 Emily W. L. Sun
Emily W. L. Sun Alyce M. Martin1
Alyce M. Martin1 Richard L. Young
Richard L. Young Damien J. Keating
Damien J. Keating- 1College of Medicine and Public Health, Flinders University, Adelaide, SA, Australia
- 2Nutrition and Metabolism, South Australian Health and Medical Research Institute, Adelaide, SA, Australia
- 3Adelaide Medical School, The University of Adelaide, Adelaide, SA, Australia
Enteroendocrine cells lining the gut epithelium constitute the largest endocrine organ in the body and secrete over 20 different hormones in response to cues from ingested foods and changes in nutritional status. Not only do these hormones convey signals from the gut to the brain via the gut-brain axis, they also act directly on metabolically important peripheral targets in a highly concerted fashion to maintain energy balance and glucose homeostasis. Gut-derived hormones released during fasting tend to be orexigenic and have hyperglycaemic potential. Conversely, gut hormones secreted postprandially generally promote satiety and facilitate glucose clearance. Although some of the metabolic benefits conferred by bariatric surgeries have been ascribed to changes in the secretory profiles of various gut hormones, the therapeutic potential of the enteroendocrine system as a viable target against metabolic diseases remain largely underexploited, except for incretin-mimetics. This review provides a brief overview of the physiological importance and highlights the therapeutic potential of the following gut hormones: serotonin, glucose-dependent insulinotropic peptide, glucagon-like peptide 1, oxyntomodulin, peptide YY, insulin-like peptide 5, and ghrelin.
Introduction
Gut enteroendocrine cells (EECs) are specialized secretory cells that are sparsely scattered throughout the mucosal epithelium of the gastrointestinal (GI) tract and which constitute the largest endocrine organ by mass in the body (1). EECs have the capacity to “sample” luminal contents on the apical membrane, and collectively release more than 20 different hormones basolaterally in response to a variety of stimuli. With each having their own specialized functions, EECs have been historically characterized by their hormonal profiles, such as glucagon-like peptide 1 (GLP-1)- and peptide YY (PYY)-secreting L-cells and serotonin (5-hydroxytryptamine, 5-HT)-secreting enterochromaffin (EC) cells. It is now accepted that there are vast overlaps in the secretory profiles of EECs (2) and the “one cell type, one hormone” dogma is widely rejected. Studies using transgenic mice expressing fluorescent reporter proteins driven by promoters of different gut hormones revealed that multiple hormones can be simultaneously expressed by an individual EEC (3, 4) while high-resolution microscopy shows that these different hormones are packaged into separate vesicles within the EEC (5–7). Expression of EEC hormones are also regionally distinct, as many gut hormones are confined to specific regions of the gut, while a subset, such as 5-HT and somatostatin, are present throughout the GI tract (8, 9). Enteroendocrine hormones are implicated in a wide range of physiological functions including gastrointestinal motility, appetite control, and glucose homeostasis (10). Mounting evidence demonstrates the importance of gut hormones in regulating peripheral metabolism in health and disease and as a result, a myriad of therapeutics against metabolic diseases that are based on the actions of specific gut hormones are currently under clinical development (11–13). As such, it is timely to review the literature regarding the metabolic actions of these gut hormones: serotonin, glucose-dependent insulinotropic peptide, glucagon-like peptide-1 (GLP-1), oxyntomodulin, peptide YY (PYY) and ghrelin. We also discuss the metabolic actions of insulin-like peptide 5, a recently characterized gut hormone that are co-secreted with GLP-1 and PYY.
Serotonin
Serotonin (5-HT) is produced by enterochromaffin (EC) cells, which constitute ~50% of the total EEC population and are scattered throughout the length of the gut, from the stomach to the distal colon (2, 8). Although better known for its actions in the CNS, more than 90% of total body 5-HT is synthesized by EC cells, the majority of this being stored in platelets (14, 15). Tryptophan hydroxylase 1 (TPH1) is the rate-limiting enzyme of 5-HT synthesis in specific non-neuronal cells and its expression in the gut mucosa is limited to EC cells. EC cells have the capacity to sense a wide range of stimuli present in the gut lumen such as glucose and fructose (16, 17), the medium chain fatty acid, lauric acid (18), various tastants and olfactants (19), and to secrete 5-HT in response. 5-HT secretion from EC cells is also regulated by mechanical stimuli (20), and neural and endocrine input such as adrenergic stimulation and GABA and somatostatin inhibition (21). In addition, microbial metabolite signals from the gut microbiome also augment colonic EC cell density, 5-HT secretion and circulating 5-HT levels (22).
Although traditionally regarded as a regulator for gastric motility (23–25) and more recently, a mediator in the pathogenesis of inflammatory intestinal disorders (14, 26), mounting evidence highlights gut-derived 5-HT as a modulator of peripheral metabolism (27, 28). Under fasting conditions, gut-derived 5-HT, together with glucagon, markedly increases hepatic glucose output, a main driver of fasting euglycaemia, by increasing hepatic gluconeogenesis and glycogenolysis (29), while inhibiting glucose uptake and glycogen synthesis in the liver (30). In conjunction, 5-HT promotes lipolysis within white adipocytes to liberate free fatty acids (FFAs) and glycerol (30) as key substrates for hepatic gluconeogenesis, and further enhance hepatic glucose output. Moreover, gut-derived 5-HT promotes energy conservation and weight gain by reducing energy expenditure, via actions to attenuate thermogenesis in brown adipose tissue (31) and inhibit the browning of white adipose tissue (32).
Gut-derived 5-HT also attenuates the release of several metabolically important blood glucose-lowering chemokines, such as adiponectin from adipose tissue (33), and bone-derived osteocalcin and lipocalin 2 (34–36), through inhibition of osteoblast proliferation (37). Significantly elevated mucosal TPH1 expression in obese humans (38, 39) and elevated levels of circulating 5-HT in individuals with type 2 diabetes (T2D) (40–42) or obesity (38) has been reported. Inhibition of intestinal TPH1 in mice, through tissue-specific ablation or pharmacological inhibition, conveys protection from high-fat diet (HFD)-induced dyslipidaemia and glucose intolerance (30–32). This confirms a causative role of elevated gut-derived 5-HT as a driver of metabolic dysfunction. TPH1 inhibition also protects mice from diet-induced obesity (DIO) (31). However, despite clear evidence that EC cell-derived 5-HT negatively impacts energy balance and glucose homeostasis, the underlying causes of elevated 5-HT levels with obesity and T2D remain unclear. Likely drivers of increased circulating 5-HT are increased density or glucose-sensitivity of duodenal EC cells, as evidenced in obese human duodenal EC cells (38), however molecular mechanisms underlying this are not understood. Due to the heterogeneity in 5-HT receptors across many tissues (43), targeting 5-HT receptor signaling pathways may not be a viable therapeutic target for treatment of metabolic disease.
Glucose-dependent Insulinotropic Peptide
Glucose-dependent Insulinotropic Peptide (GIP) is a 42-amino acid peptide hormone produced by K cells located primarily in the proximal small intestine (44). GIP is secreted in response to nutrient stimulation and exerts its actions by binding to the GIP receptor (GIPR) expressed by pancreatic islet cells (45), adipocytes (46), bone cells (47), and the CNS (48). Circulating GIP is rapidly degraded by dipeptidyl peptidase IV (DPP4), a serine protease that is widely expressed throughout the body, especially in endothelial cells (49). The insulinotropic effect of GIP, together with GLP-1, accounts for more than 70% of postprandial insulin secretion (50). GIP also increases insulin biosynthesis (49), promotes β-cell proliferation and inhibits β-cell apoptosis (51). The insulinotropic effects of GIP are dramatically attenuated in T2D patients (52, 53), and this is believed to be a major contributing factor to impaired postprandial insulin secretion in these individuals. Moreover, the insulinotropic potency of GIP is markedly reduced in non-diabetic, first-degree relatives of T2D patients (54), suggesting altered GIP signaling could be one of the many predisposing factors for T2D later in life. While the mechanism underlying the diminished insulin response to GIP in T2D has not yet been fully elucidated, receptor downregulation (55) and desensitization (56) have been suggested as potential causes. Although GIP only stimulates glucagon secretion under hypo- and euglycaemic conditions in healthy individuals (57), its glucagonotropic effect is exaggerated in T2D patients during hyperglycaemia (58). This further worsens glycaemic control in these patients, and in combination with the reduced insulinotropic potency renders GIP an undesirable therapeutic target for T2D treatment.
The anabolic properties of GIP closely resemble those of insulin, as it promotes lipid uptake and inhibits lipolysis in adipocytes (59). Several studies have reported elevated GIP levels in obese humans (60, 61). Elevated GIP levels and duodenal K cell hyperplasia (62) have also been reported in HFD-treated mice, while Gipr deficiency protects mice from HFD-, leptin deficiency- or ovariectomy-induced weight gain (63, 64). GIP also induces osteopontin expression in adipocytes (65), an adipokine associated with obesity-related systemic low grade inflammation (66, 67). Adipocyte-specific Gipr ablation protects mice from HFD-induced insulin resistance and hepatic steatosis, potentially by reducing circulating levels of pro-inflammatory cytokines (68). However, the obesogenic effects of GIP are only apparent during nutrient excess, as chow-fed Gipr and Gip knockout animals are of similar weight as their wild type counterparts (69). The role of GIP in energy balance is further complicated by paradoxical findings that mice overexpressing Gip were leaner than wild type controls, when fed either a standard-chow or HFD (70). Such observation could be attributed to the anti-apoptotic effect of GIP on osteoblasts (71), as osteoblast-derived hormones such as osteocalcin and lipocalcin 2 are implicated in regulating peripheral metabolism and modulate food intake (36, 72). Furthermore, powerful evidence has emerged to show that GIPR signaling can enhance GLP-1-induced weight loss (11, 73).
Glucagon-like Peptide 1
Glucagon-like Peptide 1 (GLP-1) is an incretin hormone secreted by enteroendocrine L cells upon ingestion of nutrients, including glucose (74), and typically within 10–15 min into the postprandial period (75). GLP-1 is subjected to rapid degradation by DPP4 (76) and acts via the GLP-1 receptor (GLP-1R) expressed on a myriad of target tissues (75). GLP-1 plays a key role in maintaining glucose homeostasis, as it markedly increases glucose-stimulated insulin secretion (GSIS) (77) and attenuates hepatic glucose production, independent of its effect on pancreatic islets (78, 79). There is growing appreciation that a considerable portion of the glucose-lowering effect of GLP-1 is underscored by its inhibitory effect on gastric motility (80–83) and its glucagonostatic action (84, 85), which are preserved in obese and T2D patients (86, 87). Unlike GIP, the potent insulinotropic effect of GLP-1 is predominantly preserved in T2D patients and, thus, has led to the development of GLP-1-based therapies for preserving blood glucose control in individuals with T2D.
In addition to its multifaceted glucose-lowering effect, GLP-1 regulates energy balance and adiposity through its effects on satiety and appetite. The acute anorectic effect of GLP-1 is mediated by GLP-1R located on vagal afferents (88), which relays the signal to appetite control centers, namely the NTS in the brainstem, to reduce food intake (89) (Figure 1). GLP-1R are also widely expressed in brainstem and hypothalamic regions implicated in appetite control (90). In humans, acute administration of pharmacological doses of GLP-1 significantly induce satiety and reduce food intake (91–93). Furthermore, exaggerated postprandial GLP-1 response is believed to contribute to the increased satiety reported by many gastric-bypass surgery patients (94–96). However, a recent clinical study reported that the infusion of exendin 9-39, a GLP-1R antagonist, did not affect ad libitum food intake in post-RYGB patients, although the authors also reported a concomitant increase in plasma levels of the anorexigenic hormone PYY (discussed below), which might offset the orexigenic effect of GLP-1R antagonism (94). The DPP4-resistant GLP-1R agonist, liraglutide, is now in clinical use as a weight-loss therapeutic in obese/overweight individuals (97). GLP-1 is also implicated in regulating hedonic eating through GLP-1Rs located elsewhere in the brainstem (98–100). Peripherally administered GLP-1R agonists may also act directly on GLP-1R at other sites in the brain, notably circumventricular organs and some hypothalamic regions with fenestrated capillaries (101–103). Indeed, Liraglutide can directly activate anorectic POMC/CART neurons in rodents and thus, indirectly inhibit orexigenic AgRP/NPY neurons in the arcuate nucleus (ARC) to reduce food intake (101). As endogenous GLP-1 has a very short half-life, these central actions are likely to be more relevant during therapeutic use of DPP4-resistant GLP-1R analogs, or in post-gastric bypass surgeries, in which GLP-1 “equivalent” levels, or postprandial GLP-1, respectively, are augmented and sufficient to elicit anorectic responses at these CNS targets.
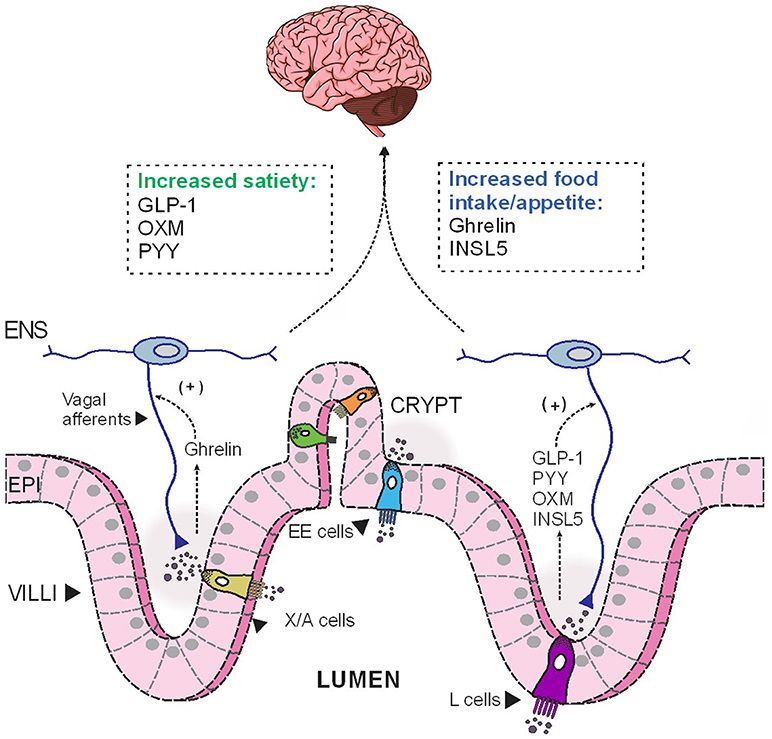
Figure 1. The opposing effects of anorectic and orexigenic gut hormones on food intake. Depending on the luminal stimulation, EE cells release different hormones basolaterally, which then diffuse across the lamina propria to act on their corresponding receptors expressed on nerves endings of vagal and enteric afferents. These hormonal cues are subsequently relayed to the CNS to modulate food intake. EE, enteroendocrine; ENS, enteric nervous system; EPI, epithelial cells; INSL5, insulin-like peptide 5; GLP-1, glucagon-like peptide 1; OXM, oxyntomodulin; PYY, peptide YY.
Oxyntomodulin
Oxyntomodulin (OXM) is a 37-amino acid peptide that contains the entire amino acid sequence of glucagon (104) and is co-secreted with GLP-1 by enteroendocrine L cells at an equimolar ratio (105). Although an endogenous OXM receptor has not been identified, OXM exerts weak agonist activity on GLP-1R (106) and the glucagon receptor (GCGR) (107). Nevertheless, pharmacological levels of OXM (sufficient to activate GLP-1R and GCGR) have shown anti-obesity effects in humans, by significantly reducing appetite (108, 109) and increasing energy expenditure (110). In addition, OXM treatment improved glucose tolerance in high-fat fed mice by potentiating GSIS (111), in a glucose-dependent manner (112), and has an anti-apoptotic effect on β cells (112). OXM infusion significantly reduced glycaemic excursion by augmenting GSIS in obese subjects with or without T2D (113). Such observations prompted the investigation into the potential metabolic benefits of GLP-1R and GCGR co-activation (114, 115), which led to the subsequent development of GLP-1R/GCGR co-agonists (73, 116) and, later, GIPR/GLP-1R/GCGR tri-agonists (117). These agonists have shown impressive anti-obesity effects in preclinical models and are currently being evaluated in phase 2 clinical trials (118).
Peptide YY
Peptide YY (PYY) is co-localized with GLP-1 in enteroendocrine L cells (7, 119) and is co-released with GLP-1 postprandially, in proportion to caloric intake (119, 120). In contrast to GLP-1, which is present in sufficient amount in the duodenum to account for the immediate postprandial surge, PYY abundance is very low in the upper gut and increases distally from the ileum toward to colon (121, 122). Thus, postprandial PYY release under normal physiological conditions is likely to be mediated through paracrine and neural mechanisms (123). An exaggerated postprandial PYY response is observed in gastric bypass patients, and is likely attributed to the increased flow of nutrients into the PYY-rich distal gut, which can directly stimulate L cells (124, 125). Human PYY circulates in two active forms: PYY1−36 and PYY3−36, the latter being an active cleavage product of the former by DPP4 (126). Both are key mediators of the “ileal brake,” a local feedback mechanism triggered by the arrival of nutrients in the ileum that inhibits gastric and pancreatic secretions and proximal intestinal motility (127). The physiological effects of PYY are mediated through a family of NPY receptors (termed Y1, Y2, Y3, Y4, and Y5), which are differentially expressed in a wide range of tissue including enterocytes, myenteric and submucosal neurons and extrinsic primary afferent nerve fibers (123).
Exogenous PYY administration significantly reduces food intake in both obese and lean subjects (128, 129). Pyy-deficient mice are hyperphagic and obese (130) while Pyy overexpression protects mice against obesity induced by HFDs or leptin deficiency (131). Although the “ileal brake” mechanism contributes to its satiating effect (132), PYY3−36 induces satiety primarily by targeting the hypothalamus. The role of PYY as a satiety hormone has been debated, as several independent research groups did not reproduce the anorectic effect in humans reported in the original study by Batterham et al. (133). Moreover, due to its nauseating effect at higher doses (134–136), PYY has not been pursued as an anti-obesity target.
PYY infusion in humans had limited effects on plasma glucose, insulin or glucagon levels on its own (128, 137), nor did it affect glucose excursion and insulin levels during intravenous (138) or oral glucose challenge (136). PYY has trophic effects on pancreatic β cells (139), but such effects are believed to be mediated by islet-derived, rather than gut-derived PYY (140). However, as postprandial PYY levels after gastric bypass surgeries are elevated several folds, it may be possible for gut-derived PYY to exert protective effect on β cells in these settings.
Ghrelin
Ghrelin is an orexigenic hormone secreted by X/A cells present in the mucosa throughout the length of the GI tract, with the highest abundance in the gastric fundus. Circulating ghrelin is significantly elevated during fasting and attenuated upon meal initiation. Post-translational acylation of the ghrelin peptide by ghrelin O-acyl-transferase (GOAT) is crucial for its activity at its endogenous receptor, growth hormone (GH) secretagogue receptor (GHSR1a) (13). GHSR1a is highly expressed in the CNS and is capable of stimulating GH release from the anterior pituitary (13), and lower levels of expression are found in the periphery including the small intestine and pancreatic islets (141). Exogenous ghrelin reliably increases food intake in various species, including humans (142). The orexigenic action of ghrelin is mediated through direct stimulation of the orexigenic AgRP/NPY neurons and concomitant inhibition of the anorectic POMC/CART neurons in the ARC (143, 144). Weight loss achieved through caloric restriction is accompanied by marked elevation in circulating ghrelin (145), which increases feeding drive and has therefore been ascribed as a natural defense against weight loss. Ghrelin is also an anabolic hormone that drives lipogenesis, independent of its effect on appetite (146). Altogether, the orexigenic and anabolic properties of ghrelin renders the ghrelin-GOAT-GHSR1a axis an attractive anti-obesity target. Pharmacological blockade of GOAT or GHSR1a have yielded promising results in preclinical models of obesity (147–150). However, genetic disruption of different components of the ghrelin-GOAT-GHSR1a axis in mice did not have the anticipated anorectic or anti-obesity effects (151–154). Neither Ghrelin nor GOAT deficiency rescue the obese and hyperphagic phenotype of ob/ob mice (152, 155). As such, these data indicate a dispensable role for ghrelin in the regulation of feeding and bodyweight, and that the role of ghrelin in increasing feeding drive may be limited to fasting conditions.
Contrary to its limited role in feeding behavior, ghrelin is a key regulator of glucose homeostasis. Exogenous ghrelin markedly increases blood glucose levels in humans, while genetic ablation of ghrelin or its receptor improve glucose tolerance in HFD-fed and ob/ob mice (152, 156). Ghrelin receptor signaling, specifically in hypothalamic AgRP/NPY neurons, is a critical countermeasure to prevent hypoglycaemia (143). Mice with attenuated ghrelin signaling, due to GOAT-deficiency or ghrelin cell ablation, have a blunted counter-regulatory GH response, and display profound fasting-induced hypoglycaemia (157, 158). Ghrelin protects against hypoglycaemia by triggering the direct release of GH from the anterior pituitary (159), increasing glucagon secretion (160) and inhibiting insulin secretion (161, 162). Ghrelin can protect mice from hypoglycaemia in the absence of intact GCGR signaling (163). Thus, ghrelin may be a potential treatment for acute insulin-induced hypoglycaemia in type 1 diabetes patients.
Insulin-like Peptide 5
Insulin-like peptide 5 (INSL-5) is predominantly expressed in the brain and colonic L cells (164, 165), with immunohistochemical staining and FACS analysis revealing that INSL-5 is overwhelmingly co-expressed with GLP-1 (164). Belonging to the Relaxin-peptide superfamily, INSL-5 has recently been identified as anorexigenic hormone. Secreted INSL-5 acts on the Relaxin/Insulin-like family peptide receptor 4 (RXFP4) (166), which is expressed along the GI tract, the nodose ganglion and the enteric nervous system (164), and inhibits adenylyl cyclase activity (167). Intraperitoneal, but not intracerebroventricular, administration of INSL-5 dose-dependently increases food intake in mice, indicating the peptide may exert its orexigenic effect by acting on peripheral targets, rather than via the CNS (164).
Strong evidence supports the role of INSL-5 as an energy sensor within the colon. Colonic Insl5 and plasma INSL-5 levels are elevated during fasting in calorie-restricted mice and normalize upon refeeding (164). Increased colonic expression of Isnl5 is also observed in germ-free (GF) mice, which lack a gut microbiome (168) and microbial-produced colonic short-chain fatty acids (SCFAs). As a consequence, GF mice have energy-depleted colonocytes due to the absence of their SCFA energy source, butyrate (169). Indeed, the introduction of a functional gut microbiome, which increases luminal SCFA availability, leads to reduced Insl5 expression, in a manner similar to refeeding calorie-restricted mice (169). The role of INSL-5 as an energy sensor within the colon is not restricted to the availability of SCFAs, as Insl5 expression in GF mice can also be reduced following HFD consumption, in which unabsorbed lipids provide an alternative energy source to colonocytes (168). As such, INSL-5 may serve as an important link between the gut microbiota and host in the context of metabolism.
The effect of INSL-5 on glucose homeostasis is less clear. While it was initially reported that mice deficient in Insl5 were mildly glucose-intolerant (170), this appears to be age (170) and strain-dependent (164, 168). Insl5−/− mice have impaired intraperitoneal glucose tolerance but superior insulin sensitivity and moderately reduced hepatic glucose production (168). The impact of INSL-5 on glucose control in mice also appears dependent on the mode of glucose delivery, as blood glucose or insulin levels were similar in Insl5−/− mice compared to WT following an oral glucose test (164, 168). As oral but not intraperitoneal glucose administration stimulates the parasympathetic aspects of the gut-brain axis to centrally mediate hepatic glucose production (168), these findings suggest that INSL-5 may influence glucose homeostasis via direct actions on hepatocytes to influence hepatic gluconeogenesis. Studies on the insulinotropic action of INSL-5 have produced conflicting results (167, 171). As Insl5 is not expressed in pancreatic islets (164, 168), any direct effects of endogenous INSL-5 on islets would appear to occur in an endocrine fashion. Circulating INSL-5 levels are estimated to be in the picomolar range (164, 172), which is several orders of magnitude lower than the EC50 of INSL-5 on RXRP4 (166) and the supraphysiological concentrations used in the majority of insulin secretion experiments may have contributed to the conflicting results.
Concluding Remarks
Although enteroendocrine cells make up only 1% of the epithelial cell population along the GI tract (9), the hormones they secrete in response to one's nutritional status have profound impacts on peripheral metabolism (Figure 2). We have provided an overview of the metabolic actions of some of these gut hormones, including their role in maintaining glucose homeostasis and energy balance. Under fasting conditions, ghrelin and INSL5 levels are elevated to induce hunger and to prevent hypoglycaemia. Conversely, during the postprandial period, elevated GIP and GLP-1 levels augment postprandial insulin secretion to prevent hyperglycaemia. In addition to its insulinotropic effect, GLP-1 also act in concert with PYY and OXM to induce satiety (Figure 1). Moreover, some of the impressive metabolic gains from bariatric surgeries have been ascribed to alterations in the secretory profile of gut hormones. Altogether, the enteroendocrine system represents an attractive therapeutic target for treating metabolic disease as the pleiotropic effects of different gut hormones can be exploited individually.
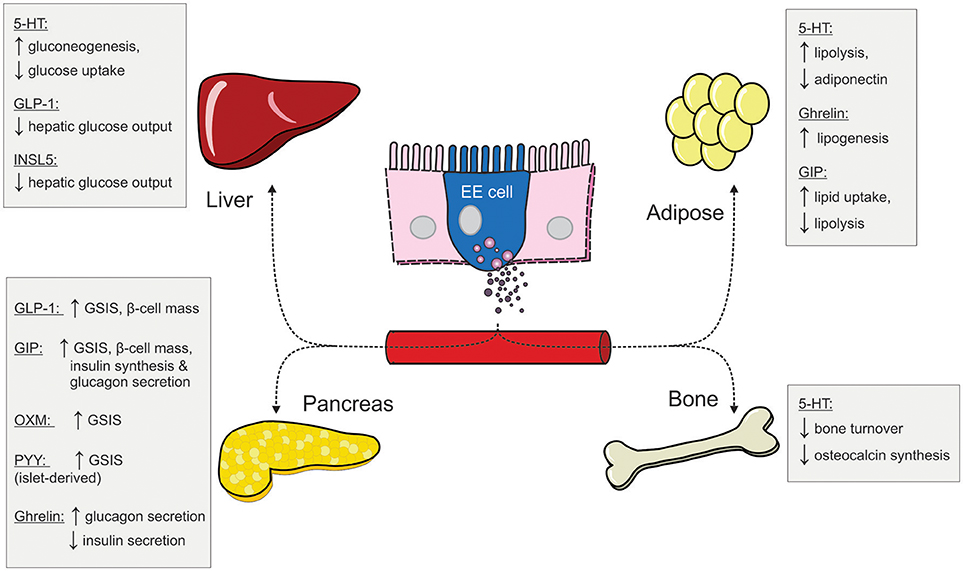
Figure 2. The peripheral metabolic effects of different gut hormones (5-HT, serotonin; EE, enteroendocrine; GIP, glucose-dependent insulinotropic hormone; GLP-1, glucagon-like peptide 1; INSL5, insulin-like peptide 5; OXM, oxyntomodulin; PYY, peptide YY; GSIS, glucose-stimulated insulin secretion).
Author Contributions
ES and AM wrote the manuscript. DK and RY critically reviewed the manuscript. All authors approved the final version for publication.
Funding
DK was supported by a research fellowship from the National Health and Medical Research Council (NHMRC) of Australia.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Engelstoft MS, Egerod KL, Lund ML, Schwartz TW. Enteroendocrine cell types revisited. Curr Opin Pharmacol. (2013) 13:912–21. doi: 10.1016/j.coph.2013.09.018
3. Egerod KL, Engelstoft MS, Grunddal KV, Nohr MK, Secher A, Sakata I, et al. A major lineage of enteroendocrine cells coexpress CCK, secretin, GIP, GLP-1, PYY, and neurotensin but not somatostatin. Endocrinology (2012) 153:5782–95. doi: 10.1210/en.2012-1595
4. Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CA, Parker HE, et al. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology (2012) 153:3054–65. doi: 10.1210/en.2011-2170
5. Fothergill LJ, Callaghan B, Hunne B, Bravo DM, Furness JB. Costorage of enteroendocrine hormones evaluated at the cell and subcellular levels in male mice. Endocrinology (2017) 158:2113–23. doi: 10.1210/en.2017-00243
6. Grunddal KV, Ratner CF, Svendsen B, Sommer F, Engelstoft MS, Madsen AN, et al. Neurotensin is coexpressed, coreleased, and acts together with GLP-1 and PYY in enteroendocrine control of metabolism. Endocrinology (2016) 157:176–94. doi: 10.1210/en.2015-1600
7. Cho HJ, Robinson ES, Rivera LR, McMillan PJ, Testro A, Nikfarjam M, et al. Glucagon-like peptide 1 and peptide YY are in separate storage organelles in enteroendocrine cells. Cell Tissue Res. (2014) 357:63–9. doi: 10.1007/s00441-014-1886-9
8. Raghupathi R, Duffield MD, Zelkas L, Meedeniya A, Brookes SJ, Sia TC, et al. Identification of unique release kinetics of serotonin from guinea-pig and human enterochromaffin cells. J Physiol. (2013) 591:5959–75. doi: 10.1113/jphysiol.2013.259796
9. Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol. (2016) 78:277–99. doi: 10.1146/annurev-physiol-021115-105439
10. Gribble FM. The gut endocrine system as a coordinator of postprandial nutrient homoeostasis. Proc Nutr Soc. (2012) 71:456–62. doi: 10.1017/S0029665112000705
11. Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet (2018) 392:2180–93. doi: 10.1016/S0140-6736(18)32260-8
12. Matthes S, Bader M. Peripheral serotonin synthesis as a new drug target. Trends Pharmacol Sci. (2018) 39:560–572. doi: 10.1016/j.tips.2018.03.004
13. Muller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, et al. Ghrelin. Mol Metab. (2015) 4:437–60. doi: 10.1016/j.molmet.2015.03.005
14. Spiller R. Serotonin and GI clinical disorders. Neuropharmacology (2008) 55:1072–80. doi: 10.1016/j.neuropharm.2008.07.016
15. Gershon MD. 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr Opin Endocrinol Diabetes Obes. (2013) 20:14–21. doi: 10.1097/MED.0b013e32835bc703
16. Martin AM, Lumsden AL, Young RL, Jessup CF, Spencer NJ, Keating DJ. Regional differences in nutrient-induced secretion of gut serotonin. Physiol Rep. (2017) 5:e13199. doi: 10.14814/phy2.13199
17. Zelkas L, Raghupathi R, Lumsden AL, Martin AM, Sun E, Spencer NJ, et al. Serotonin-secreting enteroendocrine cells respond via diverse mechanisms to acute and chronic changes in glucose availability. Nutr Metab. (2015) 12:55. doi: 10.1186/s12986-015-0051-0
18. Symonds EL, Peiris M, Page AJ, Chia B, Dogra H, Masding A, et al. Mechanisms of activation of mouse and human enteroendocrine cells by nutrients. Gut (2015) 64:618–26. doi: 10.1136/gutjnl-2014-306834
19. Kidd M, Modlin IM, Gustafsson BI, Drozdov I, Hauso O, Pfragner R. Luminal regulation of normal and neoplastic human EC cell serotonin release is mediated by bile salts, amines, tastants, and olfactants. Am J Physiol Gastrointest Liver Physiol. (2008) 295:G260–72. doi: 10.1152/ajpgi.00056.2008
20. Wang F, Knutson K, Alcaino C, Linden DR, Gibbons SJ, Kashyap P, et al. Mechanosensitive ion channel Piezo2 is important for enterochromaffin cell response to mechanical forces. J Physiol. (2017) 595:79–91. doi: 10.1113/JP272718
21. Modlin IM, Kidd M, Pfragner R, Eick GN, and Champaneria MC. The functional characterization of normal and neoplastic human enterochromaffin cells. J Clin Endocrinol Metab. (2006) 91:2340–8. doi: 10.1210/jc.2006-0110
22. Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell (2015) 161:264–76. doi: 10.1016/j.cell.2015.02.047
23. Spencer NJ, Sia TC, Brookes SJ, Costa M, Keating DJ. CrossTalk opposing view: 5-HT is not necessary for peristalsis. J Physiol. (2015) 593:3229–31. doi: 10.1113/JP270183
24. Spencer NJ, Nicholas SJ, Robinson L, Kyloh M, Flack N, Brookes SJ, et al. Mechanisms underlying distension-evoked peristalsis in guinea pig distal colon: is there a role for enterochromaffin cells? Am J Physiol Gastrointest Liver Physiol. (2011) 301:G519–27. doi: 10.1152/ajpgi.00101.2011
25. Keating DJ, Spencer NJ. Release of 5-hydroxytryptamine from the mucosa is not required for the generation or propagation of colonic migrating motor complexes. Gastroenterology (2010) 138:659–70 670 e1-2. doi: 10.1053/j.gastro.2009.09.020
26. Ghia JE, Li N, Wang H, Collins M, Deng Y, El-Sharkawy RT, et al. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology (2009) 137:1649–60. doi: 10.1053/j.gastro.2009.08.041
27. Martin AM, Young RL, Leong L, Rogers GB, Spencer NJ, Jessup CF, et al. The diverse metabolic roles of peripheral serotonin. Endocrinology (2017) 158:1049–63. doi: 10.1210/en.2016-1839
28. Young RL, Lumsden AL, Keating DJ. Gut serotonin is a regulator of obesity and metabolism. Gastroenterology (2015) 149:253–5. doi: 10.1053/j.gastro.2015.05.020
29. Levine RA, Pesch LA, Klatskin G, Giarman NJ. Effect of serotonin on glycogen metabolism in isolated rat liver. J Clin Invest. (1964) 43:797–809. doi: 10.1172/JCI104966
30. Sumara G, Sumara O, Kim JK, Karsenty G. Gut-derived serotonin is a multifunctional determinant to fasting adaptation. Cell Metab. (2012) 16: 588–600. doi: 10.1016/j.cmet.2012.09.014
31. Crane JD, Palanivel R, Mottillo EP, Bujak AL, Wang H, Ford RJ, et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat Med. (2015) 21:166–72. doi: 10.1038/nm.3766
32. Oh CM, Namkung J, Go Y, Shong KE, Kim K, Kim H, et al. Regulation of systemic energy homeostasis by serotonin in adipose tissues. Nat Commun. (2015) 6:6794. doi: 10.1038/ncomms7794
33. Uchida-Kitajima S, Yamauchi T, Takashina Y, Okada-Iwabu M, Iwabu M, Ueki K, et al. 5-Hydroxytryptamine 2A receptor signaling cascade modulates adiponectin and plasminogen activator inhibitor 1 expression in adipose tissue. FEBS Lett (2008) 582:3037–44. doi: 10.1016/j.febslet.2008.07.044
34. Zoch ML, Clemens TL, Riddle RC. New insights into the biology of osteocalcin. Bone (2016) 82:42–9. doi: 10.1016/j.bone.2015.05.046
35. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell (2007) 130:456–69. doi: 10.1016/j.cell.2007.05.047
36. Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z, et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature (2017) 543:385–90. doi: 10.1038/nature21697
37. Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell (2008) 135:825–37. doi: 10.1016/j.cell.2008.09.059
38. Young RL, Lumsden AL, Martin AM, Schober G, Pezos N, Thazhath SS, et al. Augmented capacity for peripheral serotonin release in human obesity. Int J Obes. (2018). 42:1880–9. doi: 10.1038/s41366-018-0047-8
39. Le Beyec J, Pelletier AL, Arapis K, Hourseau M, Cluzeaud F, Descatoire V, et al. Overexpression of gastric leptin precedes adipocyte leptin during high-fat diet and is linked to 5HT-containing enterochromaffin cells. Int J Obes. (2014) 38:1357–64. doi: 10.1038/ijo.2014.14
40. Takahashi T, Yano M, Minami J, Haraguchi T, Koga N, Higashi K, et al. Sarpogrelate hydrochloride, a serotonin2A receptor antagonist, reduces albuminuria in diabetic patients with early-stage diabetic nephropathy. Diabetes Res Clin Pract. (2002) 58:123–9. doi: 10.1016/S0168-8227(02)00105-5
41. Malyszko J, Urano T, Knofler R, Taminato A, Yoshimi T, Takada Y, et al. Daily variations of platelet aggregation in relation to blood and plasma serotonin in diabetes. Thromb Res. (1994) 75:569–76. doi: 10.1016/0049-3848(94)90231-3
42. Barradas MA, Gill DS, Fonseca VA, Mikhailidis DP, Dandona P. Intraplatelet serotonin in patients with diabetes mellitus and peripheral vascular disease. Eur J Clin Invest. (1988) 18: 399–404.
43. Nichols DE, Nichols CD. Serotonin receptors. Chem Rev. (2008) 108 :614–41. doi: 10.1021/cr078224o
44. Mortensen K, Petersen LL, ØRskov C. Colocalization of GLP-1 and GIP in human and porcine intestine. Ann NY Acad Sci. (2006) 921:469–72. doi: 10.1111/j.1749-6632.2000.tb07017.x
45. Gremlich S, Porret A, Hani EH, Cherif D, Vionnet N, Froguel P, et al. Cloning, functional expression, and chromosomal localization of the human pancreatic islet glucose-dependent insulinotropic polypeptide receptor. Diabetes (1995) 44:1202–8. doi: 10.2337/diab.44.10.1202
46. Yip RG, Boylan MO, Kieffer TJ, Wolfe MM. Functional GIP receptors are present on adipocytes. Endocrinology (1998) 139:4004–7. doi: 10.1210/endo.139.9.6288
47. Bollag RJ, Zhong Q, Phillips P, Min L, Zhong L, Cameron R, et al. Isales, osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology (2000) 141:1228–35. doi: 10.1210/endo.141.3.7366
48. Faivre E, Gault VA, Thorens B, Holscher C. Glucose-dependent insulinotropic polypeptide receptor knockout mice are impaired in learning, synaptic plasticity, and neurogenesis. J Neurophysiol. (2011) 105:1574–80. doi: 10.1152/jn.00866.2010
49. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
50. Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. (1986) 63:492–8. doi: 10.1210/jcem-63-2-492
51. Widenmaier SB, Ao Z, Kim SJ, Warnock G, McIntosh CH. Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J Biol Chem. (2009) 284:30372–82. doi: 10.1074/jbc.M109.060178
52. Vilsboll T, Krarup T, Madsbad S, Holst JJ. Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia (2002) 45:1111–9. doi: 10.1007/s00125-002-0878-6
53. Mentis N, Vardarli I, Kothe LD, Holst JJ, Deacon CF, Theodorakis M, et al. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes (2011) 60:1270–6. doi: 10.2337/db10-1332
54. Meier JJ, Hucking K, Holst JJ, Deacon CF, Schmiegel WH, Nauck MA. Reduced insulinotropic effect of gastric inhibitory polypeptide in first-degree relatives of patients with type 2 diabetes. Diabetes (2001) 50:2497–504. doi: 10.2337/diabetes.50.11.2497
55. Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CH, Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum Mol Genet. (2009) 18:2388–99. doi: 10.1093/hmg/ddp178
56. Tseng CC, Zhang XY. Role of G protein-coupled receptor kinases in glucose-dependent insulinotropic polypeptide receptor signaling*. Endocrinology (2000) 141:947–52. doi: 10.1210/endo.141.3.7365
57. Christensen M, Vedtofte L, Holst JJ, Vilsboll T, Knop FK. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes (2011) 60:3103–9. doi: 10.2337/db11-0979
58. Lund A, Vilsboll T, Bagger JI, Holst JJ, Knop FK. The separate and combined impact of the intestinal hormones, GIP, GLP-1, and GLP-2, on glucagon secretion in type 2 diabetes. Am J Physiol Endocrinol Metab. (2011) 300:E1038–46. doi: 10.1152/ajpendo.00665.2010
59. Gogebakan O, Andres J, Biedasek K, Mai K, Kuhnen P, Krude H, et al. Glucose-dependent insulinotropic polypeptide reduces fat-specific expression and activity of 11beta-hydroxysteroid dehydrogenase type 1 and inhibits release of free fatty acids. Diabetes (2012) 61:292–300. doi: 10.2337/db10-0902
60. Calanna S, Christensen M, Holst JJ, Laferrere B, Gluud LL, Vilsboll T, et al. Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta-analysis of clinical studies. Diabetes Care (2013) 36:3346–52. doi: 10.2337/dc13-0465
61. Theodorakis MJ, Carlson O, Muller DC, Egan JM. Elevated plasma glucose-dependent insulinotropic polypeptide associates with hyperinsulinemia in impaired glucose tolerance. Diabetes Care (2004) 27:1692–8. doi: 10.2337/diacare.27.7.1692
62. Gniuli D, Calcagno A, Dalla Libera L, Calvani R, Leccesi L, Caristo ME, et al. High-fat feeding stimulates endocrine, glucose-dependent insulinotropic polypeptide (GIP)-expressing cell hyperplasia in the duodenum of Wistar rats. Diabetologia (2010) 53:2233–40. doi: 10.1007/s00125-010-1830-9
63. Isken F, Pfeiffer AF, Nogueiras R, Osterhoff MA, Ristow M, Thorens B, et al. Deficiency of glucose-dependent insulinotropic polypeptide receptor prevents ovariectomy-induced obesity in mice. Am J Physiol Endocrinol Metab. 295 (2008) E350–5. doi: 10.1152/ajpendo.00008.2008
64. Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. (2002) 8:738–42. doi: 10.1038/nm727
65. Ahlqvist E, Osmark P, Kuulasmaa T, Pilgaard K, Omar B, Brons C, et al. Link between GIP and osteopontin in adipose tissue and insulin resistance. Diabetes (2013) 62:2088–94. doi: 10.2337/db12-0976
66. Chen S, Okahara F, Osaki N, Shimotoyodome A. Increased GIP signaling induces adipose inflammation via a HIF-1alpha-dependent pathway and impairs insulin sensitivity in mice. Am J Physiol Endocrinol Metab. (2015) 308:E414–25. doi: 10.1152/ajpendo.00418.2014
67. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. (2007) 117:2877–88. doi: 10.1172/JCI31986
68. Joo E, Harada N, Yamane S, Fukushima T, Taura D, Iwasaki K, et al. Inhibition of gastric inhibitory polypeptide receptor signaling in adipose tissue reduces insulin resistance and hepatic steatosis in high-fat diet-fed mice. Diabetes (2017) 66:868–79. doi: 10.2337/db16-0758
69. Nasteska D, Harada N, Suzuki K, Yamane S, Hamasaki A, Joo E, et al. Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high-fat diet conditions. Diabetes (2014) 63:2332–43. doi: 10.2337/db13-1563
70. Kim SJ, Nian C, Karunakaran S, Clee SM, Isales CM, McIntosh CH. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS ONE (2012) 7:e40156. doi: 10.1371/journal.pone.0040156
71. Tsukiyama K, Yamada Y, Yamada C, Harada N, Kawasaki Y, Ogura M, et al. Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Mol Endocrinol. (2006) 20:1644–51. doi: 10.1210/me.2005-0187
72. Mera P, Ferron M, Mosialou I. Regulation of energy metabolism by bone-derived hormones. Cold Spring Harb Perspect Med. (2018) 8:a031666 doi: 10.1101/cshperspect.a031666
73. Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. (2013) 5:209ra151. doi: 10.1126/scitranslmed.3007218
74. Sun EW, de Fontgalland D, Rabbitt P, Hollington P, Sposato L, Due SL, et al. Mechanisms controlling glucose-induced GLP-1 secretion in human small intestine. Diabetes (2017) 66:2144–9. doi: 10.2337/db17-0058
75. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. (2007) 87:1409–39. doi: 10.1152/physrev.00034.2006
76. Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology (1999) 140:5356–63. doi: 10.1210/endo.140.11.7143
77. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet (1987) 2:1300–4. doi: 10.1016/S0140-6736(87)91194-9
78. Dardevet D, Moore MC, DiCostanzo CA, Farmer B, Neal DW, Snead W, et al. Insulin secretion-independent effects of GLP-1 on canine liver glucose metabolism do not involve portal vein GLP-1 receptors. Am J Physiol Gastrointest Liver Physiol. (2005) 289:G806–14. doi: 10.1152/ajpgi.00121.2005
79. Ahren B. Hepato-incretin function of GLP-1: novel concept and target in type 1 diabetes. Diabetes (2015) 64:715–7. doi: 10.2337/db14-1671
80. Meier JJ, Kemmeries G, Holst JJ, Nauck MA. Erythromycin antagonizes the deceleration of gastric emptying by glucagon-like peptide 1 and unmasks its insulinotropic effect in healthy subjects. Diabetes (2005) 54:2212–8. doi: 10.2337/diabetes.54.7.2212
81. Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. (1997) 273:E981–8. doi: 10.1152/ajpendo.1997.273.5.E981
82. Hellstrom PM. GLP-1: broadening the incretin concept to involve gut motility. Regul Pept. (2009) 156:9–12. doi: 10.1016/j.regpep.2009.04.004
83. Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7-36) amide in type 2 (noninsulin-dependent) diabetic patients. J Clin Endocrinol Metab. (1996) 81:327–32.
84. Hare KJ, Vilsboll T, Asmar M, Deacon CF, Knop FK, Holst JJ. The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes (2010) 59:1765–70. doi: 10.2337/db09-1414
85. Creutzfeldt WO, Kleine N, Willms B, Orskov C, Holst JJ, Nauck M.A. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon-like peptide I(7-36) amide in type I diabetic patients. Diabetes Care (1996) 19:580–6.
86. Hare KJ, Knop FK, Asmar M, Madsbad S, Deacon CF, Holst JJ, et al. Preserved inhibitory potency of GLP-1 on glucagon secretion in type 2 diabetes mellitus. J Clin Endocrinol Metab. (2009) 94:4679–87. doi: 10.1210/jc.2009-0921
87. Marathe CS, Rayner CK, Jones KL, Horowitz M. Effects of GLP-1 and incretin-based therapies on gastrointestinal motor function. Exp Diabetes Res. (2011) 2011:279530. doi: 10.1155/2011/279530
88. Plamboeck A, Veedfald S, Deacon CF, Hartmann B, Wettergren A, Svendsen LB, et al. The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G1117–27. doi: 10.1152/ajpgi.00035.2013
89. Baraboi ED, St-Pierre DH, Shooner J, Timofeeva E, Richard D. Brain activation following peripheral administration of the GLP-1 receptor agonist exendin-4. Am J Physiol Regul Integr Comp Physiol. (2011) 301:R1011–24. doi: 10.1152/ajpregu.00424.2010
90. Kanse SM, Kreymann B, Ghatei MA, Bloom SR. Identification and characterization of glucagon-like peptide-1 7-36 amide-binding sites in the rat brain and lung. FEBS Lett. (1988) 241:209–12. doi: 10.1016/0014-5793(88)81063-9
91. Verdich C, Flint A, Gutzwiller JP, Naslund E, Beglinger C, Hellstrom PM, et al. A meta-analysis of the effect of glucagon-like peptide-1 (7-36) amide on ad libitum energy intake in humans. J Clin Endocrinol Metab. (2001) 86:4382–9. doi: 10.1210/jc.86.9.4382
92. Flint A, Raben A, Astrup A, Holst JJ, Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. (1998) 101:515–20.
93. Gutzwiller JP, Drewe J, Goke B, Schmidt H, Rohrer B, Lareida J, et al. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol. (1999) 276:R1541–4. doi: 10.1152/ajpregu.1999.276.5.R1541
94. Svane MS, Jorgensen NB, Bojsen-Moller KN, Dirksen C, Nielsen S, Kristiansen VB, et al. Peptide YY and glucagon-like peptide-1 contribute to decreased food intake after Roux-en-Y gastric bypass surgery. Int J Obes. (2016) 40:1699–706 doi: 10.1038/ijo.2016.121
95. Ten Kulve JS, Veltman DJ, Gerdes VEA, van Bloemendaal L, Barkhof F, Deacon CF, et al. Elevated postoperative endogenous GLP-1 levels mediate effects of roux-en-Y gastric bypass on neural responsivity to food cues. Diabetes Care (2017) 40:1522–29. doi: 10.2337/dc16-2113
96. Madsbad S, Dirksen C, Holst JJ. Mechanisms of changes in glucose metabolism and bodyweight after bariatric surgery. Lancet Diabetes Endocrinol. (2014) 2:152–64. doi: 10.1016/S2213-8587(13)70218-3
97. Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. (2015) 373:11–22. doi: 10.1056/NEJMoa1411892
98. Alhadeff AL, Grill HJ. Hindbrain nucleus tractus solitarius glucagon-like peptide-1 receptor signaling reduces appetitive and motivational aspects of feeding. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R465–70. doi: 10.1152/ajpregu.00179.2014
99. Dickson SL, Shirazi RH, Hansson C, Bergquist F, Nissbrandt H, Skibicka KP. The glucagon-like peptide 1 (GLP-1) analogue, exendin-4, decreases the rewarding value of food: a new role for mesolimbic GLP-1 receptors. J Neurosci. (2012) 32:4812–20. doi: 10.1523/JNEUROSCI.6326-11.2012
100. Alhadeff AL, Mergler BD, Zimmer DJ, Turner CA, Reiner DJ, Schmidt HD, et al. Endogenous glucagon-like peptide-1 receptor signaling in the nucleus tractus solitarius is required for food intake control. Neuropsychopharmacology (2017) 42:1471–9. doi: 10.1038/npp.2016.246
101. Secher A, Jelsing J, Baquero AF, Hecksher-Sorensen J, Cowley MA, Dalboge LS, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest. (2014) 124:4473–88. doi: 10.1172/JCI75276
102. Orskov C, Poulsen SS, Moller M, Holst JJ. Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide I. Diabetes (1996) 45:832–5. doi: 10.2337/diab.45.6.832
103. Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci (2002) 18:7–14. doi: 10.1385/JMN:18:1-2:07
104. Bataille D, Gespach C, Tatemoto K, Marie JC, Coudray AM, Rosselin G, et al. Bioactive enteroglucagon (oxyntomodulin): present knowledge on its chemical structure and its biological activities. Peptides (1981) 2 (Suppl. 2):41–4. doi: 10.1016/0196-9781(81)90008-5
105. Wewer Albrechtsen NJ, Hornburg D, Albrechtsen R, Svendsen B, Torang S, Jepsen SL, et al. Oxyntomodulin identified as a marker of Type 2 diabetes and gastric bypass surgery by mass-spectrometry based profiling of human plasma. EBioMed. (2016) 7:112–20. doi: 10.1016/j.ebiom.2016.03.034
106. Gros L, Thorens B, Bataille D, Kervran A. Glucagon-like peptide-1-(7-36) amide, oxyntomodulin, and glucagon interact with a common receptor in a somatostatin-secreting cell line. Endocrinology (1993) 133:631–8. doi: 10.1210/endo.133.2.8102095
107. Baldissera FG, Holst JJ, Knuhtsen S, Hilsted L, Nielsen O.V. Oxyntomodulin (glicentin-(33-69)): pharmacokinetics, binding to liver cell membranes, effects on isolated perfused pig pancreas, and secretion from isolated perfused lower small intestine of pigs. Regul Pept. (1988) 21:151–66.
108. Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsboll T. Effect of oxyntomodulin, glucagon, GLP-1, and combined glucagon +GLP-1 infusion on food intake, appetite, and resting energy expenditure. J Clin Endocrinol Metab. (2015) 100:4541–52. doi: 10.1210/jc.2015-2335
109. Wynne K, Park AJ, Small CJ, Patterson M, Ellis SM, Murphy KG, et al. Bloom, subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double-blind, randomized, controlled trial. Diabetes (2005) 54:2390–5. doi: 10.2337/diabetes.54.8.2390
110. Wynne K, Park AJ, Small CJ, Meeran K, Ghatei MAG.S., Frost et al. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes. (2006) 30:1729–36. doi: 10.1038/sj.ijo.0803344
111. Parlevliet ET, Heijboer AC, Schroder-van der Elst JP, Havekes LM, Romijn JA, Pijl H, et al. Oxyntomodulin ameliorates glucose intolerance in mice fed a high-fat diet. Am J Physiol Endocrinol Metab. (2008) 294:E142–7. doi: 10.1152/ajpendo.00576.2007
112. Maida A, Lovshin JA, Baggio LL, Drucker DJ. The glucagon-like peptide-1 receptor agonist oxyntomodulin enhances beta-cell function but does not inhibit gastric emptying in mice. Endocrinology (2008) 149:5670–8. doi: 10.1210/en.2008-0336
113. Shankar SS, Shankar RR, Mixson LA, Miller DL, Pramanik B, O'Dowd AK, et al. Native oxyntomodulin has significant glucoregulatory effects independent of weight loss in obese humans with and without Type 2 diabetes. Diabetes (2018) 67:1105–12 doi: 10.2337/db17-1331
114. Pocai A. Unraveling oxyntomodulin, GLP1's enigmatic brother. J Endocrinol. (2012) 215:335–46. doi: 10.1530/JOE-12-0368
115. Pocai A. Action and therapeutic potential of oxyntomodulin. Mol Metab. (2014) 3:241–51. doi: 10.1016/j.molmet.2013.12.001
116. Clemmensen C, Chabenne J, Finan B, Sullivan L, Fischer K, Kuchler D, et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes (2014) 63:1422–7. doi: 10.2337/db13-1609
117. Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. (2015) 21:27–36. doi: 10.1038/nm.3761
118. Tschop MH, Finan B, Clemmensen C, Gelfanov V, Perez-Tilve D, Muller TD, et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab (2016) 24:51–62. doi: 10.1016/j.cmet.2016.06.021
119. Habib AM, Richards P, Rogers GJ, Reimann F, Gribble FM. Co-localisation and secretion of glucagon-like peptide 1 and peptide YY from primary cultured human L cells. Diabetologia (2013) 56:1413–6. doi: 10.1007/s00125-013-2887-z
120. Rozengurt N, Wu SV, Chen MC, Huang C, Sternini C, Rozengurt E. Colocalization of the alpha-subunit of gustducin with PYY and GLP-1 in L cells of human colon. Am J Physiol Gastrointest Liver Physiol. (2006) 291:G792–802. doi: 10.1152/ajpgi.00074.2006
121. Cho HJ, Kosari S, Hunne B, Callaghan B, Rivera LR, Bravo DM, et al. Differences in hormone localisation patterns of K and L type enteroendocrine cells in the mouse and pig small intestine and colon. Cell Tissue Res. (2015) 359:693–8. doi: 10.1007/s00441-014-2033-3
122. Ekblad E, Sundler F. Distribution of pancreatic polypeptide and peptide YY. Peptides (2002) 23:251–61. doi: 10.1016/S0196-9781(01)00601-5
123. Holzer P, Reichmann F, Farzi A. Neuropeptide Y, peptide YY and pancreatic polypeptide in the gut-brain axis. Neuropeptides (2012) 46:261–74. doi: 10.1016/j.npep.2012.08.005
124. Pournaras DJ, Aasheim ET, Bueter M, Ahmed AR, Welbourn R, Olbers T, et al. Effect of bypassing the proximal gut on gut hormones involved with glycemic control and weight loss. Surg Obes Relat Dis. (2012) 8:371–4. doi: 10.1016/j.soard.2012.01.021
125. Dirksen C, Damgaard M, Bojsen-Moller KN, Jorgensen NB, Kielgast U, Jacobsen SH, et al. Fast pouch emptying, delayed small intestinal transit, and exaggerated gut hormone responses after Roux-en-Y gastric bypass. Neurogastroenterol Motil. (2013) 25:346–e255. doi: 10.1111/nmo.12087
126. Mentlein R, Dahms P, Grandt D, Kruger R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul Pept. (1993) 49:133–44. doi: 10.1016/0167-0115(93)90435-B
127. Savage AP, Adrian TE, Carolan G, Chatterjee VK, Bloom SR. Effects of peptide YY (PYY) on mouth to caecum intestinal transit time and on the rate of gastric emptying in healthy volunteers. Gut (1987) 28:166–70. doi: 10.1136/gut.28.2.166
128. Batterham RL, Cohen MA, Ellis SM, Le Roux CW, Withers DJ, Frost GS, et al. Inhibition of food intake in obese subjects by peptide YY3-36. N Engl J Med. (2003) 349:941–8. doi: 10.1056/NEJMoa030204
129. Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature (2002) 418:650–4. doi: 10.1038/nature00887
130. Batterham RL, Heffron H, Kapoor S, Chivers JE, Chandarana K, Herzog H, et al. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. (2006) 4:223–33. doi: 10.1016/j.cmet.2006.08.001
131. Boey D, Lin S, Enriquez RF, Lee NJ, Slack K, Couzens MPA., et al. PYY transgenic mice are protected against diet-induced and genetic obesity. Neuropeptides (2008) 42:19–30. doi: 10.1016/j.npep.2007.11.003
132. Maljaars PW, Peters HP, Mela DJ, Masclee AA. Ileal brake: a sensible food target for appetite control. a review. Physiol Behav. (2008) 95:271–81. doi: 10.1016/j.physbeh.2008.07.018
133. Boggiano MM, Chandler PC, Oswald KD, Rodgers RJ, Blundell JE, Ishii Y., et al. PYY3-36 as an anti-obesity drug target. Obes Rev. (2005) 6:307–22. doi: 10.1111/j.1467-789X.2005.00218.x
134. Gantz I, Erondu N, Mallick M, Musser B, Krishna R, Tanaka WKK., et al. Amatruda, efficacy and safety of intranasal peptide YY3-36 for weight reduction in obese adults. J Clin Endocrinol Metab. (2007) 92:1754–7. doi: 10.1210/jc.2006-1806
135. le Roux CW, Borg CM, Murphy KG, Vincent RP, Ghatei MA, Bloom SR. Supraphysiological doses of intravenous PYY3-36 cause nausea, but no additional reduction in food intake. Ann Clin Biochem. (2008) 45:93–5. doi: 10.1258/acb.2007.007068
136. Sloth B, Holst JJ, Flint A, Gregersen NT, Astrup A. Effects of PYY1-36 and PYY3-36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in obese and lean subjects. Am J Physiol Endocrinol Metab. (2007) 292:E1062–8. doi: 10.1152/ajpendo.00450.2006
137. Adrian TE, Sagor GR, Savage AP, Bacarese-Hamilton AJ, Hall GM, Bloom SR. Peptide YY kinetics and effects on blood pressure and circulating pancreatic and gastrointestinal hormones and metabolites in man. J Clin Endocrinol Metab. (1986) 63:803–7. doi: 10.1210/jcem-63-4-803
138. Ahren B, Larsson H. Peptide YY does not inhibit glucose-stimulated insulin secretion in humans. Eur J Endocrinol. (1996) 134:362–5. doi: 10.1530/eje.0.1340362
139. Shi YC, Loh K, Bensellam M, Lee K, Zhai L, Lau J, et al. Pancreatic PYY is critical in the control of insulin secretion and glucose homeostasis in female mice. Endocrinology (2015) 156:3122–36. doi: 10.1210/en.2015-1168
140. Sam AH, Gunner DJ, King A, Persaud SJ, Brooks L, Hostomska K, et al. Bewick, selective ablation of peptide YY cells in adult mice reveals their role in beta cell survival. Gastroenterology (2012) 143:459–68. doi: 10.1053/j.gastro.2012.04.047
141. Sun Y, Garcia JM, Smith RG. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology (2007) 148:1323–9. doi: 10.1210/en.2006-0782
142. Druce MR, Wren AM, Park AJ, Milton JE, Patterson M, Frost G, et al. Ghrelin increases food intake in obese as well as lean subjects. Int J Obes. (2005) 29:1130–6. doi: 10.1038/sj.ijo.0803001
143. Wang Q, Liu C, Uchida A, Chuang JC, Walker A, Liu T, et al. Arcuate AgRP neurons mediate orexigenic and glucoregulatory actions of ghrelin. Mol Metab. (2014) 3:64–72. doi: 10.1016/j.molmet.2013.10.001
144. Chen HY, Trumbauer ME, Chen AS, Weingarth DT, Adams JR, Frazier EG, et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology (2004) 145:2607–12. doi: 10.1210/en.2003-1596
145. Cummings DE, Weigle DS, Frayo RS, Breen PA, Ma MK, Dellinger EP, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. (2002) 346:1623–30. doi: 10.1056/NEJMoa012908
146. Perez-Tilve D, Heppner K, Kirchner H, Lockie SH, Woods SC, Smiley DL, et al. Ghrelin-induced adiposity is independent of orexigenic effects. FASEB J. (2011) 25:2814–22. doi: 10.1096/fj.11-183632
147. Asakawa A, Inui A, Kaga T, Katsuura G, Fujimiya M, Fujino MA, et al. Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut (2003) 52:947–52. doi: 10.1136/gut.52.7.947
148. Esler WP, Rudolph J, Claus TH, Tang W, Barucci N, Brown SE, et al. Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology (2007) 148:5175–85. doi: 10.1210/en.2007-0239
149. Ge X, Yang H, Bednarek MA, Galon-Tilleman H, Chen P, Chen M, et al. LEAP2 is an endogenous antagonist of the ghrelin receptor. Cell Metab. (2018) 27:461–9e6. doi: 10.1016/j.cmet.2017.10.016
150. Azegami T, Yuki Y, Sawada S, Mejima M, Ishige K, Akiyoshi K, et al. Nanogel-based nasal ghrelin vaccine prevents obesity. Mucosal Immunol. (2017) 10:1351–60. doi: 10.1038/mi.2016.137
151. McFarlane MR, Brown MS, Goldstein JL, Zhao TJ. Induced ablation of ghrelin cells in adult mice does not decrease food intake, body weight, or response to high-fat diet. Cell Metab. (2014) 20:54–60. doi: 10.1016/j.cmet.2014.04.007
152. Sun Y, Asnicar M, Saha PK, Chan L, Smith RG. Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell Metab. (2006) 3:379–86. doi: 10.1016/j.cmet.2006.04.004
153. Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, et al. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci USA. (2004) 101:8227–32. doi: 10.1073/pnas.0402763101
154. Sun YX, Ahmed S, Smith R.G. Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol. (2003) 23:7973–81. doi: 10.1128/MCB.23.22.7973-7981.2003
155. Kirchner H, Heppner KM, Holland J, Kabra D, Tschop MH, Pfluger PT. Ablation of ghrelin O-acyltransferase does not improve glucose intolerance or body adiposity in mice on a leptin-deficient ob/ob background. PLoS ONE (2013) 8:e61822. doi: 10.1371/journal.pone.0061822
156. Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest. (2005) 115:3564–72. doi: 10.1172/JCI26002
157. Zhao TJ, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci USA. (2010) 107:7467–72. doi: 10.1073/pnas.1002271107
158. Mani BK, Osborne-Lawrence S, Vijayaraghavan P, Hepler C, Zigman JM. beta1-Adrenergic receptor deficiency in ghrelin-expressing cells causes hypoglycemia in susceptible individuals. J Clin Invest. (2016) 126:3467–78. doi: 10.1172/JCI86270
159. Yamazaki M, Nakamura K, Kobayashi H, Matsubara M, Hayashi Y, Kangawa K, et al. Regulational effect of ghrelin on growth hormone secretion from perifused rat anterior pituitary cells. J Neuroendocrinol. (2002) 14:156–62. doi: 10.1046/j.0007-1331.2001.00757.x
160. Chuang JC, Sakata I, Kohno D, Perello M, Osborne-Lawrence S, Repa JJ, et al. Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Mol Endocrinol. (2011) 25:1600–11. doi: 10.1210/me.2011-1001
161. Reimer MK, Pacini G, Ahren B. Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology (2003) 144:916–21. doi: 10.1210/en.2002-220819
162. Tong J, Prigeon RL, Davis HW, Bidlingmaier M, Kahn SE, Cummings DE, et al. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes (2010) 59:2145–51. doi: 10.2337/db10-0504
163. Mani BK, Uchida A, Lee Y, Osborne-Lawrence S, Charron MJ, Unger RH, et al. Hypoglycemic effect of combined ghrelin and glucagon receptor blockade. Diabetes (2017) 66:1847–57. doi: 10.2337/db16-1303
164. Grosse J, Heffron H, Burling K, Akhter Hossain M, Habib AM, Rogers GJ, et al. Gribble, Insulin-like peptide 5 is an orexigenic gastrointestinal hormone. Proc Natl Acad Sci USA. (2014) 111:11133–8. doi: 10.1073/pnas.1411413111
165. Mashima H, Ohno H, Yamada Y, Sakai T, Ohnishi H. INSL5 may be a unique marker of colorectal endocrine cells and neuroendocrine tumors. Biochem Biophys Res Commun. (2013):586–92. doi: 10.1016/j.bbrc.2013.02.042
166. Liu C, Kuei C, Sutton S, Chen J, Bonaventure P, Wu J, et al. INSL5 is a high affinity specific agonist for GPCR142 (GPR100). J Biol Chem. (2005) 280:292–300. doi: 10.1074/jbc.M409916200
167. Ang SY, Hutchinson DS, Patil N, Evans BA, Bathgate RAD, Halls ML, et al. Signal transduction pathways activated by insulin-like peptide 5 at the relaxin family peptide RXFP4 receptor. Br J Pharmacol. (2017) 174:1077–89. doi: 10.1111/bph.13522
168. Lee YS, De Vadder F, Tremaroli V, Wichmann A, Mithieux G, Backhed F. Insulin-like peptide 5 is a microbially regulated peptide that promotes hepatic glucose production. Mol Metab. (2016) 5:263–70. doi: 10.1016/j.molmet.2016.01.007
169. Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. (2011) 13:517–26. doi: 10.1016/j.cmet.2011.02.018
170. Burnicka-Turek O, Mohamed BA, Shirneshan K, Thanasupawat T, Hombach-Klonisch S, Klonisch T, et al. INSL5-deficient mice display an alteration in glucose homeostasis and an impaired fertility. Endocrinology (2012) 153:4655–65. doi: 10.1210/en.2012-1161
171. Luo X, Li T, Zhu Y, Dai Y, Zhao J, Guo ZY, et al. The insulinotrophic effect of insulin-like peptide 5 in vitro and in vivo. Biochem J. (2015) 466:467–73. doi: 10.1042/BJ20141113
Keywords: GLP-1, PYY, serotonin, GIP-glucose-dependent insulinotropic peptide, oxyntomodulin, ghrelin, enteroendocine cells, insulin-like peptide 5 (INSL5)
Citation: Sun EWL, Martin AM, Young RL and Keating DJ (2019) The Regulation of Peripheral Metabolism by Gut-Derived Hormones. Front. Endocrinol. 9:754. doi: 10.3389/fendo.2018.00754
Received: 31 October 2018; Accepted: 27 November 2018;
Published: 04 January 2019.
Edited by:
Maximilian Bielohuby, Sanofi, FranceReviewed by:
Yoshio Fujitani, Gunma University, JapanAndreas Hoeflich, Leibniz Institute for Farm Animal Biology, Germany
Copyright © 2019 Sun, Martin, Young and Keating. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Damien J. Keating, ZGFtaWVuLmtlYXRpbmdAZmxpbmRlcnMuZWR1LmF1