 Guillaume Vial1,2†
Guillaume Vial1,2† Dominique Detaille
Dominique Detaille Bruno Guigas
Bruno Guigas
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 07 May 2019
Sec. Clinical Diabetes
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00294
This article is part of the Research TopicMetformin: Beyond DiabetesView all 12 articles
Metformin is a drug from the biguanide family that is used for decades as the first-line therapeutic choice for the treatment of type 2 diabetes. Despite its worldwide democratization, owing to its clinical efficacy, high safety profile and cheap cost, the exact mechanism(s) of action of this anti-hyperglycemic molecule with pleiotropic properties still remains to be fully elucidated. The concept that metformin would exert some of its actions though modulation of the mitochondrial bioenergetics was initially forged in the 50s but undeniably revived at the beginning of the twenty-first century when it was shown to induce a weak but specific inhibition of the mitochondrial respiratory-chain complex 1. Furthermore, metformin has been reported to reduce generation of reactive oxygen species at the complex 1 and to prevent mitochondrial-mediated apoptosis, suggesting that it can protect against oxidative stress-induced cell death. Nevertheless, despite some recent progress and the demonstration of its key role in the inhibition of hepatic gluconeogenesis, the exact nature of the mitochondrial interaction between the drug and the complex 1 is still poorly characterized. Recent studies reported that metformin may also have anti-neoplastic properties by inhibiting cancer cell growth and proliferation, at least partly through its mitochondrial action. As such, many trials are currently conducted for exploring the repositioning of metformin as a potential drug for cancer therapy. In this mini-review, we discuss both historical and more recent findings on the central role played by the interaction between metformin and the mitochondria in its cellular mechanism of action.
Historically, the origins of metformin (dimethylbiguanide) came from the Middle Age where medieval doctors used extract from the French Lilac Galega officinalis to treat various diseases (1). At the beginning of the twentieth century, the plant was found to be rich in guanidine, an active ingredient that was later reported to have potent anti-hyperglycemic properties. Guanidine derivatives gave rise to the biguanide family, among which metformin is currently the only therapeutic survivor for the treatment of type 2 diabetes. Indeed, after withdrawal of buformin and phenformin at the end of the 70′s, metformin hydrochloride gradually became the most widely prescribed oral antidiabetic agent, due to its efficient glucose-lowering effect, weight-neutral characteristic, high safety profile associated with low risk of hypoglycemia, and cost-effectiveness as a generic drug (2). Since then, metformin is well recognized for its ability to lower hyperglycemia by decreasing hepatic glucose production while reducing glucotoxicity in different tissues, a feature that might explain some of its cardioprotective benefits (2, 3). However, despite its worldwide democratization, the exact mechanism(s) of action of this molecule with apparent pleiotropic properties still remains to be fully elucidated. As many drugs, the cellular effects of metformin rely on its unique physicochemical characteristics, which include a high hydrophilicity, some metal-binding properties and a pKa within the physiological pH range, implying that the molecule exists solely in its positively charged cationic form (4). Due to its poor lipophilicity, metformin does not cross cell membranes by simple passive diffusion and its bio-distribution relies on tissue-specific transporters, including plasma membrane monoamine transporter (PMAT) in the intestine, organic cation transporter 1 (OCT1) in the liver, and both organic cation transporter 2 (OCT2) and multidrug and toxin extruder (MATE)1/2 in the kidneys (4, 5). By contrast, phenformin exhibits a higher lipophilicity than metformin, owing to its larger phenylethyl side chain, and is therefore crossing more easily lipid membrane bilayer, a property that might explain their differences in terms of selectivity and potency. Various underlying mechanisms have been suggested for metformin throughout the six decades following its first commercialization but a consensus only started to emerge during the last years, placing mitochondria at the heart of metformin's cellular actions.
At the beginning of 2000, the group of Xavier Leverve was the first to report that metformin selectively inhibits the mitochondrial respiratory-chain complex 1 and, as a result, decreases NADH oxidation, reduces proton gradient across the inner mitochondrial membrane, and decreases oxygen consumption rate (6) (Figure 1). This major breakthrough was rapidly complemented by a supportive study from Halestrap's group published couple of months later (7). Although the inhibitory effect of metformin on complex 1 was first evidenced in rat hepatocytes in these two seminal studies, it was thereafter confirmed in various species and plenty of biological models, including lately in cancer cells (Table 1). Importantly, metformin only exerts a weak and reversible selective inhibition of complex 1 (IC50 ~20 mM), making it a peculiar type of inhibitor that does not resemble the canonical ones like rotenone and piericidin A (IC50 ~2 μM), which are both uncharged and highly hydrophobic molecules (24). It is worth mentioning that, although the discovery of complex 1 inhibition by metformin undoubtedly constituted a major advance in the understanding of its cellular mode of action, some inhibitory effects of biguanides on mitochondrial oxidative phosphorylation (OXPHOS) were already reported by Gunnar Hollunger, a Swedish scientist at the University of Lund, as early as 1955 (25), and by the German biochemist Günter Schäfer in the 60's (26).
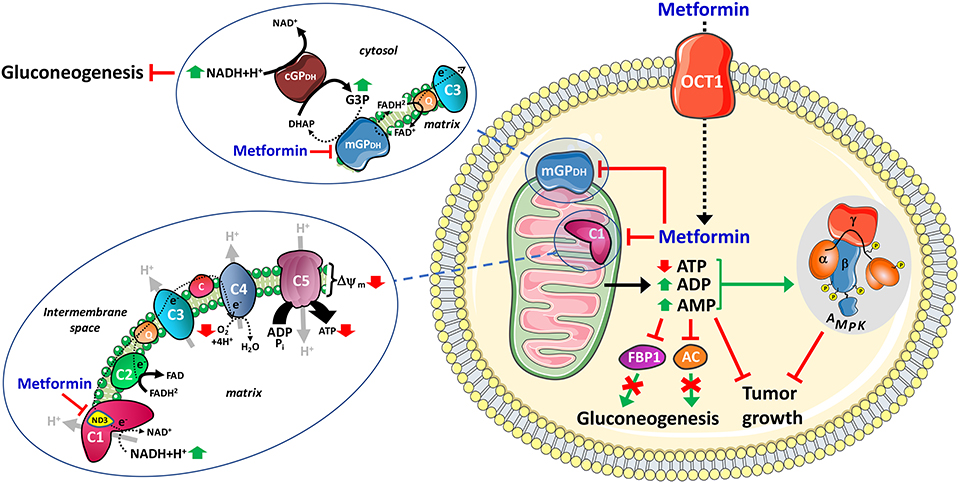
Figure 1. Mitochondrial mechanisms of action of metformin. After cellular uptake, mainly through OCT1 in hepatocytes, the mitochondria is the primary target of metformin which exerts specific inhibition on the respiratory-chain complex 1, presumably through direct interaction with the ND3 core subunit, and on mitochondrial glycerophosphate dehydrogenase (mGPDH). The inhibition of complex 1 decreases NADH oxidation, proton pumping across the inner mitochondrial membrane and oxygen consumption rate, resulting in lower proton gradient (Δψ) and reduction of proton-driven ATP synthesis from ADP and inorganic phosphate (Pi). The inhibition of mGPDH modulates cytosolic and mitochondrial redox state resulting in increased cytosolic NADH. FBP1, fructose-1,6-bisphosphatase-1. AC, adenylate cyclase.
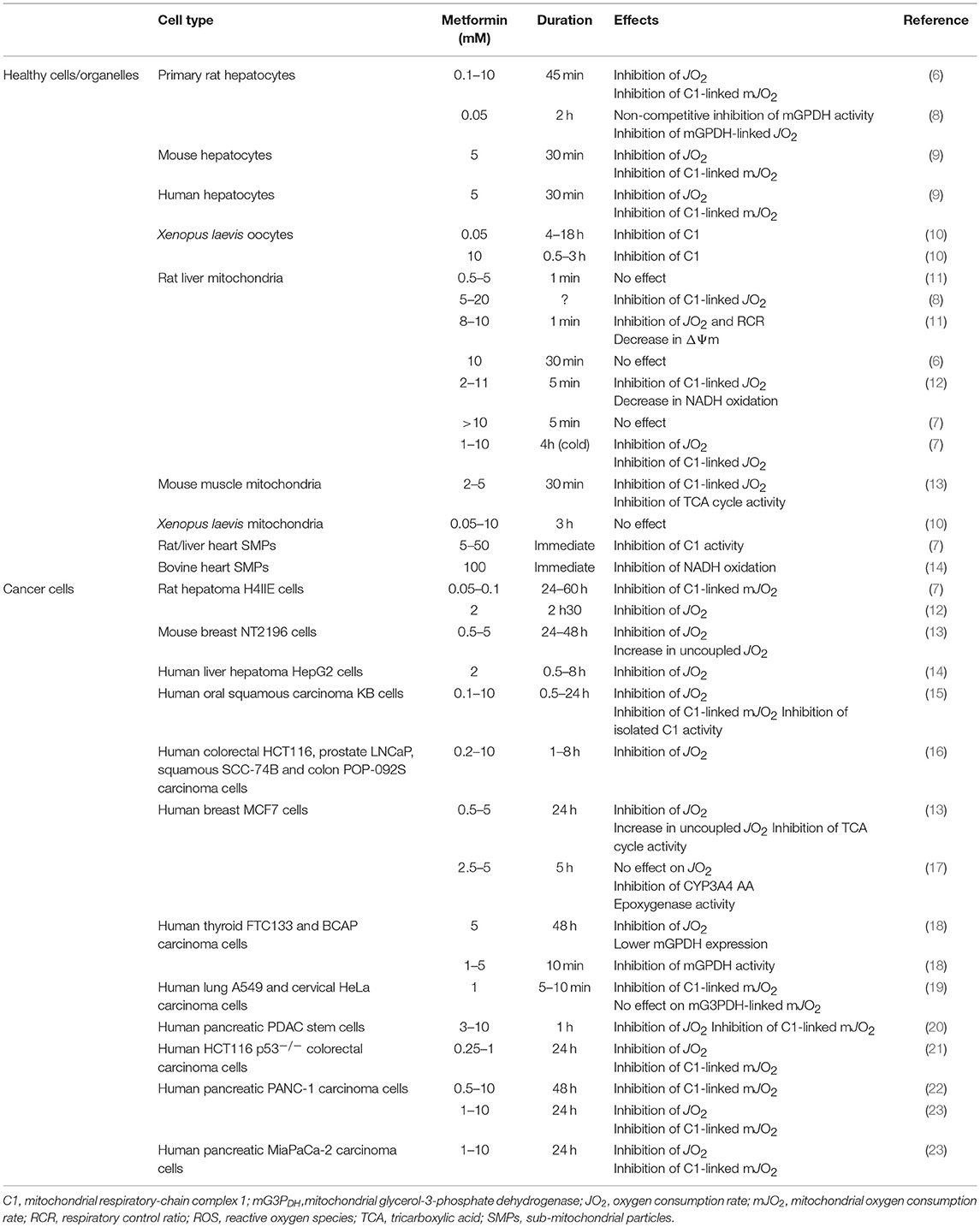
Table 1. Ex vivo and in vitro mitochondrial effects of metformin.
How exactly metformin gets into the mitochondria and whether it inhibits complex 1 directly or not remains unclear and is still a matter of debate (27). Very high concentrations of metformin (20–100 mM) were reported to directly inhibit complex 1 activity in isolated mitochondria or in inside-out structured sub-mitochondrial particles (SMPs) whereas clinically relevant drug concentrations (<100 μM) did not (Table 1). By contrast, micromolar concentrations of the drug are sufficient to achieve a dose- and time-dependent in situ inhibition of mitochondrial complex 1 in various cell types (6, 10, 15, 28, 29) or in vivo in skeletal muscle from healthy and diabetic rats (30). Among the possible explanations, the positive charge of metformin was proposed to account for its slow accumulation within the matrix of energized mitochondria of intact cells, driven by their transmembrane electrochemical potential (ΔΨ) (7, 14). Indeed, according to thermodynamic laws and the Nernst equation, a ~1,000-fold accumulation of a positively charged molecule might theoretically occur in energized mitochondria with a physiologically relevant ΔΨ, suggesting that metformin could reach millimolar concentration in the organelle despite a cytoplasmic level within the micromolar range (27). Furthermore, a ΔΨ-driven mitochondrial import of the biguanide might also provide an explanation for its weak inhibitory effect on complex 1, the reduction in mitochondrial membrane potential induced by the drug limiting its subsequent buildup. However, no accumulation of radioactively-labeled metformin was observed in mitochondria isolated from Xenopus laevis oocytes and exposed to concentrations of the drug that inhibit complex 1 (10). Therefore, even if a direct effect of metformin on complex 1 turns out possible, it seems to be highly facilitated in intact cells regardless of the exact mechanism involved in this process. Although the low accumulation of metformin into mitochondria could primarily be explained by the slow permeation of the drug across the plasma membrane, some studies have also suggested the existence of a specific transport system mediating its mitochondrial import. As such, the observation that the inhibitory effect of metformin on complex 1 was temperature-dependent in intact Xenopus laevis oocytes and that low concentration (50 μM) was able to directly inhibit complex 1 activity in isolated mitochondria when delivered as a liposomal-encapsulated form that can eventually fuse with the organelle led to the hypothesis of an endocytic vesicular routing of the drug from the plasma membrane to the mitochondria (10). However, the molecular components involved in this putative process still remain obscure and would deserve extensive investigation, including in mammalian cells. On the other hand, it has also been reported that intra-mitochondrial accumulation of phenformin, another biguanide, could at least partly be mediated by the mitochondrial organic cation/carnitine transporter 1 (OCTN1) (31). More recently, a protein-mediated mitochondrial import of the biguanide was also suggested based on the fact that direct conjugation of a phenyl group and bis-substitution of the biguanide moiety on the molecule prevent its uptake into mitochondria, irrespective of the compound hydrophobicity (32). However, whether this could also occur for metformin was not assessed and remains therefore to be investigated.
The mammalian respiratory-chain complex 1 (NADH:ubiquinone oxidoreductase) is a large L-shaped membrane-bound redox enzyme composed of at least 45 different subunits that couples the transfer of electrons from NADH to the ubiquinone pool with a transfer of protons from the mitochondrial matrix toward the intermembrane space (33). The complex 1 exists in two distinct forms: a fully competent active one and a so-called “deactive” D-form where the enzyme is catalytically incompetent but can be activated by a slow reaction of NADH oxidation coupled to ubiquinone reduction (34). As metformin inhibits NADH oxidation by complex 1 in isolated mitochondria from bovine heart, yeast Pichia pastoris, bacterium Escherichia coli (14, 32), as well as from C. elegans (35), it is likely that the molecule binds to some of the phylogenetically conserved “core” subunits of the complex rather than to mammalian-specific accessory ones (33). While it has been shown that metformin did not alter the structural integrity of the whole complex (14), the exact molecular interactions between the drug and the complex 1 remain to be elucidated. In order to investigate how metformin, and other biguanides, could interact with complex 1 for regulating its activity, the group of Judy Hirst has elegantly dissected the effects of the drug at different levels of the catalytic cycle of the enzyme. They demonstrated that metformin is a reversible non-competitive inhibitor that probably binds to some amphipathic regions of the enzyme, i.e., where some hydrophilic and hydrophobic amino acids are in close proximity, and inhibits a rate-limiting step coupled to ubiquinone reduction, but does not competitively bind to the ubiquinone-binding site on complex 1 (14). Moreover, metformin rather stimulates the NADH:FeCN oxidoreduction reaction and does not alter the thermal stability of the flavin site, except at extremely high non-relevant concentration (200 mM), indicating that NADH oxidation occurring at the flavin site is probably not involved in the inhibition of complex 1 by the drug. Similarly, metformin does not modulate the FeS cluster of the NADH-reduced complex 1, suggesting that the intramolecular electron transfer is not impaired (14). However, using SMPs, the authors showed that inhibition of NADH oxidation by metformin is immediate when the drug is added prior to the initiation of catalysis but is delayed once catalysis has already started (14). Altogether, this strongly suggests that the inhibition depends on the catalytic status of complex 1, occurring primarily when the enzyme is in its “deactive” conformation with redox and proton transfer domains no longer efficiently coupled (14). Ultimately, the authors proposed that the Cys39-containing matrix loop of subunit ND3 located within the amphipathic region between the redox and proton-transfer domains might be the binding site for metformin on complex 1, stabilizing the enzyme in an open-loop deactive conformation state (14). It is worth mentioning that most of the above-mentioned mechanistic studies were performed using isolated organelles and high concentrations of the drug and that such experimental in vitro conditions might not always reflect the physiological in situ environment. For instance, complex 1 forms respiratory-chain supercomplexes together with complexes 3 and 4 (36), a supramolecular organization that is lost in SMPs and may affect the interaction of metformin with complex 1 and/or the regulatory effect of the drug on mitochondrial OXPHOS.
Besides their central role in cellular energy homeostasis, mitochondria are also the main source of reactive oxygen species (ROS) which, on top of potentially causing oxidative damages, could also play a key role as signaling molecules in various pathways (37). Superoxide anions are primarily generated by the mitochondria, mostly at complexes 1 and 3 of the electron transfer chain (ETC) where electrons are leaking and could react with oxygen. It is now well recognized that complex 1 can produce superoxide by both forward (site IF) and reverse electron (site IQ) fluxes, depending on substrates used to fuel the ETC (38). As such, rotenone can either increase or decrease mitochondrial ROS production at complex 1, depending on whether glutamate-malate (forward) or succinate (reverse) are provided as respiratory-chain substrates, respectively. By contrast, it has been shown that metformin specifically decreases the ROS production driven by the reverse electron transfer (RET) but without increasing ROS generation through the forward direction (39). Interestingly, a similar lowering effect on ROS production at the complex 1 than the one observed with metformin was also recently reported for imeglimin, a molecule belonging to the tetrahydrotriazine-containing novel class of oral glucose-lowering agents (40), suggesting that inhibition of this RET-mediated ROS production may play a role in the mechanisms of action of the two antidiabetic drugs, notably by conferring protection against oxidative stress-related cell death (15, 28, 29). In line with this, a new generation of oxidative stress inhibitors that specifically neutralize ROS produced via RET at the IQ site within complex 1 has been shown to lower oxidative damage, inhibit cellular stress signaling and protect against ischemia-reperfusion heart injury (41). Furthermore, it has also been suggested that some of the anti-inflammatory effect of metformin observed in lipopolysaccharide-stimulated bone-marrow derived macrophages could result from the specific inhibition of RET-derived ROS production at the complex 1 (42). Taken together, these findings suggest that targeting RET-linked ROS occurring at the mitochondrial respiratory-chain complex 1 using metformin or metformin-like molecules might be therapeutically relevant in the context of both cardiometabolic and inflammatory diseases.
Metformin exerts its anti-hyperglycemic action primarily through reduction of hepatic glucose production (3). A major breakthrough occurred in 2001 when Zhou and colleagues reported that metformin increased the AMP-activated protein kinase (AMPK) activity in hepatocytes, a feature associated with inhibition of gluconeogenesis (43). AMPK is a protein kinase that functions as energy gauge which constantly senses the cellular energy status by monitoring AMP, ADP, and ATP levels (44, 45). Once activated in response to decrease in ATP and concomitant rise in intracellular ADP and AMP levels, AMPK inhibits ATP-consuming anabolic processes and promotes ATP-generating catabolic pathways by direct phosphorylation of a broad range of downstream effectors that are involved in the regulation of various metabolic processes, ultimately leading to restoration of cellular energy balance (44). It took a decade of controversy before the general acceptance that AMPK activation by metformin results from increased ADP:ATP and AMP:ATP ratios secondary to inhibition of the mitochondrial respiratory-chain complex 1 (3). Of note, only biguanides with physicochemical characteristics allowing them to enter the mitochondria and to inhibit complex 1 were shown to activate AMPK (32). However, using liver-specific AMPK knockout mice, Foretz and colleagues demonstrated that metformin lowers gluconeogenesis by an AMPK-independent mechanism involving a decrease in cellular energy state, a strong correlation being observed between the increase in cellular [AMP]:[ATP] and the inhibition of gluconeogenesis (46). Altogether, although metformin can activate AMPK, it is therefore neither necessary nor sufficient for inducing acute inhibition of gluconeogenesis (Figure 1). In line with this, two other studies also demonstrated that metformin can inhibit hepatic glucose production through AMPK-independent mechanisms: one by AMP-mediated inhibition of adenylate cyclase and subsequent reduction in glucagon-increased cyclic adenosine monophosphate (cAMP) levels (47); the other one through modulation of cytosolic redox state via direct inhibition of the mitochondrial glycerol-3-phosphate dehydrogenase (mGPDH) (8) (Figure 1). mGPDH is a flavin-linked respiratory-chain dehydrogenase belonging to the glycerol phosphate shuttle that couples the oxidation of glycerol-3-phosphate to dihydroxyacetone with reduction of FAD to FADH2 and the transfer of electrons to coenzyme Q of the ETC, contributing as such to the maintenance of the redox potential across the inner mitochondrial membrane (48). Remarkably, Shulman's group reported that metformin exerts an in vitro non-competitive inhibition of the enzyme, with a Ki value (~40 μM) within the clinical range of drug concentrations, leading to increased hepatic cytosolic NADH/NAD+ ratio and impaired gluconeogenesis from redox-dependent substrates, such as lactate and glycerol, in rats (8). In a recent follow-up study, they showed that both acute and chronic treatment with metformin also inhibit hepatic gluconeogenesis in a redox-dependent manner in diabetic rats, without apparent changes in mitochondrial citrate synthase flux and hepatic nucleotide concentrations (49). By contrast, Sakamoto's group recently provided new supportive elements strengthening the key role of mitochondria-mediated modulation of cellular energy homeostasis in the inhibition of hepatic gluconeogenesis by metformin. Indeed, in an elegant study using knock-in mice, the authors demonstrated that a point mutation in the gluconeogenic enzyme fructose-1,6-bisphosphatase-1 (F1BP) which impairs its allosteric inhibition by AMP reduced the anti-hyperglycemic effect of metformin in diabetic mice (50). Altogether, this strongly suggests that the transient rise in intracellular AMP levels resulting from the weak and reversible inhibition of the respiratory-chain complex 1 by metformin is crucial for inhibiting hepatic gluconeogenesis, either by modulating adenylate cyclase or FBP1 activity (Figure 1).
A growing body of epidemiological and clinical studies reported that metformin reduces cancer risk in patients with type 2 diabetes and improves survival outcome of cancer patients with breast, ovarian, liver and colorectal tumors (51). Although an extensive overview on this topic can be found elsewhere [for recent reviews see (51–54)], it is striking that the mitochondrial effect of metformin could again play a crucial role in the anti-tumorigenic effect of the drug. Indeed, the inhibition of complex 1 was observed in many cancer cells (Table 1) and usually leads to reduced mitochondrial OXPHOS and ATP depletion, ultimately resulting in AMPK-mediated activation of catabolic pathways and inhibition of anabolic processes through its regulation of mechanistic target of rapamycin complex 1 (mTORC1) (54). While some AMPK- and mTORC1-independent mechanisms can also co-exist (55), this metabolic reprogramming lowers growth and proliferation of cancer cells, at least partly due to inhibition of protein and lipid synthesis. It also promotes cell cycle arrest and apoptosis in cells that cannot cope with the energetic stress (54). Wheaton and colleagues clearly showed that the reduction of tumor growth by metformin was prevented in cancer cells expressing NDI1, a metformin-resistant yeast analog of complex 1, highlighting the central role played by inhibition of this mitochondrial target in the antineoplastic effect of the drug (21). This is also consistent with another study showing that phenformin exerts its anti-tumorigenic effects by inhibiting complex 1 (56). Nevertheless, most of the effects of metformin were generally observed at supratherapeutic concentrations and the drug bioavailability in cancer cells is still questionable. Interestingly, more lipophilic derivatives of metformin targeting the mitochondria are currently under investigation with the aim of developing analogs with higher bioavailability and antitumor activity than metformin. Remarkably, some of these newly synthetized molecules were recently reported to be nearly 1,000-fold more potent than metformin in inhibiting mitochondrial complex 1 activity and to exert both anti-proliferative and radiosensitizing effects in pancreatic cancer cells (23). Altogether, developing such kind of mitochondria-targeted metformin-like drugs could pave the way for promising new therapeutic strategies that might also be relevant for various other pathologies than cancer (57).
Although the interest around metformin has been significantly revived during the last years, principally due to the potential repositioning of this antidiabetic drug for the treatment of cancer, it still remains crucial to better decipher the mechanism by which it inhibits the mitochondrial respiratory-chain complex 1, notably the exact nature of their interaction. Elucidating this aspect may advance our understanding of how metformin regulates cellular energetics and be decisive for optimizing future drug development and therapeutic interventions, notably for cancer patients.
GV performed literature search and drafted the manuscript. DD performed literature search and drafted the manuscript. BG performed literature search, designed the figure, wrote, and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BGs' group was/is supported by funding from the Société Francophone du Diabète (SFD), the European Federation for the Study of Diabetes (EFSD), the DiabetesFonds, and the Dutch Organization for Scientific Research (ZonMW).
1. Bailey CJ. Metformin: historical overview. Diabetologia. (2017) 60:1566–76. doi: 10.1007/s00125-017-4318-z
2. Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. (2017) 60:1586–93. doi: 10.1007/s00125-017-4336-x
3. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. (2014) 20:953–66. doi: 10.1016/j.cmet.2014.09.018
4. Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. (2011) 50:81–98. doi: 10.2165/11534750-000000000-00000
5. Gormsen LC, Sundelin EI, Jensen JB, Vendelbo MH, Jakobsen S, Munk OL, et al. In vivo imaging of human 11C-metformin in peripheral organs: dosimetry, biodistribution, and kinetic analyses. J Nucl Med. (2016) 57:1920–6. doi: 10.2967/jnumed.116.177774
6. El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
7. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. (2000) 348(Pt 3):607–14. doi: 10.1042/bj3480607
8. Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. (2014) 510:542–6. doi: 10.1038/nature13270
9. Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. (2011) 54:3101–110. doi: 10.1007/s00125-011-2311-5
10. Detaille D, Guigas B, Leverve X, Wiernsperger N, Devos P. Obligatory role of membrane events in the regulatory effect of metformin on the respiratory chain function. Biochem Pharmacol. (2002) 63:1259–72. doi: 10.1016/S0006-2952(02)00858-4
11. Carvalho C, Correia S, Santos MS, Seica R, Oliveira CR, Moreira PI. Metformin promotes isolated rat liver mitochondria impairment. Mol Cell Biochem. (2008) 308:75–83. doi: 10.1007/s11010-007-9614-3
12. Cameron AR, Logie L, Patel K, Erhardt S, Bacon S, Middleton P, et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. (2018) 14:187–97. doi: 10.1016/j.redox.2017.08.018
13. Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. (2014) 2:12. doi: 10.1186/2049–3002-2-12
14. Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. (2014) 462:475–87. doi: 10.1042/BJ20140620
15. Guigas B, Detaille D, Chauvin C, Batandier C, De Oliveira F, Fontaine E, et al. Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem J. (2004) 382:877–84. doi: 10.1042/BJ20040885
16. Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res. (2013) 19:6741–50. doi: 10.1158/1078-0432.CCR-13-1787
17. Guo Z, Sevrioukova IF, Denisov IG, Zhang X, Chiu TL, Thomas DG, et al. Heme binding biguanides target cytochrome P450-dependent cancer cell mitochondria. Cell Chem Biol. (2017) 24:1259–75 e1256. doi: 10.1016/j.chembiol.2017.08.009
18. Thakur S, Daley B, Gaskins K, Vasko VV, Boufraqech M, Patel D, et al. Metformin targets mitochondrial glycerophosphate dehydrogenase (mGPDH) to control rate of oxidative phosphorylation and growth of thyroid cancer in vitro and in vivo. Clin Cancer Res. (2018) 24:4030–43. doi: 10.1158/1078-0432.CCR-17-3167
19. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. (2016) 24:716–27. doi: 10.1016/j.cmet.2016.09.006
20. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. (2015) 22:590–605. doi: 10.1016/j.cmet.2015.08.015
21. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. (2014) 3:e02242. doi: 10.7554/eLife.02242
22. Boukalova S, Stursa J, Werner L, Ezrova Z, Cerny J, Bezawork-Geleta A, et al. Mitochondrial targeting of metformin enhances its activity against pancreatic cancer. Mol Cancer Ther. (2016) 15:2875–86. doi: 10.1158/1535-7163.MCT-15-1021
23. Cheng G, Zielonka J, Ouari O, Lopez M, McAllister D, Boyle K, et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. (2016) 76:3904–15. doi: 10.1158/0008-5472.CAN-15-2534
24. Degli Esposti M. Inhibitors of NADH-ubiquinone reductase: an overview. Biochim Biophys Acta. (1998) 1364:222–35. doi: 10.1016/S0005-2728(98)00029-2
25. Hollunger G. Guanidines and oxidative phosphorylations. Acta Pharmacol Toxicol (Copenh). (1955) 11:1–84. doi: 10.1111/j.1600-0773.1955.tb02972.x
26. Schafer G. Site-specific uncoupling and inhibition of oxidative phosphorylation by biguanides. II. Biochim Biophys Acta. (1969) 172:334–7. doi: 10.1016/0005-2728(69)90077-2
27. Fontaine E. Metformin and respiratory chain complex I: the last piece of the puzzle? Biochem J. (2014) 463:e3–5. doi: 10.1042/BJ20141020
28. Detaille D, Guigas B, Chauvin C, Batandier C, Fontaine E, Wiernsperger N, et al. Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes. (2005) 54:2179–87. doi: 10.2337/diabetes.54.7.2179
29. El-Mir MY, Detaille D, R-Villanueva G, Delgado-Esteban M, Guigas B, Attia S, et al. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J Mol Neurosci. (2008) 34:77–87. doi: 10.1007/s12031-007-9002-1
30. Wessels B, Ciapaite J, van den Broek NM, Nicolay K, Prompers JJ. Metformin impairs mitochondrial function in skeletal muscle of both lean and diabetic rats in a dose-dependent manner. PLoS ONE. (2014) 9:e100525. doi: 10.1371/journal.pone.0100525
31. Shitara Y, Nakamichi N, Norioka M, Shima H, Kato Y, Horie T. Role of organic cation/carnitine transporter 1 in uptake of phenformin and inhibitory effect on complex I respiration in mitochondria. Toxicol Sci. (2013) 132:32–42. doi: 10.1093/toxsci/kfs330
32. Bridges HR, Sirvio VA, Agip AN, Hirst J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC Biol. (2016) 14:65. doi: 10.1186/s12915-016-0287-9
33. Hirst J. Mitochondrial complex I. Annu Rev Biochem. (2013) 82:551–75. doi: 10.1146/annurev-biochem-070511-103700
34. Vinogradov AD. Catalytic properties of the mitochondrial NADH-ubiquinone oxidoreductase (complex I) and the pseudo-reversible active/inactive enzyme transition. Biochim Biophys Acta. (1998) 1364:169–85. doi: 10.1016/S0005-2728(98)00026-7
35. Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, et al. An ancient, unified mechanism for metformin growth inhibition in C. elegans and Cancer. Cell. (2016) 167:1705–18 e1713. doi: 10.1016/j.cell.2016.11.055
36. Letts JA, Sazanov LA. Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat Struct Mol Biol. (2017) 24:800–8. doi: 10.1038/nsmb.3460
37. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. (2012) 48:158–67. doi: 10.1016/j.molcel.2012.09.025
38. Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem. (2017) 292:16804–9. doi: 10.1074/jbc.R117.789271
39. Batandier C, Guigas B, Detaille D, El-Mir MY, Fontaine E, Rigoulet M, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr. (2006) 38:33–42. doi: 10.1007/s10863-006-9003-8
40. Detaille D, Vial G, Borel AL, Cottet-Rouselle C, Hallakou-Bozec S, Bolze S, et al. Imeglimin prevents human endothelial cell death by inhibiting mitochondrial permeability transition without inhibiting mitochondrial respiration. Cell Death Discov. (2016) 2:15072. doi: 10.1038/cddiscovery.2015.72
41. Brand MD, Goncalves RL, Orr AL, Vargas L, Gerencser AA, Borch Jensen M, et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. (2016) 24:582–92. doi: 10.1016/j.cmet.2016.08.012
42. Kelly B, Tannahill GM, Murphy MP, O'Neill LA. Metformin inhibits the production of reactive oxygen species from NADH: ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem. (2015) 290:20348–59. doi: 10.1074/jbc.M115.662114
43. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI200113505
44. Hardie DG. AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu Rev Nutr. (2014) 34:31–55. doi: 10.1146/annurev-nutr-071812-161148
45. Guigas B, Viollet B. Targeting AMPK: from ancient drugs to new small-molecule activators. Exp Suppl. (2016) 107:327–350. doi: 10.1007/978-3-319-43589-3_13
46. Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. (2010) 120:2355–69. doi: 10.1172/JCI40671
47. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. (2013) 494:256–60. doi: 10.1038/nature11808
48. Mracek T, Drahota Z, Houstek J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim Biophys Acta. (2013) 1827:401–10. doi: 10.1016/j.bbabio.2012.11.014
49. Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang XM, Zhang D, et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat Med. (2018) 24:1384–94. doi: 10.1038/s41591-018-0125-4
50. Hunter RW, Hughey CC, Lantier L, Sundelin EI, Peggie M, Zeqiraj E, et al. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat Med. (2018) 24:1395–406. doi: 10.1038/s41591-018-0159-7
51. Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. (2015) 66:17–29. doi: 10.1146/annurev-med-062613-093128
52. Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe VV, Ford LG. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia. (2017) 60:1639–47. doi: 10.1007/s00125-017-4372-6
53. Mallik R, Chowdhury TA. Metformin in cancer. Diabetes Res Clin Pract. (2018) 143:409–19. doi: 10.1016/j.diabres.2018.05.023
54. Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I. Metformin as an anticancer agent. Trends Pharmacol Sci. (2018) 39:867–78. doi: 10.1016/j.tips.2018.07.006
55. Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. (2010) 11:390–401. doi: 10.1016/j.cmet.2010.03.014
56. Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. (2014) 508:108–12. doi: 10.1038/nature13110
Keywords: biguanides, respiratory-chain complex 1, bioenergetics, AMPK, cancer
Citation: Vial G, Detaille D and Guigas B (2019) Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 10:294. doi: 10.3389/fendo.2019.00294
Received: 12 December 2018; Accepted: 24 April 2019;
Published: 07 May 2019.
Edited by:
Frederic Bost, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Bilikere S. Dwarakanath, Shanghai Proton and Heavy Ion Center (SPHIC), ChinaCopyright © 2019 Vial, Detaille and Guigas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno Guigas, Yi5nLmEuZ3VpZ2FzQGx1bWMubmw=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.