 Raquel Buj
Raquel Buj Katherine M. Aird
Katherine M. Aird- Department of Cellular and Molecular Physiology, Penn State College of Medicine, Hershey, PA, United States
The maintenance of a healthy deoxyribonucleotide triphosphate (dNTP) pool is critical for the proper replication and repair of both nuclear and mitochondrial DNA. Temporal, spatial, and ratio imbalances of the four dNTPs have been shown to have a mutagenic and cytotoxic effect. It is, therefore, essential for cell homeostasis to maintain the balance between the processes of dNTP biosynthesis and degradation. Multiple oncogenic signaling pathways, such as c-Myc, p53, and mTORC1 feed into dNTP metabolism, and there is a clear role for dNTP imbalances in cancer initiation and progression. Additionally, multiple chemotherapeutics target these pathways to inhibit nucleotide synthesis. Less is understood about the role for dNTP levels in metabolic disorders and syndromes and whether alterations in dNTP levels change cancer incidence in these patients. For instance, while deficiencies in some metabolic pathways known to play a role in nucleotide synthesis are pro-tumorigenic (e.g., p53 mutations), others confer an advantage against the onset of cancer (G6PD). More recent evidence indicates that there are changes in nucleotide metabolism in diabetes, obesity, and insulin resistance; however, whether these changes play a mechanistic role is unclear. In this review, we will address the complex network of metabolic pathways, whereby cells can fuel dNTP biosynthesis and catabolism in cancer, and we will discuss the potential role for this pathway in metabolic disease.
Introduction
The maintenance of deoxyribonucleotide triphosphate (dNTP) pools is critical for multiple cellular pathways. For instance, imbalances in dNTPs are associated with genomic instability (1). Likewise, they have also been shown to disturb mitochondrial DNA (mtDNA) and consequently mitochondrial fitness, which may lead to mitochondrial diseases (MDs), such as diabetes, obesity, and cancer (2). Additionally, disorders of purine and pyrimidine metabolism (DPPM) profoundly affect cell metabolism, underlying the importance of nucleotides for cell behavior (3). Thus, both nucleotide synthesis and degradation must be exquisitely fine-tuned. In this review, we will focus on synthesis of dNTPs and the consequences of dNTP pool imbalances in cancer and MDs.
Healthy dNTP Pools
A correct balance of dNTPs is necessary for the prevention of multiple pathologies. A healthy cell must maintain two asymmetric and spatial-temporal dNTP pools; one for nuclear DNA synthesis and repair and another for mtDNA replication and repair. Disruptions in dNTP balance are associated with enhanced mutagenesis, leading to genomic instability, which promotes cancer (4), and may have a role in metabolic disease (5).
Cytosolic dNTP pool concentrations positively correlate with the cell cycle. In fact, the amount of dNTPs at the beginning of S-phase is not enough for a complete DNA duplication (6). The S-phase increase in dNTPs is necessary for faithful nuclear DNA replication. mtDNA is replicated continuously in post-mitotic cells, and faithful maintenance of mtDNA also depends on correctly balanced dNTPs (7). Thus, both proliferating and non-proliferating cells need to fine-tune nucleotide and dNTP synthesis to allow for both nuclear and mtDNA replication and repair to maintain the health of the cell.
Anabolism and Catabolism of Nucleotides
Cells possess two biosynthetic pathways to produce dNTPs: de novo and salvage (8). Purines and pyrimidines arise from two different de novo pathways that generate nucleotides starting from raw material (glucose, glutamine, aspartate, and HCO3) (9). The de novo nucleotide synthesis pathway is highly energy-intensive (9). Therefore, cells have developed a more energy-efficient route to synthesize nucleotides, termed the salvage pathway (10). The salvage pathway acts as a recycling plant taking free nitrogen bases and nucleosides arising from nucleic acid breakdown and diet (9). Nucleosides are hydrophilic compounds, thus proper function of nucleoside transporters (SLC29 and SLC28 families) is an essential requirement for salvage pathway function (11). Ribonucleotides obtained by either pathway can be reduced to their deoxyribonucleotide counterpart in a reaction catalyzed by ribonucleotide reductase (RNR) (12).
Turnover of RNA and other nucleotides occurs regularly to maintain homeostasis. Human cells cannot break down the purine ring. Purine catabolism involves a sequence of three reactions in which nucleotides are stripped step-by-step from their phosphates and sugar to finally become oxidized to the end product uric acid (UA), which is excreted into the urine (13). Conversely, uracil and thymidine rings can be completely degraded to β-alanine and β-aminoisobutyrate, respectively. Subsequently, both metabolites can be excreted or transformed into intermediates of the tricarboxylic acid (TCA) cycle (14). Biosynthesis and catabolism of nucleotides and dNTPs are highlighted in Figure 1.
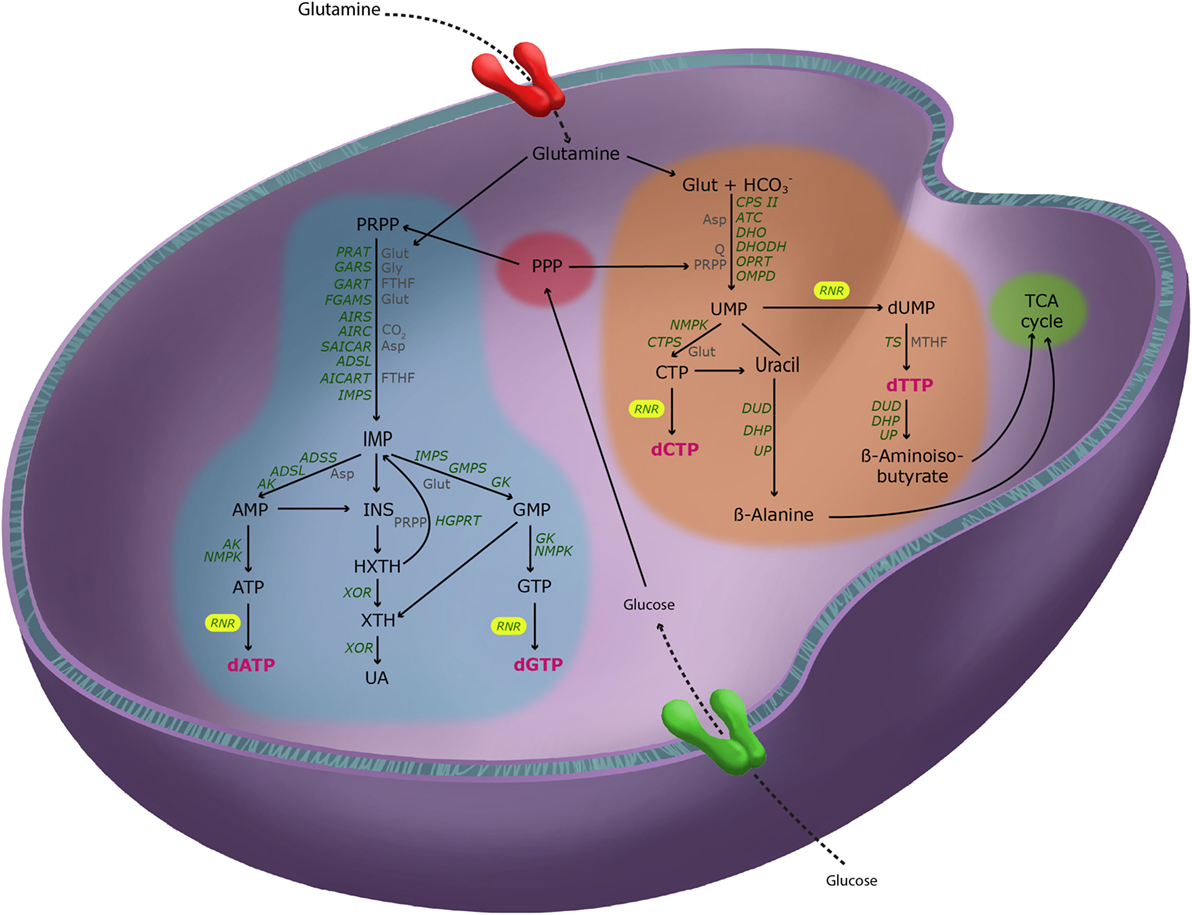
Figure 1. Pathways of deoxyribonucleotide metabolism in mammalian cells. Simplified representation of purine (blue) and pyrimidine (orange) metabolism and their crosstalk with the major metabolic pathways, the pentose phosphate pathway [(PPP), red] and the tricarboxylic acid cycle (green). Key metabolic enzymes (green), their principal reactive substrates (gray), and the four deoxyribonucleotide triphosphate (dNTP) end-products (magenta) are shown. Glucose and glutamine feed into both purine and pyrimidine metabolism to donate carbons and nitrogens to all dNTPs. Abbreviations: RAT, phosphoribosylpyrophosphate amidotransferase; GARS, glycinamide ribonucleotide synthetase; GART, glycinamide ribonucleotide transformylase; FGAMS, phosphoribosylformyl-glycineamide synthetase; AIRS, phosphoribosylaminoimidazole synthetase; AIRC, phosphoribosylaminoimidazole carboxylase; SAICAR, phosphoribosylaminoimidazole-succinocarboxamide; ADSL, adenylosuccinate lyase; AICART, phosphoribosylaminoimidazolecarboxamide formyltransferase; IMPS, inosine monophosphate synthase; ADSS, adenylosuccinate synthetase; AK, adenylate kinase; NMPK, nucleotide monophosphate kinase; IMPS, inosine monophosphate dehydrogenase; GMPS, guanosine monophosphate synthetase; GK, guanylate kinase; XOR, xanthine oxidoreductase; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; RNR, ribonucleotide reductase; CPS II, carbamoyl phosphate synthetase II; ATC, aspartate carbamoyltransferase; DHO, dihydroorotase; DHOD, dihydroorotase dehydrogenase; OPRT, orotate phosphoribosyltransferase; OMPD, orotidine monophosphate decarboxylase; CPTS, cytidine triphosphate synthetase; TS, thymidylate synthase; DUD, dihydrouracil dehydrogenase; DHP, dihydropyrimidinase; UP, ureidopropionase; Glut, glutamine; Gly, glycine; FTHF, N10-formyltetrahydrofolate; Asp, aspartate; PRPP, phosphoribosylpyrophosphate; Q, ubiquinone; MTHF, N5,N10-methylenetetrahydrofolate.
Impaired Nucleotide Metabolism in Cancer and Metabolic Disease
Deregulation of nucleotide metabolism is associated with a broad spectrum of pathological conditions, including cancer and MDs (15–17). Virtually all metabolic pathways have been implicated in dNTP biosynthesis. Thus, de novo and salvage pathways, as well as all involved anapleurotic reactions (Figure 1), need to be highly cross-regulated.
It is well known that cancer cells must increase dNTP biosynthesis (18) to ensure rapid replication of the genome (17). This occurs through a variety of pathways (discussed below). In contrast, MDs are caused by congenital or acquired genetic defects in metabolic enzymes. DPPM are due to abnormalities in the biosynthesis, interconversion, and degradation of nucleotides (19). DPPM have a wide variety of clinical presentations, highlighting the importance of proper nucleotide metabolism for cell and organism function (15). Alterations in nucleotide metabolism are also present in other metabolic-related pathological conditions, such as diabetes, obesity, and insulin resistance (20–22) (Table 1). In this section, we will summarize some important features affecting nucleotide metabolism in cancer and MDs.
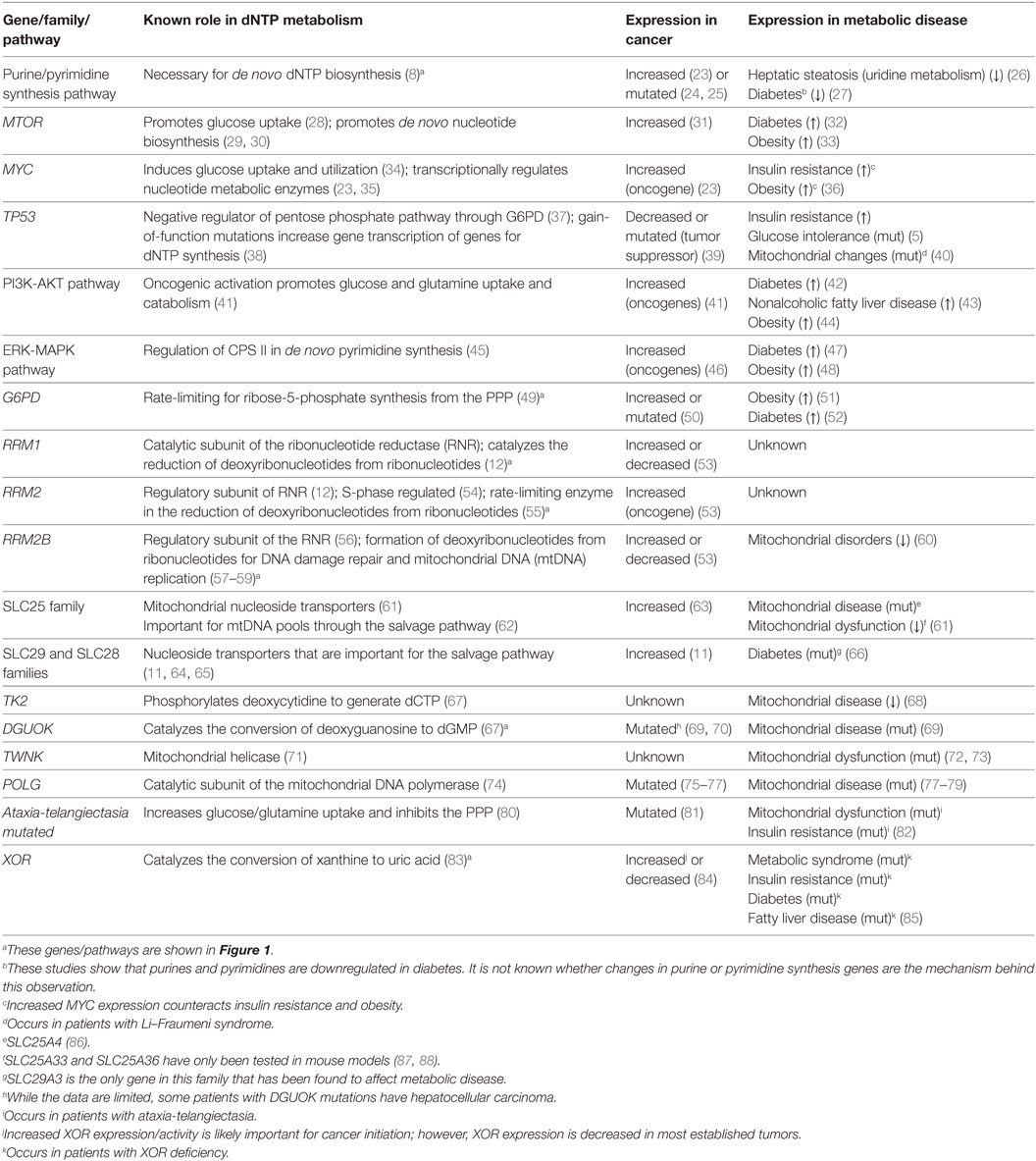
Table 1. Genes, protein families, and pathways discussed in this review: role in deoxyribonucleotide triphosphate (dNTP) metabolism and expression in cancer and metabolic disease.
Deregulation of Major Growth Signaling Pathways Leads to Nucleotide Pool Imbalances in Cancer and Metabolic Disease
The main growth signaling pathways (PI3K-AKT and ERK1/2-MAPK) are induced and maintained during metabolic reprogramming of cancer (18). Additionally, deregulation of these pathways may contribute to different MDs, including diabetes, obesity, or steatosis resistance (33, 89, 90). These pathways sense and orchestrate nutrient utilization; therefore, is not surprising that alterations in these pathways affect energy and biomass production and cause a broad variety of diseases.
mTOR is a central signaling pathway that integrates environmental inputs (e.g., nutrients and hormones) into downstream pathways to control many cellular processes (91). This includes regulation of metabolism, growth, and survival (32). Indeed, the mTORC1/2 pathway not only promotes glucose uptake and protein and lipid biosynthesis, but also promotes nucleotide biosynthesis (29, 30) and uptake of nucleosides through transporters (88). At least one member of this pathway is altered in 38% of human cancer (92). Altered metabolism induced by aberrant mTORC1 activation has also been shown to play a role in diabetes and obesity (32, 93).
c-Myc, one of the most commonly altered proteins in human cancer, is also regulated by PI3K-AKT and ERK1/2-MAPK pathways (94). c-Myc is a highly pleiotropic transcription factor considered a master regulator of cell metabolism (34, 35) through regulation of glycolysis, glutamine metabolism, and mitochondrial biogenesis (95, 96). Indeed, c-Myc has been shown to induce hepatic glucose uptake and utilization, while blocking gluconeogenesis and ketogenesis, suggesting a counteracting effect of c-Myc in obesity and insulin resistance (36, 97). In addition to regulating glucose and glutamine, substrates for purine and pyrimidine biosynthesis (Figure 1) (98), c-Myc also transcriptionally regulates nucleotide metabolic enzyme gene expression (35). Thus, deregulation in c-Myc acutely alters nucleotide homeostasis in cancer (99), and it is interesting to speculate that the role of c-Myc in MDs is also related to nucleotide metabolism.
Previous publications from our laboratory and others have shown that DNA damage and DNA damage response (DDR) proteins regulate dNTP biosynthesis in the context of cancer (80, 100, 101). Interestingly, upregulation of p53, a key player in the DDR, in adipose tissue is associated with increased inflammation and insulin resistance (102). Notably, wild-type p53 negatively regulates G6PD activity (37), the rate-limiting enzyme of the pentose phosphate pathway and one of the most important sources of nucleotides (103). Upregulation of G6PD correlates with functional defects in liver, heart, and pancreas of obese and diabetic animals (104). Although the relationship between G6PD upregulation and increased oxidative stress has been studied in MD (105), the implication for nucleotide metabolism has not yet been addressed. More research is needed to understand the contribution of dNTP imbalances due to G6PD deregulation in diabetes and obesity.
An imbalance in nucleotides has been shown in two different studies related to diabetes (106, 107). Additionally, pyrimidine metabolism has been linked to fatty liver (26). Interestingly, increasing evidence suggests a link between obesity, a risk factor for non-alcoholic fatty liver disease (108), and cancer. Obese patients show many cancer-promoting features, such as chronic low-level inflammation (109), insulin-resistance/diabetes (110), and deregulation of mTORC1 (111). Although the contribution of deregulated nucleotide pools promoting cancer has been extensively demonstrated (18, 112–115), their role in MD and metabolic-related diseases has not yet been elucidated. Based on these recent studies, we speculate that deregulation of nucleotide pools may in part contribute to the altered metabolic landscape promoting obesity and diabetes. Studying the implications of altered nucleotide pools in these diseases would open a therapeutic window based on modulation of nucleotide metabolism.
RNR in Cancer and Metabolic Disease
Ribonucleotide reductase reduces ribonucleotides to the corresponding deoxyribonucleotides (116, 117). In mammals, RNR is a tetrameric enzyme composed of two homodimeric subunits, RRM1 and RRM2. Whereas, RRM1 is continuously expressed throughout the cell-cycle, expression of RRM2 is activated upon entry into S-phase (54, 118). Additionally, RRM2 is rapidly degraded via the proteasome in G2 (12, 119). Thus, RRM2 is considered rate-limiting for RNR activity. RRM2B (RNR subunit M2B) is an alternative M2 subunit that is induced by p53 activation in response to DNA damage (56). RRM2B is not cell-cycle regulated per se, but it plays key roles in enhancing dNTP synthesis in cells under stress (120–122) and mediating mtDNA synthesis and repair (57–59).
The role of RNR in cancer is clear as it was one of the first identified DNA damage-induced enzymes (123). While RRM2 overexpression is tumorigenic, leading to lung neoplasms in vivo, RRM1 reduces tumor formation, migration, and metastasis [reviewed in Ref. (53)]. Previous studies from our lab and others have shown the potential of RRM2 as a prognostic and diagnostic biomarker in multiple cancers (112, 124–127). However, the utility of RRM1 and RRM2B as a tumor biomarker is still unclear [reviewed in Ref. (53)].
Although there is no study directly linking RNR with MD, RRM2B is required for mtDNA synthesis and healthy mitochondrial function (57). Deregulated mitochondria are associated with a higher risk of diabetes and obesity (discussed below). Therefore, it is possible that RNR function is linked to these MDs (Table 1). More mechanistic studies will be needed to determine the role for RNR in obesity and diabetes.
Mitochondrial Dysfunction in dNTP Pool Disruption During Cancer and Metabolic Disease
The mitochondria are one of the most important organelles for eukaryotic function (128). In addition to the production of ATP through oxidative phosphorylation, mitochondria are also the scaffold of several metabolic reactions for cellular building block synthesis (e.g., fatty acid beta-oxidation, one-carbon/folate cycle, TCA cycle, amino acid metabolism, etc.) (129). Hence, altered mitochondrial behavior has a broad impact on cellular metabolism.
Maintenance of mitochondrial dNTP pools is critical for proper mtDNA function. Alterations in nuclear genes involved in transport of cytosolic dNTPs (e.g., SLC25A4), the salvage nucleotide biosynthesis in the inner mitochondrial membrane (e.g., TK2 and DGUOK), and genes involved in mtDNA replication (e.g., TWNK and POLG) are implicated in both cancer and metabolic syndromes (63, 68, 77–79, 130–133). Moreover, dysfunction in the electron transport chain induces oxidative stress, which has been associated with impaired one-carbon metabolism (134, 135), an essential anapleurotic pathway for both purine and pyrimidine nucleotides. Mitochondrial genomic instability due to increased levels of reactive oxygen species (ROS) and/or mutations in mtDNA or nuclear genes involved in mitochondria function are underlying factors of MDs, and contribute to cancer and diabetes (136). Alterations in genes discussed above that are important for dNTP homeostasis and mitochondrial function are highlighted in Table 1.
Although the link between mitochondrial dysfunction and MD has been studied for the past two decades, the results are contradictory (137). These contradictory results mainly arise from the complex relationship between mitochondria and metabolism, but also from the lack of global and standardized methodological strategies to phenotype insulin-resistance in humans (138). Dysregulation of nucleotide metabolism is an important aspect of mitochondrial dysfunction; therefore, their role in MDs should not be ignored.
Relationship Between DPPM and Cancer
It is clear that cancer is a metabolic disease; however, a predisposition to cancer is not a foregone conclusion in patients with DPPM, who by definition have alterations in nucleotide supplies. Interestingly, while deficiencies in some metabolic pathways known to play a role in nucleotide synthesis are pro-tumorigenic, others confer an advantage against the onset of cancer. This highlights the large variability in the clinical presentation of these disorders.
Alterations in p53 or ataxia-telangiectasia mutated (ATM) lead to metabolic changes and predispose patients to cancer. Patients with germline TP53 (encoding for p53) mutations have Li–Fraumeni syndrome and are predisposed to cancer (139, 140). Interestingly, a recent report showed that nucleotide metabolism is regulated by the gain-of-function activity of mutant p53 (38). Consistently, wild-type p53 negatively regulates G6PD and PPP activity to decrease dNTP synthesis (37). Similarly, our group has previously shown that ATM (mutated in some ataxia-telangiectasia patients) inactivation increases glucose uptake and enhances glucose flux through the PPP and ultimately increases dNTP biosynthesis (Figure 1) (80, 141). Indeed, patients with ATM mutations show alterations in glucose homeostasis (142, 143). It is well-known that these patients have an increased susceptibility to cancer (144). It is interesting to speculate that alterations in dNTP metabolism may play a role in the cancer predisposition in these patients; however, further studies are needed to support this notion.
Other DPPM confer a tumor suppressive benefit. For instance, patients with G6PD deficiency have a reduced risk of some cancers (145–147) (Table 1). This suggests that hyperactivity of dNTP synthesis is more likely to increase cancer risk than deficiencies in synthesis.
Finally, some DPPM have both a pro- and anti-tumorigenic effect. Deficiency in xanthine oxidoreductase (XOR), the enzyme that catalyzes the last step in purine catabolism (Figure 1), increases UA (148). There is a dual role for UA in cancer, the so-called the oxidant–antioxidant UA paradox (149). On one hand, extracellular UA is a potent ROS scavenger, thus protecting cells against oxidative stress (150). On the other hand, high intracellular levels of UA in a XOR-deficient cellular background promote dNTP biosynthesis and tumor growth by shuttling XOR precursors (xanthine and hypoxanthine) into the purine salvage pathway (149). Additionally, intracellular UA is pro-inflammatory by inducing NADPH-oxidases that lead to oxidative stress and cancer (151, 152). This again emphasizes the complex nature of these disorders in relation to cancer (Table 1).
Together, the lack of consensus in predisposition to cancer in DPPM patients points to the significant redundancy in the dNTP biosynthetic pathways. This should not be surprising due to the fact that dNTP synthesis is critical for organismal survival and, therefore, we have evolved to have multiple metabolic arms feeding into the same pathway. Understanding whether these patients are predisposed or not to cancer will be incredibly important for the clinical management of these patients.
Therapeutic Modulation of Deoxyribonucleotide Metabolism in Cancer and Metabolic Disease
As described in this review, the balance of dNTPs must be tightly regulated in the cell. Many cancer types show alterations in dNTP levels, supporting their rapid proliferation. Likewise, defective mutations in anabolic and catabolic nucleotide enzymes, causing imbalances in the dNTP pools or in their precursors, are associated with different grades of disease severity in DPPM. Thus, it is not surprising that therapies for both cancer and DPPM focus on restoration of the normal balance of intracellular nucleotides.
Some of the first chemotherapeutic agents were cytotoxic nucleoside analogs and nucleobases (e.g., thiopurines and fluoropyrimidines) (153). These antimetabolites have a similar molecular structure to endogenous nucleotides and interfere with nucleotide metabolic pathways and DNA/RNA synthesis (154). Inhibitors of RNR were one of the first cancer therapies [reviewed in Ref. (53)] and are still used today. For instance, gemcitabine, a chemotherapeutic nucleoside analog, is used in pancreatic adenocarcinoma, but also in breast, bladder, and non-small cell lung cancer (155). Unfortunately, resistance to gemcitabine in common, often through an increase in nucleotide synthesis pathways or transport of nucleosides (156). Other successful chemotherapeutic regimens include methotrexate, which reduces substrates for purine and pyrimidine biosynthesis (157). Finally, specific inhibition of enzymes in the de novo pathway and/or in anapleurotic reactions (glucose and glutamine metabolism) has also been used as adjuvant therapies in cancer (154).
The spectrum of nucleotide therapies for DPPM is much broader in scope due to the high variability of deficiencies (3). Thus, deficiencies resulting in the overproduction of UA are treated with allopurinol, an inhibitor of xanthine oxidase (16). In other cases, patients can be treated with oral supplements of specific nucleotides they are lacking (16). What is clear is that cancer patients with DPPM cannot be treated with antimetabolites such as 5-fluoro-uracil due to severe side effects (19). This suggests that cancer patients, DPPM must remain above a certain threshold of nucleotide pools to remain healthy. Finally, no nucleotide therapies are currently used for MDs, such as diabetes or obesity. More studies will need to be performed to determine whether nucleotide metabolism plays a contributing role to these pathologies before these types of therapies can be tested.
Conclusion
For decades researchers and clinicians alike have recognized the importance of fine-tuned dNTP levels for cellular homeostasis, as shown by the number of anti-cancer therapies based on the abolishment of nucleotide synthesis. In addition, the broad range of pathologies associated with congenital defects in nucleotide metabolic enzymes further demonstrates the importance of healthy intracellular dNTP levels. However, the association between cancer and MD and whether nucleotide pools are interconnected in these pathologies remains unclear. Future work will need to focus on mechanistic and population-based studies to determine whether nucleotide pool imbalances in MD lead to changes in cancer predisposition and whether targeting these pathways for cancer therapy affects metabolic homeostasis and function in normal cells.
Author Contributions
RB and KA conceived of and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the members of the Aird lab for their thoughtful comments. We would also like to thank Fran Vazquez for help with the dNTP metabolism schematic. RB and KA are supported by an NIH/NCI grant (R00CA194309).
References
1. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol (2014) 16(1):2–9. doi:10.1038/ncb2897
2. Gilkerson R. Commentary: mitochondrial DNA damage and loss in diabetes. Diabetes Metab Res Rev (2016) 32:672–4. doi:10.1002/dmrr.2833
3. Van Den Berghe G, Vincent MF, Marie S. Disorders of purine and pyrimidine metabolism. In: Saudubray JM, Berghe GD, Walter JH, editors. Inborn Metabolic Diseases: Diagnosis and Treatment. Berlin, Heidelberg: Springer-Verlag (2012). p. 499–518.
4. Mathews CK. DNA precursor metabolism and genomic stability. FASEB J (2006) 20:1300–14. doi:10.1096/fj.06-5730rev
5. Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab (2014) 20:967–77. doi:10.1016/j.cmet.2014.10.008
6. Kohalmi SE, Glattke M, McIntosh EM, Kunz BA. Mutational specificity of DNA precursor pool imbalances in yeast arising from deoxycytidylate deaminase deficiency or treatment with thymidylate. J Mol Biol (1991) 220:933–46. doi:10.1016/0022-2836(91)90364-C
7. Pai CC, Kearsey SE. A critical balance: DNTPs and the maintenance of genome stability. Genes (Basel) (2017) 8(2):E57. doi:10.3390/genes8020057
8. Blakley RL, Vitols E. The control of nucleotide biosynthesis. Annu Rev Biochem (1968) 37:201–24. doi:10.1146/annurev.bi.37.070168.001221
9. Lane AN, Fan TWM. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res (2015) 43(4):2466–85. doi:10.1093/nar/gkv047
10. Moffatt BA, Ashihara H. Purine and pyrimidine nucleotide synthesis and metabolism. Arab B (2002) 1:e0018. doi:10.1199/tab.0018
11. Young JD, Yao SYM, Baldwin JM, Cass CE, Baldwin SA. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med (2013) 34:529–47. doi:10.1016/j.mam.2012.05.007
12. Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat Struct Mol Biol (2011) 18:316–22. doi:10.1038/nsmb.2007
13. Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol (2016) 213:8–14. doi:10.1016/j.ijcard.2015.08.109
14. Wasternack C. Degradation of pyrimidines and pyrimidine analogs-pathways and mutual influences. Pharmacol Ther (1980) 8:629–51. doi:10.1016/0163-7258(80)90079-0
15. Van Gennip AH. Defects in metabolism of purines and pyrimidines. Ned Tijdschr Klin Chem (1999) 24:171–5.
16. Jurecka A. Inborn errors of purine and pyrimidine metabolism. J Inherit Metab Dis (2009) 32:247–63. doi:10.1007/s10545-009-1094-z
17. Kohnken R, Kodigepalli KM, Wu L. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer (2015) 14:176. doi:10.1186/s12943-015-0446-6
18. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23:27–47. doi:10.1016/j.cmet.2015.12.006
19. Bakker JA, Bierau J. Purine and pyrimidine metabolism: still more to learn. Ned Tijdschr voor Klin Chemie en Lab (2012) 37:3–6.
20. Choi Y-J, Yoon Y, Lee K-Y, Kang Y-P, Lim DK, Kwon SW, et al. Orotic acid induces hypertension associated with impaired endothelial nitric oxide synthesis. Toxicol Sci (2015) 144:307–17. doi:10.1093/toxsci/kfv003
21. Kunjara S, Sochor M, Ali M, Drake A, Greenbaum AL, McLean P. Pyrimidine nucleotide synthesis in the rat kidney in early diabetes. Biochem Med Metab Biol (1991) 46:215–25. doi:10.1016/0885-4505(91)90069-W
22. Tsushima Y, Nishizawa H, Tochino Y, Nakatsuji H, Sekimoto R, Nagao H, et al. Uric acid secretion from adipose tissue and its increase in obesity. J Biol Chem (2013) 288:27138–49. doi:10.1074/jbc.M113.485094
23. Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci U S A (2017) 114:E7697–706. doi:10.1073/pnas.1710366114
24. Berg M, Ågesen TH, Thiis-Evensen E, Merok MA, Teixeira MR, Vatn MH, et al. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol Cancer (2010) 9:100. doi:10.1186/1476-4598-9-100
25. Jo YS, Oh HR, Kim MS, Yoo NJ, Lee SH. Frameshift mutations of OGDH, PPAT and PCCA genes in gastric and colorectal cancers. Neoplasma (2016) 63:681–6. doi:10.4149/neo_2016_504
26. Le TT, Ziemba A, Urasaki Y, Hayes E, Brotman S, Pizzorno G. Disruption of uridine homeostasis links liver pyrimidine metabolism to lipid accumulation. J Lipid Res (2013) 54:1044–57. doi:10.1194/jlr.M034249
27. Pillwein K, Reardon MA, Jayaram HN, Natsumeda Y, Elliott WL, Faderan MA, et al. Insulin regulatory effects on purine- and pyrimidine metabolism in alloxan diabetic rat liver. Padiatr Padol (1988) 23:135–44.
28. Cornu M, Albert V, Hall MN. MTOR in aging, metabolism, and cancer. Curr Opin Genet Dev (2013) 23(1):53–62. doi:10.1016/j.gde.2012.12.005
29. Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science (2016) 351:728–33. doi:10.1126/science.aad0489
30. Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science (2013) 339:1323–8. doi:10.1126/science.1228792
31. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell (2007) 12(1):9–22. doi:10.1016/j.ccr.2007.05.008
32. Ardestani A, Lupse B, Kido Y, Leibowitz G, Maedler K. mTORC1 signaling: a double-edged sword in diabetic β cells. Cell Metab (2018) 27:314–31. doi:10.1016/j.cmet.2017.11.004
33. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology (2005) 146:1473–81. doi:10.1210/en.2004-0921
35. Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One (2008) 3(7):e2722. doi:10.1371/journal.pone.0002722
36. Riu E, Ferre T, Hidalgo A, Mas A, Franckhauser S, Otaegui P, et al. Overexpression of c-myc in the liver prevents obesity and insulin resistance. FASEB J (2003) 17:1715–7. doi:10.1096/fj.02-1163fje
37. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol (2011) 13:310–6. doi:10.1038/ncb2172
38. Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun (2015) 6:7389. doi:10.1038/ncomms8389
39. Liu J, Zhang C, Feng Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim Biophys Sin (Shanghai) (2014) 46(3):170–9. doi:10.1093/abbs/gmt144
40. Wang P-Y, Ma W, Park J-Y, Celi FS, Arena R, Choi JW, et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N Engl J Med (2013) 368:1027–32. doi:10.1056/NEJMoa1214091
41. Tong X, Zhao F, Thompson CB. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr Opin Genet Dev (2009) 19(1):32–7. doi:10.1016/j.gde.2009.01.002
42. Mackenzie RW, Elliott BT. Akt/PKB activation and insulin signaling: a novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metab Syndr Obes (2014) 7:55–64. doi:10.2147/DMSO.S48260
43. Matsuda S, Kobayashi M, Kitagishi Y. Roles for PI3K/AKT/PTEN pathway in cell signaling of nonalcoholic fatty liver disease. ISRN Endocrinol (2013) 2013:472432. doi:10.1155/2013/472432
44. Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, et al. Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab (2008) 7:159–72. doi:10.1016/J.CMET.2007.11.003
45. Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, et al. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature (2000) 403:328–32. doi:10.1038/35002111
46. Burotto M, Chiou VL, Lee J-M, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer (2014) 120(22):3446–56. doi:10.1002/cncr.28864
47. Carlson CJ, Koterski S, Sciotti RJ, Poccard GB, Rondinone CM. Enhanced basal activation of mitogen-activated protein kinases in adipocytes from type 2 diabetes: potential role of p38 in the downregulation of GLUT4 expression. Diabetes (2003) 52:634–41. doi:10.2337/diabetes.52.3.634
48. Bost F, Aouadi M, Caron L, Binétruy B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie (2005) 87:51–6. doi:10.1016/J.BIOCHI.2004.10.018
49. Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem (1998) 273:10609–17. doi:10.1074/JBC.273.17.10609
50. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39:347–54. doi:10.1016/j.tibs.2014.06.005
51. Park J, Rho HK, Kim KH, Choe SS, Lee YS, Kim JB. Overexpression of glucose-6-phosphate dehydrogenase is associated with lipid dysregulation and insulin resistance in obesity. Mol Cell Biol (2005) 25:5146–57. doi:10.1128/MCB.25.12.5146-5157.2005
52. Carette C, Dubois-Laforgue D, Gautier J-F, Timsit J. Diabetes mellitus and glucose-6-phosphate dehydrogenase deficiency: from one crisis to another. Diabetes Metab (2011) 37:79–82. doi:10.1016/j.diabet.2010.09.004
53. Aird KM, Zhang R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett (2015) 356:204–10. doi:10.1016/j.canlet.2014.01.017
54. Engström Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem (1985) 260:9114–6.
55. Kolberg M, Strand KR, Graff P, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta Proteins Proteomics (2004) 1699(1–2):1–34. doi:10.1016/j.bbapap.2004.02.007
56. Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature (2000) 404:42–9. doi:10.1038/35003506
57. Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet (2007) 39:776–80. doi:10.1038/ng2040
58. Håkansson P, Hofer A, Thelander L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem (2006) 281:7834–41. doi:10.1074/jbc.M512894200
59. Pontarin G, Ferraro P, Bee L, Reichard P, Bianchi V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc Natl Acad Sci U S A (2012) 109:13302–7. doi:10.1073/pnas.1211289109
60. Bornstein B, Area E, Flanigan KM, Ganesh J, Jayakar P, Swoboda KJ, et al. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul Disord (2008) 18:453–9. doi:10.1016/j.nmd.2008.04.006
61. Palmieri F, Monne M. Discoveries, metabolic roles and diseases of mitochondrial carriers: a review. Biochim Biophys Acta (2016) 1863:2362–78. doi:10.1016/j.bbamcr.2016.03.007
62. Gutierrez-Aguilar M, Baines CP. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem J (2013) 454:371–86. doi:10.1042/BJ20121753
63. Clemencon B, Babot M, Trezeguet V. The mitochondrial ADP/ATP carrier (SLC25 family): pathological implications of its dysfunction. Mol Aspects Med (2013) 34:485–93. doi:10.1016/j.mam.2012.05.006
64. Baldwin SA, Beal PR, Yao SYM, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch (2004) 447:735–43. doi:10.1007/s00424-003-1103-2
65. Gray JH, Owen RP, Giacomini KM. The concentrative nucleoside transporter family, SLC28. Pflugers Arch (2004) 447:728–34. doi:10.1007/s00424-003-1107-y
66. Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Abu Libdeh A, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet (2008) 83:529–34. doi:10.1016/j.ajhg.2008.09.013
67. Arner ES, Eriksson S. Mammalian deoxyribonucleoside kinases. Pharmacol Ther (1995) 67:155–86. doi:10.1016/0163-7258(95)00015-9
68. El-Hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. Biochim Biophys Acta (2017) 1863:1539–55. doi:10.1016/j.bbadis.2017.02.017
69. Freisinger P, Fütterer N, Lankes E, Gempel K, Berger TM, Spalinger J, et al. Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations. Arch Neurol (2006) 63:1129–34. doi:10.1001/archneur.63.8.1129
70. Ronchi D, Garone C, Bordoni A, Gutierrez Rios P, Calvo SE, Ripolone M, et al. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain (2012) 135:3404–15. doi:10.1093/brain/aws258
71. Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet (2001) 28:223–31. doi:10.1038/90058
72. Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu Rev Med (2008) 59:131–46. doi:10.1146/annurev.med.59.053006.104646
73. Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, et al. Mutant mitochondrial helicase twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A (2005) 102:17687–92. doi:10.1073/pnas.0505551102
74. Krasich R, Copeland WC. DNA polymerases in the mitochondria: a critical review of the evidence. Front Biosci (Landmark Ed) (2017) 22:692–709. doi:10.2741/4510
75. Singh B, Owens KM, Bajpai P, Desouki MM, Srinivasasainagendra V, Tiwari HK, et al. Mitochondrial DNA polymerase POLG1 disease mutations and germline variants promote tumorigenic properties. PLoS One (2015) 10:e0139846. doi:10.1371/journal.pone.0139846
76. Singh KK, Ayyasamy V, Owens KM, Koul MS, Vujcic M. Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. J Hum Genet (2009) 54:516–24. doi:10.1038/jhg.2009.71
77. Tervasmaki A, Mantere T, Hartikainen JM, Kauppila S, Lee H-M, Koivuluoma S, et al. Rare missense mutations in RECQL and POLG associate with inherited predisposition to breast cancer. Int J Cancer (2018). doi:10.1002/ijc.31259
78. Linkowska K, Jawien A, Marszalek A, Malyarchuk BA, Tonska K, Bartnik E, et al. Mitochondrial DNA polymerase gamma mutations and their implications in mtDNA alterations in colorectal cancer. Ann Hum Genet (2015) 79(5):320–8. doi:10.1111/ahg.12111
79. Long X, Wang X, Chen Y, Guo X, Zhou F, Fan Y, et al. Polymorphisms in POLG were associated with the prognosis and mtDNA content in hepatocellular carcinoma patients. Bull Cancer (2017) 104:500–7. doi:10.1016/j.bulcan.2017.02.005
80. Aird KM, Worth AJ, Snyder NW, Lee JV, Sivanand S, Liu Q, et al. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep (2015) 11:893–901. doi:10.1016/j.celrep.2015.04.014
81. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther (2016) 15:1781–91. doi:10.1158/1535-7163.MCT-15-0945
82. Guleria A, Chandna S. ATM kinase: much more than a DNA damage responsive protein. DNA Repair (Amst) (2016) 39:1–20. doi:10.1016/j.dnarep.2015.12.009
83. Parks DA, Granger DN. Xanthine oxidase: biochemistry, distribution and physiology. Acta Physiol Scand Suppl (1986) 548:87–99.
84. Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med (2016) 5:546–57. doi:10.1002/cam4.601
85. Chen C, Lu J-M, Yao Q. Hyperuricemia-related diseases and xanthine oxidoreductase (XOR) inhibitors: an overview. Med Sci Monit (2016) 22:2501–12. doi:10.12659/MSM.899852
86. Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science (2000) 289:782–5. doi:10.1126/science.289.5480.782
87. Favre C, Zhdanov A, Leahy M, Papkovsky D, O’Connor R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene (2010) 29:3964–76. doi:10.1038/onc.2010.146
88. Floyd S, Favre C, Lasorsa FM, Leahy M, Trigiante G, Stroebel P, et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol Biol Cell (2007) 18:3545–55. doi:10.1091/mbc.E06-12-1109
89. Kenerson HL, Subramanian S, McIntyre R, Kazami M, Yeung RS. Livers with constitutive mTORC1 activity resist steatosis independent of feedback suppression of akt. PLoS One (2015) 10(2):e0117000. doi:10.1371/journal.pone.0117000
90. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature (2004) 431:200–5. doi:10.1038/nature02866
91. Kim LC, Cook RS, Chen J. MTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene (2017) 36(16):2191–201. doi:10.1038/onc.2016.363
92. Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol (2016) 2:1565–73. doi:10.1001/jamaoncol.2016.0891
93. Malley CO, Pidgeon GP. The mTOR pathway in obesity driven gastrointestinal cancers: potential targets and clinical trials. BBA Clin (2016) 5:29–40. doi:10.1016/j.bbacli.2015.11.003
94. Sears RC. The life cycle of c-Myc: from synthesis to degradation. Cell Cycle (2004) 3(9):1133–7. doi:10.4161/cc.3.9.1145
95. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res (2012). doi:10.1158/1078-0432.CCR-12-0977
96. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A (2008) 105:18782–7. doi:10.1073/pnas.0810199105
97. Riu E, Ferre T, Mas A, Hidalgo A, Franckhauser S, Bosch F. Overexpression of c-myc in diabetic mice restores altered expression of the transcription factor genes that regulate liver metabolism. Biochem J (2002) 368:931–7. doi:10.1042/BJ20020605
98. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng (2017) 19:163–94. doi:10.1146/annurev-bioeng-071516-044546
99. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A (2007) 104:19345–50. doi:10.1073/pnas.0709747104
100. Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell (2003) 112:391–401. doi:10.1016/S0092-8674(03)00075-8
101. Le TM, Poddar S, Capri JR, Abt ER, Kim W, Wei L, et al. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat Commun (2017) 8:241. doi:10.1038/s41467-017-00221-3
102. Shimizu I, Yoshida Y, Katsuno T, Tateno K, Okada S, Moriya J, et al. P53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab (2012) 15:51–64. doi:10.1016/j.cmet.2011.12.006
103. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 8(8):592–602. doi:10.1007/s13238-014-0082-8
104. Lee J-W, Choi AH, Ham M, Kim J-W, Choe SS, Park J, et al. G6PD up-regulation promotes pancreatic beta-cell dysfunction. Endocrinology (2011) 152(3):793–803. doi:10.1210/en.2010-0606
105. Ham M, Choe SS, Shin KC, Choi G, Kim JW, Noh JR, et al. Glucose-6-phosphate dehydrogenase deficiency improves insulin resistance with reduced adipose tissue inflammation in obesity. Diabetes (2016) 65:2624–38. doi:10.2337/db16-0060
106. Wang L, Pi Z, Liu S, Liu Z, Song F. Targeted metabolome profiling by dual-probe microdialysis sampling and treatment using Gardenia jasminoides for rats with type 2 diabetes. Sci Rep (2017) 7:10105. doi:10.1038/s41598-017-10172-w
107. Xia J-F, Liang Q-L, Liang X-P, Wang Y-M, Hu P, Li P, et al. Ultraviolet and tandem mass spectrometry for simultaneous quantification of 21 pivotal metabolites in plasma from patients with diabetic nephropathy. J Chromatogr B Analyt Technol Biomed Life Sci (2009) 877:1930–6. doi:10.1016/j.jchromb.2009.05.047
108. Li L, Liu DW, Yan HY, Wang ZY, Zhao SH, Wang B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: evidence from a meta-analysis of 21 cohort studies. Obes Rev (2016) 17:510–9. doi:10.1111/obr.12407
109. Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol (2012) 56:704–13. doi:10.1016/j.jhep.2011.09.020
110. Gallagher EJ, LeRoith D. Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol Rev (2015) 95:727–48. doi:10.1152/physrev.00030.2014
111. Dann SG, Selvaraj A, Thomas G. mTOR complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med (2007) 13(6):252–9. doi:10.1016/j.molmed.2007.04.002
112. Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep (2013) 3:1252–65. doi:10.1016/j.celrep.2013.03.004
113. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell (2011) 145:435–46. doi:10.1016/j.cell.2011.03.044
114. Fatkhutdinov N, Sproesser K, Krepler C, Liu Q, Brafford PA, Herlyn M, et al. Targeting RRM2 and mutant BRAF is a novel combinatorial strategy for melanoma. Mol Cancer Res (2016) 14:767–75. doi:10.1158/1541-7786.mcr-16-0099
115. Xu X, Page JL, Surtees JA, Liu H, Lagedrost S, Lu Y, et al. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res (2008) 68:2652–60. doi:10.1158/0008-5472.CAN-07-5873
116. Hofer A, Crona M, Logan DT, Sjöberg BM. DNA building blocks: keeping control of manufacture. Crit Rev Biochem Mol Biol (2012) 47(1):50–63. doi:10.3109/10409238.2011.630372
117. Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem (2006) 75:681–706. doi:10.1146/annurev.biochem.75.103004.142443
118. Eriksson S, Graslund A, Skog S, Thelander L, Tribukait B. Cell cycle-dependent regulation of mammalian ribonucleotide reductase. The S phase-correlated increase in subunit M2 is regulated by de novo protein synthesis. J Biol Chem (1984) 259:11695–700.
119. D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell (2012) 149:1023–34. doi:10.1016/j.cell.2012.03.043
120. Foskolou IP, Jorgensen C, Leszczynska KB, Olcina MM, Tarhonskaya H, Haisma B, et al. Ribonucleotide reductase requires subunit switching in hypoxia to maintain DNA replication. Mol Cell (2017) 66:206–220.e9. doi:10.1016/j.molcel.2017.03.005
121. Kimura T, Takeda S, Sagiya Y, Gotoh M, Nakamura Y, Arakawa H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat Genet (2003) 34:440–5. doi:10.1038/ng1212
122. Liu X, Xue L, Yen Y. Redox property of ribonucleotide reductase small subunit M2 and p53R2. Methods Mol Biol (2008) 477:195–206. doi:10.1007/978-1-60327-517-0_15
123. Elledge SJ, Zhou Z, Allen JB, Navas TA. DNA damage and cell cycle regulation of ribonucleotide reductase. Bioessays (1993) 15:333–9. doi:10.1002/bies.950150507
124. Aird KM, Li H, Xin F, Konstantinopoulos PA, Zhang RG. Identification of ribonucleotide reductase M2 as a potential target for pro-senescence therapy in epithelial ovarian cancer. Cell Cycle (2014) 13:199–207. doi:10.4161/cc.26953
125. Dressman HK, Hans C, Bild A, Olson JA, Rosen E, Marcom PK, et al. Gene expression profiles of multiple breast cancer phenotypes and response to neoadjuvant chemotherapy. Clin Cancer Res (2006) 12:819–26. doi:10.1158/1078-0432.CCR-05-1447
126. Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T, Nakata K, et al. Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine-based adjuvant chemotherapy. Neoplasia (2010) 12:807–17. doi:10.1593/neo.10458
127. Liu X, Zhou B, Xue L, Yen F, Chu P, Un F, et al. Ribonucleotide reductase subunits M2 and p53R2 are potential biomarkers for metastasis of colon cancer. Clin Color Cancer (2007) 6:374–81. doi:10.3816/CCC.2007.n.007
128. Sagan L. On the origin of mitosing cells. J Theor Biol (1967) 14:225–IN6. doi:10.1016/0022-5193(67)90079-3
129. Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res (2012) 111(9):1198–207. doi:10.1161/CIRCRESAHA.112.268946
130. Gandhi VV, Samuels DC. Correlated tissue expression of genes of cytoplasmic and mitochondrial nucleotide metabolisms in normal tissues is disrupted in transformed tissues. Nucleosides Nucleotides Nucleic Acids (2012) 31:112–29. doi:10.1080/15257770.2011.644101
132. Lee W, Johnson J, Gough DJ, Donoghue J, Cagnone GLM, Vaghjiani V, et al. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis (2015) 6:e1664. doi:10.1038/cddis.2015.34
133. Zhou X, Kannisto K, Curbo S, von Dobeln U, Hultenby K, Isetun S, et al. Thymidine kinase 2 deficiency-induced mtDNA depletion in mouse liver leads to defect beta-oxidation. PLoS One (2013) 8:e58843. doi:10.1371/journal.pone.0058843
134. Bao XR, Ong S, Goldberger O, Peng J, Sharma R, Thompson DA, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife (2016) 5:e10575. doi:10.7554/eLife.10575
135. Nikkanen J, Forsström S, Euro L, Paetau I, Kohnz RA, Wang L, et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab (2016) 23:635–48. doi:10.1016/j.cmet.2016.01.019
136. Wallace DC. Mitochondrial diseases in man and mouse. Science (1999) 283(5407):1482–8. doi:10.1126/science.283.5407.1482
137. Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect (2014) 4:R1–15. doi:10.1530/EC-14-0092
138. Koliaki C, Roden M. Alterations of mitochondrial function and insulin sensitivity in human obesity and diabetes mellitus. Annu Rev Nutr (2016):1–31. doi:10.1146/annurev-nutr-071715-050656
139. Kamihara J, Rana HQ, Garber JE. Germline TP53 mutations and the changing landscape of Li-Fraumeni syndrome. Hum Mutat (2014) 35(6):654–62. doi:10.1002/humu.22559
141. Dahl ES, Aird KM. Ataxia-telangiectasia mutated modulation of carbon metabolism in cancer. Front Oncol (2017) 7:291. doi:10.3389/fonc.2017.00291
142. Bar RS, Levis WR, Rechler MM, Harrison LC, Siebert C, Podskalny J, et al. Extreme insulin resistance in ataxia telangiectasia: defect in affinity of insulin receptors. N Engl J Med (1978) 298:1164–71. doi:10.1056/NEJM197805252982103
143. McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol (2012) 7:303–21. doi:10.1146/annurev-pathol-011811-132509
144. McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep (2004) 5:772–6. doi:10.1038/sj.embor.7400210
145. Dore MP, Davoli A, Longo N, Marras G, Pes GM. Glucose-6-phosphate dehydrogenase deficiency and risk of colorectal cancer in Northern Sardinia: a retrospective observational study. Medicine (Baltimore) (2016) 95:e5254. doi:10.1097/MD.0000000000005254
146. Manganelli G, Masullo U, Passarelli S, Filosa S. Glucose-6-phosphate dehydrogenase deficiency: disadvantages and possible benefits. Cardiovasc Hematol Disord Drug Targets (2013) 13:73–82. doi:10.2174/1871529X11313010008
147. Pes GM, Bassotti G, Dore MP. Colorectal cancer mortality in relation to glucose-6-phosphate dehydrogenase deficiency and consanguinity in Sardinia: a spatial correlation analysis. Asian Pac J Cancer Prev (2017) 18:2403–7. doi:10.22034/APJCP.2017.18.9.2403
148. Fini MA, Elias A, Johnson RJ, Wright RM. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin Transl Med (2012) 1:16. doi:10.1186/2001-1326-1-16
149. Sautin YY, Johnson RJ. Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids (2008) 27(6):608–19. doi:10.1080/15257770802138558
150. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact (2006) 160(1):1–40. doi:10.1016/j.cbi.2005.12.009
151. Lu W, Xu Y, Shao X, Gao F, Li Y, Hu J, et al. Uric acid produces an inflammatory response through activation of NF-κB in the hypothalamus: implications for the pathogenesis of metabolic disorders. Sci Rep (2015) 5. doi:10.1038/srep12144
152. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med (2010) 49(11):1603–16. doi:10.1016/j.freeradbiomed.2010.09.006
153. Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol (2002) 3(7):415–24. doi:10.1016/S1470-2045(02)00788-X
154. Muñoz-Pinedo C, El Mjiyad N, Ricci J-E. Cancer metabolism: current perspectives and future directions. Cell Death Dis (2012) 3:e248. doi:10.1038/cddis.2011.123
155. de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol (2014) 741:8–16. doi:10.1016/j.ejphar.2014.07.041
156. Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to gemcitabine. Cancers (Basel) (2017) 9. doi:10.3390/cancers9110157
Keywords: purines, pyrimidines, c-Myc, p53, mTORC1, diabetes, obesity
Citation: Buj R and Aird KM (2018) Deoxyribonucleotide Triphosphate Metabolism in Cancer and Metabolic Disease. Front. Endocrinol. 9:177. doi: 10.3389/fendo.2018.00177
Received: 09 February 2018; Accepted: 03 April 2018;
Published: 18 April 2018
Edited by:
Che-Pei Kung, Washington University in St. Louis, United StatesReviewed by:
Marco Falasca, Curtin University, AustraliaEva Surmacz, Temple University, United States
Copyright: © 2018 Buj and Aird. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine M. Aird, a2FpcmRAcHN1LmVkdQ==