between the beta-adrenergic and an orphan nuclear hormone receptor pathway. J Biol Chem. (2005) 280:12573–84. doi: 10.1074/jbc.M409580200
69. Amoasii L, Holland W, Sanchez-Ortiz E, Baskin KK, Pearson M, Burgess SC, et al. A MED13-dependent skeletal muscle gene program controls systemic glucose homeostasis and hepatic metabolism. Genes Dev. (2016) 30:434–46. doi: 10.1101/gad.273128.115
70. Pearen MA, Myers SA, Raichur S, Ryall JG, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, a target of beta-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology. (2008) 149:2853–65. doi: 10.1210/en.2007-1202
71. Pearen MA, Eriksson NA, Fitzsimmons RL, Goode JM, Martel N, Andrikopoulos S, et al. The nuclear receptor, Nor-1, markedly increases type II oxidative muscle fibers and resistance to fatigue. Mol Endocrinol. (2012) 26:372–84. doi: 10.1210/me.2011-1274
72. Ponnio T, Conneely OM. nor-1 regulates hippocampal axon guidance, pyramidal cell survival, and seizure susceptibility. Mol Cell Biol. (2004) 24:9070–8. doi: 10.1128/MCB.24.20.9070-9078.2004
73. Pal M, Febbraio MA, Whitham M. From cytokine to myokine: the emerging role of interleukin-6 in metabolic regulation. Immunol Cell Biol. (2014) 92:331–9. doi: 10.1038/icb.2014.16
74. Belizario JE, Fontes-Oliveira CC, Borges JP, Kashiabara JA, Vannier E. Skeletal muscle wasting and renewal: a pivotal role of myokine IL-6. Springerplus. (2016) 5:619. doi: 10.1186/s40064-016-2197-2
75. Zhou J, Liu B, Liang C, Li Y, Song YH. Cytokine signaling in skeletal muscle wasting. Trends Endocrinol Metab. (2016) 27:335–47. doi: 10.1016/j.tem.2016.03.002
76. Allen DL, Uyenishi JJ, Cleary AS, Mehan RS, Lindsay SF, Reed JM. Calcineurin activates interleukin-6 transcription in mouse skeletal muscle in vivo and in C2C12 myotubes in vitro. Am J Physiol Regul Integr Comp Physiol. (2010) 298:R198–210. doi: 10.1152/ajpregu.00325.2009
77. Kolmus K, Van Troys M, Van Wesemael K, Ampe C, Haegeman G, Tavernier J, et al. beta-agonists selectively modulate proinflammatory gene expression in skeletal muscle cells via non-canonical nuclear crosstalk mechanisms. PLoS ONE. (2014) 9:e90649. doi: 10.1371/journal.pone.0090649
78. Nakajima K, Kusafuka T, Takeda T, Fujitani Y, Nakae K, Hirano T. Identification of a novel interleukin-6 response element containing an Ets-binding site and a CRE-like site in the junB promoter. Mol Cell Biol. (1993) 13:3027–41. doi: 10.1128/MCB.13.5.3027
79. Puntschart A, Wey E, Jostarndt K, Vogt M, Wittwer M, Widmer HR, et al. Expression of fos and jun genes in human skeletal muscle after exercise. Am J Physiol. (1998) 274(1 Pt 1):C129-37. doi: 10.1152/ajpcell.1998.274.1.C129
80. Trenerry MK, Carey KA, Ward AC, Cameron-Smith D. STAT3 signaling is activated in human skeletal muscle following acute resistance exercise. J Appl Physiol. (2007) 102:1483–9. doi: 10.1152/japplphysiol.01147.2006
81. Coletta DK, Balas B, Chavez AO, Baig M, Abdul-Ghani M, Kashyap SR, et al. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo. Am J Physiol Endocrinol Metab. (2008) 294:E910–7. doi: 10.1152/ajpendo.00607.2007
82. Raffaello A, Milan G, Masiero E, Carnio S, Lee D, Lanfranchi G, et al. JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J Cell Biol. (2010) 191:101–13. doi: 10.1083/jcb.201001136
83. Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. (2005) 437:1109–11. doi: 10.1038/nature03967
84. Lin X, Takemori H, Katoh Y, Doi J, Horike N, Makino A, et al. Salt-inducible kinase is involved in the ACTH/cAMP-dependent protein kinase signaling in Y1 mouse adrenocortical tumor cells. Mol Endocrinol. (2001) 15:1264–76. doi: 10.1210/mend.15.8.0675
85. Wein MN, Foretz M, Fisher DE, Xavier RJ, Kronenberg HM. Salt-inducible kinases: physiology, regulation by cAMP, and therapeutic potential: (Trends Endocrinol. Metab. 29, 723-735, 2018). Trends Endocrinol Metab. (2019) 29:723–35. doi: 10.1016/j.tem.2018.08.004
86. Takemori H, Katoh Hashimoto Y, Nakae J, Olson EN, Okamoto M. Inactivation of HDAC5 by SIK1 in AICAR-treated C2C12 myoblasts. Endocr J. (2009) 56:121–30. doi: 10.1507/endocrj.K08E-173
87. Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, Shaw RJ, et al. A hormone-dependent module regulating energy balance. Cell. (2011) 145:596–606. doi: 10.1016/j.cell.2011.04.013
88. Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. (2011) 145:607–21. doi: 10.1016/j.cell.2011.03.043
89. Montoya A, Amaya CN, Belmont A, Diab N, Trevino R, Villanueva G, et al. Use of non-selective beta-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget. (2017) 8:6446–60. doi: 10.18632/oncotarget.14119
90. Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. (2017) 358:321–6. doi: 10.1126/science.aah5072
91. Huang Q, Tan Q, Mao K, Yang G, Ma G, Luo P, et al. The role of adrenergic receptors in lung cancer. Am J Cancer Res. (2018) 8:2227–37.
92. Coelho M, Soares-Silva C, Brandao D, Marino F, Cosentino M, Ribeiro L. beta-Adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives. J Cancer Res Clin Oncol. (2017) 143:275–91. doi: 10.1007/s00432-016-2278-1
93. Chen A, Brar B, Choi CS, Rousso D, Vaughan J, Kuperman Y, et al. Urocortin 2 modulates glucose utilization and insulin sensitivity in skeletal muscle. Proc Natl Acad Sci USA. (2006) 103:16580–5. doi: 10.1073/pnas.0607337103
94. Hinkle RT, Donnelly E, Cody DB, Bauer MB, Isfort RJ. Urocortin II treatment reduces skeletal muscle mass and function loss during atrophy and increases nonatrophying skeletal muscle mass and function. Endocrinology. (2003) 144:4939–46. doi: 10.1210/en.2003-0271
95. Hinkle RT, Donnelly E, Cody DB, Bauer MB, Sheldon RJ, Isfort RJ. Corticotropin releasing factor 2 receptor agonists reduce the denervation-induced loss of rat skeletal muscle mass and force and increase non-atrophying skeletal muscle mass and force. J Muscle Res Cell Motil. (2004) 25:539–47. doi: 10.1007/s10974-004-4088-3
96. Hinkle RT, Donnelly E, Cody DB, Samuelsson S, Lange JS, Bauer MB, et al. Activation of the CRF 2 receptor modulates skeletal muscle mass under physiological and pathological conditions. Am J Physiol Endocrinol Metab. (2003) 285:E889–98. doi: 10.1152/ajpendo.00081.2003
97. Hinkle RT, Lefever FR, Dolan ET, Reichart DL, Dietrich JA, Gropp KE, et al. Corticortophin releasing factor 2 receptor agonist treatment significantly slows disease progression in mdx mice. BMC Med. (2007) 5:18. doi: 10.1186/1741-7015-5-18
98. Tezval H, Atschekzei F, Peters I, Waalkes S, Hennenlotter J, Stenzl A, et al. Reduced mRNA expression level of corticotropin-releasing hormone-binding protein is associated with aggressive human kidney cancer. BMC Cancer. (2013) 13:199. doi: 10.1186/1471-2407-13-199
99. Wang J, Jin L, Chen J, Li S. Activation of corticotropin-releasing factor receptor 2 inhibits the growth of human small cell lung carcinoma cells. Cancer Invest. (2010) 28:146–55. doi: 10.3109/07357900903179617
100. Hao Z, Huang Y, Cleman J, Jovin IS, Vale WW, Bale TL, et al. Urocortin2 inhibits tumor growth via effects on vascularization and cell proliferation. Proc Natl Acad Sci USA. (2008) 105:3939–44. doi: 10.1073/pnas.0712366105
101. von Maltzahn J, Bentzinger CF, Rudnicki MA. Wnt7a-Fzd7 signalling directly activates the Akt/mTOR anabolic growth pathway in skeletal muscle. Nat Cell Biol. (2011) 14:186–91. doi: 10.1038/ncb2404
102. Phesse T, Flanagan D, Vincan E. Frizzled7: a promising achilles' heel for targeting the wnt receptor complex to treat cancer. Cancers. (2016) 8:50. doi: 10.3390/cancers8050050
103. Sasaki T, Kuboyama A, Mita M, Murata S, Shimizu M, Inoue J, et al. The exercise-inducible bile acid receptor Tgr5 improves skeletal muscle function in mice. J Biol Chem. (2018) 293:10322–32. doi: 10.1074/jbc.RA118.002733
104. Guo C, Chen WD, Wang YD. TGR5, not only a metabolic regulator. Front Physiol. (2016) 7:646. doi: 10.3389/fphys.2016.00646
105. Cao W, Tian W, Hong J, Li D, Tavares R, Noble L, et al. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G322–7. doi: 10.1152/ajpgi.00263.2012
106. Pang C, LaLonde A, Godfrey TE, Que J, Sun J, Wu TT, et al. Bile salt receptor TGR5 is highly expressed in esophageal adenocarcinoma and precancerous lesions with significantly worse overall survival and gender differences. Clin Exp Gastroenterol. (2017) 10:29–37. doi: 10.2147/CEG.S117842
107. Su J, Zhang Q, Qi H, Wu L, Li Y, Yu D, et al. The G-protein-coupled bile acid receptor Gpbar1 (TGR5) protects against renal inflammation and renal cancer cell proliferation and migration through antagonizing NF-kappaB and STAT3 signaling pathways. Oncotarget. (2017) 8:54378–87. doi: 10.18632/oncotarget.17533
108. Joassard OR, Durieux AC, Freyssenet DG. beta2-Adrenergic agonists and the treatment of skeletal muscle wasting disorders. Int J Biochem Cell Biol. (2013) 45:2309–21. doi: 10.1016/j.biocel.2013.06.025
109. Ryall JG, Lynch GS. The potential and the pitfalls of beta-adrenoceptor agonists for the management of skeletal muscle wasting. Pharmacol Ther. (2008) 120:219–32. doi: 10.1016/j.pharmthera.2008.06.003
110. Koziczak-Holbro M, Rigel DF, Dumotier B, Sykes DA, Tsao J, Nguyen NH, et al. Pharmacological characterization of a novel 5-hydroxybenzothiazolone-derived beta 2-adrenoceptor agonist with functional selectivity for anabolic effects on skeletal muscle resulting in a wider cardiovascular safety window in preclinical studies. J Pharmacol Exp Ther. (2019) 369:188–99. doi: 10.1124/jpet.118.255307
111. Jamieson PM, Cleasby ME, Kuperman Y, Morton NM, Kelly PA, Brownstein DG, et al. Urocortin 3 transgenic mice exhibit a metabolically favourable phenotype resisting obesity and hyperglycaemia on a high-fat diet. Diabetologia. (2011) 54:2392–403. doi: 10.1007/s00125-011-2205-6
112. O'Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. (2013) 13:412–24. doi: 10.1038/nrc3521
113. Ohtsuka T, Tomosugi T, Kimura R, Nakamura S, Miyasaka Y, Nakata K, et al. Clinical assessment of the GNAS mutation status in patients with intraductal papillary mucinous neoplasm of the pancreas. Surg Today. (2019). doi: 10.1007/s00595-019-01797-7. [Epub ahead of print].
114. Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun. (2016) 7:13176. doi: 10.1038/ncomms13176
115. Yoon YS, Seo WY, Lee MW, Kim ST, Koo SH. Salt-inducible kinase regulates hepatic lipogenesis by controlling SREBP-1c phosphorylation. J Biol Chem. (2009) 284:10446–52. doi: 10.1074/jbc.M900096200
116. Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest. (2010) 120:4316–31. doi: 10.1172/JCI41624
117. Uebi T, Itoh Y, Hatano O, Kumagai A, Sanosaka M, Sasaki T, et al. Involvement of SIK3 in glucose and lipid homeostasis in mice. PLoS ONE. (2012) 7:e37803. doi: 10.1371/journal.pone.0037803
118. Park J, Yoon YS, Han HS, Kim YH, Ogawa Y, Park KG, et al. SIK2 is critical in the regulation of lipid homeostasis and adipogenesis in vivo. Diabetes. (2014) 63:3659–73. doi: 10.2337/db13-1423
119. Tasoulas J, Rodon L, Kaye FJ, Montminy M, Amelio AL. Adaptive transcriptional responses by CRTC coactivators in cancer. Trends Cancer. (2019) 5:111–27. doi: 10.1016/j.trecan.2018.12.002
120. Cheng H, Liu P, Wang ZC, Zou L, Santiago S, Garbitt V, et al. SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci Signal. (2009) 2:ra35. doi: 10.1126/scisignal.2000369
121. Yang L, Xie N, Huang J, Huang H, Xu S, Wang Z, et al. SIK1-LNC represses the proliferative, migrative, and invasive abilities of lung cancer cells. Onco Targets Ther. (2018) 11:4197–206. doi: 10.2147/OTT.S165278
122. Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. (2018) 20:811–22. doi: 10.1038/s41556-018-0122-3
123. Hong B, Zhang J, Yang W. Activation of the LKB1SIK1 signaling pathway inhibits the TGFbetamediated epithelialmesenchymal transition and apoptosis resistance of ovarian carcinoma cells. Mol Med Rep. (2018) 17:2837–44. doi: 10.3892/mmr.2017.8229
124. Ahmed AA, Lu Z, Jennings NB, Etemadmoghadam D, Capalbo L, Jacamo RO, et al. SIK2 is a centrosome kinase required for bipolar mitotic spindle formation that provides a potential target for therapy in ovarian cancer. Cancer Cell. (2010) 18:109–21. doi: 10.1016/j.ccr.2010.06.018
125. Miranda F, Mannion D, Liu S, Zheng Y, Mangala LS, Redondo C, et al. Salt-inducible kinase 2 couples ovarian cancer cell metabolism with survival at the adipocyte-rich metastatic niche. Cancer Cell. (2016) 30:273–89. doi: 10.1016/j.ccell.2016.06.020
Keywords: CREB, CRTC, SIK, cAMP, muscle hypertrophy, muscle atrophy, type 2 diabetes, cancer cachexia
Citation: Berdeaux R and Hutchins C (2019) Anabolic and Pro-metabolic Functions of CREB-CRTC in Skeletal Muscle: Advantages and Obstacles for Type 2 Diabetes and Cancer Cachexia. Front. Endocrinol. 10:535. doi: 10.3389/fendo.2019.00535
Received: 18 April 2019; Accepted: 18 July 2019;
Published: 02 August 2019.
Edited by:
Debbie C. Thurmond, Department of Molecular and Cellular Endocrinology, Beckman Research Institute, City of Hope, United StatesReviewed by:
Marta Letizia Hribal, Università degli studi Magna Græcia di Catanzaro, ItalyJanice M. Huss, City of Hope National Medical Center, United States
Copyright © 2019 Berdeaux and Hutchins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebecca Berdeaux, cmViZWNjYS5iZXJkZWF1eEB1dGgudG1jLmVkdQ==