 Xiaolan Deng
Xiaolan Deng Rui Su1
Rui Su1 Jianjun Chen
Jianjun Chen- 1Department of Systems Biology and The Gehr Family Center for Leukemia Research, Beckman Research Institute of City of Hope, Monrovia, CA, United States
- 2School of Pharmacy, China Medical University, Shenyang, China
Fat mass and obesity-associated protein (FTO) single-nucleotide polymorphisms (SNPs) have been linked to increased body mass and obesity in humans by genome-wide association studies (GWAS) since 2007. Although some recent studies suggest that the obesity-related SNPs in FTO influence obesity susceptibility likely through altering the expression of the adjacent genes such as IRX3 and RPGRIP1L, rather than FTO itself, a solid link between the SNP risk genotype and the increased FTO expression in both human blood cells and fibroblasts has been reported. Moreover, multiple lines of evidence have demonstrated that FTO does play a critical role in the regulation of fat mass, adipogenesis, and body weight. Epidemiology studies also showed a strong association of FTO SNPs and overweight/obesity with increased risk of various types of cancers. As the first identified messenger RNA N6-methyladenosine (m6A) demethylase, FTO has been shown recently to play m6A-dependent roles in adipogenesis and tumorigenesis (especially in the development of leukemia and glioblastoma). Given the critical roles of FTO in cancers, the development of selective and effective inhibitors targeting FTO holds potential to treat cancers. This mini review discusses the roles and underlying molecular mechanisms of FTO in both obesity and cancers, and also summarizes recent advances in the development of FTO inhibitors.
Introduction
As the first genome-wide association studies (GWAS)-identified obesity susceptibility gene, the fat mass and obesity-associated gene (FTO) has been well known for the strong association of the multiple single-nucleotide polymorphisms (SNPs) located in its intron 1 with risk of obesity (1–10). Although there are some controversial reports regarding the association between FTO SNPs and FTO expression (11–13), mouse model studies have shown the pivotal role of FTO in the regulation of fat mass, adipogenesis, and body weight (14–20). The link between the SNP risk genotype and increased FTO expression in human fibroblasts and blood cells has also been demonstrated (21–23). Studies have demonstrated that a strong association exists between FTO SNPs and/or overweight/obesity with the increased risk of various types of cancers (24–29), implying a role of FTO in the pathogenesis of cancers. Indeed, the oncogenic role of FTO has been reported in leukemia and glioblastoma (GBM), where FTO is highly expressed (30–32). More importantly, FTO was reported as the first N6-methyladenosine (m6A) demethylase of eukaryotic messenger RNA (mRNA) (33), and the functions of FTO in adipogenesis and tumorigenesis have been linked to its m6A demethylase activity (30–32, 34). As the most abundant internal modification in eukaryotic mRNAs, m6A usually occurs at the consensus motif of RRm6ACH ([G/A/U][G>A]m6AC[U>A>C]); enriched in 3′ untranslated region (UTR), gene coding regions, and especially near stop codons (35, 36). The m6A modification is deposited by the METTL3-METTL14-WTAP methyltransferase complex (i.e., writer) (37–39) and can be removed by m6A demethylases (i.e., erasers) such as FTO and ALKBH5 (33, 40). The m6A modification functions as a post-transcriptional modulator of gene expression by decreasing or increasing mRNA stability, or promoting mRNA translation efficiency through its recognition of different m6A reader proteins (41–48). The roles of m6A modification and the associated machinery in the pathogenesis of various types of cancers have been reported recently (30–32, 48–59). This review focuses on the functions of FTO in both adipogenesis and tumorigenesis and on the underlying m6A-dependent mechanisms, along with a brief discussion of recent advance in the development of FTO inhibitors and their therapeutic potential to treat cancers.
Association of FTO With Overweight/Obesity and Its Role in Adipogenesis
Obesity and overweight populations have become a global crisis, with the numbers increasing every year in adults and children. In 2015, there were 603 million adults and 108 million children who were diagnosed obese in 195 countries, and the population suffering with obesity has increased two-fold in over 70 countries during 25 years (60). Obesity is commonly caused by inherited or behavioral factors (food intake, physical activities, etc.), and it may induce other chronic diseases: diabetes, heart disease, chronic kidney disease, bone disorders, and many types of cancer (10, 26, 60). SNPs of FTO in intron 1 was first found to be associated with human obesity in European populations in 2007 (1–3), and subsequently validated by different groups in other populations including Asians (4–6), Africans (7), Hispanics (8), and Native Americans (9, 10), demonstrating a strong association between FTO SNPs in intron 1 (rs9939609, rs17817449, rs3751812, rs1421085, rs9930506, and rs7202116) and overweight or obesity (61) (see Figure 1). People carrying FTO risk alleles typically have a high body mass index (BMI), which may be due to a higher food intake (62, 63) and diminished food satiety (64), but not related to energy expenditure (62). Meta-analysis studies (65–67) have validated and confirmed that the influence of FTO variants on obesity risk is attenuated through physical activities as well as dietary and drug-based interventions (68, 69), although the underlying mechanism remains elusive. Some recent studies have suggested that the association between FTO SNPs in intron 1 and obesity might be owing to their potential influence on expression of IRX3, IRX5, and RPGRIP1L, rather than on their expression of FTO (11–13). However, there is also compelling evidence showing that such FTO SNPs are associated with increased expression of FTO (21–23, 70, 71). Moreover, animal model studies have shown that FTO plays a critical role in regulating fat mass, adipogenesis, and total body weight (14–20). For instance, FTO-deficient mice develop postnatal growth retardation and show a reduction in both adipose tissue and lean body mass (14). Conversely, overexpression of FTO in mice develops obesity by increased food intake (15), demonstrating the pivotal role of FTO expression itself in obesity (58). Therefore, there is no doubt that there is still a robust association of the FTO expression level/function with obesity and increased body mass, though the underlying mechanism has yet to be fully elucidated.
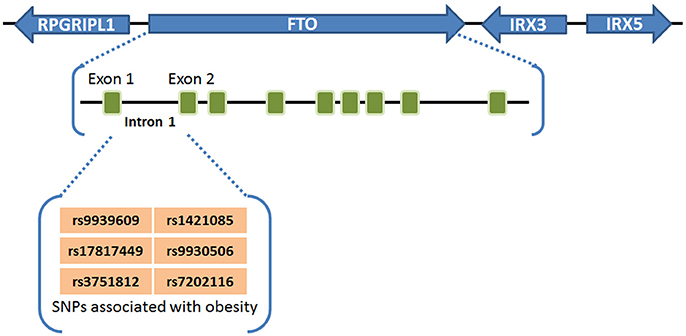
Figure 1. FTO SNPs associated with obesity. FTO SNPs in intron 1 (rs9939609, rs17817449, rs3751812, rs1421085, rs9930506, and rs7202116) have a strong association with overweight or obesity (61).
The recent discovery of FTO acting as an m6A eraser paved a novel way to reveal the molecular mechanism that links FTO with the increased susceptibility to overweight and obesity. A study in 2013 showed that the FTO obesity-risk allele (rs9939609 T/A) is associated with increased FTO expression, reduced m6A ghrelin mRNA methylation, and increased ghrelin expression (22). Ghrelin, the “hunger hormone,” is a key mediator of ingestive behavior, and its increased expression results in increased food intake and a preference for energy-dense foods, tending to lead to overweight and obesity (22, 72). A later study also reported that the FTO genotype (the AA (risk) genotype at the rs9939609 locus of FTO) impacts food intake and corticolimbic activation (73).
Excessive accumulation of adipose tissue under obese condition is a main mechanism for storage of excess energy (61). It has been reported that a positive correlation exists between the FTO level in subcutaneous adipose tissue and BMI, with a higher FTO mRNA level in adipose tissue from obese individuals than that in control populations (61, 74, 75). Zhao et al. demonstrated that FTO-mediated m6A demethylation regulates mRNA splicing and plays a critical role in the regulation of adipogenesis (34). They showed that FTO expression is inversely correlated with the m6A level during adipogenesis, and FTO depletion blocks differentiation and wild-type FTO (but not FTO mutant) restores adipogenesis; mechanistically, FTO mediates differentiation through the regulation of m6A levels around splice sites, thereby controlling the exonic splicing of the adipogenic regulator factor RUNX1T1 (34, 76). Similarly, another study also revealed that the demethylase activity of FTO is functionally required for pre-adipocyte (3T3-L1) differentiation (77). Furthermore, Merkestein et al. showed FTO regulates adipocyte differentiation in vivo, and further revealed that FTO enhances adipocyte numbers during mitotic clonal expansion at an early stage of adipogenesis (19). The compelling evidence of these studies supports FTO-mediated m6A demethylation playing a pivotal role on adipogenesis regulatory.
Association of FTO With Cancers and Its Oncogenic Role in Both Tumorigenesis and Drug Response
Epidemiology studies show that FTO SNPs (including rs9939609, rs17817449, rs8050136, rs1477196, rs6499640, rs16953002, rs11075995, and rs1121980) and overweight/obesity are strongly associated with an increased risk of various types of cancers, including breast cancer, prostate cancer, kidney cancer, endometrial cancer, pancreatic cancers, lymphoma, and leukemia (24–29). For instance, several SNPs of intron 1 of FTO (including rs7206790, rs8047395, rs9939609, and rs1477196) are all significantly associated with breast cancer risk, and rs1477196 shows the strongest association (29). Notably, SNPs outside of intron 1 of FTO could also be associated with cancer risk. For example, rs16953002 of intron 8 of FTO has been identified to be significantly associated with melanoma risk (28). It is possible that the obesity-associated SNPs lead to increased expression of FTO, which in turn contributes (at least to some extent) to an increased susceptibility to overweight and obese, as well as an increased risk of cancer development (30). Indeed, several recent studies have suggested that FTO plays an oncogenic role in various types of cancers such as leukemia, brain tumor, breast cancer, gastric cancer, endometrial carcinoma, and cervical squamous cell carcinoma (CSCC) where it is overexpressed (30–32, 78–82). Li et al. provided the first in vivo animal model study demonstrating a critical oncogenic role of FTO in cancer (30). They reported that FTO is highly expressed in certain subtypes of acute myeloid leukemias (AMLs) such as those carrying t(11q23)/MLL-rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutation (30). They further showed that forced expression of FTO significantly promoted human AML cell survival and proliferation and inhibited human AML cell differentiation and apoptosis, and forced expression of FTO significantly promoted leukemogenesis in mice (30). The opposite was true when endogenous expression of FTO was depleted (30). Subsequently, Su et al. reported that by the inhibition of FTO's oncogenic role, R-2-hydroxyglutarate (R-2HG), a previously well-recognized oncometabolite (83–90), actually exhibits a broad and intrinsic antitumor activity in AML and GBM (31). Cui et al. reported that targeting glioblastoma stem(-like) cells (GSCs) with a FTO inhibitor in mice could significantly inhibit the development of GSC-initiated tumor in vivo (32). It was also reported that the depletion of FTO expression significantly inhibited cell proliferation, migration, and invasion of human gastric cancer cell lines, and the opposite phenomenon was observed when FTO was forced expressed (80).
FTO has also been reported to affect the response of cancer cells to drug treatment. Li et al. showed that a knockdown of FTO could significantly enhance the response of human AML cells to all-trans retinoic acid (ATRA) treatment and promote ATRA-induced AML cell differentiation (30). Su et al. reported that analogous to FTO depletion, R-2HG treatment also sensitized human AML cells to standard chemotherapeutic agents such as ATRA, azacitidine (AZA), Decitabine, and Daunorubicin in vitro (31). They further showed that R-2HG treatment also sensitized human AML cells to Decitabine and Daunorubicin in vivo in immunodeficient xenotransplantation recipient mice (31). Similarly, Zhou et al. reported that FTO enhanced the resistance of CSCC cells to chemo-radiotherapy (82). Consistent with the function of FTO in drug resistance, it was reported that overexpression of FTO is a marker for poor prognosis in cancers such as gastric cancer and endometrial carcinoma (80, 81).
Mechanistically, the roles of FTO in tumorigenesis and drug response have been linked to its m6A demethylase activity. Li et al. reported that FTO negatively regulates expression of a set of tumor suppressor target genes, such as ASB2 and RARA [two genes implicated in leukemia cell proliferation and drug response (91–93)], through post-transcriptionally modulating m6A abundance of the target mRNA transcripts and thereby affecting their stability (30). Su et al. further reported that FTO also positively regulates expression of a set of oncogenic targets such as MYC and CEBPA through an m6A-dependent mechanism (31). The suppression effect of the FTO inhibitor on GSC growth/proliferation and survival is also believed to be owing to the inhibition of the m6A demethylase activity of FTO (32). In CSCC, FTO has been reported to enhance chemo-radiotherapy both in vitro and in vivo through positively regulating expression of β-catenin (CTNNB1) via an m6A-dependent mechanism (82). Collectively, evidence is emerging that FTO plays critical oncogenic roles in various types of cancers as an m6A demethylase, and post-transcriptionally regulates expression of a number of functionally important target genes through m6A-dependent mechanisms.
Identification of Small Molecule Inhibitors Targeting FTO
Since the discovery of FTO as an m6A demethylase in 2011 (33), efforts have been made to identify selective small-molecule inhibitors targeting FTO's m6A demethylase activity (94–98). FTO belongs to the AlkB family, and the crystal structure of FTO resolved in 2010 (99) shows a strong Fe (II) and α-ketoglutarate (αKG) dependent activity as a dioxygenase, at N-terminals. Chen et al. reported in 2012 that rhein, a natural product, competitively binds to an FTO active site, and exerts an inhibitory activity on FTO-dependent m6A demethylation in cells, through directly disrupting the bindings between FTO and the m6A substrate (94). In 2014, Zheng et al. developed a selective FTO inhibitor that also selectively inhibits the m6A demethylase activity of FTO and increases the m6A levels in cells (95); a later study showed that this FTO inhibitor (i.e., MO-I-500) could significantly inhibit the survival and/or colony formation of human SUM149 cells, a triple-negative inflammatory breast cancer cell line (97). Meclofenamic acid (MA), a nonsteroidal anti-inflammatory drug, was discovered to specifically inhibit FTO's m6A demethylase activity, while paring ALKBH5 (96). MA has been further proved to effectively inhibit the survival and growth of GBM cells through suppression of the m6A demethylase activity of FTO (32). In addition, Compound 12 has been developed based on a α-KG tethering strategy, which could selectively inhibit FTO over other AlkB subfamilies (including ALKBH5) and α-KG oxygenases (98). Su et al. showed that R-2HG is also an inhibitor of FTO that binds direct to FTO protein and significantly inhibits the m6A demethylase activity of FTO in a dose-dependent manner, leading to a significant increase of global m6A abundance in R-2HG-treated sensitive leukemia cells (31).
Discussion and Conclusions
A growing body of evidence suggests that FTO plays critical roles in both overweight/obesity and cancers. As the first m6A demethylase identified, FTO has been shown to regulate expression of a number of important target genes through post-transcriptionally reducing their m6A levels and thereby affecting the stability and/or splicing of target mRNAs, in turn leading to promoting adipogenesis, tumorigenesis, and drug resistance of cancer cells. Therefore, although FTO may regulate expression of distinct sets of target mRNAs in different cell types, it affects overweight/obesity and cancers likely through similar, m6A demethylase activity-dependent mechanisms (see Figure 2). The strong association between FTO SNPs or overweight/obesity with an increased risk of cancers suggests that the obesity-associated function of FTO in metabolism may also contribute to its effects in cancers (Figure 2). Indeed, the FTO gene variant related to cancer risk is unlikely independent of adiposity (100). In addition, it was reported that by targeting the PI3K/AKT signaling, FTO influences breast cancer cell energy metabolism including lactic acid, ATP, pyruvate kinase activity, and hexokinase activity (79).
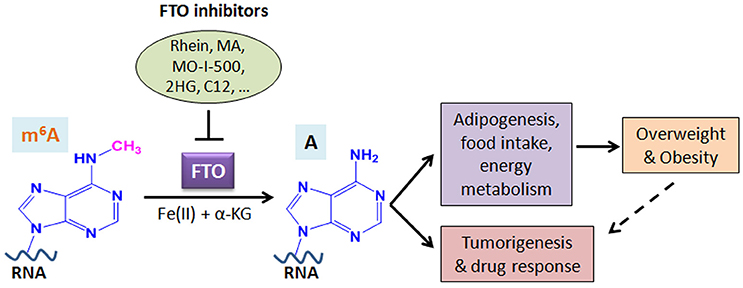
Figure 2. Schematic illustration of the roles of FTO in RNA m6A modification, overweight/obesity, and tumorigenesis/drug response. As an m6A demethylase, FTO post-transcriptionally regulates expression of its critical target genes and thereby contributes to overweight/obesity (likely through affecting adipogenesis, food intake, and energy metabolism) and cancers (including tumorigenesis and drug response). The obesity-associated function of FTO in metabolism may also contribute to cancers. Inhibition of FTO-mediated m6A demethylation by various inhibitors holds therapeutic potential to treat FTO-overexpressing cancers. MA, meclofenamic acid; 2HG, 2-hydroxyglutarate; C12, Compound 12 (98).
Given the essential role of FTO in cancer development and drug resistance, targeting FTO holds therapeutic potential in treating cancers in which FTO is overexpressed. Thus far, FTO inhibitors have been tested in vitro and in vivo, and show potent antitumor effects in treating both GBM and breast cancer (32, 97). Similarly, Su et al. showed that by targeting FTO directly, R-2HG exhibits a strong antitumor effect in both leukemia and GBM, especially when in combination with standard chemotherapeutic agents (31). These studies provide proof-of-concept evidence demonstrating that FTO is a realistic druggable target in treating cancers. In the near future, when more effective and selective inhibitors of FTO are developed, they could be applied, especially in combination with other therapeutic agents, into the clinic to treat various types of cancers. On the other hand, although FTO also plays a role in obesity, it was argued that FTO might not be a good pharmaceutical target to treat obesity, because the factors leading to obesity might be more complex (101, 102). Thus, a deeper understanding of the factors contributing to obesity could lead to the development of therapeutics targeting obesity.
Author Contributions
XD and JC drafted and revised the manuscript, while RS and SS contributed to the revision of the manuscript.
Conflict of Interest Statement
A patent has been filed by JC and RS based on their work on R-2HG/FTO.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors apologize to colleagues whose work could not be included due to space limitations. This work was supported in part by grants NO.81603149 (XD) from National Nature Science Foundation of China, as well as the National Institutes of Health (NIH) R01 Grants CA214965 (JC), CA211614 (JC), and CA178454 (JC). JC is a Leukemia & Lymphoma Society (LLS) Scholar.
Abbreviations
FTO, the fat mass and obesity-associated protein; SNP, single-nucleotide polymorphism; GWAS, genome-wide association study; mRNA, messenger RNA; m6A, N6-methyladenosine; GBM, glioblastoma; UTR, untranslated region; BMI, body mass index; CSCC, cervical squamous cell carcinoma; AML, acute myeloid leukemia; R-2HG, R-2-hydroxyglutarate; GSCs, glioblastoma stem(-like) cells; ATRA, all-trans-retinoic acid; AZA, azacitidine; αKG, α-ketoglutarate; MA, meclofenamic acid.
References
1. Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. (2007) 39:724–6. doi: 10.1038/ng2048
2. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science (2007) 316:889–94. doi: 10.1126/science.1141634
3. Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet (2007) 3:e115. doi: 10.1371/journal.pgen.0030115
4. Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban HJ, et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nature Genetics (2009) 41:527–34. doi: 10.1038/ng.357
5. Wen WQ, Cho YS, Zheng W, Dorajoo R, Kato N, Qi L, et al. Meta-analysis identifies common variants associated with body mass index in east Asians. Nat Genet. (2012) 44:307–11. doi: 10.1038/ng.1087
6. Okada Y, Kubo M, Ohmiya H, Takahashi A, Kumasaka N, Hosono N, et al. Common variants at CDKAL1 and KLF9 are associated with body mass index in east Asian populations. Nat Genet. (2012) 44:302–6. doi: 10.1038/ng.1086
7. Monda KL, Chen GK, Taylor KC, Palmer C, Edwards TL, Lange LA, et al. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat Genet (2013) 45:690–6. doi: 10.1038/ng.2608
8. Dong CH, Beecham A, Slifer S, Wang LY, McClendon M, Blanton S, et al. Genome-wide linkage and peak-wide association study of obesity-related quantitative traits in Caribbean Hispanics. Hum Genet. (2011) 129:209–19. doi: 10.1007/s00439-010-0916-2
9. Rong R, Hanson RL, Ortiz D, Wiedrich C, Kobes S, Knowler WC, et al. Association Analysis of Variation in/Near FTO, CDKAL1, SLC30A8, HHEX, EXT2, IGF2BP2, LOC387761, and CDKN2B With Type 2 diabetes and related quantitative traits in pima Indians. Diabetes (2009) 58:478–88. doi: 10.2337/db08-0877
10. Merkestein M, Sellayah D Role of FTO in adipocyte development and function: recent insights. Int J Endocrinol. (2015) 2015:521381. doi: 10.1155/2015/521381
11. Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature (2014) 507:371–5. doi: 10.1038/nature13138
12. Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. (2015) 373:895–907. doi: 10.1056/NEJMoa1502214
13. Stratigopoulos G, Martin Carli JF, O'Day DR, Wang L, Leduc CA, Lanzano P, et al. Hypomorphism for RPGRIP1L, a ciliary gene vicinal to the FTO locus, causes increased adiposity in mice. Cell Metab. (2014) 19:767–79. doi: 10.1016/j.cmet.2014.04.009
14. Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, et al. Inactivation of the Fto gene protects from obesity. Nature (2009) 458:894–8. doi: 10.1038/nature07848
15. Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. (2010) 42:1086–92. doi: 10.1038/ng.713
16. Church C, Lee S, Bagg EA, McTaggart JS, Deacon R, Gerken T, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. (2009) 5:e1000599. doi: 10.1371/journal.pgen.1000599
17. McMurray F, Church CD, Larder R, Nicholson G, Wells S, Teboul L, et al. Adult onset global loss of the fto gene alters body composition and metabolism in the mouse. PLoS Genet. (2013) 9:e1003166. doi: 10.1371/journal.pgen.1003166
18. Gao X, Shin YH, Li M, Wang F, Tong Q, Zhang P. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS ONE (2010) 5:e14005. doi: 10.1371/journal.pone.0014005
19. Merkestein M, Laber S, McMurray F, Andrew D, Sachse G, Sanderson J, et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat Commun. (2015) 6:6792. doi: 10.1038/ncomms7792
20. Ronkainen J, Mondini E, Cinti F, Cinti S, Sebert S, Savolainen MJ, et al. FTO-deficiency affects the gene and microRNA expression involved in brown adipogenesis and browning of white adipose tissue in mice. Int J Mol Sci. (2016) 17:1851. doi: 10.3390/ijms17111851
21. Berulava T, Horsthemke B. The obesity-associated SNPs in intron 1 of the FTO gene affect primary transcript levels. Eur J Hum Genet. (2010) 18:1054–56. doi: 10.1038/ejhg.2010.71
22. Karra E, O'Daly OG, Choudhury AI, Yousseif A, Millership S, Neary MT, et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Invest. (2013) 123:3539–51. doi: 10.1172/JCI44403
23. Villalobos-Comparan M, Teresa Flores-Dorantes M, Teresa Villarreal-Molina M, Rodriguez-Cruz M, Garcia-Ulloa AC, Robles L, et al. The FTO gene is associated with adulthood obesity in the Mexican population. Obesity (2008) 16:2296–301. doi: 10.1038/oby.2008.367
24. Soderberg KC, Kaprio J, Verkasalo PK, Pukkala E, Koskenvuo M, Lundqvist E, et al. Overweight, obesity and risk of haematological malignancies: a cohort study of Swedish and Finnish twins. Eur J Cancer (2009) 45:1232–8. doi: 10.1016/j.ejca.2008.11.004
25. Castillo JJ, Mull N, Reagan JL, Nemr S, Mitri J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: a meta-analysis of observational studies. Blood (2012) 119:4845–50. doi: 10.1182/blood-2011-06-362830
26. Hernandez-Caballero ME, Sierra-Ramirez JA. Single nucleotide polymorphisms of the FTO gene and cancer risk: an overview. Mol Biol Rep. (2015) 42:699–704. doi: 10.1007/s11033-014-3817-y
27. Huang X, Zhao J, Yang M, Li M, Zheng J. Association between FTO gene polymorphism (rs9939609 T/A) and cancer risk: a meta-analysis. Eur J Cancer Care (2017) 26. doi: 10.1111/ecc.12464
28. Iles MM, Law MH, Stacey SN, Han J, Fang S, Pfeiffer R, et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat Genet. (2013) 45:428–32:432e421. doi: 10.1038/ng.2571
29. Kaklamani V, Yi N, Sadim M, Siziopikou K, Zhang K, Xu Y, et al. The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med Genet (2011) 12:52. doi: 10.1186/1471-2350-12-52
30. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell (2017) 31:127–41. doi: 10.1016/j.ccell.2016.11.017
31. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell (2018) 172:90–105 e123. doi: 10.1016/j.cell.2017.11.031
32. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. (2017) 18:2622–34. doi: 10.1016/j.celrep.2017.02.059
33. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
34. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. (2014) 24:1403–19. doi: 10.1038/cr.2014.151
35. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell (2017) 169:1187–200. doi: 10.1016/j.cell.2017.05.045
36. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev. Mol Cell Biol. (2017) 18:31–42. doi: 10.1038/nrm.2016.132
37. Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA (1997) 3:1233–47.
38. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–95. doi: 10.1038/nchembio.1432
39. Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. (2014) 16:191–8. doi: 10.1038/ncb2902
40. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
41. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature (2014) 505:117–20. doi: 10.1038/nature12730
42. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
43. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. (2017) 27:315–28. doi: 10.1038/cr.2017.15
44. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A Reader YTHDC1 regulates mRNA Splicing. Mol Cell (2016) 61:507–19. doi: 10.1016/j.molcel.2016.01.012
45. Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. (2017) 27:444–7. doi: 10.1038/cr.2017.10
46. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. (2017) 27:1115–27. doi: 10.1038/cr.2017.99
47. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 Mediates Nuclear Export of N6-methyladenosine methylated mRNAs. Elife (2017) 6:31311. doi: 10.7554/eLife.31311
48. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
49. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell (2018) 22:191–205 e199. doi: 10.1016/j.stem.2017.11.016
50. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. (2017) 23:1369–76. doi: 10.1038/nm.4416
51. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature (2017) 552:126–31. doi: 10.1038/nature24678
52. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A Methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell (2016) 62:335–45. doi: 10.1016/j.molcel.2016.03.021
53. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m6A Demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell (2017) 31:591–606 e596. doi: 10.1016/j.ccell.2017.02.013
54. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. (2016) 113:E2047–56. doi: 10.1073/pnas.1602883113
55. Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene (2018) 37:522–33. doi: 10.1038/onc.2017.351
56. Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology (2017) 65:529–43. doi: 10.1002/hep.28885
57. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase METTL3 promotes liver cancer progression through YTHDF2 dependent post-transcriptional silencing of SOCS2. Hepatology (2017) 67:2254–70. doi: 10.1002/hep.29683
58. Deng X, Su R, Feng X, Wei M, Chen J. Role of N(6)-methyladenosine modification in cancer. Curr Opin Genet Dev. (2017) 48:1–7.
59. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. (2018) 28:507–17. doi: 10.1038/s41422-018-0034-6
60. GBD 2015 Obesity Collaborators, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 Countries over 25 Years. N Engl J Med. (2017) 377:13–27. doi: 10.1056/NEJMoa1614362
61. Zhao X, Yang Y, Sun BF, Zhao YL, Yang YG. FTO and obesity: mechanisms of association. Curr Diab Rep. (2014) 14:486. doi: 10.1007/s11892-014-0486-0
62. Haupt A, Thamer C, Staiger H, Tschritter O, Kirchhoff K, Machicao F, et al. Variation in the FTO gene influences food intake but not energy expenditure. Exp Clin Endocrinol Diab. (2009) 117:194–7. doi: 10.1055/s-0028-1087176
63. Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CN. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med (2008) 359:2558–66. doi: 10.1056/NEJMoa0803839
64. Wardle J, Carnell S, Haworth CM, Farooqi IS, O'Rahilly S, Plomin R. Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab. (2008) 93:3640–3. doi: 10.1210/jc.2008-0472
65. Graff M, Scott RA, Justice AE, Young KL, Feitosa MF, Barata L, et al. Genome-wide physical activity interactions in adiposity - A meta-analysis of 200,452 adults. PLoS Genet (2017) 13:e1006528. doi: 10.1371/journal.pgen.1006528
66. Graff M, Scott RA, Justice AE, Young KL, Feitosa MF, Barata L, et al. Correction: genome-wide physical activity interactions in adiposity - A meta-analysis of 200,452 adults. PLoS Genet. (2017) 13:e1006972. doi: 10.1371/journal.pgen.1006972
67. Livingstone KM, Celis-Morales C, Papandonatos GD, Erar B, Florez JC, Jablonski KA, et al. FTO genotype and weight loss: systematic review and meta-analysis of 9563 individual participant data from eight randomised controlled trials. BMJ (2016) 354:i4707. doi: 10.1136/bmj.i4707
68. Kilpelainen TO, Qi L, Brage S, Sharp SJ, Sonestedt E, Demerath E, et al. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med. (2011) 8:e1001116. doi: 10.1371/journal.pmed.1001116
69. Li S, Zhao JH, Luan J, Ekelund U, Luben RN, Khaw KT, et al. Physical activity attenuates the genetic predisposition to obesity in 20,000 men and women from EPIC-Norfolk prospective population study. PLoS Med. (2010) 7:100332. doi: 10.1371/journal.pmed.1000332
70. Yang J, Loos RJ, Powell JE, Medland SE, Speliotes EK, Chasman DI, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature (2012) 490:267–72. doi: 10.1038/nature11401
71. Melnik BC. Milk: an epigenetic amplifier of FTO-mediated transcription? Implications for Western diseases. J Transl Med. (2015) 13:385. doi: 10.1186/s12967-015-0746-z
72. Eissing L. Metabolism: FTO-associated obesity risk is linked to brain food responses via modulation of ghrelin levels. Nat Rev Endocrinol (2013) 9:564. doi: 10.1038/nrendo.2013.155
73. Melhorn SJ, Askren MK, Chung WK, Kratz M, Bosch TA, Tyagi V, et al. FTO genotype impacts food intake and corticolimbic activation. Am J Clin Nutr. (2018) 107:145–154. doi: 10.1093/ajcn/nqx029
74. Tews D, Fischer-Posovszky P, Wabitsch M. Regulation of FTO and FTM expression during human preadipocyte differentiation. Horm Metab Res. (2011) 43:17–21. doi: 10.1055/s-0030-1265130
75. Lappalainen T, Kolehmainen M, Schwab U, Pulkkinen L, de Mello VD, Vaittinen M, et al. Gene expression of FTO in human subcutaneous adipose tissue, peripheral blood mononuclear cells and adipocyte cell line. J Nutrigenet Nutrigen. (2010) 3:37–45. doi: 10.1159/000320732
76. Ben-Haim MS, Moshitch-Moshkovitz S, Rechavi G. FTO: linking m6A demethylation to adipogenesis. Cell Res. (2015) 25:3–4. doi: 10.1038/cr.2014.162
77. Zhang MZ, Zhang Y, Ma J, Guo FM, Cao Q, Zhang Y, et al. The demethylase activity of FTO (fat mass and obesity associated protein) is required for preadipocyte differentiation. PLoS ONE (2015) 10:133788. doi: 10.1371/journal.pone.0133788
78. Tan A, Dang Y, Chen G, Mo Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int J Clin Exp Pathol. (2015) 8:13405–10.
79. Liu Y, Wang R, Zhang L, Li J, Lou K, Shi B. The lipid metabolism gene FTO influences breast cancer cell energy metabolism via the PI3K/AKT signaling pathway. Oncol Lett. (2017) 13:4685–90. doi: 10.3892/ol.2017.6038
80. Xu D, Shao W, Jiang Y, Wang X, Liu Y, Liu X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol Rep. (2017) 38:2285–92. doi: 10.3892/or.2017.5904
81. Zhu Y, Shen J, Gao L, Feng Y. Estrogen promotes fat mass and obesity-associated protein nuclear localization and enhances endometrial cancer cell proliferation via the mTOR signaling pathway. Oncol Rep. (2016) 35:2391–97. doi: 10.3892/or.2016.4613
82. Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. (2018) 57:590–7. doi: 10.1002/mc.22782
83. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science (2013) 340:626–30. doi: 10.1126/science.1236062
84. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science (2013) 340:622–6. doi: 10.1126/science.1234769
85. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
86. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science (2009) 324:261–5. doi: 10.1126/science.1170944
87. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
88. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483:474–8. doi: 10.1038/nature10860
89. Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. (2015) 11:878–86. doi: 10.1038/nchembio.1930
90. Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science (2013) 339:1621–5. doi: 10.1126/science.1231677
91. Kohroki J, Fujita S, Itoh N, Yamada Y, Imai H, Yumoto N, et al. ATRA-regulated Asb-2 gene induced in differentiation of HL-60 leukemia cells. FEBS Lett. (2001) 505:223–8. doi: 10.1016/S0014-5793(01)02829-0
92. Guibal FC, Moog-Lutz C, Smolewski P, Di Gioia Y, Darzynkiewicz Z, Lutz PG, et al. ASB-2 inhibits growth and promotes commitment in myeloid leukemia cells. J Biol Chem. (2002) 277:218–24. doi: 10.1074/jbc.M108476200
93. Glasow A, Prodromou N, Xu K, von Lindern M, Zelent A. Retinoids and myelomonocytic growth factors cooperatively activate RARA and induce human myeloid leukemia cell differentiation via MAP kinase pathways. Blood (2005) 105:341–9. doi: 10.1182/blood-2004-03-1074
94. Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. (2012) 134:17963–71. doi: 10.1021/ja3064149
95. Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME, et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. (2014) 5:658–65. doi: 10.1021/cn500042t
96. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. (2015) 43:373–84. doi: 10.1093/nar/gku1276
97. Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS ONE (2016) 11:e0159072. doi: 10.1371/journal.pone.0159072
98. Toh JDW, Sun L, Lau LZM, Tan J, Low JJA, Tang CWQ, et al. A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N(6)-methyladenosine demethylase FTO. Chem Sci. (2015) 6:112–22. doi: 10.1039/C4SC02554G
99. Han Z, Niu T, Chang J, Lei X, Zhao M, Wang Q, et al. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature (2010) 464:1205–9. doi: 10.1038/nature08921
100. Kang Y, Liu F, Liu Y. Is FTO gene variant related to cancer risk independently of adiposity? An updated meta-analysis of 129,467 cases and 290,633 controls. Oncotarget (2017) 8:50987–96. doi: 10.18632/oncotarget.16446
101. Loos RJ, Yeo GS. The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol. (2014) 10:51–61. doi: 10.1038/nrendo.2013.227
Keywords: fat mass and obesity-associated protein (FTO), obesity, cancer, mRNA N6-methyladenosine (m6A), m6A demethylase, AML, GBM, FTO inhibitors
Citation: Deng X, Su R, Stanford S and Chen J (2018) Critical Enzymatic Functions of FTO in Obesity and Cancer. Front. Endocrinol. 9:396. doi: 10.3389/fendo.2018.00396
Received: 04 May 2018; Accepted: 27 June 2018;
Published: 30 July 2018.
Edited by:
Che-Pei Kung, Washington University in St. Louis, United StatesReviewed by:
Eric Kalkhoven, Utrecht University, NetherlandsLuca De Toni, Università degli Studi di Padova, Italy
Copyright © 2018 Deng, Su, Stanford and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianjun Chen, amlhbmNoZW5AY29oLm9yZw==