 Veronica Maria Tagi
Veronica Maria Tagi Cosimo Giannini
Cosimo Giannini Francesco Chiarelli
Francesco Chiarelli
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 04 June 2019
Sec. Pediatric Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00342
This article is part of the Research TopicMetabolic Syndrome in Children and AdolescentsView all 5 articles
Insulin resistance (IR) is a pathological condition strongly associated with obesity. However, corticosteroids or growth hormone therapy and genetic diseases may affect insulin sensitivity lifelong. In obese children and adolescents of any age there is an evident association between IR and an increased prevalence of type 2 diabetes (T2D) and other elements contributing to the metabolic syndrome, leading to a higher cardiovascular risk. Therefore, early diagnosis and interventions in the attempt to prevent T2D when glycemia values are still normal is fundamental. The gold standard technique used to evaluate IR is the hyperinsulinemic euglycemic clamp, however it is costly and difficult to perform in clinical and research sets. Therefore, several surrogate markers have been proposed. Although the treatment of insulin resistance in children is firstly targeted to lifestyle interventions, in selected cases the integration of a pharmacological intervention might be taken into consideration. The aim of this review is to present the current knowledge on IR in children, starting with an outline of the recent evidences about the congenital forms of deficiency in insulin functioning and therefore focusing on the physiopathology of IR, its appropriate measurement, consequences, treatment options and prevention strategies.
Insulin resistance (IR) is a shared pathological condition supporting several dysmetabolic status including obesity and type 2 diabetes (T2D), dyslipidemia, atherosclerosis, polycystic ovarian syndrome (PCOS), and non-alcoholic fatty liver disease (NAFLD) (1, 2). An increased degree of IR is common in children and adolescents and is strongly associated with obesity (3). In obese children and adolescents of any age, a strong association between IR and a higher prevalence of the components of the metabolic syndrome (MS) has been observed, therefore a higher cardiovascular risk is predicted in these subjects (4) 1,2. The pathogenesis of this correlation has been found in the β-cell dysfunction which occurs in obese children and adolescents, due to the increased ectopic fat deposition. Unfortunately, appreciable β-cell destruction may occur before glucose tolerance or fasting glucose levels become impaired (5). Therefore, the advantage to prevent T2D in conditions of euglycemia appears evident. Thus, early recognition of insulin-resistant youths favors both population-based research and clinical practice (6).
The aim of this review is to present the current knowledge on IR in children, starting with an outline of the recent evidences about the congenital forms of deficiency in insulin functioning and therefore focusing on the physiopathology of IR, its appropriate measurement, consequences, treatment options and prevention strategies.
IR is a decreased tissue response to insulin-mediated cellular actions (3). It may be due to several causes, including the excess of adipose tissue, which is physiologically insulin resistant.
Although IR is more often related to obesity, also normal-weight children may be affected, suggesting that an increased adiposity is not its unique determinant (7). On the other hand, it has been demonstrated that obesity does not always lead to this pathological condition (8–10).
Other factors responsible for IR are the prolonged use of corticosteroids or growth hormone therapy and some uncommon genetic diseases, due to mutations of the insulin receptor or proteins involved in the transduction of the insulin signal (11). Moreover, puberty is a physiological condition which may be responsible for IR itself (12).
According to euglycemic hyperinsulinemic clamp studies in adult populations, IR is primarily related to the response of skeletal muscle. In fact, it is well-known that the uptake of infused glucose in the muscle is roughly of 75 vs. 2–3% taken by the adipose tissue (9).
The prevalence of obesity is increasing worldwide in children of all ages. In developing countries the prevalence of overweight and obese children aged <5 years previously reported to be 6.1 and 11.7%, was, respectively, drastically increased at an annual rate of 0.5 and 1.1% until 2013 (13). Consequently, a contemporary increasing of the incidence of IR and T2D occurs.
A recent study conducted by Arslanian et al. comparing obese adolescents and adults with impaired glucose tolerance (IGT), showed a greater insulin resistance in adolescents than adults, despite similar degrees of adiposity and glycemic status (14). This finding could give an explanation to the less improvement in insulin sensitivity in response to metformin and faster decline of β-cell function registered in youth than in adults with T2DM.
Different hypotheses have been proposed to understand the physiological mechanisms involved in this substantial different: a stronger impact of obesity on insulin sensitivity in youth, although it has been reported that insulin sensitivity is worsen by aging, a major visceral abdominal fat distribution although total body fat is similar, a superior glucotoxic or inflammatory effect induced by IGT in youth in comparison with adults, or the lower level of HDL in adolescents, considering the theory according to which HDL plays a role in inducing glucose disposal through skeletal muscle.
The clinical presentation of IR is variable and depends on its etiology and severity. The mechanisms responsible for its different signs and symptoms are still unknown. It has been hypothesized that high insulin blood levels, due to the high concentration of glucose, may excessively stimulate specific insulin-dependent pathways, resulting in acanthosis nigricans, ovarian hyperandrogenism (PCOS), lipodystrophy, accelerated or impaired linear growth, autoimmunity, and muscle cramps3. The distribution of adipose tissue depends on the type of impaired insulin sensitivity. In fact, central and abdominal obesity, generally associated with the ectopic deposition of fat (e.g., mainly into the muscle and liver) are common, while in most genetic syndromes an ectopic deposition in muscle and liver is associated with a reduced content of fat in the usual fat depot sites.
In presence of children with severe phenotypes, an inherited alteration of the action of insulin should be suspected and fasting serum insulin should be measured. If insulin is elevated with normal or high blood glucose, additional studies should be directed to look for insulin receptor mutations, circulating anti-insulin receptor antibodies, or other disorders.
Inherited lipodystrophies are rare disorders in which selective loss of adipose tissue and a predisposition to insulin resistance are typical (15). Among them, some manifest at birth, such as congenital generalized lipodystrophy (CGL) and neonatal progeroid syndrome, while others have a later onset, such as familial partial lipodystrophies (FPLD) and mandibulosacral dysplasia manifest later in life. The higher is the extent of body fat loss in these patients, the more severe is the metabolic syndrome by which they are affected. Table 1 lists all subtypes of genetic lipodystrophies classifying them according to their possible inheritance patterns or their possibility to be caused by a de novo heterozygous mutation. The two most prevalent variations are CGL and FPLD. For its complex clinical presentation and severe metabolic complications we will focus on CGL, which has an estimated prevalence of 1 in 10 million.
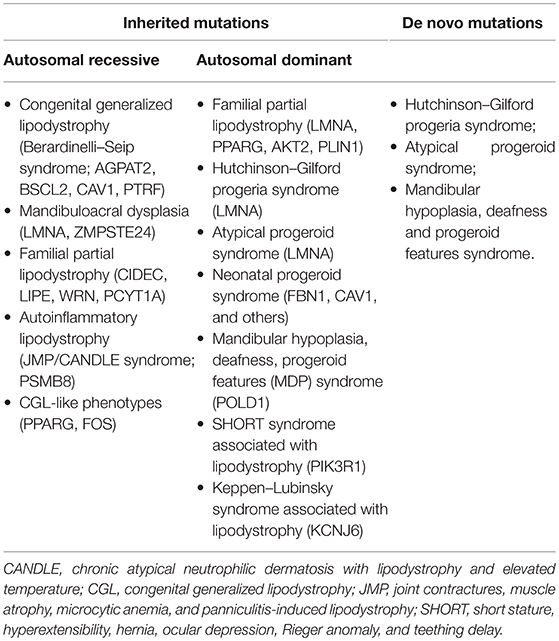
Table 1. Genetic lipodystrophies listed according to their possible inheritance patterns.
Essential diagnostic criteria for CGL are lack of fat tissue involving the whole body and excessive muscular tissue representation at birth or soon thereafter. RMI should be performed to confirm the typical body fat distribution characterizing this syndrome. Molecular diagnosis is the last step which provides certain diagnosis (15). Other clinical features that may be observed are prominent veins, secondary to body fat lack, accelerated growth, due to increased appetite, prominence of umbilicus or umbilical hernias, probably caused by hepatomegaly and/or splenomegaly, fatty liver, which can evolve in fibrosis and cirrhosis requiring liver transplantation in severe cases, acanthosis nigricans, cardio myopathy, focal segmental glomerulosclerosis, mild hirsutism, and clitoromegaly in females, irregular menstrual periods with polycystic ovaries, focal lytic bone lesions, due to the lack of normal bone marrow fat, and advanced bone age. Subjects affected by CGL develop insulin resistance in early childhood and almost half of them manifest diabetes mellitus at puberty. In these patients diabetes mellitus is resistant to ketosis, maybe because of their endogenous hyperinsulinaemia and some of them need very high doses of insulin to control glycaemia, up to 3,000 units per day. Most CGL patients present high triglycerides blood levels from late childhood or adolescence and when it is severe xanthomas and frequent pancreatitis occur, in particular when DM is not well-controlled. Low HDL cholesterol is another frequent finding.
The lack of fat tissue also induces hypoadiponectinemia and hypoleptinaemia, leading to increased appetite and therefore to worsen metabolic complications.
Four types of CGL are known. Type 1 is due to mutations in AGPAT2, encoding lysophosphatidic acid acyltransferase-β, which is involved in triglyceride biosynthesis. Subjects with this subtype of CGL can present typical acromegalic features. Type 2 CGL is secondary to mutations in BSCL2, encoding seipin, a transmembrane protein which seems to play a role in lipid droplet assembly, adipocyte differentiation and fusion of lipid droplets. In subjects with this subtype a higher prevalence of cardiomyopathy and mild mental retardation has been observed. Type 3 CGL is caused by mutations in CAV1, encoding caveolin 1, the main component of caveolae, microdomains of the plasma membrane responsible for promoting the stability and function of lipid droplets, transporting and storing fatty acids and cholesterol and increasing insulin signaling. In this case mechanical adipose tissue and fat in the bone marrow seem preserved (15). In type 4 mutations occur in PTRF, encoding cavin-1, an e substance which is essential for the synthesis of caveolae. In this case lipodystrophy is not severe at birth but might be progressive in childhood. In addition to the classical features, children present congenital myopathy with high blood creatine kinase levels, distal metaphyseal deformation with joint stiffness, pyloric stenosis, atlanto-axial instability, percussion induced, local protracted muscle contractions, and a predisposition to serious arrhythmias which can cause sudden death.
The Seip-Berardinelli syndrome is a rare congenital disorder included in the generalized lipodystrophies, whose estimated prevalence is lower than one case per one million people.
Congenital generalized lipodystrophies are inherited as an autosomal recessive trait and in 95% of reported cases are due to the mutation of AGPAT2 and BSCL2 (16). A relevant subcutaneous fat lack is the main sign in early life. Adipose tissue appears very poor in the subcutaneous areas of the abdomen and thorax and in the bone marrow, whereas it is normally represented in the orbits, mouth and tongue, palms and soles, scalp, perineum, and periarticular regions (17). Children affected by Seip-Berardinelli syndrome present with voracious appetite, accelerated growth, increased metabolic rate, and advanced bone age, with a usually normal final height. In addition, these patients may have acanthosis nigricans and hepatic steatosis, which increases the risk to develop cirrhosis, prominent musculature, precocious secondary sexual development, and, in some cases, intellectual impairment (16, 18). IR has an early onset in these patients (19). Studies conducted on the correlation between IR and lipoatrophy do not have an univocal answer concerning an hypothetical altered insulin receptor expression, function and signaling involved in its pathogenesis (20).
Experimental researches (21, 22) suggest that leptin has an important role of leptin and adiponectin in the pathogenesis of IR in Seip-Berardinelli Syndrome, but their mechanism of action in humans is still unknown.
Donohue syndrome, or leprechaunism, is a rare autosomal recessive disorder with a reported incidence of 1 in 4 million live births. It is caused by the mutation of a gene located on chromosome 19p13, altering the correct binding of the insulin receptor to insulin (23). Two milder forms of insulin resistance in which the same mechanism is impaired are Rabson Mendenhall Syndrome and Type A Insulin Resistance syndrome (23).
Donohue syndrome is characterized by severe intrauterine growth retardation and severe failure to thrive, marasmus and malnutrition despite an adequate alimentation in childhood (23). The triad including severe hyperinsulinism, fasting hypoglycaemia, and postprandial hyperglycemia in association with the presence of clinical features involving face (Elfin like pointed chin Microcephaly Low set prominent ears Orbital hypertelorism Broad nose Thick lips Facial hair), skin (Hypertrichosis Acanthosis nigricans Excessive thick, hyperelastic skin Decreased subcutaneous fat Muscle wasting), and other body districts (Large hands and feet (relative to body) Low body weight for age Hypotonia Abdominal distension Reduced lateral thoracic dimensions Hyperplasia of nipples, genitals, other organs) are sufficient for the diagnosis (23).
Low plasma glucose, secondary to the acceleration of fasting metabolism, and reduce response to exogenous insulin in these patients are associated with hypertrophy of the pancreatic islets of Langerhans. Other findings are splenomegaly and hepatomegaly (23), with glycogen and iron accumulation and bile duct cholestasis, enlarged kidneys, nephrocalcinosis, adrenal glands atrophy, delayed bone age, with deformation of metaphysics and epiphysis, and hypertrophic cardiomyopathy, which generally manifests at 1–2 months of age and seems to be caused by supraphysiological hyperinsulinism.
Insulin regulates glucose homeostasis acting mostly on hepatic, muscular and fatty tissues. In the hepatic tissue, insulin inhibits gluconeogenesis and glycogenolysis, therefore reducing the production of glucose and induces glycogen storage. In muscular and fatty tissues, it favors the uptake, storage, and use of glucose. Moreover, insulin is responsible for the induction of potassium transport in muscle, of the differentiation of cells into adipocytes, and of the production of androgens by ovaries and retention of sodium by the kidney. Insulin performs all these functions binding with a specific transmembrane protein receptor, which is encoded by a single gene localized on chromosome 19. This interaction induces the processing of a precursor of 1,382 with the final realization of a mature receptor, consisting of two α and two β subunits. The extracellular α subunit is essential for a high-affinity binding of insulin, while the transmembrane part of the β subunit in involved in the transduction of the signal into the cell. Therefore, tyrosine residues of the β subunit located intracellularly undergo phosphorylation, leading to the activation of the intracellular tyrosine kinase (24). Once tyrosine kinase is activated, it phosphorylates tyrosine residues outside the kinase domain of the receptor creating binding sites for signaling proteins with src-homology 2 (SH2) domains, or phosphotyrosine- binding (PTB) domains. The signaling network through which insulin exerts its effects is made of so-called “critical nodes.” Among them, the three best known are the phosphorylation of insulin receptor substrate (IRS) proteins by the activated insulin receptor, the recruitment of class 1A PI3K to the phosphorylated IRS proteins, and therefore the activation of the AKT serine threonine kinases by 2 phosphoinositide-dependent kinases (PDK1 and−2) (25).
In order to understand the pathogenesis of insulin resistance it should considered whether, in addition to the impairment of insulin's glucose-lowering action, its other functions are compromised too. In fact, for a well-known negative feedback loop, the blood glucose concentration regulates insulinemia. For this reason, pathologies involving the only glucose-lowering action determine compensatory increase of blood insulin concentration, determining the exposition of any other less insulin-resistant pathway or tissue to higher insulin action (25). Recent studies on humans have emphasized this concept showing that even severe genetic mutations involving the insulin receptor resulted in hyperglycemia, hyperinsulinemia, ovulatory dysfunction, hyperandrogenism, acanthosis nigricans, and soft tissue overgrowth, but did not show manifestations of impairment of other insulin functions, presenting normal triglycerides, HDL- cholesterol, absence of liver steatosis, and adiponectin within the physiological limits. In a family affected by a defect in the AKT serine threonine kinases, which is more distal in insulin signaling, subjects presented severe liver steatosis, altered lipid profile and low adiponectin concentrations.
Individuals with genetic mutations in C-terminal SH2 domain of class 1A PI3K, located between the insulin receptor and AKT2 in the insulin signaling pathway, have been shown preserved liver fat, lipid profile and plasma adiponectin, although sever insulin resistance is present. Moreover, SHORT syndrome, including short stature, joint hyperextensibility, ocular depression, altered development of the iris (Rieger anomaly), and teething delay (25–27), as well as lipodystrophy, have been found in these patients.
The gold standard for the assessment of IR is the hyperinsulinemic euglycemic clamp; however the intravenous glucose tolerance test (IVGTT) and/or the insulin tolerance test (ITT)/insulin suppression test are more frequently used because they are easier to perform (26, 27).
During the hyperinsulinemic-euglycemic clamp insulin is administered intravenously at a constant rate which increases and maintains systemic insulinemia, while a glucose intravenous infusion at variable rates occurs, in order to maintain glucose levels within the normal range.
The glucose infusion rate during the steady state is directly related to insulin sensitivity; in fact, in case of insulin sensitivity, glucose is rapidly consumed by tissues in a condition of hyperinsulinism, therefore high doses of glucose must be infused in order to maintain euglycemia. In contrast, insulin resistant subjects have a low need of glucose infusion to maintain euglycemia since they are characterized by impaired glucose uptake and consumption (28).
The glucose tolerance test (GTT) analyses the effects of exogenous glucose, administered orally, intraperitoneally, or intravenously, on the systemic clearance of glucose. This method is not applicable to individuals with altered pancreatic functioning (28).
Intravenous GTT (IVGTT) consists of measurement of basal insulinemia and afterwards injection of glucose into vein for 3 min, followed by the measurement of blood insulin levels at 1 and 3 min after the injection. IVGTT gives more reliable results, since it avoids variations due to gastro-intestinal factors which occur in case of oral glucose administration4.
The insulin tolerance test (ITT) evaluates the systemic glucose clearance in response to intraperitoneal administration of insulin. Severe hypoglycaemia is a frequent adverse effect of ITT and its results may be not reliable in case of systemic counter regulatory responses (28).
The previously mentioned tests are invasive, expensive and complex to use in the daily clinical practice, as well as difficult to perform in population based research studies (29). Therefore, several surrogate tools of measurement have been proposed. In Table 2 the most used methods to evaluate IR in both clinical and research settings are synthesized. The most common surrogate markers are the fasting plasma insulin (FPI), the homeostasis model assessment insulin resistance (HOMA-IR) and the quantitative insulin sensitivity check index (QUICKI), which have been demonstrated to correlate favorably and have been validated in childhood with the hyperinsulinemic-euglycemic clamp (30, 31).

Table 2. Main surrogate indexes of Insulin Resistance in children and adolescents.
The FPI is commonly used as surrogate marker of insulin sensitivity by characterizing insulin levels during a fasting state. Although fasting insulin is not considered an adequate method for the evaluation of insulin sensitivity, it may represent a valid index of compensatory hyperinsulinemia and liver insulin metabolism. Moreover, FPI does not always correlate well with IR in pediatric patients (32, 33). In addition, in contrast to other markers such as HOMA-IR, this index does not evaluate insulin concentration in regards to fasting glucose values. HOMA-IR is a paradigm model which allows to determine the IR rate using only the fasting glucose and insulin values. A higher value of HOMA-IR corresponds to a more severe IR (34). Therefore, its great advantage is that it requires a single blood sample. In contrast, obtained data need to be interpreted carefully, especially in subjects with relevant alteration of glucose metabolism (35). It is used to calculate the correlation between steady-state insulinemia and glycemia in order to estimate β-cell function and IR (36). The interaction between fasting glucose and insulin represents an index of the equilibrium the output of glucose by the liver and the insulin secretion, regulated by a feedback loop between hepatic cells and pancreatic β-cells (37, 38).
In comparison with fasting plasma insulin (FPI), HOMA-IR is a more accurate tool for the assessment of insulin sensitivity for several reasons. First, it gives the possibility to assess the relationship between the functioning of β-cells and insulin sensitivity in individuals with impaired glucose tolerance. Moreover, it allows to perform repeated measurements over long periods of time in subjects who continue to present abnormal glucose tolerance.
During the last decades several HOMA-IR indexes have been proposed according to the different formulas. HOMA1-IR, the original HOMA model by Matthews et al. (36), contains an easier approximation of the original non-linear solution to the interactive equations (39). HOMA2-IR is the updated computer model, which has non-linear solutions; this is the preferred model when HOMA is compared with other models, such as the minimal model. The updated of the HOMA model considers the variations in hepatic and peripheral glucose resistance5.
Although different studies have tried to identify normal values of this model for children and adolescents, reliable reference ranges of HOMA-IR are not available yet (40, 41). In 2015 Shashai et al. conducted a study on 2,573 Caucasian children and adolescents in order to define the specific percentiles of HOMA-IR in relation to age, gende-, and BMI and to establish suitable cut-offs to distinguish between low and high cardiometabolic risk. Their findings demonstrate that, even though age, gender and body adiposity are responsible for IR physiological changes, values higher than 1.68 in normal-weight subjects defines a “non-physiological state” and may pose the patient at an increased risk for cardiovascular disease. Instead, if subjects are overweight and obese, the cut-off rises to 3.42 (34).
The QUICKI model is like the simple equation used in the HOMA models with the exception that it is derived from the inverse of the sum of the logarithms of the fasting insulin and fasting glucose. Therefore, there is a perfect inverse correlation between QUICKI and HOMA. Thus, the disadvantages or limitations of the two methods are identical.
The main disadvantages of HOMA-IR and QUICKI are that changes occur in β-cell function over time and there is not an universal insulin assay standardization. Moreover, no data are available to prove the efficacy of markers of IR to predict response to treatment. In addition, in case of mutations of the insulin gene, the circulating insulin has subnormal bioactivity but normal immunoactivity (33, 42–44).
Some clinicians use serum sex hormone-binding globulin (SHBG) as a surrogate index of IR, but it has not been fully validated in the clinical setting, particularly in children (45).
In asymptomatic patients, serum triglyceride concentrations, and particularly the ratio of triglyceride to high-density lipoprotein (HDL) cholesterol concentrations are also useful markers for IR in children6.
The triglyceride-to-HDL cholesterol (TG/HDL-C) ratio has been observed to be associated with IR in white obese boys and girls. In fact, a study conducted on a population of 1,452 obese youths undertaking an oral glucose tolerance test and a fasting lipid profile, showed that the TG/HDL-C ratio is strongly related to insulin secretion and sensitivity and might therefore be used in a clinical setting in order to defined children and adolescent with insulin resistance in different ethnic groups (46).
The Mc Auley Index represents another surrogate index which has been demonstrated to have a higher sensitivity with a similar specificity in predicting insulin sensitivity if compared with fasting insulin alone, however in study mainly performed in young adults (47).
Two other insulin sensitivity markers obtained during an oral glucose tolerance test (OGTT) have been validated in adults. These indexes, which are well-correlated with the M-values for stimulated insulin sensitivity derived from the euglycemic-hyperinsulinemic clamp, are defined as: the whole body insulin sensitivity index (WBISI), conceived by Matsuda and De Fronzo (48), and the insulin sensitivity index (ISI), introduced by Soonthornpun et al. (49). Regarding these indexes, Yeckel et al. have demonstrated that the effects on glycemia and insulinemia derived from the OGTT in obese youths can be used to estimate insulin sensitivity and that the WBISI and ISI indexes may be potential good instruments for more complex studies evaluating insulin sensitivity in larger samples (50).
During the last decades several studies have defined several and well-characterized risk factors for IR including ethnicity, puberty, adipose tissue variant depots, polycystic ovary syndrome (PCOS), gene variants, family history of Diabetes or gestational diabetes, and fetal growth pattern during pregnancy.
The two most important unchangeable risk factors for IR in children are ethnicity and puberty (3). Several studies have demonstrated that Caucasian children are affected by IR more often than African, American, Hispanic, Pima Indian and Asian children (51–53).
Puberty is physiologically responsible for IR; in fact, during this period of life, insulin sensitivity undergoes a decline of around 25–50% and improves when puberty ends (51–53).
There is a strong association between IR and abdominal obesity, which is known to represent one of the main elements of the MS (54). According to large-population based studies, subcutaneous adipose tissue (SAT), and visceral adipose tissue (VAT) seem to be both related to HOMA-IR. In addition, VAT correlates more strongly to insulin variables than SAT. The pathogenesis on the basis of this evident correlation is still not known exactly but several hypotheses have been considered. SAT and VAT secrete free fatty acids into blood, and higher plasmatic free fatty acid levels seem to be associated with IR. Moreover, VAT presents a strong correlation with endothelial dysfunction and higher blood C-reactive protein values, which may give an explanation to inflammation secondary to higher VAT depots.
Since adipose tissue acts as an endocrine organ, these two tissues play a fundamental role in this field. VAT is more strongly associated with adiponectin levels and releases interleukin-6 (IL-6) and plasminogen activator inhibitor-1 (PAI-1) to a greater extent than SAT. These main factors may be the cause of an higher IR in patients with elevated VAT, in fact IL-6, and PAI-1 reduce in case of weight loss and properly correlate with a parallel improvement of insulin sensitivity (55).
Adolescents affected by PCOS often present IR, whose severity has been observed to be higher in obese patients than in lean ones (56, 57).
Risk factors include genetics, which is considered a great determinant in the incidence of IR. The principal variants which increase the risk to develop type 2 DM, that could represent the final effect of IR, are listed in Table 3. The peroxisome proliferator-activated receptor gamma (PPARγ) variant Pro12Ala was one of the first genetic variants found to be related to a decreased risk of developing T2D (58–63). Gene variants of the transcription factor 7-like 2 (TCF7L2) are associated to the risk of developing diabetes more than any other gene7. rs972283 is located near KLF14 (kruppel-like factor 14). KLF14 gene and protein expression have been observed to be significantly decreased in both muscle and adipose tissue in individuals affected by T2D. The insulin receptor substrate 1 (IRS1) is one of the loci responsible for the insulin signaling pathway. The allele C at rs2943641 adjacent to IRS1 was found to be related to IR and hyperinsulinaemia in a European population, whereas the SNP, rs2943650, near IRS1, has found to be related to a lower percentage of body fat, higher triglycerides and IR, and decreased HDL-cholesterol. Glucokinase regulator (GCKR) encodes a protein which inhibits glucokinase, which is involved in the regulation glucose storage in the liver.
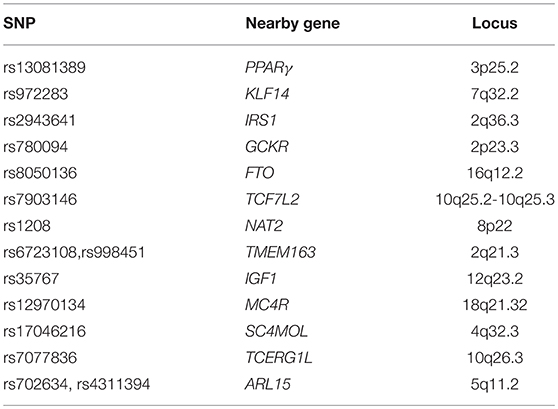
Table 3. Main gene variants associated to insulin resistance.
Variants within the first intron of FTO gene are strongly associated with both BMI and insulin sensitivity. Mutations in N-acetyltransferase 1 (NAT1) or 2 (NAT2) impair insulin sensitivity. Low concentrations of IGF-1 in the blood sample are associated with a reduction in insulin sensitivity. Protein phosphatase 1 regulatory subunit 3B (PPP1R3B) is a recent discovered locus which seems to be involved in the glycogen synthesis in skeletal muscle, whereas growth factor receptor bound protein 14 (GRB14) has an interaction with receptor tyrosine kinases (64).
Being born from a mother with pre-existing diabetes mellitus (DM) or gestational DM (GDM) is another risk factor for obesity and impaired insulin sensitivity, even in offspring with normal birth weight (65–67). A major component of body fat has been observed in these newborns (68), although the association between the excess of adiposity and future development of IR remains controversial (3).
Even if the criteria for diagnosing GDM are absent, hyperglycaemia during pregnancy is a relevant risk factor for obesity and IR in the offspring (69).
It has been noticed that children who gain weight too fast have higher risk to develop IR in childhood and to present long-term effects of IR in adulthood (70–76).
Increased IR has been also found in preterm children, with a persistence in adulthood in association with truncal obesity (77).
The adverse effects of IR are primarily due to the hyperinsulinemia which occurs in case of IR (26, 27). Reduced insulin sensitivity represents an important risk factor for the development of T2D (3), being one of the two fundamental elements involved in the pathogenesis of T2D, together with β-cell dysfunction (78). IR is strongly correlated with hypertriglyceridemia and hypertension, high C-reactive protein levels, T2D and low plasma HDL-cholesterol. In addition, MS and cardiometabolic risk factors are well-known outcomes of IR in several ethnic groups (10). In fact, IR has been demonstrated to be a reliable marker in the prediction of cardiovascular risk (79, 80).
An observational study on a Japanese adult population from the Kyushu-Okinawa Population Study (KOPS) and on an American sample of adult Caucasian individuals from the Framingham Offspring Study revealed that the prevalence rate of cardiovascular diseases in Japanese population was much lower than the one observed in the USA8. More importantly, these differences could not be considered secondary to standard cardiovascular disease risk factors, but were related to significant population differences in IR (81).
In another study on a community-based sample exclusive of diabetes from the Framingham Heart Study, the incidence of coronary heart disease (CHD) events associated with low HDL-cholesterol (HDL-C), or low triglycerides concentrations was markedly increased only if IR was present. In contrast, if IR was absent, triglycerides or HDL-C, did not show to be significant risk factors for CHD. Moreover, this study confirmed the concept derived from more general studies that IR might represent an independent predictor of cardiovascular risk in either general populations and diabetic populations9 (82).
It has been experimentally shown that increase blood triglycerides concentrations, as well as decreased levels of HDL-C, can be secondary to IR; in fact, IR improved the hepatic synthesis and secretion of triglyceride-rich VLDL particles. These evidences support the concept that the risk for CHD associated with HDL-cholesterol and triglycerides may be better determined in conjunction with IR measurement. Even though tools for the measurement of fasting plasma insulin still need to be defined, it has been demonstrated that even a locally defined measure of IR, combined with decreased HDL-C and increased triglycerides concentrations, is more accurate in the identification of the CHD risk than the use of triglycerides and HDL-C blood concentrations (82)10.
In a community-based cohort from the Framingham Heart Study no relation between IR and incident atrial fibrillation (AF) has been observed (83). Lower natriuretic peptide levels have been registered in both non-obese and obese individuals. A relative natriuretic peptide deficiency, probably due to the IR found in obese individuals may represent one reason of the induction of hypertension by IR (84).
One of the mechanisms through which MS and IR are involved in the pathogenesis of cardiovascular diseases seems to be found in the impairment of vascular function by influencing the component metabolic abnormalities that comprise MS. In fact, in a large community-based cohort study using age- and sex-adjusted models, a relation between IR and larger baseline brachial artery diameter and higher baseline flow was observed.
The components of the MS are strictly linked to endothelial dysfunction, in particular with a reduced endothelium-dependent vasodilation. This endothelial dysfunction seems to be associated also to IR in selected patients, such as young obese individuals; in addition, insulin resistant offspring of diabetic individuals have a reduced brachial artery flow-mediated dilation. Other studies observed a lack of association between IR and vascular dysfunction when concurrent risk factors, including the MS components, were adjusted, suggesting a strong relation between the components of MS and IR in the pathogenesis of cardiovascular diseases (85).
It has been suggested that IR may be involved in the pathogenesis of atherosclerosis, according to the evidence that the more pronounced IR in youths is, the more circulating biomarkers of endothelial dysfunction are elevated, while adiponectin, which plays an antiatherogenic role, is reduced (86).
In a study conducted on the German population involved in the Kooperative Gesundheitsforschung in der Region Augsburg (KORA) S4/F4 study, lower values of adiponectin have been demonstrated to be associated with higher risk of T2D in insulin resistant individuals, estimated by homeostasis model assessment (HOMA-IR), but not in insulin sensible ones; this evidence suggests that low adiponectin may have a deleterious effect only in presence of IR. Moreover, an association between high ceramides with saturated fatty acids in the blood sample and HOMA-IR and between higher sphingomyelins with saturated fatty acids and lower HOMA-IR in individuals with normal BMI was observed. This study prompts lowering of circulating ceramides with saturated fatty acids as a possible target in pre-diabetes (87, 88).
Laboratory studies have shown that in β-cells in culture (inducing an increase of sphingomyelin by inhibiting its hydrolysis) prevents palmitate-induced lipotoxicity (89), while the inhibition of sphingomyelin synthase in myotubes (causing a reduced sphingomyelin level) leads to an impaired insulin signaling (90). Conversely, studies conducted on knock-out mice show the protective role of lowering sphingomyelin high-fat induced obesity and IR (91, 92).
Clamp studies in adolescents and young adults showed that non-alcoholic fatty liver disease (NAFLD) is associated with IR, probably because of the increased abdominal visceral adiposity (93, 94).
The relevant association between IR and cardiovascular risk in the pediatric population is well known, especially in obese children (95–97), even though further research is needed to clarify this association. However, the available tools for the measurement of insulin sensitivity are costly, complex and long-lasting and there is no indication for treating isolated IR (3). Therefore, the preconditions for the fulfillment of a screening project in children are absent, even in obese patients (3, 98, 99).
Among the tools proposed for the screening of IR, the insulin sensitivity index (ISI0,120), proposed by Gutt et al. may represent a useful alternative predictor of both DM and CVD events, even if it deserves further evaluation, particularly in children.
A cohort study on 2,898 from the Framingham Offspring Study without diabetes or cardiovascular diseases at baseline demonstrated that the ISI0,120 and the MS phenotype are independent predictors of CVD. In fact, as shown by the National Cholesterol Education Program (NCEP), MS phenotype seems not to be enough complete for the detection of the cardiovascular disease risk associated with IR (100)11.
The evidence offered by the Prevention of Renal and Vascular End Stage Disease (PREVEND) study on a Dutch cohort from the general population of the city of Groningen, suggest that the Lipoprotein Insulin Resistance Index (LP-IR) may represent a valid tool for the detection of individuals with IR, who risk the development of T2DM even if no clinical signs, such as overweight or impaired glycemia, are manifest12. In addition, LP-IR considers early lipoprotein alterations in insulin resistant subjects completely reversable with intervention with diet and exercise, making primary prevention easier and more efficient. Independent population studies, such as the Multi-Ethnic Study of Atherosclerosis (MESA) and the Women's Health Study (WHS), have confirmed the association between LP-IR and secondary development of T2DM, even in individuals who have the prerequisites to be considered at low risk. These studies suggest that LP-IR may add significant information to the Framingham Offspring Study (FOS) risk score due to the early occurrence of the lipoprotein changes in comparison to dysglycemia (101).
Primary prevention of IR consists in avoiding the most common changeable risk factors including maternal obesity, gestational diabetes, maternal undernutrition and smoking during pregnancy (65, 102–106). Moreover, although a direct association between breast-feeding and improved insulin sensitivity is unknown, breast-feeding should be promoted because it is involved in preventing obesity in children (107, 108).
Finally, physical activity should be promoted in all children, in association with combined intervention programs aimed at avoiding excessive weight gain (3).
The first approach to IR in children consists of lifestyle interventions, including dietary modifications and increased physical activity.
Some studies suggest that exercise may have a major impact on the improvement of insulin sensitivity than the isolated reduction of body mass index (109). The Framingham Heart Study gives evidence that there is a significant association between physical activity and sedentary time and insulin sensitivity and adipokine blood levels. In particular, physical activity is associated with higher insulin sensitivity, while sedentary time is positively associated with leptin and fatty acid binding protein (FABP) levels. In addition, it has been observed that the number of steps per day is directly related to IGF-1 levels and inversely related to high sensitivity C-reactive protein (hsCRP) values.
Despite the evidence of the role of exercise training programmes in the improvement of insulin sensitivity, the mechanisms through which physical activity reduces this pathological condition and whether this response is simply the effect of changes in body composition is still unknown (110).
It has been demonstrated that a reduced fat intake through diet improve insulin sensitivity in adolescents (3, 111). In fact, a diet based on a high intake of whole-grain and fibers seems to promote weight loss and lower IR (112).
A pharmacologic intervention in obese children is sometimes needed to implement the effects of these primary prevention interventions. However, rare but serious side effects have been observed with all the available medications, therefore they should be used in selected cases. Patients' age, weight and comorbidities should be listed when considering pharmacological therapies and a close monitoring is needed once they are introduced. Moreover, longer-term (≥1 year) evidence of benefits in youth is still missing (113, 114).
Metformin, a biguanide derivate, has been demonstrated to have beneficial results in further decreasing BMI (115). It reduces insulin resistance by decreasing fasting plasma glucose and insulin concentrations in adults, as demonstrated in large, randomized, clinical trials. Metformin has been approved by the US Food and Drug Administration for the treatment of T2D in 10-year-old or older children and it is the only treatment evaluated in formal clinical trials concerning prediabetes in children. In non-diabetic obese adults it reduces food intake, causing weight loss and reduction of fasting plasma glucose, cholesterol, and insulin concentrations. In addition, metformin has been demonstrated to improve BMI, body fat composition, fasting glucose, insulin, glycated hemoglobin (HbA1c), IR expressed by the HOMA-IR, blood pressure and lipid profile in short trials conducted on small sample sizes of children and adolescents (115)13. Although it has been observed that metformin improves insulin sensitivity in adolescents with T2D and PCOS, metformin is not yet indicated as a treatment for isolated IR (3).
Long-term and consistent data are still missing to establish its role in the pediatric population.
Gastro-intestinal adverse drug reactions, among which abdominal pain, nausea, metallic taste, bloating, and diarrhea, are commonly observed in patients treated with metformin and can be prevented or intensely reduced by starting its administration with low doses and increasing the dose gradually or using extended-release formulations.
Infrequent side effects observed in clinical trials are lactic acidosis, especially in case of overdose, and vitamin B12 deficiency, requiring monitoring, and when necessary adequately supplementing this vitamin when necessary.
In conclusion, metformin is safe and presents evident positive effects on insulin sensitivity, but it should be introduced only in selected patients, as suggested by the 2017 Pediatric Obesity Clinical Guidelines from The Endocrine Society. Furthermore, its long-term benefits in insulin-resistant children are still to be analyzed.
Glucagon-like peptide-1 (GLP-1) is a gut hormone, which interferes with the inflammatory processes by reducing the release of inflammatory cytokines and inhibiting the infiltration of macrophages into the adipose tissue, the liver and the blood vessel wall (116). Since chronic inflammation is involved in the pathogenesis of IR, the pharmacodynamics studies conducted on GLP-1, suggest that GLP-1 analogs, such as Liraglutide, could improve insulin sensitivity in insulin resistant patients (116). Danne et al. (117) conducted a 5 week randomized, double-blind, placebo-controlled trial on 21 obese adolescents from 12 to 17 years old and with a Tanner stage of 2 to 5. Gastrointestinal side effects of liraglutide, especially abdominal pain, were observed in 96.5% of cases and mild hypoglycaemia occurred in 8 patients, in one-half of cases after prolonged fast. No severe hypoglycaemia was reported. No severe treatment emergent adverse effects were observed.
Despite the favorable effects of liraglutide, such as improvement of BMI z score, body weight, FPG, HbA1c and fasting serum insulin observed, none was statistically significant, probably due to the short duration of the trial and small number of participants (117).
Dipeptidylpeptidase-4(DPP4) inhibitors are novel oral glucose-lowering drugs which decreases the inhibition of endogenous incretins to induce the secretion of insulin in relation to glucose blood levels, resulting in improved fasting plasma glucose, postprandial glucose, and HbA1c level. Although there is no evidence in children, DPP4 inhibitors, in combination with insulin, have been demonstrated to decrease HbA1c blood concentrations with a minor weight gain and incidence of hypo- glycemia in adults affected by T2D (118).
The sodium–glucose cotransporter type 1 (SGLT1) is the main transporter involved in the absorption of glucose and galactose in the gastrointestinal tract, while sodium–glucose cotransporter type 2 (SGLT2) is located in the kidney, where it is responsible for the reabsorption of 90% filtered glucose (119, 120). Phase 2 and 3 clinical studies conducted on Sotagliflozin, an oral potent dual inhibitor of SGLT1 and SGLT2, have shown an improved glycaemic control with lower HbA1c, less side effects such as body weight and hypertension, and a persistent efficacy in lower estimated glomerular filtration rate levels in adults with type 1 and 2 diabetes. However, more consistent data are needed to establish its real benefits and side effects on insulin-resistant adults and children (120).
Weight loss drugs such as sibutramine and orlistat are able to improve insulin sensitivity in children and adolescents, although their use need to be properly and selectively adopted in this age group (121–123).
The management of congenital generalized lipodystrophy is multidisciplinary and must be adapted to the characteristics of each patient. It may include psychological support, cosmetic surgery, the promotion of a high carbohydrate, low-fat diet and regular exercise for type 1 CGL, the avoidance of strenuous exercise and use of β-adrenergic blockers and other anti-arrhythmic medications for patients with type 4 CGL. It is unclear whether patients with type 2 CGL and cardiomyopathy should avoid exercise (15). For subjects who present extremely high blood triglycerides levels treatment with fibric acid derivatives are suggested, adding, in specific cases, low-dose statins in order to reduce non-HDL cholesterol.
Metformin and sulphonylureas are the first-line therapy for diabetes mellitus in patients with CGL.
Metreleptin, a recombinant analog of human leptin, seems to be effective in the treatment of metabolic complications in type 1 and 2 CGL. This molecule has its main effect on hypothalamus reducing appetite. In 2014, the FDA approved the use of metreleptin, in addition to dietary changes, in patients with CGL and AGL (15).
The only therapy available for patients with leprechaunism to date is recombinant IGF1, aiming to prevent compensatory hyperinsulinemia (23). IGF has about the 6% of the glucose-lowering action effect of insulin and has a similar structure to it, therefore it is able to bind to insulin receptors triggering peripheral glucose uptake and glycogen synthesis and inhibiting protein catabolism. The efficacy of this treatment is still controversial and further studies are needed but evidence form the small number of in vivo studies available at the moment seems promising (23).
IR is a pathological condition strongly associated with obesity and involved in the pathogenesis of T2D. Obesity is not the only determinant of IR; prolonged use of corticosteroids or growth hormone therapy and genetic diseases may be responsible for this condition too.
The gold standard for the assessment of IR is the hyperinsulinemic euglycemic clamp, nevertheless its costs and difficult management in clinical and research activity have determined the need of surrogate methods. HOMA-IR and QUICKI present a favorable correlation with the hyperinsulinemic-euglycemic clamp, however they are not considered a valid test for the evaluation of IR, therefore in the clinical practice the diagnosis of IR in obese patients is based on clinical features, including hyperglycemia, dyslipidemia, abdominal obesity, and hypertension. The main unchangeable risk factors for IR in children are the Caucasian ethnicity and puberty. At present, the preconditions for the fulfillment of a screening project in children are absent, even in obese patients. The prevention of IR in children consists in avoiding maternal obesity, gestational diabetes, maternal undernutrition, and smoking during pregnancy and encouraging breast-feeding and physical activity.
The treatment of insulin resistant children is firstly targeted to lifestyle interventions. In selected cases, the integration of a pharmacological intervention is needed. However, adequate data concerning the safety and long-term efficiency of drugs in patients with IR are not available yet.
Despite several studies have been conducted in children and adolescent with IR, many questions remain open. Further studies, and above all large-population long-lasting observational studies should be undertaken in order to unravel the unsolved issues related to its pathogenesis, diagnosis and treatment.
All the authors have made a substantial, direct and intellectual contribution to the work. CF revised it critically. All authors listed approved the work for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^Available online at: www.ncbi.nlm.nih.gov (accessed January 5, 2019).
2. ^Available online at: www.science.gov (accessed January 15, 2019).
3. ^Available online at:https://www.pl.scribd.com
4. ^Available online at: https://www.ucsfhealth.org/tests/003466.html (accessed May 2, 2019).
5. ^Available online at: https://www.care.diabetesjournals.org (accessed January 10, 2018).
6. ^Available online at: https://www.diabetesincontrol.com (accessed January 20, 2019).
7. ^Available online at: https://www.helmholtz-muenchen.de (accessed January 20, 2019).
8. ^Available online at: https://www.jcb.rupress.org (accessed January 20, 2010).
9. ^Available online at: https://www.atvb.ahajournals.org (accessed January 8, 2019).
10. ^Available online at: https://www.atvb.ahajournals.org (accessed January 8, 2019).
11. ^Available online at: https://www.diabetes.diabetesjournals.org (accessed January 15, 2019).
12. ^Available online at: https://www.pdfs.semanticscholar.org (accessed January, 15 2019).
13. ^Available online at: https://www.eprints.ucl.ac.uk (accessed January 1, 2019).
1. Volovelsky O, Weiss R. Fatty liver disease in obese children relation to other metabolic risk factors. Int J Pediatric Obes. (2011) 6:59–64. doi: 10.3109/17477166.2011.583661
2. Kozusko K, Patel S, Savage DB. Human congenital perilipin deficiency and insulin resistance. Hormone Resist Hypersensi. (2013) 24:150–5. doi: 10.1159/000342511
3. Levy-Marchal C, Arslanian S, Cutfield W, Sinaiko A, Druet C, Marcovecchio ML, et al. Insulin resistance in children: Consensus, perspective, and future directions. J Clin Endocrinol Metabol. (2010) 95:5189–98. doi: 10.1210/jc.2010-1047
4. Juárez-López C, Klünder-Klünder M, Medina-Bravo P, Madrigal-Azcárate A, Mass-Díaz E, Flores-Huerta S. Insulin resistance and its association with the components of the metabolic syndrome among obese children and adolescents. BMC Public Health. (2010) 10:318. doi: 10.1186/1471-2458-10-318
5. Ferrannini E. Insulin resistance is central to the burden of diabetes. Diab Metab Rev. (1997) 13:81–6.
6. Giannini C, Caprio S. Islet function in obese adolescents. Diab Obes Metabo. (2012) 14:40–5. doi: 10.1111/j.1463-1326.2012.01643.x
7. McLaughlin T, Allison G, Abbasi F, Lamendola C, Reaven G. Prevalence of insulin resistance and associated cardiovascular disease risk factors among normal weight, overweight, and obese individuals. Metab Clin Exp. (2004) 53:495–9. doi: 10.1016/j.metabol.2003.10.032
8. Hollenbeck C, Reaven GM. Variations in insulin-stimulated glucose uptake in healthy individuals with normal glucose tolerance. J Clin Endocrinol Metabol. (1987) 64:1169–73. doi: 10.1210/jcem-64-6-1169
9. Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G. Insulin resistance and hypersecretion in obesit. J Clin Invest. (1997) 100:1166–73. doi: 10.1172/JCI119628
10. Sinaiko AR, Steinberger J, Moran A, Prineas RJ, Vessby B, Basu S, et al. Relation of body mass index and insulin resistance to cardiovascular risk factors, inflammatory factors, and oxidative stress during adolescence. Circulation. (2005) 111:1985–91. doi: 10.1161/01.CIR.0000161837.23846.57
11. Semple RK, Savage DB, Cochran EK, Gorden P, ORahilly S. Genetic syndromes of severe insulin resistance 3006. Endocr Rev. (2011) 32:498–514. doi: 10.1210/er.2010-0020
12. Goran MI, Gower BA. Longitudinal study on pubertal insulin resistance. Diabetes. (2001) 50:2444–50. doi: 10.2337/diabetes.50.11.2444
13. Aldhoon-Hainerová I, Zamrazilová H, Dušátková L, Sedláčková B, Hlavatý P, Hill M, et al. Glucose homeostasis and insulin resistance: prevalence, gender differences and predictors in adolescents. Diabetol Metabo Syndrome. (2014) 6:100. doi: 10.1186/1758-5996-6-100
14. Arslanian S, Kim JY, Nasr A, Bacha F, Tfayli H, Lee S, et al. Insulin sensitivity across the lifespan from obese adolescents to obese adults with impaired glucose tolerance : who is worse off? Pediatr Diabetes. (2017) 19:205–11. doi: 10.1111/pedi.12562
15. Patni N, Garg A. Congenital generalized lipodystrophies — new insights into metabolic dysfunction. Nat Rev Endocrinol. (2015) 11:522–34. doi: 10.1038/nrendo.2015.123
16. Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. (2009) 1791:507–13. doi: 10.1016/j.bbalip.2008.12.014
17. Chandalia M, Garg A, Vuitch F, Nizzi F. Postmortem findings in congenital generalized lipodystrophy. J Clin Endocrinol Metab. (1995) 80:3077–81. doi: 10.1210/jcem.80.10.7559900
18. Agarwal A, Garg A. Genetic disorders of adipose tissue development, differentiation, and death. Annu Rev Genomics Hum Genet. (2006) 7:175–99. doi: 10.1146/annurev.genom.7.080505.115715
19. Pardini VC, Victoria IM, Rocha SM, Andrade DG, Rocha AM, Pieroni FB, et al. Leptin levels, beta-cell function, and insulin sensitivity in families with congenital and acquired generalized lipoatropic diabetes. J Clin Endocrinol Metab. (1998) 83:503–8. doi: 10.1210/jcem.83.2.4567
20. Ahima RS, Flier JS, Spiegelman BM, Mohamed-Ali V, et al. Adipose tissue as an endocrine organ. Trends Endocrinol Metab. (2000) 11:327–32. doi: 10.1016/s1043-2760(00)00301-5
21. Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, et al. Life without white fat: A transgenic mouse. Genes Dev. (1998) 12:3168–81. doi: 10.1101/gad.12.20.3168
22. Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL, et al. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. (1998) 12:3182–94. doi: 10.1101/gad.12.20.3182
23. Kirkwood A, Stuart G, Harding L. Donohue syndrome : a review of literature, case series, and anesthetic considerations. Pediatr Anesth. (2017) 28:23–7. doi: 10.1111/pan.13273
24. Moller DE, Flier JS. Insulin resistance - mechanisms, syndromes, and implications. New Engl J Med. (1991) 325:938–48.
25. Huang-Doran I, Tomlinson P, Payne F, Gast A, Sleigh A, Bottomley W, et al. Insulin resistance uncoupled from dyslipidemia due to C-terminal PIK3R1 mutations. JCI Insight. (2016) 1:e88766. doi: 10.1172/jci.insight.88766
26. Tritos NA, Mantzoros CS. Clinical review 97: syndromes of severe insulin resistance. J Clin Endocrinol Metab. (1998) 83:3025–30. doi: 10.1210/jc.83.9.3025
27. Buchanan TA, Watanabe RM, Xiang AH. Limitations in surrogate measures of insulin resistance. J Clin Endocrinol Metab. (2010) 95:4874–6. doi: 10.1210/jc.2010-2167
28. Kim JK. Hyperinsulinemic – euglycemic clamp to assess insulin sensitivity in vivo, in type 2 diabetes. Methods Mol Biol. (2009) 560:221–38. doi: 10.1007/978-1-59745-448-3
30. Hwu C-M. Measurements of insulin resistance in hypertension: where are we now? J Human Hypertens. (2007) 21:693–6. doi: 10.1038/sj.jhh.1002202
31. Lee S, Muniyappa R, Yan X, Chen H, Yue LQ, Hong EG, et al. Comparison between surrogate indexes of insulin sensitivity and resistance and hyperinsulinemic euglycemic clamp estimates in mice. Am J Physiol Endocrinol Metab. (2008) 294:E261–70. doi: 10.1152/ajpendo.00676.2007
32. Rasmussen-Torvik LJ, Pankow JS, Jacobs DR, Steffen LM, Moran AM, Steinberger J, et al. Heritability and genetic correlations of insulin sensitivity measured by the euglycaemic clam. Diabet Med. (2007) 24:1286–9. doi: 10.1111/j.1464-5491.2007.02271.x
33. Schwartz B, Jacobs DR, Morgan A, Sreinberger J, Hong C-P, Sinaiko AR. Measurement of insulin sensitivity in children. Diabetes Care. (2008) 31:783–8. doi: 10.2337/dc07-1376
34. Shashaj B, Luciano R, Contoli B, Stefano G, Rita M, Carmela S, et al. Reference ranges of HOMA-IR in normal-weight and obese young Caucasians. Acta Diabetologica. (2015) 53:251–60. doi: 10.1007/s00592-015-0782-4
35. Wallace TM, Levy JC, Matthews DR. Use and Abuse of HOMA Modeling. Diabetes Care. (2004) 27:1487–95. doi: 10.2337/diacare.27.6.1487
36. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. (1985) 28:412–9.
37. Turner RC, Holman RR, Matthews D, Hockaday TD, Peto J. Insulin deficiency and insulin resistance interaction in diabetes: estimation of their relative contribution by feedback analysis from basal plasma insulin and glucose concentrations. Metabolism. (1979) 28:1086–96.
38. Mori K, Emoto M, Motoyama K, Lee E, Yamada S, Morioka T, et al. Undercarboxylated osteocalcin does not correlate with insulin resistance as assessed by euglycemic hyperinsulinemic clamp technique in patients with type 2 diabetes mellitus. Diabetol Metab Syndrome. (2012) 4:53. doi: 10.1186/1758-5996-4-53
39. Hsiao JY, Wang CL, Hsia PJ, Hsieh MC, Hsin SC, Lin KD, et al. Decreased insulin secretion and insulin sensitivity are associated with liver function in subjects with fasting glucose between 100 and 109 mg/dL in Taiwanese population. Pancreas. (2007) 35:343–7. doi: 10.1097/mpa.0b013e31811f44fd
40. Yin J, Li M, Xu L, Wang Y, Cheng H, Zhao X, et al. Insulin resistance determined by Homeostasis Model Assessment (HOMA) and associations with metabolic syndrome among Chinese children and teenagers. Diabetol Metab Syndrome. (2013) 5:71. doi: 10.1186/1758-5996-5-71
41. Tobisch B, Blatniczky L, Barkai L. Cardiometabolic risk factors and insulin resistance in obese children and adolescents: relation to puberty. Pediatr Obesity. (2015) 10:37–44. doi: 10.1111/j.2047-6310.2013.00202.x
42. Sinha R, Fisch G, Teague B, Tamborlane WV, Banyas B, Allen K, et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. New Engl J Med. (2002) 346:802–10. doi: 10.1056/NEJMoa012578
43. Weiss R, Dziura J, Burgert TS, Tamborlane WV, Taksali SE, Yeckel CW, et al. Obesity and the metabolic syndrome in children and adolescents. New Engl J Med. (2004) 350:2362–74. doi: 10.1056/NEJMoa031049
44. Obi K, Ramsey M, Hinton A, Stanich P, Gray DM, Krishna SG, et al. Insights into insulin resistance, lifestyle, and anthropometric measures of patients with prior colorectal cancer compared to controls: a National Health and Nutrition Examination Survey (NHANES) Study. Curr Prob Cancer. (2018) 42:276–85. doi: 10.1016/j.currproblcancer.2017.12.002
45. Wallace IR, McKinley MC, Bell PM, Hunter SJ. Sex hormone binding globulin and insulin resistance. Clin Endocrinol. (2013) 78:321–9. doi: 10.1111/cen.12086
46. Giannini C, Santoro N, Caprio S, Kim G, Lartaud D, Shaw M, et al. The triglyceride-to-HDL cholesterol ratio: association with insulin resistance in obese youths of different ethnic backgrounds. Diabetes Care. (2011) 34:1869–74. doi: 10.2337/dc10-2234
47. McAuley KA, Williams SM, Mann JI, Walker RJ, Lewis-Barned NJ, Temple LA, et al. Diagnosing insulin resistance in the general population. Diabetes Care. (2001) 24:460–4. doi: 10.2337/diacare.24.3.460
48. Matsuda M. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clam. Diabetes Care. (1999) 22:1462–70.
49. Soonthornpun S, Setasuban W, Thamprasit A, Chayanunnukul W, Rattarasarn C, Geater A. Novel insulin sensitivity index derived from oral glucose tolerance test. J Clin Endocrinol Metab. (2003) 88:1019–23. doi: 10.1210/jc.2002-021127
50. Yeckel CW, Weiss R, Dziura J, Taksali SE, Dufour S, Burgert TS, et al. Validation of insulin sensitivity indices from oral glucose tolerance test parameters in obese children and adolescents. J Clin Endocrinol Metab. (2004) 89:1096–101. doi: 10.1210/jc.2003-031503
51. Arslanian SA, Saad R, Lewy V, Danadian K, Janosky J. Hyperinsulinemia in African-American children: decreased insulin clearance and increased insulin secretion and its relationship to insulin sensitivity. Diabetes. (2002) 51:3014–9. doi: 10.2337/diabetes.51.10.3014
52. Goran MI, Bergman RN, Cruz ML, Watanabe R. Insulin resistance and associated compensatory responses in African-American and hispanic children. Diabetes Care. (2002) 25:2184–90. doi: 10.2337/diacare.25.12.2184
53. Whincup PH. Early evidence of ethnic differences in cardiovascular risk: cross sectional comparison of British South Asian and white children. BMJ. (2002) 324:635. doi: 10.1136/bmj.324.7338.635
54. Després JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E, et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. (2008) 28:1039–49. doi: 10.1161/ATVBAHA.107.159228
55. Preis SR, Massaro JM, Robins SJ, Hoffmann U, Vasan RS, Irlbeck T, et al. Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham Heart Study. Obesity. (2011) 18:2191–8. doi: 10.1038/oby.2010.59.Abdominal
56. Arslanian SA, Lewy VD, Danadian K. Glucose intolerance in obese adolescents with polycystic ovary syndrome: roles of insulin resistance and beta-cell dysfunction and risk of cardiovascular disease. J Clin Endocrinol Metab. (2001) 86:66–71. doi: 10.1210/jcem.86.1.7123
57. Silfen ME, Denburg MR, Manibo AM, Lobo RA, Jaffe R, Ferin M, et al. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between nonobese and obese adolescents. J Clin Endocrinol Metab. (2003) 88:4682–8. doi: 10.1210/jc.2003-030617
58. Goran MI, Bergman RN, Avila Q, Watkins M, Ball GDC, Shaibi GQ, et al. Impaired glucose tolerance and reduced beta-cell function in overweight Latino children with a positive family history for type 2 diabetes. J Clin Endocrinol Metab. (2004) 89:207–12. doi: 10.1210/jc.2003-031402
59. Arslanian SA, Bacha F, Saad R, Gungor N. Family history of type 2 diabetes is associated with decreased insulin sensitivity and an impaired balance between insulin sensitivity and insulin secretion in white youth. Diabetes Care. (2005) 28:115–9. doi: 10.2337/diacare.28.1.115
60. Buzzetti R, Petrone A, Caiazzo AM, Alemanno I, Zavarella S, Capizzi M, et al. PPAR-gamma2 Pro12Ala variant is associated with greater insulin sensitivity in childhood obesity. Pediatr Res. (2005) 57:138–40. doi: 10.1203/01.PDR.0000147728.62185.21
61. Poulsen P, Levin K, Petersen I, Christensen K, Beck-Nielsen H, Vaag A. Heritability of insulin secretion, peripheral and hepatic insulin action, and intracellular glucose partitioning in young and old Danish twins. Diabetes. (2005) 54:275–83. doi: 10.2337/diabetes.54.1.275
62. Souren NY, Paulussen ADC, Loos RJF, Gielen M, Beunen G, Fagard R, et al. Anthropometry, carbohydrate and lipid metabolism in the east flanders prospective twin survey: heritabilities. Diabetologia. (2007) 50:2107–16. doi: 10.1007/s00125-007-0784-z
63. Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. (2009) 41:25–34. doi: 10.1038/ng.287
64. Brown AE, Walker M. Genetics of insulin resistance and the metabolic syndrome. Curr Cardiol Rep. (2016) 18:75. doi: 10.1007/s11886-016-0755-4
65. Silverman BL, Metzger BE, Cho NH, Loeb CA. Impaired glucose tolerance in adolescent offspring of diabetic mothers: relationship to fetal hyperinsulinism. Diabetes Care. (1995) 18:611–7. doi: 10.2337/diacare.18.5.611
66. Plagemann A, Kohlhoff R, Harder T, Rohde W, Dorner G. Overweight, obesity and impaired glucose tolerance in children of mothers with diabetes during pregnancy. Diabetes Nutr Metab. (1997) 10:116–9.
67. Bozkurt L, Göbl CS, Rami-Merhar B, Winhofer Y, Baumgartner-Parzer S, Schober E, et al. The cross-link between adipokines, insulin resistance and obesity in offspring of diabetic pregnancies. Hormone Res Paediatr. (2016) 86:300–8. doi: 10.1159/000448076
68. Catalano PM, Thomas A, Huston-Presley L, Amini SB. Increased fetal adiposity: a very sensitive marker of abnormal in utero development. Am J Obstetr Gynecol. (2003) 189:1698–704. doi: 10.1016/S0002-9378(03)00828-7
69. Hillier TA, Pedula KL, Schmidt MM, Mullen JA, Charles MA, Pettitt DJ. Childhood obesity and metabolic imprinting: The ongoing effects of maternal hyperglycemia. Diabetes Care. (2007) 30:2287–92. doi: 10.2337/dc06-2361
70. Sinaiko AR, Donahue RP, Jacobs DR, Prineas RJ. Relation of weight and rate of increase in weight during childhood and adolescence to body size, blood pressure, fasting insulin, and lipids in young adults: the minneapolis childrens blood pressure study. Circulation. (1999) 99:1471–6. doi: 10.1161/01.CIR.99.11.147
71. Eriksson JG, Forsén T, Tuomilehto J, Osmond C, Barker DJ. Early adiposity rebound in childhood and risk of Type 2 diabetes in adult life. Diabetologia. (2003) 46:190–4. doi: 10.1007/s00125-002-1012-5
72. Ong KK, Petry CJ, Emmett PM, Sandhu MS, Kiess W, Hales CN, et al. Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin-like growth factor-I levels. Diabetologia. (2004) 47:1064–70. doi: 10.1007/s00125-004-1405-8
73. Mericq V, Ong KK, Bazaes R, Peña V, Avila A, Salazar T, et al. Longitudinal changes in insulin sensitivity and secretion from birth to age three years in small- and appropriate-for-gestational-age children. Diabetologia. (2005) 48:2609–14. doi: 10.1007/s00125-005-0036-z
74. Finken MJJ, Keijzer-Veen MG, Dekker FW, Frölich M, Hille ETM, Romijn JA, et al. Preterm birth and later insulin resistance: effects of birth weight and postnatal growth in a population based longitudinal study from birth into adult life. Diabetologia. (2006) 49:478–85. doi: 10.1007/s00125-005-0118-y
75. Barker DJP. The origins of the developmental origins theory. J Int Med. (2007) 261:412–7. doi: 10.1111/j.1365-2796.2007.01809.x
76. Leunissen RWJ, Kerkhof GF, Stijnen T, Hokken-Koelega A. Timing and tempo of first-year rapid growth in relation to cardiovascular and metabolic risk profile in early adulthood. JAMA. (2009) 301:2234–42. doi: 10.1001/jama.2009.761
77. Hofman PL, Regan F, Jackson WE, Jefferies C, Knight DB, Robinson EM, et al. Premature birth and later insulin resistance. New Engl J Med. (2004) 351:2179–86. doi: 10.1056/NEJMoa042275
78. Gungor N, Bacha F, Saad R, Janosky J, Arslanian S. Youth type 2 diabetes: insulin resistance, beta-cell failure, or both? Diabetes Care. (2005) 28:638–44. doi: 10.2337/diacare.28.3.638
79. Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Meigs JB, et al. Insulin resistance as estimated by homeostasis model assessment predicts incident symptomatic cardiovascular disease in caucasian subjects from the general population: the Bruneck study. Diabetes. (2007) 30:318–24. doi: 10.2337/dc06-0919
80. Jeppesen J, Hansen TW, Rasmussen S, Ibsen H, Torp-Pedersen C, Madsbad S. Insulin resistance, the metabolic syndrome, and risk of incident cardiovascular disease: a population-based study. Crit Ultrasound J. (2009) 1:27–32. doi: 10.1016/j.jacc.2007.01.088
81. Ikezaki H, Ai M, Schaefer EJ, Otokozawa S, Asztalos BF, Nakajima K, et al. Cardiovascular disease prevalence and insulin resistance in the Kyushu – Okinawa Population Study and the Framingham Offspring Study. J Clin Lipidol. (2017) 11:348–56. doi: 10.1016/j.jacl.2017.01.014
82. Robins SJ, Lyass A, Zachariah JP, Massaro JM, Vasan R. Insulin resistance and the relationship of a dyslipidemia to coronary heart disease. Framingham Heart Study Arterioscler Thromb Vasc Biol. (2011) 31:1208–14. doi: 10.1161/ATVBAHA.110.219055.Insulin
83. Fontes JD, Lyass A, Massaro JM, Rienstra M, Dallmeier D, Schnabel RB, et al. Insulin resistance and atrial fibrillation (from the Framingham Heart Study). Am J Cardiol. (2012) 109:87–90. doi: 10.1016/j.amjcard.2011.08.008
84. Khan AM, Cheng S, Magnusson M, Larson MG, Newton-cheh C, Mccabe EL, et al. Cardiac natriuretic peptides, obesity, and insulin resistance : evidence from two community-based studies. J Clin Endocrinol Metab. (2011) 96:3242–9. doi: 10.1210/jc.2011-1182
85. Hamburg NM, Larson MG, Vita JA, Vasan RS, Keyes MJ, Widlansky ME, et al. Metabolic syndrome, insulin resistance and brachial artery vasodilator function in framingham offspring participants without clinical evidence of cardiovascular disease. Am J Cardiol. (2008) 101:82–8. doi: 10.1016/j.amjcard.2007.07.053
86. Lee S, Gungor N, Bacha F, Arslanian S. Insulin resistance: Link to the components of the metabolic syndrome and biomarkers of endothelial dysfunction in youth. Diabetes Care. (2007) 30:2091–7. doi: 10.2337/dc07-0203
87. Hivert MF, Sullivan LM, Shrader P, Fox CS, Nathan DM, DAgostino R, et al. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. Diabetologia. (2011) 54:1019–24. doi: 10.1007/s00125-011-2067-y.Insulin
88. Lemaitre RN, Yu C, Hoofnagle A, Hari N, Jensen PN, Fretts A, et al. Circulating sphingolipids, insulin, HOMA-IR, and HOMA-B: the strong heart family study. Diabetes. (2018) 67:1663–72. doi: 10.2337/db17-1449
89. Boslem E, Weir JM, MacIntosh G, Sue N, Cantley J, Meikle P, et al. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J Biol Chem. (2013) 288:26569–82. doi: 10.1074/jbc.M113.489310
90. Park M, Kaddai V, Ching J, Fridianto KT, Sieli RJ, Sugii S, et al. A role for ceramides, but not sphingomyelins, as antagonists of insulin signaling and mitochondrial metabolism in C2C12 myotubes. J Biol Chem. (2016) 291:23978–88. doi: 10.1074/jbc.M116.737684
91. Li Z, Zhang H, Liu J, Liang CP, Li Y, Li Y, et al. Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol Cell Biol. (2011) 31:4205–18. doi: 10.1128/MCB.05893-11
92. Mitsutake S, Zama K, Yokota H, Yoshida T, Tanaka M, Mitsui M, et al. Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. The Journal of biological chemistry. (2011) 286:28544–55. doi: 10.1074/jbc.M111.255646
93. Perseghin G, Bonfanti R, Magni S, Lattuada G, De Cobelli F, Canu T, et al. Insulin resistance and whole body energy homeostasis in obese adolescents with fatty liver disease. Am J Physiol Endocrinol Metab. (2006) 291:E697–703. doi: 10.1152/ajpendo.00017.2006
94. Deivanayagam S, Mohammed BS, Vitola BE, Naguib GH, Keshen TH, Kirk EP, et al. Nonalcoholic fatty liver disease is associated with hepatic and skeletal muscle insulin resistance in overweight adolescents. Am J Clin Nutr. (2008) 88:257–62. doi: 10.1097/OPX.0b013e3182540562
95. Weiss R, Bremer AA, Lustig RH. What is metabolic syndrome, and why are children getting it? Ann N Y Acad Sci. (2013) 1281:123–40. doi: 10.1111/nyas.12030
96. Watkins AN, Kelly AS, Prineas RJ, Marlatt KL, Dengel DR, Sinaiko AR, et al. Childhood wrist circumference is not a predictor of insulin resistance in adulthood. J Pediatr. (2015) 166:1085–7. doi: 10.1016/j.jpeds.2014.12.011
97. Ryder JR, Dengel DR, Jacobs DR, Sinaiko A, Kelly AS, Steinberger J. Relations among adiposity and insulin resistance with flow- mediated dilation, carotid intima-media thickness, and arterial stiffness in children. J Pediatr. (2016) 168:205–11. doi: 10.1016/j.jpeds.2015.08.034
98. Ramachandran A, Snehalatha C, Yamuna A, Murugesan N, Narayan KM. Insulin resistance and clustering of cardiometabolic risk factors in urban teenagers in southern India. Diabetes Care. (2007) 30:1828–33. doi: 10.2337/dc06-2097
99. Lee S, Bacha F, Gungor N, Arslanian S. Comparison of different definitions of pediatric metabolic syndrome: relation to abdominal adiposity, insulin resistance, adiponectin, and inflammatory biomarkers. J Pediatr. (2008) 152:177–84. doi: 10.1016/j.jpeds.2007.07.053
100. Rutter MK, Meigs JB, Sullivan LM, Agostino RBD, Wilson PW. Insulin resistance, the metabolic syndrome, and incident cardiovascular events in the framingham offspring study. Diabetes. (2005) 54:3252–7. doi: 10.2337/diabetes.54.11.3252
101. Flores-guerrero JL, Connelly MA, Shalaurova I, Gruppen EG, Kieneker LM, Dullaart RPF, et al. Lipoprotein insulin resistance index, a high-throughput measure of insulin resistance, is associated with incident type II diabetes mellitus in the prevention of renal and vascular end-stage disease study. J Clin Lipidol. (2019) 13:129–137.e1. doi: 10.1016/j.jacl.2018.11.009
102. Plagemann A, Harder T, Kohlhoff R, Rohde W, Dörner G. Glucose tolerance and insulin secretion in children of mothers with pregestational IDDM or gestational diabetes. Diabetologia. (1997) 40:1094–100. doi: 10.1007/s001250050792
103. Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, et al. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. (2000) 49:2208–11. doi: 10.2337/diabetes.49.12.2208
104. Kinra S, Rameshwar Sarma KV, Mendu VV, Ravikumar R, Mohan V, Wilkinson IB, et al. Effect of integration of supplemental nutrition with public health programmes in pregnancy and early childhood on cardiovascular risk in rural Indian adolescents: long term follow-up of Hyderabad nutrition trial. BMJ. (2008) 337:a605. doi: 10.1136/bmj.a605
105. Oken E, Levitan EB, Gillman MW. Maternal smoking during pregnancy and child overweight: systematic review and meta-analysis. Int J Obes. (2008) 32:201–10. doi: 10.1038/sj.ijo.0803760
106. Thiering E, Brüske I, Kratzsch J, Thiery J, Sausenthaler S, Meisinger C, et al. Prenatal and postnatal tobacco smoke exposure and development of insulin resistance in 10 year old children. Int J Hygiene Environ Health. (2011) 214:361–8. doi: 10.1016/j.ijheh.2011.04.004
107. Harder T, Bergmann R, Kallischnigg G, Plagemann A. Duration of breastfeeding and risk of overweight: a meta-analysis. Am J Epidemiol. (2005) 162:397–403. doi: 10.1093/aje/kwi222
108. Koletzko B, Von Kries R, Monasterolo RC, Subías JE, Scaglioni S, Giovannini M, et al. Can infant feeding choices modulate later obesity risk? Am J Clin Nutr. (2009) 89:1502S−1508S doi: 10.3945/ajcn.2009.27113D
109. Allen DB, Nemeth BA, Clark RR, Peterson SE, Eickhoff J, Carrel AL, et al. Fitness is a stronger predictor of fasting insulin levels than fatness in overweight male middle-school children. J Pediatr. (2007) 150:383–7. doi: 10.1016/j.jpeds.2006.12.051
110. Spartano NL, Stevenson MD, Xanthakis V, Larson MG, Andersson C, Murabito JM. Associations of objective physical activity with insulin sensitivity and circulating adipokine profile : the Framingham Heart Study. Clin Obes. (2017) 7:59–69. doi: 10.1111/cob.12177
111. Sunehag AL, Toffolo G, Treuth MS, Butte NF, Cobelli C, Bier DM, et al. Effects of dietary macronutrient content on glucose metabolism in children. J Clin Endocrinol Metab. (2002) 87:5168–78. doi: 10.1210/jc.2002-020674
112. Steffen LM, Jacobs DR, Murtaugh MA, Moran A, Steinberger J, Hong CP, et al. Whole grain intake is associated with lower body mass and greater insulin sensitivity among adolescents. Am J Epidemiol. (2003) 158:243–50. doi: 10.1093/aje/kwg146
113. Akhlaghi F, Matson KL, Houshang A, Kelly M, Karimani A, Sciences M. Clinical pharmacokinetics and pharmacodynamics of antihyperglycemic medications in children and adolescents with type 2 diabetes mellitus. Clin Pharmacokinet. (2017) 56:561–71. doi: 10.1007/s40262-016-0472-6
114. Chao AM, Wadden TA, Berkowitz. R. I., Chao AM, Wadden TA. The safety of pharmacologic treatment for pediatric obesity. Expert Opin Drug Safety. (2018) 17:379–85. doi: 10.1080/14740338.2018.1437143
115. Khokhar A, Umpaichitra V, Chin VL, Perez-Colon S. Metformin use in children and adolescents with prediabetes. Pediatr Clin N Am. (2017) 64:1341–53. doi: 10.1016/j.pcl.2017.08.010
116. Guo C, Huang T, Chen A, Chen X, Wang L, Shen F, et al. Glucagon-like peptide 1 improves insulin resistance in vitro through anti-inflammation of macrophages. Braz J Med Biol Res. (2016) 49:e5826. doi: 10.1590/1414-431X20165826
117. Danne T, Biester T, Kapitzke K, Jacobsen SH, Jacobsen LV, Petri K, et al. Liraglutide in an adolescent population with obesity : a randomized,double-blind,placebo-controlled 5-weektrial toassess safety,tolerability,and pharmacokinetics of liraglutide in adolescentsaged 12-17 years. J Pediatr. (2016) 181:146–153.e3. doi: 10.1016/j.jpeds.2016.10.076
118. Guo H, Fang C, Huang Y, Pei Y, Chen L, Hu J. The efficacy and safety of DPP4 inhibitors in patients with type 1 diabetes : A systematic review and meta-analysis. Diab Res Clin Pract. (2016) 1:2–9. doi: 10.1016/j.diabres.2016.08.022
119. Jens J, Birkenfeld A. Cardiometabolic crosstalk in obesity-associated arterial hypertension Rev Endocr Metab Disord. (2016) 17:19–28. doi: 10.1007/s11154-016-9348-1
120. Danne T, Biester T, Kordonouri O. Combined SGLT1 and SGLT2 inhibitors and their role in diabetes care. Diab Technol Ther. (2018) 20:S2-69–77. doi: 10.1089/dia.2018.0081
121. McDuffie JR, Calis KA, Uwaifo GI, Sebring NG, Fallon EM, Hubbard VS, et al. Three-month tolerability of orlistat in adolescents with obesity-related comorbid conditions. Obes Res. (2002) 10:642–50. doi: 10.1038/oby.2002.87
122. McDuffie JR, Calis KA, Uwaifo GI, Sebring NG, Fallon EM, Frazer TE, et al. Efficacy of orlistat as an adjunct to behavioral treatment in overweight African American and Caucasian adolescents with obesity-related co-morbid conditions. J Pediatr Endocrinol Metab. (2004) 17:307–19. doi: 10.1515/JPEM.2004.17.3.307
Keywords: insulin resistance, children, adolescents, metabolic syndrome, euglycemic clamp, HOMA-IR, QUICKI, WBISI
Citation: Tagi VM, Giannini C and Chiarelli F (2019) Insulin Resistance in Children. Front. Endocrinol. 10:342. doi: 10.3389/fendo.2019.00342
Received: 28 February 2019; Accepted: 13 May 2019;
Published: 04 June 2019.
Edited by:
Fabrizio Barbetti, University of Rome Tor Vergata, ItalyReviewed by:
Gianpaolo De Filippo, Bicêtre Hospital, FranceCopyright © 2019 Tagi, Giannini and Chiarelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Chiarelli, Y2hpYXJlbGxpQHVuaWNoLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.