 Radka Cerychova
Radka Cerychova Gabriela Pavlinkova
Gabriela Pavlinkova- 1Laboratory of Molecular Pathogenetics, Institute of Biotechnology of the Czech Academy of Sciences, Prague, Czechia
- 2Faculty of Science, Charles University, Prague, Czechia
The heart is able to metabolize any substrate, depending on its availability, to satisfy its energy requirements. Under normal physiological conditions, about 95% of ATP is produced by oxidative phosphorylation and the rest by glycolysis. Cardiac metabolism undergoes reprograming in response to a variety of physiological and pathophysiological conditions. Hypoxia-inducible factor 1 (HIF-1) mediates the metabolic adaptation to hypoxia and ischemia, including the transition from oxidative to glycolytic metabolism. During embryonic development, HIF-1 protects the embryo from intrauterine hypoxia, its deletion as well as its forced expression are embryonically lethal. A decrease in HIF-1 activity is crucial during perinatal remodeling when the heart switches from anaerobic to aerobic metabolism. In the adult heart, HIF-1 protects against hypoxia, although its deletion in cardiomyocytes affects heart function even under normoxic conditions. Diabetes impairs HIF-1 activation and thus, compromises HIF-1 mediated responses under oxygen-limited conditions. Compromised HIF-1 signaling may contribute to the teratogenicity of maternal diabetes and diabetic cardiomyopathy in adults. In this review, we discuss the function of HIF-1 in the heart throughout development into adulthood, as well as the deregulation of HIF-1 signaling in diabetes and its effects on the embryonic and adult heart.
Introduction
Oxygen is a key factor in mammalian energy metabolism (1). In normoxic conditions, cells generate adenosine triphosphate (ATP) through the mitochondrial electron transport chain, which requires oxygen as a terminal electron acceptor. In low oxygen concentrations, cells adjust their oxygen consumption and switch to anaerobic glycolysis to prevent oxygen depletion. Glycolysis is a less efficient process of generating ATP but is less oxygen demanding and thus protects cells from total anoxia and cell death. Hypoxia-inducible factor 1 (HIF-1) is the main regulator of responses to hypoxia. It consists of two subunits, HIF-1α and HIF-1β, encoded by the Hif1a and Hif1b genes, respectively. Both the HIF-1α and HIF-1β subunits are expressed constitutively, but only HIF-1α is affected by oxygen levels, as is shown in Figure 1 (2). Under normoxia, HIF-1α is rapidly degraded. First, prolyl hydroxylase domain proteins (PHDs) hydroxylate prolines in the oxygen—dependent degradation domain. The hydroxylated prolines are then recognized by the von Hippel-Lindau (VHL) protein, which serves as an E3 ubiquitin ligase. Finally, polyubiquitylated HIF-1α is degraded in the 26S proteasome. In hypoxic conditions, HIF-1α is not hydroxylated and both subunits, HIF-1α, and HIF-1β, are translocated to the nucleus, where they form a heterodimer, bind to hypoxia-responsive elements (HREs) on DNA, and activate transcription of hypoxia-responsive genes. As the main regulator of responses to hypoxia, HIF-1 signaling directly or indirectly targets several hundred genes (3). It is responsible not only for the switch from oxidative phosphorylation to glycolysis, but also for other adaptive processes, such as angiogenesis, erythropoiesis, and cell survival. In addition to responses to physiological hypoxia during development and growth, and pathological hypoxia in adult life, HIF-1 also plays an important role in aerobic glycolysis (4). In aerobic glycolysis, HIF-1 upregulates genes related to glycolytic energy metabolism in normoxia, the so-called Warburg effect. The Warburg effect is most often mentioned in relation to cancer cell growth (5), but some studies suggest the importance of aerobic glycolysis in normal proliferating cells as a mechanism for minimizing oxidative stress (6).
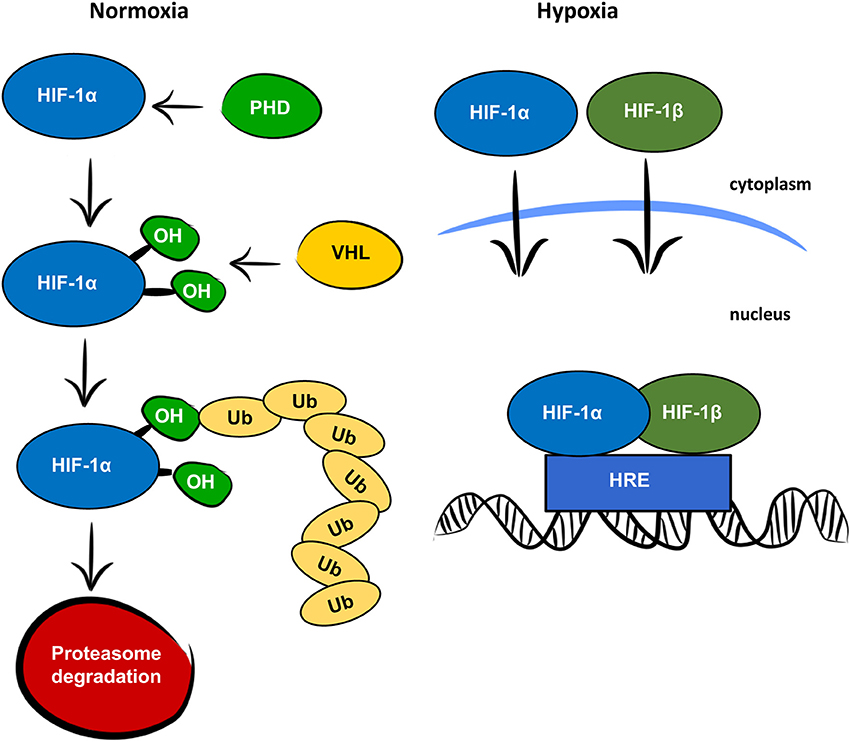
Figure 1. Oxygen dependent regulation of HIF-1α. In normoxic conditions, the HIF-1α protein is recognized by prolyl hydroxylase proteins (PHD), which hydroxylate prolines (OH) in the oxygen-dependent degradation domain. The hydroxylated prolines are recognized by the von Hippel-Lindau protein (VHL) and ubiquitinated (Ub) for degradation by the proteasome. In hypoxic conditions, HIF-1α and HIF-1β are translocated to the nucleus, where they form a heterodimer, bind to the hypoxia responsive element (HRE) and function as a transcription factor.
In this review, we will discuss the importance of HIF-1 in cardiac development, perinatal remodeling of cardiac metabolism, and in heart function in adults. We will also summarize the role of HIF-1 signaling in the responses to hypoxia in heart development and aberrant HIF-1 regulation in diabetic conditions.
Hypoxia and HIF-1 Signaling During Embryonic Development
During early development, the embryo is exposed to hypoxia, as was demonstrated by the measurement of human placental and intrauterine levels of oxygen (7, 8). During the first stages of development, the embryo uses pyruvate as the main energy substrate and glycolysis is undetectable (9). When the blastocyst stage is reached, oxygen consumption rapidly increases along with glycolysis, while the consumption of pyruvate decreases in subsequent stages. The switch to glycolytic metabolism leads to the restoration of oxygen consumption to normal levels. Physiologic hypoxic regions occur in normally developing embryos, mainly in the developing neural tube, heart, and intersomitic mesenchyme (10). There are a number of oxygen-sensing pathways, such as the energy and nutrient sensor mTOR, and the nuclear factor (NF)-κB transcriptional response, but the HIF-1 transcription system is a key feature in the cellular response to a low-oxygen environment during embryonic development (11). Although much of mammalian embryogenesis occurs at low oxygen concentrations (≤2%), it is important to note that the ability of the HIF-1 system to response enhanced and spatially extended hypoxia (nonphysiological hypoxia) is limited. Exposure to nonphysiological hypoxia (induced by environmental insult, cardiovascular defects or placental insufficiency) during early developmental stages leads to developmental defects affecting all organ systems (11). Besides intrauterine growth restriction syndrome and low birth weight, the developing heart is the most susceptible to hypoxia-induced defects.
Normal cardiac development resulting in the formation of the mature four-chambered heart involves an intricate combination of specifically timed cell migration, proliferation, and differentiation (12) (summarized in Figure 2). The embryo is especially vulnerable to death caused by hypoxia exposure at the time of septation (embryonic day E12.5–E14.5 in mice) (13), which leads to decreased proliferation, resulting in a hypoplastic myocardium. Another study establishes the critical developmental window for heart defects as E10.5–E13.5 (14). At this time point, oxygen deficiency is associated, along with myocardial thinning, with septal defects of ventricles and atria, as well as with defects in the formation of the outflow tract. Experiments with chicken embryos show that increased hypoxia in specific regions of the developing heart is associated with HIF-1α nuclear localization (15, 16). In the outflow tract, hypoxia, and thus HIF-1 signaling, is critical for normal outflow tract remodeling into the great vessels. Nuclear localization of HIF-1α occurs in the interventricular septum (IVS), atrioventricular canal, and the myocardium of the atrial wall at stage 30 when septation is complete. Hypoxic regions follow a pattern which resembles the locations of future coronary vessels, suggesting the importance of HIF-1 signaling in vascularization (17).
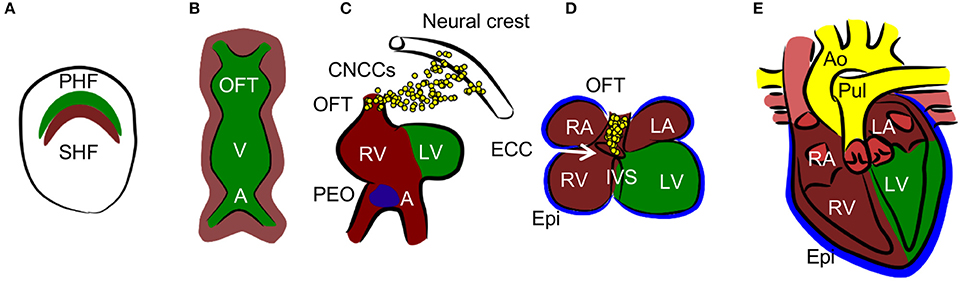
Figure 2. Heart development. (A) Cardiac progenitors form crescent shaped primary and secondary heart fields (PHF and SHF). (B)The primary heart field then forms the primary heart tube. The future outflow tract (OFT) is in the anterior part, future atria (A) are in the posterior part, and future ventricles (V) are in the middle of the tube. Cells from SHF populate the anterior and posterior parts of the tube. (C, D) The tube undergoes a process of looping, during which ventricles and atria are formed. The right ventricle (RV), left and right atria (LA and RA), and OFT are formed by SHF cells, the left ventricle (LV) is formed by PHF cells. During the process of septation of the ventricles, the muscular part of the interventricular septum (IVS) grows from the bases of the ventricles and connects with the remodeled endocardial cushion (ECC) in the atrioventricular canal. Similarly, the interatrial septum grows from the roof of the atria. Cardiac neural crest cells (CNCCs), migrating from the neural tube, contribute to the remodeling of the OFT. The proepicardial organ (PEO) is first formed on the ventral side of the future atria and through the process of looping is later positioned dorsally to the heart. The cells from the PEO migrate to the heart tube and form the epicardium (Epi). (E) The OFT is septated into the aorta (Ao) and pulmonary truncus (Pul) in the final step of heart development.
Several models of global and conditional Hif1a deletion demonstrate the importance of HIF-1 signaling in cardiac development and in embryonic development overall. Global knock-out of the Hif1a gene (Hif1a−/−) was achieved by the replacement of exon 2, encoding the bHLH domain, with a neomycin resistance cassette (18–20). This Hif1a mutation is embryonic lethal due to severe cardiovascular defects by E10.5 (18–20). Heart defects of Hif1a−/− embryos range from the improper formation of the heart tube, resulting in cardia bifida, to the defective looping of the heart tube, leading to the abnormal formation of the cardiac chambers and defective vascularization (20). Another study shows hyperplastic myocardium in Hif1a−/− mutant embryos (18). Additionally, Hif1a−/− embryos display neural tube and head fold defects, probably due to increased cell death and defective cell migration from the neural tube. Hif1a+/− embryos develop normally. Similarly, embryos with cardiac myocyte-specific deletion Mlc2v-Cre/Hif1aflox/flox develop normally (21). However, the combination of the null allele with Mlc2v-Cre (Mlc2v-Cre/Hif1a−/flox) results, similarly to the full knock-out model, in heart looping defects, a hyperplastic myocardium, and embryonic death by E12 (22). Increased mRNA levels of genes encoding proteins involved in cell cycle progression and decreased expression of cell cycle inhibitors suggest a role of HIF-1 in the regulation of proliferation. In contrast, another study used a combination of the null allele and Nkx2.5-Cre driven deletion of Hif1a (Nkx2.5-Cre/ Hif1a−/flox), which resulted in only 73% of the mutants having a complete deletion, while the remaining mutants had diminished expression of HIF-1α (23). Nkx2.5-Cre/ Hif1a−/flox mutants with efficient depletion of HIF-1α died by E17.5. Mutants displayed IVS defects and a hypoplastic myocardium due to decreased proliferation of fetal cardiomyocytes, which is in contrast with the study using the Mlc2v-Cre/Hif1a−/flox model (22) or global deletion of Hif1a (18). These contrasting results might be attributed to the different null Hif1a allele. Mouse models with the hyperplastic phenotype used Hif1atm1Jhu, affecting all cells derived from the zygote, whereas the model with the hypoplastic phenotype used Meox2-Cre for creating the null allele, which is expressed in the epiblast and does not affect the extraembryonic tissue (24). Another example of a mouse model with disrupted HIF-1 signaling resulting in a hypoplastic myocardium is a model utilizing a dominant-negative mutation (HIFdn) that specifically inhibits transcriptional responses mediated by both HIF-1 and HIF-2 in endothelial cells (25). The HIFdn mutation is embryonically lethal at E11.5; mutant embryos display hearts with a thinned myocardium and defective trabeculation. These results suggest the importance of HIF-1 signaling between embryonic and extraembryonic tissues in heart development.
Increased HIF-1α stabilization resulting from the deletion of the VHL gene is embryonically lethal at E10.5 to E12.5 due to defects in placental development (26). Even the delayed deletion of VHL using tamoxifen-induction at E10.5 results in embryonic lethality (27). Although timed tamoxifen induced deletion of VHL at E10.5 was effective in about 25% of the alleles, mutant embryos died between E14.5 and E15.5, possibly because of the impaired vasculature in the yolk sac. Affected embryos also displayed extensive hemorrhaging, liver necrosis, and dilated blood vessels. All these experiments show the importance of HIF-1 signaling during development, and that both upregulation and downregulation are fatal. The role of metabolic changes induced by the deregulation of HIF-1 signaling remains to be elucidated. For now, we can only speculate whether changes in proliferation induced by the deletion of HIF-1α are related to metabolism. However, experiments with cells lacking Hif1a show a deregulated glycolytic pathway and decreased proliferation even under normoxic conditions (18).
Our understanding of the roles of the HIF-1 system and hypoxia in embryonic development has improved. However, we still need to fully define the molecular interactions of HIF-1 in embryo morphogenesis. Examples of crosstalk between HIF-1 regulated pathways and other key regulatory pathways reveal additional molecular mechanisms through which HIF-1α and hypoxia can impact embryonic development. For example, HIF-1α interacts with β-catenin and thus connects hypoxia-induced responses to Wnt signaling (28). Another major pathway in development, Notch signaling, is positively modulated in hypoxic regions in embryos (29, 30). HIF-1α binds to the activated intracellular domain of the Notch receptor to activate transcription of target genes such as transcription repressors Hes1 and Hey2 (30). MicroRNAs (miRNAs) represent an additional regulatory mechanism in which hypoxia-HIF-1α action contributes to development. miRNAs are 21–23 nucleotide non-coding RNAs that regulate gene expression at the post-transcription level and play important roles in many biological processes and pathologies, including development and cardiovascular diseases (31). Of the known hypoxia-responsive miRNAs, miR-210 is the most consistent miRNA regulated by HIF-1α and upregulated during heart development (32–34). Another hypoxia-regulated miRNA detected early during heart development is miR-133 that regulates a cardiac-enriched transcription factor, serum response factor, responsible for the regulation of sarcomeres in the heart (35). Thus the hypoxia-HIF-1α-dependent miRNA-mediated response represents epigenetic regulation in the developing heart.
Highly proliferative cancer cells use aerobic glycolysis even in well-perfused tumors (5, 36). Although this approach is less energetically efficient, Vander Heiden et al. (36) hypothesize that proliferating cells need not only ATP but also carbohydrates as a building material. Using aerobic glycolysis allows the redirecting of some carbohydrates to the production of essential cellular building blocks, such as nucleotides and amino acids. A recent metabolomics analysis of developing embryos shows such redirection of glucose-derived carbons into the pentose phosphate pathway (37). Interestingly, the changes in cellular metabolism are not abrupt, but rather follow the anteroposterior gradient along the neural tube, which suggests an involvement of factors other than just the level of oxygen. Clearly, more research is needed in this field.
Perinatal Remodeling
After birth, cardiomyocytes undergo a number of changes (summarized in Table 1), resulting in the maturation of cardiac tissue. Most importantly, cardiac metabolism switches from the predominant use of glycolysis to the β-oxidation of lipids. Fetal glycolysis is favored not only due to the hypoxic environment in utero but also due to substrate availability. Glucose is the main substrate crossing the placenta to the fetal circuit (38). The fetal heart is adapted to glycolytic metabolism by the expression of specific metabolic gene isoforms, such as hexokinase I, pyruvate kinase 4, fetal phosphofructokinase, and M-lactate dehydrogenase (39, 40). Metabolism and gene expression gradually change after birth until weaning. Some studies show that the shift in the expression of genes involved in fatty acid metabolism begins in the second trimester, as shown in the microarray analysis of human fetal hearts (41). One of the potential regulators of this process is NEPAS (neonatal and embryonic Per-Arnt-Sim basic helix-loop-helix protein), a negative regulator of HIF-mediated gene expression, which is expressed in murine embryos and neonates from E15.5 to postnatal day 15 (P15) (42). The shift in metabolism might be not only time dependent, but also tissue-specific. At mid-gestation, HIF-1α is expressed in the compact myocardium but not in the trabeculae (43), which leads to a metabolic shift from glycolysis in the compact myocardium whereas oxidative metabolism increases in the trabeculae. The maturation of cardiac oxidative metabolism is completed after birth, although the mechanism is still not fully understood. An increased amount of oxygen, resulting in decreased HIF-1 signaling, is thought to be the main impulse for the shift. Additionally, substrate availability, and composition change after birth because breast milk also contains lipids which are not transferred across the placental barrier during prenatal development (40).
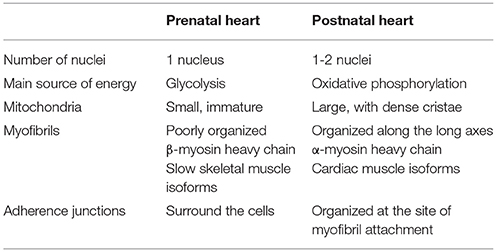
Table 1. Perinatal remodeling: a list of the main changes in cardiomyocytes during the transition from the prenatal to the postnatal phenotype.
The major part of ATP production in the adult heart (over 95%) is provided by mitochondrial oxidative phosphorylation (44). The maturation of mitochondria is an important part of perinatal cardiac remodeling. Fetal mitochondria are small and round; however, after birth, mitochondria fuse, grow bigger, and the cristae of the inner membrane become denser and more uniform (45). The total area of mitochondria per cardiomyocyte triples between E16.5 and P10.5. This process is dependent on the downregulation of HIF-1 signaling. Mitochondria in cardiomyocytes of αMHC-Cre/VHLflox/flox mice appear immature and the cells are more dependent on glycolysis than on oxidative phosphorylation. In wild type mice, perinatal cardiomyocytes are dependent on glycolysis as well as on oxidative phosphorylation.
Adult cardiomyocytes combine qualities of striated muscles and smooth muscles, so they can produce great force without any need for a pause. Similar to striated muscles, cardiomyocytes contain sarcomeres as contractile units but, unlike skeletal muscle, they do not form a syncytium and instead become binuclear via incomplete division. Cardiac beating is in fact the synchronized shortening of sarcomeres in cardiomyocytes. Synchronization is achieved by the gradual transfer of the signal from one cell to another through intercellular contacts. During embryonic development, cells are surrounded by adherence junctions and the myofibrils are poorly organized (46). At the neonatal stage, the junctions are organized at sites of myofibril attachment, which are gradually organized along the long axis of the cell. The isoform transition of sarcomeric proteins is induced by an increased force-generating capacity and Ca2+ sensitivity (47). Slow skeletal muscle isoforms of troponin-T and troponin-I expressed in the fetal heart are replaced with cardiac troponin-T and troponin-I isoforms. The beta-myosin heavy chain (β-MHC) is replaced by an α-MHC and the ratio of total MHC to actin proteins increases after birth. The β-MHC to α-MHC isoform switch is probably driven by a decrease in HIF-1 signaling. An in vitro study shows an increased expression of the Myh7 gene encoding the β-MHC in reaction to hypoxia and/or stabilized HIF-1α (48). α-MHC fibers work faster than β-MHC (49) and require more ATP molecules (50). The switch to oxidative phosphorylation allows more energy to be spent in order to produce a more efficient contraction of the heart.
HIF-1 Signaling and Adult Heart Function
The contraction of cardiomyocytes is driven by a process called excitation-contraction coupling. This means that the depolarization of the sarcolemma triggers a cascade of processes leading to contraction (51). Upon depolarization, voltage-dependent L-type Ca2+ channels are activated, so a small amount of Ca2+ is quickly released into the cell and activates ryanodine receptors (RyR) in the sarcoplasmic reticulum (SR) responsible for the release of a large amount of Ca2+ into the cytosol. Ca2+ then interacts with troponin C in the troponin-tropomyosin complex located on thin actin filaments leading to a conformational change, and binding of the globular heads of myosin filaments.
The relaxation of cardiomyocytes is mainly dependent on the active sequestration of Ca2+ by SERCA-2, an ATP-dependent Ca2+ pump, located in the membrane of SR. Per every two moles of Ca2+ transferred into the lumen of the SR, one mole of ATP is used. Under normal conditions, adult heart glycolysis represents only a minor part of energy production. The majority of ATP is produced by the oxidative phosphorylation of fatty acids (FA). However, the heart is considered to be a true omnivore, which means that under certain circumstances, lactate, ketone bodies or even acetate can be used as energy substrates (52). Since neonatal remodeling is accompanied by glycogen storage depletion and the heart has a small capacity for storage or de novo synthesis of FA, the main source of energy substrates is the blood. The metabolic adaptations to hypoxia exposure are determined by the degree and duration of hypoxia (53). Chronic exposure to moderate hypoxia, as in high-altitude inhabitants, leads to the development of defense mechanisms against acute oxidative stress. Adaptive mechanisms provide increased oxygen delivery through increased erythropoiesis and angiogenesis and metabolic remodeling in favor of carbohydrate utilization.
Acute hypoxia induces anaerobic glycolysis in order to compensate for cellular energy demands and actively suppresses oxidative phosphorylation (54). HIF-1 directly activates pyruvate dehydrogenase kinase 1 (PDK1), which inactivates pyruvate dehydrogenase, which is a part of pyruvate dehydrogenase complex that converts pyruvate into acetyl-CoA. Thus, pyruvate is redirected from the tricarboxylic acid cycle (TCA) and converted into lactate. This leads to the attenuation of mitochondrial respiration and to reactive oxygen species (ROS) production.
The functional role of HIF-1 signaling in the changes induced by hypoxia is shown by experiments with modulated HIF-1α expression. Constitutive cardiac-specific over-expression of Hif1a leads to changes in cellular metabolism and increased glucose utilization (55), resulting in cardiomyopathy in aging mice. Reversible cardiac specific introduction of the oxygen-stable form of HIF-1α also leads to changes in the expression of metabolic genes in favor of glycolysis and negatively affects heart function, with changes being detectable as early as 3 days after HIF-1α induction (56). Worsening cardiac function with time results in cardiac hypertrophy without detectable histological pathology. Heart function recovers after the Hif1a transgene is turned off, which indicates that transient expression of the oxygen-stable transgene induces only reversible changes. However, the reversible nature of these changes in this study may be explained by the fact that the expression of the Hif1a transgene was turned-off after only seven days. Mutants with altered HIF-1 signaling, transgenic mice with an αMHC-Cre driven cardiomyocyte-specific deletion of Phd2 and Phd3, and mice with a cardiomyocyte-specific deletion of Vhl, show decreased cardiac function and premature mortality (57, 58). Consistent with the effects of HIF-1 on cellular metabolism, hearts lacking PHD2 and PHD3 or lacking VHL accumulate glycogen and lipids. Similarly, cardiac myocyte-specific deletion of HIF-1α negatively affects heart contractility and leads to the decreased expression of genes involved in glucose metabolism (21).
HIF-1 Signaling in Diabetes
Diabetes is a disease affecting glucose homeostasis regardless of the etiology (59). It affects different organ systems in the body, causing severe complications, such as cardiovascular diseases, diabetic nephropathy, neuropathy, retinopathy, and impaired wound healing. The diabetic environment also negatively affects the developing fetus. Diabetic pregnancies are associated with an increased incidence of congenital malformations (diabetic embryopathy) compared with non-diabetic pregnancies. In addition to the direct teratogenicity of maternal diabetes, the intrauterine and early postnatal environments can influence the cardiovascular and metabolic health of the offspring later in life. Here, we focus on the effect of hyperglycemia on HIF-1 signaling, as summarized in Figure 3. Tissue exposure to hyperglycemia is considered to be the main factor in the development of complications, but even tight regulation of glycemia is not sufficient enough to completely prevent them. Hyperglycemia is often associated with a phenomenon called pseudohypoxia (60), which is characterized by an increased ratio of NADH/NAD+ due to an increased flux of glucose through the polyol pathway (61). In hypoxic conditions, the balance is disturbed via impaired oxidation of NADH (60). Some authors disagree with the pseudohypoxia hypothesis, since their results show no changes in the amounts of NADH and NAD+ when isolated diabetic and non-diabetic rat retinas are compared (62). Nevertheless, analyses using pimonidazole, a marker for hypoxic tissue, show increased levels of hypoxia in diabetic tissue (63, 64) and the expression of Hif1a mRNA is elevated in diabetic rat hearts (65). At the protein level, HIF-1α is stabilized not only by hypoxia but also by products of aerobic glycolysis, mainly pyruvate (4). In contrast, hyperglycemia affects HIF-1 transactivation via modification of its coactivator p300 and thus, diminishes HIF-1 transcriptional activity (66) without affecting the stability of the HIF-1α protein (67). High glucose also activates HIF-1-mediated signal transduction via a glucose-responsive carbohydrate response element binding protein (ChREBP), as shown in a model of diabetic glomerulopathy (68). ChREBP plays a functional role in glycolytic and lipogenic gene regulation (69). The next level of diabetes-induced deregulation of HIF-1 signaling involves responses to hypoxia. Experiments with cells cultured in an environment combining hyperglycemia and hypoxia show increased degradation of the HIF-1α protein (70, 71). Thus, diabetes not only causes hypoxia but also compromises HIF-1 signaling. The inability of cells/tissues to appropriately respond to hypoxia increases the risk for complications in diabetic patients. For example, the expression of Hif1a mRNA is lower and the size of the affected area is bigger in the infarcted hearts of streptozotocin-induced diabetic rats than in non-diabetic controls (65). In the following section, we discuss the effects of deregulated HIF-1 signaling in diabetes on the developing and adult heart, as summarized in Figure 4.
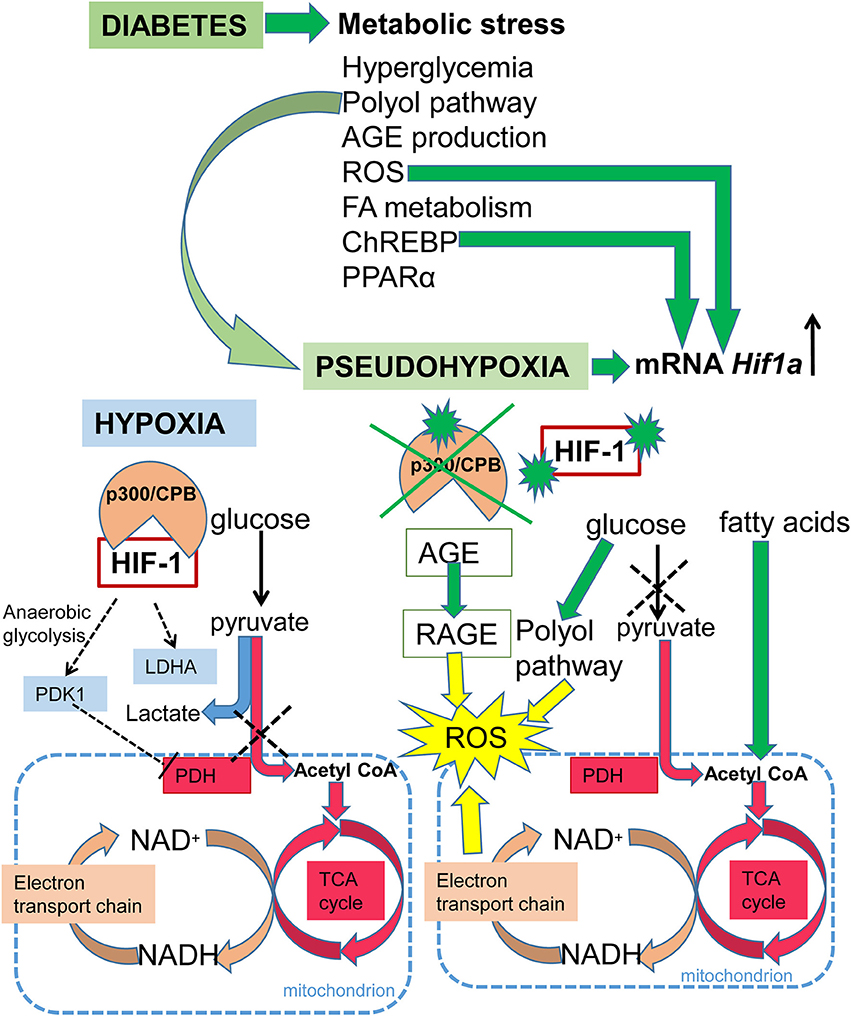
Figure 3. Schematic summary of HIF-1 signaling in hypoxia and diabetes. Under normoxia, glucose is metabolized to pyruvate and then the pyruvate dehydrogenase complex converts pyruvate to acetyl-CoA, which enters the TCA cycle, and NADH and FADH2 are generated. Electrons derived from NADH and FADH2 are utilized by the electron transport chain to generate ATP. Under hypoxia, HIF-1 activates glycolytic genes as a critical step of metabolic adaptation to hypoxia. HIF-1 induces anaerobic glycolysis by mediating the expression of pyruvate dehydrogenase kinase 1 (PDK1), which inhibits pyruvate dehydrogenase (PDH) and induces conversion of pyruvate to lactate. Under diabetes, metabolic stress is induced by: an increased ratio of NADH/NAD+ due to an increased flux of glucose through the polyol pathway resulting in pseudohypoxia; increased expression of peroxisome proliferator-activated receptor alpha (PPARα), which induces fatty acid utilization and oxidation; increased fatty acid oxidation leading to the production of acetyl-CoA; increased production of advanced glycation end products (AGE), which leads to the irreversible modification of proteins; induction of the carbohydrate response element binding protein (ChREBP); and increased ROS production. The production of ROS is induced by oxidative stress due to the increased ratio of NADH/NAD+, and by AGE via signaling through its receptor (RAGE). ROS, ChREBP, and pseudohypoxia induce transcription of Hif1a mRNA. However, in the diabetic environment, HIF-1 signaling is negatively affected via modification of its coactivator p300, and by HIF-1α protein modifications.
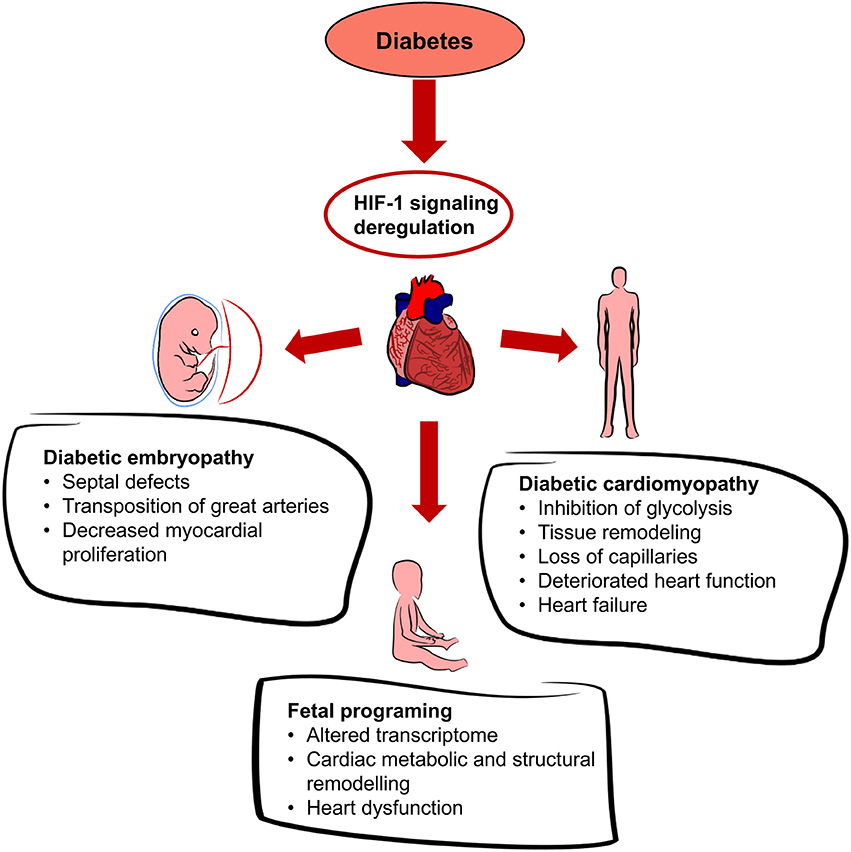
Figure 4. Effects of diabetes on the heart. Diabetes leads to systemic hyperglycemia and hyperglycemia increases tissue hypoxia. HIF-1 signaling is altered due to the changes in HIF-1α transactivation and HIF-1α protein modifications. Dysregulation of cardiac HIF-1 signaling is associated with an increased risk of diabetic embryopathy, adverse long-term outcomes for the offspring, and diabetic cardiomyopathy.
Diabetic Embryopathy
The main energy metabolic substrate in the embryo is glucose, which is obtained from maternal blood by facilitated diffusion across the placental barrier (72). Therefore, maternal hyperglycemia leads to embryonic hyperglycemia. Maternal diabetes has a teratogenic effect on the development of all organ systems; the most commonly affected are the cardiovascular, nervous, and skeletal systems (73, 74). Out of the cardiovascular defects, ventricular septal defects, and transposition of the great arteries are the most common (75). Although the mechanisms of diabetic embryopathy are not fully understood, the role of hyperglycemia is crucial. Studies using mouse models show that hyperglycemia without systemic diabetes has teratogenic effects (76). Since maternal diabetes-induced defects are similar to the defects induced by hypoxia exposure (77), the deregulation of HIF-1 signaling may play a crucial role. This idea is supported by an experiment with in vitro cultivation of embryos, which shows that hyperglycemic conditions increase oxygen depletion (78). Moreover, microarray analysis of maternal diabetes-exposed embryos showed that HIF-1 signaling pathways are significantly affected (79), and embryos with global Hif1a insufficiency display increased susceptibility to diabetic embryopathy (63). Diabetic Hif1a+/− embryos suffer from increased lethality, and an increased number of neural tube and cardiovascular defects. Additionally, the hearts of diabetic Hif1a+/− embryos are severely hypoplasic with an increased trabecular layer. The detected decrease in the proliferation of the compact myocardium suggests that cells in Hif1a+/− diabetic hearts have an altered developmental program. This has been further confirmed by mRNA analysis, which shows changes in genes crucial for embryonic heart development, such as Tbx5, Nkx2.5, Nppa, and Mef2c. Interestingly, Vegfa expression was deregulated in the diabetic hearts of both genotypes. Vegfa is a known target of HIF-1 signaling (80) and both its downregulation and upregulation is embryonically lethal (81, 82). In this context, downregulation of Vegfa in embryonic hearts with severe hyperglycemia (63) is an extremely interesting finding.
The causative role of decreased expression of the HIF-1α protein was shown in diabetic embryonic vasculopathy (83). The disadvantage of this study, in terms of applying it to heart development, lies in its focus on early development before the heart forms. Since it was shown that the kinetics of HIF-1α in response to hypoxia differ between individual organs in adult mice (84), different organs should be studied separately. Another complication for the understanding of pathologies associated with deregulated HIF-1 signaling during embryonic development is represented by rapid changes in gene expression in different developmental stages (85, 86).
Besides diabetic embryopathy, altered HIF-1 signaling may influence the long-term outcomes of the offspring from a diabetic pregnancy. Specifically, it may increase the predisposition of the offspring of diabetic mothers to cardiovascular and metabolic diseases in the process of fetal programing (87). Maternal diabetes leads to accelerated worsening of heart function in the Hif1a+/− offspring when compared to littermate controls. Many processes are affected in Hif1a+/− offspring hearts, including transcriptional changes associated with apoptosis, blood vessel physiology, inflammatory responses, and metabolism (87). Particularly, changes in the expression of Cd36, a HIF-1 target gene encoding a multifunctional receptor mediating the uptake of lipoproteins and lipoprotein-derived fatty acids, indicate changes in myocardial substrate selection and utilization in energy metabolism that play a role in the development of cardiac pathologies (87).
Diabetic Cardiomyopathy
In adults, diabetes is associated with an increased risk of cardiovascular diseases, mainly atherosclerosis (61). However, diabetes causes heart dysfunction independently from coronary artery disease and hypertension, diabetic cardiomyopathy (88, 89). Changes leading to the development of diabetic cardiomyopathy involve structural changes in the myocardium, such as increased fibrosis, lipid accumulation, formation of advanced glycation end products (AGE), and changes in myosin isoforms, as well as changes in substrate utilization, increased apoptosis, and mitochondrial dysfunction. Although, the heart is able to utilize any substrate for the generation of ATP, in diabetic conditions, utilization of glucose decreases, whereas the utilization of fatty acids increases. This is caused by the decreased expression of glucose transporter GLUT1, and glycolytic enzyme hexokinase II. These genes are targets of the HIF-1 pathway (90). In this context, the molecular interactions between HIF-1 and other nuclear partners involved in hypoxia-mediated responses, such as NF-κB, CREB, HMGA1, and HIF-2α, need to be mentioned. For example, HMGA1 is transcription factor regulating chromatin structure and gene expression (91). Defects in HMGA1 expression have been associated with insulin resistance and increased susceptibility to type 2 diabetes (92) and increased risk of acute myocardial infarction (93). HIF-2α, a paralog of HIF-1α, is the critical effector in obesity-associated cardiomyopathy (94, 95). Obesity represents an independent risk factor for cardiovascular disease [reviewed in Poirier et al. (96)] that alters the metabolic profile and induces structural cardiac adaptation by excessive lipid accumulation. Obesity affects the heart through multiple mechanisms such as dyslipidemia, glucose intolerance, hypertension, and inflammation. The inflammatory microenvironment contains small molecules such as ROS, NO, and different cytokines with the ability to activate HIF-1 and HIF-2 signaling [as reviewed in Dehne and Brune (97)]. HIF-1α activation during inflammation results in upregulation of classical target genes like VEGF, GLUT1 or metalloproteinases but also in the activation of unique transcripts such as beta 2 integrin, adenosine receptors or chemokine receptors, indicating that HIF-1 has an immune function (98). Obesity and diabetes are associated with increased levels of FA. Increased usage of FA leads to increased consumption of oxygen, which likely exceeds the FA oxidation capacity and generates tissue hypoxia (99–101). Utilization of FA is regulated by peroxisome proliferator-activated receptor α (PPARα), which targets most of the genes of the fatty acid oxidation pathway (102), is inhibited by HIF-1 (103) and its cardiac overexpression leads to inhibition of glucose uptake, mimicking the effect of diabetes (104). Increased β-oxidation of FA also leads to increased production of ROS and increased expression of mitochondrial uncoupling proteins (UCP) (105), which are thought to decrease generation of ROS (106). UCP2 and UCP3 are expressed in the heart and are targets of PPARα signaling (107, 108) which links their activity to the level of FA. Although uncoupling serves as protection against ROS, it also lowers ATP production and thus affects the efficiency of the heart (105, 109). ROS production is also triggered by AGE via signaling through its receptor (RAGE) (110). Both, ROS and AGE not only modify proteins in extra- and intracellular milieu, but also increase oxidative stress, which activates inflammatory pathways. Even without diabetes, chronic inflammation is a cause of cardiac dysfunction (111). ROS trigger HIF-1 signaling in several ways (112, 113). In general, ROS induce HIF-1α. However, as mentioned earlier, HIF-1α is increasingly degraded in hyperglycemic conditions.
miRNAs are additional important regulatory factors in cardiovascular disease, including diabetic cardiomyopathy (114). Each miRNA regulates from a few up to hundreds of target genes in normal development, growth, and function, as well as in disease. miRNAs play an important role in responses to hypoxia and/or diabetes (115–117). Most miRNAs identified with the hypoxia signature are directly regulated through HIF, as recently reviewed (115). miRNAs also regulate expression or stabilization of HIF-1α by direct binding to its mRNAs or by regulation of HIF complex units such as VHL or PHD (115). Differential expression profiles of the most abundant cardiac miRNAs, miR-1 and miR-133a, are linked to diabetic cardiomyopathy (117). miR-133a is downregulated in the diabetic heart (118), and decreased miR-133 levels induce cardiac hypertrophy (119). Moreover, experiments with isolated cardiomyocytes show increased apoptosis in connection with a decreased expression of miR-133a in hypoxic conditions (120). In line with this finding, apoptotic proteins, caspase −8, −9, and −3 are repressed, while the expression of BCL-2 increases in cells overexpressing miR-133a (120). Overexpression of miR-133a also mitigates cardiac fibrosis (119). One of the metabolic proteins targeted by miR-133 is GLUT4 (121). Furthermore, miR-133a inhibits expression of DNA methyltransferase 1 in cardiomyocytes and thus induces changes in the methylation program and overall induces a much broader effect in gene regulation (122). Downregulation of miR-1 causes hypertrophy and fibrosis in the heart, similar to the effects of miR-133a (123). Hyperglycemia induces apoptosis via increased miR-1 in cultured cardiomyocytes that blocks the protective capacity of insulin like growth factor-1 (124). In a rat cardiac model of ischemia/ reperfusion, as well as in cardiomyocytes subjected to hypoxia/reperfusion, inhibition of miR-1 increases BCL-2 expression and thus protects against hypoxia/reoxygenation injury (125). Apoptosis is also regulated by miR-195, which is upregulated in diabetic hearts (126). Its downregulation in diabetic mice decreases myocardial hypertrophy, ROS production, and apoptosis. Analysis of interfibrillar mitochondria isolated from diabetic hearts shows increased expression of miR-141, which inhibits translation of phosphate transporter SLC25A3, causing decreased activity of ATP synthase due to lack of substrate (127). Interestingly, the subsarcolemmal mitochondrial population remains unaffected. Upregulation of miR-210 is associated with cardiac hypertrophy and reactivation of the fetal transcriptional program, triggering pathological changes in the myocardium of the human failing heart (34, 35). miR-210 and miR199a/b were modulated by hypoxia and high glucose in cardiomyocytes and dysregulated in diabetic ischemic heart failure patients (114). miRNAs also represent potential diagnostic markers of diabetic complications, for example levels of circulating miR-1 and miR-133 in the serum of patients with type 2 diabetes correlate with myocardial steatosis, an early manifestation of cardiac pathology (128). The above description is only a narrow representation of miRNAs as regulators of cardiac diseases. These findings not only provide novel insights on the roles of miRNA in cardiac pathologies but also identify potential therapeutic targets for reducing cardiac injury and heart failure.
Diabetes is known to impair microvasculature (129, 130); particularly, diabetic cardiomyopathy is associated with a loss of coronary capillaries that further increases tissue hypoxia (131). Since the combination of hypoxia and hyperglycemia increases degradation of the HIF-1α protein (70, 71), the ability of the diabetic heart to respond to hypoxic conditions is compromised (101). Consistent with these data are experiments with altered HIF-1 signaling in mouse hearts. For example, the cardiac-specific overexpression of Hif1a in the streptozotocin-induced diabetic mouse model prevents the decrease in GLUT1 and hexokinase II expression as well as hexokinase activity (132). These proteins are substantial in the first two steps of glycolysis: glucose transport into the cell (133) and its phosphorylation (134). These effects of Hif1a overexpression on glycolysis may be one of the reasons for the normalization of the amount of ATP generated in Hif1a overexpressing diabetic hearts. Similar results were obtained using insulinopenic mice overexpressing hexokinase (135). Additionally, Hif1a overexpression in the diabetic heart preserves VEGF expression and angiogenesis, and prevents myocardial fibrosis and remodeling (132). Experiments using the Hif1a heterozygous deficient diabetic mouse model further underscore the importance of proper HIF-1 signaling (136). Hif1a+/− mice exposed to only 5 weeks of diabetes develop decreased fractional shortening, undetectable in control mice. Changes are also found at the molecular level, such as a decreased amount of VEGFA, an important target of HIF-1, affecting vascularization, and levels of gap-junctional protein connexin43 (Cx43) phosphorylated at serine 368. The phosphorylation of connexins influences intercellular communication (137) and is thus likely to affect heart function, in which proper signal transduction is crucial for contraction. Diabetic Hif1a+/− mice also have increased collagen type I levels, which is associated with fibrosis.
Conclusion
In this review, we focus on the role of HIF-1 in metabolism in embryonic development and in the adult heart. HIF-1 signaling regulates the cellular switch from oxidative phosphorylation to glycolysis. During embryonic development, glycolysis allows the redirection of glucose-derived carbons into the pentose phosphate pathway and thus creates anabolic building blocks for proliferating cells. Embryonic lethality of the global deletion of Hif1a due to associated cardiovascular defects underscores its importance for development. However, our knowledge of embryonic metabolism is still incomplete. A comprehensive molecular and cellular understanding of HIF activity in embryonic development will provide a basis for stem cell research and regenerative medicine. An increased level of oxygen and decreased activity of HIF-1 are key factors in the perinatal remodeling of the myocardium. During this stage, the heart undergoes a gradual transition to aerobic metabolism. In adulthood, the cardiac function of HIF-1 signaling is suppressed, but HIF-1 is still readily available for intervention in case of tissue hypoxia. The current state of knowledge suggests HIF-1 signaling is one of the crucial regulators of developmental changes in cardiac metabolism. In this review, we also addressed the effect of diabetes on HIF-1 signaling. Hyperglycemia causes hypoxia, but also affects the function of HIF-1α. The combination of hyperglycemia and hypoxia decreases the stability of HIF-1α, leading to the deregulation of HIF-1 signaling. Experiments using embryos with Hif1a global hemizygous deletion show that the deregulation of HIF-1 signaling contributes to the development of diabetic embryopathy and associated cardiovascular defects. Furthermore, Hif1a hemizygous mutant offspring of diabetic mothers have worsened heart function compared to their wild type littermates. In diabetic cardiomyopathy, consistent with the decreased function and stability of HIF-1α, the myocardium loses its ability to use glucose as a metabolic substrate. Changes in metabolism are not the only pathology induced by diabetes, but with hyperglycemia being the primary trigger in diabetes, this raises the question of whether deregulated metabolism is the origin of these dysfunctions, or just one of the consequences. Resolving this particular question may be the key to developing an efficient therapy. The greatest limitations of HIF biology research are the experimental tools used and the limitations posed by experimental models. For example, hypoxia/HIF-1 responses vary between cell lines and between models, which may affect overall conclusions. Additionally, regulation of HIF-1 signaling is a very complex process that includes regulation of gene transcription, protein stability, and protein-protein interactions. Responses to the hypoxic environment range from diversified cellular modulations to systemic responses.
Research on hypoxia and HIF-1 regulation has significantly improved our understanding of their role in health and diseases. However, given the pleiotropic and complex nature of the hypoxic response and the large number of components involved in the HIF-1 system, it is important to develop very selective HIF-1 regulators for clinical applications. Systemic administration of small-molecule inhibitors of HIF-1 activity with anticancer effects that impairs both vascular and metabolic adaptations have been successfully used in a number of preclinical studies (138). However, no selective HIF-1α inhibitor has been clinically approved up to this date, mainly due to the nonspecific mode of action of these inhibitors affecting other pathways. The topical application of PHD inhibitors has been shown to improve wound healing in diabetic animals, representing a more immediate potential for clinical applications (139). Clinical trials of the PHD inhibitors FG-4592, GSK1278863, Molidustat, and Vadadustat, for anemia in chronic kidney disease are ongoing and appear to be encouraging (140). However, it is necessary to address the tissue-specific requirements of HIF-1 activity, temporal application of therapeutics, negative side effects of systemic applications, and potential non-specificity of therapeutic agents. Deciphering the role of non-coding RNAs, epigenetics, and different genetic factors underlying the complex networks of hypoxia/HIF-1α regulation may provide novel and more specific targets for future therapies.
Author Contributions
RC wrote the first draft of the manuscript. GP finalized and conceptualized the manuscript. All authors read and approved the submitted version.
Funding
This work was supported by the Czech Science Foundation (Grant Agreement No. 16-06825S to GP); by BIOCEV CZ.1.05/1.1.00/02.0109 from the ERDF; and RVO 86652036; and by the Charles University in Prague (GA UK No. 228416 to RC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors at time of review.
Acknowledgments
We thank A. Pavlinek for editing the manuscript.
References
1. Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. (2011) 300:C385–93. doi: 10.1152/ajpcell.00485.2010
2. Kaluz S, Kaluzova M, Stanbridge EJ. Regulation of gene expression by hypoxia: integration of the HIF-transduced hypoxic signal at the hypoxia-responsive element. Clin Chim Acta (2008) 395:6–13. doi: 10.1016/j.cca.2008.05.002
3. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. (2011) 365:537–47. doi: 10.1056/NEJMra1011165
4. Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. (2002) 277:23111–5. doi: 10.1074/jbc.M202487200
5. Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg's effect. J Bioenerg Biomembr. (2007) 39:223–9. doi: 10.1007/s10863-007-9080-3
6. Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. (1997) 11:388–95.
7. Jauniaux E, Watson A, Burton G. Evaluation of respiratory gases and acid-base gradients in human fetal fluids and uteroplacental tissue between 7 and 16 weeks' gestation. Am J Obstet Gynecol. (2001) 184:998–1003. doi: 10.1067/mob.2001.111935
8. Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. (1992) 80:283–5.
9. Houghton FD, Thompson JG, Kennedy CJ, Leese HJ. Oxygen consumption and energy metabolism of the early mouse embryo. Mol Reprod Dev. (1996) 44:476–85.
10. Lee YM, Jeong CH, Koo SY, Son MJ, Song HS, Bae SK, et al. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn. (2001) 220:175–86. doi: 10.1002/1097-0177(20010201)220:2<175::AID-DVDY1101>3.0.CO;2-F
11. Dunwoodie SL. The role of hypoxia in development of the Mammalian embryo. Dev Cell (2009) 17:755–73. doi: 10.1016/j.devcel.2009.11.008
12. Moorman A, Webb S, Brown NA, Lamers W, Anderson RH. Development of the heart: (1) formation of the cardiac chambers and arterial trunks. Heart (2003) 89:806–814. doi: 10.1136/heart.89.7.806
13. Ream M, Ray AM, Chandra R, Chikaraishi DM. Early fetal hypoxia leads to growth restriction and myocardial thinning. Am J Physiol Regul Integr Comp Physiol. (2008) 295:R583–95. doi: 10.1152/ajpregu.00771.2007
14. Kenchegowda D, Liu H, Thompson K, Luo L, Martin SS, Fisher SA. Vulnerability of the developing heart to oxygen deprivation as a cause of congenital heart defects. J Am Heart Assoc. (2014) 3:e000841. doi: 10.1161/JAHA.114.000841
15. Wikenheiser J, Doughman YQ, Fisher SA, Watanabe M. Differential levels of tissue hypoxia in the developing chicken heart. Dev Dyn. (2006) 235:115–23. doi: 10.1002/dvdy.20499
16. Sugishita Y, Leifer DW, Agani F, Watanabe M, Fisher SA. Hypoxia-responsive signaling regulates the apoptosis-dependent remodeling of the embryonic avian cardiac outflow tract. Dev Biol. (2004) 273:285–96. doi: 10.1016/j.ydbio.2004.05.036
17. Wikenheiser J, Wolfram JA, Gargesha M, Yang K, Karunamuni G, Wilson DL, et al. Altered hypoxia-inducible factor-1 alpha expression levels correlate with coronary vessel anomalies. Dev Dyn. (2009) 238:2688–700. doi: 10.1002/dvdy.22089
18. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. (1998) 12:149–62.
19. Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol. (1999) 209:254–67. doi: 10.1006/dbio.1999.9253
20. Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, et al. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc Res. (2003) 60:569–79. doi: 10.1016/j.cardiores.2003.07.003
21. Huang Y, Hickey RP, Yeh JL, Liu D, Dadak A, Young LH, et al. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. (2004) 18:1138–40. doi: 10.1096/fj.04-1510fje
22. Krishnan J, Ahuja P, Bodenmann S, Knapik D, Perriard E, Krek W, et al. Essential role of developmentally activated hypoxia-inducible factor 1alpha for cardiac morphogenesis and function. Circ Res. (2008) 103:1139–46. doi: 10.1161/01.RES.0000338613.89841.c1
23. Guimaraes-Camboa N, Stowe J, Aneas I, Sakabe N, Cattaneo P, Henderson L, et al. HIF1alpha represses cell stress pathways to allow proliferation of hypoxic fetal cardiomyocytes. Dev Cell (2015) 33:507–21. doi: 10.1016/j.devcel.2015.04.021
24. Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis (2000) 26:113–5. doi: 10.1002/(SICI)1526-968X(200002)26:2<113::AID-GENE3>3.0.CO;2-2
25. Licht AH, Muller-Holtkamp F, Flamme I, Breier G. Inhibition of hypoxia-inducible factor activity in endothelial cells disrupts embryonic cardiovascular development. Blood (2006) 107:584–90. doi: 10.1182/blood-2005-07-3033
26. Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci USA. (1997) 94:9102–7.
27. Hong SB, Furihata M, Baba M, Zbar B, Schmidt LS. Vascular defects and liver damage by the acute inactivation of the VHL gene during mouse embryogenesis. Lab Invest. (2006) 86:664–75. doi: 10.1038/labinvest.3700431
28. Kaidi A, Williams AC, Paraskeva C. Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. (2007) 9:210–7. doi: 10.1038/ncb1534
29. Sainson RC, Harris AL. Hypoxia-regulated differentiation: let's step it up a Notch. Trends Mol Med. (2006) 12:141–3. doi: 10.1016/j.molmed.2006.02.001
30. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell (2005) 9:617–28. doi: 10.1016/j.devcel.2005.09.010
31. Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. (2008) 9:831–42. doi: 10.1038/nrg2455
32. Martinez SR, Ma Q, Dasgupta C, Meng X, Zhang L. MicroRNA-210 suppresses glucocorticoid receptor expression in response to hypoxia in fetal rat cardiomyocytes. Oncotarget (2017) 8:80249–64. doi: 10.18632/oncotarget.17801
33. Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation (2010) 122(11 Suppl.):S124–31. doi: 10.1161/CIRCULATIONAHA.109.928424
34. Thum T, Catalucci D, Bauersachs J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovasc Res. (2008) 79:562–70. doi: 10.1093/cvr/cvn137
35. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation (2007) 116:258–67. doi: 10.1161/CIRCULATIONAHA.107.687947
36. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
37. Miyazawa H, Yamaguchi Y, Sugiura Y, Honda K, Kondo K, Matsuda F, et al. Rewiring of embryonic glucose metabolism via suppression of PFK-1 and aldolase during mouse chorioallantoic branching. Development (2017) 144:63–73. doi: 10.1242/dev.138545
38. Herrera E, Amusquivar E. Lipid metabolism in the fetus and the newborn. Diabetes Metab Res Rev. (2000) 16:202–10. doi: 10.1385/ENDO:19:1:43
39. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation (2001) 104:2923–31. doi: 10.1161/hc4901.100526
40. Ascuitto RJ, Ross-Ascuitto NT. Substrate metabolism in the developing heart. Semin Perinatol. (1996) 20:542–63.
41. Iruretagoyena JI, Davis W, Bird C, Olsen J, Radue R, Teo Broman A, et al. Metabolic gene profile in early human fetal heart development. Mol Hum Reprod. (2014) 20:690–700. doi: 10.1093/molehr/gau026
42. Yamashita T, Ohneda O, Nagano M, Iemitsu M, Makino Y, Tanaka H, et al. Abnormal heart development and lung remodeling in mice lacking the hypoxia-inducible factor-related basic helix-loop-helix PAS protein NEPAS. Mol Cell Biol. (2008) 28:1285–97. doi: 10.1128/MCB.01332-07
43. Menendez-Montes I, Escobar B, Palacios B, Gomez MJ, Izquierdo-Garcia JL, Flores L, et al. Myocardial VHL-HIF signaling controls an embryonic metabolic switch essential for cardiac maturation. Dev Cell (2016) 39:724–39. doi: 10.1016/j.devcel.2016.11.012
44. Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation (2007) 116:434–48. doi: 10.1161/CIRCULATIONAHA.107.702795
45. Neary MT, Ng KE, Ludtmann MH, Hall AR, Piotrowska I, Ong SB, et al. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J Mol Cell Cardiol. (2014) 74:340–52. doi: 10.1016/j.yjmcc.2014.06.013
46. Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. (2006) 289:430–41. doi: 10.1016/j.ydbio.2005.10.046
47. Siedner S, Kruger M, Schroeter M, Metzler D, Roell W, Fleischmann BK, et al. Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J Physiol. (2003) 548(Pt 2):493–505. doi: 10.1113/jphysiol.2002.036509
48. Bino L, Prochazkova J, Radaszkiewicz KA, Kucera J, Kudova J, Pachernik J, et al. Hypoxia favors myosin heavy chain beta gene expression in an Hif-1alpha-dependent manner. Oncotarget (2017) 8:83684–97. doi: 10.18632/oncotarget.19016
49. Galler S, Puchert E, Gohlsch B, Schmid D, Pette D. Kinetic properties of cardiac myosin heavy chain isoforms in rat. Pflugers Arch. (2002) 445:218–23. doi: 10.1007/s00424-002-0934-6
50. Wang Y, Tanner BC, Lombardo AT, Tremble SM, Maughan DW, Vanburen P, et al. Cardiac myosin isoforms exhibit differential rates of MgADP release and MgATP binding detected by myocardial viscoelasticity. J Mol Cell Cardiol. (2013) 54:1–8. doi: 10.1016/j.yjmcc.2012.10.010
51. Walker CA, Spinale FG. The structure and function of the cardiac myocyte: a review of fundamental concepts. J Thorac Cardiovasc Surg. (1999) 118:375–82. doi: 10.1016/S0022-5223(99)70233-3
52. Kodde IF, van der Stok J, Smolenski RT, de Jong JW. Metabolic and genetic regulation of cardiac energy substrate preference. Comp Biochem Physiol A Mol Integr Physiol. (2007) 146:26–39. doi: 10.1016/j.cbpa.2006.09.014
53. Essop MF. Cardiac metabolic adaptations in response to chronic hypoxia. J Physiol. (2007) 584(Pt 3):715–26. doi: 10.1113/jphysiol.2007.143511
54. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
55. Holscher M, Schafer K, Krull S, Farhat K, Hesse A, Silter M, et al. Unfavourable consequences of chronic cardiac HIF-1alpha stabilization. Cardiovasc Res. (2012) 94:77–86. doi: 10.1093/cvr/cvs014
56. Bekeredjian R, Walton CB, MacCannell KA, Ecker J, Kruse F, Outten JT, et al. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS ONE (2010) 5:e11693. doi: 10.1371/journal.pone.0011693
57. Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, et al. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation (2010) 122:1004–16. doi: 10.1161/CIRCULATIONAHA.109.922427
58. Lei L, Mason S, Liu D, Huang Y, Marks C, Hickey R, et al. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol Cell Biol. (2008) 28:3790–803. doi: 10.1128/MCB.01580-07
59. American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care (2014) 37 (Suppl. 1):S81–90. doi: 10.2337/dc14-S081
60. Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes (1993) 42:801–13.
61. Gleissner CA, Galkina E, Nadler JL, Ley K. Mechanisms by which diabetes increases cardiovascular disease. Drug Discov Today Dis Mech. (2007) 4:131–40. doi: 10.1016/j.ddmec.2007.12.005
62. Diederen RM, Starnes CA, Berkowitz BA, Winkler BS. Reexamining the hyperglycemic pseudohypoxia hypothesis of diabetic oculopathy. Invest Ophthalmol Vis Sci. (2006) 47:2726–31. doi: 10.1167/iovs.06-0076
63. Bohuslavova R, Skvorova L, Sedmera D, Semenza GL, Pavlinkova G. Increased susceptibility of HIF-1alpha heterozygous-null mice to cardiovascular malformations associated with maternal diabetes. J Mol Cell Cardiol. (2013) 60:129–41. doi: 10.1016/j.yjmcc.2013.04.015
64. Sada K, Nishikawa T, Kukidome D, Yoshinaga T, Kajihara N, Sonoda K, et al. Hyperglycemia induces cellular hypoxia through production of mitochondrial ROS followed by suppression of aquaporin-1. PLoS ONE (2016) 11:e0158619. doi: 10.1371/journal.pone.0158619
65. Marfella R, D'Amico M, Di Filippo C, Piegari E, Nappo F, Esposito K, et al. Myocardial infarction in diabetic rats: role of hyperglycaemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia (2002) 45:1172–81. doi: 10.1007/s00125-002-0882-x
66. Thangarajah H, Yao D, Chang EI, Shi Y, Jazayeri L, Vial IN, et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc Natl Acad Sci USA. (2009) 106:13505–10. doi: 10.1073/pnas.0906670106
67. Thangarajah H, Vial IN, Grogan RH, Yao D, Shi Y, Januszyk M, et al. HIF-1alpha dysfunction in diabetes. Cell Cycle (2010) 9:75–9. doi: 10.4161/cc.9.1.10371
68. Isoe T, Makino Y, Mizumoto K, Sakagami H, Fujita Y, Honjo J, et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. (2010) 78:48–59. doi: 10.1038/ki.2010.99
69. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA. (2004) 101:7281–6. doi: 10.1073/pnas.0401516101
70. Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes (2004) 53:3226–32. doi: 10.2337/diabetes.53.12.3226
71. Ramalho AR, Toscano A, Pereira P, Girao H, Goncalves L, Marques C. Hyperglycemia-induced degradation of HIF-1alpha contributes to impaired response of cardiomyocytes to hypoxia. Rev Port Cardiol. (2017) 36:367–73. doi: 10.1016/j.repc.2016.09.018
72. Lager S, Powell TL. Regulation of nutrient transport across the placenta. J Pregnancy (2012) 2012:179827. doi: 10.1155/2012/179827
73. Gabbay-Benziv R, Reece EA, Wang F, Yang P. Birth defects in pregestational diabetes: defect range, glycemic threshold and pathogenesis. World J Diabetes (2015) 6:481–8. doi: 10.4239/wjd.v6.i3.481
74. Farrell T, Neale L, Cundy T. Congenital anomalies in the offspring of women with type 1, type 2 and gestational diabetes. Diabet Med. (2002) 19:322–6. doi: 10.1046/j.1464-5491.2002.00700.x
75. Wren C, Birrell G, Hawthorne G. Cardiovascular malformations in infants of diabetic mothers. Heart (2003) 89:1217–20. doi: 10.1136/heart.89.10.1217
76. Baack ML, Wang C, Hu S, Segar JL, Norris AW. Hyperglycemia induces embryopathy, even in the absence of systemic maternal diabetes: an in vivo test of the fuel mediated teratogenesis hypothesis. Reprod Toxicol. (2014) 46:129–36. doi: 10.1016/j.reprotox.2014.03.013
77. Ornoy A, Rand SB, Bischitz N. Hyperglycemia and hypoxia are interrelated in their teratogenic mechanism: studies on cultured rat embryos. Birth Defects Res B Dev Reprod Toxicol. (2010) 89:106–15. doi: 10.1002/bdrb.20230
78. Li R, Chase M, Jung SK, Smith PJ, Loeken MR. Hypoxic stress in diabetic pregnancy contributes to impaired embryo gene expression and defective development by inducing oxidative stress. Am J Physiol Endocrinol Metab. (2005) 289:E591–9. doi: 10.1152/ajpendo.00441.2004
79. Pavlinkova G, Salbaum JM, Kappen C. Maternal diabetes alters transcriptional programs in the developing embryo. BMC Genomics (2009) 10:274. doi: 10.1186/1471-2164-10-274
80. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood (2005) 105:659–69. doi: 10.1182/blood-2004-07-2958
81. Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature (1996) 380:435–9. doi: 10.1038/380435a0
82. Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development (2000) 127:3941–6.
83. Yang P, Reece EA. Role of HIF-1alpha in maternal hyperglycemia-induced embryonic vasculopathy. Am J Obstet Gynecol. (2011) 204:332.e331–7. doi: 10.1016/j.ajog.2011.01.012
84. Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, et al. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. Faseb J. (2001) 15:2445–53. doi: 10.1096/fj.01-0125com
85. Vijaya M, Manikandan J, Parakalan R, Dheen ST, Kumar SD, Tay SS. Differential gene expression profiles during embryonic heart development in diabetic mice pregnancy. Gene (2013) 516:218–27. doi: 10.1016/j.gene.2012.12.071
86. Bohuslavova R, Skvorova L, Cerychova R, Pavlinkova G. Gene expression profiling of changes induced by maternal diabetes in the embryonic heart. Reprod Toxicol. (2015) 57:147–56. doi: 10.1016/j.reprotox.2015.06.045
87. Cerychova R, Bohuslavova R, Papousek F, Sedmera D, Abaffy P, Benes V, et al. Adverse effects of Hif1a mutation and maternal diabetes on the offspring heart. Cardiovasc Diabetol. (2018) 17:68. doi: 10.1186/s12933-018-0713-0
88. Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. (2010) 11:31–9. doi: 10.1007/s11154-010-9131-7
89. Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech. (2009) 2:454–66. doi: 10.1242/dmm.001941
90. Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
91. Bustin M, Reeves R. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. (1996) 54:35–100.
92. Chiefari E, Ventura V, Capula C, Randazzo G, Scorcia V, Fedele M, et al. A polymorphism of HMGA1 protects against proliferative diabetic retinopathy by impairing HMGA1-induced VEGFA expression. Sci Rep. (2016) 6:39429. doi: 10.1038/srep39429
93. De Rosa S, Chiefari E, Salerno N, Ventura V, D'Ascoli GL, Arcidiacono B, et al. HMGA1 is a novel candidate gene for myocardial infarction susceptibility. Int J Cardiol. (2017) 227:331–4. doi: 10.1016/j.ijcard.2016.11.088
94. Lin Q, Huang Y, Booth CJ, Haase VH, Johnson RS, Celeste Simon M, et al. Activation of hypoxia-inducible factor-2 in adipocytes results in pathological cardiac hypertrophy. J Am Heart Assoc. (2013) 2:e000548. doi: 10.1161/JAHA.113.000548
95. Giaccia AJ. HIF-2: the missing link between obesity and cardiomyopathy. J Am Heart Assoc. (2013) 2:e000710. doi: 10.1161/JAHA.113.000710
96. Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. (2006) 26:968–76. doi: 10.1161/01.ATV.0000216787.85457.f3
97. Dehne N, Brune B. HIF-1 in the inflammatory microenvironment. Exp Cell Res. (2009) 315:1791–7. doi: 10.1016/j.yexcr.2009.03.019
98. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112:645–57. doi: 10.1038/news030303-8
99. Seferovic PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. (2015) 36:1718–27, 1727a–c. doi: 10.1093/eurheartj/ehv134
100. Schiattarella GG, Hill JA. Metabolic control and oxidative stress in pathological cardiac remodelling. Eur Heart J. (2017) 38:1399–401. doi: 10.1093/eurheartj/ehw199
101. Mansor LS, Mehta K, Aksentijevic D, Carr CA, Lund T, Cole MA, et al. Increased oxidative metabolism following hypoxia in the type 2 diabetic heart, despite normal hypoxia signalling and metabolic adaptation. J Physiol. (2016) 594:307–20. doi: 10.1113/JP271242
102. Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. (2000) 10:238–45. doi: 10.1016/S1050-1738(00)00077-3
103. Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. (2001) 166:7543–8. doi: 10.4049/jimmunol.166.12.7543
104. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. (2002) 109:121–30. doi: 10.1172/JCI14080
105. Lorenzo O, Ramirez E, Picatoste B, Egido J, Tunon J. Alteration of energy substrates and ROS production in diabetic cardiomyopathy. Mediators Inflamm. (2013) 2013:461967. doi: 10.1155/2013/461967
106. Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, et al. The biology of mitochondrial uncoupling proteins. Diabetes (2004) 53 (Suppl. 1):S130–5. doi: 10.2337/diabetes.53.2007.S130
107. Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, et al. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. FASEB J. (2001) 15:833–45. doi: 10.1096/fj.00-0351com
108. Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes (2005) 54:3496–502. doi: 10.2337/diabetes.54.12.3496
109. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes (2007) 56:2457–66. doi: 10.2337/db07-0481
110. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation (2006) 114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
111. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. (2015) 116:1254–68. doi: 10.1161/CIRCRESAHA.116.302317
112. Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol Cell Biol. (2008) 28:5106–19. doi: 10.1128/MCB.00060-08
113. Movafagh S, Crook S, Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem. (2015) 116:696–703. doi: 10.1002/jcb.25074
114. Greco S, Fasanaro P, Castelvecchio S, D'Alessandra Y, Arcelli D, Di Donato M, et al. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes (2012) 61:1633–41. doi: 10.2337/db11-0952
115. Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. (2018) 27:281–98. doi: 10.1016/j.cmet.2017.10.005
116. Shantikumar S, Caporali A, Emanueli C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res. (2012) 93:583–93. doi: 10.1093/cvr/cvr300
117. Asrih M, Steffens S. Emerging role of epigenetics and miRNA in diabetic cardiomyopathy. Cardiovasc Pathol. (2013) 22:117–25. doi: 10.1016/j.carpath.2012.07.004
118. Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW, II, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med. (2014) 18:415–21. doi: 10.1111/jcmm.12218
119. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. (2007) 13:613–18. doi: 10.1038/nm1582
120. Li AY, Yang Q, Yang K. miR-133a mediates the hypoxia-induced apoptosis by inhibiting TAGLN2 expression in cardiac myocytes. Mol Cell Biochem. (2015) 400:173–81. doi: 10.1007/s11010-014-2273-2
121. Horie T, Ono K, Nishi H, Iwanaga Y, Nagao K, Kinoshita M, et al. MicroRNA-133 regulates the expression of GLUT4 by targeting KLF15 and is involved in metabolic control in cardiac myocytes. Biochem Biophys Res Commun. (2009) 389:315–20. doi: 10.1016/j.bbrc.2009.08.136
122. Chavali V, Tyagi SC, Mishra PK. MicroRNA-133a regulates DNA methylation in diabetic cardiomyocytes. Biochem Biophys Res Commun. (2012) 425:668–72. doi: 10.1016/j.bbrc.2012.07.105
123. Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, et al. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. (2013) 2:e000078. doi: 10.1161/JAHA.113.000078
124. Yu XY, Song YH, Geng YJ, Lin QX, Shan ZX, Lin SG, et al. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem Biophys Res Commun. (2008) 376:548–52. doi: 10.1016/j.bbrc.2008.09.025
125. Zhai C, Tang G, Peng L, Hu H, Qian G, Wang S, et al. Inhibition of microRNA-1 attenuates hypoxia/re-oxygenation-induced apoptosis of cardiomyocytes by directly targeting Bcl-2 but not GADD45Beta. Am J Transl Res. (2015) 7:1952–62.
126. Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia (2015) 58:1949–58. doi: 10.1007/s00125-015-3622-8
127. Baseler WA, Thapa D, Jagannathan R, Dabkowski ER, Croston TL, Hollander JM. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am J Physiol Cell Physiol. (2012) 303:C1244–51. doi: 10.1152/ajpcell.00137.2012
128. de Gonzalo-Calvo D, van der Meer RW, Rijzewijk LJ, Smit JW, Revuelta-Lopez E, Nasarre L, et al. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep. (2017) 7:47. doi: 10.1038/s41598-017-00070-6
129. Loffler AI, Bourque JM. Coronary microvascular dysfunction, microvascular angina, and management. Curr Cardiol Rep. (2016) 18:1. doi: 10.1007/s11886-015-0682-9
130. Crea F, Camici PG, Bairey Merz CN. Coronary microvascular dysfunction: an update. Eur Heart J. (2014) 35:1101–11. doi: 10.1093/eurheartj/eht513
131. Kibel A, Selthofer-Relatic K, Drenjancevic I, Bacun T, Bosnjak I, Kibel D, et al. Coronary microvascular dysfunction in diabetes mellitus. J Int Med Res. (2017) 45:1901–29. doi: 10.1177/0300060516675504
132. Xue W, Cai L, Tan Y, Thistlethwaite P, Kang YJ, Li X, et al. Cardiac-specific overexpression of HIF-1{alpha} prevents deterioration of glycolytic pathway and cardiac remodeling in streptozotocin-induced diabetic mice. Am J Pathol. (2010) 177:97–105. doi: 10.2353/ajpath.2010.091091
133. Zhao FQ, Keating AF. Functional properties and genomics of glucose transporters. Curr Genomics (2007) 8:113–28. doi: 10.2174/138920207780368187
134. Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. (2015) 22:248–57. doi: 10.1038/cdd.2014.173
135. Ye G, Donthi RV, Metreveli NS, Epstein PN. Overexpression of hexokinase protects hypoxic and diabetic cardiomyocytes by increasing ATP generation. Cardiovasc Toxicol. (2005) 5:293–300. doi: 10.1385/CT:5:3:293
136. Bohuslavova R, Kolar F, Sedmera D, Skvorova L, Papousek F, Neckar J, et al. Partial deficiency of HIF-1alpha stimulates pathological cardiac changes in streptozotocin-induced diabetic mice. BMC Endocr Disord. (2014) 14:11. doi: 10.1186/1472-6823-14-11
137. Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys. (2000) 384:205–15. doi: 10.1006/abbi.2000.2131
138. Soni S, Padwad YS. HIF-1 in cancer therapy: two decade long story of a transcription factor. Acta Oncol. (2017) 56:503–15. doi: 10.1080/0284186X.2017.1301680
139. Botusan IR, Sunkari VG, Savu O, Catrina AI, Grunler J, Lindberg S, et al. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci USA. (2008) 105:19426–31. doi: 10.1073/pnas.0805230105
Keywords: hypoxia-inducible factor 1, embryopathy, cardiomyopathy, heart development, fetal programing
Citation: Cerychova R and Pavlinkova G (2018) HIF-1, Metabolism, and Diabetes in the Embryonic and Adult Heart. Front. Endocrinol. 9:460. doi: 10.3389/fendo.2018.00460
Received: 10 June 2018; Accepted: 26 July 2018;
Published: 15 August 2018.
Edited by:
Jan Polák, Charles University, CzechiaReviewed by:
Daniela Patrizia Foti, Università degli studi Magna Græcia di Catanzaro, ItalyBiagio Arcidiacono, Università degli studi Magna Græcia di Catanzaro, Italy
Carine Michiels, Université de Namur, Belgium
Copyright © 2018 Cerychova and Pavlinkova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Pavlinkova, Z3BhdmxpbmtvdmFAaWJ0LmNhcy5jeg==