 Rainer Girgert
Rainer Girgert Carsten Gründker
Carsten Gründker- Department of Gynecology and Obstetrics, University Medicine Göttingen, Göttingen, Germany
Estrogen receptors are important regulators of the growth of breast tumors. Three different receptors for estrogens have been identified in breast tumors, two nuclear receptors, ERα and ERβ, and a G-protein coupled estrogen receptor 1 (GPER) that initiates non-genomic effects of estrogens in the cytosol. Recent findings show that the stimulation of cytoplasmic ERα and ERβ also triggers non-genomic signaling pathways. The treatment of breast cancer with anti-estrogens depends on the presence of ERα. About 40% of all breast cancers, however, do not express ERα. One subgroup of these tumors overexpress Her-2, another important group is designated as triple-negative breast cancer, as they neither express ERα, nor progesterone receptors, nor do they overexpress Her-2. This review addresses the signaling of ERβ and GPER in ERα-negative breast tumors. In addition to the well-established EGF-receptor transactivation pathways of GPER, more recent findings of GPER-dependent activation of FOXO3a, the Hippo-pathway, and HOTAIR-activation are summarized.
Nuclear Estrogen Receptors
Estrogens, in particular 17β-estradiol, exert biological effects in a wide variety of tissues. These effects are dependent on the presence of an appropriate receptor for estrogens. The first estrogen receptor was cloned and characterized in the eighties of the last century from cDNA of the human breast cancer cell line MCF-7 (1). This receptor is a designated estrogen receptor α (ERα) in order to distinguish it from ERβ, which was discovered later. Both receptors are ligand-activated transcription factors that act by binding to DNA in the nucleus. The chromosomal location of the human ERα gene was detected on chromosome 6q25, whereas the gene for human ERβ is located at 14q22-24 (2).
Both estrogen receptors, ERα and ERβ, share common structural features. Five different domains, named A/B, C, D, E, and F, are distinguished in both receptors with varying sequence homology. The N-terminal A/B domain is the most variable and these regions of the human ERα and ERβ share only a <20% identity of the amino acid sequence. The A/B-domain contains the activation function (AF-1) that has been found to be ligand-independent and to possess promoter- and cell-specific activity (3). The C-domain that shares a 95% identity in amino acid sequence in both receptors is responsible for specific DNA-binding to promoters of the target genes. The next domain (D-domain) shares only 30% of homology between the two receptors. It forms a flexible hinge between the DNA-binding C-domain and the E-domain that binds the ligand (17β-estradiol).The D-domain is therefore a designated ligand-binding domain (LBD). This domain harbors the hormone-dependent activation function AF-2 (3).
The mechanism of action of ERα and ERβ is quite similar. Before ligand-binding, both receptors are localized in the cytosol of the cells complexed with the chaperon HSP90. The lipophilic ligands, like 17β-estradiol, enter the cells passively by diffusion through the cell membrane. The binding of the ligand leads to a conformational change of the receptors that releases HSP90 from both estrogen receptors into the cytosol. Upon the binding of estrogens, the ERs form dimers and undergo conformational changes that enable them to initiate gene transcription. The estrogen-bound receptors enter the nucleus and bind with their C-domain to the estrogen-responsive elements (ERE), specific DNA-sequences of the promoters of target genes (Figure 1A). The sequence of estrogen-response elements may vary slightly between different target genes, but all have in common a palindromic repeat of nucleotides. For example, the symmetric sequence of the ERE of the vitellogenin gene of Xenopus laevis was determined with the sequence: CAGGTCAnnnTGACCTG (4). Estrogen receptors bound to the promoters initiate the transcription of the respective estrogen receptor target gene into mRNA. To start transcription, a number of cofactors/coactivators and a RNA-polymerase have to bind to the ligand-activated ERs at the promoter. The activated RNA-polymerase synthesizes the m-RNA of the estrogen-dependent target genes and these mRNAs are exported from the nucleus to the ribosomes where they are translated to proteins. These processes are called genomic effects of estrogen and require minutes to hours until they are completed (5). But in addition a number of rapid cytosolic effects of many steroid hormones, like estrogens, androgens and corticosteroids were described in the literature as designated non-genomic effects (Figures 1B,C). These effects are already detectable within seconds or a few minutes. The most important characteristics that distinguish genomic estrogen effects from the fast non-genomic effects is the fact that the genomic effects are inhibitable by inhibitors of RNA-polymerases like cycloheximide and actinomycin D.
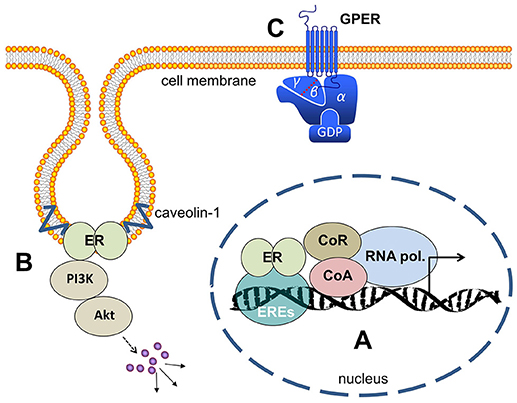
Figure 1. Estrogen receptors (A) ER's (either ERα or ERβ-homodimers, or α/β-heterodimers) bind to the ERE (estrogen responsive elements) in the nucleus and activate gene transcription. (B) At the cell membrane ER's bound to caveolin and activate cytosolic signaling like PI3-kinase/Akt and MAP-kinases. (C) GPER is a membrane-integrated 7-transmembrane receptor activating heterotrimeric G-proteins after estrogen binding and eliciting various signaling pathways as described in the main text. ER, estrogen receptor; ERE, estrogen responsive elements; CoA, co-activator; CoR, co-repressor; PI3K, phosphoinositol-3-kinase; GPER, G-protein coupled estrogen receptor 1; GDP, guanosindiphosphat.
Role of ERα in Breast Cancer
It is well established that the expression of ERα is associated with a clinical response of breast tumors to endocrine therapy, and therefore, it is also associated with a good prognosis of the treated breast cancer patients (6). Three ERα isoforms are mainly distinguished in mammals: full length ERα having a molecular weight of 66 kD and two truncated isoforms, ERα36 and ERα46 with molecular weights of 36 and 46 kD, respectively. These truncated isoforms of ERα lack the first exon of the ERα gene that encodes the activation function (AF-1) (7). Before the discovery of the G-protein coupled estrogen receptor 1 (GPER), ERα36 had been suspected to preferentially mediate most of extra nuclear effects of estrogens. ERα36 is primarily located in the plasma membrane and shows the activation of MAP-Kinases and PI3K. It has been discussed to contribute to endocrine resistance (8). The expression of ERα36 has been detected in breast, endometrial, colorectal, gastric, and hepatic cancer (9). Little is known about the distribution of ERα46, with studies focusing mostly on the vascular effects of ERα46 (10). Observations in MCF-7 breast cancer cells showed that activated ERα also directly interacts with several signaling molecules (11). For example, estradiol induces ERα nuclear export through a mechanism dependent on exportin-1 (XPO1), also known as chromosomal maintenance 1 (CRM1) (11). Several additional findings show that the stimulation of nuclear estrogen receptors located signaling pathways (12).
ERα-negative breast tumors represent 44% of all breast cancer cases. Seventeen percent of these ERα-negative breast tumors additionally overexpress Her-2, the target of trastuzumab and other anti Her-2 directed therapies. The majority of the remaining 81% of ERα-negative breast tumors that do not overexpress Her-2 nor express progesterone receptors is referred to as triple-negative breast cancers. Only two percent of the ERα(-) breast tumors not overexpressing Her-2 are positive for the progesterone-receptor (13). The second nuclear estrogen receptor ERβ is expressed in 44.4% of these triple-negative breast tumors (14).
The Role of ERβ in Breast Cancer
ERα and ERβ are differently distributed in human breast tissues. The expression of ERα is mainly restricted to nuclei of epithelial cells in lobules and ducts of the healthy breast. ERβ is also expressed in normal breast tissue (15). In addition, ERβ is detectable in myoepithelial cells as well as in surrounding stromal and endothelial cells (16). Increased levels of ERβ in the normal mammary gland were associated with a decreased risk of developing breast cancer (17).
About 60% of all breast cancers that do not express ERα were tested positively for ERβ expression (18). ERβ has been shown to possess a weaker activation function (AF1) than ERα and therefore ERβ is able to repress the transcriptional activity of ERα (19). The importance of ERβ expression on the prognosis of breast cancer is still not clear as ERβ displays different roles depending on the presence or absence of ERα. (20). Early studies supposed that ERβ is a carcinogenic factor in breast tissues (21). However, in more recent studies ERβ is described as a dominant negative regulator of estrogen signaling, since ERβ repressed ERα mediated transcription by forming heterodimers with ERα (22).
A number of isoforms of ERβ were identified as the resultant of alternative splicing of either exon 8 or exon 9 of the ERβ gene (23). The existence of these five different ERβ isoforms complicates the elucidation of the physiological role of ERβ and its involvement in the carcinogenesis of breast cancer (24). Honma et al. (25) analyzed 442 breast cancer patients treated with adjuvant tamoxifen for the impact of ERβ1 expression on overall survival rates. Patients with ERβ1 negative tumors showed a significantly worse overall survival rate than patients with tumors expressing ERβ1. Patients with ERα- and progesterone-receptor negative breast tumors expressing ERβ1 presented with a better prognosis irrespective of whether the tumors expressed Her-2 or not (25, 26).
The other four isoforms of ERβ (β2, β3, β4, and β5) turned out to be unable to bind estrogens (27). ERβ2 is the best studied isoform of all known splice variants of ERβ. It is described as a dominant-negative inhibitor of ERα, as it forms ERβ2/ERα heterodimers that cause a proteasome-dependent degradation of ERα, leading to a suppression of ERα-regulated genes (28). ERβ3 expression was found to be restricted only to the testis. ERβ4 and ERβ5 are truncated transcripts of the ERβ gene that lack ligand binding but these isoforms of ERβ dimerize with ERβ1 and enhance its transcriptional activity in a ligand-dependent manner (29). The best studied isoforms of ERβ are ERβ1 and ERβ2, and their characteristics are summarized in Table 1. The expression of ERβ1 in breast cancer is lower than the expression of ERβ2 and ERβ5 and was detected in 80% of epithelial cells (17). ERβ1 was primarily localized in the nucleus and ERβ2 was detected in the cytoplasm of the tumor cells (30). ERβ1 is able to bind estrogens eliciting transcriptional activity but ERβ2 did not. ERβ1 preferentially forms dimers with ERβ4 and ERβ5 whereas ERβ2 mainly dimerises with ERβ1 (29). ERβ1 binds stronger to the ERE of DNA than ERβ2 and ERβ2 has a higher affinity to the ERE than ERβ5 (23, 31). Several studies investigated the impact of the various ERβ-isoforms on the prognosis of breast cancer. Sugiura et al. observed a better overall survival rate of breast cancer patients that expressed high amounts of either ERβ1 and ERβ2 (32). Similar observations for ERβ1 and ERβ2 were reported from other clinical studies (33, 34). Disease-free survival rates of ERα-negative patients was also improved when tumors expressed both ERβ-isoforms (35). In contrast, Shaaban et al. (36) and Leung et al. (27) reported a longer disease-free survival when ERβ2 was highly expressed in the nucleus of breast cancer cells, and Chantzi et al. observed a poor DFS whenf nuclear ERβ2 was high (37).
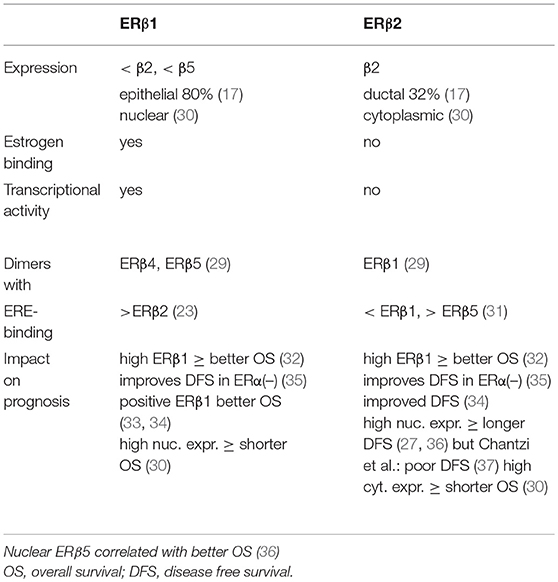
Table 1. Characteristics of ERβ isoforms ERβ1 and ERβ2.
Very scarce information is available about the impact of ERβ5 on the outcome of patient. Nuclear ERβ5 correlated with better overall survival rates in breast cancer patients (36).
In patients with stage II breast tumors, the expression of ERβ correlated with longer disease-free survival rates, because the formation of heterodimers of ERα and ERβ reduces the transcriptional activity of ERα (38). In contrast, in node-positive, breast cancer ERβ expression is a biomarker of more aggressive growth as it conveys a higher risk of relapse (39).
ERβ knock-out mice present with quite a normal ductal architecture of the mammary gland ruling out an important role of ERβ in mammary gland development (40). On the other hand, a simultaneous somatic loss of ERβ and the tumor suppressor gene p53 was shown to induce breast tumors in ERβ(−/−)/p53(−/−) transgenetic mice (41). The expression of ERβ decreases during breast cancer progression and in higher malignant tumors ERβ-expression is remarkably low (28, 42).
It has been reported that ERβ has opposing effects on the cyclin D1 promoter compared to ERα. 17β-estradiol bound to ERβ repressed cyclin D1 gene transcription and blocked ERα-mediated induction of cyclin D1 when both receptors were present in Hela cells. This is an indication that ERβ is able to modulate the proliferative effects of 17β-estradiol bound to ERα (43). As a further mechanism of the transcriptional activity of estrogens-bound ERα, it was observed that ERα is tethered to the AP-1 site and activates transcription from AP-1 sites of a number of promoters (44). In contrast, 17β-estradiol bound to ERβ had no effects on transcription from AP-1 sites (45).
Besides estrogen receptors, many breast cancer cells also express androgen receptors and become obviously detectable in cells lacking ERα effects of androgens. A comprehensive review on the role of androgen receptors was most recently published by Giovannelli et al. (46).
One particular subtype of ERα-negative breast cancer is triple-negative breast cancer (TNBC) characterized by the lack of ERα and progesterone receptor expression and the absence of Her-2 overexpression. Sixty percent of TNBC tumors were positively stained for ERβ (47). In TNBC, overexpression of the receptor for the epidermal growth factor is one hallmark for increased proliferation of these tumors (48). The expression of ERβ was shown to repress transcription of EGFR (49). In ERα-negative breast tumors, a high nuclear expression of ERβ1 was found particularly in histologically low-grade tumors when ERβ2 was also present in the nuclei. ERβ2 is a marker of early disease recurrence (50). Nuclear ERβ2 is generally associated with poor disease-free survival rates in breast cancer patients. But closer inspections of the function of ERβ lead to the conclusion that its expression seems more likely to have an activity preventing carcinogenesis. In breast cancer tissue the expression of ERβ is lower than in normal breast tissue or benign breast lesions (51). Overexpression of ERβ was shown to downregulate ERα expression by recruiting the corepressor NCoR to the promoter of ERα. In this way, ERβ was able to downregulate the growth-promoting effect of ERα (52). The adenoviral transfer of ERβ induced an increased expression of p21 and p27, causing a cell cycle arrest in G2-phase of cell division (51). In addition, the expression of cyclin D1, a major regulator of the cell cycle entry, has been shown to be inhibited in the presence of ERβ (43).
But ERβ does not only have anti-proliferative effects on breast cancer cells, it also reduces the migration and invasion of these cells, probably by means of repressing the expression of MMP9, necessary for the invasion of the extracellular matrix (53). The agonists of ERβ, liquiritigenin and ERB-041, reduced invasion of TNBC cell lines HCC1806 and HCC1937 into a synthetic extracellular matrix by up to 82%. This effect was accompanied by a significant decrease of the chemokine receptor CXCR4 (54).
Functions of Membrane-Bound Estrogen Receptor GPER
In addition to the well-characterized genomic responses to estrogen stimulation, some fast actions of estrogens that are elicited at the plasma membrane were described in a great variety of tissues. In 1986, Emons et al. provided data of rapid non-genomic effects of estradiol in pituitary gonadotrophs (55). Using a fluorescent estradiol macromolecular complex (E2-BSA-FITC) the steroid binding to membranes was analyzed on living target cells. Two types of 17β-estradiol-binding sites were detected on hormone-sensitive MCF-7 cells. One was rapidly saturated at low concentrations and a second one had a lower affinity to the macromolecular estradiol complex (56).
For a long time, a membrane-bound ERα was supposed to be responsible for extra-nuclear effects of estrogens (57). The generation of cAMP by adenylyl cyclase is considered to be a process that occurs at the plasma membrane of cells. In MCF-7 breast cancer cells, a 10-fold increase of intracellular cAMP was observed after half an hour of stimulation with 17β-estradiol (58). Estrogen is also able to increase intracellular calcium concentration. In isolated rabbit uteri, estrogen treatment doubled cellular calcium uptake (59). In cultured pituitary cells from adult female rats, the acute negative 17β-estradiol effect on GnRH-induced LH release was found to be mediated via a non-genomic mechanism (60). The authors speculated that the negative effect of 17β-estradiol might be mediated via a modulation of PKC activation. In female rat osteoblasts, 17β-estradiol increased intracellular calcium within the first 2 min of stimulation. This effect was partially inhibited by the inhibitor verapamil giving evidences for an influx of calcium from the extracellular milieu and by thapsigargin, an inhibitor of calcium stores in the endoplasmic reticulum. The initial event leading to increased cytosolic calcium is the hydrolysis of phosphatidylinositol 4,5-biphosphate by phospholipase C generating inositol 1,4,5 triphosphate (IP3) that triggers release from calcium-stores and diacylglycerol. Inhibitors of phospholipase C and pertussis toxin abolished the increase of calcium in the cytosol by 17β-estradiol (61). In addition, tamoxifen, an inhibitor of the genomic effects of ERα was not able to block the rapid increase of Ca2+, IP3 and diacylglycerol. The increased calcium concentration in the cytosol activated the mitogen-activated protein kinase (MAP-Kinase) (62).
For many years, the real nature of the estrogen receptor responsible for the rapid effects of 17β-estradiol remained unknown. First experiments detecting estrogen-binding capacities on endometrial cells were performed, using membrane-impermeable 17β-estradiol coupled to bovine serum albumin (57). On MCF-7, a breast cancer cell line expressing the nuclear estrogen receptor ERα, 17β-estradiol covalently linked to a fluorescein-labeled serum albumin bound saturably with a high affinity to the cell membrane. On MDA-MB-231 cells lacking ERα expression, no such binding sites for 17β-estradiol were detectable. This observation was initially explained by the presence of a membrane-resident ERα that might be responsible for the observed rapid estrogen signaling in breast cancer cells (56). In the Chinese hamster, ovary cell transiently transfected with ERα cDNA binding studies with labeled 17β-estradiol revealed that the receptor is detectable in the nucleus as well as in the cell membrane with almost identical dissociation constants for estradiol, but the receptor number in the cell membrane was only 3% of the nuclear receptor density (63). Using a number of ERα-specific antibodies, it was possible to detect three different epitopes of ERα on GH3/B6 rat pituitary tumor cells. It remained unknown how these ERα molecules are fixed at the plasma membrane. With the help of a computer-assisted hydrophobicity analysis of the ERα amino acid sequence, the region between AS 381 and AS 397 was identified as putative membrane-spanning domain of ERα (64).
The real nature of the membrane bound estrogen receptor is now well established. The results of Lieberherr et al. already pointed to an involvement of a receptor coupled to a heterotrimeric G-protein in the non-genomic effects of 17β-estradiol (61). Using differential cDNA library screening, a gene called GPR30, was identified, and later proved to be responsible for many of the non-genomic, membrane-initiated effects of estrogens. GPR30 (Figure 1C) is a 7-transmembrane protein belonging to the large family of G-protein coupled receptors that are embedded into cellular membranes (65, 66). In more recent literature, GPR30 has been renamed to G-protein coupled estrogen receptor 1 (GPER) to better describe this receptor's functionality. Meanwhile, a great number of natural and synthetic GPER-agonists and –antagonists were identified. Some phytoestrogens, like genistein and coumestrol, the anticancer agents 4-hydroxytamoxifen and fulvestrant, the synthetic estrogen, diethylstilbestrol, many pesticides, like DDT and DDE, herbicides, like atrazine and chemical compounds, bisphenol A and nonylphenol are known to be GPER-agonists, apart from 17β-estradiol (67). In addition to the selective estrogen receptor modulator tamoxifen, the complete ERα antagonist Fulvestrant (ICI 182,780) binds to GPER and activates signaling pathways in breast cancer cells, thus leading to the stimulation of proliferation (68).
The synthetic GPER agonist G1 that has a higher affinity to GPER than to ERα is frequently used as a GPER-agonist in experiments analyzing the signaling pathways of GPER. The discovery that 4-hydroxytamoxifen is a GPER-agonist led to the conclusion that GPER may contribute to the emergence of tamoxifen-resistant breast cancer. Ignatov et al. have shown that expression of GPER is in fact increased in breast tumors with acquired tamoxifen resistance (69).
The estrogen estriol has been identified as a natural GPER antagonist (70). A further inhibitor of GPER signaling, the substituted dihydroquinoline G15, was identified in a biomolecular screening of a compound library of small molecules (NIH-MLSMR). It binds to GPER with an affinity of 20 nM. G15 was tested in the GPER expressing breast cancer cells SKBr3 and was able to effectively block calcium mobilization by 17β-estradiol (71). Meanwhile, a compound named G36 was developed as improved antagonists with a higher affinity to GPER (72).
The localization of GPER has been controversially debated in literature for many years. GPER is known to be a G-protein coupled with the 7-transmembrane receptor this fact implies that GPER is an integral membrane protein. However, the expression of a fusion protein of GPER and green fluorescent protein in monkey kidney cells (COS7) pointed to an intracellular localization of GPER as GPER co-localized with the endoplasmic marker protein KDEL (73). Otto et al. reported that GPR30 localizes to the endoplasmic reticulum in COS7- and HEK293 cells but is not activated by estradiol (74).
On the other hand, in HeLa-cells transfected with an N-terminally FLAG-tagged GPER, expression of this protein was detected in the plasma membrane (66). The localization of GPER in the plasma membrane was confirmed through the experiments conducted by Sanden et al. (75). They showed that in Mardin-Darby canine kidney cells and in the human breast cancer cell line T47D, 17β-estradiol stimulated cAMP synthesis in a GPER dependent manner, a process defined as a plasma membrane event. In addition, they observed a GPER-dependent recruitment of β-arrestin2 to the plasma membrane of estradiol-stimulated cells. The staining of living cells showed that GPER also localized to cytoplasmic intermediate filaments that contained cytokeratins, CK7 and CK8, but not to the endoplasmic reticulum (75). This subcellular distribution of GPER between plasma membrane and cytoplasm was confirmed in the breast cancer cell line MCF-7 and in sections of a number of breast tumors (76).
In an excellent experimental series, Broselid et al. (77) highlighted the mechanisms of how GPER is localized to the plasma membrane. In HEK293 cells GPER forms a complex with MAGUK and AKAP5, where both proteins are associated with the plasma membrane. This binding was shown to depend on a C-terminal PDZ motive present in GPER. An N-terminally FLAG-tagged GPER-construct lacking this PDZ motive omitted the binding of GPER to the plasma membrane and disrupted 17β-estradiol dependent GPER signaling (77).
Signal Transduction of GPER
Many different signal transduction pathways are activated by GPER after the binding of 17β-estradiol and are described in detail below (Figure 2).
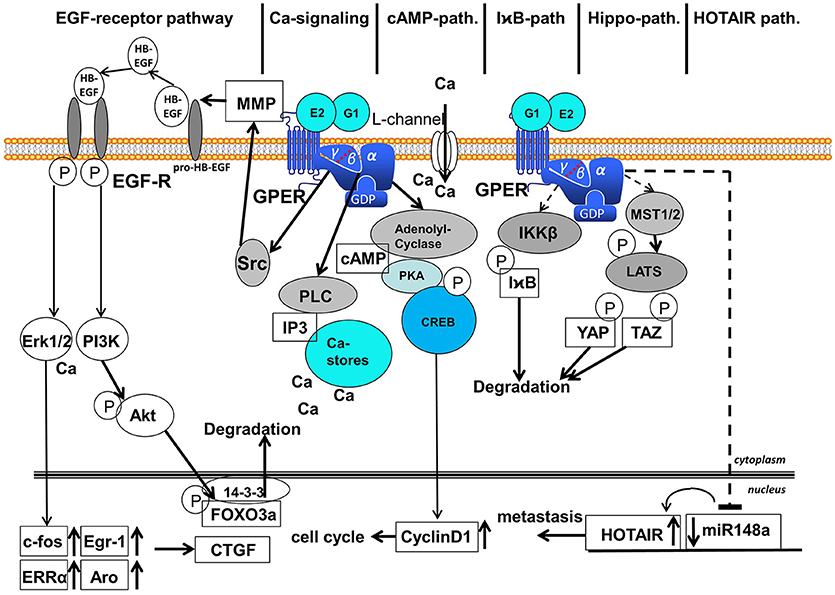
Figure 2. Signaling pathways activated after binding of E2 and G1 to GPER. Six different signaling pathways are distinguished: (1) EGF-receptor-pathway, (2) calcium-signaling, (3) cAMP-pathway, (4) IχB-pathway, (5) Hippo-pathway, and (6) HOTAIR-pathway. EGF, epidermal growth factor; MMP, matrix-metalloproteases; GPER, G-protein coupled estrogen receptor 1; Erk1/2, extracellular regulated kinase 1 and 2; PI3K, phosphoinositol-3-kinase; PLC, phospholipase C; IP3, inositol-triphosphate; cAMP, cyclic adenosine-monophosphate; PKA, protein kinase A; CREB, cAMP-responsive element binding protein; I B, inhibitor of ; IKKβ, inhibitor of kinase β; MST1/2, mammalian sterile 20-like 1 and 2; LATS, large tumor suppressor; YAP, yes-associated protein; TAZ, transcriptional coactivator with PDZ binding motif; HOTAIR, HOX-transcript antisense intergenic RNA; FOXO3a, forkhead box 3a; CTGF, connective tissue growth factor; After binding of 17β-estradiol or G1 to GPER various signaling pathways are activated in the cytosol. Ligand binding to GPER leads to the detachment of the βγ-subunit of heterotrimeric G-proteins. The EGF-receptor pathway starts with the activation of the kinase Src by βγ-G-protein that activates MMPs, liberating EGF from heparin-bound EGF. EGF binding to EGF-receptor leads to auto-phosphorylation of the EGF-receptor that activates MAP-kinase Erk1/2 and PI3-kinase. Activated Erk induces transcription of c-fos, Egr-1, ERRα and aromatase in the nucleus followed by the induction of CTGF. PI3-kinase is also activated by the phosphorylated EGF-receptor and phosphorylates the kinase Akt that phosphorylates transcription factor FOXO3a in the nucleus that is subsequently exported to the cytosol, where FOXO3a is degraded. The Calcium signaling pathway starts with the released α-subunit of heterotrimeric G-proteins that activates phospholipase C. PLC cleaves Phosphatidylinositol-4,5-bisphosphat to diacylglycerol and IP3. IP3 releases calcium ions from cytosolic calcium-stores. Ca2+ activates several enzymes in the cytosol among other things Ca2+ additionally activates Erk1/2. Binding of 17β-estradiol to GPER also opens calcium L-channels in the plasma membrane by a yet unsolved mechanism. The released α-subunit of the heterotrimeric G-protein also activates the adenolyl-cyclase (AC) in the cytosol. cAMP generated by AC activates protein kinase A (PKA) that phosphorylates the cAMP response element binding protein (CREB). Phosphorylated CREB binds as a transcription factor to promoters of genes containing a cAMP response element, for example cyclinD1, that supports the progress of the cell cycle. Less elucidated are the IB-pathway, Hippo-pathway and HOTAIR-pathway represented in this figure by hatched arrows. In the course of the IB-pathway IKKβ is activated that phosphorylates IB, an inhibitor of NF-B. Phosphorylated IB is degraded via the ubiquitination pathway allowing NFB action. In the course of the Gα-protein dependent Hippo-pathway MST1/2 are activated that phosphorylates LATS, a kinase phosphorylating the transcription factors YAP and TAZ that are subsequently degraded. In the HOTAIR-pathway expression of miR148a is inhibited in a Gα-dependent manner, but the intermediate steps leading to transcription of this microRNA are not completely elucidated, in this figure exemplified by a hatched arrow. Suppression of miR148a expression leads to an increased expression of HOTAIR that finally supports metastasis.
B, inhibitor of ; IKKβ, inhibitor of kinase β; MST1/2, mammalian sterile 20-like 1 and 2; LATS, large tumor suppressor; YAP, yes-associated protein; TAZ, transcriptional coactivator with PDZ binding motif; HOTAIR, HOX-transcript antisense intergenic RNA; FOXO3a, forkhead box 3a; CTGF, connective tissue growth factor; After binding of 17β-estradiol or G1 to GPER various signaling pathways are activated in the cytosol. Ligand binding to GPER leads to the detachment of the βγ-subunit of heterotrimeric G-proteins. The EGF-receptor pathway starts with the activation of the kinase Src by βγ-G-protein that activates MMPs, liberating EGF from heparin-bound EGF. EGF binding to EGF-receptor leads to auto-phosphorylation of the EGF-receptor that activates MAP-kinase Erk1/2 and PI3-kinase. Activated Erk induces transcription of c-fos, Egr-1, ERRα and aromatase in the nucleus followed by the induction of CTGF. PI3-kinase is also activated by the phosphorylated EGF-receptor and phosphorylates the kinase Akt that phosphorylates transcription factor FOXO3a in the nucleus that is subsequently exported to the cytosol, where FOXO3a is degraded. The Calcium signaling pathway starts with the released α-subunit of heterotrimeric G-proteins that activates phospholipase C. PLC cleaves Phosphatidylinositol-4,5-bisphosphat to diacylglycerol and IP3. IP3 releases calcium ions from cytosolic calcium-stores. Ca2+ activates several enzymes in the cytosol among other things Ca2+ additionally activates Erk1/2. Binding of 17β-estradiol to GPER also opens calcium L-channels in the plasma membrane by a yet unsolved mechanism. The released α-subunit of the heterotrimeric G-protein also activates the adenolyl-cyclase (AC) in the cytosol. cAMP generated by AC activates protein kinase A (PKA) that phosphorylates the cAMP response element binding protein (CREB). Phosphorylated CREB binds as a transcription factor to promoters of genes containing a cAMP response element, for example cyclinD1, that supports the progress of the cell cycle. Less elucidated are the IB-pathway, Hippo-pathway and HOTAIR-pathway represented in this figure by hatched arrows. In the course of the IB-pathway IKKβ is activated that phosphorylates IB, an inhibitor of NF-B. Phosphorylated IB is degraded via the ubiquitination pathway allowing NFB action. In the course of the Gα-protein dependent Hippo-pathway MST1/2 are activated that phosphorylates LATS, a kinase phosphorylating the transcription factors YAP and TAZ that are subsequently degraded. In the HOTAIR-pathway expression of miR148a is inhibited in a Gα-dependent manner, but the intermediate steps leading to transcription of this microRNA are not completely elucidated, in this figure exemplified by a hatched arrow. Suppression of miR148a expression leads to an increased expression of HOTAIR that finally supports metastasis.
Calcium-Signaling Induced by GPER
Calcium ions are known as essential intracellular messengers the regulate many cellular processes, like enzyme activity, muscle contraction or hormone secretion (78). Even the rapid increase of intracellular calcium concentration by 17β-estradiol that has initially been attributed to membrane-resident ERα has been shown to depend on the presence of GPER. Cos7-cells known to be negative for ERα were transfected with GPER. In these transfected cells, a similarly strong calcium mobilization was evoked 20 s after stimulation with 0.1 nM 17β-estradiol as in Cos7 cells expressing ERα after transfection with ERα-GFP (73). This observation pointed to the presence of a third estrogen receptor different from the previously described nuclear receptors, ERα and ERβ. But GPER does not only affect calcium mobilization from intracellular stores. In rat myometrium cells the GPER-specific agonist G1, induced a membrane depolarization by opening the nifedipine sensitive, L-type calcium channels in the cell membrane that allowed calcium influx from the extracellular environment (79).
Activation of MAP-Kinases
The signaling events triggered by the binding of 17β-estradiol to GPER were analyzed in detail by Filardo et al. (68). They observed the activation of MAP-kinase Erk after exposure to 17β-estradiol in a number of human breast cancer cell lines exhibiting different ERα expression profiles. Even in breast cancer cell line SKBr3 that expresses neither ERα nor ERβ, they determined a 6-fold increase of Erk phosphorylation after the stimulation of these cells with 1 nM 17β-estradiol for 5 min. On the other hand, in the cell line MDA-MB-231 that lacks expression of GPER, an increase of Erk phosphorylation was not induced by estrogen stimulation. After transfection of MDA-MB-231 cells with a vector containing the GPER gene, these cells became estrogen-responsive and Erk phosphorylation was clearly detectable following stimulation with 17β-estradiol. In the same experimental setting, the authors observed that this estrogen-mediated, GPER-dependent, Erk activation was sensitive to pertussis toxin pointing to an involvement of the βγ-subunits of heterotrimeric G-proteins. Additional experiments, using src family tyrosine kinase inhibitor PP2 revealed that this pertussis toxin sensitive Erk activation by 17β-estradiol requires the activity of the cytosolic kinase src. As the missing link between estrogen-activated src and the E2-activated MAP-Kinase Erk, the necessity of cleavage of heparin-bound EGF was recognized (68). From earlier work by Prenzel et al. (80), it is known that stimulation of G-protein coupled receptors in Rat-1 fibroblasts with endothelin-1 or thrombin activates membrane-bound metalloproteinases that release EGF from heparin-bound EGF (HB-EGF) to stimulate EGF-receptor, finally leading to Erk activation (68, 81).
Induction of Transcription Factors by Estrogens via GPER
In the nucleus, activated Erk increases expression of proliferation-inducing genes, like c-fos, and cyclinD1. The induction of c-fos and cyclinD1 expression by 17β-estradiol has particularly been shown in ERα/ERβ negative cell line SKBr3 and in triple-negative breast cancer cell lines (82, 83). A microarray analysis of gene expression in the GPER-expressing breast cancer cell line SKBr3, after stimulation with estradiol and with the GPER agonist tamoxifen, revealed that the connective tissue growth factor CTGF was induced nearly 10-fold and the deletion of GPER by siRNA prevented the induction of CTGF. In addition, several transcription factors, including c-fos and Egr1, were found to be upregulated by 17β-estradiol and tamoxifen using gene expression profiling of the RNA of the stimulated cells. From these experiments, CTGF appears to be a target of these transcription factors (84). CTGF is more prominently known as mediator of tissue fibrosis but has also been reported to be involved in the tumorigenesis of breast cancer (85).
Activation of PI3-Kinase
Besides Erk activation, the transactivation of the EGF-receptor by estradiol also activates PI3-kinase. The stimulation of an ERα-negative endometrial carcinoma cell line (Hec50) with 17β-estradiol or the GPER-specific agonist G1 was able to increase PI3-Kinase activity (86).
Downstream of PI3-kinase, the activation of the kinase Akt phosphorylates the transcription factor FOXO3a, a member of the forkhead-box gene family that is recognized as a tumor suppressor gene (87). In case of reduced energy supply as it is common in large solid tumors FOXO3a also acts as important inhibitor of cell cycle progression and cellular proliferation (88, 89).
Degradation of FOXO3a After Activation of GPER
The phosphorylated FOXO3a protein is subsequently bound to the nuclear export protein 14-3-3 and is then ubiquitinated and degraded by the proteasome complex in the cytosol. Nuclear exclusion of FOXO3a by activation of Akt increases cellular survival. Treatment of MCF-7 breast cancer cells with the selective GPER agonist G1 was shown to lead to an inactivation of FOXO3a (90).
In addition, the pro-survival effect of 17β-estradiol on breast cancer cells through GPER involves expression of SIRT1, a putative tumor suppressor, being a coactivator of FOXO3a, as described below. In the ERα-negative breast cancer cell line SKBr3, estradiol and GPER agonist G1 increased SIRT1 expression (91).
In triple-negative breast cancer cells MDA-MB-453 and MDA-MB-231, degradation of FOXO3a has been shown to reduce the expression of Fas-ligand, p27kip and bim, thereby preventing the induction of apoptosis. In addition, a second signaling pathway leading to FOXO3a inactivation has been discovered to be independent of Akt.
Inhibition of NFκB Following GPER-Activation
In some breast tumors devoid of p-Akt, cytoplasmic localization of FOXO3a correlates with the expression of IKKβ and is associated with poor survival rates. Phosphorylation of IκB by IκB kinase leads to the degradation IκB by the ubiquitin proteasome and thereby prevents the proliferative and antiapoptotic activity of NFκB. (92). It has further been shown that stimulation of GPER by the agonist G1 suppresses EMT of the TNBC cell line MDA-MB-231 by the down-regulation of NFκB signaling (93).
Activation of Adenylyl Cyclase
Parallel to the βγ-G-protein dependent signaling pathways of GPER, it has also been demonstrated that stimulation of GPER causes activation of the Gαs protein finally leading to adenylyl cyclase activation and cAMP accumulation in the estrogen stimulated breast cancer cells (94, 95). An elevation of intracellular cAMP by 17β-estradiol was observed in the breast cancer cell line SK-Br3 and in MDA-MB-231 cells transfected with the GPER gene. Both cell lines lack the expression of classical ERα. Surprisingly, it was also shown that the generation of cAMP in these cells attenuated the activation of Erk-1/2, thereby limiting the effects of 17β-estradiol on the EGF-receptor/Erk signaling axes (94).
Further downstream of GPER signaling the elevated cAMP activates protein kinase A. PKA phosphorylates the transcription factor CREB, leading to the transcription of genes possessing a cAMP responsive element in their promoter (96, 97). Phosphorylated CREB binds to the promoters of cyclinD1 and cyclinD2 and induces their transcription. CyclinD1 and cyclinD2 are both involved in the progression of cell cycle (82, 96).
Activation of Hippo-Signaling
For example, estrogen leads to a GPER-dependent activation of the Hippo-signaling pathway. The Hippo-pathway is involved in the control of organ size, cell proliferation, and tumor development. In basal cell carcinoma, YAP clearly increased the expression of Cyr61 (CCN1) and the connective tissue growth factor (CCN2) (98), building a bridge between activation of GPER and induction of CTGF expression, as observed by Pandey et al. (84) in SKBr3 breast cancer cells.
The Hippo-pathway involves the yes-associated protein 1 (YAP) and TAZ, a transcriptional coactivator with a PDZ-binding domain and the transcription factors of the TEA domain family (TEAD). Tumor suppression by the Hippo-pathway was reported to depend on the activity of Gαq-11, PLCβ, and Rho/ROCK. The Hippo/YAP/TAZ pathway was found to be a downstream branch of GPER signaling playing an important role in breast tumorigenesis (99).
Activation of Hippo pathway starts with MST1/2 (mammalian sterile 20-like 1), a kinase that activates LATS1/2 kinases (Large tumor suppressor). LATS1/2 inactivates the transcription factors YAP and TAZ by phosphorylation. Phosphorylated YAP and TAZ interact with the protein 14-3-3 and are retained in the cytoplasm where they are degraded by the proteasome complex (99).
Induction of Hotair
In ERα-positive as well as in triple-negative, breast cancer cell lines' expression of HOTAIR was induced by 17β-estradiol as another GPER-dependent signaling event (100). HOTAIR (HOX transcript antisense intergenic RNA) is one of many long non-coding RNAs. It is upregulated in tumors of many different cancers, including breast cancer. HOTAIR is involved in the control of apoptosis, metastasis and DNA-repair. In breast cancer metastasis HOTAIR is overexpressed more than 100-fold (101). ERα-negative tumors with a high expression of HOTAIR preferentially use the lymphatic vessels to spread to secondary sites. High levels of HOTAIR expression were associated with worse prognoses. The influence of HOTAIR expression on overall survival was analyzed in a cohort of 132 breast cancer patients. In ERα-negative breast cancer patients with high HOTAIR expression, the overall survival rate at the 100th month after diagnosis was only 46.4% compared to 62.8% in patients with low expression of HOTAIR. In contrast, in ERα-positive breast tumors no correlation was observed between overall survival and HOTAIR expression (p=0.41) (102). HOTAIR binds to promoters of silenced metastasis suppressor genes where it recruits the lysine-specific demethylase 1 (LSD1), finally leading to the induction of cancer metastasis (103). GPER-dependent migration of TNBC cell lines MDA-MB-231 and BT549 was reversed by deletion of HOTAIR in these cell lines. The upregulation of HOTAIR was shown to be achieved by the GPER-dependent suppression of microRNA miR148a (100).
Particular Role of GPER in Triple Negative Breast Cancer
In TNBC, GPER is frequently expressed very strongly and high GPER expression in this subgroup of breast cancer was found to correlate with increased recurrence. After a 36-month follow-up, 90.5% of the TNBC patients with low GPER expression were still alive, whereas in the cohort with high GPER expression only 77.8% of the patients survived after this time period (104). A knock-down of the GPER expression, using GPER-specific siRNA, was shown to prevent 17β-estradiol-dependent growth stimulation of triple negative breast cancer cell lines. In the TNBC cell lines treated with GPER-siRNA, the activation of c-src and transactivation of EGFR by 17β-estradiol and tamoxifen were completely inhibited (82). The results of these experiments provide further evidence that the non-genomic effects of 17β-estradiol, including activation of c-Src, phosphorylation of the EGF-receptor, activation of the MAP-kinase Erk1, and increased expression of c-fos are dependent on the presence of GPER.
Therapeutical Options Targeting GPER in Triple Negative Breast Cancer
Pharmacological Inhibition of GPER
Several pharmacological inhibitors of GPER have been identified. Using estriol, a natural estrogen, GPER signaling was successfully prevented in the ERα-negative breast cancer cell line SKBr3 (70). In the triple-negative breast cancer cell lines, HCC1806 and HCC70, activation of Src-kinase and EGF-receptor. The induction of c-fos by 17β-estradiol was also inhibited after pre-treatment of the cells with 10−4 M estriol (105).
In TNBC, the synthetic GPER antagonist G15 proved to be less effective in inhibiting GPER signaling than estriol (unpublished results).
Suppression of GPER Expression
(a) Inhibition of EGF-receptor
A different approach to inactivate GPER in triple-negative breast cancer is the suppression of GPER expression. In many triple-negative breast tumors, an overexpression of the EGF-receptor (Her-1) has been detected instead of an overexpression of Her2 (106). EGF is able to induce the expression of GPER in estrogen receptor negative breast cancer cells, thus facilitating a growth stimulatory effect of 17β-estradiol even in breast cancer cells lacking ERα expression (107). The fact that 17β-estradiol trans-activates the EGF-receptor via GPER and Src and so increases the expression of GPER leads to a positive feedback loop that boosts the induction of proliferation by 17β-estradiol in triple-negative breast tumors. The treatment of TNBC cells with gefitinib, an inhibitor of the tyrosine-kinase of the EGF-receptor, reduced GPER expression by up to 85%. This reduction of GPER expression successfully prevented all signaling events downstream of GPER as the activation of Src-kinase, the transactivation of the EGF-receptor, and the induction of c-fos expression by 17β-estradiol (108).
(b) Inhibition of growth hormone receptor
Growth hormone (GH) is another factor involved in the regulation of GPER expression. Antagonists of growth hormone-releasing hormone were shown to suppress in vivo growth of TNBC (109). Somavert (Pegvisomant) is a specific inhibitor of the GH-receptor that is already clinically applied in the treatment of acromegaly (110). In triple-negative breast cancer cell lines, the expression of GPER was reduced by dose-dependent treatment with Somavert. The treatment of MDA-MB-453- and HCC1806 cells with 1 μM Somavert reduced GPER-dependent p-src and EGF-receptor activation by almost 50% and induction of cyclinD1 and aromatase by estradiol was completely prevented by pretreatment of the cells with Somavert (97). The inhibition of GPER expression is a promising therapeutic intervention for TNBC.
Additional downstream pathways activated by GPER include PI3K (73, 86), PKCε (111), and voltage-gated sodium channels (112).
It has been shown that in many different cellular systems a multitude of signaling pathways are activated by 17β-estradiol or other ligands of GPER that are responsible for the induction of proliferation or increased metastasis as shown in Figure 2. In brief, signaling pathways dependent on binding of 17β-estradiol to GPER are classified as (1) EGF-receptor pathway, (2) calcium-signaling, (3) cAMP-pathway, (4) IB-pathway, (5) Hippo-pathway, and (6) HOTAIR-pathway.
After the binding of 17β-estradiol or G1 to GPER, various signaling pathways are activated in the cytosol. Ligand binding to GPER leads to the detachment of the βγ-subunit of heterotrimeric G-proteins. The EGF-receptor pathway starts with the activation of the kinase Src by βγ-G-protein that activates MMPs, liberating EGF from heparin-bound EGF. EGF binding to EGF-receptor leads to auto-phosphorylation of the EGF-receptor that activates MAP-kinase Erk1/2 and PI3-kinase. Activated Erk induces transcription of c-fos, Egr-1, ERRα and aromatase in the nucleus followed by the induction of CTGF. PI3-kinase is also activated by the phosphorylated EGF-receptor and phosphorylates the kinase Akt that phosphorylates the transcription factor FOXO3a in the nucleus, which is subsequently exported to the cytosol, where FOXO3a is degraded. The Calcium-signaling pathway starts with the released α-subunit of heterotrimeric G-proteins that activates phospholipase C. PLC cleaves Phosphatidylinositol-4,5-bisphosphat to diacylglycerol and IP3. IP3 releases calcium ions from cytosolic calcium-stores. Ca2+ activates several enzymes in the cytosol, among other things, and activates Erk1/2. The binding of 17β-estradiol to GPER also opens calcium L-channels in the plasma membrane by a yet unsolved mechanism. The released α-subunit of the heterotrimeric G-protein also activates the adenolyl-cyclase (AC) in the cytosol. cAMP generated by AC activates protein kinase A (PKA) that phosphorylates the cAMP response element binding protein (CREB). Phosphorylated CREB binds as a transcription factor to promoters of genes containing a cAMP response element, for example cyclinD1, which supports the progress of the cell cycle. Less elucidated are the IB-pathway, Hippo-pathway and HOTAIR-pathway represented in this figure by hatched arrows. In the course of the IB-pathway, IKKβ is activated and phosphorylates IB, an inhibitor of NF-B. Phosphorylated IB is degraded via the ubiquitination pathway, allowing NFB action. In the course of the Gα-protein-dependent Hippo-pathway, MST1/2 are activated and phosphorylate LATS, a kinase phosphorylating the transcription factors YAP and TAZ, which are subsequently degraded. In the HOTAIR-pathway, the expression of miR148a is inhibited in a Gα-dependent manner, but the intermediate steps leading to transcription of this microRNA are not completely elucidated, in this figure exemplified by a hatched arrow. The suppression of the miR148a expression leads to an increased expression of HOTAIR that finally supports metastasis.
Conclusion
Apart from estrogen receptor ERα that was discovered first two further estrogen receptors, ERβ and GPER are known to be important for the hormonal control of breast cancer. The complete elucidation of the function of ERβ in breast cancer is hampered by the diversity of isoforms of ERβ identified. There are still a lot of efforts to be done to clarify the impact of the various isoforms of ERβ on the transcription of target genes of estrogens in particular how the many possible heterodimers act in breast cancer cells.
For GPER, the estrogen receptor that is commonly accepted to be responsible for the extra-nuclear, non-genomic effects of estrogens a large number of signaling pathways were identified in recent years. In addition to the well-known transactivation of the EGF-receptor that leads to the activation of the MAPK-kinase- and the PI3-kinase pathway a number of further signaling events have been described to be induced by the binding of estrogens to GPER. (1) The inactivation of the transcription factor FOXO3a via the kinase Akt. (2) The activation of the Hippo-pathway that induces CTGF expression followed by enhanced metastasis. (3) Induction of HOTAIR expression that also increases the metastatic potential of breast cancer cells. The knowledge about these GPER-dependent signaling pathways will promote the development of new targeted therapies. In particular for triple negative breast tumors that strongly express GPER such therapies are urgently needed.
Author Contributions
RG and CG drafted the manuscript. GE critically revised the manuscript. All authors read and approved the final version.
Funding
This work was supported by grant GR 1895/10-2 of German Research Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, et al. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature (1986) 320:134–9. doi: 10.1038/320134a0
2. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. (1996) 93:5925–30. doi: 10.1073/pnas.93.12.5925
3. Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell (1989) 59:477–87. doi: 10.1016/0092-8674(89)90031-7
4. Gruber CJ, Gruber DM, Gruber IM, Wieser F, Huber JC. Anatomy of the estrogen response element. Trends Endocrinol Metab. (2004) 15:73–8. doi: 10.1016/j.tem.2004.01.008
5. Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. (2005) 19:833–42. doi: 10.1210/me.2004-0486
6. Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer (2011) 11:597–608. doi: 10.1038/nrc3093
7. Kampa M, Pelekanou V, Notas G, Stathopoulos EN, Castanas E. The estrogen receptor: two or more molecules, multiple variants, diverse localizations, signaling and functions. Are we undergoing a paradigm-shift as regards their significance in breast cancer? Hormones (Athens) (2013) 12:69–85. doi: 10.1007/BF03401288
8. Su X, Xu X, Li G, Lin B, Cao J, Teng L. ER-α36: a novel biomarker and potential therapeutic target in breast cancer. Onco Targets Ther. (2014) 7:1525–33. doi: 10.2147/OTT.S65345
9. Gu Y, Chen T, Lopez E, Wu W, Wang X, Cao J, et al. The therapeutic target of estrogen receptor-alpha36 in estrogen-dependent tumors. J Transl Med. (2014) 12:16. doi: 10.1186/1479-5876-12-16
10. Hisamoto K, Bender JR. Vascular cell signaling by membrane estrogen receptors. Steroids (2005) 70:382–7. doi: 10.1016/j.steroids.2005.02.011
11. Migliaccio A, Castoria G, de Falco A, Bilancio A, Giovannelli P, Di Donato M, et al. Polyproline and Tat transduction peptides in the study of the rapid actions of steroid receptors. Steroids (2012) 77:974–8. doi: 10.1016/j.steroids.2012.01.014
12. Levin ER, Hammes SR. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol. (2016) 17:783–797. doi: 10.1038/nrm.2016.122
13. Millis SZ, Gatalica Z, Winkler J, Vranic S, Kimbrough J, Reddy S, et al. Predictive biomarker profiling of >6000 breast cancer patients shows heterogeneity in TNBC, with treatment implications. Clin Breast Cancer (2015) 15:473–481 e3. doi: 10.1016/j.clbc.2015.04.008
14. Litwiniuk MM, Roznowski K, Filas V, Godlewski DD, Stawicka M, Kaleta R, et al. Expression of estrogen receptor beta in the breast carcinoma of BRCA1 mutation carriers. BMC Cancer (2008) 8:100. doi: 10.1186/1471-2407-8-100
15. Warner M, Saji S, Gustafsson JA. The normal and malignant mammary gland: a fresh look with ERβ onboard. J Mammary Gland Biol Neoplasia (2000) 5:289–94. doi: 10.1023/A:1009598828267
16. Speirs V, Skliris GP, Burdall SE, Carder PJ. Distinct expression patterns of ER alpha and ER beta in normal human mammary gland. J Clin Pathol. (2002) 55:371–4. doi: 10.1136/jcp.55.5.371
17. Huang B, Omoto Y, Iwase H, Yamashita H, Toyama T, Coombes RC, et al. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc Natl Acad Sci USA. (2014) 111:1933–8. doi: 10.1073/pnas.1323719111
18. Skliris GP, Leygue E, Curtis-Snell L, Watson PH, Murphy LC. Expression of oestrogen receptor-beta in oestrogen receptor-alpha negative human breast tumours. Br J Cancer (2006) 95:616–26. doi: 10.1038/sj.bjc.6603295
19. Ogawa S, Inoue S, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. Cross-inhibition of both estrogen receptor α and β pathways by each dominant negative mutant. FEBS Lett. (1998) 423:129–32. doi: 10.1016/S0014-5793(98)00079-9
20. Haldosen LA, Zhao C, Dahlman-Wright K. Estrogen receptor beta in breast cancer. Mol Cell Endocrinol. (2014) 382:665–72. doi: 10.1016/j.mce.2013.08.005
21. Hu YF, Lau KM, Ho SM, Russo J. Increased expression of estrogen receptor beta in chemically transformed human breast epithelial cells. Int J Oncol. (1998) 12:1225–8. doi: 10.3892/ijo.12.6.1225
22. Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv. (2003) 3:281–92. doi: 10.1124/mi.3.5.281
23. Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, et al. Cloning and characterization of human estrogen receptor beta isoforms. Biochem Biophys Res Commun. (1998) 247:75–8. doi: 10.1006/bbrc.1998.8738
24. Poola I, Fuqua SA, De Witty RL, Abraham J, Marshallack JJ, Liu A. Estrogen receptor alpha-negative breast cancer tissues express significant levels of estrogen-independent transcription factors, ERβ1 and ERβ5: potential molecular targets for chemoprevention. Clin Cancer Res. (2005) 11:7579–85. doi: 10.1158/1078-0432.CCR-05-0728
25. Honma N, Horii R, Iwase T, Saji S, Younes M, Takubo K, et al. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J Clin Oncol. (2008) 26:3727–34. doi: 10.1200/JCO.2007.14.2968
26. Zhang H, Zhang Z, Xuan L, Zheng S, Guo L, Zhan Q, et al. Evaluation of ER-alpha, ER-Beta1 and ER-Beta2 expression and correlation with clinicopathologic factors in invasive luminal subtype breast cancers. Clin Transl Oncol. (2012) 14:225–31. doi: 10.1007/s12094-012-0788-0
27. Leung YK, Lee MT, Lam HM, Tarapore P, Ho SM. Estrogen receptor-beta and breast cancer: translating biology into clinical practice. Steroids (2012) 77:727–37. doi: 10.1016/j.steroids.2012.03.008
28. Zhao C, Matthews J, Tujague M, Wan J, Strom A, Toresson G, et al. Estrogen receptor β2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res. (2007) 67:3955–62. doi: 10.1158/0008-5472.CAN-06-3505
29. Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor ER-β isoforms: a key to understanding ER-β signaling. Proc Natl Acad Sci USA. (2006) 103:13162–7. doi: 10.1073/pnas.0605676103
30. Yan M, Rayoo M, Takano EA, Fox SB. Nuclear and cytoplasmic expressions of ERβ1 and ERβ2 are predictive of response to therapy and alters prognosis in familial breast cancers. Breast Cancer Res Treat. (2011) 126:395–405. doi: 10.1007/s10549-010-0941-9
31. Peng B, Lu B, Leygue E, Murphy LC. Putative functional characteristics of human estrogen receptor-beta isoforms. J Mol Endocrinol. (2003) 30:13–29. doi: 10.1677/jme.0.0300013
32. Sugiura H, Toyama T, Hara Y, Zhang Z, Kobayashi S, Fujii Y, et al. Expression of estrogen receptor beta wild-type and its variant ERβcx/β2 is correlated with better prognosis in breast cancer. Jpn J Clin Oncol. (2007) 37:820–8. doi: 10.1093/jjco/hym114
33. Rosin G, de Boniface J, Karthik GM, Frisell J, Bergh J, Hartman J. Oestrogen receptors beta1 and betacx have divergent roles in breast cancer survival and lymph node metastasis. Br J Cancer (2014) 111:918–26. doi: 10.1038/bjc.2014.398
34. Liu J, Guo H, Mao K, Zhang K, Deng H, Liu Q. Impact of estrogen receptor-beta expression on breast cancer prognosis: a meta-analysis. Breast Cancer Res Treat. (2016) 156:149–62. doi: 10.1007/s10549-016-3721-3
35. Tan W, Li Q, Chen K, Su F, Song E, Gong C. Estrogen receptor beta as a prognostic factor in breast cancer patients: a systematic review and meta-analysis. Oncotarget (2016) 7:10373–85. doi: 10.18632/oncotarget.7219
36. Shaaban AM, Green AR, Karthik S, Alizadeh Y, Hughes TA, Harkins L, et al. Nuclear and cytoplasmic expression of ERβ1, ERβ2, and ERβ5 identifies distinct prognostic outcome for breast cancer patients. Clin Cancer Res. (2008) 14:5228–35. doi: 10.1158/1078-0432.CCR-07-4528
37. Chantzi NI, Palaiologou M, Stylianidou A, Goutas N, Vassilaros S, Kourea HP, et al. Estrogen receptor β2 is inversely correlated with Ki-67 in hyperplastic and noninvasive neoplastic breast lesions. J Cancer Res Clin Oncol. (2014) 140:1057–66. doi: 10.1007/s00432-014-1652-0
38. Gruvberger-Saal SK, Bendahl PO, Saal LH, Laakso M, Hegardt C, Eden P, et al. Estrogen receptor β expression is associated with tamoxifen response in ERα-negative breast carcinoma. Clin Cancer Res. (2007) 13:1987–94. doi: 10.1158/1078-0432.CCR-06-1823
39. Novelli F, Milella M, Melucci E, Di Benedetto A, Sperduti I, Perrone-Donnorso R, et al. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: an observational prospective study. Breast Cancer Res. (2008) 10:R74. doi: 10.1186/bcr2139
40. Forster C, Makela S, Warri A, Kietz S, Becker D, Hultenby K, et al. Involvement of estrogen receptor β in terminal differentiation of mammary gland epithelium. Proc Natl Acad Sci USA. (2002) 99:15578–83. doi: 10.1073/pnas.192561299
41. Bado I, Nikolos F, Rajapaksa G, Wu W, Castaneda J, Krishnamurthy S, et al. Somatic loss of estrogen receptor beta and p53 synergize to induce breast tumorigenesis. Breast Cancer Res. (2017) 19:79. doi: 10.1186/s13058-017-0872-z
42. Palmieri C, Cheng GJ, Saji S, Zelada-Hedman M, Warri A, Weihua Z, et al. Estrogen receptor beta in breast cancer. Endocr Relat Cancer (2002) 9:1–13. doi: 10.1677/erc.0.0090001
43. Liu MM, Albanese C, Anderson CM, Hilty K, Webb P, Uht RM, et al. Opposing action of estrogen receptors α and β on cyclin D1 gene expression. J Biol Chem. (2002) 277:24353–60. doi: 10.1074/jbc.M201829200
44. Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. (1999) 13:1672–85. doi: 10.1210/mend.13.10.0357
45. Schultz JR, Petz LN, Nardulli AM. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors α and β. J Biol Chem. (2005) 280:347–54. doi: 10.1074/jbc.M407879200
46. Giovannelli P, Di Donato M, Galasso G, Di Zazzo E, Bilancio A, Migliaccio A. The androgen receptor in breast cancer. Front Endocrinol. (2018) s9:492. doi: 10.3389/fendo.2018.00492
47. Marotti JD, Collins LC, Hu R, Tamimi RM. Estrogen receptor-β expression in invasive breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod Pathol. (2010) 23:197–204. doi: 10.1038/modpathol.2009.158
48. Chen JQ, Russo J. ERα-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochim Biophys Acta (2009) 1796:162–75. doi: 10.1016/j.bbcan.2009.06.003
49. Thomas C, Rajapaksa G, Nikolos F, Hao R, Katchy A, McCollum CW, et al. ERbeta1 represses basal breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res. (2012) 14:R148. doi: 10.1186/bcr3358
50. Chantzi NI, Tiniakos DG, Palaiologou M, Goutas N, Filippidis T, Vassilaros SD, et al. Estrogen receptor beta 2 is associated with poor prognosis in estrogen receptor alpha-negative breast carcinoma. J Cancer Res Clin Oncol. (2013) 139:1489–98. doi: 10.1007/s00432-013-1467-4
51. Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor β inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. (2004) 64:423–8. doi: 10.1158/0008-5472.CAN-03-2446
52. Bartella V, Rizza P, Barone I, Zito D, Giordano F, Giordano C, et al. Estrogen receptor beta binds Sp1 and recruits a corepressor complex to the estrogen receptor alpha gene promoter. Breast Cancer Res Treat. (2012) 134:569–81. doi: 10.1007/s10549-012-2090-9
53. Samanta S, Sharma VM, Khan A, Mercurio AM. Regulation of IMP3 by EGFR signaling and repression by ERβ: implications for triple-negative breast cancer. Oncogene (2012) 31:4689–97. doi: 10.1038/onc.2011.620
54. Hinsche O, Girgert R, Emons G, Grundker C. Estrogen receptor beta selective agonists reduce invasiveness of triple-negative breast cancer cells. Int J Oncol. (2015) 46:878–84. doi: 10.3892/ijo.2014.2778
55. Emons G, Ortmann O, Fingscheidt U, Ball P, Knuppen R. Short-term effects of oestradiol and 4-hydroxyoestradiol on gonadotrophin-releasing hormone induced luteinizing hormone secretion by rat pituitary cells in culture. Acta Endocrinol. (Copenh) (1986) 111:312–20. doi: 10.1530/acta.0.1110312
56. Berthois Y, Pourreau-Schneider N, Gandilhon P, Mittre H, Tubiana N, Martin PM. Estradiol membrane binding sites on human breast cancer cell lines. Use of a fluorescent estradiol conjugate to demonstrate plasma membrane binding systems. J Steroid Biochem. (1986) 25:963–72. doi: 10.1016/0022-4731(86)90330-4
57. Pietras RJ, Szego CM, Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature (1977) 265:69–72. doi: 10.1038/265069a0
58. Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci USA. (1994) 91:8517–21. doi: 10.1073/pnas.91.18.8517
59. Batra S. Effect of estrogen and progesterone treatment on calcium uptake by the myometrium and smooth muscle of the lower urinary tract. Eur J Pharmacol. (1986) 127:37–42. doi: 10.1016/0014-2999(86)90203-7
60. Emons G, Frevert EU, Ortmann O, Fingscheidt U, Sturm R, Kiesel L, et al. Studies on the subcellular mechanisms mediating the negative estradiol effect on GnRH-induced LH-release by rat pituitary cells in culture. Acta Endocrinol. (1989) 121:350–60. doi: 10.1530/acta.0.1210350
61. Lieberherr M, Grosse B, Kachkache M, Balsan S. Cell signaling and estrogens in female rat osteoblasts: a possible involvement of unconventional nonnuclear receptors. J Bone Miner Res. (1993) 8:1365–76. doi: 10.1002/jbmr.5650081111
62. Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci USA. (1999) 96:4686–91. doi: 10.1073/pnas.96.8.4686
63. Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol Endocrinol. (1999) 13:307–19. doi: 10.1210/me.13.2.307
64. Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. (1995) 9:404–10. doi: 10.1096/fasebj.9.5.7896011
65. Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics (1997) 45:607–17. doi: 10.1006/geno.1997.4972
66. Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. (2006) 346:904–10. doi: 10.1016/j.bbrc.2006.05.191
67. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. (2011) 7:715–26. doi: 10.1038/nrendo.2011.122
68. Filardo EJ, Quinn JA, Bland KI, Frackelton ARJr, Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog GPR30 and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
69. Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat. (2011) 128:457–66. doi: 10.1007/s10549-011-1584-1
70. Lappano R, Rosano C, De Marco P, De Francesco EM, Pezzi V, Maggiolini M. Estriol acts as a GPR30 antagonist in estrogen receptor-negative breast cancer cells. Mol Cell Endocrinol. (2010) 320:162–70. doi: 10.1016/j.mce.2010.02.006
71. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. (2009) 5:421–7. doi: 10.1038/nchembio.168
72. Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. (2011) 127:358–66. doi: 10.1016/j.jsbmb.2011.07.002
73. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (2005) 307:1625–30. doi: 10.1126/science.1106943
74. Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology (2008) 149:4846–56. doi: 10.1210/en.2008-0269
75. Sanden C, Broselid S, Cornmark L, Andersson K, Daszkiewicz-Nilsson J, Martensson UE, et al. G protein-coupled estrogen receptor 1/G protein-coupled receptor 30 localizes in the plasma membrane and traffics intracellularly on cytokeratin intermediate filaments. Mol Pharmacol. (2011) 79:400–10. doi: 10.1124/mol.110.069500
76. Samartzis EP, Noske A, Meisel A, Varga Z, Fink D, Imesch P. The G protein-coupled estrogen receptor (GPER) is expressed in two different subcellular localizations reflecting distinct tumor properties in breast cancer. PLoS ONE (2014) 9:e83296. doi: 10.1371/journal.pone.0083296
77. Broselid S, Berg KA, Chavera TA, Kahn R, Clarke WP, Olde B, et al. G protein-coupled receptor 30 (GPR30) forms a plasma membrane complex with membrane-associated guanylate kinases (MAGUKs) and protein kinase A-anchoring protein 5 (AKAP5) that constitutively inhibits cAMP production. J Biol Chem. (2014) 289:22117–27. doi: 10.1074/jbc.M114.566893
78. Hofmann F, Flockerzi V, Kahl S, Wegener JW. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev. (2014) 94:303–26. doi: 10.1152/physrev.00016.2013
79. Tica AA, Dun EC, Tica OS, Gao X, Arterburn JB, Brailoiu GC, et al. G protein-coupled estrogen receptor 1-mediated effects in the rat myometrium. Am J Physiol Cell Physiol. (2011) 301:C1262–9. doi: 10.1152/ajpcell.00501.2010
80. Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature (1999) 402:884–8. doi: 10.1038/47260
81. Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, et al. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. (2003) 278:2118–23. doi: 10.1074/jbc.M210828200
82. Girgert R, Emons G, Grundker C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: possible application in targeted therapy. Breast Cancer Res Treat. (2012) 134:199–205. doi: 10.1007/s10549-012-1968-x
83. Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17β-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. (2004) 279:27008–16. doi: 10.1074/jbc.M403588200
84. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. (2009) 28:523–32. doi: 10.1038/emboj.2008.304
85. Jacobson A, Cunningham JL. Connective tissue growth factor in tumor pathogenesis. Fibrogenesis Tissue Repair (2012) 5:S8. doi: 10.1186/1755-1536-5-S1-S8
86. Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int. (2013) 2013:472720. doi: 10.1155/2013/472720
87. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. (2008) 10:138–48. doi: 10.1038/ncb1676
88. Birkenkamp KU, Coffer PJ. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem Soc Trans. (2003) 31:292–7. doi: 10.1042/bst0310292
89. Taylor S, Lam M, Pararasa C, Brown JE, Carmichael AR, Griffiths HR. Evaluating the evidence for targeting FOXO3a in breast cancer: a systematic review. Cancer Cell Int. (2015) 15:1. doi: 10.1186/s12935-015-0156-6
90. Zekas E, Prossnitz ER. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer (2015) 15:702. doi: 10.1186/s12885-015-1699-6
91. Santolla MF, Avino S, Pellegrino M, De Francesco EM, De Marco P, Lappano R, et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis. (2015) 6:e1834. doi: 10.1038/cddis.2015.201
92. Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell (2004) 117:225–37. doi: 10.1016/S0092-8674(04)00302-2
93. Chen ZJ, Wei W, Jiang GM, Liu H, Wei WD, Yang X, et al. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol Oncol. (2016) 10:775–88. doi: 10.1016/j.molonc.2016.01.002
94. Filardo EJ, Quinn JA, Frackelton ARJ, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. (2002) 16:70–84. doi: 10.1210/mend.16.1.0758
95. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology (2005) 146:624–32. doi: 10.1210/en.2004-1064
96. Kanda N, Watanabe S, 17β-estradiol stimulates the growth of human keratinocytes by inducing cyclin D2 expression. J Invest Dermatol. (2004) 123:319–28. doi: 10.1111/j.0022-202X.2004.12645.x
97. Zucchetti AE, Barosso IR, Boaglio AC, Basiglio CL, Miszczuk G, Larocca MC, et al. G-protein-coupled receptor 30/adenylyl cyclase/protein kinase A pathway is involved in estradiol 17ss-D-glucuronide-induced cholestasis. Hepatology (2014) 59:1016–29. doi: 10.1002/hep.26752
98. Quan T, Xu Y, Qin Z, Robichaud P, Betcher S, Calderone K, et al. Elevated YAP and its downstream targets CCN1 and CCN2 in basal cell carcinoma: impact on keratinocyte proliferation and stromal cell activation. Am J Pathol. (2014) 184:937–943. doi: 10.1016/j.ajpath.2013.12.017
99. Zhou X, Wang S, Wang Z, Feng X, Liu P, Lv XB, et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest. (2015) 125:2123–35. doi: 10.1172/JCI79573
100. Tao S, He H, Chen Q. Estradiol induces HOTAIR levels via GPER-mediated miR-148a inhibition in breast cancer. J Transl Med. (2015) 13:131. doi: 10.1186/s12967-015-0489-x
101. Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature (2010) 464:1071–6. doi: 10.1038/nature08975
102. Gokmen-Polar Y, Vladislav IT, Neelamraju Y, Janga SC, Badve S. Prognostic impact of HOTAIR expression is restricted to ER-negative breast cancers. Sci Rep. (2015) 5:8765. doi: 10.1038/srep08765
103. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science (2010) 329:689–93. doi: 10.1126/science.1192002
104. Steiman J, Peralta EA, Louis S, Kamel O. Biology of the estrogen receptor, GPR30, in triple negative breast cancer. Am J Surg. (2013) 206:698–703. doi: 10.1016/j.amjsurg.2013.07.014
105. Girgert R, Emons G, Grundker C. Inhibition of GPR30 by estriol prevents growth stimulation of triple-negative breast cancer cells by 17β-estradiol. BMC Cancer (2014) 14:935. doi: 10.1186/1471-2407-14-935
106. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. (2004) 10:5367–74. doi: 10.1158/1078-0432.CCR-04-0220
107. Vivacqua A, Lappano R, De Marco P, Sisci D, Aquila S, De Amicis F, et al. G protein-coupled receptor 30 expression is up-regulated by EGF and TGF α in estrogen receptor alpha-positive cancer cells. Mol Endocrinol. (2009) 23:1815–26. doi: 10.1210/me.2009-0120
108. Girgert R, Emons G, Grundker C. 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol Rep. (2017) 37:1212–8. doi: 10.3892/or.2016.5306
109. Perez R, Schally AV, Vidaurre I, Rincon R, Block NL, Rick FG. Antagonists of growth hormone-releasing hormone suppress in vivo tumor growth and gene expression in triple negative breast cancers. Oncotarget (2012) 3:988–97. doi: 10.18632/oncotarget.634
110. Neggers SJ, Muhammad A, van der Lely AJ. Pegvisomant treatment in acromegaly. Neuroendocrinology (2016) 103:59–65. doi: 10.1159/000381644
111. Goswami C, Kuhn J, Dina OA, Fernandez-Ballester G, Levine JD, Ferrer-Montiel A, et al. Estrogen destabilizes microtubules through an ion-conductivity-independent TRPV1 pathway. J Neurochem. (2011) 117:995–1008. doi: 10.1111/j.1471-4159.2011.07270.x
Keywords: estrogen signaling, ERα-negative breast cancer, ERα, ERβ, GPER
Citation: Girgert R, Emons G and Gründker C (2019) Estrogen Signaling in ERα-Negative Breast Cancer: ERβ and GPER. Front. Endocrinol. 9:781. doi: 10.3389/fendo.2018.00781
Received: 07 November 2018; Accepted: 12 December 2018;
Published: 09 January 2019.
Edited by:
Antonio Bilancio, Università degli Studi della Campania “Luigi Vanvitelli” Naples, ItalyReviewed by:
Pia Giovannelli, Università degli Studi della Campania Luigi Vanvitelli, ItalyMarzia Di Donato, Università degli Studi della Campania Luigi Vanvitelli Caserta, Italy
Copyright © 2019 Girgert, Emons and Gründker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carsten Gründker, Z3J1bmRrZXJAbWVkLnVuaS1nb2V0dGluZ2VuLmRl