
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 12 March 2019
Sec. Experimental Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00152
 Yi Li1
Yi Li1 Meng-Chen Zhang1
Meng-Chen Zhang1 Xiao-Kang Xu1
Xiao-Kang Xu1 Yang Zhao2
Yang Zhao2 Chatoo Mahanand1
Chatoo Mahanand1 Tao Zhu3Hong Deng4
Tao Zhu3Hong Deng4 Eviatar Nevo5Ji-Zeng Du1,6,7
Eviatar Nevo5Ji-Zeng Du1,6,7 Xue-Qun Chen1,6,7*
Xue-Qun Chen1,6,7*The common understanding of p53 function is a genome guardian, which is activated by diverse stresses stimuli and mediates DNA repair, apoptosis, and cell cycle arrest. Increasing evidence has demonstrated p53 new cellular functions involved in abundant endocrine and metabolic response for maintaining homeostasis. However, TP53 is frequently mutant in human cancers, and the mutant p53 (Mut-p53) turns to an “evil” cancer-assistant. Mut-p53-induced epithelial-mesenchymal transition (EMT) plays a crucial role in the invasion and metastasis of endocrine carcinomas, and Mut-p53 is involved in cancer immune evasion by upregulating PD-L1 expression. Therefore, Mut-p53 is a valuable treatment target for malignant tumors. Targeting Mut-p53 in correcting sequence and conformation are increasingly concerned. Interestingly, in wild animals, p53 variations contribute to cancer resistant and high longevity. This review has discussed the multiple functions of p53 in health, diseases, and nature evolution, summarized the frequently mutant sites of p53, and the mechanisms of Mut-p53-mediated metastasis and immune evasion in endocrine cancers. We have provided a new insight for multiple roles of p53 in human and wild animals.
Since 1979, 40 years of research on p53 have shown it regulates complex and adaptable target gene networks to control broad biological processes (1–3). p53 was first discovered as a 53 kDa protein in SV40-transformed cells (1, 2). p53 protein has five discrete domains with different functions: two N-terminal transactivation domain (TAD) involved in the recruitment of transcriptional co-factors, a central sequence-specific DNA-binding domain (DBD) directly interacting with DNA, a C-terminal oligomerisation domain (OD), and a tetramerization domain (TET) that mediated p53 working in a tetramer manner (4). p53 directly binds to p53 response element (p53REs) that consist of two copies of a 10 base pair motif with the consensus 5′-RRRCWWGYYY-3′ (R = A/G, W = A/T, and Y = C/T) (5, 6). Because of the high diversity of p53REs, p53 controls abundant sets of target genes (3). Initially, p53 is defined as a tumor suppressor, induces cell cycle arrest, apoptosis, and senescence programs to maintain genome integrity in response to genotoxic and oncogenic stresses via regulating target gene expression, such as Cdkn1a, Puma, and Noxa (3, 7, 8). Recently, novel functions of p53 have been found in regulating metabolism (9–11), stemness (12), antioxidant (13), autophagy (14, 15), ferroptosis (16), differentiation (17), and embryo implantation (18)/fitness selection (19) processes. p53 is frequently mutant in human cancers, which induces aggressive tumors proliferation, invasion, metastasis and immune evasion (20–23). The TP53 mutation is listed as a potential candidate in disease risk prediction, personalized, and prognostic treatment (24, 25). Wild animals harbor p53 variations (same as mutants in human cancers) that contribute to anticancer and environmental adaptation, which may provide insights into understanding targeting p53 (26–28).
p53 is a crucial regulation hub in many physiological processes including metabolism (10), autophagy (14), apoptosis (29), and cell arrest (5) that maintain normal cellular homeostasis (30). Its target genes also play important roles in these processes, for instance, Glut1/4 (glucose transporter type 1 and 4), Pltp (phospholipid transfer protein), and Dhrs3 (dehydrogenase/reductase 3) are involved in the formation and transfer of lipid in mice liver (10); Lpin1 facilitates fatty acids oxidation (FAO) in low glucose environment but suppresses in normal conditions (31); Dram (damage-regulated autophagy modulator 1) and Aen (apoptosis enhancing nuclease) promote autophagy, while Tiger (TP53-induced glycolysis and apoptosis regulator) inhibits autophagy in colon carcinoma cell line (14). In individual cells, the levels of p53 protein exhibit dynamic accumulation with distinct pulses after DNA damage, both p53 pulses and target genes mRNA half-life lead to diversified expression pulses of genes (32, 33). A mathematical model has been proposed to predict the expression patterns of target genes under different p53 inputs, which provides a better understanding of p53 and its target genes interaction (34). These p53 dynamics can control cell fates. Fast-accumulating p53 can reach apoptosis threshold level and triggers cell apoptosis, whereas cells with slow-accumulating p53 are unable to execute apoptosis because they cannot exceed the threshold that increases with time by the activation of anti-apoptotic genes (32, 35), this suggests that the dynamics of p53 and target genes may impact cellular homeostasis.
Furthermore, p53 activates mTOR (downstream mammalian target of rapamycin) and AMPK (AMP-activated protein kinase) signal pathways, indirectly modulates lipid anabolism/catabolism (10, 11), carbohydrate metabolism (11), and autophagy (15). But, AMPK triggers p53-S15 phosphorylation as an upstream activator in response to glucose deprivation in primary mouse embryonic fibroblasts (MEFs) (11). p53 is an obesity regulator, lack of p53 increases lipid accumulation by restraining aromatase expression, which leads to high testosterone levels and obesity in male mice (30). In agouti-related peptide (AgRP) neurons of mice hypothalamus, knockout TP53 promotes food-induced adiposity and decreases the thermogenesis in brown adipose tissue but overexpressed p53 results in more body weight loss than TP53 KO mice, and c-Jun N-terminal kinase (JNK) is indispensable in both processes (36).
Endocrine and metabolic disorders are common but complex, which has aroused much concern. There is evidence that wild-type p53 (Wt-p53) involves diabetes, liver steatosis and endocrine tumors (30, 37, 38). Wt-p53 impairs insulin secretion signals in pancreatic β cell via inhibiting autophagic clearance in damaged mitochondria of diabetes (38). In type 2 diabetes, Wt-p53 upregulation leads to β cell failure in hyperglycemia and congenital hyperinsulinism, and promotes insulin resistance in adipose tissue (37, 39). Furthermore, Wt-p53 is associated with male-related tumors, the C-terminal lysine methylation of Wt-p53 repressed its transcriptional activity upon DNA damage and prevented cycle arrest in testicular germ cell tumor (40). Androgen restricts Wt-p53 function and causes p53 trans-localization by activating p53 SUMOylation in prostate cancer (41).
Genomic data from more than 20,000 patients have confirmed that the TP53 is the most commonly mutated gene in all human cancers (42–44), such as hepatocellular carcinoma (45), colorectal cancer (46), lymphoma (47), and mucosal melanoma (48). Data in this review has collected seven endocrine cancers data from The Cancer Genome Atlas (TCGA) in cBioPortal (http://www.cbioportal.org/) (49, 50), and analyzed amino acid (AA) sites of Mut-p53 in these endocrine carcinomas (Figure 1). The frequency of p53 mutations in endocrine cancers is varying, it is ~55% in pancreatic adenocarcinoma and ovarian serous cystadenocarcinoma, only 0.4% in thyroid carcinoma and 3.2% in thymoma (Figure 1A). R175, R213, Y220, R248, R273, and R282 of p53 DBD (residue 98-289) are frequently mutant AA sites in these endocrine cancers, and it is striking that R273 site is mutant in most of the endocrine cancers (Figure 1A). Atlas of location for collected mutated sites from endocrine cancers is displayed in 3D-p53 structure (Figures 1B,C). TP53 mutations show a preference for missense mutations rather than frameshift or non-sense that are frequently occurred in other tumor suppressor genes (42). From more than 80,000 human cancer cases in the Universal Mutation Database (UMD) (http://p53.fr/the-database), over 70% TP53 are missense mutations (51, 52). p53 with point mutations commonly produce a full-length protein with one single AA substitution (53). The spectrum of p53 missense mutations contains over 2,000 different AA changes, which affect the interaction between p53 and DNA (42). R175, G245, R248, R273, and R282 are part of hotspot mutations that causing DNA binding loss, which can be divided into two categories: “DNA-contact mutation” (R248 and R273) that contact DNA directly and “conformation mutation” (R175, G245, and R282) that perturb the structure of DBD (42, 53, 54).
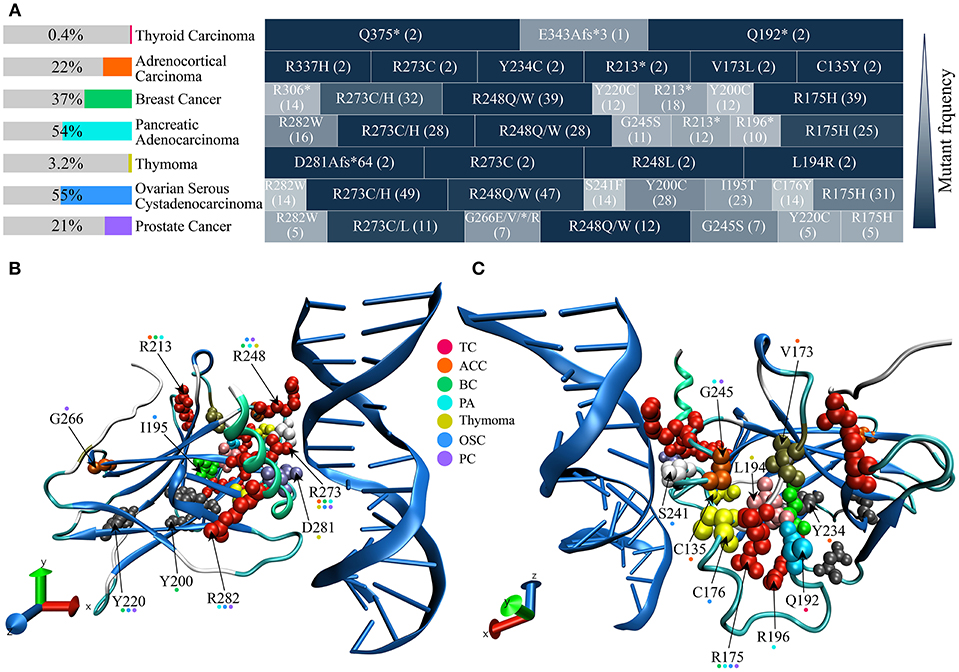
Figure 1. The frequency of p53 mutation, mutated sites, and site location in 3D structure from common endocrine cancers. p53 missense mutation data are obtained from The Cancer Genome Atlas (TCGA) in cBioPortal (http://www.cbioportal.org/). (A) p53 mutant frequencies and high frequency mutation sites of seven common endocrine cancers. p53 mutant frequencies of each cancer are shown in the left color bar. The right side indicates p53 high frequency mutation sites in every cancer. Different shades of block depicting the relative mutant frequency of p53 site, compared with the highest mutant rate site (dark blue) in the same cancer. Cases number of respective mutation is shown in parenthesis. (B,C) Mutation sites structural position analysis of p53 DBD. Cancers involved in each mutant site are indicated in color dot above AA mark. DBD, DNA binding domain; TC, Thyroid Carcinoma; ACC, Adrenocortical Carcinoma; BC, Breast Cancer; PA, Pancreatic Adenocarcinoma; OSC, Ovarian Serous Cystadenocarcinoma; PC, Prostate Cancer.
Mut-p53 can stably accumulate from escaping the degradation of negative regulators and forming aggregates with p63 and p73 (55, 56).There are three types of p53 mutation consequence, loss-of-function (LOF), dominant-negative (DN), and gain-of-function (GOF), have been concluded (55). First, LOF-p53 abolish the transcriptional activation of partially innate target genes, such as mdm2 (murine double minute 2), puma, and p21 (42). Second, tetramer Mut-p53 exert a DN effect on Wt-p53 (42). Third, GOF-p53 can regulate novel target genes and turn to be an oncogenic role in inducing tumorigenesis, tumor proliferation, invasion and metastasis, tumor inflammation, tumor tissue remodeling, and evading growth suppresses (55, 57). Moreover, these consequences can occur simultaneously (55).
High invasion and metastatic risk are the fatal hallmark of endocrine adenocarcinomas, such as 60~80% breast and prostate cancer have developed bone metastasis (58), about 70% pancreatic cancer patients die from extensive metastatic diseases (59). Epithelial-mesenchymal transition (EMT) is prerequisite for primary endocrine adenocarcinomas metastasizing to blood, bone, and other organs (58, 60), Mut-p53 is essential to EMT process. This review summarized the pivotal molecular pathways and inhibitors in Mut-p53-mediated EMT/non-EMT process related invasion and metastasis (Figure 2).
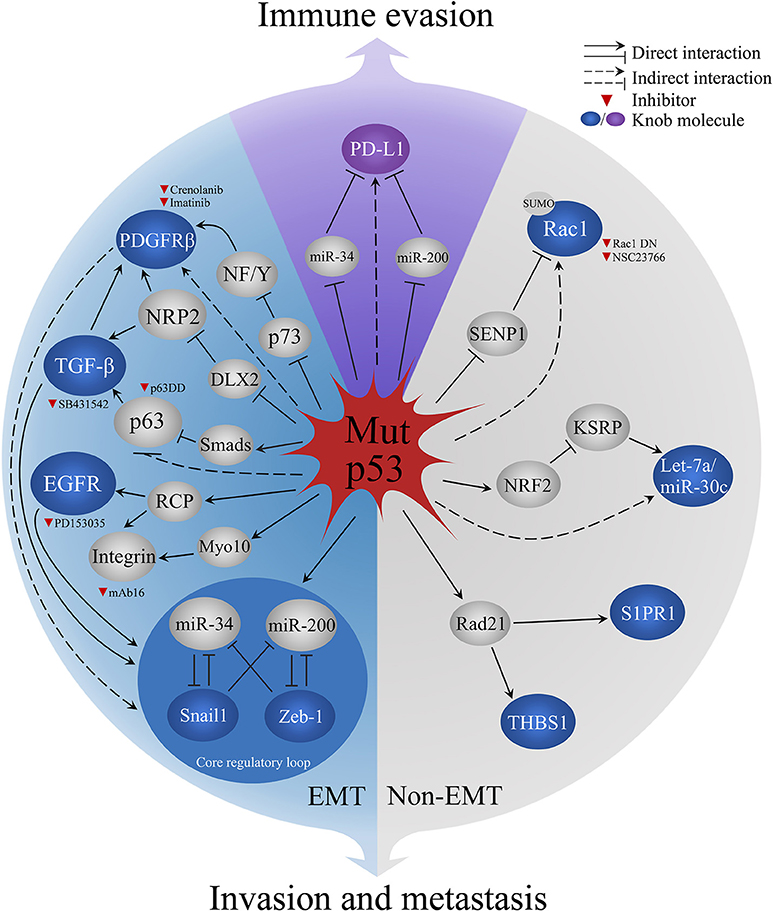
Figure 2. Mechanisms of Mut-p53 aggravate malignant behaviors in endocrine cancers. Blue sector represents Mut-p53-mediated EMT process. Gray sector represents Non-EMT metastasis. Purple sector indicates Mut-p53-regulated PD-L1 immune evasion in cancer cells. Red triangle indicates the inhibitors for current research. PDGFRb, platelet-derived growth factor receptor b; NF/Y, nuclear factors; NRP2, Neuropilin-2; DLX2, Distal-less homeobox 2; TGF-β, transforming growth factor β; EGFR, Epidermal growth factor receptor; RCP, Rab-coupling protein; Myo10, Myosin-X; Rac1, Ras-related C3 botulinum toxin substrate 1; SENP1, SUMO-specific protease 1; miR-34, miRNA 34; miR-200, miRNA 200; NRF2, Nuclear factor-like 2; miR-30c, miRNA 30c; KSRP, KH-type splicing regulatory protein; Rad21, Double-strand-break repair protein rad21; S1PR1, Sphingosine-1-phosphate receptor 1; THBS1, Thrombospodin 1; PD-L1, programmed death-ligand 1.
Mut-p53 mediates the functions of EMT inducers, including three crucial inducers, transforming growth factor-β (TGF-β), platelet-derived growth factor receptor (PDGFR), and epidermal growth factor receptor (EGFR) (Figure 2). Mut-p53 empowers TGF-β to trigger metastasis of breast cancer cells via forming the ternary complex of Mut-p53, p63, and Smads that prevents the inhibition function of p63 to TGF-β (61). In pancreatic cancer mouse model (KPC model, LSL-KrasG12D; P53R172H/+; Pdx1-cre), PDGFRβ is upregulated and induces tumor metastasis by Mut-p53-mediated p73/NF-Y complex disruption (62). Furthermore, neuropilin-2 (NRP2) is a co-receptor of TGF-β receptor and PDGFR, which can be upregulated by Mut-p53 R273H and enhance the EMT inducing signals in breast cancer cells (63). Mut-p53 promotes the sustained activation of EGFR via suppressing miR-27a/EGFR axis in breast cancer cells, which increases the mRNA of Zeb-1 and Slug to promote EMT (64, 65). Moreover, Mut-p53 (R175H and R273H) enhance the recycling of integrins and EGFR via rab-coupling protein (RCP), which results in the activation of EGFR/integrin signaling that leads to the invasion behavior in vivo and in vitro (53). The recycled integrin move to an invasion membrane protrusion to drive cell migration by binding filopodia-inducing motor protein Myosin-X (Myo10) in a Mut-p53 dependent manner (66). Mut-p53 also controls the expression of crucial EMT regulators (Figure 2), including EMT-inducing transcription factors (EMT-TFs; like Snail, Twist, Zeb1, and Zeb2) and post-transcriptional regulators micro-RNA200 (miR-200) and micro-RNA34 (miR-34). Mut-p53 breaks the balance of EMT core regulatory mechanism, two inhibitory loops (Zeb1/miR-200 or Snail1/miR-34), via losing the activity of binding and promoting miR-200/miR-34, which causes the upregulation of Zeb-1/Snail1 that activates the EMT program (67, 68). But, the role of EMT-TFs in metastasis of endocrine carcinomas was challenged recently. Fischer et al. (69) and Zheng et al. (70) have shown the overexpression of miR-200 or depletion of Snai1/Twist1 had no effect on metastasis in breast-to-lung metastasis models and KPC model. However, the depletion of Zeb-1 significantly reduced tumor metastasis in the same KPC model (71), which suggested various EMT-TFs have different sub-functions of tumor metastasis in cancers.
Mut-p53 aggravates malignancy of tumor in the non-EMT mechanism (Figure 2). Mut-p53 competes with SUMO-specific protease 1 (SENP1) to combine Rac1, a small GTPase, and sustains its SUMOylation to promote tumor progression in two breast cancer cell lines [SK-BR-3 (R175H) and MDA-MB468 (R273H)] (72). Mut-p53 (R248) is overexpressed in ovarian cancer cell lines, which can interact with double-strand-break repair protein rad21 (Rad21) and upregulate the expression of Rad21 target genes, sphingosine-1-phosphate receptor 1 (S1PR1), and thrombospodin 1 (THBS1), that related to cell migration in intact cells or cancer cell lines (73). Furthermore, Mut-p53 interacts with nuclear factor-like 2 (Nrf2), a proteasome activator, resulting in the proteasome-mediated degradation of KH-type splicing regulatory protein (KSRP, miRNA maturation factor) in triple-negative breast cancer cells. Thus, downregulated tumor suppressive miRNAs let-7a and miR-30c lead to tumor metastasis (74). There is growing evidence to support that mesenchymal-epithelial transition (MET), the reverse process of EMT, is associated with metastatic colonization (75). Current research suggests reactivating cell signaling pathways and facilitating attachment to heterologous cells are the mechanism of MET. Blocking MET process has been proven to restrain tumor progression in glioma (76). However, the role of Mut-p53 in MET is unclear. Both EMT and MET are critical for embryos/tissues development and wound healing (77), inhibiting general EMT/MET regulators, such as Zeb1, Snail1, or Twist may have adverse effects in other normal biological process. Therefore, blocking p53 negative properties is an attractive strategy for basic and clinical research.
Increasing evidence has shown that Mut-p53 regulates immune evasion by increasing the expression of programmed death-ligand 1 (PD-L1) in tumor cells (78). Based on the genomic, transcriptomic, proteomic, and clinical cancer database in non-small cell lung cancer and lung adenocarcinoma, patients with TP53 mutations, or Mut-p53-related EMT phenotypes have higher PD-L1 mRNA expression and low levels of miR-34/miR-200 (23, 78, 79). Both miR-34 and miR-200 suppress PD-L1 by specifically binding to PD-L1 3′-UTR (80). Mut-p53 decreases the expression of miR-34/miR-200 in EMT, which leads to high levels of PD-L1 in non-small cell lung cancer. However, the higher PD-L1 show a significant benefit to PD-1/PD-L1 blockade therapy, p53 may also be a potential guilder for immunotherapy selecting (23, 79).
Previous research in targeting p53 mostly is to block p53 accumulation or gain of oncogenic function (81–84). However, correcting Mut-p53 gene sequence and protein conformation are emerging as novel strategies in targeting p53. In re-editing p53 sequence, the CRISPR/Cas9 system is a preferred genetic engineering technique that has a promising prospect (85). CRISPR/Cas9 editing with hydrodynamic injection can specifically trigger TP53 (229 site indel) and pten (125 site insertion) mutation in adult mouse liver and directly induce tumors in 2 months that is similar to phenocopies of the Cre-loxP method (86). Recently found CRISPR/Cas9 also successfully modified a genomic fragment as large as 65kb length (TP53 locus about 20.5kb) in mice zygotes and embryos (87). CRISPR/Cas9 technique already can efficiently relieve disease phenotypes of hereditary tyrosinemia type I (HT-1) in mice by correcting fumarylacetoacetate hydrolase (FAH) mutation (causing HT-1) (88). These findings suggested that CRISPR/Cas9 could correct Mut-p53 sequence by replacing with a Wt-p53 functional copy, which is possible to be utilized in clinical research. In modifying the conformation of Mut-p53, various compounds have been reported to reconvert Mut-p53 to Wt-like-p53 structure, such as PRIMA-1 [2,2-bis(hydroxymethyl)quinuclidin-3-one] and APR-246 (PRIMA-1MET) (89). Recently found a brief exposure of ZMC1 (NSC319726), one of the Zinc metallochaperones compounds from thiosemicarbazone family, is sufficient to reactivate p53 R175H and exhibit Wt-p53 transcriptional activity with few toxicity (90). ZMC1 raises intracellular Zn2+ levels to return p53 R175H mutant (zinc-deficient) to “Wt-like-p53” conformation via the recombination of Zn2+ and p53 R175H (91), which induces apoptosis in murine cell lines and significantly increases the median survival of KPC mice model (90, 91). It is surprising that a natural product cruciferous vegetable-derived phenethyl isothiocyanate (PEITC) can reactivate p53 R175H function by restoring Wt-like conformation and stimulating canonical Wt-p53 target genes expression. PEITC is already used in anti-Mut-p53 research and clinical trials (https://clinicaltrials.gov/ct2/results?cond=&term$=+PEITC), which explores a new example of targeting p53 by dietary compounds (92). Therefore, targeting p53 with CRISPR/Cas9 re-editing and conformation remodeling compounds are attractive strategies in translation medicine.
Wild animals have numerous p53 variations that contribute to environmental adaptation and cancer resistance, however, these variations also have been found in human cancer, which implies special mechanisms exist behind p53 variations in wild animals (26–28, 93). Israel Spalax is an anticancer blind subterranean mole rat, there are two specific p53 variations exist in DBD, K172, and K207, compared with human and mouse sequences (R174/R209 in human). R174 and R209 mutations in human p53 have been found in esophageal carcinoma, uterine cervix tumor, colorectal cancer, and endocrine cancers (breast cancer, pancreatic adenocarcinoma, and ovarian cancer) [Figure 1, (93)]. But, p53 variations in Spalax is associated with adaptive evolution, which overactives cell cycle arrest genes (p21/cycG) and downregulates proapoptosis genes (puma and Noxa) with no apaf1 expression (93). And, in two subspecies of Spalax galili with sharply divergent abutting ecologies, the different methylation modifications of p53 lead to adaptive regulation of p53 pathway and cell-cycle arrest, which indicates p53 epigenetic changes contribute to sympatric speciation (94). Further research reveals that in response to hyperproliferation, released IFN-β induces a p53-triggered anticancer mechanism via inducing necrotic cell death rather than apoptosis in lung primary fibroblasts of Spalax judaei and Spalax golani (28). p53 codon 104 variations exist in underground mole rat Myospalax baileyi (M.b), and root vole Microtus oeconomus (M.o) in Qinghai-Tibet plateau of China, which is also found in fishes and giant tortoise (26, 95). The corresponding human mutation S106 was discovered in one multiple primary cancer case. These specific p53 variations in M.b and M.o display an extreme environmental adaptation strategy in transcriptional regulation, for instance, M.b p53 elicits increased expression of proapoptotic genes under hypoxia/cold and antiapoptotic genes under acidic stress, but M.o suppresses all the apoptotic genes and displays a remarkable sensitive to hypoxia (26). In naked mole rat (Heterocephalus glaber, NMR), p53 with four PXXP (P = proline, X = any amino acid) motifs in proline-rich domain (five motifs in human and one in rat) increase the convergent evolution possibility of NMR and human, which evolved enhanced DNA damage response and extended lifespan (96). The African and Asia elephant genomes contain a single TP53 gene and 19 TP53 retrogenes (1 copy TP53 in human) (27). The TP53RTG proteins are encoded by 14 retrogenes, and W23G variation exists in all TP53RTGs. TP53RTGs escape MDM2-medicated ubiquitination by the interaction breaking of TP53RTGs and MDM2. Meanwhile, p53 is stabilized by forming TP53RTG/p53 dimer that blocks the degradation by MDM2 (97). High levels of p53 upregulate a re-functionalized retrogene, leukemia inhibitory factor 6 (LIF6), that induces apoptosis in Asian elephant dermal fibroblasts cells (98). The multicopy TP53, variations in 14 TP53RTGs, and retrogene reactivation by Wt-p53 are part of the outcome in the evolutionary selection, which enhances the sensitivity in responding to DNA damage, and induces apoptosis rather than DNA repair in elephant (27, 97). Wt-p53 in wild animal elephants and mole rats induces entirely wipe out injured cells by necrosis/strong apoptosis rather than DNA repair like human. Natural wild animals with high longevity and anti-cancer provide an environmental adaptation and cellular homeostasis model for exploring the mechanisms of molecular variations and evolution.
This mini-review summarizes various roles of p53 in human and animals. As a cellular sensor for homeostasis, p53 involves in metabolism, autophagy regulation, insulin resistance or secretion, and food intake. Wt-p53 is also responsible for the occurrence and developing of endocrine diseases and tumors. p53 is frequently mutant in endocrine cancers and Mut-p53 promotes malignant behaviors by mediating EMT and non-EMT in metastasis and upregulating PD-L1 in immune evasion. Recovering Mut-p53 gene sequence and protein structure with CRISPR-Cas9 or dietary compounds already showed a great research value. Mut-p53 is also a potential candidate for PD-L1 immunotherapy selecting and health risk prediction. In wild animals, p53 variations contribute to environmental adaptation and cancer resistance. p53 is a multifunctional molecule, exploring the new p53 functions, investigating the p53 variations in wild animals, and returning the “evil” Mut-p53 to an “angel” in physiological system are fascinating.
Publicly available datasets were analyzed in this study. This data can be found here: http://www.cbioportal.org.
YL, M-CZ, and X-QC drafted the manuscript. X-QC and J-ZD supervised the project and conceived of the student. X-KX, CM, YZ, and EN contributed to manuscript revisions. TZ and HD contributed to discussions and suggestion. YL and M-CZ contributed to figure and manuscript editing.
This work was supported by the Ministry Science and Technology of China, National Basic Research Program (973) of the Ministry of Science and Technology of China Grants 2012CB518200 and National Natural Science Foundation of China Grants 31471140, 81741120, 30870971, and NSF of Zhejiang Province (LY12H16027). EN received financial support from the Ancell-Teicher Research Foundation for Genetics and Molecular Evolution.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. (1979) 278:261–3. doi: 10.1038/278261a0
2. Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. (1979) 17:43–52. doi: 10.1016/0092-8674(79)90293-9
3. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. (2017) 170:1062–78. doi: 10.1016/j.cell.2017.08.028
4. Marcel V, Nguyen Van Long F, Diaz JJ. 40 years of research put p53 in translation. Cancers. (2018) 10:152. doi: 10.3390/cancers10050152
5. Nguyen TT, Grimm SA, Bushel PR, Li J, Li Y, Bennett BD, et al. Revealing a human p53 universe. Nucleic Acids Res. (2018) 46:8153–67. doi: 10.1093/nar/gky720
6. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. (2008) 9:402–12. doi: 10.1038/nrm2395
7. Uehara I, Tanaka N. Role of p53 in the regulation of the inflammatory tumor microenvironment and tumor suppression. Cancers. (2018) 10:219. doi: 10.3390/cancers10070219
8. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. (2009) 9:749–58. doi: 10.1038/nrc2723
9. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. (2015) 356(2 Pt A):197–203. doi: 10.1016/j.canlet.2013.12.025
10. Goldstein I, Rotter V. Regulation of lipid metabolism by p53-fighting two villains with one sword. Trends Endocrinol Metab. (2012) 23:567–75. doi: 10.1016/j.tem.2012.06.007
11. Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. (2013) 18:617–33. doi: 10.1016/j.cmet.2013.06.019
12. Tao L, Roberts AL, Dunphy KA, Bigelow C, Yan H, Jerry DJ. Repression of mammary stem/progenitor cells by p53 is mediated by Notch and separable from apoptotic activity. Stem Cells. (2011) 29:119–27. doi: 10.1002/stem.552
13. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. (2010) 107:7455–60. doi: 10.1073/pnas.1001006107
14. Sui X, Jin L, Huang X, Geng S, He C, Hu X. p53 signaling and autophagy in cancer: a revolutionary strategy could be developed for cancer treatment. Autophagy. (2014) 7:565–71. doi: 10.4161/auto.7.6.14073
15. Balaburski GM, Hontz RD, Murphy ME. p53 and ARF: unexpected players in autophagy. Trends Cell Biol. (2010) 20:363–9. doi: 10.1016/j.tcb.2010.02.007
16. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
17. Wang Q, Zou Y, Nowotschin S, Kim SY, Li QV, Soh CL, et al. The p53 family coordinates wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell. (2017) 20:70–86. doi: 10.1016/j.stem.2016.10.002
18. Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol. (2011) 12:259–65. doi: 10.1038/nrm3086
19. Bowling S, Di Gregorio A, Sancho M, Pozzi S, Aarts M, Signore M, et al. P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat Commun. (2018) 9:1763. doi: 10.1038/s41467-018-04167-y
20. Bates SE. Endocrine cancers: defying the paradigms. Clin Cancer Res. (2016) 22:4980. doi: 10.1158/1078-0432.CCR-16-0366
21. Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. (2015) 523:352–6. doi: 10.1038/nature14430
22. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. (2013) 15:2–8. doi: 10.1038/ncb2641
23. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. (2016) 108:djv303. doi: 10.1093/jnci/djv303
24. Hof J, Krentz S, van Schewick C, Korner G, Shalapour S, Rhein P, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. (2011) 29:3185–93. doi: 10.1200/JCO.2011.34.8144
25. Parkinson CA, Gale D, Piskorz AM, Biggs H, Hodgkin C, Addley H, et al. Exploratory analysis of TP53 mutations in circulating tumour DNA as biomarkers of treatment response for patients with relapsed high-grade serous ovarian carcinoma: a retrospective study. PLoS Med. (2016) 13:e1002198. doi: 10.1371/journal.pmed.1002198
26. Zhao Y, Ren JL, Wang MY, Zhang ST, Liu Y, Li M, et al. Codon 104 variation of p53 gene provides adaptive apoptotic responses to extreme environments in mammals of the Tibet plateau. Proc Natl Acad Sci USA. (2013) 110:20639–44. doi: 10.1073/pnas.1320369110
27. Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, et al. Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA. (2015) 314:1850–60. doi: 10.1001/jama.2015.13134
28. Gorbunova V, Hine C, Tian X, Ablaeva J, Gudkov AV, Nevo E, et al. Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc Natl Acad Sci USA. (2012) 109:19392–6. doi: 10.1073/pnas.1217211109
29. Zhao Y, Wang MY, Hao K, Chen XQ, Du JZ. CRHR1 mediates p53 transcription induced by high altitude hypoxia through ERK 1/2 signaling in rat hepatic cells. Peptides. (2013) 44:8–14. doi: 10.1016/j.peptides.2013.03.023
30. Wang X, Zhao X, Gao X, Mei Y, Wu M. A new role of p53 in regulating lipid metabolism. J Mol Cell Biol. (2013) 5:147–50. doi: 10.1093/jmcb/mjs064
31. Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. (2011) 44:491–501. doi: 10.1016/j.molcel.2011.08.038
32. Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 dynamics control cell fate. Science. (2012) 336:1440–4. doi: 10.1126/science.1218351
33. Porter JR, Fisher BE, Batchelor E. p53 pulses diversify target gene expression dynamics in an mRNA half-life-dependent manner and melineate co-regulated target gene subnetworks. Cell Syst. (2016) 2:272–82. doi: 10.1016/j.cels.2016.03.006
34. Hafner A, Stewart-Ornstein J, Purvis JE, Forrester WC, Bulyk ML, Lahav G. p53 pulses lead to distinct patterns of gene expression albeit similar DNA-binding dynamics. Nat Struct Mol Biol. (2017) 24:840–7. doi: 10.1038/nsmb.3452
35. Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell. (2016) 165:631–42. doi: 10.1016/j.cell.2016.03.025
36. Quinones M, Al-Massadi O, Folgueira C, Bremser S, Gallego R, Torres-Leal L, et al. p53 in AgRP neurons is required for protection against diet-induced obesity via JNK1. Nat Commun. (2018) 9:3432. doi: 10.1038/s41467-018-05711-6
37. Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. (2009) 15:1082–7. doi: 10.1038/nm.2014
38. Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, et al. Inhibition of p53 preserves parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc Natl Acad Sci USA. (2014) 111:3116–21. doi: 10.1073/pnas.1318951111
39. Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Schyr-Ben Haroush R, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab. (2014) 19:109–21. doi: 10.1016/j.cmet.2013.11.007
40. Zhu JJ, Dou ZX, Sammons MA, Levine AJ, Berger SL. Lysine methylation represses p53 activity in teratocarcinoma cancer cells. Proc Natl Acad Sci USA. (2016) 113:9822–7. doi: 10.1073/pnas.1610387113
41. Ashikari D, Takayama K, Tanaka T, Suzuki Y, Obinata D, Fujimura T, et al. Androgen induces G3BP2 and SUMO-mediated p53 nuclear export in prostate cancer. Oncogene. (2017) 36:6272–81. doi: 10.1038/onc.2017.225
42. Stiewe T, Haran TE. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist Updat. (2018) 38:27–43. doi: 10.1016/j.drup.2018.05.001
43. Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. (2016) 85:375–404. doi: 10.1146/annurev-biochem-060815-014710
44. Stracquadanio G, Wang X, Wallace MD, Grawenda AM, Zhang P, Hewitt J, et al. The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat Rev Cancer. (2016) 16:251–65. doi: 10.1038/nrc.2016.15
45. Cancer Genome Atlas Research Network Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. (2017) 169:1327–41 e23. doi: 10.1016/j.cell.2017.05.046
46. Liu Y, Zhang X, Han C, Wan G, Huang X, Ivan C, et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature. (2015) 520:697–701. doi: 10.1038/nature14418
47. Jethwat A, Slabicki M, Huellein J, Jentzsch M, Dala V, Rabe S, et al. TRRAP is essential for regulating the accumulation of mutant and wild-type p53 in lymphoma. Blood. (2018) 131:2789–802. doi: 10.1182/blood-2017-09-806679
48. Wong K, van der Weyden L, Schott CR, Foote A, Constantino-Casas F, Smith S, et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat Commun. (2019) 10:353. doi: 10.1038/s41467-018-08081-1
49. Gao JJ, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. (2013) 6:pl1. doi: 10.1126/scisignal.2004088
50. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
51. Leroy B, Ballinger ML, Baran-Marszak F, Bond GL, Braithwaite A, Concin N, et al. Recommended guidelines for validation, quality control, and reporting of TP53 variants in clinical practice. Cancer Res. (2017) 77:1250–60. doi: 10.1158/0008-5472.CAN-16-2179
52. Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat. (2014) 35:672–88. doi: 10.1002/humu.22552
53. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. (2009) 139:1327–41. doi: 10.1016/j.cell.2009.11.026
54. Walerych D, Lisek K, Del Sal G. Mutant p53: one, no one, and one hundred thousand. Front Oncol. (2015) 5:289. doi: 10.3389/fonc.2015.00289
55. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. (2010) 2:a001107. doi: 10.1101/cshperspect.a001107
56. Silva JL, Cino EA, Soares IN, Ferreira VF, de Oliveira GAP. Targeting the prion-like aggregation of mutant p53 to combat cancer. Acc Chem Res. (2018) 51:181–90. doi: 10.1021/acs.accounts.7b00473
57. Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ. (2018) 25:161–8. doi: 10.1038/cdd.2017.185
58. Hofbauer LC, Rachner TD, Coleman RE, Jakob F. Endocrine aspects of bone metastases. Lancet Diabetes Endocrinol. (2014) 2:500–12. doi: 10.1016/S2213-8587(13)70203-1
59. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. (2014) 371:1039–49. doi: 10.1056/NEJMra1404198
60. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
61. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A mutant-p53/smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. (2009) 137:87–98. doi: 10.1016/j.cell.2009.01.039
62. Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. (2014) 157:382–94. doi: 10.1016/j.cell.2014.01.066
63. Lv T, Wu X, Sun L, Hu Q, Wan Y, Wang L, et al. p53-R273H upregulates neuropilin-2 to promote cell mobility and tumor metastasis. Cell Death Dis. (2017) 8:e2995. doi: 10.1038/cddis.2017.376
64. Claperon A, Mergey M, Nguyen Ho-Bouldoires TH, Vignjevic D, Wendum D, Chretien Y, et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J Hepatol. (2014) 61:325–32. doi: 10.1016/j.jhep.2014.03.033
65. Wang W, Cheng B, Miao L, Mei Y, Wu M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR expression. Cell Death Dis. (2013) 4:e574. doi: 10.1038/cddis.2013.97
66. Arjonen A, Kaukonen R, Mattila E, Rouhi P, Hognas G, Sihto H, et al. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J Clin Invest. (2014) 124:1069–82. doi: 10.1172/JCI67280
67. Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. (2011) 13:317–23. doi: 10.1038/ncb2173
68. Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. (2011) 195:417–33. doi: 10.1083/jcb.201103097
69. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. (2015) 527:472–6. doi: 10.1038/nature15748
70. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. (2015) 527:525–30. doi: 10.1038/nature16064
71. Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. (2017) 19:518–29. doi: 10.1038/ncb3513
72. Yue X, Zhang C, Zhao Y, Liu J, Lin AW, Tan VM, et al. Gain-of-function mutant p53 activates small GTPase Rac1 through SUMOylation to promote tumor progression. Genes Dev. (2017) 31:1641–54. doi: 10.1101/gad.301564.117
73. Ahn JH, Kim TJ, Lee JH, Choi JH. Mutant p53 stimulates cell invasion through an interaction with Rad21 in human ovarian cancer cells. Sci Rep. (2017) 7:9076. doi: 10.1038/s41598-017-08880-4
74. Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol. (2016) 18:897–909. doi: 10.1038/ncb3380
75. Gunasinghe NP, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. (2012) 31:469–78. doi: 10.1007/s10555-012-9377-5
76. Wu Y, Fan Q, Zeng F, Zhu J, Chen J, Fan D, et al. Peptide-functionalized nanoinhibitor restrains brain tumorgrowth by abrogating mesenchymal-epithelial transition factor (MET) signaling. Nano Lett. (2018) 18:5488–98. doi: 10.1021/acs.nanolett.8b01879
77. Owusu-Akyaw A, Krishnamoorthy K, Goldsmith LT, Morelli SS. The role of mesenchymal-epithelial transition in endometrial function. Hum Reprod Update. (2019) 25:114–33. doi: 10.1093/humupd/dmy035
78. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2017) 23:3012–24. doi: 10.1158/1078-0432.CCR-16-2554
79. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. (2014) 5:5241. doi: 10.1038/ncomms6241
80. Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem Sci. (2018) 43:1014–32. doi: 10.1016/j.tibs.2018.09.004
81. Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. (2011) 18:1904–13. doi: 10.1038/cdd.2011.71
82. Chowdhury P, Lin GE, Liu K, Song Y, Lin FT, Lin WC. Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nat Commun. (2014) 5:5476. doi: 10.1038/ncomms6476
83. Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. (2012) 18:64–76. doi: 10.1158/1078-0432.CCR-11-1000
84. Wei S, Kozono S, Kats L, Nechama M, Li W, Guarnerio J, et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat Med. (2015) 21:457–66. doi: 10.1038/nm.3839
85. Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. (2015) 15:387–95. doi: 10.1038/nrc3950
86. Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. (2014) 514:380–4. doi: 10.1038/nature13589
87. Zhang L, Jia R, Palange NJ, Satheka AC, Togo J, An Y, et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS ONE. (2015) 10:e0120396. doi: 10.1371/journal.pone.0120396
88. Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. (2014) 32:551–3. doi: 10.1038/nbt.2884
89. Duffy MJ, Synnott NC, Crown J. Mutant p53 as a target for cancer treatment. Eur J Cancer. (2017) 83:258–65. doi: 10.1016/j.ejca.2017.06.023
90. Yu X, Kogan S, Chen Y, Tsang AT, Withers T, Lin H, et al. Zinc metallochaperones reactivate mutant p53 using an ON/OFF switch mechanism: a new paradigm in cancer therapeutics. Clin Cancer Res. (2018) 43: 1014–32. doi: 10.1158/1078-0432.CCR-18-0822
91. Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. (2012) 21:614–25. doi: 10.1016/j.ccr.2012.03.042
92. Aggarwal M, Saxena R, Sinclair E, Fu Y, Jacobs A, Dyba M, et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. (2016) 23:1615–27. doi: 10.1038/cdd.2016.48
93. Ashur-Fabian O, Avivi A, Trakhtenbrot L, Adamsky K, Cohen M, Kajakaro G, et al. Evolution of p53 in hypoxia-stressed Spalax mimics human tumor mutation. Proc Natl Acad Sci USA. (2004) 101:12236–41. doi: 10.1073/pnas.0404998101
94. Zhao Y, Tang JW, Yang Z, Cao YB, Ren JL, Ben-Abu Y, et al. Adaptive methylation regulation of p53 pathway in sympatric speciation of blind mole rats, Spalax. Proc Natl Acad Sci USA. (2016) 113:2146–51. doi: 10.1073/pnas.1522658112
95. Quesada V, Rodríguez SF, Miller J, Silva JP, Jiang ZF, Tapia W. Giant tortoise genomes provide insights into longevity and age-related disease. Nat Ecol Evol. (2018) 3:87–95. doi: 10.1038/s41559-018-0733-x
96. Keane M, Craig T, Alfoldi J, Berlin AM, Johnson J, Seluanov A, et al. The naked mole rat genome resource: facilitating analyses of cancer and longevity-related adaptations. Bioinformatics. (2014) 30:3558–60. doi: 10.1093/bioinformatics/btu579
97. Sulak M, Fong L, Mika K, Chigurupati S, Yon L, Mongan NP, et al. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. Elife. (2016) 5:e11994. doi: 10.7554/eLife.11994
Keywords: p53, mutation, metastasis, transcriptional factor, stress, variation
Citation: Li Y, Zhang M-C, Xu X-K, Zhao Y, Mahanand C, Zhu T, Deng H, Nevo E, Du J-Z and Chen X-Q (2019) Functional Diversity of p53 in Human and Wild Animals. Front. Endocrinol. 10:152. doi: 10.3389/fendo.2019.00152
Received: 06 January 2019; Accepted: 20 February 2019;
Published: 12 March 2019.
Edited by:
Yong Zhu, East Carolina University, United StatesReviewed by:
Taisen Iguchi, National Institute for Basic Biology, JapanCopyright © 2019 Li, Zhang, Xu, Zhao, Mahanand, Zhu, Deng, Nevo, Du and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-Qun Chen, Y2hld3lnQHpqdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.