 Kook Hwan Kim
Kook Hwan Kim Myung-Shik Lee
Myung-Shik Lee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 24 August 2018
Sec. Cellular Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00485
This article is part of the Research Topic Disease Mechanisms and Diagnostic/Therapeutic Approaches in NAFLD View all 6 articles
Non-alcoholic fatty liver disease (NAFLD) is an emerging global health problem and a potential risk factor for type 2 diabetes, cardiovascular disease, and chronic kidney disease. Nonalcoholic steatohepatitis (NASH), an advanced form of NAFLD, is a predisposing factor for development of cirrhosis and hepatocellular carcinoma. The increasing prevalence of NASH emphasizes the need for novel therapeutic approaches. Although therapeutic drugs against NASH are not yet available, fundamental insights into the pathogenesis of NASH have been made during the past few decades. Multiple therapeutic strategies have been developed and are currently being explored in clinical trials or preclinical testing. The pathogenesis of NASH involves multiple intracellular/extracellular events in various cell types in the liver or crosstalk events between the liver and other organs. Here, we review current findings and knowledge regarding the pathogenesis of NASH, focusing on the most recent advances. We also highlight hormone-based therapeutic approaches for treatment of NASH.
The liver is a central metabolic organ that coordinates whole-body energy homeostasis by regulating glucose, lipid, and protein metabolism. The liver is also the main organ of detoxification and processes pharmaceutical products or environmental xenobiotics absorbed from the intestine. Therefore, liver diseases can cause systemic metabolic abnormalities; conversely, the liver is an important target organ of diverse metabolic disorders, which may lead to the development of non-alcoholic fatty liver disease (NAFLD), alcoholic fatty liver disease (AFLD), cirrhosis, and hepatocellular carcinoma (HCC). Among these diseases, NAFLD is one of the most prevalent chronic liver diseases and is an emerging global public health threat. NAFLD affects about 1.8 billion people worldwide with a prevalence of ~20–30% (1). The pathological spectrum of NAFLD ranges from simple steatosis to advanced stages including non-alcoholic steatohepatitis (NASH), hepatic fibrosis, and cirrhosis. The prevalence of simple steatosis with lipid accumulation exceeding 5% of liver weight ranges from 15 to 40% in the general population. Among patients with simple steatosis, 10–20% develop NASH which is defined as steatosis with hepatic inflammation and fibrosis (1). NASH can progress to more severe stages such as cirrhosis and HCC (2). NAFLD/NASH is an emerging risk factor for type 2 diabetes, cardiovascular disease, and chronic kidney disease (3). In particular, NAFLD/NASH is closely associated with several metabolic disorders such as obesity, dyslipidemia, and type 2 diabetes (3). It is estimated that 70–80% of obese or diabetic subjects have NAFLD, with a NASH prevalence of 10–20%. Despite the clinical importance of NAFLD/NASH, therapeutic drugs against these diseases have not yet been developed. However, numerous studies suggest that NASH develops by multiple intracellular/extracellular events in different liver cell types such as hepatocytes, hepatic stellate cells (HSCs), Kupffer cells (resident macrophages in the liver), and infiltrating macrophages (4) and by inter-organ crosstalk between the liver and other tissues including adipose tissue or the intestine (5, 6). This “multiple-parallel hit” model has recently been considered as a more adequate hypothesis to understand the pathogenesis of NASH than the “two-hit” model in which hepatic steatosis, the “first hit,” increases susceptibility to NASH caused by a “second hit” such as oxidative stress and inflammatory cytokines (7). Here, we briefly highlight molecular mechanisms of the pathogenesis of NASH, focusing on recent findings supporting the “multiple-parallel hit” hypothesis. We also describe potential therapeutic strategies based on hormones for treatment of NASH.
Hepatocytes, Kupffer cells/infiltrating macrophages, and HSCs play key roles in hepatic steatosis, inflammation, and fibrosis, respectively. In this section, we discuss intracellular events in individual cells and intercellular crosstalk between different cell types within the liver in the pathogenesis of NASH, focusing on recent advances in the potential role of hepatocytes. We also briefly describe the importance of the adipose tissue-liver axis and intestine-liver axis in the pathogenesis of NASH, emphasizing the most recent findings (Figure 1).
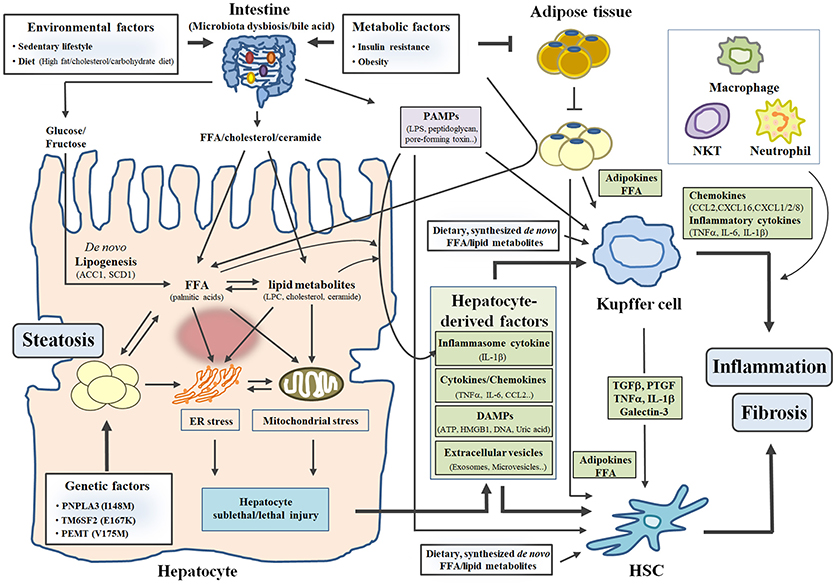
Figure 1. The “mutiple-parallel hit” model in the pathogenesis of NASH. Three factors (environmental, metabolic and genetic factors) contribute to the development and progression of NASH by affecting diverse organs such as the liver, the intestine, and adipose tissue. In particular, excess caloric or inappropriate intake (environmental factor) increases toxic free fatty acid (FFA) and lipid metabolites (LPC, cholesterol and ceramide) in hepatocytes, leading to hepatic steatosis and hepatocyte sublethal/lethal injuries. Subsequently, hepatocyte-derived factors (such as cytokines/chemokines, DAMPs and extracellular vesicles) stimulate inflammatory response in Kupffer cell and fibrotic response in HSC, which leads to the development of inflammation and fibrosis in the liver. FFA and lipid metabolites derived from diets or synthesized de novo also activates Kupffer cell and HSC. In addition, insulin resistance and obesity (metabolic factor) influence organ-crosstalk between the intestine/adipose tissue and the liver, contributing to the development and progression of NASH.
Hepatic steatosis develops by interactions among genetic, metabolic, and environmental factors (8). Sedentary lifestyle and excess caloric or inappropriate intake (high glucose, high fructose, high fat, or high cholesterol diet) are well-known environmental factors associated with hepatic steatosis and NAFLD/NASH. Excess dietary glucose or fructose enters the liver through the bloodstream after uptake in the small intestine and is subsequently utilized for the production of hepatic triacylglycerol (TG) via de novo fatty acid synthesis, as well as being a source for hepatic glycogen storage. Dietary fatty acids absorbed in the small intestine also contribute to hepatic TG formation by two different pathways. In the first, free short-chain fatty acids transported into the blood from the small intestine are taken up by the liver via fatty acid transporter [cluster of differentiation 36 (CD36), fatty acid transporter protein (FATP)] together with free fatty acid (FFA) “spillover” generated from lipoprotein lipase (LPL)-mediated chylomicron hydrolysis. In the second pathway, chylomicron TGs containing middle/long-chain fatty acids are hydrolyzed by LPL in peripheral tissues such as skeletal muscle and adipose tissue and consequently become chylomicron remnants, which are taken up by the liver through their receptors [low density lipoprotein receptor (LDLR) and LDL receptor-related protein (LRP)].
In addition to environmental factors, metabolic factors such as insulin resistance, obesity, and dyslipidemia increase susceptibility to hepatic steatosis and NAFLD/NASH (9, 10). In particular, insulin resistance in adipose tissue impairs the ability of insulin to suppress lipolysis; consequently, FFAs released from adipose tissue are transported into the liver. FFAs from adipose tissue contribute to ~60% of hepatic lipid accumulation together with FFA “spillover” (11). Hyperinsulinemia or hyperglycemia, a key feature of insulin resistance, also increases hepatic de novo fatty acid synthesis via upregulation of SREBP1 and ChREBP, impairs hepatic β-oxidation, and enhances CD36-mediated fatty acid uptake, thereby contributing to the development of hepatic steatosis (12, 13). Genome-wide association studies suggest that variation in several genes [e.g., PNPLA3 (patatin-like phospholipase domain-containing protein 3), TM6SP2 (transmembrane 6 superfamily member 2), and FADS (fatty acid desaturase)] related to hepatic lipid metabolism are associated with increased hepatic steatosis in human NAFLD subjects (14–16). A more detailed discussion regarding the contribution of genetic (or epigenetic) factors to the pathogenesis of NASH has been recently reviewed elsewhere (17). Thus, various risk factors contribute to the development of hepatic steatosis and NALFD/NASH by affecting hepatic lipid metabolism through multiple pathways (Figure 1).
Excessive lipid accumulation in the liver causes hepatocellular lipotoxicity via cellular and organelle oxidative stresses including endoplasmic reticulum (ER) stress and mitochondrial dysfunction, eventually leading to hepatocyte cell death (Figure 1) (18, 19). In particular, ER stress is implicated in the development and progression of NAFLD (20, 21). When cells are exposed in ER stress, the integrated stress signal mediated by three ER stress sensors (PERK, IRE1α, and ATF6) triggers unfolded protein response (UPR), which serves as a compensatory mechanism to maintain ER homeostasis. However, prolonged or excessive lipotoxic ER stress overwhelms the capacity of UPR and induces hepatic cell death via two main pathways (a mitochondria-dependent intrinsic pathway and a death receptor-mediated extrinsic pathway) (22). Moreover, chronic ER stress increases the production of reactive oxygen species (ROS) and activates the NF-κB or c-Jun N-terminal kinase (JNK) pathway, leading to hepatic inflammation (21). In addition, chronic ER stress results in hepatic lipid accumulation via activation of de novo fatty acid synthesis in a manner dependent on ER stress-sensing pathways (23), suggesting that a vicious cycle between ER stress and hepatic steatosis may promote the development and progression of NAFLD/NASH.
The roles of saturated fatty acids and other lipid metabolites [lysophosphatidylcholine (LPC), ceramide, and free cholesterol] as potential mediators of hepatocellular lipotoxicity in NAFLD/NASH have recently emerged (6, 19). Palmitic acid, the most abundant long-chain saturated fatty acid in vivo, has been reported to trigger oxidative stress and ER/lysosomal/mitochondrial stresses, causing lipotoxicity-related cell death (24–26). LPC and ceramide have been also reported to act as lipid intermediates mediating the effect of palmitic acid on hepatocellular lipotoxicity (27). In particular, LPC generated from palmitic acid triggers mitochondria-dependent apoptotic machinery via activation of the G-protein-coupled receptor Galpha(i)-JNK pathway (27) or of the CCAAT/enhancer binding homologous protein (CHOP)/JNK pathway (28). Ceramide synthesized via a de novo pathway from palmitic acid also causes mitochondrial dysfunction (such as impairment of mitochondrial respiratory chain and increase of mitochondrial membrane permeability), leading to hepatocyte cell death (29). In addition, ceramide generated from tumor necrosis factor alpha (TNFα)-induced activation of the acid sphingomyelinase (ASMase) pathway contributes to hepatocyte apoptosis (30). Another lipid metabolite, free cholesterol, can also induce hepatocyte necrosis/apoptosis by depleting mitochondrial glutathione (31) and can induce hepatocyte pyroptosis/necrosis by generating cholesterol crystals within lipid droplets (32). Importantly, concentrations of saturated fatty acid (e.g., palmitic acid), ceramide, LPC, or free cholesterol are increased in the livers of human subjects with NASH (27, 33, 34) and of animals with NASH (35), suggesting that free saturated fatty acids and other lipid metabolites as mentioned above contribute to the development and progression of NASH. Numerous studies suggest that therapeutic approaches to inhibit hepatic lipid accumulation, lipid-induced oxidative stress, and lipotoxicity-mediated cell death are promising strategies for treatment of NAFLD/NASH (36, 37). Phase 1, 2a, 2b, and 3 trials are being undertaken to test peroxisome proliferator activated receptor alpha/delta (PPARα/δ) activator (Elafibranor/GFT505; Clinical Trials NCT02704403), liver X receptor alpha (LXRα) inhibitor (Oltipraz; NCT02068339), acetyl-CoA carboxylase (ACC) inhibitors (GS-0976; NCT02856555 and PF-05221304; NCT03248882), stearoyl-CoA desaturase 1 (SCD1) inhibitor (Aramchol; NCT02279524), diacylglycerol acyltransferase 1 (DGAT1) inhibitor (Pradigastat/LCQ908; NCT01811472), and DGAT2 inhibitor (PF-06865571; NCT03513588) as targeting strategies to reduce hepatic steatosis in NASH patients via enhancement of β-oxidation or inhibition of fatty acid/TG synthesis. Natural antioxidants such as vitamin E and resveratrol have been used (or are being investigated) as therapeutic compounds to attenuate oxidative stress (36, 38). Caspase inhibitor (Emricasan/IDN-6556; NCT02686762) and apoptosis signaling kinase-1 (ASK-1) inhibitor (Selonsertib/GS-4997; NCT03053050), which target hepatic cell death, are also being evaluated in phase 2b and 3 studies of NASH patients, respectively.
In particular, ASK1 inhibitor can reduce hepatic steatosis, inflammation, and fibrosis (39, 40). In the unstimulated state, inactive ASK1 forms a high-molecular protein complex through homotypic interaction between two adjacent carboxy-terminal coiled-coil domains and through binding of its N-terminal region to thioredoxin (TRX). In an oxidative stressed state, ASK1 dissociated from TRX is homo-oligomerized in association with TNF receptor-associated factor (TRAF) and is subsequently auto-phosphorylated on threonine residue Thr838, which ultimately leads to the formation of active ASK1 signalosome. Then, this complex phosphorylates and activates p38 and JNK, resulting in increase of hepatocyte injury via the BAX-caspase pathway, impairment of hepatic insulin resistance via Ser307 phosphorylation of insulin receptor substrate, increase of hepatic inflammation via pro-inflammatory cytokine/chemokine production, and increase of hepatic fibrosis via collagen production (39, 40). Recent emerging evidence suggests that several ASK1-interacting proteins such as Dickkopf-3 (DKK3), CASP8 and FADD-like apoptosis regulator (CFLAR, known as cFlip), and TNFα-induced protein 3 (TNFAIP3, known as A20) represent attractive therapeutic targets for the prevention and treatment of NAFLD/NASH (41–43). Further understanding of molecular mechanisms underlying the regulation of ASK1 activity will provide opportunities to identify novel therapeutic targets and to develop promising therapeutic compounds for treatment of NAFLD/NASH.
Several studies suggest that hepatocyte-derived factors (or molecules) can act on non-parenchymal cells such as Kupffer cells and HSCs, which can in turn contribute to the progression of NASH (Figure 1) (44–46). In response to various stimuli such as excessive lipids and lipopolysaccharide (LPS), hepatocytes can directly produce chemokines [chemokine (C-C motif) ligand 2 (CCL2)] and pro-inflammatory cytokines (TNFα, IL-6 and IL-1β), although the amount of chemokines/cytokines released from hepatocytes is lower compared to that released from non-parenchymal cells. Subsequently, increased chemokine/cytokine production results in infiltration of macrophages into the liver and activation of Kupffer cells/HSCs (44, 46). In addition, sublethal or lethal hepatocellular injuries (apoptosis, necrosis, necroptosis, or pyroptosis) trigger signals for NASH promotion through releases of inflammatory cytokines, damage-associated molecular patterns (DAMPs) such as high mobility group box 1(HMGB1), ATP, uric acid, or mitochondrial DNA, and extracellular vesicles (18, 44, 47). DAMPs can cause hepatic inflammation and fibrosis by activating Kupffer cells and HSCs via paracrine or endocrine actions (44, 47).
It has been reported that circulating levels of extracellular vesicles such as microvesicles (microparticles) and exosomes are increased in NASH mouse models (48, 49) and patients with NASH (50). Extracellular vesicles released from hepatocytes treated with lipotoxic fatty acids can act as messenger signals mediating intercellular communication between hepatocytes and non-parenchymal cells, which contributes to aggravation of inflammation and fibrosis in NASH (48, 49, 51). For example, saturated fatty acids stimulate the release of extracellular vesicles including vanin-1, a surface cargo protein found on hepatocytes, leading to increases in chemotaxis and migration of endothelial cells (48). Consequently, enhanced angiogenesis promotes recruitment of inflammatory cells and fibrogenesis of HSCs, contributing to the progression of NASH. In addition, fatty acid-induced release of exosomes containing microRNAs (miR-128-3p) contributes to the activation of HSCs in fibrosis (49). Palmitic acid and LPC also induce the release of extracellular vesicles from hepatocytes via the death receptor 5 (DR5) signaling pathway or rho-associated coiled-coil-containing protein kinase 1 (ROCK1) (51). In this process, TRAIL-bearing extracellular vesicles activate inflammatory responses in macrophages via the DR5 signaling pathway in a receptor-interacting serine/threonine-protein kinase 1 (RIPK1)-dependent manner (51). Furthermore, mass spectrometry analysis demonstrated the presence of many DAMPs in extracellular vesicles released from hepatocytes (52). All these findings suggest that extracellular vesicles play key roles in intercellular communication between different cell types in the liver and in the development and progression of NASH. Due to the importance of extracellular vesicles in NASH pathogenesis, strategies to block the release of extracellular vesicles and to inhibit specific molecules (or cargos) within extracellular vesicles have been considered as therapeutic interventions for NASH.
In addition to hepatocyte-derived factors, lipotoxic fatty acids such as palmitic acid and ceramide can activate Kupffer cells and subsequently promote the production of pro-inflammatory cytokines (TNFα and IL-6) (Figure 1) (53). Kupffer cells also secrete several chemokines such as CCL2, chemokine (C-X-C motif) ligand 16 (CXCL16), and CXCL1/2/8 to recruit peripheral macrophages, natural killer T cells, and neutrophils, respectively (Figure 1) (54–56). Consequently, Kupffer cells and recruited innate immune cells can coordinately aggravate inflammation in the liver. Moreover, several cytokines such as TGFβ, lectin galactose binding soluble 3 (LGALS3, known as Galectin-3), platelet-derived growth factor (PDGF), and TNFα/IL-1β produced from Kupffer cells/infiltrating macrophages are able to activate quiescent HSCs and increase proliferation or survival of HSCs (Figure 1) (57–60). In addition, palmitic acid-induced TNFα and IL-1β in infiltrating macrophages can cause lipid accumulation and insulin resistance in hepatocytes (61).
Inflammasome is a cytoplasmic multiprotein complex that is activated by two distinct signals: the first signal for upregulation of inflammasome-related genes [pro-IL-1β, NLR family pyrin domain containing 3 (NLRP3), and Caspase 1] and a second signal for functional inflammasome assembly and activation of Caspase 1 (62). Inflammasome-mediated IL-1β secretion is initiated by activation of toll-like receptors (TLR) as a priming signal and stimulated by diverse second signals such as DAMPs, pathogen-associated molecular patterns (PAMPs) (e.g., pore-forming toxin), and toxic lipids (e.g., palmitic acids and cholesterol crystals) (63). NLRP3 inflammasome-dependent IL-1β secretion from activated Kupffer cells/infiltrating macrophages and hepatocytes plays a crucial role in progression of NAFLD/NASH (64, 65). Mice with global overexpression of mutant NLRP3 (constitutive active) showed severe hepatocyte pyroptotic cell death and severe hepatic inflammation/fibrosis, while myeloid cell (Kupffer cells)-specific mutant NLRP3-overexpressing mice exhibited mild hepatic inflammation/fibrosis in the absence of hepatocyte pyroptotsis (64), suggesting that both Kupffer cells and hepatocytes are important in NLRP3 inflammation-mediated liver injury. In parallel, NLRP3 knockout mice showed improved hepatic injury, inflammation, and fibrosis in the liver after a choline-deficient amino acid-defined (CDAA) diet feeding (65). Furthermore, treatment with MCC950 (CP-456773), a small molecule NLRP3 inhibitor, attenuates liver inflammation and fibrosis in mice fed a methionine-choline-deficient (MCD) diet (66). IL-1β knockout mice also displayed reduced steatohepatitis and hepatic fibrosis after an atherogenic cholesterol-rich diet feeding (67). In contrast, IL-1β receptor antagonist (IL-1Ra) knockout mice showed aggravated hepatic steatosis, inflammation, and fibrosis after the same diet feeding (68). All these results suggest that inhibition of NLRP3 or IL-1β could be an attractive therapeutic strategy for treatment of NALFD/NASH.
Due to the crucial role of Kupffer cell/infiltrating macrophages-derived cytokines/chemokines in steatosis, inflammation, and fibrosis, functional inhibition of these secretory proteins is considered as a promising therapeutic approach for treatment of NASH. An orally available drug capable of inhibiting recruitment of monocyte/macrophage and activation of HSCs, dual chemokine (C-C motif) receptor 2 (CCR2)/CCR5 antagonist (Cenicriviroc: NCT03028740) is currently being investigated in a phase 3 clinical trial of human NASH patients with liver fibrosis. An oral inhibitor (BI1467335/PXS-4728A; NCT03166735) of amine oxidase copper-containing 3 (AOC3) is being evaluated in a phase 2a NASH study as an anti-inflammatory compound to block the recruitment of immune cells in the liver. Galectin-3, a Kupffer cell/macrophage-derived lectin, is required for TGFβ-mediated activation of HSCs (59), and an inhibitor of Galectin 3 (GR-MD-02; NCT02462967) is also being evaluated as an anti-fibrotic drug in a phase 2b trial in NASH patients.
HSCs, the major source of type I collagen in the liver, play a crucial role in NASH-related fibrosis (69). Hepatocyte-derived factors (cytokines, DAMPs, or extracellular vesicles) and Kupffer/macrophage-released cytokines/chemokines trigger signaling cascades to transform “quiescent” HSCs into “activated” HSCs (myofibroblast-like cells) (69). Consequently, activated HSCs increase the secretion of collagen and other extracellular matrix proteins, leading to fibrotic scarring and ultimately chronic fibrosis in the liver (69). Thus, one of the potential strategies to inhibit NASH-related fibrosis is to suppress accumulation of extracellular matrix proteins including collagen or to directly inhibit HSC activation. Lysyl oxidase-like 2 (LOXL2), an enzyme promoting collagen cross-linking, is upregulated in the livers of animals with fibrosis (70) and of diabetic patients with NAFLD (71) and is essential in hepatic fibrogenesis (72). Phase 2b clinical studies in NASH patients using an antibody against LOXL2 (Simtuzumab/GS-6624; NCT01672866 and NCT01672879) were recently terminated. Simtuzumab had no effect on hepatic fibrosis in NASH patients with bridging fibrosis or cirrhosis (73). Heat shock protein 47 (HSP47) is a molecular chaperone which plays a crucial role in the maturation and secretion of collagen. While vitamin A-coupled lipid nanoparticle (BMS-986263/ND-LO2-s0201) containing siRNA against HSP47 is being investigated in preclinical stage for treatment of NASH, efficacy of BMS-986263 in human fibrosis patients (NCT02227459) and human cirrhosis patients (NCT03420768) are being evaluated in phase 1 and 2 clinical trials, respectively.
Recent knowledge regarding the roles and contributions of adipose tissue and intestine in the pathogenesis of NASH have been extensively highlighted in previous reviews (5, 6). Here, we briefly describe the relationships between the adipose tissue-liver axis or intestine-liver axis and NASH.
Obesity-associated insulin resistance in adipose tissue contributes to the development and progression of hepatic steatosis and NASH (5). As mentioned previously, impaired suppression of insulin-mediated lipolysis in white adipose tissue (WAT) leads to hepatic steatosis through increased fatty acid uptake of hepatocytes (Figure 1). In addition, inflammation in WAT systemically affects hepatic inflammation (74). Lean mice transplanted with visceral adipose tissue from obese mice exhibited elevated infiltration of neutrophils and macrophages in the liver and ultimately suffered from aggravated liver damage after NASH diet feeding (74). However, transplantation of visceral fat from obese mice with depletion of adipose tissue macrophage (ATM) by treatment with clodronate liposomes did not cause hepatic inflammation in lean mice (74), suggesting that ATM directly contributes to hepatic inflammation and NASH progression. Furthermore, adipokines (such as adiponectin and leptin) secreted from WAT have reported to affect lipid accumulation, inflammation, and fibrosis in the liver (Figure 1) (75).
In addition to the role of WAT, BAT (brown adipose tissue) is associated with the development and progression of NAFLD. A couple of previous papers suggest that transplantation of BAT alleviates hepatic steatosis in HFD-fed obese mice and leptin-deficient ob/ob mice (76, 77). Conversely, treatment with propranolol, a β-adrenergic receptor antagonist, worsens liver injury in mice fed a half-methionine and choline-deficient diet supplemented with ethionine (HMCDE) due to increased hepatocyte cell death (78). Furthermore, a recent paper suggests that thermoneutral housing exacerbates HFD-driven NAFLD in mice, which is related to reduced activation of BAT, although additional events (augmented intestinal permeability, dysbiosis of the microbiome, and altered immune responsiveness caused by decreased norepinephrine/corticosterone) could also contribute to these phenotypes (79). Despite the presence of severe steatosis and inflammation in HFD-fed mice housed at a thermoneutral temperature, hepatic fibrosis did not develop in these mice (79). Thus, it will be interesting to investigate whether activation of thermogenic adipocytes ameliorates the development and progression of NASH in mice fed NASH diets such as MCD diet, high-fat/high-cholesterol diet, or high-fat/high-fructose diet.
Growing evidence suggests that the intestine-liver axis plays a crucial role in the maintenance of metabolic homeostasis, and that its impairment is an important causal factor in the pathogenesis of diverse liver diseases such as obesity-related steatosis, NAFLD/NASH, and liver cancer (80, 81). Feeding of HFD or NASH diets causes impairment of intestinal barriers, dysbiosis of the microbiota, and alterations of intestinal immunity, leading to increased translocation of bacteria or bacterial products into the systemic circulation (82, 83). Consequently, bacteria or bacterial products are able to reach the liver through the portal vein. In the liver, conserved motifs/structures of bacteria and bacterial products (PAMPs) are recognized by pathogen recognition receptors (PPRs) of various cell types (hepatocytes, Kupffer cells/infiltrating macrophages, and HSCs). In particular, PAMPs such as LPS, peptidoglycan, and bacterial DNA stimulate multiple signaling cascades via interactions with PAMP-specific TLRs and NOD-like receptors (NLRs), leading to hepatic steatosis, inflammation, and fibrosis in the liver (Figure 1) (6, 84).
Emerging evidence suggests that lipid intermediates derived from the intestine trigger the development of NAFLD/NASH and insulin resistance (85, 86). In mice fed diets supplemented with palmitic acid or palm oils, concentrations of ceramide were increased in the intestine and serum/plasma (87). Ceramide derived from the intestines of mice fed HFD systemically causes ER/mitochondrial stresses and increases fatty acid synthesis in hepatocytes, which leads to hepatic lipid accumulation, hepatic cell death, and inflammation (Figure 1) (85, 86). Intriguingly, these phenotypes are attenuated in intestine-specific hypoxia-inducible factor 2α (HIF2α)- or farnesoid X receptor (FXR)-knockout mice which showed decreased intestinal ceramide level due to reduced expression of HIF2α- or FXR-target genes involved in ceramide synthesis (85, 86). Furthermore, intestinal ceramide production and hepatic steatosis are attenuated in HFD-fed mice treated with antibiotics (85), suggesting that HFD-induced alterations of the microbiota contribute to increased intestinal ceramide production and hepatic lipid accumulation. In particular, taurocholic acid (TCA) and tauro-β-muricholic acid (T-β-MCA) produced from the liver competitively act as agonist and antagonist for FXR signaling in intestinal epithelial cells, respectively (85). In mice fed HFD, T-β-MCA is converted to MCA by bile salt hydrolase (BSH), a microbial enzyme, which results in aggravated hepatic steatosis and hepatic injury due to increased intestinal ceramide production via activation of TCA-mediated agonistic action for FXR and inhibition of T-β-MCA-mediated antagonistic action (85). In HFD-fed mice treated with antibiotics, however, accumulated T-β-MCA inhibits intestinal FXR signaling and subsequently suppresses intestinal ceramide synthesis, leading to improvements in hepatic steatosis and hepatic injury (85). In addition, hormones derived from the intestine influence the development and progression of NAFLD/NASH (88), which will be discussed in next section. In view of the importance of impaired intestinal barrier-induced penetration of microbial products and microbial dysbiosis-induced changes of intestinal signaling in the pathogenesis of NAFLD/NASH, therapeutic modulation of the intestine-liver axis represents an attractive strategy for treatment of NAFLD/NASH. A phase 2a clinical study using IMM-124E (composed of anti-LPS antibody and glycosphingolipid adjuvants) is currently underway in human NASH patients (NCT02316717). Further understanding of intestine-liver interactions will help identify novel therapeutic targets and molecules to prevent and treat NAFLD/NASH.
Based on the “multiple-parallel hit” model of the pathogenesis of NASH, therapeutic approaches such as reduction of steatosis, blockade of hepatic cell death, suppression of hepatic immune cells, and inhibition of fibrogenic action of HSCs are considered as attractive strategies for treatment of NASH. Since hormones systemically influence diverse tissues (or cell types) in the body, they can have effects on multiple steps in the pathogenesis of NASH. Thus, hormone-based therapy is an attractive strategy for treatment of NASH. In this section, we briefly review current hormone-based NASH therapies.
FGF19 (FGF15 in rodents) is a postprandial endocrine hormone that is produced in the intestine by bile acid-induced FXR activation (89), and that plays a key role in the regulation of bile acid and lipid metabolism in the liver (89). FGF19 also inhibits gluconeogenesis and stimulates hepatic glycogen and protein synthesis via insulin-independent action (90). In addition to its physiological effects, therapeutic administration of FGF19 or genetic overexpression of FGF19 also have pharmacological effects such as decreased hepatosteatosis/adiposity and improved insulin sensitivity via enhancement of β-oxidation/thermogenesis, inhibition of lipogenesis, or amelioration of lipotoxicity-induced ER stress (91–93). In contrast, HFD-fed FGF15 knockout mice showed increased adiposity or exacerbated ER stress and hepatosteatosis (93). Recent emerging studies suggest that FGF19 ameliorates muscle wasting via direct action on skeletal muscle (94) and corrects type 1 diabetes via inhibition of the hypothalamic–pituitary–adrenal axis (95). Intriguingly, circulating FGF19 level is decreased in human NAFLD/NASH subjects, and hepatic response to FGF19 is impaired in human NAFLD subjects with insulin resistance (96, 97). Therapeutic administration of an engineered FGF19 (NGM282/M70, a nontumorigenic FGF19 variant) eliminates lipotoxicity and bile acid toxicity, leading to improvements of steatohepatitis and fibrosis in a NASH mouse model (98). However, endogenous FGF15 deletion had no effect on steatosis, inflammation, or fibrosis in mice fed HFD for 6 months (99). In a phase 2a clinical study (NCT02443116), treatment of human NASH patients with NGM282 resulted in reduced hepatic steatosis and decreased markers of hepatic inflammation/fibrosis with acceptable safety (100).
FGF21 is expressed as an endocrine hormone predominantly in the liver and other metabolic tissues such as adipose tissue, muscle, and pancreas. FGF21 plays physiologically important roles in the regulation of glucose/lipid metabolism and maintenance of energy balance in response to changes in nutritional status such as starvation (101, 102) and environmental stimuli such as cold exposure or exercise (103, 104). FGF21 can exert beneficial effects on obesity and related metabolic diseases (105, 106). Pharmacological treatment with FGF21 or overexpression of FGF21 improves diet-induced obesity and insulin resistance by enhancing insulin-mediated glucose uptake and β-oxidation/thermogenesis (105, 106). Furthermore, FGF21 ameliorates obesity-induced ER stress, increased serum levels of liver enzymes, and insulin resistance (107). Emerging evidence suggests that FGF21 is also implicated in the pathogenesis and treatment of NASH (108, 109). FGF21 is increased in the livers of NASH animal models (108) and of human patients with NASH (109). Therapeutic treatment with FGF21 or overexpression of FGF21 causes improvements of MCD diet-induced steatosis, inflammation, and fibrosis in mice by reducing hepatic lipotoxicity and increasing β-oxidation (108). Moreover, treatment with FGF21 suppresses PDGF-induced activation of HSCs in vitro (110), implying that direct anti-fibrogenic action of FGF21 in HSCs may contribute to FGF21-induced improvement of fibrosis in vivo. In line with preclinical studies, a recent phase 2a clinical study using a pegylated analog of FGF21 (PEG-FGF21, BMS-986036) suggests reduction of steatosis and improvement of markers of fibrosis and liver injury in human NASH patients (111). Phase 2b clinical studies (NCT03486899 and NCT03486912) to evaluate the safety and efficacy of BMS-986036 in human NASH patients with severe stage 3 fibrosis or cirrhosis were recently started.
GLP-1 is a proglucagon-derived hormone that is secreted from the intestine in response to changes in nutrients (112). GLP-1 plays a crucial role in the regulation of glucose metabolism by enhancing insulin release, suppressing glucagon secretion, and inhibiting gastric emptying (112). Furthermore, pharmacological treatment with GLP-1 leads to improvements of diet-induced obesity and insulin resistance by suppression of food intake and enhancement of thermogenesis (113, 114). Seven synthetic GLP-1 receptor agonists (exenatide, exenatide long-acting release, liraglutide, albiglutide, dulaglutide, lixisenatide, and semaglutide) are approved and available for treatment of type 2 diabetes. Intriguingly, GLP-1 analogs also ameliorate not only hepatic steatosis, but also hepatic inflammation and fibrosis in mice (115). Furthermore, in a phase 2 clinical study (NCT01237119) for efficacy of liraglutide in human NASH patients, patients receiving liraglutide for 48 weeks showed significant reductions of hepatic ballooning, steatosis, and serum alanine aminotransferase (ALT) level compared to patients who received placebo (116). Liraglutide-induced reduction of hepatic lipid accumulation is probably due to decreased hepatic de novo fatty acid synthesis (117). A phase 2 clinical study using semaglutide in human NASH patients (NCT02970942) is currently underway.
In line with the therapeutic effect of GLP-1, pharmacological treatment with sitagliptin or linagliptin, inhibitors of dipeptidylpeptidase 4 (DPP4), an enzyme that degrades GLP-1, improves hepatic steatosis, inflammation, and fibrosis in NASH mouse models (118, 119). Furthermore, vildagliptin, another DPP4 inhibitor, ameliorates serum liver enzyme levels and hepatic steatosis in human NAFLD patients with dyslipidemia (120). In contrast, sitagliptin treatment for 24 weeks had no beneficial effects on serum liver enzyme levels, fatty liver, or fibrosis in human NASH or NAFLD patients (NCT01260246 and NCT01963845) (121, 122). Further large-scale studies are needed to evaluate the efficacy and clinical importance of DDP4 inhibitor for treatment of NAFLD/NASH.
Glucagon/GLP-1 receptor dual agonists and glucagon/glucose-dependent insulinotropic polypeptide (GIP)/GLP-1 receptor triple agonists are attractive therapeutic agents to treat NAFLD/NASH as well as obesity-related diabetes (123, 124). Some preclinical studies have shown improvement of obesity-related metabolic deterioration in mice treated with glucagon/GLP-1 receptor dual agonist (125) or glucagon/GIP/GLP-1 receptor triple agonist (126). Pharmacological treatment with a pegylated analog of oxyntomodulin (PEG-OXM, G49), a natural agonist of the glucagon/GLP-1 receptor, improves steatohepatitis in MCD diet-fed mice, probably due to reductions of ER/mitochondrial stresses and hepatocyte apoptosis (127). Treatment with glucagon/GIP/GLP-1 receptor triple agonists also leads to significant improvement of steatohepatitis in female mice fed high-fat/high-sucrose diets and also in male mice, albeit to a lesser extent (128). While several phase 1 or 2 clinical studies using glucagon/GLP-1 receptor dual agonists and glucagon/GIP/GLP-1 receptor triple agonists are being conducted in human obese diabetic patients (123, 124), clinical studies using glucagon/GLP-1 receptor dual agonist (SAR425899; NCT03437720) and glucagon/GIP/GLP-1 receptor triple agonist (HM15211) for NASH patients have recently been registered.
GDF15, an endocrine hormone belonging to the TGFβ superfamily, is ubiquitously expressed in various tissues, with the highest levels in the liver, placenta, and macrophages (129–131). Numerous studies suggest that GDF15 is induced by diverse stress stimuli such as fatty acids, ER/mitochondrial stressors, and LPS (132–134), and that serum GDF15 level is increased in human subjects with diseases such as cardiovascular disease, chronic kidney disease, obesity, diabetes, and cancer (135, 136). Intriguingly, recent emerging evidence suggests that GDF15 exerts beneficial effects on obesity-related insulin resistance in mice and monkeys through the suppression of food intake via GDNF family receptor α-like (GFRAL)-dependent anorexic action (137–140). GDF15 also stimulates oxidative metabolism in macrophages or metabolic tissues such as adipose tissue and liver, leading to the improvement of insulin resistance and hepatic steatosis in obese mice (133, 141). In addition to its anti-obesity and anti-diabetic effects, GDF15 has been implicated in the development and progression of NAFLD/NASH (142, 143). Serum GDF15 level is elevated in NASH animal models (143) and in human subjects with NASH (142, 143). In two dietary NASH models using MCD and amylin liver NASH (AMLN) diets, GDF15 knockout mice showed deteriorated steatohepatitis and fibrosis in the liver, while GDF15 transgenic mice were resistant to diet-induced NASH phenotypes (143). Furthermore, treatment with recombinant GDF15 ameliorates lipid accumulation, inflammation, and fibrosis in mice caused by alcohol feeding (144), suggesting that GDF15 is a promising therapeutic candidate to treat alcoholic steatohepatitis (ASH) as well as NASH. While preclinical studies using GDF15 analogs have been conducted, clinical studies aimed at treating obesity-related type 2 diabetes or NAFLD/NASH have yet to be performed.
NAFLD/NASH is a public health problem worldwide, but there are no therapeutic drugs approved for its treatment. Numerous preclinical animal models based on genetic or dietary manipulation with relevance to human NAFLD/NASH have been developed to explore the mechanisms underlying the development of NAFLD/NASH and identify novel targets for its treatment (145, 146). The studies conducted using these models have made important steps forward in our understanding of the pathogenesis of NASH and in the discovery of potential therapeutic candidates. As discussed above, sophisticated communication between parenchymal and non-parenchymal cells in the liver or between the liver and other organs contributes to the development and progression of NAFLD/NASH (Figure 1). Thus, multiple components (hepatic lipid accumulation, oxidative stress, ER/mitochondrial stress, hepatocyte cell death, hepatocyte-released DAMPs/extracellular vesicles, Kupffer cell activation, inflammatory cell recruiting, HSC activation, insulin resistance, adipose tissue inflammation, and microbiota dysbiosis) are attractive therapeutic targets to treat NAFLD/NASH. In this review, we summarize preclinical or clinical therapeutic efficacies of several pharmacological agents targeting these multiple components of NASH. Pharmacological agents under development for treatment of NAFLD/NASH (including some candidates not discussed in this review) are listed in Table 1. Given the value of a “multiple-parallel model” in the pathogenesis of NASH, monotherapy with a single agent targeting multiple components (e.g., BMS-986036/PEG-FGF21 for targeting insulin resistance, hepatic lipid accumulation, oxidative stress, and ER stress) or combined therapy with agents targeting a single component (e.g., ASK1 inhibitor [Selonsertib] and ACC inhibitor [GS-0976]) will be efficacious therapeutic approaches for treatment of NASH, in addition to monotherapy with an agent targeting a single component. Thus, further understanding of NASH pathogenesis and preclinical/clinical studies to evaluate the efficacy of candidate agents will accelerate novel therapeutic innovations for treatment of NAFLD/NASH and related metabolic disorders.
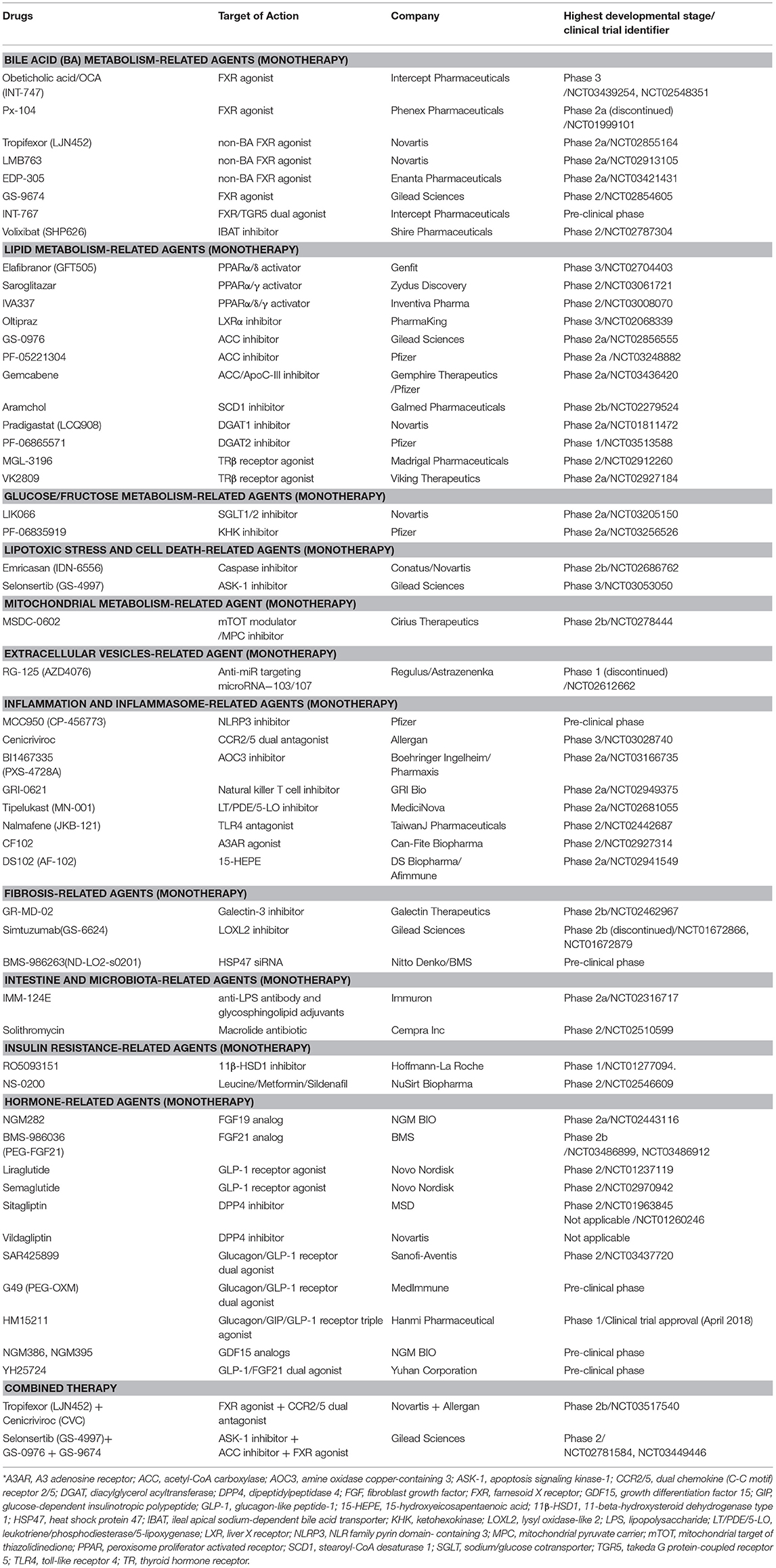
Table 1. Pharmacological agents under development for treatment of NAFLD/NASH.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (2013R1A6A3A04065825 to KK) and the Global Research Laboratory Grant of the National Research Foundation of Korea (NRF-2010-00347 to M-SL).
1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
2. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. (2013) 10:656–65. doi: 10.1038/nrgastro.2013.183
3. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. (2013) 10:330–44. doi: 10.1038/nrgastro.2013.41
4. Magee N, Zou A, Zhang Y. Pathogenesis of nonalcoholic steatohepatitis: interactions between liver parenchymal and nonparenchymal cells. BioMed Res Int. (2016) 2016:5170402. doi: 10.1155/2016/5170402
5. Vonghia L, Francque S. Cross talk of the immune system in the adipose tissue and the liver in non-alcoholic steatohepatitis: pathology and beyond. World J Hepatol. (2015) 7:1905–12. doi: 10.4254/wjh.v7.i15.1905
6. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
7. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology (2010) 52:1836–46. doi: 10.1002/hep.24001
8. Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers (2015) 1:15080. doi: 10.1038/nrdp.2015.80
9. Luyckx FH, Lefebvre PJ, Scheen AJ. Non-alcoholic steatohepatitis: association with obesity and insulin resistance, and influence of weight loss. Diabetes Metab. (2000) 26:98–106.
10. Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes (2001) 50:1844–50. doi: 10.2337/diabetes.50.8.1844
11. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. (2005) 115:1343–51. doi: 10.1172/jci23621
12. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. (2008) 118:829–38. doi: 10.1172/jci34275
13. Steneberg P, Sykaras AG, Backlund F, Straseviciene J, Soderstrom I, Edlund H. Hyperinsulinemia enhances hepatic expression of the fatty acid transporter Cd36 and provokes hepatosteatosis and hepatic insulin resistance. J Biol Chem. (2015) 290:19034–43. doi: 10.1074/jbc.M115.640292
14. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2008) 40:1461–5. doi: 10.1038/ng.257
15. Sookoian S, Castano GO, Scian R, Mallardi P, Fernandez Gianotti T, Burgueno AL, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology (2015) 61:515–25. doi: 10.1002/hep.27556
16. Wang L, Athinarayanan S, Jiang G, Chalasani N, Zhang M, Liu W. Fatty acid desaturase 1 gene polymorphisms control human hepatic lipid composition. Hepatology (2015) 61:119–28. doi: 10.1002/hep.27373
17. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol. (2018) 68:268–79. doi: 10.1016/j.jhep.2017.09.003
18. Hirsova P, Ibrahim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. (2016) 57:1758–70. doi: 10.1194/jlr.R066357
19. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism (2016) 65:1049–61. doi: 10.1016/j.metabol.2016.02.014
20. Pagliassotti MJ. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu Rev Nutr. (2012) 32:17–33. doi: 10.1146/annurev-nutr-071811-150644
21. Zhang XQ, Xu CF, Yu CH, Chen WX, Li YM. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. (2014) 20:1768–76. doi: 10.3748/wjg.v20.i7.1768
22. Akazawa Y, Nakao K. Lipotoxicity pathways intersect in hepatocytes: Endoplasmic reticulum stress, c-Jun N-terminal kinase-1, and death receptors. Hepatol Res. (2016) 46:977–84. doi: 10.1111/hepr.12658
23. Baiceanu A, Mesdom P, Lagouge M, Foufelle F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol. (2016) 12:710–22. doi: 10.1038/nrendo.2016.124
24. Wei Y, Wang D, Gentile CL. Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem. (2009) 331:31–40. doi: 10.1007/s11010-009-0142-1
25. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology (2004) 40:185–94. doi: 10.1002/hep.20283
26. Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology (2008) 47:1495–503. doi: 10.1002/hep.22183
27. Han MS, Park SY, Shinzawa K, Kim S, Chung KW, Lee JH, et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res. (2008) 49:84–97. doi: 10.1194/jlr.M700184-JLR200
28. Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, et al. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G77–84. doi: 10.1152/ajpgi.00301.2011
29. Arora AS, Jones BJ, Patel TC, Bronk SF, Gores GJ. Ceramide induces hepatocyte cell death through disruption of mitochondrial function in the rat. Hepatology (1997) 25:958–63. doi: 10.1002/hep.510250428
30. Osawa Y, Uchinami H, Bielawski J, Schwabe RF, Hannun YA, Brenner DA. Roles for C16-ceramide and sphingosine 1-phosphate in regulating hepatocyte apoptosis in response to tumor necrosis factor-alpha. J Biol Chem. (2005) 280:27879–87. doi: 10.1074/jbc.M503002200
31. Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. (2006) 4:185–98. doi: 10.1016/j.cmet.2006.07.006
32. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res. (2017) 58:1067–79. doi: 10.1194/jlr.M072454
33. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology (2007) 46:1081–90. doi: 10.1002/hep.21763
34. Chiappini F, Coilly A, Kadar H, Gual P, Tran A, Desterke C, et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci Rep. (2017) 7:46658. doi: 10.1038/srep46658
35. Tu LN, Showalter MR, Cajka T, Fan S, Pillai VV, Fiehn O, et al. Metabolomic characteristics of cholesterol-induced non-obese nonalcoholic fatty liver disease in mice. Sci Rep. (2017) 7:6120. doi: 10.1038/s41598-017-05040-6
36. Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. (2016) 15:249–74. doi: 10.1038/nrd.2015.3
37. Konerman MA, Jones JC, Harrison SA. Pharmacotherapy for NASH: Current and emerging. J Hepatol. (2018) 68:362–75. doi: 10.1016/j.jhep.2017.10.015
38. Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA (2011) 305:1659–68. doi: 10.1001/jama.2011.520
39. Kovalic AJ, Satapathy SK, Chalasani N. Targeting incretin hormones and the ASK-1 pathway as therapeutic options in the treatment of non-alcoholic steatohepatitis. Hepatol Int. (2018) 12:97–106. doi: 10.1007/s12072-018-9854-1
40. Schuster S, Feldstein AE. NASH: Novel therapeutic strategies targeting ASK1 in NASH. Nat Rev Gastroenterol Hepatol. (2017) 14:329–30. doi: 10.1038/nrgastro.2017.42
41. Xie L, Wang PX, Zhang P, Zhang XJ, Zhao GN, Wang A, et al. DKK3 expression in hepatocytes defines susceptibility to liver steatosis and obesity. J Hepatol. (2016) 65:113–24. doi: 10.1016/j.jhep.2016.03.008
42. Wang PX, Ji YX, Zhang XJ, Zhao LP, Yan ZZ, Zhang P, et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat Med. (2017) 23:439–49. doi: 10.1038/nm.4290
43. Zhang P, Wang PX, Zhao LP, Zhang X, Ji YX, Zhang XJ, et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat Med. (2018) 24:84–94. doi: 10.1038/nm.4453
44. Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut (2018) 67:963–72. doi: 10.1136/gutjnl-2017-315691
45. Greuter T, Malhi H, Gores GJ, Shah VH. Therapeutic opportunities for alcoholic steatohepatitis and nonalcoholic steatohepatitis: exploiting similarities and differences in pathogenesis. JCI Insight (2017) 2:e95354. doi: 10.1172/jci.insight.95354
46. Hirsova P, Gores GJ. Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. (2015) 1:17–27. doi: 10.1016/j.jcmgh.2014.11.005
47. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. (2013) 10:627–36. doi: 10.1038/nrgastro.2013.149
48. Povero D, Eguchi A, Niesman IR, Andronikou N, de Mollerat du Jeu X, Mulya A, et al. Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci Signal. (2013) 6:ra88. doi: 10.1126/scisignal.2004512
49. Povero D, Panera N, Eguchi A, Johnson CD, Papouchado BG, de Araujo Horcel L, et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell Mol Gastroenterol Hepatol. (2015) 1:646–63.e4. doi: 10.1016/j.jcmgh.2015.07.007
50. Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res. (2016) 57:233–45. doi: 10.1194/jlr.M063412
51. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology (2016) 150:956–67. doi: 10.1053/j.gastro.2015.12.037
52. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology (2016) 63:731–44. doi: 10.1002/hep.28252
53. Tang T, Sui Y, Lian M, Li Z, Hua J. Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PLoS ONE (2013) 8:e81949. doi: 10.1371/journal.pone.0081949
54. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1310–21. doi: 10.1152/ajpgi.00365.2011
55. Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C, et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J Immunol. (2013) 190:5226–36. doi: 10.4049/jimmunol.1202909
56. Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology (2014) 147:577–94.e1. doi: 10.1053/j.gastro.2014.06.043
57. Friedman SL. Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest. (1989) 84:1780–5. doi: 10.1172/jci114362
58. Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. (2000) 204:19–28. doi: 10.1006/cimm.2000.1687
59. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA. (2006) 103:5060–5. doi: 10.1073/pnas.0511167103
60. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (2013) 58:1461–73. doi: 10.1002/hep.26429
61. Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun. (2017) 8:2247. doi: 10.1038/s41467-017-02325-2
62. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
63. Del Campo JA, Gallego P, Grande L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J Hepatol. (2018) 10:1–7. doi: 10.4254/wjh.v10.i1.1
64. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (2014) 59:898–910. doi: 10.1002/hep.26592
65. Wree A, McGeough MD, Pena CA, Schlattjan M, Li H, Inzaugarat ME, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med. (2014) 92:1069–82. doi: 10.1007/s00109-014-1170-1
66. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
67. Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. (2011) 55:1086–94. doi: 10.1016/j.jhep.2011.01.048
68. Isoda K, Sawada S, Ayaori M, Matsuki T, Horai R, Kagata Y, et al. Deficiency of interleukin-1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. J Biol Chem. (2005) 280:7002–9. doi: 10.1074/jbc.M412220200
69. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
70. Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. (2010) 16:1009–17. doi: 10.1038/nm.2208
71. Dongiovanni P, Meroni M, Baselli GA, Bassani GA, Rametta R, Pietrelli A, et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin Sci. (2017) 131:1301–15. doi: 10.1042/cs20170175
72. Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut (2017) 66:1697–708. doi: 10.1136/gutjnl-2016-312473
73. Sanyal A, Abdelmalek MF, Diehl AM, Caldwell S, Shiffman ML, Ghalib R, et al. Efficacy and safety of simtuzumab for the treatment of NASH with bridging fibrosis or cirrhosis: results of two phase 2b, dose-ranging, randomized, placebo-controlled trials [Abstract]. J Hepatol. (2017) 66(Suppl):S54. doi: 10.1016/S0168-8278(17)30370-7
74. Bijnen M, Josefs T, Cuijpers I, Maalsen CJ, van de Gaar J, Vroomen M, et al. Adipose tissue macrophages induce hepatic neutrophil recruitment and macrophage accumulation in mice. Gut (2018) 67:1317–27. doi: 10.1136/gutjnl-2016-313654
75. Boutari C, Perakakis N, Mantzoros CS. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol Metab. (2018) 33:33–43. doi: 10.3803/EnM.2018.33.1.33
76. Liu X, Zheng Z, Zhu X, Meng M, Li L, Shen Y, et al. Brown adipose tissue transplantation improves whole-body energy metabolism. Cell Res. (2013) 23:851–4. doi: 10.1038/cr.2013.64
77. Liu X, Wang S, You Y, Meng M, Zheng Z, Dong M, et al. Brown adipose tissue transplantation reverses obesity in Ob/Ob Mice. Endocrinology (2015) 156:2461–9. doi: 10.1210/en.2014-1598
78. McKee C, Soeda J, Asilmaz E, Sigalla B, Morgan M, Sinelli N, et al. Propranolol, a beta-adrenoceptor antagonist, worsens liver injury in a model of non-alcoholic steatohepatitis. Biochem Biophys Res Commun. (2013) 437:597–602. doi: 10.1016/j.bbrc.2013.07.005
79. Giles DA, Moreno-Fernandez ME, Stankiewicz TE, Graspeuntner S, Cappelletti M, Wu D, et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat Med. (2017) 23:829–38. doi: 10.1038/nm.4346
80. Brandi G, De Lorenzo S, Candela M, Pantaleo MA, Bellentani S, Tovoli F, et al. Microbiota, NASH, HCC and the potential role of probiotics. Carcinogenesis (2017) 38:231–40. doi: 10.1093/carcin/bgx007
81. Yang BG, Hur KY, Lee MS. Alterations in gut microbiota and immunity by dietary fat. Yonsei Med J. (2017) 58:1083–91. doi: 10.3349/ymj.2017.58.6.1083
82. Lee JC, Lee HY, Kim TK, Kim MS, Park YM, Kim J, et al. Obesogenic diet-induced gut barrier dysfunction and pathobiont expansion aggravate experimental colitis. PLoS ONE (2017) 12:e0187515. doi: 10.1371/journal.pone.0187515
83. Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol. (2015) 1:222–32. doi: 10.1016/j.jcmgh.2015.01.001
84. Chassaing B, Etienne-Mesmin L, Gewirtz AT, Microbiota-liver axis in hepatic disease. Hepatology (2014) 59:328–39. doi: 10.1002/hep.26494
85. Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. (2015) 125:386–402. doi: 10.1172/jci76738
86. Xie C, Yagai T, Luo Y, Liang X, Chen T, Wang Q, et al. Activation of intestinal hypoxia-inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat Med. (2017) 23:1298–308. doi: 10.1038/nm.4412
87. Tran TT, Postal BG, Demignot S, Ribeiro A, Osinski C, Pais de Barros JP, et al. Short term palmitate supply impairs intestinal insulin signaling via ceramide production. J Biol Chem. (2016) 291:16328–38. doi: 10.1074/jbc.M115.709626
88. Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R. Targeting the gut-liver axis in liver disease. J Hepatol. (2017) 67:1084–103. doi: 10.1016/j.jhep.2017.05.007
89. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. (2005) 2:217–25. doi: 10.1016/j.cmet.2005.09.001
90. Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science (2011) 331:1621–4. doi: 10.1126/science.1198363
91. Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology (2002) 143:1741–7. doi: 10.1210/endo.143.5.8850
92. Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology (2004) 145:2594–603. doi: 10.1210/en.2003-1671
93. Alvarez-Sola G, Uriarte I, Latasa MU, Fernandez-Barrena MG, Urtasun R, Elizalde M, et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut (2017) 66:1818–28. doi: 10.1136/gutjnl-2016-312975
94. Benoit B, Meugnier E, Castelli M, Chanon S, Vieille-Marchiset A, Durand C, et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat Med. (2017) 23:990–96. doi: 10.1038/nm.4363
95. Perry RJ, Lee S, Ma L, Zhang D, Schlessinger J. Shulman GI, FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Commun. (2015) 6:6980. doi: 10.1038/ncomms7980
96. Schreuder TC, Marsman HA, Lenicek M, van Werven JR, Nederveen AJ, Jansen PL, et al. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G440–5. doi: 10.1152/ajpgi.00322.2009
97. Alisi A, Ceccarelli S, Panera N, Prono F, Petrini S, De Stefanis C, et al. Association between serum atypical fibroblast growth factors 21 and 19 and pediatric nonalcoholic fatty liver disease. PLoS ONE (2013) 8:e67160. doi: 10.1371/journal.pone.0067160
98. Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun. (2017) 1:1024–42. doi: 10.1002/hep4.1108
99. Schumacher JD, Kong B, Pan Y, Zhan L, Sun R, Aa J, et al. The effect of fibroblast growth factor 15 deficiency on the development of high fat diet induced non-alcoholic steatohepatitis. Toxicol Appl Pharmacol. (2017) 330:1–8. doi: 10.1016/j.taap.2017.06.023
100. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet (2018) 391:1174–85. doi: 10.1016/s0140-6736(18)30474-4
101. Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. (2007) 5:415–25. doi: 10.1016/j.cmet.2007.05.003
102. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. (2007) 5:426–37. doi: 10.1016/j.cmet.2007.05.002
103. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. (2012) 26:271–81. doi: 10.1101/gad.177857.111
104. Kim KH, Kim SH, Min YK, Yang HM, Lee JB, Lee MS. Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS ONE (2013) 8:e63517. doi: 10.1371/journal.pone.0063517
105. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. (2005) 115:1627–35. doi: 10.1172/jci23606
106. Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes (2009) 58:250–9. doi: 10.2337/db08-0392
107. Kim SH, Kim KH, Kim HK, Kim MJ, Back SH, Konishi M, et al. Fibroblast growth factor 21 participates in adaptation to endoplasmic reticulum stress and attenuates obesity-induced hepatic metabolic stress. Diabetologia (2015) 58:809–18. doi: 10.1007/s00125-014-3475-6
108. Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, et al. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology (2014) 147:1073–83.e6. doi: 10.1053/j.gastro.2014.07.044
109. Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology (2010) 139:456–63. doi: 10.1053/j.gastro.2010.04.054
110. Xu P, Zhang Y, Liu Y, Yuan Q, Song L, Liu M, et al. Fibroblast growth factor 21 attenuates hepatic fibrogenesis through TGF-beta/smad2/3 and NF-kappaB signaling pathways. Toxicol Appl Pharmacol. (2016) 290:43–53. doi: 10.1016/j.taap.2015.11.012
111. Sanyal A, Charles ED, Neuschwander-Tetri B, Loomba R, Harrison S, Abdelmalek M, et al. BMS-986036 (pegylated FGF21) in patients with non-alcoholic steatohepatitis: a phase 2 study [Abstract]. J Hepatol. (2017) 66(Suppl):S89. doi: 10.1016/S0168-8278(17)30443-9
112. Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. (2012) 8:728–42. doi: 10.1038/nrendo.2012.140
113. Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature (1996) 379:69–72. doi: 10.1038/379069a0
114. Lockie SH, Heppner KM, Chaudhary N, Chabenne JR, Morgan DA, Veyrat-Durebex C, et al. Direct control of brown adipose tissue thermogenesis by central nervous system glucagon-like peptide-1 receptor signaling. Diabetes (2012) 61:2753–62. doi: 10.2337/db11-1556
115. Trevaskis JL, Griffin PS, Wittmer C, Neuschwander-Tetri BA, Brunt EM, Dolman CS, et al. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G762–72. doi: 10.1152/ajpgi.00476.2011
116. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet (2016) 387:679–90. doi: 10.1016/s0140-6736(15)00803-x
117. Armstrong MJ, Hull D, Guo K, Barton D, Hazlehurst JM, Gathercole LL, et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol. (2016) 64:399–408. doi: 10.1016/j.jhep.2015.08.038
118. Klein T, Fujii M, Sandel J, Shibazaki Y, Wakamatsu K, Mark M, et al. Linagliptin alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis. Med Mol Morphol. (2014) 47:137–49. doi: 10.1007/s00795-013-0053-9
119. Jung YA, Choi YK, Jung GS, Seo HY, Kim HS, Jang BK, et al. Sitagliptin attenuates methionine/choline-deficient diet-induced steatohepatitis. Diabetes Res Clin Pract. (2014) 105:47–57. doi: 10.1016/j.diabres.2014.04.028
120. Hussain M, Majeed Babar MZ, Hussain MS, Akhtar L. Vildagliptin ameliorates biochemical, metabolic and fatty changes associated with non alcoholic fatty liver disease. Pak J Med Sci. (2016) 32:1396–401. doi: 10.12669/pjms.326.11133
121. Joy TR, McKenzie CA, Tirona RG. Summers K, Seney S, Chakrabarti S, et al. Sitagliptin in patients with non-alcoholic steatohepatitis: a randomized, placebo-controlled trial. World J Gastroenterol. (2017) 23:141–50. doi: 10.3748/wjg.v23.i1.141
122. Cui J, Philo L, Nguyen P, Hofflich H, Hernandez C, Bettencourt R, et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: a randomized controlled trial. J Hepatol. (2016) 65:369–76. doi: 10.1016/j.jhep.2016.04.021
123. Sanchez-Garrido MA, Brandt SJ, Clemmensen C, Muller TD, DiMarchi RD, Tschop MH. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia (2017) 60:1851–61. doi: 10.1007/s00125-017-4354-8
124. Brandt SJ, Gotz A, Tschop MH, Muller TD. Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides (2018) 100:190–201. doi: 10.1016/j.peptides.2017.12.021
125. Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes (2009) 58:2258–66. doi: 10.2337/db09-0278
126. Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. (2015) 21:27–36. doi: 10.1038/nm.3761
127. Valdecantos MP, Pardo V, Ruiz L, Castro-Sanchez L, Lanzon B, Fernandez-Millan E, et al. A novel glucagon-like peptide 1/glucagon receptor dual agonist improves steatohepatitis and liver regeneration in mice. Hepatology (2017) 65:950–68. doi: 10.1002/hep.28962
128. Jall S, Sachs S, Clemmensen C, Finan B, Neff F, DiMarchi RD, et al. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol Metab. (2017) 6:440–46. doi: 10.1016/j.molmet.2017.02.002
129. Hsiao EC, Koniaris LG, Zimmers-Koniaris T, Sebald SM, Huynh TV, Lee SJ. Characterization of growth-differentiation factor 15, a transforming growth factor beta superfamily member induced following liver injury. Mol Cell Biol. (2000) 20:3742–51. doi: 10.1128/MCB.20.10.3742-3751.2000
130. Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci USA. (1997) 94:11514–9. doi: 10.1073/pnas.94.21.11514
131. Lawton LN, Bonaldo MF, Jelenc PC, Qiu L, Baumes SA, Marcelino RA, et al. Identification of a novel member of the TGF-beta superfamily highly expressed in human placenta. Gene (1997) 203:17–26. doi: 10.1016/S0378-1119(97)00485-X
132. Lee SH, Yamaguchi K, Kim JS, Eling TE, Safe S, Park Y, et al. Conjugated linoleic acid stimulates an anti-tumorigenic protein NAG-1 in an isomer specific manner. Carcinogenesis (2006) 27:972–81. doi: 10.1093/carcin/bgi268
133. Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J Cell Biol. (2017) 216:149–65. doi: 10.1083/jcb.201607110
134. Kim JM, Kosak JP, Kim JK, Kissling G, Germolec DR, Zeldin DC, et al. NAG-1/GDF15 transgenic mouse has less white adipose tissue and a reduced inflammatory response. Med Inflamm. (2013) 2013:641851. doi: 10.1155/2013/641851
135. Fujita Y, Taniguchi Y, Shinkai S, Tanaka M, Ito M. Secreted growth differentiation factor 15 as a potential biomarker for mitochondrial dysfunctions in aging and age-related disorders. Geriatr Gerontol Int. (2016) 16 (Suppl.1):17–29. doi: 10.1111/ggi.12724
136. Adela R, Banerjee SK. GDF-15 as a Target and biomarker for diabetes and cardiovascular diseases: a translational prospective. J Diabetes Res. (2015) 2015:490842. doi: 10.1155/2015/490842
137. Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med. (2017) 23:1215–9. doi: 10.1038/nm.4393
138. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjaer SB, et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med. (2017) 23:1158–66. doi: 10.1038/nm.4394
139. Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nature medicine (2017) 23:1150–57. doi: 10.1038/nm.4392
140. Hsu JY, Crawley S, Chen M, Ayupova DA, Lindhout DA, Higbee J, et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature (2017) 550:255–59. doi: 10.1038/nature24042
141. Jung SB, Choi MJ, Ryu D, Yi HS, Lee SE, Chang JY, et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat Commun. (2018) 9:1551. doi: 10.1038/s41467-018-03998-z
142. Koo BK, Um SH, Seo DS, Joo SK, Bae JM, Park JH, et al. Growth differentiation factor 15 predicts advanced fibrosis in biopsy-proven non-alcoholic fatty liver disease. Liver Int. (2018) 38:695–705. doi: 10.1111/liv.13587
143. Kim KH, Kim SH, Han DH, Jo YS, Lee YH, Lee MS, Growth differentiation factor 15 ameliorates nonalcoholic steatohepatitis and related metabolic disorders in mice. Sci Rep. (2018) 8:6789. doi: 10.1038/s41598-018-25098-0
144. Chung HK, Kim JT, Kim HW, Kwon M, Kim SY, Shong M, et al. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci Rep. (2017) 7:17238. doi: 10.1038/s41598-017-17574-w
145. Lau JK, Zhang X, Yu J. Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J Pathol. (2017) 241:36–44. doi: 10.1002/path.4829
Keywords: NAFLD, NASH, steatosis, inflammation, fibrosis, multiple-parallel hit
Citation: Kim KH and Lee M-S (2018) Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front. Endocrinol. 9:485. doi: 10.3389/fendo.2018.00485
Received: 30 April 2018; Accepted: 06 August 2018;
Published: 24 August 2018.
Edited by:
Kouichi Miura, Jichi Medical University, JapanReviewed by:
Won-Il Jeong, College of Life Science and Bioengineering, KAIST, South KoreaCopyright © 2018 Kim and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kook Hwan Kim, a29va2h3YW5raW1AZ21haWwuY29t; a2hraW0xOTc4QHl1aHMuYWM=
Myung-Shik Lee, bXNsZWUwOTIzQHl1aHMuYWM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.