 Christopher D. Kassotis*
Christopher D. Kassotis* Heather M. Stapleton
Heather M. Stapleton- Nicholas School of the Environment, Duke University, Durham, NC, United States
Obesity and metabolic disorders are of great societal concern and generate substantial human health care costs globally. Interventions have resulted in only minimal impacts on disrupting this worsening health trend, increasing attention on putative environmental contributors. Exposure to numerous environmental contaminants have, over decades, been demonstrated to result in increased metabolic dysfunction and/or weight gain in cell and animal models, and in some cases, even in humans. There are numerous mechanisms through which environmental contaminants may contribute to metabolic dysfunction, though certain mechanisms, such as activation of the peroxisome proliferator activated receptor gamma or the retinoid x receptor, have received considerably more attention than less-studied mechanisms such as antagonism of the thyroid receptor, androgen receptor, or mitochondrial toxicity. As such, research on putative metabolic disruptors is growing rapidly, as is our understanding of molecular mechanisms underlying these effects. Concurrent with these advances, new research has evaluated current models of adipogenesis, and new models have been proposed. Only in the last several years have studies really begun to address complex mixtures of contaminants and how these mixtures may disrupt metabolic health in environmentally relevant exposure scenarios. Several studies have begun to assess environmental mixtures from various environments and study the mechanisms underlying their putative metabolic dysfunction; these studies hold real promise in highlighting crucial mechanisms driving observed organismal effects. In addition, high-throughput toxicity databases (ToxCast, etc.) may provide future benefits in prioritizing chemicals for in vivo testing, particularly once the causative molecular mechanisms promoting dysfunction are better understood and expert critiques are used to hone the databases. In this review, we will review the available literature linking metabolic disruption to endocrine-mediated molecular mechanisms, discuss the novel application of environmental mixtures and implications for in vivo metabolic health, and discuss the putative utility of applying high-throughput toxicity databases to answering complex organismal health outcome questions.
Endocrine Disruptors as Causative Factor in Metabolic Disruption
Endocrine disrupting chemicals (EDCs) have been demonstrated to directly modulate metabolism in vivo and/or triglyceride accumulation in vitro through various receptor-mediated pathways (1–5), suggesting a potential causative link between exposure to EDCs and the increasing global prevalence of metabolic disorders, including obesity (6). Chronic metabolic health conditions are rapidly increasing in prevalence and cost to society worldwide: in the US, 39.6 and 9.7% of adults aged 20 and older are currently classified as obese or have been diagnosed with diabetes, respectively, with increasing occurrence in younger age groups as well (7–10). These conditions contribute to a rising share of health care costs; in the US, >$600 million is directed to obesity-related and diabetes-related illnesses in adults (10, 11). These effects are mirrored in animal populations, with an analysis of >20,000 animals from 24 populations reporting increased weight gain in numerous species including monkeys, both laboratory and urban mice, cats, dogs, etc. (12). Notably, attempted interventions have yielded minimal effects, and analyses have determined that activity, caloric intake, and genetics are insufficient to explain the magnitude and speed of this change (13, 14). As fat cell development is driven and modulated by nuclear hormone receptor signaling (2, 15–17), EDCs that activate or inhibit these hormone pathways may be causative agents in promoting modulation of fat cell development, energy homeostasis, basal metabolic rate, hormonal control of appetite and satiety, and brain circuitry controlling food intake and energy expenditure and ultimately contributing to the development of Metabolic Syndrome (Figure 1) (18).
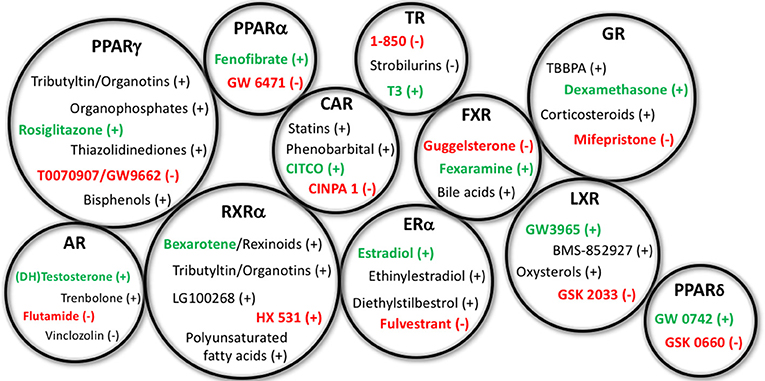
Figure 1. Representative EDCs Capable of Affecting Adipogenesis. Representative endocrine disrupting chemicals (EDCs) capable of affecting adipogenesis and/or metabolic health through the specified nuclear receptor pathways listed above. Gross circle size intended to express a general sense of the reported research into assessing these varying mechanisms; for example, PPARγ, RXRα, and GR have previously received the bulk of the research, whereas others have received less. Agonists for the receptors are depicted with a (+) following the chemicals, whereas antagonists are denoted with the (−). Standard positive and negative control chemicals for each receptor (for evaluating these pathways) are bolded to distinguish from the other EDC examples. PPAR, peroxisome proliferator activated receptor; RXR, retinoid X receptor; AR, androgen receptor; ER, estrogen receptor; CAR, constitutive androstane receptor; TR, thyroid receptor; FXR, farnesoid X receptor; LXR, liver X receptor; GR, glucocorticoid receptor.
Numerous environmental toxicants have been demonstrated as metabolic disruptors in vivo, supporting EDCs as a causative factor in these adverse health trends (14). There is a rich literature demonstrating effects of antibiotics on weight gain in humans and diverse animal species. Experiments demonstrating their efficacy in promoting weight gain in agricultural species were published by the 1950's, presumably operating through effects on gut microbiota impacting the processing of carbohydrates in the diet (19, 20). More recent publications have demonstrated that several weeks of subtherapeutic antibiotics increase fat mass and weight, particularly when begun during gestation (21, 22), and human epidemiological studies have demonstrated increased risk of becoming overweight when children were exposed early in life (23, 24). Other notable examples include diethylstilbestrol (DES), a pharmaceutical provided to pregnant women in the 1940's through 1970's in the mistaken assumption it would reduce rates of abortion, miscarriage, and premature labor (25); it was later determined to induce a variety of adverse health effects in both males and females exposed during gestation (26–29). DES has been demonstrated to promote triglyceride accumulation in vitro, seemingly through an estrogen-receptor mediated mechanism (30), and both gestational and perinatal DES exposure increases body weight, body fat, and alters serum lipid profiles in rodent models throughout life (31–33). Increased risks of obesity in human adults exposed prenatally to DES have also been reported (34), delineating apparent translational effects. Our lab has recently demonstrated that common chemicals and environmental mixtures associated with unconventional oil and gas (UOG) operations can activate the peroxisome proliferator activated receptor gamma (PPARγ) and promote triglyceride accumulation and pre-adipocyte proliferation in vitro (35), and that gestational exposure to a mixture of UOG chemicals resulted in increased body weights through weaning in a rodent model (36, 37). UOG development has also been associated with increased prevalence of low birth weight and small for gestational age births in the Northeast US (38), and decreased prevalence of low birth weights and increased risk of higher birth weight babies in Colorado (39); both low (40, 41) and high (42, 43) birth weights are associated with greater risks for obesity later in life.
As costs associated with in vivo screening of putative metabolism disruptors are prohibitively high, utilizing lower-order testing, and screening is essential to narrow higher-order testing to chemicals most likely to be active. Various pre-adipocyte and mesenchymal stem cell models (both rodent and human, primarily) have been utilized to assess putative in vivo metabolic disruptors in vitro; 3T3-L1 mouse pre-adipocytes have proven reliable as an in vitro screen for identifying likely obesogenic chemicals in vivo, and other models such as the OP9 mouse bone marrow-derived stromal pre-adipocyte cell line (44, 45) allow for assessments of varying molecular pathways important for the process of differentiation. Additionally, various multipotent mesenchymal cells and cell lines (46, 47) offer the additional ability to assess commitment to the adipocyte lineage as a distinct process from adipocyte differentiation (48). However, these assays are lengthy and their relative abilities to correctly identify chemicals may depend on both cell line and cell source. As such, there is a critical need to develop better methods for correctly predicting metabolic disruptors. Several high-throughput (HTP) screening programs now exist (Tox21, ToxCast) that report activity across mechanisms known to modulate metabolic health for thousands of chemicals. Harnessing these data sets to broadly assess high-scoring chemicals (across these molecular pathways) for more targeted in vitro and in vivo testing could provide a valuable tool for reducing research costs and more broadly assessing the tens of thousands of commercial chemicals for potential contributions to adverse health outcomes in humans and/or animals.
In addition to high-throughput screening, assessments of mixtures have become more commonplace in recent years. Tools to evaluate the chemical constituents and biological activities associated with complex environmental mixtures have vastly increased the capabilities within this sphere, though standard approaches to mixtures are still lacking in many respects, particularly in terms of relevance to human and animal exposure. One notable mixture that has received increasing attention is indoor house dust; our laboratory and others have collected and analytically characterized house dust from different environments around the world and routinely report numerous classes of EDCs (known to be hormonally active), including flame retardants, phthalates, pesticides, perfluoroalkyl substances (PFAS), and others that span a wide range of concentrations (49–51). Humans, and perhaps most importantly small children, are chronically exposed to household dust, and thus receive exposure to EDCs present in the dust. The EPA estimates children ingest 60–100 mg of dust per day from indoor environments (52), contributing to chronic oral and inhalation exposures to EDCs (49, 53, 54), and compounded by other routes of exposure. Notably, numerous studies have demonstrated clear links between levels of indoor semi-volatile indoor contaminants (SVOCs) on hand wipes with levels in house dust (55, 56), with other studies demonstrating clear links with urinary and serum levels (55, 57, 58), providing evidence for this exposure route contributing to an increased body burden of specific chemicals. As such, environmental matrices such as this may represent a clear exposure route for humans and could provide critical information on biological effects of summed mixture exposures.
Nuclear Receptor Mechanisms Mediating Metabolic Disruption
While activation of the peroxisome proliferator activated receptor gamma (PPARγ) is likely the best-described mechanism through which adipogenesis is initiated/promoted, activation or inhibition of numerous other receptor systems have been described to directly or indirectly modulate adipocyte lineage commitment and/or differentiation of pre-adipocytes and subsequent accumulation of triglycerides, including thyroid receptor-beta (TRβ), glucocorticoid receptor (GR), estrogen receptor (ER), androgen receptor (AR), liver X receptor (LXR), retinoid X receptor (RXR), and others (59) (Table 1). Several studies have assessed the expression of nuclear receptors throughout the differentiation process, reporting that 30 nuclear receptors were expressed throughout the differentiation process to varying degrees and at varying timepoints (15, 60). Recent work by Chappell et al. demonstrated putative GR-mediated effects prior to PPARγ activation after exposure of 3T3-L1 cells to tetrabrominated bisphenol A (TBBPA) (61). Notably, EDCs capable of acting through each of these pathways have been described previously to modulate metabolic health in vitro, in vivo, or in human epidemiological studies; though importantly, certain molecular mechanisms have received far greater research attention than others (Figure 2).
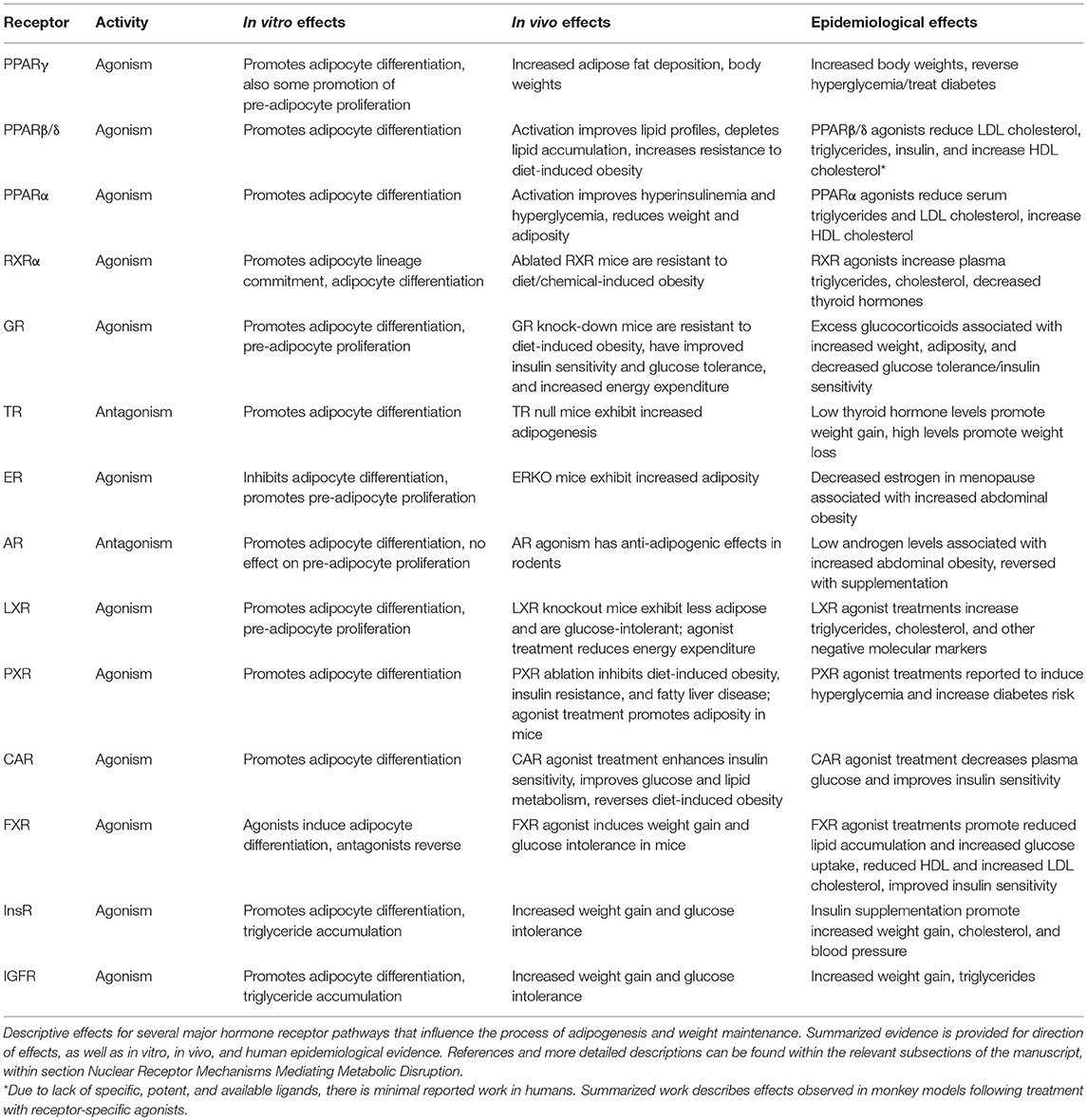
Table 1. Major hormone receptor pathways capable of promoting adipogenesis.
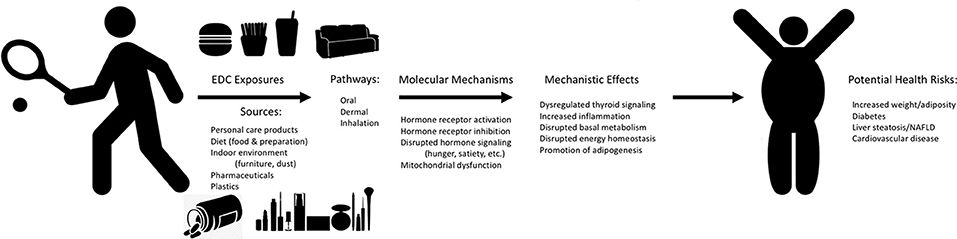
Figure 2. Mechanisms of EDC Exposure and Potential Human Metabolic Health Effects. Graphical depiction of the potential sources and exposure pathways for humans to endocrine disrupting chemicals (EDCs), the molecular mechanisms related to metabolic health through which these EDCs may act to drive specific mechanistic effects, all of which may contribute to potential adverse health risks for humans. Effects reported are representative and are not comprehensive to all molecular mechanisms and mechanistic effects.
Peroxisome Proliferator Activated Receptors (PPARs)
PPARγ is often considered the only nuclear receptor whose activation is necessary and sufficient to initiate adipogenesis (62, 63). Treatment of 3T3-L1 cells as well as other pre-adipocyte and/or other committed adipocyte lineage cells with PPARγ agonists induces a potent and efficacious increase in triglyceride accumulation, which has long been realized (64, 65); as such, PPARγ agonists such as rosiglitazone and/or troglitazone are routinely utilized as positive control ligands for these assays (63). Utilized as therapeutic agents to treat type 2 diabetes, these thiazolidinediones may act to improve insulin sensitivity via induction of PPARγ in diverse tissue types, proliferation of smaller adipocytes that are more insulin-sensitive, or via mediation of the tumor necrosis factor alpha (TNF-α), leptin, or fatty acid signaling pathways [reviewed in (66)]. To establish the necessity of this pathway to adipogenesis, Rosen et al. utilized embryonic stem cell and chimeric mouse models. They demonstrated that PPARγ-null cells tended to not generate adipocytes, suggesting an essential role for this receptor in their formation. They further demonstrated gene dosage effects in vitro; cells lacking both copies of PPARγ could not be induced to differentiate, cells with one copy exhibited an intermediate degree of differentiation, and wild-type cells exhibited robust differentiation and efficacious expression of adipocyte-specific molecular markers (62). Clonal expansion and growth arrest occurs concurrently with expression of two proteins, PPARγ and the CCAAT enhancer binding protein alpha (C/EBPα), and these markers are both important for the differentiation of pre-adipocytes to adipocytes (63). Further experiments in this laboratory demonstrated that while C/EBPα is also a primary marker for the initiation of differentiation, it operates within a single initiating pathway with PPARγ (67). In cells deficient of PPARγ, C/EBPα was not capable of promoting adipogenesis by itself, suggesting an important but non-essential role in inducing and maintaining PPARγ expression, as well as an accessory role in mediating insulin sensitivity via direct induction of the insulin receptor (67).
The other PPAR isoforms, α and β/δ, have received considerably less attention as it relates to adipogenesis, though gain/loss-of-function experiments suggest putative roles. Experiments in vitro demonstrated that induction with a PPARβ/δ-specific ligand induced robust triglyceride accumulation in wild type cells (68) [and in vivo (69)], while PPARβ/δ-null cells differentiate and accumulate triglycerides less efficaciously following PPARγ-mediated induction (68). This suggests that while PPARβ/δ is not necessary for adipogenesis, the interplay of these isoforms is necessary to induce maximal differentiation and triglyceride accumulation in adipocytes. This was supported by other work reporting that PPARβ/δ activation promotes PPARγ expression, potentially bolstering adipogenesis, and providing a supportive role (70, 71). When examining the isoforms in isolation, Brun et al. reported that receptor isoform-specific activation via ligands failed to induce adipogenesis and triglyceride accumulation for PPARβ/δ, but did for PPARα, if to a lesser extent and over a longer time-course than via activation of PPARγ (72). RNA isolations on day five demonstrated that PPARα-treated cells had minimal or no induction of various adipocyte markers, relative to robust induction in the PPARγ-treated cells; however, both had robust expression by day seven post-induction, while PPARβ/δ exhibited only minimal expression at much later time points (72). Further experiments demonstrated that C/EBPα acted cooperatively with PPARγ to stimulate adipogenesis as expected, but not with PPARα or β/δ (72), suggesting distinct mechanisms. PPARβ/δ activation in mice has anti-adipogenic effects, improving lipid profiles, reducing lipid accumulation, and increasing resistance to diet-induced obesity (73, 74). PPARα activation in mice has similarly been demonstrated to result in anti-adipogenic effects, including improved hyperinsulinemia and hyperglycemia, lowered triglycerides, increased resistance to diet-induced obesity, and decreased weight and adiposity (75–77). Fenofibrates (PPARα agonists) administered to humans have similarly been demonstrated to decrease serum triglycerides and LDL cholesterol and increase HDL cholesterol (78, 79). While absence of good selective PPARβ/δ agonists has hindered human therapeutic examination, limited work with a selective and potent PPARβ/δ agonist in rhesus monkeys reported lowered LDL cholesterol, triglycerides, insulin, and increased HDL cholesterol (80).
Retinoid X Receptor (RXRα)
PPARγ functions as a heterodimer with RXR, suggesting that this receptor might also have dependent and/or independent roles in adipogenesis (62). Indeed, more than a decade ago, it was reported that organotins, potent activators of both PPARγ and RXRα, were also extremely potent inducers of adipogenesis (81, 82). Other studies have confirmed that receptor-specific activation of RXRα promotes both adipogenic differentiation and pre-adipocyte proliferation (60, 83, 84). Mechanistic experiments have further determined that of more than 5000 PPARγ:RXR DNA-binding sites in adipocytes, most are occupied by non-PPARγ:RXR heterodimers during the early stages of differentiation and transition to PPARγ:RXR in the later stages of differentiation (85). Mice with ablated adipocyte-RXRα are resistant to diet and chemical-induced obesity and exhibit impaired lipolysis during fasting (86); RXR agonists have also been demonstrated to sensitize diabetic and obese mice to insulin (87) and decrease hyperglycemia, hypertriglyceridemia, hyperinsulinemia, and both weight gain and food intake in several rodent models (87–89). More recent work from the Blumberg lab elegantly described RXR activation as an essential signal for commitment of mesenchymal stem cells to the adipocyte cell lineage, as well as separately promoting subsequent differentiation (48). Follow-up investigation determined that RXR activation-induced adipocyte differentiation created a functionally distinct adipocyte relative to those induced by PPARγ activation; RXR activation resulted in decreased glucose uptake, expression of adiponectin, and did not induce molecular pathways involved in adipocyte browning, suggesting a dysfunctional white adipose tissue that could potentially contribute to elevated obesity and/or diabetes risk (90). Therapeutic treatment by rexinoids in humans has reported increased plasma triglycerides, increased plasma cholesterol, and decreased thyroid hormones (91–93).
Liver X Receptor (LXR), Constitutive Androstane Receptor (CAR), Pregnane X Receptor (PXR), and Farnesoid X Receptor (FXR)
LXR, CAR, FXR, AND PXR are permissive binding partners with RXR, forming receptor heterodimers that can be activated by ligands for either receptor or both (potentially resulting in a synergistic effect), reviewed in Shulman et al. (94). LXRα is expressed primarily in the adipose, liver, intestine, and kidney, while the β isoform is ubiquitously expressed; LXRs mediate cholesterol transport, stimulating cholesterol efflux from macrophages, promoting transport in serum and uptake into liver, increase degradation of cholesterol into bile acids, inhibit absorption in the intestine, and synthesize fatty acids and triglycerides (94). Some disparate results have been reported in vitro: Hummasti et al. reported that LXR agonists failed to promote triglyceride accumulation and/or adipocyte differentiation in 3T3-L1 cells and 3T3-F442A cells, though did regulate adipocyte-specific gene expression (95). However, other studies have described LXR-mediated promotion of triglyceride accumulation, adipocyte differentiation, adipocyte-specific gene expression, and pre-adipocyte proliferation both in vitro and in vivo (60, 96, 97), potentially via activation of PPARγ (96, 97). These disparate results could be explained by cells lines and/or cell sources, as we previously reported different LXR expression and responsiveness in varying pre-adipocyte sources (60). Selective knockdown experiments have demonstrated LXRα as the primary regulator of lipolysis (98), with the β isoform more involved in cholesterol regulation (99). LXRβ-specific knockout mice have less adipose, but normal insulin sensitivity and adipocyte hormones; however, they are glucose-intolerant and accumulate lipid in pancreatic islets, putatively mediated by regulation of cholesterol transporters (99). Adipocytes have been demonstrated to be smaller in LXR deficient mice (97), and energy expenditure is increased, with reduced triglyceride accumulation in brown adipose (100); in parallel, energy expenditure is reduced in LXR agonist-treated wild type mice, and triglyceride accumulation was increased in brown adipose (100). In humans, LXR expression is higher in obese individuals, and receptor isoform polymorphisms have been associated with increased risks of obesity (101). Therapeutic treatment with LXR agonists resulted in increased plasma and hepatic triglycerides, cholesterol, and other negative metabolic markers in humans as well as primate and rodent models (102), despite some beneficial effects.
CAR and PXR are two closely-related liver-enriched receptors that have also been associated with metabolic function, and were reviewed in detail previously (103, 104). While originally appreciated as regulating xenobiotic metabolizing enzymes, they have also been demonstrated to help regulate energy homeostasis, immune function, lipid metabolism, and glucose homeostasis (103, 104). PXR appears to mediate effects through PPARγ, with PXR activation directly inducing PPARγ and other lipogenic gene expression such as Cd36, though potentially in a species-specific manner (104). CAR may promote effects on energy homeostasis through crosstalk with PPARα, or similarly to PXR, through activation of the free fatty acid uptake transporter Cd36 and inhibition of sterol regulatory element-binding protein (SREBP) (105). In animals, PXR ablation inhibits diet-induced obesity, insulin resistance, and fatty liver disease in various rodent models, suggesting PXR antagonism as a putative anti-obesogenic and anti-diabetic pathway (104, 106). PXR agonist treatment in mice promotes hepatic triglyceride accumulation, and constitutively active PXR mice exhibit enlarged and fatty liver disease, reviewed in (107). Treatment with CAR agonists, in contrast, enhances insulin sensitivity, improves glucose and lipid metabolism, and reverses diet-induced obesity in rodents, reviewed in (103). In humans, the CAR agonist phenobarbital has been reported to decrease plasma glucose levels and improve insulin sensitivity in patients with diabetes (103, 108, 109), and though PXR is particularly promiscuous, activation of PXR by rifampicin, statins, and other pharmaceuticals have been reported to induce hyperglycemia in patients and increase the risk of developing diabetes (106). While activation of CAR is seemingly more therapeutically beneficial relative to PXR, it also carries with it side effects such as liver hyperplasia and carcinogenesis (103), among other effects.
Modulation of FXR has also been assessed as it relates to adipogenesis and a potential therapeutic target in treating metabolic syndrome, reviewed in (110). Endogenously activated by bile acids, FXR regulates bile acid synthesis, enterohepatic circulation, lipid metabolism, and thus indirectly regulates other bile acid associated receptors, discussed in Prawitt et al. (111). Researchers have described that FXR is expressed in adipocytes from adult mice and in differentiated 3T3-L1 cells, but not in the undifferentiated pre-adipocytes (112). Treatment with an FXR agonist increased adipocyte differentiation in 3T3-L1 cells, whereas treatment with an FXR antagonist reversed this (112); FXR agonist treatment also enhanced insulin signaling and insulin-stimulated glucose uptake (113). Pro-apoptotic and anti-adipogenic effects of guggelsterone (FXR antagonist) have also been reported by other researchers (114). Treatment with an FXR agonist in mice with diet-induced obesity worsened weight gain and glucose intolerance, seemingly mediated through reduction of the bile acid pool size and energy expenditure (115). However, other research in mouse models suggests beneficial effects for FXR agonist (GW4064) treatment (111, 116). FXR knockout/deficient mice exhibit decreased adipose tissue, lower leptin concentrations, elevated plasma free fatty acids, resistance to rosiglitazone-induced obesity, and their embryonic fibroblasts are also resistant to rosiglitazone-induced triglyceride accumulation and differentiation due to increased lipolysis and decreased lipogenesis (113, 117, 118); despite these apparent positive metabolic effects, FXR deficient animals (both mice and rabbits) also exhibit impaired glucose tolerance and insulin resistance, which are corrected with FXR agonist supplementation (113, 119). FXR expression has also been demonstrated to be downregulated and/or dysfunctional in obese humans (110, 120), suggesting downregulation may play a potential role in human obesity. PXR mice exhibit FXR agonist therapeutic trials in humans have reported reduced liver lipid accumulation and increased glucose uptake [reviewed in (110)], reduced HDL cholesterol and increased LDL cholesterol, and improvements in insulin resistance [reviewed in (111)], suggesting that FXR antagonists and/or selective FXR receptor modulators might promote more beneficial effects in some tissues and for specific metabolic endpoints (116).
Thyroid Receptor (TR)
TR also forms a heterodimer with RXR, though in contrast to other receptors discussed above, it is considered a non-permissive heterodimer (can be activated only by thyroid receptor ligands and not RXR ligands), reviewed in Shulman et al. (94). While less frequently assessed as a contributory molecular pathway for adipogenesis, one of the defining characteristics of thyroid hormone action is maintenance of metabolic health and maintenance of lipid and carbohydrate metabolism, blood pressure, and body mass [reviewed in (94, 121)]. Hypothyroidism (low thyroxine (T4) and triiodothyronine (T3), high thyroid stimulating hormone (TSH)) is characterized by weight gain, while hyperthyroidism (high T4 and T3, low TSH) is characterized by weight loss (122, 123). As such, thyroid hormones are generally considered anti-obesogenic, and hypothyroid-associated adiposity can be reduced with supplementation (124–126). TRα primarily regulates thermogenesis and TRβ primarily regulates cholesterol metabolism and lipogenesis, as well as a number of genes and enzymes necessary for pre-adipocyte proliferation and adipocyte differentiation, either directly or via PPARγ [reviewed in (121)]. Studies have demonstrated some disparate findings regarding the role of TR in adipogenesis itself. For example, antagonism of TR has been demonstrated to efficaciously modulate adipocyte differentiation, purportedly via PPARγ, reviewed in (16); however, we've previously demonstrated that 3T3-L1 treatment with 1–850 (TR antagonist) resulted in efficacious triglyceride accumulation (60), and TR-null mice exhibit increased adipogenesis (127). Others, in contrast, have reported that treatment with triiodothyronine (T3; TR agonist) promoted adipocyte gene expression and decreased pre-adipocyte proliferation in Ob L771 mouse pre-adipocytes (128) or triglyceride accumulation and lipogenic gene expression in 3T3-L1 pre-adipocytes (60, 129). Other work suggested differing roles at varying levels of treatment; when 3T3-F442A cells were treated with hyperthyroid T3 levels, the proportion of adipocytes was increased but expression of lipogenic enzymes and triglyceride accumulation were decreased, whereas lower levels stimulated adipose conversion, expression of lipogenic enzymes, and pre-adipocyte proliferation (130).
Glucocorticoid Receptor (GR)
The GR is intimately connected to lipid metabolism, with a wealth of in vitro, in vivo, and human epidemiological evidence supporting its role in adipose formation and maintenance [reviewed in (131)]. Treatment with dexamethasone induces a potent and efficacious triglyceride accumulation and pre-adipocyte proliferation response in various mesenchymal and pre-adipocyte models, often to greater extents and at lower concentrations than through direct activation of PPARγ (60, 132), potentially mediated at least in part through activation of PPARγ (133), though in other cases without meaningful activation of PPARγ (134); in support, treatment with GR antagonists inhibits differentiation in various mesenchymal and pre-adipocyte models (135). Other studies have reported that glucocorticoids alone were insufficient to promote adipogenesis either in 3T3-L1 cells (136) or in other models (137), though stimulated robust differentiation in combination with insulin (137). As mentioned above, Chappell et al. demonstrated putative GR-mediated effects prior to PPARγ activation after exposure to tetrabrominated bisphenol A (TBBPA) (61), which may explain why isobutylmethylxanthine (IBMX; PPARγ ligand) treatment prior to dexamethasone (GR agonist) failed to induce significant differentiation using the same cell model in another lab, while dexamethasone treatment before IBMX promoted robust differentiation (138). The authors posited that glucocorticoid activation may be necessary for an intermediate commitment state prior to differentiation via PPARγ (138); however, this could also be due to differing responsiveness to PPARγ and GR ligands based on 3T3-L1 cell source, which we have reported on previously (60).
Other research has evaluated the putative role of the mineralocorticoid receptor (MR), an additional high-affinity binder of glucocorticoids; treatment of 3T3-F442A and 3T3-L1 cells with the mineralocorticoid agonist aldosterone promoted adipocyte differentiation, which appeared to be mediated through PPARγ activation; inhibition and knock-down of the MR inhibited adipogenesis, whereas knock-down of the GR did not (139). More recent work, however, demonstrated that silencing GR, but not MR, inhibited the pro-adipogenic activity of cortisol, and also decreased leptin and adiponectin, whereas MR knock-down actually increased leptin (140). Research in mice investigated knocking out local glucocorticoid action via 11β-hydroxysteroid dehydrogenase (glucocorticoid inactivator) overexpression exhibited resistance to diet-induced obesity/reduced fat accumulation, decreased food intake, improved insulin sensitivity and glucose tolerance, and increased energy expenditure (141). Glucocorticoid excess in mice in contrast resulted in decreased osteogenic gene expression and mineralization and increased expression of adipogenic genes (142). Cushing's syndrome (excess cortisol production) is associated with increased weight gain, hypertension, type 2 diabetes, and fatty tissue deposits (143, 144), suggesting a pro-adipogenic effect of glucocorticoids in humans as well. Further, prenatal/antenatal dexamethasone (GR agonist) is often utilized to promote development of lungs in infants at risk of being born premature (145, 146). Epidemiological studies have reported that dexamethasone treatment is associated with reduced birth weight in infants, even after correcting for weeks of gestation (145, 146), and exhibited hypertension and greater subsequent administration of insulin for hyperglycemia (146).
Estrogens and Androgens
Often considered opposing sex steroids, androgens, and estrogens have also been described to have opposing effects on adipogenesis, reviewed in Cooke and Naaz (147). Experiments comparing differentiation extent in rat pre-adipocytes determined no effects for either androgens or estrogens in promoting differentiation in male pre-adipocytes; however, estrogens elicited a pro-adipogenic effect (via pre-adipocyte proliferation) and androgens elicited an anti-androgenic effect in female cells, potentially mediated by modulation of insulin growth factor 1 receptor (IGF1R) and PPARγ expression (148). This promotion of pre-adipocyte proliferation by estrogens has been successfully replicated in both male and female omental pre-adipocytes (149), while the inhibitory effect of estrogens on differentiation/triglyceride accumulation may be dose-dependent (150). Related work has determined some of the inhibitory effects of estrogens on adipogenesis appear to occur through the G-protein-coupled estrogen receptor 1 (GPER) rather than the classical estrogen receptor itself (151), and that inhibitory effects on adipogenesis are concurrent with enhancement of osteogenesis (152). Interestingly, estrogen receptor knock-out (ERKO) mice exhibit increased fat pad weights, adipocyte size, and adipocyte numbers relative to wild type control animals, as well as insulin resistance and impaired glucose tolerance (153). This is mirrored in humans, as decreased estrogen levels at menopause are associated with increased abdominal obesity that is ameliorated with estrogen replacement therapy [reviewed in (154)], an effect also observed in ovariectomized female mice (155).
Androgens are generally considered anti-obesogenic [reviewed in (156, 157)], and treatment with androgens has been demonstrated to inhibit adipogenesis in adipose tissue samples from both sexes (158) and reduce fat mass in humans [reviewed in (159)]. Dihydrotestosterone inhibits triglyceride accumulation and adipocyte gene expression in human mesenchymal stem cells and pre-adipocytes from various depots, whereas anti-androgen co-treatment attenuated those effects, and had no apparent impact on pre-adipocyte proliferation in either model (160). Other research has replicated these findings, suggesting some of the effects occur through inhibition of the multipotent stem cell to pre-adipocyte commitment (161, 162). In contrast, anti-androgens have been suggested to act as obesogens; androgen receptor knock out (ARKO) mice exhibit increased obesity (163), flutamide has been demonstrated to modulate lipid profiles in women (164), and hypogonadism (characterized by testosterone deficiency) is associated with obesity, hypertension, dyslipidemia, insulin resistance, and other metabolic effects, which may be corrected with androgen supplementation (159).
Other Receptors
A variety of other receptors, from the nuclear receptor family, receptor tyrosine kinase family, and others, have described roles in adipogenesis and/or lipogenesis. For example, both the insulin and IGF-1 receptors have widely accepted roles in growth, tissue-specific hypertrophy, and weight maintenance (165–168). Many others, including the aryl hydrocarbon, retinoic acid, low density lipoprotein receptors, among others, have established roles in adipogenesis but could not be discussed in detail within the scope of this review. Importantly, while the bulk of study has assessed activation of PPARγ and RXR, numerous other receptor systems interplay to promote and maintain adipocytes, and must be taken into account when evaluating environmental mixtures.
Mitochondrial Toxicity as a Contributory Factor to Metabolic Disruption
Mitochondria are the major location of fatty acid oxidation, making them essential in lipid metabolism; as such, dysfunction can contribute to numerous adverse metabolic health consequences, including altered lipid accumulation, metabolism, and insulin resistance (169, 170). Mitochondrial function is intimately connected with metabolic health, as it helps regulate energy expenditure, production of ATP, and removal of reactive oxygen species (ROS); ROS reduce oxygen consumption and inhibit fatty acid oxidation in adipocytes, promoting lipid accumulation [reviewed in (169)]. ROS production mainly occurs at complex I and III in mitochondria, and is increased when excess electrons are provided to the mitochondrial respiratory chains (when proton gradient is high and ATP demand is low), as described in Kim et al. (171). Excess electrons are transferred to oxygen, converted to superoxide, and subsequently to hydrogen peroxide; this ROS acts to damage proteins, DNA, and lipids, and activates pathways (via activation of serine kinases) that phosphorylate insulin receptor substrate proteins and inhibit insulin signaling, thus promoting insulin resistance and ultimately resulting in metabolic dysfunction (171, 172). Mitochondrial dysfunction and resultant lipid accumulation in accessory tissues is also capable of further impeding insulin signaling and glucose metabolism, promoting further dysfunction (173); indeed, maternal obesity during pregnancy in rodents contributes to a transgenerational mitochondrial dysfunction phenotype (inhibited insulin signaling for three generations) (174). Notably, chronic oxidative stress has been well-described in obese individuals, suggesting a link between ROS production/management and hyperplasia [reviewed in (175)]. To minimize damage from these ROS, cells require a balance between ATP synthesis through oxidative phosphorylation and dissipation of the proton gradient (169). Mitochondrial dysfunction can also directly contribute to cardiovascular disease, another hallmark disease of metabolic syndrome, and myocardial metabolic function is intimately connected to obesity, diabetes, and altered insulin signaling [reviewed in (176)]. Research suggests that decreases in ATP production due to inhibited mitochondrial respiration, increased oxidative stress, and inhibited calcium signaling can all contribute to diastolic dysfunction via reduced velocity of myocardial relaxation velocity and myocardial compliance (173, 176).
Adipocytes are capable of regulating metabolic insults by altering their number, morphology, as well as the intracellular mitochondrial distribution (169). Mitochondrial biogenesis is an essential component of adipogenesis, with mitochondrial numbers increasing markedly after initiation of pre-adipocyte differentiation and reaching a maximum toward the end; this can be noted via treatment with the PPARγ agonist rosiglitazone, wherein treated cells demonstrate increased mitochondrial content and function, with increased basal oxygen consumption, ATP respiration, and proton leak (173, 177, 178). ATP levels are naturally reduced with increasing degree of adipocyte differentiation, putatively due to increased ATP demands for lipogenesis (179), and reduced levels are further exacerbated when electron transport chain inhibition occurs (178). Reduced mitochondrial biogenesis, ATP levels, and dysfunctional mitochondrial electron transport have been reported in both humans and animals with metabolic syndrome (173, 179). PPARγ co-activator 1α (PGC-1α) is a master regulator of mitochondrial biogenesis and gene expression and is a potent co-activator of PPAR isoforms: expression in fat or muscle cells increases mtDNA content, expression of mitochondrial genes, and mitochondrial respiration (176). PGC-1α-stimulated biogenesis in the heart ultimately promotes overt heart failure, another mechanism through which metabolic dysfunction can lead to cardiac dysfunction (176). Biogenesis appears closely linked to adipocyte differentiation, as up-regulation of mitochondrial biogenesis is well-reported following induction of adipogenesis and for up to 10 days post-differentiation (177, 180), suggesting that mitochondria are needed to supply the substrates and factors necessary to support adipogenesis-driven lipogenesis. Mitochondria also have functionally distinct roles in white vs. brown adipose tissue. White adipose tissue is composed of numerous depots of large lipid droplet adipocytes throughout the body and is essential for maintenance of metabolic health. Brown adipose tissue, in contrast, is smaller and localized to the neck and upper-chest in adult humans, and is composed of adipocytes with large numbers of smaller lipid droplets and more numerous mitochondria [reviewed in (180)]. In brown adipose tissue, the heat derived from thermogenesis is produced primarily by the high mitochondrial content of these cells, via oxidation of fatty acids and other components (180). Uncoupling proteins (UCPs) play a key role in this process, serving to uncouple mitochondrial respiration from ATP generation by inducing a proton leak, which subsequently allows for energy dissipation as heat (180).
Numerous environmental toxicants have been demonstrated to promote mitochondrial dysfunction, and these contaminants may also advance metabolic dysfunction, leading to obesity, and diabetes. Certain mitochondrial disorders that are characterized by impaired oxidative phosphorylation are also associated with disrupted lipid homeostasis: myoclonic epilepsy with ragged red fibers is associated with triglyceride accumulation in muscles and multiple symmetrical lipomatosis, a condition characterized by abnormally small white adipocytes containing numerous small lipid droplets rather than the classical large central droplet that displaces the nucleus (170). Mitochondrial oxidative phosphorylation inhibitors and protein synthesis inhibitors impair mitochondrial respiration and promote triglyceride accumulation in 3T3-L1 cells, which retain their precursor fibroblastic morphology and do not express adipocyte-specific markers (170, 180). Previous research has demonstrated that treatment of 3T3-L1 cells with rotenone (a complex I inhibitor), antimycin A, stigmatellin, and myxothiazol (complex III inhibitors), and oligomycin (ATP synthase inhibitor) promoted triglyceride accumulation in a dose-dependent manner (170). Interestingly, these mitochondrial respiration inhibitors promoted triglyceride accumulation in numerous small lipid droplets, cells retained their fibroblastic morphology, and classical adipocyte-specific genes were not expressed in these cells (170), suggesting a differentiation-independent mechanism of triglyceride accumulation. Specifically, antimycin A, which inhibits complex III, induces triglyceride accumulation in pre-adipocytes via a putative differentiation-independent mechanism (170); these cells exhibit multi-vesicular lipid accumulation, reduced expression of standard differentiation markers (FABP4, C/EBP), and suppression of PPARγ and RXR, supporting other studies suggesting mitochondrial dysfunction may inhibit adipocyte differentiation (178).
We recently demonstrated a similar phenotype in experiments with pyraclostrobin, a strobilurin-class fungicide used on strawberries, spinach, and other produce items, with production of >2 million pounds per year (181, 182). Pyraclostrobin and other strobilurin fungicides have been demonstrated to inhibit complex III (183), suggesting a potential mechanism for metabolic disruption. We previously reported that pyraclostrobin, azoxystrobin, fluoxastrobin, and trifloxystrobin all induced both triglyceride accumulation and pre-adipocyte proliferation in 3T3-L1 cells (184). Previous research in 3T3-L1 cells and a human adipose-derived stem cell model suggested this did not occur through activation of PPARγ and that standard differentiation markers were lacking (47, 185), supporting the case for a differentiation-independent mechanism. Mechanism was further interrogated in our laboratory through co-exposure experiments in 3T3-L1 cells; we reported that PPARγ antagonists did not protect against pyraclostrobin-mediated triglyceride accumulation (177). Instead, pyraclostrobin promoted mitochondrial dysfunction, including reduced ATP, mitochondrial membrane potential, basal mitochondrial respiration, ATP-linked respiration, and spare respiratory capacity (177). In addition, pyraclostrobin-treated cells exhibited reduced expression of genes regulating glucose transport, glycolysis, fatty acid oxidation, and lipogenesis (177). Lastly, co-treatment with a cAMP responsive element binding protein (CREB) inhibitor reduced pyraclostrobin-mediated triglyceride accumulation (177). These results all suggest that toxicants capable of disrupting mitochondrial function may also have the potential to affect metabolic health, via modulation of lipogenesis and other metabolic processes.
Similarly, a recent study reported that several samples of oil sands process-affected water (OSPW), wastewater produced during the extraction of bitumen from oil sands, exhibited PPARγ agonist activity and promoted triglyceride accumulation in 3T3-L1 cells (186). Causative ligand characterization identified several hydroxylated/polyoxygenated carboxylic acids and hydroxylated sulfates as the major PPARγ ligands (186); naphthenic acids, a mixture of carboxylic acids and natural component of petroleum, are a major component of OSPW. Interestingly, while these are posited as promoting adipogenesis via PPARγ activation, a recent publication demonstrated that naphthenic acids isolated from oil sands water acted to uncouple oxidative phosphorylation, inhibit respiration, and increase the production of ROS (187). As noted above, these are mechanisms that can promote triglyceride accumulation in cells, suggesting that this may be an additional mechanism for the observed adipogenic effects of these waters in the previous publication (186). Notably, this occurred at environmentally-relevant concentrations for OSPW, suggesting that this or a combination of these mechanisms may promote the environmental sample-induced adipogenicity.
Mitochondrial ROS is produced by pre-adipocytes during and throughout differentiation, and its presence activates several early-stage differentiation markers, including C/EBP, PPAR, and CREB (63, 188). Direct impacts on adipogenesis appear less certain, which has been delineated in greater detail previously (189); research suggests that ROS may be essential for adipogenesis, but also may perturb the process. Some research has demonstrated that ROS promoted mitotic clonal expansion in 3T3-L1 cells (190), a necessary step prior to induction of differentiation. Other research has described inhibitory effects on differentiation, with ROS inhibiting both pre-adipocyte proliferation and adipocyte differentiation/triglyceride accumulation (180, 191, 192). Still other researchers, via co-treatment experiments utilizing antioxidants, demonstrated that ROS impacted differentiation but not pre-adipocyte proliferation: treatment with an antioxidant (reducing ROS) reduced lipid accumulation in mesenchymal stem cells (193). However, the varying cell models used for these experiments may mediate these apparent differences; all studies agree that ROS appear to modulate early-stage differentiation, though the mechanisms of this modulation appear to vary based on cell lines, sources, and experimental details.
Several mitochondrial toxicants have been demonstrated to promote insulin resistance and/or metabolic syndrome in epidemiological studies, reviewed in detail previously (172, 194), though this research area has as of yet received limited attention in the context of metabolic disruption. Several organochlorine pesticides have been implicated in metabolic effects via mitochondrial dysfunction. Specifically, atrazine has been demonstrated to directly inhibit complexes I and III, reducing oxygen consumption and leading to accumulation of superoxides; chronic exposure in rats has been demonstrated to decrease basal metabolic rate, and increase body weight, intra-abdominal fat, and promote insulin resistance independent of food intake or activity levels (172, 195). Much of the remaining literature has focused on the role of polychlorinated biphenyls (PCBs). Several congeners have been demonstrated to promote mitochondrial dysfunction in vitro (196, 197), exposure resulted in/exacerbated obesity, insulin resistance, and hyperinsulinemia in mice (198), and higher exposure to PCBs has been linked to increased risk of obesity, dyslipidemia, and/or insulin resistance in a number of epidemiological studies (199–202).
Available in vitro Models of Adipogenesis and Metabolic Disruption
Numerous in vitro models have been developed and utilized for the purpose of identifying potential metabolic disrupting chemicals, reviewed in detail previously (203, 204). Generally, these models can be described as assessing two key parameters of adipocyte development: commitment to the adipocyte lineage from multipotent precursor cells (generally through the use of mesenchymal stem cell (MSC) models) and differentiation into mature adipocytes (generally through the use of pre-adipocyte models). MSC models have the additional benefit of being capable of assessing both endpoints, though are seemingly less frequently utilized than the available pre-adipocyte models. In addition, several research groups have begun to report on the three-dimensional culture of pre-adipocytes, which may shed additional light on mechanisms in a more physiologically relevant system. All of these assays are lengthy and their relative abilities to correctly identify chemicals may depend on both cell line and cell source. As such, there is a critical need to develop better methods for correctly predicting metabolic disruptors. While murine models have historically been used preferentially, a growing number of species utilized and a growing movement toward utilization of human models may help expand our understanding of translational mechanisms and potential environmental contaminant impacts on human health.
Perhaps the best known pre-adipocyte model is the 3T3-L1 mouse cell line. First described in the 1970's, it has proven reliable as an in vitro screen over several decades for identifying likely obesogenic chemicals in vivo (205, 206). These cells are already committed to the adipocyte lineage and cannot develop into other cell types; however, they generally require activation of particular signaling pathways to promote further development. Following exposure to adipogenic chemicals, these cells differentiate into adipocytes, accumulate triglycerides, and come to resemble a mature human white fat cell (44, 46, 205, 206). While 3T3-L1 cells have seemingly come to be considered the de facto model of adipogenesis, some inherent concerns remain about their utility. As we have described recently (60), while this line has been well-characterized (207), it is somewhat unreliable in sourcing. For example, while we know much about the molecular mechanisms underpinning the development of mature adipocytes based on this cell line, nuclear receptor expression related to adipogenesis is markedly different between different lots and sources of this cell line (60). Moreover, on investigation into this apparent discord in source, we discovered that the American Type Culture Collection (ATCC) maintains five distinct lots of 3T3-L1 cells, which all seemingly have differing degrees of differentiation success. This issue with cell line integrity was highlighted in a recent paper (208), suggesting that these differences can contribute to real discrepancies in the ability to replicate findings across laboratories. As the current ATCC cells are meaningfully different in the expression of key adipogenic pathways from the Zenbio-sourced cells (which are sourced from the isolating laboratory), it is unclear whether our understanding of the mechanisms underlying adipogenesis are from the original cells, the ATCC cells, or where these research paths diverge. Care needs to be taken to assess reproducibility across stocks and between laboratories and carefully untangle where the research underlying this cell line belongs. Other pre-adipocyte models also exist, including the OP9 mouse bone marrow-derived stromal pre-adipocyte cell line (44, 45), a line that allows for considerably faster differentiation, though which we have demonstrated to exhibit different nuclear receptor expression and differing degrees of responsiveness to adipogenic chemicals (60). These varying pre-adipocyte models allow for assessments of varying molecular pathways important for the process of differentiation via both source of the cells and species [discussed further in (204)].
Various multipotent mesenchymal cells and cell lines (46, 47) offer the additional ability to assess commitment to the adipocyte lineage as a distinct process from adipocyte differentiation (48). MSC use and applicability in adipogenesis research has been reviewed in detail previously (209). The variability of these cell lines are reportedly lower than the pre-adipocyte models, they are purportedly easier to isolate and culture, and they have additional utility in that they can be utilized to assess both differentiation of adipocytes but also commitment to the adipocyte lineage vs. other cell lineages. For example, many researchers have utilized these cell lines to evaluate the interplay between commitment to the osteogenic vs. adipogenic lineages following exposure to specific environmental contaminants (210–213). Recent work elegantly described a novel protocol for evaluating both adipogenic lineage commitment and subsequent differentiation as distinct processes in primary MSCs (48), which has been described previously for the C3H10T1/2 stem cell model (214, 215). These advancements raise the utility of this model and warrants further investigation into replicability, reproducibility of this model across laboratories, and comparisons of translation to human health relative to the pre-adipocyte models currently utilized.
Lastly, a number of research labs have begun to describe spheroid cell cultures of adipocyte models (216–220), which may carry some inherent benefits over the standard adherent monolayer cultures. These studies have suggested that spheroid culture improves the efficiency, extent, and/or speed of differentiation (216–221), retains the multipotent potential of these cells (217, 222), and transcriptomic analyses have suggested a potentially more representative model of adipocyte gene expression relative to known in vivo mechanisms (216). These models may allow for a more comprehensive understanding of adipose physiology than was possible via interrogation of the monolayer cell cultures, and should be evaluated further for replicability and translation potential relative to the standard monolayer cultures.
Metabolic Disruption Potential of Environmental Mixtures
As noted above, the assessments of environmental samples have proven an interesting new approach to evaluating potential mixture toxicity. With tens of thousands of chemicals in use and new chemicals regularly added, there are too many to characterize individually, and certainly no capabilities to assess all potential combinations of them (223, 224). Body burden studies have and continue to report human exposure to hundreds of chemicals on a regular basis (225, 226), demonstrating the problem of realistic mixture exposure studies. To add to the complexity, research has reported additive effects on several hormone receptors both in vitro and in vivo (227–231), demonstrating that mixtures can induce effects at levels below those induced by individual chemicals. From a toxicological perspective, evaluating whole environmental samples: wastewater, surface/groundwater, indoor house dust, air samples, etc. for biological activities has emerged as a promising tact to assess potential adverse health concerns from exposure to actual mixtures present in the environment, given that it can evaluate more realistic environmentally relevant exposures (Figure 3).
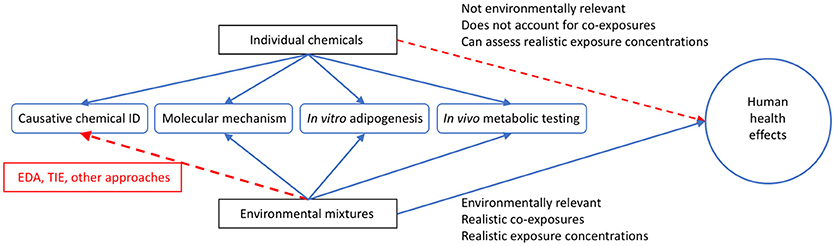
Figure 3. Utility of Utilizing Environmental Mixtures for Human Health Assessments. Graphical depiction comparing individual chemicals and environmental mixtures for the assessment of potential human health effects. While environmental mixtures have less use in identifying causative chemicals in all cases (though tools like effect directed analysis (EDA) and toxicity identification evaluation (TIE) can be used to elucidate this), these mixtures are more relevant in reflecting the suite of chemicals that people are exposed to on a daily basis than utilizing single chemicals alone, and more often reflect actual environmental exposure concentrations. In this figure, the blue lines indicate positive relationships and the red lines indicate difficulty for single chemicals or mixtures in assessing the related outcome.
Numerous natural and exogenous contaminants can contribute to human exposure; as such, measuring the total receptor bioactivities has proven useful for assessing the total magnitude of potential effects (232–239). While analytical chemistry techniques and equipment have drastically improved, allowing for more precise measurements of contaminants at lower concentrations, recent research has suggested we lack complete information on all causative bioactive chemicals present in the environment (240, 241). While non-targeted analytical efforts have improved, we still lack sufficient software and comprehensive protocols to enable robust and reproducible non-targeted assessments of contaminants across laboratories. To address this need of addressing mixture toxicity without necessarily understanding the full chemical complexity, bioassays have been utilized to assess biological activities of actual environmental samples. Reporter gene assays are one such commonly-utilized tool, assessing total receptor activities (agonism and antagonism), and valued due to their low cost, ease of use, reliability, high sensitivity, and ease of adapting for multiple receptors (227–244). These assays provide the capability to assess the total receptor activity of potentially numerous low-concentration EDCs (without identifying each causative chemical) rather than assessing each constituent chemical individually.
Applying this method to human epidemiological research has shown great potential; a number of researchers have rigorously characterized how in vivo mixtures of contaminants correspond with total hormone receptor bioactivities of human and animal matrices (serum, tissues, etc.) (245–251). Moreover, some researchers have begun to utilize bioactivities directly to assess human health outcomes. For example, researchers have correlated the total placental estrogenic activity with increased reproductive malformations (252) and impaired motor development (253), total adipose estrogenic activity with increased risk for breast cancer (254), and placental estrogenic activity with increased birth weight in boys (255). Other research has failed to report significant associations, including a lack of any association between adipose estrogenic activity and risk for type-2 diabetes (256), potentially due to a greater role for other receptors in pathogenesis (257). These studies demonstrate the potential utility of this method, particularly when targeted based on a comprehensive understanding of etiology and molecular mechanisms.
Several studies have begun to apply these techniques to metabolic endpoints, assessing pertinent receptor bioactivities (GR, PPARγ, and others) as well as utilizing less high-throughput adipogenesis or other assays for predicting in vivo metabolic disruption potentials. Some of these environmental case studies are discussed in greater detail below:
Metabolic Disruption Potential of Indoor House Dust
As noted above, numerous studies have documented the detection of EDCs from diverse chemical classes in indoor house dust samples from a variety of sources. A number of studies have assessed the bioactivities for solvent-extracted house dust, reporting PPARγ, GR, and ER agonist activities as well as AR and TR antagonist activities, at concentrations ≥ 15 μg dust equivalence per mL (DEQ/mL, mass of extracted dust per volume of assay medium) (258–260). Our laboratory also assessed the modulation of PPARγ by house dust extracts, reporting that 21 of 24 examined indoor house dust extracts exhibited significant PPARγ binding at 3 mg DEQ/mL (120 μg dust per assay well) using a relative binding affinity assay (261) and 15 of 25 extracts activated PPARγ at ≤ 50% of the maximal positive control response at concentrations ≥100 μg DEQ/mL (4 μg/well) using a commercially-available reporter assay (262, 263). This work demonstrated activation of pathways known to regulate adipogenesis at very low concentrations, and subsequently informed our follow-up studies examining higher-order effects on adipogenesis.
We recently evaluated >40 common SVOCs that are routinely detected in indoor house dust samples for adipogenic activity in the 3T3-L1 murine pre-adipocyte cell model. We found that > two-thirds of these chemicals independently induced significant triglyceride accumulation and/or pre-adipocyte proliferation (184). Specifically, pyraclostrobin (strobilurin fungicide), dibutyl phthalate (DBP), tert-butyl-phenyl diphenyl phosphate (TBPDP), and the isopropylated triaryl phosphates (ITPs, mixture of isomers) exhibited near or supra-maximal triglyceride accumulation relative to the rosiglitazone (positive control)-induced maximum (184). We further assessed eleven house dust extracts collected from central North Carolina (NC), USA households; we found that ten of these 11 extracts exhibited significant triglyceride accumulation and/or pre-adipocyte proliferation at < 20 μg of dust/well (184). This activity occurred at orders of magnitude lower concentrations than those the EPA estimates children to consume each day. As such, this raises concerns for potential impacts on in vivo metabolic health.
A recent follow-up to this study evaluated the adipogenic activity of 137 house dust extracts from central NC households and attempted to determine putative causative chemicals, molecular mechanisms, and potential impacts on human metabolic health (264). We reported that 90% of the dust extracts exhibited significant adipogenic activity, < 60% via significant triglyceride accumulation, and >70% of samples via significant pre-adipocyte proliferation, with >40% of effects occurring at < 10 μg dust/well (264). Increasing dust-induced triglyceride accumulation was positively correlated with serum thyroid stimulating hormone levels in adult residents, and negatively correlated with serum free triiodothyronine (T3) and thyroxine (T4) (264). Interestingly, proliferation tended to be positively correlated with residents' body mass index (BMI; p < 0.10), potentially suggesting adipogenic chemicals present in the dust are associated with the weights of residents, but further research with larger sample sizes are needed to substantiate this. We further assessed TR antagonism as a potential contributory causative mechanism in these effects, and found that TRβ antagonism of these extracts (265) was positively correlated with triglyceride accumulation (264). Both T3 co-treatment and siRNA knock-down of TR inhibited the dust-induced triglyceride accumulation of these extracts, supporting the role of TR antagonism as a contributory molecular mechanism.
Metabolic Disruption Potential of Oil and Gas-Associated Wastewaters
Three separate sets of studies have assessed different aspects of oil and gas operations and metabolic disruption, reporting in vitro and/or in vivo evidence of metabolic disruption by oil and gas associated environmental mixtures. The first assessed three replicate samples of oil sands process-affected water (OSPW), wastewater produced during the extraction of bitumen from oil sands (186). They reported that an OSPW sample activated PPARγ at concentrations as low as 0.025x relative water concentration (40-fold dilution relative to pure water). This sample was further fractionated, with the majority of PPARγ activity in fractions two and five (five fractions), and fractions three through five exhibited significant triglyceride accumulation and induction of adipogenic genes (fatty acid binding protein and lipoprotein lipase). A pull-down assay and chemical analysis was further utilized to identify the causative ligands present in fraction five that were inducing the adipogenic effects; this analysis revealed hydroxylated/polyoxygenated carboxylic acids and hydroxylated sulfates as the major PPARγ ligands inducing adipogenesis in these samples (186), though the small sample size requires further substantiation.
Another set of studies assessed the metabolic disruption potential of crude oil singly or mixed with Corexit oil dispersant mixture (266, 267). To distinguish these mixtures, they utilized several simpler mixtures in culture media, including: Corexit 9500 + MC252 oil, varying dilutions of MC252 oil, and varying dilutions of Corexit with corn oil; they found that the Corexit + oil treatments stimulated PPARγ, while the MC252 oil alone did not, suggesting a component of Corexit promoting the observed effects (267). The Corexit + oil mixture was further fractionated to determine causative ligands, with Tween 80 and dioctyl sodium sulfosuccinate (DOSS) identified as highly abundant chemicals in the active fraction (267). DOSS was further demonstrated to be active in PPAR response element-luciferase transgenic mice and stimulate triglyceride accumulation and expression of fatty acid binding protein (Fabp4) in 3T3-L1 cells (267). Follow-up work assessed the Corexit + oil mixture and Corexit alone for activation of RXRα, finding dose-dependent activation, presumably mediated by Corexit constituents (266). Constituent chemicals were further evaluated, and DOSS, Span 80, and Tween 80 all demonstrated some degree of RXRα activity, with Span 80 also stimulating triglyceride accumulation and adipocyte gene expression in 3T3-L1 cells. Interestingly, a combination of DOSS and Span 80 resulted in putative synergistic effects on adipocyte differentiation, potentially due to diverging molecular mechanisms (Span 80 exhibited a much more efficacious response for RXRα than PPARγ, while DOSS exhibited no RXRα activity but did activate PPARγ) (266).
The last set of studies, from our laboratory, evaluated unconventional oil and gas associated wastewater and chemicals. Our work on this topic began with receptor activity testing for 24 common hydraulic fracturing chemicals, reporting that 21 and 7 chemicals antagonized AR and TR in two cell-based assays, and that mixtures of these chemicals appeared to act synergistically for TR and additively for AR (36, 268). We further documented AR and TR antagonist activities in surface, ground, and/or drinking water near UOG operations in several regions, including CO, WY, WV, and ND [(268), Kassotis et al., in preparation, (269–271)], and evaluated a mixture of 23 common UOG chemicals via a gestational exposure experiment in C57 mice, reported putative metabolic effects (offspring exhibited increased body weights, among other effects) (36, 37). We further interrogated this by evaluating the ability of this 23-mix, several UOG wastewater samples, and several UOG wastewater-impacted surface water samples to stimulate adipogenesis in 3T3-L1 cells and activate PPARγ in a reporter gene assay (35). We demonstrated that UOG wastewater samples exhibited significant triglyceride accumulation and/or pre-adipocyte proliferation at relative water concentrations as low as 0.001x, UOG-impacted surface water extracts at concentrations as low as 0.04x, and the 23-mix at 1 μM; these effects co-occurred with PPARγ activation for some samples but not others (35), suggesting differing mechanisms. Related work demonstrated highly efficacious triglyceride accumulation for various non-ionic alkylphenol and alcohol polyethoxylates in the absence of PPARγ activation and potentially mediated by TR antagonism (272). These compounds are reportedly found at high concentrations in UOG wastewater (273–275) and may be responsible for some of the observed non-PPARγ-mediated effects observed in the UOG samples.
Potential Utility of High-Throughput Databases to Predicting Metabolic Disruption
The costs and time investments associated with in vivo examination of putative metabolism disruptors are prohibitively high; as such, utilizing lower-order testing and screening is critical to target higher-order testing on chemicals most likely to be active. Application of numerous in vitro models for assessing putative “obesogens” or “metabolic disruptors” over the last several decades has revealed numerous contaminants capable of affecting metabolic health (18), with recent publications suggesting that these contaminants are likely common in indoor and outdoor environments (2, 184, 272). While these pre-adipocyte and mesenchymal stem cell models are useful in determining potential in vivo metabolic disruptors, they are also time and energy intensive and their relative abilities to correctly identify chemicals may depend on both cell line and source. Further, their mechanisms of assessing adipogenic commitment, adipocyte differentiation, adipocyte proliferation, and/or lipid accumulation may not capture the full spectrum of endpoints that compose metabolic dysfunction more broadly, particularly endpoints related to “diabetogens”. As such, there is a critical need to develop better methods for correctly predicting metabolic disruptors, and while more simplistic models such as activation of PPARγ are often applied, the vast suite of mechanisms influencing this process (discussed above) require a more holistic approach to integrating causative molecular mechanisms. Several high-throughput (HTP) screening programs now exist (Tox21, ToxCast) that report activity across numerous molecular mechanisms for thousands of chemicals, many that are known to be relevant to metabolic health. Harnessing these data sets to broadly assess high-scoring chemicals (across relevant molecular pathways for select endpoints of interest) for more targeted higher-order testing may provide a valuable tool for reducing time and research costs and achieving a more broad assessment of the tens of thousands of commercial chemicals for potential contribution to adverse health outcomes in humans and/or animals.
This issue of utilizing HTP data in predictive models is not new and has been applied by a number of researchers to various in vivo endpoints, with varying degrees of success (276). Most of these methods have utilized ToxCast Phase I data, due to the more recent release (October 2015) of Phase II results, and as a result, some of the inherent issues reported by these studies have since been addressed. For example, Schwarzman et al. attempted to build a model to predict breast carcinogens, though had insufficient data on particular endpoints critical to altered mammary development (277). Many of the pathways missing, including prolactin, progesterone, and estrogen receptor beta effects, among others, are now pathways with associated assays in the Phase II database. Russell et al. applied a broad approach to predicting 60 in vivo endpoints, 56 of which were predicted at < 55% accuracy (278), though notably did not aggregate assays to predict in vivo endpoints. Given that health outcomes are nearly always driven by overlapping molecular pathways, this is not altogether surprising. Other researchers utilized assay aggregation and were more successful in building predictive models that performed with promising accuracy (>70%). Martin et al. utilized a suite of ToxCast assays to develop a predictive model for rat reproductive toxicity, achieving ~75% accuracies for training and test sets (279). Notably, this model incorrectly predicted five of 21 external validation chemicals as predicted negatives, all of which reduced early offspring survival with limited accompanying effects on reproductive performance or reproductive tract development, suggesting a gap in assays targeting these endpoints. Another model applied ToxCast data to rat prenatal developmental toxicity, with >70% accuracy with species-specific models (280), and found that if they further refined this to more specific developmental outcomes, they got even better predictive success (80–90%). Liu et al. utilized both Phase I and Phase II data to predict hepatotoxicity (hypertrophy, injury, and proliferative lesions), and reported 53–61% accuracy using only Phase I data, but >80% when utilizing the expanded Phase II data (281).
Recently, Auerbach et al. presented predictive models of putative obesogenic and/or diabetogenic chemicals through analyzing ToxCast HTP results (282). The researchers, utilizing experts in a diversity of metabolic health disciplines, selected known molecular pathways that had been previously demonstrated to modulate metabolic health, and combined them into a combined score metric for predicting likely vs. less-likely metabolic disrupting chemicals. Janesick et al. recently tested a portion of this method, utilizing a suite of assays deemed relevant for adipocyte differentiation (16 assays across 8 molecular mechanisms) to assess 24 chemicals (11 with highest activation scores across the selected assays, 6 with medium activation scores, and 7 presumed negative controls with low activation scores) for activation of RXRα, PPARγ, and triglyceride accumulation in 3T3-L1 cells (47). They reported that 7 of 17 high and medium-scoring and 2 of 7 low-scoring chemicals were active in 3T3-L1 cells, suggesting poor predictivity (high rates of both false positives and false negatives). The authors suggested several potential hypotheses for the poor performance, including: poor performance of PPARγ assays, incorrect selection of assays for the predictive model, and improper weighting of endpoints (rather than based on mechanism importance) and assays within each endpoint (rather than based on assay performance).
We recently undertook an effort to improve the predictive utility of this model by expanding the pathways and attempting to incorporate some of the suggestions made by Janesick et al. (47). Among these, we expanded the outcome by performing a targeted literature search on all chemicals and any evidence of effects on metabolic health. This model performed best when used as a gross metabolic disruption prediction model, using literature searches to identify any in vitro or in vivo evidence of adipogenesis or disrupted metabolic health (weight gain, adipose development, insulin/glucose signaling, effects on appetite/satiety, etc.). When applied to a novel set of chemicals for which we had assessed adipogenic activities in 3T3-L1 cells (60, 184), the original prediction model performed well at predicting gross metabolic disruption; we observed low rates of both false negatives (7.9%) and false positives (7.9%), and an apparent accuracy of 84% (283).
We also attempted to bolster this model through inclusion of additional pathways known to modulate metabolic health, in hopes of reducing false negatives, though discovered that expanding the model to incorporate all of these pathways would produce an inappropriately large and unwieldy model with a considerably inflated false positive detection rate. Nonetheless, we determined that additional pathways could be incorporated into the model if there were a better method for de-selecting less important or artifactual pathways. Z score corrections were designed to address this by removing the bioactivities nearest cytotoxicity as presumed false negatives/non-specific effects. In our analysis, utilizing the cytotoxicity-derived z score values to remove putative cytotoxicity-impacted pathways was effective at reducing false positives, but at the expense of increasing false negatives. We determined that utilizing Z score corrections (even with a low threshold) was not an effective option to clarify important pathways and reduce false positives.
Results from these publications suggest that further improvements should focus on bolstering molecular pathways with poor-performing assays or where replicate experiments and/or assays are not available for a given endpoint within ToxCast. Ensuring data integrity and robustness is of profound importance to correct predictions. Efforts such as this have tremendous putative utility, as screening all chemicals and mixtures of chemicals for all endpoints is not feasible, and determining a screen of HTP assays could save tremendous time and cost and allow for a dramatically narrowed scope of testing in vivo. Further testing is required to substantiate this adipogenic prediction model for predicting in vivo metabolic disruption across a larger chemical space, but these preliminary results and success with other complex biological effects demonstrate a clear potential for implementation into predicting metabolic disruption and potentially helping reduce and better target in vitro and in vivo chemical assessments in the future.
Author Contributions
CK and HS planned and outlined the proposed review. CK wrote the review, and HS read and bolstered the review via feedback and guidance.
Funding
Project supported by a grant (R01 ES016099) and a fellowship (F32 ES027320; CK) from the National Institute of Environmental Health Sciences.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chamorro-Garcia R, Sahu M, Abbey RJ, Laude J, Pham N, Blumberg B. Transgenerational inheritance of increased fat depot size, stem cell reprogramming, and hepatic steatosis elicited by prenatal exposure to the obesogen tributyltin in mice. Environ Health Perspect. (2013) 121:359–66. doi: 10.1289/ehp.1205701
2. Heindel JJ, vom Saal FS, Blumberg B, Bovolin P, Calamandrei G, Ceresini G, et al. Parma consensus statement on metabolic disruptors. Environ Health (2015) 14:54. doi: 10.1186/s12940-015-0042-7
3. Li X, Ycaza J, Blumberg B. The environmental obesogen tributyltin chloride acts via peroxisome proliferator activated receptor gamma to induce adipogenesis in murine 3T3-L1 preadipocytes. J Steroid Biochem Mol Biol. (2011) 127:9–15. doi: 10.1016/j.jsbmb.2011.03.012
4. Patisaul HB, Roberts SC, Mabrey N, McCaffrey KA, Gear RB, Braun J, et al. Accumulation and endocrine disrupting effects of the flame retardant mixture Firemaster(R) 550 in rats: an exploratory assessment. J Biochem Mol Toxicol. (2013) 27:124–36. doi: 10.1002/jbt.21439
5. Riu A, McCollum CW, Pinto CL, Grimaldi M, Hillenweck A, Perdu E, et al. Halogenated bisphenol-A analogs act as obesogens in zebrafish larvae (Danio rerio). Toxicol Sci. (2014) 139:48–58. doi: 10.1093/toxsci/kfu036
6. The Endocrine Disruption Exchange (TEDX). TEDX List of Potential Endocrine Disruptors (2018). Available online at: https://endocrinedisruption.org/interactive-tools/tedx-list-of-potential-endocrine-disruptors/search-the-tedx-list
7. Skinner AC, Ravanbakht SN, Skelton JA, Perrin EM, Armstrong SC. Prevalence of obesity and severe obesity in US children, 1999–2016. Pediatrics (2018) 24:1116–23. doi: 10.1542/peds.2017-3459
8. Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of Obesity Among Adults Youth: United States, 2015–2016. Hyattsville, MD: National Center for Health Statistics (2017). Available online at: https://www.cdc.gov/nchs/data/databriefs/db288.pdf
9. Ruiz D, Becerra M, Jagai JS, Ard K, Sargis RM. Disparities in environmental exposures to endocrine-disrupting chemicals and diabetes risk in vulnerable populations. Diabetes Care (2018) 41:193–205. doi: 10.2337/dc16-2765
10. Yang W, Dall TM, Beronjia K, Lin J, Semilla AP, Chakrabarti R, et al. Economic Costs of Diabetes in the U.S. in 2017. Diabetes Care (2018) 41:917–28.
11. Biener A, Cawley J, Meyerhoefer C. The impact of obesity on medical care costs and labor market outcomes in the US. Clin Chem. (2018) 64:108–17. doi: 10.1373/clinchem.2017.272450
12. Klimentidis YC, Beasley TM, Lin HY, Murati G, Glass GE, Guyton M, et al. Canaries in the coal mine: a cross-species analysis of the plurality of obesity epidemics. Proc Biol Sci. (2011) 278:1626–32. doi: 10.1098/rspb.2010.1890
13. Brown TJ, Todd A, O'Malley C, Moore HJ, Husband AK, Bambra C, et al. Community pharmacy-delivered interventions for public health priorities: a systematic review of interventions for alcohol reduction, smoking cessation and weight management, including meta-analysis for smoking cessation. BMJ Open (2016) 6:e009828. doi: 10.1136/bmjopen-2015-009828
14. Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. (2002) 8:185–92. doi: 10.1089/107555302317371479
15. Fu M, Sun T, Bookout AL, Downes M, Yu RT, Evans RM, et al. A nuclear receptor atlas: 3T3-L1 adipogenesis. Mol Endocrinol. (2005) 19:2437–50. doi: 10.1210/me.2004-0539
16. Lu C, Cheng SY. Thyroid hormone receptors regulate adipogenesis and carcinogenesis via crosstalk signaling with peroxisome proliferator-activated receptors. J Mol Endocrinol. (2010) 44:143–54. doi: 10.1677/JME-09-0107
17. Niemela SM, Miettinen S, Konttinen Y, Waris T, Kellomaki M, Ashammakhi NA, et al. Fat tissue: views on reconstruction and exploitation. J Craniofac Surg. (2007) 18:325–35. doi: 10.1097/scs.0b013e3180333b6a
18. Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol. (2017) 68:3–33. doi: 10.1016/j.reprotox.2016.10.001
19. Coates ME, Dickinson CD, Harrison GF, Kon SK, Cummins SH, Cuthbertson WF. Mode of action of antibiotics in stimulating growth of chicks. Nature (1951) 168:332. doi: 10.1038/168332a0
20. Barber RS, Braude R, Mitchell KG. Antibiotic and copper supplements for fattening pigs. Br J Nutr. (1955) 9:378–81. doi: 10.1079/BJN19550054
21. Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell (2014) 158:705–21. doi: 10.1016/j.cell.2014.05.052
22. Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature (2012) 488:621–6. doi: 10.1038/nature11400
23. Ajslev TA, Andersen CS, Gamborg M, Sorensen TI, Jess T. Childhood overweight after establishment of the gut microbiota: the role of delivery mode, pre-pregnancy weight and early administration of antibiotics. Int J Obes. (2011) 35:522–9. doi: 10.1038/ijo.2011.27
24. Trasande L, Blustein J, Liu M, Corwin E, Cox LM, Blaser MJ. Infant antibiotic exposures and early-life body mass. Int J Obes. (2013) 37:16–23. doi: 10.1038/ijo.2012.132
25. Barnes AB, Colton T, Gundersen J, Noller KL, Tilley BC, Strama T, et al. Fertility and outcome of pregnancy in women exposed in utero to diethylstilbestrol. N Engl J Med. (1980) 302:5. doi: 10.1056/NEJM198003133021105
26. Herbst AL, Hubby MM, Blough RR, Azizi F. A comparison of pregnancy experience in DES-exposed and DES-unexposed daughters. J Reprod Med. (1980) 24:62–9. doi: 10.1097/00006254-198007000-00023
27. Herbst AL, Scully RE, Robboy SJ. Prenatal diethylstilbestrol exposure and human genital tract abnormalities. Natl Cancer Inst Monogr. (1979) 51:11.
28. Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, et al. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med. (2011) 365:1304–14. doi: 10.1056/NEJMoa1013961
29. Newbold RR, Bullock BC, Mc Lachlan JA. Exposure to diethylstilbestrol during pregnancy permanently alters the ovary and oviduct. Biol Reprod. (1983) 28:735–44. doi: 10.1095/biolreprod28.3.735
30. Hao CJ, Cheng XJ, Xia HF, Ma X. The endocrine disruptor diethylstilbestrol induces adipocyte differentiation and promotes obesity in mice. Toxicol Appl Pharmacol. (2012) 263:102–10. doi: 10.1016/j.taap.2012.06.003
31. Newbold RR, Padilla-Banks E, Jefferson WN. Environmental estrogens and obesity. Mol Cell Endocrinol. (2009) 304:84–9. doi: 10.1016/j.mce.2009.02.024
32. Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Perinatal exposure to environmental estrogens and the development of obesity. Mol Nutr Food Res. (2007) 51:912–7. doi: 10.1002/mnfr.200600259
33. Newbold RR, Padilla-Banks E, Snyder RJ, Phillips TM, Jefferson WN. Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod Toxicol. (2007) 23:290–6. doi: 10.1016/j.reprotox.2006.12.010
34. Hatch EE, Troisi R, Palmer JR, Wise LA, Titus L, Strohsnitter WC, et al. Prenatal diethylstilbestrol exposure and risk of obesity in adult women. J Dev Orig Health Dis. (2015) 6:201–7. doi: 10.1017/S2040174415000033
35. Kassotis CD, Nagel SC, Stapleton HM. Unconventional oil and gas chemicals and wastewater-impacted water samples promote adipogenesis via PPARγ-dependent and independent mechanisms in 3T3-L1 cells. Sci Total Environ. (2018) 640–1:1601–10. doi: 10.1016/j.scitotenv.2018.05.030
36. Kassotis CD, Bromfield JJ, Klemp KC, Meng CX, Wolfe A, Zoeller RT, et al. Adverse reproductive and developmental health outcomes following prenatal exposure to a hydraulic fracturing chemical mixture in female C57Bl/6 mice. Endocrinology (2016) 157:3469–81. doi: 10.1210/en.2016-1242
37. Kassotis CD, Klemp KC, Vu DC, Lin CH, Meng CX, Besch-Williford CL, et al. Endocrine-disrupting activity of hydraulic fracturing chemicals and adverse health outcomes after prenatal exposure in male mice. Endocrinology 156 (2015):4458–73. doi: 10.1210/en.2015-1375
38. Stacy SL, Brink LL, Larkin JC, Sadovsky Y, Goldstein BD, Pitt BR, et al. Perinatal outcomes and unconventional natural gas operations in southwest pennsylvania. PLoS ONE (2015) 10:e0126425. doi: 10.1371/journal.pone.0126425
39. McKenzie LM, Guo R, Witter RZ, Savitz DA, Newman LS, Adgate JL. Birth outcomes and maternal residential proximity to natural gas development in rural Colorado. Environ Health Perspect. (2014) 122:412–7. doi: 10.1289/ehp.1306722
40. Jornayvaz FR, Vollenweider P, Bochud M, Mooser V, Waeber G, Marques-Vidal P. Low birth weight leads to obesity, diabetes and increased leptin levels in adults: the CoLaus study. Cardiovasc Diabetol. (2016) 15:73. doi: 10.1186/s12933-016-0389-2
41. Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ. Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation (1996) 94:3246–50. doi: 10.1161/01.CIR.94.12.3246
42. Hirschler V, Bugna J, Roque M, Gilligan T, Gonzalez C. Does low birth weight predict obesity/overweight and metabolic syndrome in elementary school children? Arch Med Res. (2008) 39:796–802. doi: 10.1016/j.arcmed.2008.08.003
43. Danielzik S, Czerwinski-Mast M, Langnase K, Dilba B, Muller MJ. Parental overweight, socioeconomic status and high birth weight are the major determinants of overweight and obesity in 5–7 y-old children: baseline data of the Kiel Obesity Prevention Study (KOPS). Int J Obes Relat Metab Disord. (2004) 28:1494–502. doi: 10.1038/sj.ijo.0802756
44. Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Park C, Choi K, et al. OP9 mouse stromal cells rapidly differentiate into adipocytes: characterization of a useful new model of adipogenesis. J Lipid Res. (2006) 47:450–60. doi: 10.1194/jlr.D500037-JLR200
45. Lane JM, Doyle JR, Fortin JP, Kopin AS, Ordovas JM. Development of an OP9 derived cell line as a robust model to rapidly study adipocyte differentiation. PLoS ONE (2014) 9:e112123. doi: 10.1371/journal.pone.0112123
46. Pillai HK, Fang M, Beglov D, Kozakov D, Vajda S, Stapleton HM, et al. Ligand binding and activation of PPARgamma by Firemaster(R) 550: effects on adipogenesis and osteogenesis in vitro. Environ Health Perspect. (2014) 122:1225–32. doi: 10.1289/ehp.1408111
47. Janesick AS, Dimastrogiovanni G, Vanek L, Boulos C, Chamorro-García R, Tang W, et al. On the utility of ToxCast and ToxPi as methods for identifying new obesogens. Environ Health Perspect. (2016) 124:1214–26. doi: 10.1289/ehp.1510352
48. Shoucri BM, Martinez ES, Abreo TJ, Hung VT, Moosova Z, Shioda T, et al. Retinoid X receptor activation alters the chromatin landscape to commit mesenchymal stem cells to the adipose lineage. Endocrinology (2017) 158:3109–25. doi: 10.1210/en.2017-00348
49. Rudel RA, Camann DE, Spengler JD, Korn LR, Brody JG. Phthalates, alkylphenols, pesticides, polybrominated diphenyl ethers, and other endocrine-disrupting compounds in indoor air and dust. Environ Sci Technol. (2003) 37:4543–53. doi: 10.1021/es0264596
50. Stapleton HM, Harner T, Shoeib M, Keller JM, Schantz MM, Leigh SD, et al. Determination of polybrominated diphenyl ethers in indoor dust standard reference materials. Anal Bioanal Chem. (2006) 384:791–800. doi: 10.1007/s00216-005-0227-y
51. Stapleton HM, Klosterhaus S, Eagle S, Fuh J, Meeker JD, Blum A, et al. Detection of organophosphate flame retardants in furniture foam and U.S. house dust. Environ Sci Technol. (2009) 43:7490–5. doi: 10.1021/es9014019
52. US Environmental Protection Agency (EPA). Exposure Factors Handbook Chapter 5 (Update): Soil and Dust Ingestion. Washington, DC: US EPA Office of Research and Development (2017).
53. Fraser AJ, Webster TF, Watkins DJ, Nelson JW, Stapleton HM, Calafat AM, et al. Polyfluorinated compounds in serum linked to indoor air in office environments. Environ Sci Technol. (2012) 46:1209–15. doi: 10.1021/es2038257
54. Watkins DJ, McClean MD, Fraser AJ, Weinberg J, Stapleton HM, Webster TF. Associations between PBDEs in office air, dust, and surface wipes. Environ Int. (2013) 59:124–32. doi: 10.1016/j.envint.2013.06.001
55. Hoffman K, Garantziotis S, Birnbaum LS, Stapleton HM. Monitoring indoor exposure to organophosphate flame retardants: hand wipes and house dust. Environ Health Perspect. (2015) 123:160–5. doi: 10.1289/ehp.1408669
56. Stapleton HM, Misenheimer J, Hoffman K, Webster TF. Flame retardant associations between children's handwipes and house dust. Chemosphere (2014) 116:54–60. doi: 10.1016/j.chemosphere.2013.12.100
57. Phillips AL, Hammel SC, Hoffman K, Lorenzo AM, Chen A, Webster TF, et al. Children's residential exposure to organophosphate ester flame retardants and plasticizers: Investigating exposure pathways in the TESIE study. Environ Int. (2018) 116:176–85. doi: 10.1016/j.envint.2018.04.013
58. Hoffman K, Webster TF, Sjodin A, Stapleton HM. Toddler's behavior and its impacts on exposure to polybrominated diphenyl ethers. J Expo Sci Environ Epidemiol. (2017) 27:193–7. doi: 10.1038/jes.2016.11
59. Niemelä S, Miettinen S, Sarkanen JR, Ashammakhi N. Adipose tissue and adipocyte differentiation: molecular and cellular aspects and tissue engineering applications. In: Ashammakhi N, Reis R, Chiellini F, editors. Topics in Tissue Engineering (2008). p. 1–26. Available online at: https://www.oulu.fi/spareparts/ebook_topics_in_t_e_vol4/
60. Kassotis CD, Masse L, Kim S, Schlezinger JJ, Webster TF, Stapleton HM. Characterization of adipogenic chemicals in three different cell culture systems: implications for reproducibility based on cell source and handling. Sci Rep. (2017) 7:42104. doi: 10.1038/srep42104
61. Chappell VA, Janesick A, Blumberg B, Fenton SE. Tetrabromobisphenol-A promotes early adipogenesis and lipogenesis in 3T3-L1 cells. Toxicol Sci. (2018) 166:332–44. doi: 10.1093/toxsci/kfy209
62. Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell (1999) 4:611–7. doi: 10.1016/S1097-2765(00)80211-7
63. Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol. (2000) 16:145–71. doi: 10.1146/annurev.cellbio.16.1.145
64. Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA. Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology (1994) 135:798–800. doi: 10.1210/endo.135.2.8033830
65. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor y (PPARy). J Biol Chem. (1995) 270:12953–6. doi: 10.1074/jbc.270.22.12953
66. Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes (1998) 47:507–14. doi: 10.2337/diabetes.47.4.507
67. Rosen ED, C.-Hsu H, Wang X, Sakai S, Freeman MW, Gonzalez FJ, et al. C/EBP-alpha induces adipogenesis through PPAR-gamma: a unified pathway. Genes Dev. (2001) 16:22–6. doi: 10.1101/gad.948702
68. Matsusue K, Peters JM, Gonzalez FJ. PPARbeta/delta potentiates PPARgamma-stimulated adipocyte differentiation. FASEB J. (2004) 18:1477–9. doi: 10.1096/fj.04-1944fje
69. Schmuth M, Haqq CM, Cairns WJ, Holder JC, Dorsam S, Chang S, et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J Invest Dermatol. (2004) 122:971–83. doi: 10.1111/j.0022-202X.2004.22412.x
70. Bastie C, Holst D, Gaillard D, Jehl-Pietri C, Grimaldi PA. Expression of peroxisome proliferator-activated receptor PPARdelta promotes induction of PPARgamma and adipocyte differentiation in 3T3C2 fibroblasts. J Biol Chem. (1999) 274:21920–5. doi: 10.1074/jbc.274.31.21920
71. Bastie C, Luquet S, Holst D, Jehl-Pietri C, Grimaldi PA. Alterations of peroxisome proliferator-activated receptor δ activity affect fatty acid-controlled adipose differentiation. J Biol Chem. (2000) 275:38768–73. doi: 10.1074/jbc.M006450200
72. Brun RP, Tontonoz P, Forman BM, Ellis R, Chen J, Evans RM, et al. Differential activation of adipogenesis by multiple PPAR isoforms. Genes Dev. (1996) 10:974–84. doi: 10.1101/gad.10.8.974
73. Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell (2003) 113:159–70. doi: 10.1016/S0092-8674(03)00269-1
74. Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol Cell Biol. (2000) 20:5119–28. doi: 10.1128/MCB.20.14.5119-5128.2000
75. Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem. (2000) 275:16638–42. doi: 10.1074/jbc.275.22.16638
76. Jeong S, Yoon M. Fenofibrate inhibits adipocyte hypertrophy and insulin resistance by activating adipose PPARalpha in high fat diet-induced obese mice. Exp Mol Med. (2009) 41:397–405. doi: 10.3858/emm.2009.41.6.045
77. Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, et al. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes (2005) 54:3358–70. doi: 10.2337/diabetes.54.12.3358
78. Das M, Irvin MR, Sha J, Aslibekyan S, Hidalgo B, Perry RT, et al. Lipid changes due to fenofibrate treatment are not associated with changes in DNA methylation patterns in the GOLDN study. Front Genet. (2015) 6:304. doi: 10.3389/fgene.2015.00304
79. Forcheron F, Cachefo A, Thevenon S, Pinteur C, Beylot M. Mechanisms of the triglyceride- and cholesterol-lowering effect of fenofibrate in hyperlipidemic type 2 diabetic patients. Diabetes (2002) 51:3486–91. doi: 10.2337/diabetes.51.12.3486
80. Oliver WR Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Nat Acad Sci USA. (2001) 98:5306–11. doi: 10.1073/pnas.091021198
81. Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, Nishikawa J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol. (2005) 67:766–74. doi: 10.1124/mol.104.008409
82. Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, et al. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. (2006) 20:2141–55. doi: 10.1210/me.2005-0367
83. Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Nat Acad Sci USA. (1997) 94:237–41. doi: 10.1073/pnas.94.1.237
84. Canan Koch SS, Dardashti LJ, Cesario RM, Croston GE, Boehm MF, Heyman RA, et al. Synthesis of retinoid X receptor-specific ligands that are potent inducers of adipogenesis in 3T3-L1 cells. J Med Chem. (1999) 42:742–50. doi: 10.1021/jm980621r
85. Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. (2008) 22:2953–67. doi: 10.1101/gad.501108
86. Imai T, Jiang M, Chambon P, Metzger D. Impaired adipogenesis and lipolysis in the mouse upon selective ablation of the retinoid X receptor alpha mediated by a tamoxifen-inducible chimeric Cre recombinase (Cre-ERT2) in adipocytes. Proc Nat Acad Sci USA. (2001) 98:224–8. doi: 10.1073/pnas.011528898
87. Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cesario RM, Jow L, et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature (1997) 386:407–10. doi: 10.1038/386407a0
88. Sadasivuni MK, Reddy BM, Singh J, Anup MO, Sunil V, Lakshmi MN, et al. CNX-013-B2, a unique pan tissue acting rexinoid, modulates several nuclear receptors and controls multiple risk factors of the metabolic syndrome without risk of hypertriglyceridemia, hepatomegaly and body weight gain in animal models. Diabetol Metab Syndr. (2014) 6:83. doi: 10.1186/1758-5996-6-83
89. Emilsson V, O'Dowd J, Wang S, Liu YL, Sennitt M, Heyman R, et al. The effects of rexinoids and rosiglitazone on body weight and uncoupling protein isoform expression in the Zucker fa/fa rat. Metabolism (2000) 49:1610–5. doi: 10.1053/meta.2000.18692
90. Shoucri BM, Hung VT, Chamorro-Garcia R, Shioda T, Blumberg B. Retinoid X receptor activation during adipogenesis of female mesenchymal stem cells programs a dysfunctional adipocyte. Endocrinology (2018) 159:2863–83. doi: 10.1210/en.2018-00056
91. Farol LT, Hymes KB. Bexarotene: a clinical review. Expert Rev Anticancer Ther. (2004) 4:180–8. doi: 10.1586/14737140.4.2.180
92. de Vries-van der Weij J, de Haan W, Hu L, Kuif M, Oei HL, van der Hoorn JW, et al. Bexarotene induces dyslipidemia by increased very low-density lipoprotein production and cholesteryl ester transfer protein-mediated reduction of high-density lipoprotein. Endocrinology (2009) 150:2368–75. doi: 10.1210/en.2008-1540
93. Pinaire JA, Reifel-Miller A. Therapeutic potential of retinoid x receptor modulators for the treatment of the metabolic syndrome. PPAR Res. (2007) 2007:94156. doi: 10.1155/2007/94156
94. Shulman AI, Mangelsdorf DJ. Retinoid x receptor heterodimers in the metabolic syndrome. N Engl J Med. (2005) 353:604–15. doi: 10.1056/NEJMra043590
95. Hummasti S, Laffitte BA, Watson MA, Galardi C, Chao LC, Ramamurthy L, et al. Liver X receptors are regulators of adipocyte gene expression but not differentiation: identification of apoD as a direct target. J Lipid Res. (2004) 45:616–25. doi: 10.1194/jlr.M300312-JLR200
96. Seo JB, Moon HM, Kim WS, Lee YS, Jeong HW, Yoo EJ, et al. Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor gamma expression. Mol Cell Biol. (2004) 24:3430–44. doi: 10.1128/MCB.24.8.3430-3444.2004
97. Juvet LK, Andresen SM, Schuster GU, Dalen KT, Tobin KA, Hollung K, et al. On the role of liver X receptors in lipid accumulation in adipocytes. Mol Endocrinol. (2003) 17:172–82. doi: 10.1210/me.2001-0210
98. Stenson BM, Ryden M, Venteclef N, Dahlman I, Pettersson AM, Mairal A, et al. Liver X receptor (LXR) regulates human adipocyte lipolysis. J Biol Chem. (2011) 286:370–9. doi: 10.1074/jbc.M110.179499
99. Gerin I, Dolinsky VW, Shackman JG, Kennedy RT, Chiang SH, Burant CF, et al. LXRbeta is required for adipocyte growth, glucose homeostasis, and beta cell function. J Biol Chem. (2005) 280:23024–31. doi: 10.1074/jbc.M412564200
100. Korach-Andre M, Archer A, Barros RP, Parini P, Gustafsson JA. Both liver-X receptor (LXR) isoforms control energy expenditure by regulating brown adipose tissue activity. Proc Nat Acad Sci USA. (2011) 108:403–8. doi: 10.1073/pnas.1017884108
101. Dahlman I, Nilsson M, Jiao H, Hoffstedt J, Lindgren CM, Humphreys K, et al. Liver X receptor gene polymorphisms and adipose tissue expression levels in obesity. Pharmacogenet Genomics (2006) 16:881–9. doi: 10.1097/01.fpc.0000236334.49422.48
102. Kirchgessner TG, Sleph P, Ostrowski J, Lupisella J, Ryan CS, Liu X, et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab. (2016) 24:223–33. doi: 10.1016/j.cmet.2016.07.016
103. Gao J, Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol Sci. (2012) 33:552–8. doi: 10.1016/j.tips.2012.07.003
104. Moreau A, Vilarem MJ, Maurel P, Pascussi JM. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol Pharm. (2008) 5:35–41. doi: 10.1021/mp700103m
105. Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology (2008) 134:556–67. doi: 10.1053/j.gastro.2007.11.037
106. He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O'Doherty RM, et al. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes (2013) 62:1876–87. doi: 10.2337/db12-1039
107. Wada T, Gao J, Xie W. PXR and CAR in energy metabolism. Trends Endocrinol Metabol. (2009) 20:273–9. doi: 10.1016/j.tem.2009.03.003
108. Lahtela JT, Arranto AJ, Sotaniemi EA. Enzyme inducers improve insulin sensitivity in non-insulin-dependent diabetic subjects. Diabetes (1985) 34:911–6. doi: 10.2337/diab.34.9.911
109. Sotaniemi EA, Karvonen I. Glucose tolerance and insulin response to glucose load before and after enzyme inducing therapy in subjects with glucose intolerance and patients with NIDDM having hyperinsulinemia or relative insulin deficiency. Diabetes Res. (1989) 11:131–9.
110. Jiao Y, Lu Y, Li XY. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol Sin. (2015) 36:44–50. doi: 10.1038/aps.2014.116
111. Prawitt J, Caron S, Staels B. How to modulate FXR activity to treat the metabolic syndrome. Drug Discov Today Dis Mech. (2009) 6:e55–64. doi: 10.1016/j.ddmec.2010.05.002
112. Rizzo G, Disante M, Mencarelli A, Renga B, Gioiello A, Pellicciari R, et al. The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Mol Pharmacol. (2006) 70:1164–73. doi: 10.1124/mol.106.023820
113. Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. (2006) 281:11039–49. doi: 10.1074/jbc.M510258200
114. Yang JY, Della-Fera MA, Baile CA. Guggulsterone inhibits adipocyte differentiation and induces apoptosis in 3T3-L1 cells. Obesity (2008) 16:16–22. doi: 10.1038/oby.2007.24
115. Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, et al. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem. (2011) 286:26913–20. doi: 10.1074/jbc.M111.248203
116. Rader DJ, Liver X. Receptor and farnesoid X receptor as therapeutic targets. Am J Cardiol. (2007) 100:15N−9N. doi: 10.1016/j.amjcard.2007.08.008
117. Abdelkarim M, Caron S, Duhem C, Prawitt J, Dumont J, Lucas A, et al. The farnesoid X receptor regulates adipocyte differentiation and function by promoting peroxisome proliferator-activated receptor-gamma and interfering with the Wnt/beta-catenin pathways. J Biol Chem. (2010) 285:36759–67. doi: 10.1074/jbc.M110.166231
118. Prawitt J, Abdelkarim M, Stroeve JH, Popescu I, Duez H, Velagapudi VR, et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes (2011) 60:1861–71. doi: 10.2337/db11-0030
119. Maneschi E, Vignozzi L, Morelli A, Mello T, Filippi S, Cellai I, et al. FXR activation normalizes insulin sensitivity in visceral preadipocytes of a rabbit model of MetS. J Endocr. (2013) 218:215–31. doi: 10.1530/JOE-13-0109
120. Lu Y, Ma Z, Zhang Z, Xiong X, Wang X, Zhang H, et al. Yin Yang 1 promotes hepatic steatosis through repression of farnesoid X receptor in obese mice. Gut (2014) 63:170–8. doi: 10.1136/gutjnl-2012-303150
121. Obregon MJ. Thyroid hormone and adipocyte differentiation. Thyroid (2008) 18:185–95. doi: 10.1089/thy.2007.0254
122. Kolyvanos Naumann U, Furer J, Kaser L, Vetter W. Hypothyroidism. main symptoms: fatigue, weight gain, depression, myalgia, edema. Praxis (2007) 96:1411–7. doi: 10.1024/1661-8157.96.38.1411
123. Bratusch-Marrain P, Schmid P, Waldhausl W, Schlick W. Specific weight loss in hyperthyroidism. Horm Metab Res. (1978) 10:412–5. doi: 10.1055/s-0028-1093403
124. Bryzgalova G, Effendic S, Khan A, Rehnmark S, Barbounis P, Boulet J, et al. Anti-obesity, anti-diabetic, and lipid lowering effects of the thyroid receptor beta subtype selective agonist KB-141. J Steroid Biochem Mole Biol. (2008) 111:262–7. doi: 10.1016/j.jsbmb.2008.06.010
125. Dale J, Daykin J, Holder R, Sheppard MC, Franklyn JA. Weight gain following treatment of hyperthyroidism. Clin Endocrinol. (2001) 55:233–9. doi: 10.1046/j.1365-2265.2001.01329.x
126. Lonn L, Stenlof K, Ottosson M, Lindroos AK, Nystrom E, Sjostrom L. Body weight and body composition changes after treatment of hyperthyroidism. J Clin Endocr Metabol. (1998) 83:4269–73. doi: 10.1210/jc.83.12.4269
127. Kindblom JM, Gevers EF, Skrtic SM, Lindberg MK, Gothe S, Tornell J, et al. Increased adipogenesis in bone marrow but decreased bone mineral density in mice devoid of thyroid hormone receptors. Bone (2005) 36:607–16. doi: 10.1016/j.bone.2005.01.017
128. Darimont C, Gaillard D, Ailhaud G, Negrel R. Terminal differentiation of mouse preadipocyte cells: adipogenic and antimitogenic role of triiodothyronine. Mol Cell Endocr. (1993) 98:67–73. doi: 10.1016/0303-7207(93)90238-F
129. Jiang W, Miyamoto T, Kakizawa T, Sakuma T, Nishio S, Takeda T, et al. Expression of thyroid hormone receptor alpha in 3T3-L1 adipocytes; triiodothyronine increases the expression of lipogenic enzyme and triglyceride accumulation. J Endocr. (2004) 182:295–302. doi: 10.1677/joe.0.1820295
130. Flores-Delgado G, Marsch-Moreno M, Kuri-Harcuch W. Thyroid hormone stimulates adipocyte differentiation of 3T3 cells. Mol Cell Biochem. (1987) 76:35–43. doi: 10.1007/BF00219396
131. John K, Marino JS, Sanchez ER, Hinds TD, Jr. The glucocorticoid receptor: cause of or cure for obesity? Am J Physiol. Endocr Metabol. (2016) 310:E249–57. doi: 10.1152/ajpendo.00478.2015
132. Contador D, Ezquer F, Espinosa M, Arango-Rodriguez M, Puebla C, Sobrevia L, et al. Dexamethasone and rosiglitazone are sufficient and necessary for producing functional adipocytes from mesenchymal stem cells. Exp Biol Med. (2015) 240:1235–46. doi: 10.1177/1535370214566565
133. Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest (1997) 99:2416–22. doi: 10.1172/JCI119424
134. Sargis RM, Johnson DN, Choudhury RA, Brady MJ. Environmental endocrine disruptors promote adipogenesis in the 3T3-L1 cell line through glucocorticoid receptor activation. Obesity (2010) 18:1283–8. doi: 10.1038/oby.2009.419
135. Asada M, Rauch A, Shimizu H, Maruyama H, Miyaki S, Shibamori M, et al. DNA binding-dependent glucocorticoid receptor activity promotes adipogenesis via Kruppel-like factor 15 gene expression. Lab Invest. (2011) 91:203–15. doi: 10.1038/labinvest.2010.170
136. Greenberger JS. Corticosteroid-dependent differentiation of human marrow preadipocytes in vitro. in vitro (1979) 15:823–8. doi: 10.1007/BF02618309
137. Hauner H, Schmid P, Pfeiffer EF. Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. J Clin Endocrinol Metabol. (1987) 64:832–5. doi: 10.1210/jcem-64-4-832
138. Pantoja C, Huff JT, Yamamoto KR. Glucocorticoid signaling defines a novel commitment state during adipogenesis in vitro. Mol Biol Cell. (2008) 19:4032–41. doi: 10.1091/mbc.e08-04-0420
139. Caprio M, Feve B, Claes A, Viengchareun S, Lombes M, Zennaro MC. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. (2007) 21:2185–94. doi: 10.1096/fj.06-7970com
140. Lee MJ, Fried SK. The glucocorticoid receptor, not the mineralocorticoid receptor, plays the dominant role in adipogenesis and adipokine production in human adipocytes. Int J Obes. (2014) 38:1228–33. doi: 10.1038/ijo.2014.6
141. Kershaw EE, Morton NM, Dhillon H, Ramage L, Seckl JR, Flier JS. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes (2005) 54:1023–31. doi: 10.2337/diabetes.54.4.1023
142. Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum. (2008) 58:1674–86. doi: 10.1002/art.23454
143. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of cushing's syndrome: an endocrine society clinical practice guideline. J Clin Endocr Metabol. (2008) 93:1526–40. doi: 10.1210/jc.2008-0125
144. Krotkiewski M, Blohme B, Lindholm N, Bjorntorp P. The effects of adrenal corticosteroids on regional adipocyte size in man. J Clin Endocr Metabol. (1976) 42:91–7. doi: 10.1210/jcem-42-1-91
145. Bloom SL, Sheffield JS, McIntire DD, Leveno KJ. Antenatal dexamethasone and decreased birth weight. Obstetr Gynecol. (2001) 97:485–90. Available online at: https://journals.lww.com/greenjournal/Fulltext/2001/04000/Antenatal_Dexamethasone_and_Decreased_Birth_Weight.1.aspx
146. Halliday H. Adverse effects of early dexamethasone treatment in extremely-low-birth-weight infants. J Pediatr. (2001) 139:163–4. doi: 10.1056/NEJM200101113440203
147. Cooke PS, Naaz A. Role of estrogens in adipocyte development and function. Exp Biol Med. (2004) 229:1127–35. doi: 10.1177/153537020422901107
148. Dieudonne MN, Pecquery R, Leneveu MC, Giudicelli Y. Opposite effects of androgens and estrogens on adipogenesis in rat preadipocytes: evidence for sex and site-related specificities and possible involvement of insulin-like growth factor 1 receptor and peroxisome proliferator-activated receptor gamma2. Endocrinology (2000) 141:649–56. doi: 10.1210/endo.141.2.7293
149. Roncari DA, Van RL. Promotion of human adipocyte precursor replication by 17beta-estradiol in culture. J Clin Invest. (1978) 62:503–8. doi: 10.1172/JCI109153
150. Hong L, Colpan A, Peptan IA. Modulations of 17-beta estradiol on osteogenic and adipogenic differentiations of human mesenchymal stem cells. Tissue Eng. (2006) 12:2747–53. doi: 10.1089/ten.2006.12.2747
151. Zhu P, Yuen JM, Sham KW, Cheng CH. GPER mediates the inhibitory actions of estrogen on adipogenesis in 3T3-L1 cells through perturbation of mitotic clonal expansion. General Comp Endocrinol. (2013) 193:19–26. doi: 10.1016/j.ygcen.2013.07.004
152. Okazaki R, Inoue D, Shibata M, Saika M, Kido S, Ooka H, et al. Estrogen promotes early osteoblast differentiation and inhibits adipocyte differentiation in mouse bone marrow stromal cell lines that express estrogen receptor (ER) alpha or beta. Endocrinology (2002) 143:2349–56. doi: 10.1210/endo.143.6.8854
153. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Nat Acad Sci USA. (2000) 97:12729–34. doi: 10.1073/pnas.97.23.12729
154. Davis SR, Castelo-Branco C, Chedraui P, Lumsden MA, Nappi RE, Shah D, et al. Understanding weight gain at menopause. Climacteric (2012) 15:419–429. doi: 10.3109/13697137.2012.707385
155. Stubbins RE, Holcomb VB, Hong J, Nunez NP. Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance. Eur J Nutr. (2012) 51:861–70. doi: 10.1007/s00394-011-0266-4
156. Blouin K, Boivin A, Tchernof A. Androgens and body fat distribution. J Steroid Biochem Mol Biol. (2008) 108:272–80. doi: 10.1016/j.jsbmb.2007.09.001
157. O'Reilly MW, House PJ, Tomlinson JW. Understanding androgen action in adipose tissue. J Steroid Biochem Mol Biol. (2014) 143:277–84. doi: 10.1016/j.jsbmb.2014.04.008
158. Blouin K, Nadeau M, Perreault M, Veilleux A, Drolet R, Marceau P, et al. Effects of androgens on adipocyte differentiation and adipose tissue explant metabolism in men and women. Clin Endocrinol. (2010) 72:176–88. doi: 10.1111/j.1365-2265.2009.03645.x
159. Mammi C, Calanchini M, Antelmi A, Cinti F, Rosano GM, Lenzi A, et al. Androgens and adipose tissue in males: a complex and reciprocal interplay. Int J Endocrinol. (2012) 2012:789653. doi: 10.1155/2012/789653
160. Gupta V, Bhasin S, Guo W, Singh R, Miki R, Chauhan P, et al. Effects of dihydrotestosterone on differentiation and proliferation of human mesenchymal stem cells and preadipocytes. Mol Cel Endocrinol. (2008) 296:32–40. doi: 10.1016/j.mce.2008.08.019
161. Chazenbalk G, Singh P, Irge D, Shah A, Abbott DH, Dumesic DA. Androgens inhibit adipogenesis during human adipose stem cell commitment to preadipocyte formation. Steroids (2013) 78:920–6. doi: 10.1016/j.steroids.2013.05.001
162. Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology (2003) 144:5081–8. doi: 10.1210/en.2003-0741
163. Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. (2003) 300:167–71. doi: 10.1016/S0006-291X(02)02774-2
164. Diamanti-Kandarakis E, Mitrakou A, Raptis S, Tolis G, Duleba AJ. The effect of a pure antiandrogen receptor blocker, flutamide, on the lipid profile in the polycystic ovary syndrome. J Clin Endocrinol Metabol. (1998) 83:2699–705. doi: 10.1210/jcem.83.8.5041
165. Cusin I, Sainsbury A, Doyle P, Rohner-Jeanrenaud F, Jeanrenaud B. The ob gene and insulin. a relationship leading to clues to the understanding of obesity. Diabetes (1995) 44:1467–70. doi: 10.2337/diab.44.12.1467
166. Cusin I, Rohner-Jeanrenaud F, Terrettaz J, Jeanrenaud B. Hyperinsulinemia and its impact on obesity and insulin resistance. Int J Obes Relat Metab Disord. (1992) 16 (Suppl. 4):S1–11.
168. Garten A, Schuster S, Kiess W. The insulin-like growth factors in adipogenesis and obesity. Endocrinol Metab Clin North Am. (2012) 41:283–95. doi: 10.1016/j.ecl.2012.04.011
169. Bournat JC, Brown CW. Mitochondrial dysfunction in obesity. Curr Opin Endocrinol Diabet Obes. (2010) 17:446–52. doi: 10.1097/MED.0b013e32833c3026
170. Vankoningsloo S, Piens M, Lecocq C, Gilson A, De Pauw A, Renard P, et al. Mitochondrial dysfunction induces triglyceride accumulation in 3T3-L1 cells: role of fatty acid beta-oxidation and glucose. J Lipid Res. (2005) 46:1133–49. doi: 10.1194/jlr.M400464-JLR200
171. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circul Res. (2008) 102:401–14. doi: 10.1161/CIRCRESAHA.107.165472
172. Lim S, Cho YM, Park KS, Lee HK. Persistent organic pollutants, mitochondrial dysfunction, and metabolic syndrome. Ann NY Acad Sci. (2010) 1201:166–76. doi: 10.1111/j.1749-6632.2010.05622.x
173. Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med. (2010) 88:993–1001. doi: 10.1007/s00109-010-0663-9
174. Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, et al. Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep. (2016) 16:1–8. doi: 10.1016/j.celrep.2016.05.065
175. De Pauw A, Tejerina S, Raes M, Keijer J, Arnould T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am J Pathol. (2009) 175:927–39. doi: 10.2353/ajpath.2009.081155
176. Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci. (2008) 114:195–210. doi: 10.1042/CS20070166
177. Luz AL, Kassotis CD, Stapleton HM, Meyer JN. The high-production volume fungicide pyraclostrobin induces triglyceride accumulation associated with mitochondrial dysfunction, and promotes adipocyte differentiation independent of PPARgamma activation, in 3T3-L1 cells. Toxicol. (2018) 393:150–9. doi: 10.1016/j.tox.2017.11.010
178. Lu RH, Ji H, Chang ZG, Su SS, Yang GS. Mitochondrial development and the influence of its dysfunction during rat adipocyte differentiation. Mol Biol Rep. (2010) 37:2173–82. doi: 10.1007/s11033-009-9695-z
179. Kusminski CM, Scherer PE. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol Metabol. (2012) 23:435–43. doi: 10.1016/j.tem.2012.06.004
180. Medina-Gomez G. Mitochondria and endocrine function of adipose tissue. Best Prac Res. Clin. Endocrinol Metabol. (2012) 26:791–804. doi: 10.1016/j.beem.2012.06.002
181. U. Survey SG. Estimated annual agricultural pesticide use - pyraclostrobin. In: U.S.D.o. Interior, editor. Pesticide National Synthesis Project (2016). Available online at: https://water.usgs.gov/nawqa/pnsp/usage/maps/show_map.php?year=2016&map=PYRACLOSTROBIN&hilo=L
182. Pearson BL, Simon JM, McCoy ES, Salazar G, Fragola G, Zylka MJ. Identification of chemicals that mimic transcriptional changes associated with autism, brain aging and neurodegeneration. Nature Commun. (2016) 7:11173. doi: 10.1038/ncomms11173
183. Bartlett DW, Clough JM, Godwin JR, Hall AA, Hamer M, Parr-Dobrzanski B. The strobilurin fungicides. Pest Manag Sci. (2002) 58:649–62. doi: 10.1002/ps.520
184. Kassotis CD, Hoffman K, Stapleton HM. Characterization of adipogenic activity of semi-volatile indoor contaminants and house dust. Environ Sci Technol In (2017) 51:8735–45. doi: 10.1021/acs.est.7b01788
185. Foley B, Doheny DL, Black MB, Pendse SN, Wetmore BA, Clewell RA, et al. Screening toxcast prioritized chemicals for PPARG function in a human adipose-derived stem cell model of adipogenesis. Toxicol Sci. (2017) 155:85–100. doi: 10.1093/toxsci/kfw186
186. Peng H, Sun J, Alharbi HA, Jones PD, Giesy JP, Wiseman S. Peroxisome proliferator-activated receptor γ is a sensitive target for oil sands process-affected water: effects on adipogenesis and identification of ligands. Environ Sci Technol. (2016) 50:7816–24. doi: 10.1021/acs.est.6b01890
187. Rundle K, Sharaf M, Stevens D, Kamunde C, Van Den Heuvel MR. Oil sands-derived naphthenic acids are oxidative uncouplers and impair electron transport in isolated mitochondria. Environ Sci Technol. (2018) 52:10803–11. doi: 10.1021/acs.est.8b02638
188. Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, et al. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. (2011) 14:537–44. doi: 10.1016/j.cmet.2011.08.007
189. Wang X, Hai C. Redox modulation of adipocyte differentiation: hypothesis of “Redox Chain” and novel insights into intervention of adipogenesis and obesity. Free Radic Biol Med. (2015) 89:99–125. doi: 10.1016/j.freeradbiomed.2015.07.012
190. Lee H, Lee YJ, Choi H, Ko EH, Kim JW. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J Biol Chem. (2009) 284:10601–9. doi: 10.1074/jbc.M808742200
191. Carriere A, Carmona MC, Fernandez Y, Rigoulet M, Wenger RH, Penicaud L, et al. Mitochondrial reactive oxygen species control the transcription factor CHOP-10/GADD153 and adipocyte differentiation: a mechanism for hypoxia-dependent effect. J Biol Chem. (2004) 279:40462–9. doi: 10.1074/jbc.M407258200
192. Carriere A, Fernandez Y, Rigoulet M, Penicaud L, Casteilla L. Inhibition of preadipocyte proliferation by mitochondrial reactive oxygen species. FEBS Lett. (2003) 550:163–7. doi: 10.1016/S0014-5793(03)00862-7
193. Kanda Y, Hinata T, Kang SW, Watanabe Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci. (2011) 89:250–8. doi: 10.1016/j.lfs.2011.06.007
194. Lee HK, Cho YM, Kwak SH, Lim S, Park KS, Shim EB. Mitochondrial dysfunction and metabolic syndrome-looking for environmental factors. Biochim Biophys Acta (2010) 1800:282–9. doi: 10.1016/j.bbagen.2009.11.010
195. Lim S, Ahn SY, Song IC, Chung MH, Jang HC, Park KS, et al. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS ONE (2009) 4:e5186. doi: 10.1371/journal.pone.0005186
196. Shen K, Shen C, Yu J, Yu C, Chen L, Shi D, et al. PCB congeners induced mitochondrial dysfunction in Vero cells. J Hazard Mater. (2011) 185:24–8. doi: 10.1016/j.jhazmat.2010.08.061
197. Aly HA, Domenech O. Aroclor 1254 induced cytotoxicity and mitochondrial dysfunction in isolated rat hepatocytes. Toxicology (2009) 262:175–83. doi: 10.1016/j.tox.2009.05.018
198. Gray SL, Shaw AC, Gagne AX, Chan HM. Chronic exposure to PCBs (Aroclor 1254) exacerbates obesity-induced insulin resistance and hyperinsulinemia in mice. J Toxicol Environ Health. Part A (2013) 76:701–15. doi: 10.1080/15287394.2013.796503
199. Donat-Vargas C, Gea A, Sayon-Orea C, Carlos S, Martinez-Gonzalez MA, Bes-Rastrollo M. Association between dietary intakes of PCBs and the risk of obesity: the SUN project. J Epidemiol Commun Health (2014) 68:834–41. doi: 10.1136/jech-2013-203752
200. Lee DH, Steffes MW, Sjodin A, Jones RS, Needham LL, Jacobs DR Jr. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS ONE (2011) 6:e15977. doi: 10.1371/journal.pone.0015977
201. Tanaka T, Morita A, Kato M, Hirai T, Mizoue T, Terauchi Y, et al. Congener-specific polychlorinated biphenyls and the prevalence of diabetes in the Saku Control Obesity Program (SCOP). Endocr J. (2011) 58:589–96. doi: 10.1507/endocrj.K10E-361
202. Dirinck E, Jorens PG, Covaci A, Geens T, Roosens L, Neels H, et al. Obesity and persistent organic pollutants: possible obesogenic effect of organochlorine pesticides and polychlorinated biphenyls. Obesity (2011) 19:709–14. doi: 10.1038/oby.2010.133
203. Poulos SP, Dodson MV, Hausman GJ. Cell line models for differentiation: preadipocytes and adipocytes. Exp Biol Med. (2010) 235:1185–93. doi: 10.1258/ebm.2010.010063
204. Ruiz-Ojeda FJ, Ruperez AI, Gomez-Llorente C, Gil A, Aguilera CM. Cell models and their application for studying adipogenic differentiation in relation to obesity: a review. Int J Mol Sci. (2016) 17:E1040. doi: 10.3390/ijms17071040
205. Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell (1975) 5:19–27. doi: 10.1016/0092-8674(75)90087-2
206. Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell (1974) 3:127–33. doi: 10.1016/0092-8674(74)90116-0
207. Guo X, Liao K. Analysis of gene expression profile during 3T3-L1 preadipocyte differentiation. Gene (2000) 251:45–53. doi: 10.1016/S0378-1119(00)00192-X
208. Kleensang A, Vantangoli MM, Odwin-DaCosta S, Andersen ME, Boekelheide K, Bouhifd M, et al. Genetic variability in a frozen batch of MCF-7 cells invisible in routine authentication affecting cell function. Sci Rep. (2016) 6:28994. doi: 10.1038/srep28994
209. Janderova L, McNeil M, Murrell AN, Mynatt RL, Smith SR. Human mesenchymal stem cells as an in vitro model for human adipogenesis. Obes Res. (2003) 11:65–74. doi: 10.1038/oby.2003.11
210. Muruganandan S, Roman AA, Sinal CJ. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cel Mol Life Sci. (2009) 66:236–53. doi: 10.1007/s00018-008-8429-z
211. Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell (2016) 18:782–96. doi: 10.1016/j.stem.2016.02.015
212. Csaki C, Matis U, Mobasheri A, Ye H, Shakibaei M. Chondrogenesis, osteogenesis and adipogenesis of canine mesenchymal stem cells: a biochemical, morphological and ultrastructural study. Histochem Cell Biol. (2007) 128:507–20. doi: 10.1007/s00418-007-0337-z
213. Kim S, Li A, Monti S, Schlezinger JJ. Tributyltin induces a transcriptional response without a brite adipocyte signature in adipocyte models. Arch Toxicol. (2018) 92:2859–74. doi: 10.1007/s00204-018-2268-y
214. Tang QQ, Otto TC, Lane MD. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Nat Acad Sci USA. (2004) 101:9607–11. doi: 10.1073/pnas.0403100101
215. Katagiri T, Yamaguchi A, Ikeda T, Yoshiki S, Wozney JM, Rosen V, et al. The non-osteogenic mouse pluripotent cell line, C3H10T1/2, is induced to differentiate into osteoblastic cells by recombinant human bone morphogenetic protein-2. Biochem Biophys Res Commun. (1990) 172:295–9. doi: 10.1016/S0006-291X(05)80208-6
216. Wang W, Itaka K, Ohba S, Nishiyama N, Chung UI, Yamasaki Y, et al. 3D spheroid culture system on micropatterned substrates for improved differentiation efficiency of multipotent mesenchymal stem cells. Biomaterials (2009) 30:2705–15. doi: 10.1016/j.biomaterials.2009.01.030
217. Baraniak PR, McDevitt TC. Scaffold-free culture of mesenchymal stem cell spheroids in suspension preserves multilineage potential. Cell Tissue Res. (2012) 347:701–11. doi: 10.1007/s00441-011-1215-5
218. Cheng NC, Wang S, Young TH. The influence of spheroid formation of human adipose-derived stem cells on chitosan films on stemness and differentiation capabilities. Biomaterials (2012) 33:1748–58. doi: 10.1016/j.biomaterials.2011.11.049
219. Turner PA, Gurumurthy B, Bailey JL, Elks CM, Janorkar AV. Adipogenic differentiation of human adipose-derived stem cells grown as spheroids. Process Biochem. (2017) 59:312–20. doi: 10.1016/j.procbio.2017.02.003
220. Turner PA, Harris LM, Purser CA, Baker RC, Janorkar AV. A surface-tethered spheroid model for functional evaluation of 3T3-L1 adipocytes. Biotechnol Bioeng. (2014) 111:174–83. doi: 10.1002/bit.25099
221. Miyagawa Y, Okita H, Hiroyama M, Sakamoto R, Kobayashi M, Nakajima H, et al. A microfabricated scaffold induces the spheroid formation of human bone marrow-derived mesenchymal progenitor cells and promotes efficient adipogenic differentiation. Tissue Eng Part A. (2011) 17:513–21. doi: 10.1089/ten.tea.2009.0810
222. Kapur SK, Wang X, Shang H, Yun S, Li X, Feng G, et al. Human adipose stem cells maintain proliferative, synthetic and multipotential properties when suspension cultured as self-assembling spheroids. Biofabrication (2012) 4:025004. doi: 10.1088/1758-5082/4/2/025004
224. Markell D. An overview of TSCA, its history and key underlying assumptions, and its place in environmental regulation. Washington Univ J Law Pol. (2010) 32:42. Available online at: https://openscholarship.wustl.edu/law_journal_law_policy/vol32/iss1/11/
225. D.o.H.a.Services H. National Report on Human Exposure to Environmental Chemicals. Centers for Disease Control and Prevention (2015).
226. Houlihan J, Kropp T, Wiles R, Gray S, Campbell C. BodyBurden: The Pollution in Newborns. Environmental Working Group (2005).
227. Silva E, Rajapakse N, Kortenkamp A. Something from “nothing”–eight weak estrogenic chemicals combined at concentrations below NOECs produce significant mixture effects. Environ Sci Technol. (2002) 36:1751–6. doi: 10.1021/es0101227
228. Rajapakse N, Silva E, Kortenkamp A. Combining xenoestrogens at levels below individual no-observed-effect concentrations dramatically enhances steroid hormone action. Environ Health Perspect. (2002) 110:917–21. doi: 10.1289/ehp.02110917
229. Christiansen S, Scholze M, Axelstad M, Boberg J, Kortenkamp A, Hass U. Combined exposure to anti-androgens causes markedly increased frequencies of hypospadias in the rat. Int J Androl. (2008) 31:241–8. doi: 10.1111/j.1365-2605.2008.00866.x
230. Orton F, Rosivatz E, Scholze M, Kortenkamp A. Competitive androgen receptor antagonism as a factor determining the predictability of cumulative antiandrogenic effects of widely used pesticides. Environ Health Perspect. (2012) 120:1578–84. doi: 10.1289/ehp.1205391
231. Thrupp TJ, Runnalls TJ, Scholze M, Kugathas S, Kortenkamp A, Sumpter JP. The consequences of exposure to mixtures of chemicals: Something from ‘nothing' and ‘a lot from a little' when fish are exposed to steroid hormones. Sci Total Environ. (2018) 619–20:1482–92. doi: 10.1016/j.scitotenv.2017.11.081
232. Zhao LJ, Ying GG, Yang B, Liu SL, Zhou J, Chen FZ, et al. Screening of multiple hormonal activities in surface water and sediment from the Pearl River system, South China, using effect-directed in vitro bioassays. Environ Toxicol Chem. (2011) 30:2208–15. doi: 10.1002/etc.625
233. Soto AM, Calabro JM, Prechtl NV, Yau AY, Orlando EF, Daxenberger A, et al. Androgenic and estrogenic activity in water bodies receiving cattle feedlot effluent in Eastern Nebraska, USA. Environ Health Perspect. (2003) 112:346–52. doi: 10.1289/ehp.6590
234. Pawlowski S, Ternes TA, Bonerz M, Rastall AC, Erdinger L, Braunbeck T. Estrogenicity of solid phase-extracted water samples from two municipal sewage treatment plant effluents and river Rhine water using the yeast estrogen screen. Toxicol in vitro (2004) 18:129–38. doi: 10.1016/j.tiv.2003.08.006
235. Shi W, Hu X, Zhang F, Hu G, Hao Y, Zhang X, et al. Occurrence of thyroid hormone activities in drinking water from eastern China: contributions of phthalate esters. Environ Sci Technol. (2012) 46:1811–8. doi: 10.1021/es202625r
236. Schriks M, Van Leerdam JASC, Van Der Linden B, Van Der Burg, Van Wezel AP, De Voogt P. High-resolution mass spectrometric identification and quantification of glucocorticoid compounds in various Wastewaters in The Netherlands. Environ Sci Technol. (2010) 44:4766–74. doi: 10.1021/es100013x
237. Van der Linden SC, Heringa MB, Man HY, Sonneveld E, Puijker LM, Brouwer A, et al. Detection of multiple hormonal activities in wastewater effluents and surface water, using a panel of steroid receptor CALUX bioassays. Environ Sci Technol. (2008) 42:5814–20. doi: 10.1021/es702897y
238. Stavreva DA, George AA, Klausmeyer P, Varticovski L, Sack D, Voss TC, et al. Prevalent glucocorticoid and androgen activity in US water sources. Sci Rep. (2012) 2:937. doi: 10.1038/srep00937
239. Liu S, Ying GG, Zhou LJ, Zhang RQ, Chen ZF, Lai HJ. Steroids in a typical swine farm and their release into the environment. Water Res. (2012) 46:3754–68. doi: 10.1016/j.watres.2012.04.006
240. Rostkowski P, Horwood J, Shears JA, Lange A, Oladapo FO, Besselink HT, et al. Bioassay-Directed identification of novel antiandrogenic compounds in bile of fish exposed to wastewater effluents. Environ Sci Technol. (2011) 45:10660–7. doi: 10.1021/es202966c
241. Kortenkamp A, Scholze M, Ermler S. Mind the gap: can we explain declining male reproductive health with known antiandrogens? Reproduction (2014) 147:515–27. doi: 10.1530/REP-13-0440
242. Office of Prevention, Toxic Substances. OPPTS 890.1300: Estrogen Receptor Transcriptional Activation (Human Cell Line (HeLa-9903)) (2009). p. 23.
243. Naylor LH. Reporter gene technology: the future looks bright. Biochem Pharmacol. (1999) 58:749–57. doi: 10.1016/S0006-2952(99)00096-9
244. Soto AM, Maffini MV, Schaeberle CM, Sonnenschein C. Strengths and weaknesses of in vitro assays for estrogenic and androgenic activity. Best Pract Res Clin Endocrinol Metabol. (2006) 20:15–33. doi: 10.1016/j.beem.2005.09.001
245. Kruger T, Hjelmborg PS, Jonsson BA, Hagmar L, Giwercman A, Manicardi GC, et al. Xenoandrogenic activity in serum differs across European and Inuit populations. Environ Health Perspect. (2007) 115 (Suppl. 1):21–7. doi: 10.1289/ehp.9353
246. Lopez-Espinosa MJ, Silva E, Granada A, Molina-Molina JM, Fernandez MF, Aguilar-Garduno C, et al. Assessment of the total effective xenoestrogen burden in extracts of human placentas. Biomarkers (2009) 14:271–7. doi: 10.1080/13547500902893744
247. Fernandez MF, Aguilar-Garduno C, Molina-Molina JM, Arrebola JP, Olea N. The total effective xenoestrogen burden, a biomarker of exposure to xenoestrogen mixtures, is predicted by the (anti)estrogenicity of its components. Reprod Toxicol. (2008) 26:8–12. doi: 10.1016/j.reprotox.2008.06.002
248. Arrebola JP, Fernandez MF, Molina-Molina JM, Martin-Olmedo P, Exposito J, Olea N. Predictors of the total effective xenoestrogen burden (TEXB) in human adipose tissue. A pilot study. Reprod Toxicol. (2012) 33:45–52. doi: 10.1016/j.reprotox.2011.10.015
249. Fernandez MF, Molina-Molina JM, Lopez-Espinosa MJ, Freire C, Campoy C, Ibarluzea J, et al. Biomonitoring of environmental estrogens in human tissues. Int J Hyg Environ Health (2007) 210:429–32. doi: 10.1016/j.ijheh.2007.01.014
250. Fernandez MF, Santa-Marina L, Ibarluzea JM, Exposito J, Aurrekoetxea JJ, Torne P, et al. Analysis of population characteristics related to the total effective xenoestrogen burden: a biomarker of xenoestrogen exposure in breast cancer. Eur J Cancer (2007) 43:1290–9. doi: 10.1016/j.ejca.2007.03.010
251. Yordy JE, Mollenhauer MA, Wilson RM, Wells RS, Hohn A, Sweeney J, et al. Complex contaminant exposure in cetaceans: a comparative E-Screen analysis of bottlenose dolphin blubber and mixtures of four persistent organic pollutants. Environ Toxicol Chem. (2010) 29:2143–53. doi: 10.1002/etc.279
252. Fernandez MF, Olmos B, Granada A, Lopez-Espinosa MJ, Molina-Molina JM, Fernandez JM, et al. Human exposure to endocrine-disrupting chemicals and prenatal risk factors for cryptorchidism and hypospadias: a nested case-control study. Environ Health Perspect. (2007) 115 (Suppl. 1):8–14. doi: 10.1289/ehp.9351
253. Vilahur N, Fernandez MF, Bustamante M, Ramos R, Forns J, Ballester F, et al. In utero exposure to mixtures of xenoestrogens and child neuropsychological development. Environ Res. (2014) 134C:98–104. doi: 10.1016/j.envres.2014.07.002
254. Ibarluzea Jm J, Fernandez MF, Santa-Marina L, Olea-Serrano MF, Rivas AM, Aurrekoetxea JJ, et al. Breast cancer risk and the combined effect of environmental estrogens. Cancer Causes Control (2004) 15:591–600. doi: 10.1023/B:CACO.0000036167.51236.86
255. Vilahur N, Molina-Molina JM, Bustamante M, Murcia M, Arrebola JP, Ballester F, et al. Male specific association between xenoestrogen levels in placenta and birthweight. Environ Int. (2013) 51:174–81. doi: 10.1016/j.envint.2012.10.004
256. Arrebola JP, Pumarega J, Gasull M, Fernandez MF, Martin-Olmedo P, Molina-Molina JM, et al. Adipose tissue concentrations of persistent organic pollutants and prevalence of type 2 diabetes in adults from Southern Spain. Environ Res. (2013) 122:31–7. doi: 10.1016/j.envres.2012.12.001
257. Ruzzin J, Lee DH, Carpenter DO, Jacobs DR, Jr. Reconsidering metabolic diseases: the impacts of persistent organic pollutants. Atherosclerosis (2012) 224:1–3. doi: 10.1016/j.atherosclerosis.2012.02.039
258. Chou HP, Lee HC, Ko CF, Lin JY, Kawanishi M, Yagi T, et al. Detection of hormone-like and genotoxic activities in indoor dust from taiwan using a battery of in vitro bioassays. Aerosol Air Qual Res. (2015) 15:1412–21. doi: 10.4209/aaqr.2015.06.0404
259. Suzuki G, Takigami H, Nose K, Takahashi S, Asari M, Sakai S. Dioxin-like and transthyretin-binding compounds in indoor dusts collected from Japan: average daily dose and possible implications for children. Environ Sci Technol. (2007) 41:1487–93. doi: 10.1021/es061907l
260. Suzuki G, Tue NM, Malarvannan G, Sudaryanto A, Takahashi S, Tanabe S, et al. Similarities in the endocrine-disrupting potencies of indoor dust and flame retardants by using human osteosarcoma (U2OS) cell-based reporter gene assays. Environm Sci Technol. (2013) 47:2898–908. doi: 10.1021/es304691a
261. Fang M, Webster TF, Ferguson PL, Stapleton HM. Characterizing the peroxisome proliferator-activated receptor (PPARgamma) ligand binding potential of several major flame retardants, their metabolites, and chemical mixtures in house dust. Environ Health Perspect. (2015) 123:166–72. doi: 10.1289/ehp.1408522
262. Fang M, Webster TF, Stapleton HM. Activation of human peroxisome proliferator-activated nuclear receptors (PPARgamma1) by semi-volatile compounds (SVOCs) and chemical mixtures in indoor dust. Environ Sci Technol. (2015) 49:10057–64. doi: 10.1021/acs.est.5b01523
263. Fang M, Webster TF, Stapleton HM. Effect-directed analysis of human peroxisome proliferator-activated nuclear receptors (PPARgamma1) ligands in indoor dust. Environ Sci Technol. (2015) 49:10065–73. doi: 10.1021/acs.est.5b01524
264. Kassotis, CD, Kollitz, EM, Hoffman, K, Sosa, JA, and Stapleton, HM. Thyroid receptor antagonism as a contributory mechanism for adipogenesis induced by environmental mixtures in 3T3-L1 cells. (in press).
265. Kollitz EM, Kassotis CD, Hoffman K, Ferguson PL, Sosa JA, Stapleton HM. Chemical mixtures isolated from house dust disrupt thyroid receptor β (TRβ) signaling. Environ Sci Technol. (2018) 52:11857–64. doi: 10.1021/acs.est.8b03283
266. Bowers RR, Temkin AM, Guillette LJ, Baatz JE, Spyropoulos DD. The commonly used nonionic surfactant Span 80 has RXRalpha transactivation activity, which likely increases the obesogenic potential of oil dispersants and food emulsifiers. General Comparat Endocrinol. (2016) 238:61–8. doi: 10.1016/j.ygcen.2016.04.029
267. Temkin AM, Bowers RR, Magaletta ME, Holshouser S, Maggi A, Ciana P, et al. Effects of crude oil/dispersant mixture and dispersant components on ppargamma activity and : identification of dioctyl sodium sulfosuccinate (DOSS; CAS #577–11–7) as a probable obesogen. Environ Health Perspect. (2015) 124:112–19. doi: 10.1289/ehp.1409672
268. Kassotis CD, Tillitt DE, Davis JW, Hormann AM, Nagel SC. Estrogen and androgen receptor activities of hydraulic fracturing chemicals and surface and ground water in a drilling-dense region. Endocrinology (2014) 155:897–907. doi: 10.1210/en.2013-1697
269. Kassotis CD, Iwanowicz LR, Akob DM, Cozzarelli IM, Mumford AC, Orem WH, et al. Endocrine disrupting activities of surface water associated with a West Virginia oil and gas industry wastewater disposal site. Sci Total Environ. (2016) 557–8:901–10. doi: 10.1016/j.scitotenv.2016.03.113
270. Kassotis CD, Vu DC, Vo PH, Lin HC, Cornelius-Green JN, Patton S, et al. Endocrine disrupting activities and organic contaminants associated with oil and gas operations in wyoming groundwater. Arch Environ Contamin Toxicol. (2018) 75:247–58. doi: 10.1007/s00244-018-0521-2
271. Cozzarelli IM, Skalak KJ, Kent DB, Engle MA, Benthem A, Mumford AC, et al. Environmental signatures and effects of an oil and gas wastewater spill in the Williston Basin, North Dakota. Sci Total Environ. (2017) 579:1781–93. doi: 10.1016/j.scitotenv.2016.11.157
272. Kassotis CD, Kollitz EM, Ferguson PL, Stapleton HM. Nonionic ethoxylated surfactants induce adipogenesis in 3T3-L1 cells. Toxicol Sci. (2018) 162:124–36. doi: 10.1093/toxsci/kfx234
273. Getzinger GJ, O'Connor MP, Hoelzer K, Drollette BD, Karatum O, Deshusses MA, et al. Natural gas residual fluids: sources, endpoints, and organic chemical composition after centralized waste treatment in Pennsylvania. Environ Sci Technol. (2015) 49:8347–55. doi: 10.1021/acs.est.5b00471
274. Ferrer I, Thurman EM. Chemical constituents and analytical approaches for hydraulic fracturing waters. Trends Environ Anal Chem. (2015) 5:18–25. doi: 10.1016/j.teac.2015.01.003
275. Thurman EM, Ferrer I, Blotevogel J, Borch T. Analysis of hydraulic fracturing flowback and produced waters using accurate mass: identification of ethoxylated surfactants. Anal Chem. (2014) 86:9653–61. doi: 10.1021/ac502163k
276. Knudsen TB, Keller DA, Sander M, Carney EW, Doerrer NG, Eaton DL, et al. FutureTox II: in vitro data and in silico models for predictive toxicology. Toxicol Sci. (2015) 143:256–67. doi: 10.1093/toxsci/kfu234
277. Schwarzman MR, Ackerman JM, Dairkee SH, Fenton SE, Johnson D, Navarro KM, et al. Screening for chemical contributions to breast cancer risk: a case study for chemical safety evaluation. Environ Health Perspect. (2015) 123:1255–64. doi: 10.1289/ehp.1408337
278. Thomas RS, Black MB, Li L, Healy E, Chu TM, Bao W, et al. A comprehensive statistical analysis of predicting in vivo hazard using high-throughput in vitro screening. Toxicol Sci. (2012) 128:398–417. doi: 10.1093/toxsci/kfs159
279. Martin MT, Knudsen TB, Reif DM, Houck KA, Judson RS, Kavlock RJ, et al. Predictive model of rat reproductive toxicity from ToxCast high throughput screening. Biol Reprod. (2011) 85:327–39. doi: 10.1095/biolreprod.111.090977
280. Sipes NS, Martin MT, Reif DM, Kleinstreuer NC, Judson RS, Singh AV, et al. Predictive models of prenatal developmental toxicity from ToxCast high-throughput screening data. Toxicol Sci. (2011) 124:109–27. doi: 10.1093/toxsci/kfr220
281. Liu J, Mansouri K, Judson RS, Martin MT, Hong H, Chen M, et al. Predicting hepatotoxicity using ToxCast in vitro bioactivity and chemical structure. Chem Res Toxicol. (2015) 28:738–51. doi: 10.1021/tx500501h
Keywords: endocrine disrupting chemicals, obesogen, diabetogen, adipogenesis, 3T3-L1, obesity, diabetes
Citation: Kassotis CD and Stapleton HM (2019) Endocrine-Mediated Mechanisms of Metabolic Disruption and New Approaches to Examine the Public Health Threat. Front. Endocrinol. 10:39. doi: 10.3389/fendo.2019.00039
Received: 28 September 2018; Accepted: 17 January 2019;
Published: 07 February 2019.
Edited by:
Robert Sargis, University of Illinois at Chicago, United StatesReviewed by:
Jacqueline M. Stephens, Louisiana State University, United StatesMark Andrew Lawson, University of California, San Diego, United States
Copyright © 2019 Kassotis and Stapleton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher D. Kassotis, Y2hyaXN0b3BoZXIua2Fzc290aXNAZHVrZS5lZHU=