 Mattia Barbot
Mattia Barbot Filippo Ceccato
Filippo Ceccato
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 05 June 2018
Sec. Pituitary Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00284
This article is part of the Research TopicDiabetes Secondary to Pituitary DiseasesView all 7 articles
Associated with important comorbidities that significantly reduce patients’ overall wellbeing and life expectancy, Cushing’s disease (CD) is the most common cause of endogenous hypercortisolism. Glucocorticoid excess can lead to diabetes, and although its prevalence is probably underestimated, up to 50% of patients with CD have varying degrees of altered glucose metabolism. Fasting glycemia may nevertheless be normal in some patients in whom glucocorticoid excess leads primarily to higher postprandial glucose levels. An oral glucose tolerance test should thus be performed in all CD patients to identify glucose metabolism abnormalities. Since diabetes mellitus (DM) is a consequence of cortisol excess, treating CD also serves to alleviate impaired glucose metabolism. Although transsphenoidal pituitary surgery remains the first-line treatment for CD, it is not always effective and other treatment strategies may be necessary. This work examines the main features of DM secondary to CD and focuses on antidiabetic drugs and how cortisol-lowering medication affects glucose metabolism.
Cushing’s disease (CD) is a rare pathology characterized by uncontrolled ACTH secretion from a pituitary adrenocorticotroph adenoma that leads to an increase in cortisol production by the adrenal glands (1). It is a serious condition characterized by metabolic derangements that may include visceral adiposity, hepatic steatosis, dyslipidaemia, and diabetes mellitus (DM) (2, 3). The prevalence of DM in CD patients is thought to fall between 20 and 45%, although the figure may be underestimated, as an oral glucose tolerance test may not be performed when a patient’s fasting glycemia is normal (4). Approximately 10–30% of patients have impaired glucose tolerance, and the overall prevalence of glucose metabolism impairments reaches nearly 70% of cases (4). No gender-related differences in prevalence have been noted (5). Generally speaking, the severity of hypercortisolism is correlated with insulin resistance and DM (6), although the correlation has not always been confirmed (7, 8). This discrepancy might depend on the wide inter-individual susceptibility to glucocorticoids. Age, genetic predisposition, and lifestyle variables combined with the duration and degree of hypercortisolism may all strongly contribute to glucose tolerance impairment in CD patients (9).
Cortisol, which is a steroid hormone, regulates a wide range of body processes, but it displays its main effect after food intake. It seems to contribute to glucose intolerance and to reduce insulin sensitivity (10). In the liver, chronic hypercortisolism impairs fasting and postprandial glucose (Figure 1). Cortisol exacerbates gluconeogenesis and hepatic glucose output through both direct and indirect effects (4). It regulates glucocorticoid-responsive target genes by upregulating key gluconeogenic enzymes such as phosphoenolpyruvate carboxykinase and glucose-6-phosphatase (4). Chronic glucocorticoid exposure also induces selective insulin resistance that impedes the inhibitory effect of insulin on hepatic glucose output (11). Skeletal muscles account for 70–80% of the body’s use of glucose. CD increases the rate of proteolysis and of muscle mass loss (12) with consequent decreased muscle insulin responsiveness and impaired glucose uptake (13). Abdominal obesity is closely associated with metabolic syndrome and CD; adipocyte-derived lipids in visceral adipose tissue can contribute to the onset of peripheral insulin resistance and DM by promoting altered insulin signaling in adipocytes, increased lipolysis, aberrant adipokine secretion, and low-grade inflammation (4). Although human studies have uncovered normal incretin secretion and a reduction in its insulin-releasing effects on β-cells, the incretin system seems to be affected, in animals, by hypercortisolemia since a blunted GLP-1 secretion has been observed in response to glucocorticoid administration (14, 15). Cortisol is necessary for the normal development of the adrenal medulla that is specialized in the synthesis, storage, and secretion of catecholamines from chromaffin cells; its excess enhances the adrenergic-mediated increase in gluconeogenesis (16). Cortisol excess also has an important impact on GH/IGF-1 leading to an increase in visceral fat and insulin resistance (17, 18). Bone has also been found to be involved in glucose homeostasis (19); prolonged glucocorticoid exposure causes a reduction in circulating osteocalcin which in turn can enhance insulin resistance (20). Cortisol’s biological effects can be modulated in target tissues by the activity of 11β-hydroxysteroid dehydrogenase (11βHSD) enzymes; isoform 1 converts cortisone into cortisol, amplifying its action in the liver and adipose tissue where it is primarily expressed (21). The overexpression of 11βHSD1 in adipose tissue is correlated with the development of insulin resistance and obesity (22). Finally, glucocorticoid receptor polymorphisms may also play a role in the development of metabolic complications (23). The A3669G polymorphism has been found to play a protective role in CD (24) by producing glucocorticoid resistance that increases the expression and stabilization of the dominant-negative GR-β splice variant (24, 25).
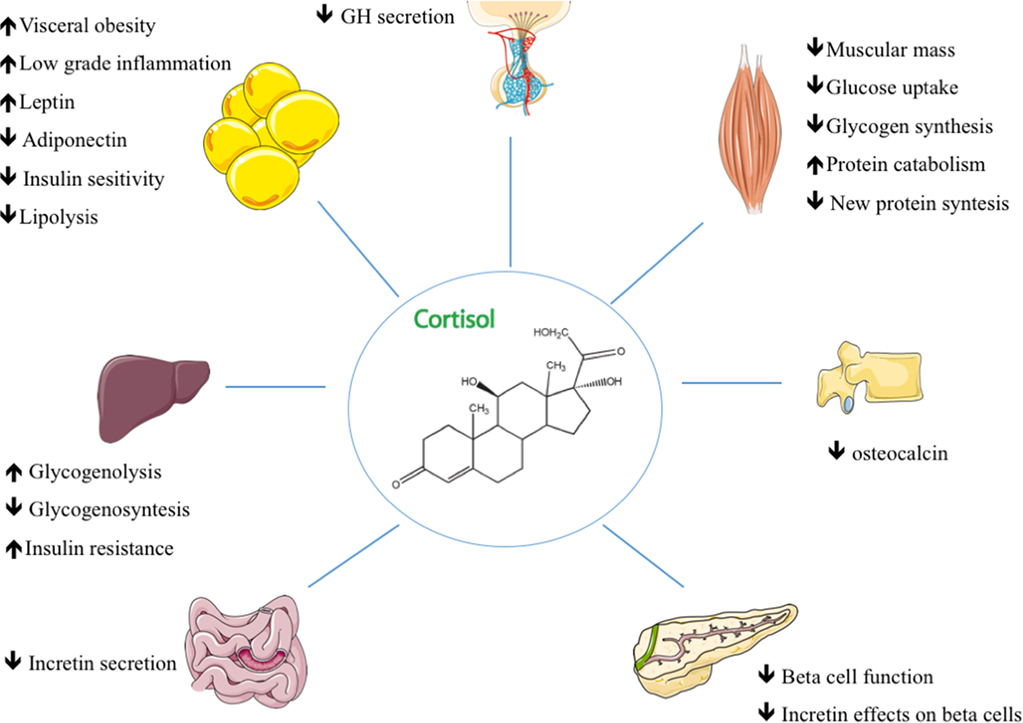
Figure 1. The main mechanisms of action in glucocorticoid-induced diabetes and their effects on target tissues in Cushing’s disease.
As diabetes is a consequence of cortisol excess, treating the underlying disease is unquestionably indispensable in these patients. Despite increasing interest in medical therapy, the first-line approach still remains transsphenoidal surgery (26) which provides, when it is performed by an experienced neurosurgeon, a mean remission rate of 77.8% (3, 27). Normalized cortisol levels after surgery is generally followed by an improvement in the patient’s glucose metabolism, but insulin resistance and cardiovascular risks may persist (28), particularly in patients with a genetic predisposition and/or persistent visceral adiposity (9). This means that specific, although usually at lower doses, hypoglycemic treatment may be required even after remission following surgery (4).
Although correcting cortisol hypersecretion leads to an improvement in glucose homeostasis, patients with CD and DM need to achieve glycemic control regardless of normalized cortisol levels. Treating diabetes in patients with CD does not essentially differ from treating it in ordinary circumstances (29). There are nevertheless some pathophysiological features that should be considered. Physical activity can be recommended in CD patients although resistance exercises should be limited in patients with glucocorticoid-induced myopathy (30). Metformin remains the first-line therapy of hyperglycemia in CD (31). While metformin lowers fasting plasma glucose concentrations by reducing hepatic glucose production (32) without causing hypoglycemia or weight gain (33), it may have undesired gastrointestinal side effects, that can be worsened by the concomitant use of pasireotide (34). Extended-release formulas can improve gastrointestinal tolerability (35). Acarbose inhibits the enzymatic cleavage of complex carbohydrates, delaying their absorption and thereby reducing postprandial glycemic excursions (36). Although it does not seem to have negative effects on body weight, it often produces gastrointestinal side effects (37). Peroxisome proliferator-activated receptor-γ (PPARγ) agonists interact with the nuclear receptor PPARγ ameliorating insulin resistance and improving hepatic and muscle insulin sensitivity (38). They may also potentiate the effects of metformin (39) on glycemic control and trigger anti-proliferative effects on tumor cells. Pioglitazone treatment has not, however, been found to reduce ACTH or cortisol levels in CD (40, 41) and may cause weight gain and/or edema which may worsen the CD patient’s clinical condition (4, 42, 43).
Sulfonylureas and glinides stimulate insulin secretion in a glucose-independent manner triggering both the immediate and sustained release of insulin from intracellular granules (44). They are rarely used independently, but are mainly recommended for short-term periods to manage postprandial glycemia (45). Other drugs that produce good postprandial glucose control are dipeptidyl peptidase-4 inhibitors, which do not seem to affect weight (46), and since they are well tolerated, they are usually associated with metformin in cases of treatment intensification (47). GLP-1 receptor agonists act by enhancing glucose-induced biosynthesis and insulin secretion, inhibiting glucagon secretion, delaying gastric emptying, and reducing appetite (48, 49). Other positive effects such as weight loss and blood pressure reduction have also been noted (50). Incretin-based therapy may be the best treatment option for CD given its rapid action and positive effect on postprandial glycemia (51). Sodium-glucose co-transporter 2 inhibitors are a new class of diabetic drugs (52) that cause a reduction in body weight and blood pressure (53). In the light of reports linking them to a higher risk of urinary and genital infections (54), the decision to use them should be carefully evaluated in CD patients who show a high risk of infections and systemic dissemination (55). Another possible, although rare, side effect that has been reported in diabetic patients is the development of euglycaemic ketoacidosis probably caused by an increase in glucagon levels and consequently enhanced ketogenesis (56). But given their proven capacity to improve glycemic control and their association to a low rate of cardiovascular events (57), their use should be considered in appropriate cases. Insulin may become necessary in patients in whom uncontrolled diabetes persists (29). Basal-bolus insulin therapy including basal, prandial, and a supplemental correction-factor insulin is considered the most flexible option for patients with hypercortisolism (58).
Pasireotide, a multi-somatostatin receptor ligand that is able to bind four of the five known SSTR subtypes (SSTRs1–3 and SSTR5) with a 40-fold greater binding affinity for SSTR5 with respect to octreotide (59, 60), was the first agent to be approved for the treatment of adult CD patients. Pasireotide has the same security profile of first-generation somatostatin analogs although it is associated with a relatively high incidence of hyperglycemia (61, 62) due to the expression of SSTR5 even in pancreatic β-cells (63). When the pathophysiology of pasireotide-induced hyperglycemia was investigated in healthy volunteers (64, 65), short-term administration was found to be followed by an increase in blood glucose levels associated with reduced insulin secretion (66) and no significant alteration in glucagon output (60). In addition, pasireotide can potentially reduce the secretion of other pituitary hormones (67). Although a decrease in the counterregulatory action of GH can reduce insulin resistance and hepatic gluconeogenesis, its deficiency can further increase metabolic complications as a result of body composition changes (68).
Clinical practice has shown that glucose alterations are more common and serious at the time therapy is begun but tend to stabilize over time. In some cases, an improvement in glucose control has been reported following long-term treatment, probably as a result of an improvement in insulin resistance (69). Interestingly, pasireotide-induced hyperglycemia seems to be independent of the doses that are assumed (65). Besides reducing insulin secretion, pasireotide also has a significant inhibitory effect on GLP-1 and on glucose-dependent insulinotropic peptide secretion (65).
In a phase II, proof-of-concept, open-label multicenter study, 39 patients with CD were treated with 600 µg twice daily pasireotide for 15 days. There was a significant reduction in urinary free cortisol (UFC) values in 2/3 of the patients and complete normalization in 17%. Hyperglycemia was found in 14 patients; 5 of whom with a history of DM or impaired fasting glucose. Hyperglycemia, which was mild in most of the cases, was managed with diet and oral hypoglycemic agents (61).
The effectiveness of this treatment was confirmed by a phase III clinical trial in which 15 and 26% of patients achieved UFC normalization after 6 months of treatment with, respectively, 600 or 900 µg of pasireotide twice daily (62). Hyperglycemia was reported in up to 73% of cases; the rate was significantly higher than that observed for first-generation SST analogs. Treatment needed to be suspended because of uncontrolled DM in 6% of the patients (62). A rise in HbA1c levels from 5.8 to 7.3% was recorded after 12 months of pasireotide. Blood glucose and HbA1c levels increased rapidly after pasireotide treatment was begun despite a decline in cortisol levels (62).
A recent phase III study examining a long-acting formulation of pasireotide confirmed the advantage of its convenient once-monthly administration schedule. In addition, long-acting pasireotide normalized UFC in 40% of the patients and its safety profile was found to be similar to that of the subcutaneous formulation (67). Mean fasting plasma glucose and HbA1c concentrations increased within 1–2 months and antidiabetic medication needed to be initiated or adjusted in approximately in 50% of cases (67). It is therefore crucial to carefully check blood glucose concentrations in patients initiating pasireotide and to begin glucose-lowering therapy promptly whenever it proves necessary (47).
Since dopamine receptor agonist is able to reduce hypothalamic stimulation that increases during liver gluconeogenesis, lipid synthesis, and insulin resistance (70, 71) bromocriptine has been used to treat metabolic disorders associated with insulin resistance and obesity (72). When cabergoline, a potent dopamine agonist was tested in patients with type 2 diabetes, its ability to reduce both fasting glycemia and HbA1c was confirmed (71). Cabergoline may also improve glycemic control through weight loss, although this effect has not been consistently found by all studies (73). Since dopamine D2 receptor expression was found in almost 80% of adenocorticotroph cells, cabergoline could be an efficacious treatment for CD (74); some studies have reported that cabergoline is effective in controlling hypercortisolism in 30–45% of patients (75–77). Diabetes and glucose intolerance ameliorated in 60 and 46%, respectively, irrespective of cortisol levels (75). A recent large retrospective multicenter study on 62 patients confirmed already published results reporting long-term cortisol normalization in 20–25% of cases and a significant improvement in glycemic control in 40% of cases (78).
After it was shown to be involved in reducing ACTH secretion and tumor growth in in vitro and animal models via inhibition of POMC expression in corticotroph tumors, RA, a nuclear receptor ligand, has been considered another potential option for CD treatment (79). The first clinical study examining its effect on seven CD patients demonstrated a UFC reduction ≥50% in five out of seven patients after 6 months of treatment and a complete response in three of the cases. There was a significant improvement in glycemia and HbA1c (decreased by 0.4–1.2%) in all five patients with DM at baseline (80). The 13-cis isomer of RA was recently examined by an open-label trial; UFC normalization was reached in 4 out of the 16 patients at 12 months, with up to a 52% reduction in UFC in the rest. There was also an overall significant reduction in fasting glycemia (81). RA’s ameliorative effect on glucose metabolism can be attributed to both its cortisol-lowering action and its direct effects. In fact, retinol active metabolites are able to stimulate insulin secretion, enhance mRNA expression of glucose transporter GLUT2, and promote lipolysis in adipocytes by activating PPARγ (82, 83) (Table 1).
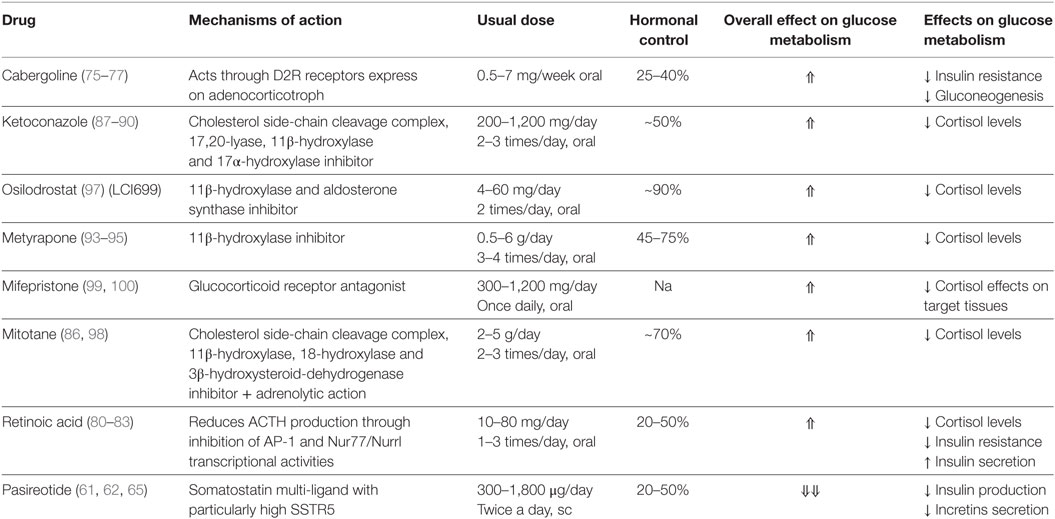
Table 1. Medications available to treat Cushing’s disease and their effects on glucose metabolism.
Ketoconazole is an imidazole derivative that reduces adrenal steroid production by inhibiting numerous steroidogenic enzymes (84, 85). Although it has been used for decades, no perspective studies are as yet available (86). At doses of 200–1,200 mg/day, it is able to improve glucose metabolism in CD patients (87–90). Ketoconazole enantiomer (DIO-902) has been found to be effective at lowering HbA1c, fasting glucose, total cholesterol levels, and LDL cholesterol levels (91).
A French study retrospectively assessed 38 CD patients receiving ketoconazole (200–1,200 mg/day) for a median of 23 months; the 5 patients who had DM all achieved cortisol regulation and an improvement in metabolic control (88). Another retrospective study examining 62 CD patients receiving steroidogenesis inhibitors as pre-surgical treatment (ketoconazole, metyrapone, or their combination) reported that HbA1C levels fell in those patients whose cortisol levels were entirely or partially controlled, but it became necessary to gradually increase insulin or prescribe oral antidiabetic drugs for the non-controlled patients (89).
A large retrospective multicenter study by Castinetti et al. reviewing data on 200 CD patients treated with ketoconazole monotherapy; at baseline, 31.8% of the patients had DM. Glycemic control improved in more than half of the diabetic patients after ketoconazole therapy (90).
Metyrapone inhibits the final step in cortisol synthesis, namely the conversion of 11-deoxycortisol into cortisol by 11β-hydroxylase (86). Due to its rapid action, it is particularly suitable to achieve cortisol control within a few days’ time and thus to improve glucose metabolism over a short time period (92). Jeffcoate et al. evaluated the efficacy of metyrapone at doses ranging from 500 to 4,000 mg/day in 13 CD patients after a mean of 21 months. Of the seven who had an abnormal glucose tolerance at the baseline, five showed improvement after 3 months of treatment (93). Another study reported a significant improvement in glucose metabolism in more than 80% of CD patients (94). Daniel et al. recently conducted a large retrospective study on metyrapone in 195 CD patients, 35% of whom were diabetic. The agent’s effects on glucose metabolism were not analyzed by the study, but hypoglycemia was reported in three patients taking antidiabetic drugs following an improvement in hypercortisolism (95).
Osilodrostat (LCI699), an adrenal steriodogenesis inhibitor developed for the treatment of CD, is currently undergoing investigation. Its mechanism of action is similar to that of metyrapone, it is a potent inhibitor of 11β-hydroxylase and aldosterone synthase (86). A 10-week, proof-of-concept study examining 12 CD patients receiving osilodrostat, reported no important changes in insulin levels, although a nearly significant decrease in HbA1c was noted (96).
A subsequent 22-week, multicenter, prospective, open-label, phase II study examined the agent’s effect on 19 CD patients. Although the proportion of responders reached 90% as far as glucose metabolism was concerned, fasting plasma glucose and HbA1c levels fell from baseline to week 22; the greatest reduction was noted in patients with previous DM (97).
Given its strong adrenolytic effect, mitotane has been widely used as an adjuvant treatment for adrenal carcinoma (26). Since its activity is long lasting, the medication is also prescribed to patients with CD (86). Baudry et al. retrospectively reviewed the clinical charts of 76 patients from a single center who were treated with mitotane. Remission with a statistically significant improvement in both fasting and postprandial serum glucose levels was achieved in 48 (72%) of those receiving the drug for at least 6 months (98).
Mifepristone is a glucocorticoid receptor antagonist that was approved by the U.S. Food and Drug Administration in 2012 for the treatment of hyperglycemia in CS patients who are not candidates for surgery (4). An open-label, multicenter, prospective, 6-month study was conducted on 50 patients with endogenous CS (43 CD) who were refractory to other therapies. Study results showed that 29 had DM or IGT. After 24 weeks of mifepristone treatment, fasting plasma glucose and HbA1c decreased, respectively, from 8.3 ± 4.1 to 5.8 ± 2.1 mmol/L and from 7.43 ± 1.52 to 6.29 ± 0.99%, leading to a reduction in the number of antidiabetic medications the patients were taking (99). A large percentage of the patients showed improved insulin resistance, with the greatest amelioration taking place during the first 6 weeks of treatment, suggesting that the early rapid improvement was linked to the direct effects of glucocorticoid blockade, while the later one depended on weight loss (100).
As no single drug has shown complete efficacy, combining drugs with additive, synergistic actions, is a strategy that has been used to increase the possibility of controlling hypercortisolism using lower doses (101) and more effectively managing glucose metabolism. A small prospective trial examining 14 patients with CD found that combining cabergoline and ketoconazole was more efficacious than using either of the two drugs alone, not only with regard to hormonal control but also as far as glucose metabolism was concerned (102). Other combinations such as the association of pasireotide-cabergoline and ketoconazole have also been utilized. One small trial found a complete response in 88% of patients receiving pasireotide subcutaneously and cabergoline. Glucose homeostasis alterations, which have been linked to pasireotide treatment, were, nevertheless, common (glycated hemoglobin level, 5.8 ± 0.2 to 6.7 ± 0.3%). Since another drug was being used, it was possible to use lower doses of pasireotide and thus to reduce its detrimental effect on glycemia (103). A metyrapone, ketoconazole, and mitotane combination was utilized in 11 patients with severe CD (4 cases of CD) as an alternative to rescue adrenalectomy. All the patients showed a rapid clinical improvement; five of the eight diabetic patients showed improved glycemic control (104).
Early diagnosis can reduce disease-related complications and improve life expectancy in CD patients, and DM is one of its most frequent although underestimated complications. Appropriate treatment is based on antidiabetic medication and, first and foremost, treating the underlying disease. Transsphenoidal surgery remains the most effective treatment to control both cortisol and glucose metabolism as it can guarantee long-term remission in a high percentage of patients, but other options need to be considered when it is ineffective or unfeasible. With the exception of pasireotide, all cortisol-lowering medications have been shown to be effective in reducing to some degree the severity of hyperglycemia. Due to its action on peripheral insulin sensitivity, which is the primary mechanism responsible for glucose intolerance in CD, metformin represents the mainstay of antidiabetic treatment. When treatment intensification becomes necessary, incretin-based therapies may represent a useful option. Beyond glucocorticoid excess, other factors implicated in DM development such as age, genetic predisposition, and lifestyle variables combined with the duration and degree of hypercortisolism, may contribute to impaired glucose tolerance.
MB: literature revision and drafting of the article. FC: drafting of the article. CS: critical revision of the article and final approval.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with the authors.
1. Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet (2015) 386(9996):913–27. doi:10.1016/S0140-6736(14)61375-1
2. van Raalte DH, Ouwens DM, Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: towards expansion of therapeutic options? Eur J Clin Invest (2009) 39(2):81–93. doi:10.1111/j.1365-2362.2008.02067.x
3. Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BM, Colao A. Complications of Cushing’s syndrome: state of the art. Lancet Diabetes Endocrinol (2016) 4(7):611–29. doi:10.1016/S2213-8587(16)00086-3
4. Scaroni C, Zilio M, Foti M, Boscaro M. Glucose metabolism abnormalities in Cushing syndrome: from molecular basis to clinical management. Endocr Rev (2017) 38(3):189–219. doi:10.1210/er.2016-1105
5. Zilio M, Barbot M, Ceccato F, Camozzi V, Bilora F, Casonato A, et al. Diagnosis and complications of Cushing’s disease: gender-related differences. Clin Endocrinol (Oxf) (2014) 80(3):403–10. doi:10.1111/cen.12299
6. Mancini T, Kola B, Mantero F, Boscaro M, Arnaldi G. High cardiovascular risk in patients with Cushing’s syndrome according to 1999 WHO/ISH guidelines. Clin Endocrinol (Oxf) (2004) 61(6):768–77. doi:10.1111/j.1365-2265.2004.02168.x
7. Biering H, Knappe G, Gerl H, Lochs H. Prevalence of diabetes in acromegaly and Cushing syndrome. Acta Med Austriaca (2000) 27(1):27–31. doi:10.1046/j.1563-2571.2000.00106.x
8. Pecori Giraldi F, Moro M, Cavagnini F; Study Group on the Hypothalamo-Pituitary-Adrenal Axis of the Italian Society of Endocrinology. Gender-related differences in the presentation and course of Cushing’s disease. J Clin Endocrinol Metab (2003) 88(4):1554–8. doi:10.1210/jc.2002-021518
9. Giordano C, Guarnotta V, Pivonello R, Amato MC, Simeoli C, Ciresi A, et al. Is diabetes in Cushing’s syndrome only a consequence of hypercortisolism? Eur J Endocrinol (2013) 170(2):311–9. doi:10.1530/EJE-13-0754
10. Suh S, Park MK. Glucocorticoid-induced diabetes mellitus: an important but overlooked problem. Endocrinol Metab (Seoul) (2017) 32(2):180–9. doi:10.3803/EnM.2017.32.2.180
11. Pivonello R, De Leo M, Vitale P, Cozzolino A, Simeoli C, De Martino MC, et al. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology (2010) 92(Suppl 1):77–81. doi:10.1159/000314319
12. Bodine SC, Furlow JD. Glucocorticoids and skeletal muscle. Adv Exp Med Biol (2015) 872:145–76. doi:10.1007/978-1-4939-2895-8_7
13. Kuo T, Harris CA, Wang JC. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol (2013) 380:79–88. doi:10.1016/j.mce.2013.03.003
14. Kappe C, Fransson L, Wolbert P, Ortsater H. Glucocorticoids suppress GLP-1 secretion: possible contribution to their diabetogenic effects. Clin Sci (Lond) (2015) 129:405–14. doi:10.1042/CS20140719
15. Eriksen M, Jensen DH, Tribler S, Holst JJ, Madsbad S, Krarup T. Reduction of insulinotropic properties of GLP-1 and GIP after glucocorticoid-induced insulin resistance. Diabetologia (2015) 58:920–8. doi:10.1007/s00125-015-3522-y
16. Sherwin RS, Shamoon H, Hendler R, Saccà L, Eigler N, Walesky M. Epinephrine and the regulation of glucose metabolism: effect of diabetes and hormonal interactions. Metabolism (1980) 29(11 Suppl 1):1146–54. doi:10.1016/0026-0495(80)90024-4
17. Mazziotti G, Giustina A. Glucocorticoids and the regulation of growth hormone secretion. Nat Rev Endocrinol (2013) 9:265–76. doi:10.1038/nrendo.2013.5
18. Abs R, Mattsson AF, Thunander M, Verhelst J, Góth MI, Wilton P, et al. Prevalence of diabetes mellitus in 6050 hypopituitary patients with adult-onset GH deficiency before GH replacement: a KIMS analysis. Eur J Endocrinol (2013) 168(3):297–305. doi:10.1530/EJE-12-0807
19. Oldknow KJ, MacRae VE, Farquharson C. Endocrine role of bone: recent and emerging perspectives beyond osteocalcin. J Endocrinol (2015) 225:R1–19. doi:10.1530/JOE-14-0584
20. Brennan-Speranza TC, Henneicke H, Gasparini SJ, Blankenstein KI, Heinevetter U, Cogger VC, et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. J Clin Invest (2012) 122:4172–89. doi:10.1172/JCI63377
21. Seckl JR. 11beta-hydroxysteroid deydrogenases: changing glucocorticoid action. Curr Opin Pharmacol (2004) 4(6):597–602. doi:10.1016/j.coph.2004.09.001
22. Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science (2001) 294(5549):2166–70. doi:10.1126/science.1066285
23. Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol (2007) 275(1–2):43–61. doi:10.1016/j.mce.2007.05.015
24. Trementino L, Appolloni G, Concettoni C, Cardinaletti M, Boscaro M, Arnaldi G. Association of glucocorticoid receptor polymorphism A3669G with decreased risk of developing diabetes in patients with Cushing’s syndrome. Eur J Endocrinol (2012) 166(1):35–42. doi:10.1530/EJE-11-0722
25. Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest (1995) 95(6):2435–41. doi:10.1172/JCI117943
26. Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2015) 100(8):2807–31. doi:10.1210/jc.2015-1818
27. Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab (2008) 93(7):2454–62. doi:10.1210/jc.2007-2734
28. Colao A, Pivonello R, Spiezia S, Faggiano A, Ferone D, Filippella M, et al. Persistence of increased cardiovascular risk in patients with Cushing’s disease after five years of successful cure. J Clin Endocrinol Metab (1999) 84(8):2664–72. doi:10.1210/jcem.84.8.5896
29. Baroni MG, Giorgino F, Pezzino V, Scaroni C, Avogaro A. Italian Society for the Study of Diabetes (SID)/Italian Endocrinological Society (SIE) guidelines on the treatment of hyperglycemia in Cushing’s syndrome and acromegaly. J Endocrinol Invest (2016) 39(2):235–55. doi:10.1007/s40618-015-0404-6
30. Scillitani A, Mazziotti G, Di Somma C, Moretti S, Stigliano A, Pivonello R, et al. Treatment of skeletal impairment in patients with endogenous hypercortisolism: when and how? Osteoporos Int (2014) 25(2):441–6. doi:10.1007/s00198-013-2588-y
31. Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia (2012) 55(6):1577–96. doi:10.1007/s00125-012-2534-0
32. DeFronzo RA, Barzilai N, Simonson DC. Mechanism of metformin action in obese and lean noninsulin-dependent diabetic subjects. J Clin Endocrinol Metab (1991) 73(6):1294–301. doi:10.1210/jcem-73-6-1294
33. Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med (2008) 359(15):1577–89. doi:10.1056/NEJMoa0806470
34. Breitschaft A, Hu K, Hermosillo Reséndiz K, Darstein C, Golor G. Management of hyperglycemia associated with pasireotide (SOM230): healthy volunteer study. Diabetes Res Clin Pract (2014) 103(3):458–65. doi:10.1016/j.diabres.2013.12.011
35. Blonde L, Dailey GE, Jabbour SA, Reasner CA, Mills DJ. Gastrointestinal tolerability of extended release metformin tablets compared to immediate-release metformin tablets: results of a retrospective cohort study. Curr Med Res Opin (2004) 20(4):565–72. doi:10.1185/030079904125003278
36. Van de Laar FA, Lucassen PL, Akkermans RP, Van de Lisdonk EH, Rutten GE, Van Weel C. Alpha glucosidase inhibitors for type 2 diabetes mellitus. Cochrane Database Syst Rev (2005) 18(2):CD003639. doi:10.1002/14651858.CD003639.pub2
37. Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M, et al. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised trial. Lancet (2002) 359(9323):2072–7. doi:10.1016/S0140-6736(02)08905-5
38. Yau H, Rivera K, Lomonaco R, Cusi K. The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr Diab Rep (2013) 13(3):329–41. doi:10.1007/s11892-013-0378-8
39. Miyazaki Y, Mahankali A, Matsuda M, Glass L, Mahankali S, Ferrannini E, et al. Improved glycemic control and enhanced insulin sensitivity in type 2 diabetic subjects treated with pioglitazone. Diabetes Care (2001) 24(4):710–9. doi:10.2337/diacare.24.4.710
40. Suri D, Weiss RE. Effect of pioglitazone on adrenocorticotropic hormone and cortisol secretion in Cushing’s disease. J Clin Endocrinol Metab (2005) 90(3):1340–6. doi:10.1210/jc.2004-1746
41. Heaney AP. PPAR-gamma in Cushing’s disease. Pituitary (2004) 7(4):265–9. doi:10.1007/s11102-005-1430-8
42. Loke YK, Singh S, Furberg CD. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. CMAJ (2009) 180(1):32–9. doi:10.1503/cmaj.080486
43. Mazziotti G, Gazzaruso C, Giustina A. Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol Metab (2011) 22(12):499–506. doi:10.1016/j.tem.2011.09.001
44. Rendell M. The role of sulphonylureas in the management of type 2 diabetes mellitus. Drugs (2004) 64(12):1339–58. doi:10.2165/00003495-200464120-00006
45. Rendell MS, Jovanovic L. Targeting postprandial hyperglycemia. Metabolism (2006) 55(9):1263–81. doi:10.1016/j.metabol.2006.05.012
46. Rosenstock J, Rendell MS, Gross JL, Fleck PR, Wilson CA, Mekki Q. Alogliptin added to insulin therapy in patients with type 2 diabetes reduces HbA(1C) without causing weight gain or increased hypoglycaemia. Diabetes Obes Metab (2009) 11(12):1145–52. doi:10.1111/j.1463-1326.2009.01124.x
47. Colao A, De Block C, Gaztambide MS, Kumar S, Seufert J, Casanueva FF. Managing hyperglycemia in patients with Cushing’s disease treated with pasireotide: medical expert recommendations. Pituitary (2014) 17(2):180–6. doi:10.1007/s11102-013-0483-3
48. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet (2006) 368(9548):1696–705. doi:10.1016/S0140-6736(06)69705-5
49. Drucker DJ. The biology of incretin hormones. Cell Metab (2006) 3(3):153–65. doi:10.1016/j.cmet.2006.01.004
50. Robinson LE, Holt TA, Rees K, Randeva HS, O’Hare JP. Effects of exenatide and liraglutide on heart rate, blood pressure and body weight: systematic review and meta-analysis. BMJ Open (2013) 3(1):e001986. doi:10.1136/bmjopen-2012-001986
51. Yanai H, Masui Y, Yoshikawa R, Kunimatsu J, Kaneko H. Dipeptidyl peptidase-4 inhibitor for steroid-induced diabetes. World J Diabetes (2010) 1(3):99–100. doi:10.4239/wjd.v1.i3.99
52. Monica Reddy RP, Inzucchi SE. SGLT2 inhibitors in the management of type 2 diabetes. Endocrine (2016) 53(2):364–72. doi:10.1007/s12020-016-0943-4
53. Chen LH, Leung PS. Inhibition of the sodium glucose co-transporter-2: its beneficial action and potential combination therapy for type 2 diabetes mellitus. Diabetes Obes Metab (2013) 15(5):392–402. doi:10.1111/dom.12064
54. Li D, Wang T, Shen S, Fang Z, Dong Y, Tang H. Urinary tract and genital infections in patients with type 2 diabetes treated with sodium-glucose co-transporter 2 inhibitors: a meta-analysis of randomized controlled trials. Diabetes Obes Metab (2017) 19(3):348–55. doi:10.1111/dom.12825
55. Fareau GG, Vassilopoulou-Sellin R. Hypercortisolemia and infection. Infect Dis Clin North Am (2007) 21(3):639–57,viii. doi:10.1016/j.idc.2007.06.001
56. Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab (2015) 100(8):2849–52. doi:10.1210/jc.2015-1884
57. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med (2015) 373(22):2117–28. doi:10.1056/NEJMoa1504720
58. Burt MG, Drake SM, Aguilar-Loza NR, Esterman A, Stranks SN, Roberts GW. Efficacy of a basal bolus insulin protocol to treat prednisolone-induced hyperglycaemia in hospitalised patients. Intern Med J (2015) 45(3):261–6. doi:10.1111/imj.12680
59. Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol (2002) 146:707–16. doi:10.1530/eje.0.1460707
60. Ben-Shlomo A, Schmid H, Wawrowsky K, Pichurin O, Hubina E, Chesnokova V, et al. Differential ligand-mediated pituitary somatostatin receptor subtype signaling: implications for corticotroph tumor therapy. J Clin Endocrinol Metab (2009) 94(11):4342–50. doi:10.1210/jc.2009-1311
61. Boscaro M, Ludlam WH, Atkinson B, Glusman JE, Petersenn S, Reincke M, et al. Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. J Clin Endocrinol Metab (2009) 94(1):115–22. doi:10.1210/jc.2008-1008
62. Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med (2012) 366(10):914–24. doi:10.1056/NEJMoa1105743
63. Mitra SW, Mezey E, Hunyady B, Chamberlain L, Hayes E, Foor F, et al. Colocalization of somatostatin receptor sst5 and insulin in rat pancreatic beta-cells. Endocrinology (1999) 140(8):3790–6. doi:10.1210/endo.140.8.6937
64. Petersenn S, Unger N, Hu K, Weisshaar B, Zhang Y, Bouillaud E, et al. Pasireotide (SOM230), a novel multireceptor-targeted somatostatin analogue, is well tolerated when administered as a continuous 7-day subcutaneous infusion in healthy male volunteers. J Clin Pharmacol (2012) 52(7):1017–27. doi:10.1177/0091270011408727
65. Henry RR, Ciaraldi TP, Armstrong D, Burke P, Ligueros-Saylan M, Mudaliar S. Hyperglycemia associated with pasireotide: results from a mechanistic study in healthy volunteers. J Clin Endocrinol Metab (2013) 98(8):3446–53. doi:10.1210/jc.2013-1771
66. Pedroncelli AM. Medical treatment of Cushing’s disease: somatostatin analogues and pasireotide. Neuroendocrinology (2010) 2010(92 Suppl 1):120–4. doi:10.1159/000314352
67. Lacroix A, Gu F, Gallardo W, Pivonello R, Yu Y, Witek P, et al. Efficacy and safety of once-monthly pasireotide in Cushing’s disease: a 12 month clinical trial. Lancet Diabetes Endocrinol (2018) 6(1):17–26. doi:10.1016/S2213-8587(17)30326-1
68. Møller N, Jørgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev (2009) 30(2):152–77. doi:10.1210/er.2008-0027
69. Trementino L, Cardinaletti M, Concettoni C, Marcelli G, Boscaro M, Arnaldi G. Up-to 5-year efficacy of pasireotide in a patient with Cushing’s disease and pre-existing diabetes: literature review and clinical practice considerations. Pituitary (2015) 18(3):359–65. doi:10.1007/s11102-014-0582-9
70. Luo S, Meier AH, Cincotta AH. Bromocriptine reduces obesity, glucose intolerance and extracellular monoamine metabolite levels in the ventromedial hypothalamus of Syrian hamsters. Neuroendocrinology (2008) 68:1–0. doi:10.1159/000054344
71. Bahar A, Kashi Z, Daneshpour E, Akha O, Ala S. Effects of cabergoline on blood glucose levels in type 2 diabetic patients: a double-blind controlled clinical trial. Medicine (Baltimore) (2016) 95(40):e4818. doi:10.1097/MD.0000000000004818
72. Holt RI, Barnett AH, Bailey CJ. Bromocriptine: old drug, new formulation and new indication. Diabetes Obes Metab (2010) 12(12):1048–57. doi:10.1111/j.1463-1326.2010.01304.x
73. Lamos EM, Levitt DL, Munir KM. A review of dopamine agonist therapy in type 2 diabetes and effects on cardio-metabolic parameters. Prim Care Diabetes (2016) 10(1):60–5. doi:10.1016/j.pcd.2015.10.008
74. Pivonello R, Waaijers M, Kros JM, Pivonello C, de Angelis C, Cozzolino A, et al. Dopamine D2 receptor expression in the corticotroph cells of the human normal pituitary gland. Endocrine (2017) 57(2):314–25. doi:10.1007/s12020-016-1107-2
75. Pivonello R, De Martino MC, Cappabianca P, De Leo M, Faggiano A, Lombardi G, et al. The medical treatment of Cushing’s disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab (2009) 94(1):223–30. doi:10.1210/jc.2008-1533
76. Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing’s disease. Eur J Endocrinol (2010) 163(5):709–16. doi:10.1530/EJE-10-0382
77. Lila AR, Gopal RA, Acharya SV, George J, Sarathi V, Bandgar T, et al. Efficacy of cabergoline in uncured (persistent or recurrent) Cushing disease after pituitary surgical treatment with or without radiotherapy. Endocr Pract (2010) 16(6):968–76. doi:10.4158/EP10031.OR
78. Ferriere A, Cortet C, Chanson P, Delemer B, Caron P, Chabre O, et al. Cabergoline for Cushing’s disease: a large retrospective multicenter study. Eur J Endocrinol (2017) 176(3):305–14. doi:10.1530/EJE-16-0662
79. Páez-Pereda M, Kovalovsky D, Hopfner U, Theodoropoulou M, Pagotto U, Uhl E, et al. Retinoic acid prevents experimental Cushing syndrome. J Clin Invest (2001) 108(8):1123–31. doi:10.1172/JCI11098
80. Pecori Giraldi F, Ambrogio AG, Andrioli M, Sanguin F, Karamouzis I, Corsello SM, et al. Potential role for retinoic acid in patients with Cushing’s disease. J Clin Endocrinol Metab (2012) 97(10):3577–83. doi:10.1210/jc.2012-2328
81. Vilar L, Albuquerque JL, Lyra R, Trovão Diniz E, Rangel Filho F, Gadelha P, et al. The role of isotretinoin therapy for Cushing’s disease: results of a prospective study. Int J Endocrinol (2016) 2016:8173182. doi:10.1155/2016/8173182
82. Blumentrath J, Neye H, Verspohl EJ. Effects of retinoids and thiazolidinediones on proliferation, insulin release, insulin mRNA, GLUT 2 transporter protein and mRNA of INS-1 cells. Cell Biochem Funct (2001) 19(3):159–69. doi:10.1002/cbf.907
83. Berry DC, Noy N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor beta/delta and retinoic acid receptor. Mol Cell Biol (2009) 29(12):3286–96. doi:10.1128/MCB.01742-08
84. Engelhardt D, Weber MM, Miksch T, Abedinpour F, Jaspers C. The influence of ketoconazole on human adrenal steroidogenesis: incubation studies with tissue slices. Clin Endocrinol(Oxf) (1991) 35:163–8. doi:10.1111/j.1365-2265.1991.tb03516.x
85. Sonino N, Boscaro M. Medical therapy for Cushing’s disease. Endocrinol Metab Clin North Am (1999) 28:211–22. doi:10.1016/S0889-8529(05)70064-5
86. Ceccato F, Barbot M, Zilio M, Albiger N, Mantero F, Scaroni C. Therapeutic strategies for Cushing’s syndrome: an update. Exp Opin Orphan Drugs (2015) 3(1):45–56. doi:10.1517/21678707.2015.991714
87. Sonino N, Boscaro M, Paoletta A, Mantero F, Ziliotto D. Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients. Clin Endocrinol (Oxf) (1991) 35(4):347–52. doi:10.1111/j.1365-2265.1991.tb03547.x
88. Castinetti F, Morange I, Jaquet P, Conte-Devolx B, Brue T. Ketoconazole revisited: a preoperative or postoperative treatment in Cushing’s disease. Eur J Endocrinol (2008) 158(1):91–9. doi:10.1530/EJE-07-0514
89. Valassi E, Crespo I, Gich I, Rodríguez J, Webb SM. A reappraisal of the medical therapy with steroidogenesis inhibitors in Cushing’s syndrome. Clin Endocrinol (Oxf) (2012) 77(5):735–42. doi:10.1111/j.1365-2265.2012.04424.x
90. Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab (2014) 99(5):1623–30. doi:10.1210/jc.2013-3628
91. Arakaki R, Welles B. Ketoconazole enantiomer for the treatment of diabetes mellitus. Expert Opin Investig Drugs (2010) 19(2):185–94. doi:10.1517/13543780903381411
92. Munir A, Newell-Price J. Management of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology (2010) 92(Suppl 1):82–5. doi:10.1159/000314316
93. Jeffcoate WJ, Rees LH, Tomlin S, Jones AE, Edwards CR, Besser GM. Metyrapone in long-term management of Cushing’s disease. Br Med J (1977) 2(6081):215–7. doi:10.1136/bmj.2.6081.215
94. Verhelst JA, Trainer PJ, Howlett TA, Perry L, Rees LH, Grossman AB, et al. Short and long-term responses to metyrapone in the medical management of 91 patients with Cushing’s syndrome. Clin Endocrinol (Oxf) (1991) 35(2):169–78. doi:10.1111/j.1365-2265.1991.tb03517.x
95. Daniel E, Aylwin S, Mustafa O, Ball S, Munir A, Boelaert K, et al. Effectiveness of metyrapone in treating Cushing’s syndrome: a retrospective multicenter study in 195 patients. J Clin Endocrinol Metab (2015) 100(11):4146–54. doi:10.1210/jc.2015-2616
96. Bertagna X, Pivonello R, Fleseriu M, Zhang Y, Robinson P, Taylor A, et al. LCI699, a potent 11β-hydroxylase inhibitor, normalizes urinary cortisol in patients with Cushing’s disease: results from a multicenter, proof-of-concept study. J Clin Endocrinol Metab (2014) 99(4):1375–83. doi:10.1210/jc.2013-2117
97. Fleseriu M, Pivonello R, Young J, Hamrahian AH, Molitch ME, Shimizu C, et al. Osilodrostat, a potent oral 11β-hydroxylase inhibitor: 22-week, prospective, phase II study in Cushing’s disease. Pituitary (2016) 19(2):138–48. doi:10.1007/s11102-015-0692-z
98. Baudry C, Coste J, Bou Khalil R, Silvera S, Guignat L, Guibourdenche J, et al. Efficiency and tolerance of mitotane in Cushing’s disease in 76 patients from a single center. Eur J Endocrinol (2012) 167(4):473–81. doi:10.1530/EJE-12-0358
99. Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C, et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab (2012) 97(6):2039–49. doi:10.1210/jc.2011-3350
100. Wallia A, Colleran K, Purnell JQ, Gross C, Molitch ME. Improvement in insulin sensitivity during mifepristone treatment of Cushing syndrome: early and late effects. Diabetes Care (2013) 36(9):e147–8. doi:10.2337/dc13-0246
101. Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of Cushing’s disease. Endocr Rev (2015) 36(4):385–486. doi:10.1210/er.2013-1048
102. Barbot M, Albiger N, Ceccato F, Zilio M, Frigo AC, Denaro L, et al. Combination therapy for Cushing’s disease: effectiveness of two schedules of treatment: should we start with cabergoline or ketoconazole? Pituitary (2014) 17(2):109–17. doi:10.1007/s11102-013-0475-3
103. Feelders RA, de Bruin C, Pereira AM, Romijn JA, Netea-Maier RT, Hermus AR, et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing’s disease. N Engl J Med (2010) 362(19):1846–8. doi:10.1056/NEJMc1000094
104. Kamenický P, Droumaguet C, Salenave S, Blanchard A, Jublanc C, Gautier JF, et al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab (2011) 96(9):2796–804. doi:10.1210/jc.2011-0536
Keywords: Cushing’s disease, diabetes, glucocorticoids, insulin resistance, cortisol-lowering medication
Citation: Barbot M, Ceccato F and Scaroni C (2018) Diabetes Mellitus Secondary to Cushing’s Disease. Front. Endocrinol. 9:284. doi: 10.3389/fendo.2018.00284
Received: 13 February 2018; Accepted: 14 May 2018;
Published: 05 June 2018
Edited by:
Carla Giordano, Università degli Studi di Palermo, ItalyReviewed by:
Anton Luger, Medizinische Universität Wien, AustriaCopyright: © 2018 Barbot, Ceccato and Scaroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mattia Barbot, bWF0dGlhYmFyYm90QGFsaWNlLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.