 Jessie M. Barra
Jessie M. Barra Hubert M. Tse
Hubert M. Tse- Department of Microbiology, Comprehensive Diabetes Center, University of Alabama at Birmingham, Birmingham, AL, United States
Type 1 diabetes is an autoimmune disease that results in the progressive destruction of insulin-producing pancreatic β-cells inside the islets of Langerhans. The loss of this vital population leaves patients with a lifelong dependency on exogenous insulin and puts them at risk for life-threatening complications. One method being investigated to help restore insulin independence in these patients is islet cell transplantation. However, challenges associated with transplant rejection and islet viability have prevented long-term β-cell function. Redox signaling and the production of reactive oxygen species (ROS) by recipient immune cells and transplanted islets themselves are key players in graft rejection. Therefore, dissipation of ROS generation is a viable intervention that can protect transplanted islets from immune-mediated destruction. Here, we will discuss the newly appreciated role of redox signaling and ROS synthesis during graft rejection as well as new strategies being tested for their efficacy in redox modulation during islet cell transplantation.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by chronic inflammation where self-reactive immune responses selectively target and destroy β-cells within the pancreas. In a majority of patients, insulin therapies can help regulate the rapid variations in blood glucose levels that result from this autoimmune attack, however, this is not a cure and for a relatively large number of patients, exogenous insulin treatment is not enough for them to maintain stable blood glucose levels (1). A major hurdle for insulin therapy is the ability to optimally sense and respond to glucose fluctuations as rapidly or precisely as a living β-cell. Therefore, the constant struggle to achieve efficient glucose homeostasis still persists. The death of these vital insulin-secreting cells within the islets of Langerhans and the resulting glucose dysregulation leaves patients at risk for developing serious life-threatening complications including cardiovascular disease, neuropathy, and renal failure (2).
Poor glucose control and the prevalence of secondary risks associated with T1D have lead researchers to investigate alternative treatment options for these patients. Unfortunately, in humans, there are currently no adequate metrics to detect these diabetic patients before the onset of autoimmunity. Usually, the presentation of symptoms, such as fatigue, extreme thirst, polyuria, or weight loss prompts a visit to a health professional, and only then does the presence of autoantibodies in their blood lead to their diagnosis. However, even at the time of diagnosis these patients still have some functioning β-cell mass remaining. Attempts have been made to delay or reverse the severity of T1D after diagnosis and to prevent further β-cell loss by utilizing immunotherapies to dampen autoreactive responses. A few of these therapies include the inhibitory cytotoxic T lymphocyte associated antigen-4 (CTLA-4)-Ig to block effector T cells (Teffs) (3), anti-CD20 therapy to deplete functional B cell responses and increase regulatory T cell (Treg) responses (4), and interleukin 1 beta (IL-1β)/IL-1R antagonists including anakinra and canakinumab to neutralize inflammatory signaling cascades including MAPK and NF-κB pathways (5). Clinical trials utilizing these treatments in early onset T1D patients displayed variable efficacy for maintaining higher C peptide levels with less reliance on exogenous insulin, however, any benefits were only temporary, and treatment was not successful in halting the progression of disease (3–5). The persistent challenges in early detection and the minimal effectiveness of immunotherapies have lead to a search for alternative treatment options to restore the functionality of insulin regulation in individuals after the destruction of β-cells has already occurred. One such attractive therapy is islet transplantation.
Islet cell transplantation is a more permanent alternative to exogenous insulin therapies with fewer long-term complications. By restoring functional β-cells into these patients, they can more accurately modulate their blood glucose levels and diminish the risks associated with glucose fluctuations. Unfortunately, as with any other organ or tissue transplant, immune-mediated graft rejection as well as an initial loss in islet graft viability induced by oxidative stress and inflammation continue to pose challenges for the long-term success of this strategy. There is also damage associated with the recurrence of autoimmunity toward the graft in the T1D patient as well as islet-specific risks of immunosuppression. In addition to the low survivability of the islet graft, other barriers to widespread utilization of this therapy include the sensitivity of islets toward hypoxia, redox-associated mechanical and chemical damage during isolation, and poor viability and islet yield from human cadaveric donors (6, 7). Subsequently, efficient human islet transplantation normally requires more than one cadaveric donor per recipient and some patients require consecutive transplants to prolong adequate blood glucose regulation (8).
Despite the hurdles that still need to be overcome, islet transplantation has come a long way in the last three decades. Prior to the late 1990s, islet transplantation into human patients had very little success in maintaining euglycemia and preventing hypoglycemic events, with few patients being insulin-independent beyond 1 week after transplantation (9). Since the 1980s, digestive enzymes and a mechanical shaking process known as the Ricordi method have been used to isolate islets (10, 11). Then, in 2000, a group led by Dr. James Shapiro at the University of Alberta published what would come to be known as the Edmonton protocol (12). This small clinical trial followed seven patients beyond 1 year after intraportal islet transplantation. The Edmonton protocol revolutionized the way human islets were isolated by utilizing xenoprotein-free isolation media and transplanting the purified islets directly after isolation, eliminating the dangers of islet culture. The islets were infused into the portal vein and utilized a novel combination of immunosuppression including sirolimus, low dose tacrolimus, and daclizumab, a monoclonal antibody that recognizes CD25. All recipients attained insulin independence for nearly 5 months after transplantation. This marked a profound improvement in patient outcomes compared to previous reports, and the protocol was soon adopted worldwide. In the nearly two decades since the Edmonton protocol was reported, advancements in our understanding of islet biology, islet graft viability, methods to protect islets following isolation, in vitro culture, and islet transplantation has improved. According to the 2016 Collaborative Islet Transplant Registry 9th Annual Report, 50% of recipients maintain insulin independence beyond 1 year posttransplantation, and around 20% of islet transplant recipients are insulin-independent after 5 years.
Ultimately, one challenge that still persists is the harmful side effects of immunosuppressive drugs to the patient as well as the islet graft (13). These anti-rejection medications inhibit the adaptive immune response; however, most of them do not protect the graft from redox-mediated destruction or direct autoimmune inflammatory interactions. In fact, the use of corticosteroids and tacrolimus can cause serious adverse effects including diabetogenicity and elevated extracellular reactive oxygen species (ROS) production in the islets themselves (14–17). It has been shown that immunosuppression with tacrolimus, sirolimus, and anti-IL-2R can even promote the proliferation of autoreactive memory T cells due to a chronic increase in serum IL-7 and IL-15 levels (18), potentially leading to a recurrence of autoimmunity. Tacrolimus and sirolimus have also been shown to impair mitochondrial calcium uptake and ATP production (19, 20), which are key steps in the glucose responsiveness of β-cells (21, 22).
Although the mechanisms that contribute to autoreactive immune responses in T1D and islet transplantation are not fully understood, what has become clear is the significant impact inflammation and oxidative stress can have on immune responses, β-cell function, and β-cell survival. Genetic attenuation of superoxide synthesis in the non-obese diabetic (NOD) mouse model through a point mutation in the nicotinamide adenine dinucleotide phosphate [NAD(P)H] oxidase (NOX) complex can influence innate and adaptive immune responses necessary for spontaneous diabetes progression (23–25). The inability to produce superoxide through the NOX complex highlights the important role of ROS generation and inflammation in disease progression, induction of β-cell death, and β-cell dysfunction (26). The generation of free radicals is not inherently a detrimental biological process, as ROS control apoptotic pathways within the cell, and the NOX complex is involved in eradicating microbial infections. While both of these responses are vital to cellular turnover and health, elevated ROS levels can influence cellular proliferation, survival, and the induction of inflammatory signaling cascades to mediate cellular damage (27). The dysregulation of ROS synthesis in an autoimmune setting can contribute to inappropriate activation of the immune system to recognize healthy tissue as foreign. This problem is particularly dangerous if an elevated level of ROS production overwhelms antioxidant defenses, which can result in oxidative stress, ROS-mediated damage, and eventual cell death (28).
In the context of islet transplantation, the role for redox signaling is even more vital due to the relatively low levels of native antioxidant defenses within the β-cell including superoxide dismutase (SOD), catalase, and glutathione peroxidase (Gpx-1), leaving them highly susceptible to ROS-mediated damage (6, 7). The impact of redox signaling within the context of islet destruction is twofold. The presence of oxidative species such as hydrogen peroxide (H2O2) and superoxide anions can impact glucose sensing within the β-cell (29), but they can also serve as a third signal to promote the maturation and expansion of β-cell-specific autoreactive T cell subsets (30–32). These autoreactive immune responses can initiate the destruction of β-cells though either the induction of apoptosis using the FAS pathway or by necrosis through the release of pro-inflammatory cytokines, perforin, granzyme B, and ROS (33, 34).
As scientists begin to appreciate the role of ROS in mediating inflammation and promoting transplant rejection, dissipating oxidative stress is a prime target for immunotherapies during islet cell transplantation to reduce islet vulnerability, boost patient outcomes, and prolong insulin independence (35). One proposed method to address these persistent challenges is to target the production of these reactive species during different stages of islet transplantation. The hope is that attenuating the redox status of the islets themselves or the surrounding microenvironment will promote islet function and prolong graft viability without the need for toxic immunosuppressive drugs.
Immune Mechanisms Involved in Islet Transplantation Rejection
Islet transplantation into patients with T1D comes with a risk for alloimmune responses as well as recurrent autoimmunity. Both responses can utilize redox signaling to facilitate their damaging effects on the islet graft. During allogeneic graft rejection, host immune responses can become activated through either direct or indirect recognition of donor tissues (Figure 1). Direct graft recognition involves the interaction of donor tissue-resident antigen-presenting cells (APCs) and host T cells through an MHC-mismatch interaction (36, 37). Indirect recognition involves the processing of donor graft peptides by host APCs to stimulate host T cells through the corresponding MHC interactions. Both direct and indirect recognition pathways require the involvement of co-stimulatory molecules to trigger and activate T cell responses. To understand why these aberrant signaling pathways and the corresponding redox responses are vital at various stages of islet transplantation, it is necessary to acknowledge the interplay between redox signaling and inflammatory responses. While others have examined certain specific pathways in great depth (38, 39), this review will highlight pathways involved in redox-dependent inflammation.
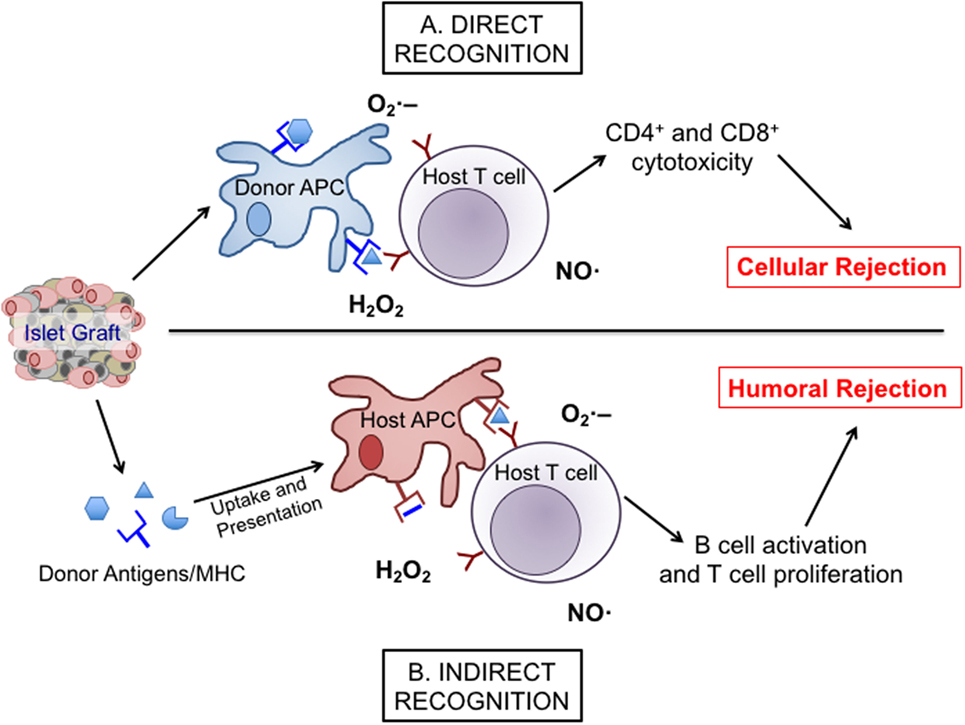
Figure 1. The direct and indirect recognition pathways of islet allograft destruction. After transplantation, islet-resident antigen-presenting cells (APCs) from the donor graft can present directly to host T cells with stimulatory signals provided by reactive oxygen species, such as , •OH, and H2O2 to promote the expansion and activation of alloreactive cytotoxic T cell subsets. This stimulation can then activate inducible nitric oxide synthase (iNOS), NAD(P)H oxidase, and mitochondrial oxidative pathways within the T cell to produce more nitric oxide (NO), hydrogen peroxide, and superoxide, eventually leading to cellular rejection via the direct pathway (A). Alternatively, islet antigens can be shed into the surrounding environment following transplantation to be engulfed and presented by host APCs and then indirectly presented to host T cells. Through co-stimulation and the release of , •OH, H2O2, and NO, those APCs promote a classical antibody response involving the activation and expansion of alloreactive T cells and humoral rejection (B).
Direct Recognition and Redox Signaling
In allogeneic transplantation, the direct recognition pathway involves donor APCs interacting with host effector CD4 and CD8 T cells to facilitate contact-mediated allograft rejection (40, 41). During islet transplantation, the direct recognition pathway stimulates a cellular rejection response in which direct killing of the islet graft by T cells is the primary endpoint (37). Several studies indicate that there are two requirements to execute direct islet allograft recognition and damage: the production of interferon gamma (IFN-γ) by T cells (42) and the initiation of apoptotic pathways through perforin and/or the use of Fas/FasL interactions between activated T cells and target tissues (43). Both of these mechanisms involved in islet allograft destruction have redox-dependent components that are intimately connected to their inflammatory responses.
The production of IFN-γ as well as other inflammatory mediators by APCs and by T cells is a tightly controlled process. One key regulator of inflammatory cytokine production involves the redox status of intracellular thiols. Reduced glutathione (GSH) is the most abundant free thiol in mammalian cells and is an important regulator of multiple cellular processes (44, 45). During stress conditions, GSH is oxidized into glutathione disulfide, leading to the activation of damage responses within the cell including the unfolded protein response (UPR) and apoptosis (45). Dendritic cells and macrophages with elevated levels of intracellular GSH produce more IFN-γ than those with low intracellular GSH levels (46, 47). This increased inflammatory profile by APCs can skew T cell responses through the synthesis of T cell polarizing cytokines such as IL-12 (47) and in an autocrine fashion to further promote APC activation (48). Once macrophages are activated, they produce large amounts of ROS as well as IFN-γ and IL-1β (38). These inflammatory signaling molecules aid in macrophage killing of target pathogens or facilitate islet graft destruction by mediating phagocytosis of β-cell debris.
Interferon gamma, tumor necrosis factor alpha (TNF-α), and IL-1β not only perpetuate damaging innate and adaptive immune responses but they also interact with their cognate cytokine receptors on the surface of the β-cell. Engagement of these β-cell surface receptors can initiate the activation of the RAS signaling cascade (49). Through a string of downstream phosphorylation events, the RAS pathway leads to the activation of MAPK and Myc, which can enter the nucleus and induce the transcription of genes involved in cell division, survival, and the production of inflammatory mediators (50). This pathway is redox-mediated through mitochondrial hydrogen peroxide activation of the Jun N-terminal protein kinase (JNK), which activates MAPK and stress pathways to further propagate inflammatory cytokine synthesis and apoptotic cell death (51).
The presence of ROS such as superoxide and hydrogen peroxide can also play a role during contact-dependent damage within the islet graft since the maturation of CD8 T cells to become cytolytic is a redox-dependent process (31). As a consequence of CD8 T cell activation by donor APCs, contact-mediated production of perforin and granzyme B by cytotoxic lymphocytes can permeabilize cells within the islet graft (52). Once these toxic molecules engage and enter the cell membrane, they initiate caspase-signaling cascades, which lead to either the direct initiation of cellular apoptosis through caspases-3 and -7 or to the cleavage of pro-apoptotic Bcl-2 family member, Bid, by caspase-8 (53). Bid then binds to the mitochondrial membrane and activates mitochondrial outer membrane permeabilization, stimulating the release of cytochrome c to kill the cell (54).
Redox regulation of apoptotic pathways has many facets that are detailed elsewhere (45), however, it is important to note that the primary role for ROS during apoptotic cascades involves intrinsic pathways. Mitochondria inside the β-cell can initiate stress-induced production of superoxide, which can then be converted into hydrogen peroxide and hydroxyl radicals (•OH) through the Fenton reaction (55). These radical ions are potent inducers of further redox-mediated DNA and protein damage inside the cell (56) and can lead to further apoptosis or inflammatory processes that negatively impact the viability of the graft.
Indirect Recognition and Redox Signaling
Indirect recognition of allogeneic transplants involves the interaction between host APCs with host T cells. The host APC will process and present graft antigens on MHC II molecules to activate host CD4 T cells (40, 41). The indirect recognition pathway can promote two major forms of immune responses: the humoral B cell/antibody response through the interaction between CD4 follicular helper T cells with naïve B cells and the continual activation of innate responses, namely macrophage-associated killing (37).
The transition of a B cell to a terminally differentiated plasma cell requires various cellular and metabolic changes, some of which have redox components. H2O2 is involved in B cell receptor signaling and activation (57). In addition, as a B cell transitions to a differentiated plasma cell, the endoplasmic reticulum (ER) undergoes drastic reorganization and expansion. During ER stress, a host of ER-based enzymes generate ROS as byproducts, leading to multiple changes in antibody production, i.e., the switch to IgM (58) and proliferation within these newly formed plasma cells (59, 60). Specifically, it has been demonstrated that oxidation of Keap1, a negative regulator of the antioxidant response, allows for the nuclear internalization of Nrf2 and transcriptional activation of various target genes involved in B cell differentiation and antioxidant defenses (61).
After differentiation, antibodies produced by activated plasma cells can bind to the islet graft and activate the complement system to induce apoptosis within the target cell and facilitate islet graft destruction by cytotoxic lymphocytes through Fc binding (62). Antibody responses by B cells are not the only redox-dependent mechanism that can contribute to islet graft destruction. B cells are also capable of producing inflammatory cytokines including IL-6 that is redox regulated (63, 64). When a B cell receives co-stimulation through the CD40 surface receptor, the cross-linking of this receptor leads to the generation of ROS and subsequent activation of JNK pathways, resulting in the increased secretion of IL-6 into the surrounding environment (65, 66). IL-6 interacting with the cognate IL-6R can promote activation and proliferation of other immune cells by signaling through JAK2. This protein can initiate the MAPK cascade described above, or interact with STAT3, forming the JAK/STAT complex (67, 68). STAT3 is vital for optimal activation and effector function in T cells because it can directly enter the nucleus and initiate the transcription of inflammatory genes, or activate the NF-κB pathway and affect the cell cycle (69, 70). Therefore, redox regulation of B cell responses including antibody production and secretion of inflammatory cytokines can perpetuate damaging T cell responses to further destroy the islet graft.
Role of Free Radicals and Pro-Inflammatory Mediators Involved in Islet Cell Transplantation Rejection
The interplay between free radicals and inflammatory molecules modulates β-cell dysfunction and death during multiple stages of purification from the pancreas and transplantation. Islets are sensitive to hypoxic stress or damage signals that occur during isolation and culture including pro-inflammatory cytokines and free radicals. Stress or damage caused by hypoxia, cold ischemia, and reperfusion can activate downstream inflammatory cascades including the NF-κB signaling pathway (71–73). After transplantation, immune effector cells including macrophages, neutrophils, B cells, and T cells migrate to the transplant site and target the islet graft for destruction by releasing pro-inflammatory cytokines, antibodies, and free radicals (74–76). Understanding the redox-dependent signaling pathways during islet isolation and following transplantation is vital to the development of novel interventions to improve transplantation success and prevent β-cell dysfunction.
Redox Signaling in Islet Isolation and Culture
Pancreatic islets in their natural setting have rather high oxygen tension, with islets receiving more than 15% of the total pancreatic blood supply (77). This massive influx of blood and nutrients plays a key role in their rapid ability to regulate glucose levels, however, linked to their relatively low levels of antioxidant defenses, it also leaves them highly susceptible to ROS-mediated damage (6, 7). In addition, this sensitivity also further exacerbates their susceptibility to oxidative damage during isolation when they are deprived of that elevated oxygen supply, leaving them in a hypoxic state (78–80).
To separate islet cells from the surrounding tissue of the pancreas, harsh digestive enzymes and mechanical separation techniques are utilized to break down exocrine tissue while leaving the islets mostly intact. These methods, while efficient, induce a level of oxidative and mechanical stress as vascularization and in turn, the nutritional stores of the islets are severed (81). This detachment from the extracellular matrix leaves islets reliant on passive diffusion to survive the isolation and transplantation process (82, 83). Consecutive days incubated under hypoxic conditions in vitro can have serious impacts on islet function and survival. The increase in hypoxia and oxidative stress within in vitro cultured islets can induce DNA damage and the peroxidation of proteins and lipids (84, 85). Mitochondrial-derived stress can cause larger islets to develop a necrotic core as less oxygen is able to diffuse to the cells in the center (86) as well as impacting insulin secretion through stress-associated decreases in mitochondrial Ca2+ uptake (87). Islets compensate for the low availability of oxygen in culture by upregulating transcription factors including hypoxia-inducible factors, which induce transcription of multiple genes including toll-like receptors (TLRs) and genes involved in vascular endothelial growth factor (VEGF) signaling (88, 89). Hypoxic conditions can also activate NF-κB to induce the upregulation of inducible nitric oxide synthase (iNOS) and monocyte chemoattractant protein-1 (MCP-1) expression (71), which can have significant impacts on local inflammation after the islets are transplanted.
Strides have been made in recent decades in an attempt to combat oxidative stress with the advent of less damaging enzymatic digestion methods (90) and isolation techniques to improve cadaveric human islet yield (91), but challenges persist that motivate researchers to find alternative strategies to dampen oxidative stress and hypoxia. One method to protect islets from redox-mediated damage following isolation is to increase expression of detoxifying or antioxidant enzymes. Under normal circumstances, redox scavengers are upregulated in response to inflammatory signals released from cells during times of damage or stress. Unfortunately, the low levels of these scavenging enzymes in islets make it difficult for them to combat redox stress. A few of these key enzymes include SOD, manganese superoxide dismutase (MnSOD), and Gpx-1, antioxidant enzymes that are present at lower levels in islets than in other rodent tissues (7). Therefore, increasing endogenous antioxidant defenses or supplementing with exogenous scavengers could protect isolated islets from oxidative stress.
Strategies to Dampen Redox-Mediated Damage in Isolated Islets
Mechanical and metabolic stress during islet isolation can significantly reduce the number of viable islets available for transplantation. To combat this early loss in islet mass, various groups have attempted to protect purified islets by targeting the redox mechanisms underlying sources of cellular stress. There are two primary techniques utilized to dampen oxidative stress and redox-mediated damage in purified islets: either supplementing culture media with exogenous redox scavengers like MnSOD or genetic manipulation of the islets themselves.
The first method to decrease the oxidative damage that islets endure in culture is to treat with antioxidants after isolation. The activation of both NF-κB and poly (ADP-ribose) polymerase pathways contribute to islet damage during the isolation process (92, 93). Dissipating oxidative stress through the use of a SOD mimetic can decrease NF-κB activation, reduce the production of inflammatory MCP-1 and IL-6 by human islet cells during stress conditions, and reduce and production by macrophages (93, 94). This same antioxidant demonstrated protection from STZ-induced apoptosis during in vitro human islet cultures as well as prolonged islet allotransplant survival in MHC-mismatched mouse models after purified islets were cultured in the presence of the SOD mimetic (95). Systemic administration of the SOD mimetic through the use of sustained release pellets prolonged the viability of allogeneic islet grafts by reducing immune migration to the site of transplantation. The reduction in inflammation and increase in graft viability observed in the above studies is a key step in protecting islets from oxidative stress produced by immune cells and can promote long-term survival of an islet graft. Similarly, a naturally occurring antioxidant from the extract of Chinese bayberries, cyanidin-3-O-glucoside (C3G), was shown to increase expression of heme oxygenase-1, Bcl-2, and survivin, antioxidant, and anti-apoptotic regulators that protect islet cells from oxidative stress in vitro (96, 97). In addition, C3G treatment of isolated islets prior to transplantation demonstrated prolonged graft survival with fewer islet numbers required to induce euglycemia when transplanted either under the kidney capsule or into the hepatic portal vein (97). The use of soluble antioxidants, while somewhat protective, is a short-term treatment option, and once these islets are transplanted, they are still susceptible to immune-mediated damage. Genetic modifications of purified islets may provide a more permanent solution and supply antioxidant protection that can persist long after transplantation.
Viral transduction of isolated islets to overexpress antioxidant genes provides benefits to islet survival not only during in vitro culture but also following transplantation. Transgenic mice overexpressing SOD and Gpx-1 within islets displayed a marked resistance to redox-mediated damage in vitro and improved glycemic control after transplantation under the renal capsule of syngeneic mouse recipients (98). One group genetically altered isolated murine islets to overexpress MnSOD and found that upon transplantation into immunodeficient recipients, the transgenic islets displayed a marked delay in graft failure following adoptive transfer with diabetogenic T cells (99). Similarly, transfection of islets with a lentiviral vector containing thioredoxin, an ROS scavenger, reduced the toxic effects of H2O2 in vitro and prolonged graft viability after transplantation into the kidney capsule of spontaneously diabetic NOD mice (100). Other groups have also shown protective effects of glutamylcysteine ligase and SOD overexpression on islet function (101, 102), further supporting the important role of antioxidant defenses and oxidative stress for the maintenance of islet graft function.
These studies have focused on the treatment of islets after isolation, however, in human islet isolation, another hurdle also exists. Most human islets isolated for transplantation are obtained from cadaveric or brain dead organ donors. Unfortunately, these donor conditions create an elevation of inflammatory and redox-mediated damage to human tissues that can negatively impact islet yield. Therefore, it is not surprising that human islet transplant recipients can require three or more donors to obtain sufficient islet equivalents for a single transplant (103). If human islets will continue to be used for transplantation, the state of the donor before isolation cannot be ignored. One group found that treatment of brain dead rats with exendin-4, a glucagon-like peptide-1 analog that acts to increase insulin secretion and decrease glucagon production (104), served to prevent islet viability loss induced by brain death-related inflammation as well as increasing glucose-stimulated insulin secretion of these isolated islets (105). In addition, exendin-4 has also been shown to reduce hypoxia-related islet injury, reduce oxidative stress, and improved both syngeneic and xenotransplantation survival in mouse transplants (106).
While dissipation of oxidative stress during isolation and culture is important to improve islet yield, viability, and function from donors for transplantation, there are numerous other redox-dependent insults transplanted islets have to withstand to delay graft failure including immune-mediated inflammation. Therefore, defining how the two arms of the immune system facilitate islet transplant rejection, graft failure, and synergize with oxidative stress is important to prolong the survival of transplanted islets into patients with T1D.
Acute Responses and Redox Signaling After Islet Transplantation
Following transplantation, islets are susceptible to acute mechanisms of stress that lead to the loss of a large portion of islet cell mass and function (107). One such stress includes ischemia reperfusion injury as these islet cells are placed back into living tissue. The rapid influx of a multitude of nutrients as well as soluble factors not seen in culture media induces an inflammatory response involving oxidative stress known as the instant blood-mediated inflammatory reaction (IBMIR). This reaction is a nonspecific response by the innate immune system that causes robust coagulation and immune infiltration into and around the islets (88, 108), which leads to the induction of cellular apoptotic signaling pathways and internal activation of oxidative stress within the β-cell.
During allogeneic transplantation into the hepatic portal vein, IBMIR-associated responses cause an instantaneous activation of complement pathways that can lead to thrombosis and significant loss of the islet graft (109, 110). One of the major initiating factors in this response is the expression of tissue factor (TF) by islet endothelial cells (ECs). This factor can lead to the activation of thrombin, platelet activation, and secretion of other inflammatory mediators that can perpetuate inflammation and induce macrophage-directed killing (111–113). Once this cascade has begun, upward of 60% of the islet graft is lost within the first few days (82, 107). This local inflammatory cascade at the site of transplantation can induce tissue-resident macrophages to produce superoxide and hydrogen peroxide to damage surrounding tissues (114, 115). The outflow of these reactive molecules can directly lead to DNA strand breakage and peroxidation of proteins or lipids while also activating a number of signaling pathways shown to induce apoptosis in the vulnerable islet graft (116). Not only can local redox signaling originating from the site of transplantation during IBMIR impact islet survival but also the functionality and glucose responsiveness of the β-cells. Therefore, targeting this reaction immediately after islet transplantation is a good technique to prevent the early loss of islet mass.
Another key innate immune mechanism of inflammation during this early stage of islet transplantation is the release of danger signals known as danger-associated molecular patterns into the extracellular space. These danger signals are highly pro-inflammatory, being recognized by pattern recognition receptors (PRRs) on innate immune cells as well as by epithelial cells (117). One key subset of PRRs are TLRs, which recognize specific pathogen-associated molecular patterns, including lipopolysaccharide, dsRNA, flagellin, and unmethylated CpG. Signaling through these TLRs can lead to downstream activation of MyD88, a myeloid differentiation adaptor protein that plays a key role in signal transduction associated with the activation of immune responses (118, 119). Once activated, MyD88 can initiate NF-κB-dependent transcription, one of the major transcription factors involved in the inflammatory response toward the islet graft (120). One group found that inhibition of MyD88 dimerization with small molecule TJ-M2010-6 in NOD mice displayed reduced onset of diabetes, inhibited insulitis, and suppressed T cell activation (121).
Targeting Redox-Mediated Acute Responses After Islet Transplantation
There are multiple ways to target these acute interactions and prolong graft survival. One of the major stages of IBMIR is coagulation and platelet aggregation around the islet graft. To diminish this damaging reaction, one group utilized α-1 antitrypsin, a serine protease inhibitor to reduce IBMIR coagulation and cytokine-induced inflammation in human islets transplanted into the portal vein of NOD. scid mice (122). Administration of α-1 antitrypsin reduced TF expression by the islets, inhibited neutrophil infiltration, and protected islet grafts from IBMIR-mediated damage. Another strategy involves developmental endothelial locus-1 (Del-1), an endothelial-derived homeostatic factor that has anti-inflammatory properties due to its involvement in leukocyte adhesion (123, 124). The overexpression of Del-1 reduced leukocyte-platelet aggregation, which protected islets from IBMIR-associated damage (124). Alternatively, targeting the production of TF by the islets themselves can be inhibited by nicotinamide treatment (113).
The damage from IBMIR can cause an outflow of cytokines from these early immune effectors that can activate redox-sensitive signaling cascades within the islets. These redox-dependent pathways induce apoptosis within the islet graft and compromise insulin secretion from β-cells (125). In human islets, it has been shown that IFN-α can participate in the early stages of T1D progression by triggering ER stress responses to reduce insulin production (126). This change in insulin production was linked to a functional delay in the rate of proinsulin to insulin conversion within the ER. The role of oxidative stress in this ER response has also been investigated. In particular, it has been suggested that the production of iNOS and nitric oxide (NO) within isolated islets after cytokine exposure can lead to the activation of UPR within the ER (127, 128). During times of cellular stress, misfolded proteins can accumulate within the ER lumen. When this build up occurs, it can cause damage to cellular functions as well as disrupt cell division and survival (129). The UPR cascade is designed to protect the cell by increasing protein degradation, upregulating transcription of protein folding machinery, and reestablishing proper ER function. However, if the UPR is incapable of compensating for the amount of cellular stress, such as in the case of chronic inflammation following islet transplantation, the cell can undergo apoptosis. This has been shown to occur during spontaneous T1D progression in the NOD mouse model. ER stress responses were demonstrated to precede the onset of hyperglycemia in the NOD mouse, establishing a link between redox signaling, ER stress, and the early wave of islet dysfunction seen in T1D models (130). Pre-diabetic NOD mice displayed β-cells with fewer secretory granules and a more fragmented ER when compared to β cells from diabetes resistant mouse strains.
It has been shown that UPR defects in β-cells from both animal models of T1D as well as from human patients can contribute to the pathogenesis of autoimmune diabetes (131). Blocking UPR hyperactivation through the use of tyrosine kinase inhibitors such as KIRA8 and imatinib displayed reductions in ER stress-induced apoptosis and even reversed autoimmune diabetes in the NOD mouse (132, 133). In addition to autoimmune diabetes, targeting the UPR during ER stress may be a potential therapeutic target to delay islet transplant rejection. Negi et al. provided evidence of ER stress being implicated in the high degree of human islet loss during isolation and during the early posttransplantation period when these islets were engrafted into a chronic hyperglycemic environment (134). To circumvent ER stress and apoptosis, one group found that pre-treatment of human islets with glial cell line-derived neurotrophic factor reduced ER stress and improved graft function after transplantation into the kidney capsule of diabetic immunodeficient mice (132, 133, 135).
In addition to ER stress, one of the major problems during islet transplantation is the early loss of functional insulin-producing cells due to hypoxia-related injury (107, 136). Redox reactions are tightly linked to hypoxic and reoxygenation conditions as the cellular electron transport chain of the mitochondria become damaged (136, 137). Due to the intimate link between mitochondrial function and insulin secretion, the mitochondrial stress induced by hypoxic or reoxygenation conditions can induce β-cell dysfunction. It has been demonstrated that even transient exposure to H2O2 can reduce β-cell glucose responsiveness by upward of 40% long after the stress has been removed (138). This decrease in responsiveness correlated with increased mitochondrial ROS and decreased mitochondrial biogenesis, solidifying the link between internal sources of oxidative stress and β-cell dysfunction. In addition, dissipating mitochondrial ROS through antioxidants such as MitoTempo or Mitoquinone can protect β-cells from oxidative damage and increase insulin responsiveness in diabetic conditions (139). Not only can mitochondrial ROS impact insulin secretion but it can also induce further DNA and protein damage (55). Therefore, potentially reducing mitochondrial ROS-mediated damage through the use of redox regulators could protect islets from this initial loss of functional β-cell mass in the few weeks after implantation (85).
To address this problem of reoxygenation, researchers have attempted to use gene delivery or co-culture methods to promote revascularization of these islets in the days after transplantation, thereby limiting oxidative damage and loss of early graft function. One popular method to achieve this is the use of VEGF. This particular growth factor gained attention due to its limited ability to cause secondary side effects as compared to cytokines like transforming growth factor-β (140). Using an adenovirus-based delivery system, the induction of elevated levels of VEGF in human islets resulted in a protective effect from TNF-α and IFN-γ induced apoptosis (141). Researchers also found that the addition of VEGF promoted revascularization in human islets transplanted under the kidney capsule of mouse recipients by promoting the growth of new blood vessel formation (142). Co-expression of VEGF and an IL-1R agonist demonstrated suppressive effects on cytokine- and consequently redox-mediated necrosis and apoptosis (143). Therefore, combinatorial therapies including VEGF expression and IL-1β-dependent signaling blockade demonstrate promise in maintaining stable islet engraftment and function.
Transplantation studies targeting these early immune responses demonstrate some protection for the islet graft; however, the islet graft is still susceptible to immune-mediated damage from adaptive immune effectors. Therefore, targeting one pathway may not be sufficient to prevent redox-mediated islet destruction. Dissipation of only one subset of free radicals provided a modest protective effect and negligible improvement in islet function (144, 145). Perhaps a more comprehensive blockade of redox signaling mechanisms during islet transplantation would improve the duration of islet viability. Support for this hypothesis is demonstrated through the use of cell-permeable catalytic antioxidants, which are effective in delaying streptozocin-induced islet cell death, and decreasing the synthesis of inflammatory cytokines and free radical production by immune cells (31, 94, 95). Treatment of rat islets with metallothionein, a broad antioxidant involved in a wide array of protective stress responses (146), can restore and maintain euglycemia after subcutaneous islet transplantation, a result not seen in untreated control transplants due to the challenge of revascularization under the skin (147). An even more drastic effect was seen with the use of fusion proteins combining metallothionein and SOD to target multiple sources of free radical damage, leading to improved graft survival of syngeneic transplantation models in mice (148).
Taken together, redox-meditated destruction of islet cell grafts can be initiated and perpetuated by a multitude of different signaling pathways induced by immune cells and β-cells. With its multiple intersections with inflammatory responses as well as the production of redox molecules such as an increase in iNOS expression and production of , the NF-κB pathway has become a major target for therapeutic intervention (Figure 2). Using two common NF-κB inhibitors, withaferin A, which inhibits IKKβ and NEMO complex formation (149), or an analog of resveratrol, which blocks the phosphorylation and subsequent nuclear localization of the p65 NF-κB subunit (150), Kanak et al. found that NF-κB blockade reduced the release of C-peptide and proinsulin as well as the production of pro-inflammatory cytokines and chemokines including TNF-α, MCP-1, IL-8, and IL-6 in in vitro human islet and blood co-cultures (88). The use of a natural NF-κB inhibitor, withaferin A, is another good example. The addition of withaferin A induced a decreased expression of five key inflammatory genes, RANTES (CCL5), IP10 (CXCL10), MIG (CXCL9), IL1B, and NOS2 when islets were cultured in the presence of a cytokine cocktail as compared to controls (151), indicating a strong anti-inflammatory response in addition to a reduction in redox mediators involved in this vital signaling cascade.
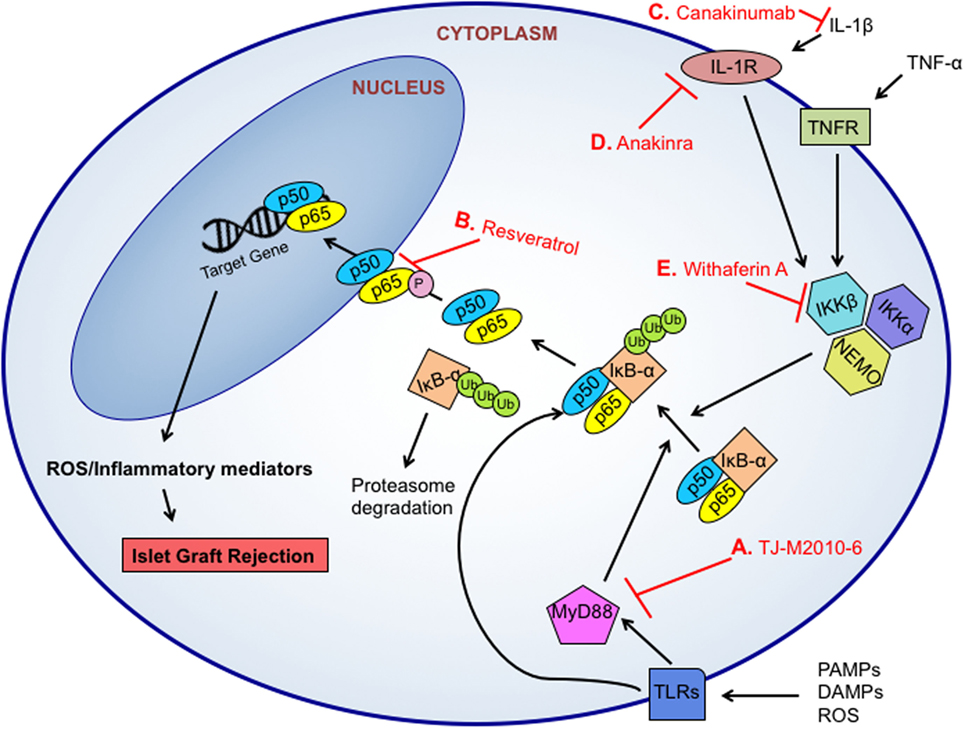
Figure 2. Targeted therapeutic approaches for NF-κB inhibition. NF-κB can induce the transcription of various inflammatory and oxidative molecules to facilitate islet graft rejection. The NF-κB pathway can be triggered by pro-inflammatory cytokine signaling, pathogen-associated molecular patterns (PAMPs)-, danger associated molecular patterns (DAMPs)-, or reactive oxygen species (ROS)-initiated toll-like receptor (TLR)-dependent signals. TLR-signaling activates the MyD88-dependent or MyD88-independent signaling pathways, which results in IKK phosphorylation, IκB-α degradation in the proteasome, and NF-κB (p50/p65) nuclear translocation. Small molecule inhibitors like TJ-M2010-6 can prevent MyD88 activation and IκB-α degradation (A). Resveratrol inhibits the phosphorylation and subsequent nuclear localization of the NF-κB p65 subunit to prevent transcription (B). Signaling from cytokines like interleukin 1 beta (IL-1β) have been targeted through the use of monoclonal antibodies including anti-IL-1 canakinumab (C), which blocks binding of IL-1β to its receptor, and the use of IL-1R antagonist anakinra to block signaling through the receptor (D). The IKK complex is a target for NF-κB inhibition. Specifically, inhibition of IKKβ and NEMO complex formation by withaferin A can prevent the phosphorylation and release of IκB-α (E).
While the blockade of this vital pathway has shown some potential at reducing inflammatory responses, understanding the functional and redox-dependent mechanisms involved in activating these pathways at various stages of islet transplantation is critical to understanding the immune-mediated pathology of islet cell destruction and graft failure. With these overlapping mechanisms in mind, it is not surprising that targeting a single pathway may not be efficacious in eliminating the challenges facing the field of islet cell transplantation. One example is the use of imatinib, which hinders the non-receptor tyrosine kinase c-Abl. This drug was initially used to treat chronic myeloid leukemia; however, several clinical trials also demonstrated improvement or reversal of diabetes phenotypes (152, 153). In animal models, imatinib demonstrated protection from both spontaneous and drug-induced islet death and dysfunction (154–156), and when investigated further, imatinib treatment of human islets demonstrated a decrease in islet inflammation following cytokine exposure (157). There has also been some data indicating that imatinib treatment may be capable of reversing autoimmune diabetes in NOD mice by blunting the ER stress responses within pancreatic β-cells (132, 133). With these biological roles, researchers believed imatinib would be a potent inhibitor of redox-mediated apoptotic pathways. However, when used for in vivo transplantation studies, pre-treatment of islet cells or treatment of recipients posttransplantation did not improve islet transplant outcomes (158). The failure of imatinib treatment to protect islet grafts in vivo serves as a reminder that oxidative damage and redox signaling is complex and a potent mediator of multiple pathways involved in graft failure.
While the above therapies are promising techniques for the reduction of inflammatory reactions, transplanting antigenic islets and delaying graft rejection into a recipient with established autoimmune diabetes is a herculean task. Not only will there be issues of MHC incompatibility in these allotransplant settings, which will potentially mark the graft for destruction, but also an inherent autoimmune response primed and ready to produce signaling molecules and oxidative species to immediately attack the transplanted β-cells is also present. The development of novel therapies that can efficiently decrease adaptive immune responses involved in graft destruction is necessary if there is hope for diminishing graft rejection without the use of immunosuppressants.
Therapies Targeting Adaptive Immune Rejection of Islet Grafts
There are multiple strategies being investigated to suppress the adaptive immune responses that contribute to islet graft destruction, however, two in particular have gained more attention in the last few decades: islet encapsulation strategies to provide a barrier between the sensitive islet graft and the immune system and co-transplantation methods using accessory cells to dampen inflammatory immune responses. Both seek to provide immuoprotection to the islet graft while maintaining the ability for the β-cells to respond to environmental stimuli.
Current strategies for protecting the islet graft against adaptive immune rejection utilize inhibitors of some of the most commonly utilized pathways for inflammatory responses.
One method to provide protection from immune-mediated damage without perpetual dependency is encapsulation of isolated islets with materials designed to delay immune rejection. These materials have gained attention for their potential to provide an immunoprotective and physical barrier between the immune system and newly transplanted islets. Encapsulation aims to produce a semi-permeable membrane around islet cells that allows insulin and other nutrients access across the membrane while excluding larger proteins like antibodies or interactions with immune cells (159). There are three common methods used for encapsulation: (1) an intravascular device, (2) macroencapsulation, and (3) microencapsulation (160–162).
The first category requires the use of a small chamber containing multiple islets, that is, then directly connected to a blood supply (160), and while this type of intravascular device was capable of restoring euglycemia in mouse models (163), the threat of thrombosis made this method unreasonable for clinical use. Macroencapsulation of islets does not require direct attachment to a blood supply and is more attractive for clinical application. However, the thickness of these capsules can impede the transfer of insulin, oxygen, and other nutrients, potentially harming the islets and limiting possible transplantation sites (164, 165). More recently, the development of new technologies including the subcutaneous implantation of islets held within a thin membrane-bound device by TheraCyte can protect insulin-producing cells from the immune system and delay islet allograft rejection (166, 167). In addition, a device by ViaCyte utilizing PEC-01 precursor insulin-producing cells and a subcutaneous transplantation site is currently in a phase 1 clinical trial. Finally, microencapsulation is the encapsulation of a single islet, attempting to address the porosity and mass issues that plagued the earlier methods. Reducing the width and the number of encapsulated islets improves porosity and reduces redox-related injury, however, retrieval of transplanted islets is more difficult (161, 168). Finally, alginate is typically used for islet micro- and macroencapsulation, but is also innately immunogenic due to an inability to generate a completely pure form of this algae-derived compound (169, 170).
While each method above has shown some success in restoring euglycemia in animal models and clinical trials (171), the inability to consistently control the size, shape, and thickness of these capsules continue to hamper long-term success. In addition, addressing the issue of reactive species, which may be small enough to cross these semi-permeable membranes, continues to pose a challenge and is a source of much debate (168, 172). In an attempt to address the setbacks associated with these encapsulation materials, the congregation of microspheres containing suppressive materials with or around these islets may provide a solution. Microspheres are specialized structures comprised from thin layers of cross-linked polymers, which can then be optimized for porosity to suit the desired cellular effect (173, 174). Because this method relies less on a bulky shell, microspheres offer the flexibility to address larger issues such as the cellular microenvironment, both within and outside the capsule. For example, the congregation of curcumin, an anti-apoptotic drug containing free radical scavenging capabilities, with the polymer poly(lactic-co-glycolic acid) to form heterospheroids can decrease oxidative stress and bolster insulin release in rat islets when used as an encapsulation material (174). This technique allows for a localized release of the redox-modulating drug directly at the site of transplantation without degradation in circulation.
Another novel method of redox-dependent protection that does not compromise size, permeability, or charge of the islets is the use of a layer-by-layer (LbL) polymer ultrathin coating. These nanothin layers allow for the manipulation of surface area, permeability, and bioreactivity of the encapsulation material while still providing protection to the encapsulated islets (175). This technique allows for the potential aggregation of different materials into a single, confluent capsule and opens the door for addressing multiple mechanisms of islet transplant destruction. Furthermore, in contrast to other methods, the LbL technique does not require a priming step for adherence of the biomaterial to the islet surface, which has been shown to be detrimental to the stability and viability of islet cells (176). Instead, these ultrathin coatings rely on hydrogen bonding between the lipid polymer and the lipid bilayer of the membrane to form an anchor point which binds the polymer to the surface of the cell (177).
Utilizing this LbL technique, capsules composed of poly(N-vinylpyrrolidone) and tannic acid (TA), a powerful antioxidant, can suppress the production of IFN-γ and TNF-α, pro-inflammatory cytokines, which are key players in islet cell destruction (178, 179). These capsules can scavenge ROS as well as reactive nitrogen species produced by immune cells, demonstrating their redox-modulation capacity. The TA-containing capsules are also efficacious in suppressing pro-inflammatory chemokine production by innate cells, leading to a decrease in T cell trafficking to the site of inflammation and a decrease in T cell activation (180). By reducing immune cell trafficking, these capsules not only serve as a physical barrier to immune destruction but also serve in a localized manner to suppress immune responses without eliciting global immunosuppression.
Other strategies to reduce early loss of β-cell mass include co-transplantation with accessory cells that can enhance islet function, prevent apoptosis, promote vascularization, and provide immunoprotection, including mesenchymal stem cells (MSCs), ECs, Tregs, and myeloid-derived suppressor cells (MDSCs) (181). MSCs are mesodermal multipotent cells that have self-renewing properties and can be isolated from almost every adult tissue (182). They can surround purified islets in culture due to their strong adhesive capabilities and improve islet graft viability and revascularization in both rodent and non-human primate models of co-transplantation (183–185). MSCs can suppress inflammatory immune responses including the proliferation of cytotoxic T cell subsets in part through NO synthesis and inhibition of STAT5 phosphorylation (186). In islet transplantation, Mohammadi Ayenehdeh et al. demonstrated that congregation of adipose tissue-derived MSCs with isolated islets in a hydrogel could maintain euglycemia for more than 30 days during intraperitoneal allotransplant (187). This prolonged islet survival was in part due to an increase in Treg populations as well as a reduction in the inflammatory cytokines IFN-γ and IL-17A.
Another population being investigated for its resistance to IBMIR reactions is ECs. Co-culture of human ECs with isolated pig islets was shown to prevent IBMIR-mediated islet damage both in vitro (188) as well as after co-transplantation into the kidney capsule of diabetic immunodeficient mice (189). Another group found that co-culturing isolated human islets with primary human ECs produced a protective coating that would surround the islets and protect them from IBMIR upon transplantation into the portal vein (190). This co-transplantation strategy also induced a reduction in CD11b+ innate immune cell infiltration into these islet grafts, indicating that the presence of ECs served as an immunoprotective barrier for transplanted islets.
An innate immune cell type that has gained attention for its contact-dependent immunosuppression is the MDSCs. Through their production of superoxide, iNOS, and elevated arginase activity, MDSCs can suppress Teff activation and function while promoting Treg development (191, 192). During allogeneic islet transplantation into the kidney capsule of diabetic mice, co-transplantation of MDSCs increased the presence of Treg cells through the B7-H1/PD-1 pathway (192) and reduced CD8 T cell infiltration by activating iNOS (193).
Regulatory T cells are an immune cell population with the potential to prevent islet graft rejection due to their suppressive effects on immune responses. A recent clinical trial demonstrated that ex vivo expansion and subsequent infusion of autologous human Treg cells in 12 patients with newly diagnosed T1D lowered the patients’ exogenous insulin requirements and prolonged endogenous islet survival (194). The use of Treg cells also show some promise in animal models of islet transplantation, however, due to the short half-life of expanded Tregs as well as challenges in migration from peripheral blood to the site of engraftment (195), alternative strategies to improve Treg localization have been attempted. The combination of CTLA-4, a key protein receptor that downregulates immune responses, and reparixin, which blocks against inflammatory neutrophil infiltration, resulted in lower serum IFN-γ as well as decreased T cell infiltration into the islet graft after transplantation under the kidney capsule (196). Other groups have investigated the use of fusion proteins and antibodies to promote graft survival. Zhang et al. utilized CTLA-4/Fc and demonstrated reduced local inflammation, a concomitant increase in Foxp3+ Treg cells, and improved engraftment (197). Treg cells have also been utilized in co-transplantation strategies where co-aggregation of syngeneic Treg cells with purified allogeneic islets within an agarose hydrogel displayed prolonged allograft survival after transplantation into the portal vein of mice (198).
Conclusion
Due to the comprehensive role of oxidative stress on islet transplantation, targeting redox-dependent inflammatory responses during islet isolation, in vitro culture, and after transplantation has the potential to increase islet viability and function (Figure 3). Utilization of a broad range of antioxidants including SOD mimetics to dissipate ROS synthesis can prolong islet viability and maintain function during islet isolation (93). The addition of a physical barrier as well as an immune modulator is likely the most promising combinatorial approach to protect islet transplants from immune-mediated rejection. Interventions including islet encapsulation with TA may be efficacious and safer in delaying allograft rejection than global immunosuppressive therapies (12, 15, 162, 180). In an effort to achieve stable islet engraftment in patients following islet transplantation, therapies that specifically target the removal of free radicals and redox-dependent signaling are highly warranted. It is apparent that synergistic interactions between redox biology and immune responses following islet transplantation are an underrepresented area of research. Future strategies implementing the LbL encapsulation approach in combination with potent antioxidants can enhance the viability and yield of isolated islets, making islet cell transplantation a realistic and curative treatment option for patients with T1D.
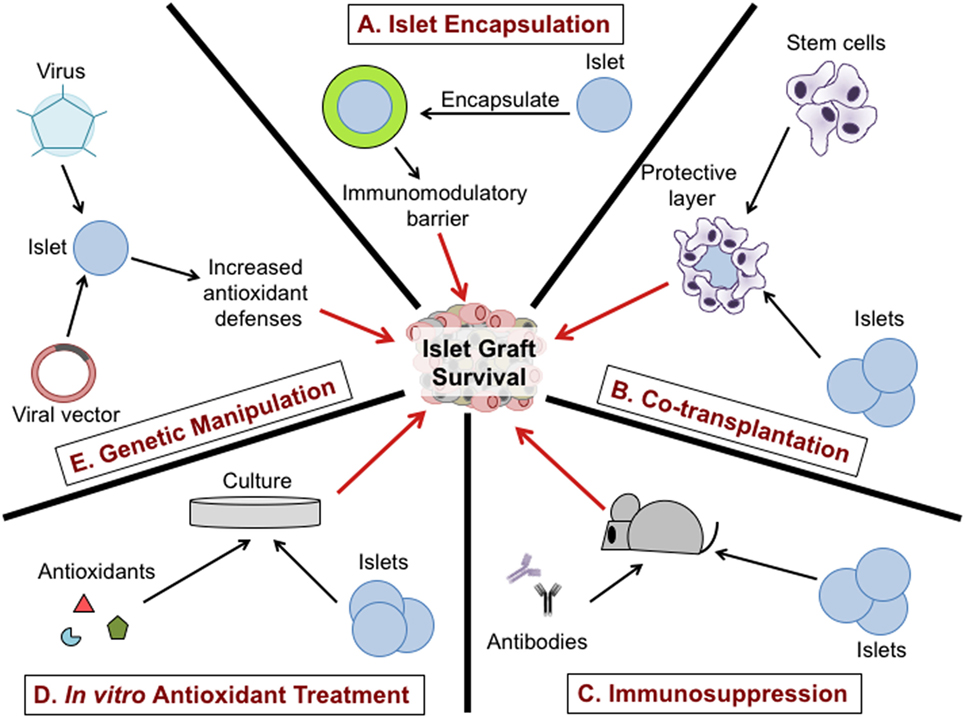
Figure 3. Therapeutic approaches to protect islet graft viability. Encapsulation of purified islets provides a physical and potentially immunomodulatory barrier between the islet graft and the host immune system (A). Co-transplantation of stem cells or regulatory immune cells with the islet graft can reduce immune infiltration to the graft (B). The use of immunosuppressive drugs such as monoclonal antibodies specific for immune cell subsets can suppress inflammatory responses in the host and prolong graft survival (C). Treatment of purified islets with antioxidants during culture can dissipate free radicals involved in islet dysfunction and viability, may enhance engraftment and delay graft rejection (D). Transfection of purified islets using a virus or viral vector to increase antioxidant defenses can promote islet graft viability and prolong survival after transplantation (E).
Author Contributions
JB and HT wrote the manuscript and reviewed/edited manuscript. HT is the guarantor of this review article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to apologize to researchers whose primary observations that form the basis of current knowledge in the field could not be cited due to space limitations, or have been acknowledged indirectly, by referring to current reviews. Our work was supported by grants from the American Diabetes Association (7-12-CD-11), Juvenile Diabetes Research Foundation (1-SRA-2015-42-A-N), and the National Institutes of Health (DK099550, T32.GM109780).
Abbreviations
APC, antigen-presenting cell; CITR, Collaborative Islet Transplant Registry; CTLA-4, cytotoxic T lymphocyte associated antigen-4; DAMPs, danger-associated molecular patterns; DCs, dendritic cells; Del-1, developmental endothelial locus-1; ECs, endothelial cells; ER, endoplasmic reticulum; Gpx-1, glutathione peroxidase; GSH, glutathione; GSSG, glutathione disulfide; HIF, hypoxia-inducible factor; HO-1, heme oxygenase-1; H2O2, hydrogen peroxide; IBMIR, instant blood-mediated inflammatory reaction; IFN-γ, interferon gamma; IL-1β, interleukin 1 beta; iNOS, inducible nitric oxide synthase; JNK, Jun N-terminal protein kinase; LbL, layer-by-layer; MCP-1, monocyte chemoattractant protein-1; MDSCs, myeloid-derived suppressor cells; MnSOD, manganese superoxide dismutase; MOMP, mitochondrial outer membrane permeabilization; MSCs, mesenchymal stem cells; NAD(P)H, nicotinamide adenine dinucleotide phosphate; NOD, non-obese diabetic; NOX, NAD(P)H oxidase; , superoxide; •OH, hydroxyl radical; PAMPs, pathogen-associated molecular patterns; PARP, poly (ADP-ribose) polymerase; PRRs, pattern recognition receptors; PVPON, poly(N-vinylpyrrolidone); RNS, reactive nitrogen species; ROS, reactive oxygen species; SOD, superoxide dismutase; TA, tannic acid; Teff, effector T cell; TF, tissue factor; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor alpha; Treg, regulatory T cell; T1D, type 1 diabetes; UPR, unfolded protein response; VEGF, vascular endothelial growth factor.
References
1. Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care (2003) 26:832–6. doi:10.2337/diacare.26.3.832
2. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature (2010) 464:1293–300. doi:10.1038/nature08933
3. Orban T, Bundy B, Becker DJ, Dimeglio LA, Gitelman SE, Goland R, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet (2011) 378:412–9. doi:10.1016/S0140-6736(11)60886-6
4. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med (2009) 361:2143–52. doi:10.1056/NEJMoa0904452
5. Moran A, Bundy B, Becker DJ, Dimeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet (2013) 381:1905–15. doi:10.1016/S0140-6736(13)60023-9
6. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med (1996) 20:463–6. doi:10.1016/0891-5849(96)02051-5
7. Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes (1997) 46:1733–42. doi:10.2337/diabetes.46.11.1733
8. Pepper AR, Gala-Lopez B, Ziff O, Shapiro AJ. Current status of clinical islet transplantation. World J Transplant (2013) 3:48–53. doi:10.5500/wjt.v3.i4.48
9. Agarwal A, Brayman KL. Update on islet cell transplantation for type 1 diabetes. Semin Intervent Radiol (2012) 29(2):90–8. doi:10.1055/s-0032-1312569.
10. Saravanan PB, Kanak MA, Chang CA, Darden C, Yoshimatsu G, Lawrence MC, et al. Islet damage during isolation as assessed by miRNAs and the correlation of miRNA levels with posttransplantation outcome in islet autotransplantation. Am J Transplant (2017) 18(4):982–9. doi:10.1111/ajt.14615
11. Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW. Automated method for isolation of human pancreatic islets. Diabetes (1988) 37:413–20. doi:10.2337/diab.37.4.413
12. Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med (2000) 343:230–8. doi:10.1056/NEJM200007273430401
13. Harlan DM, Kenyon NS, Korsgren O, Roep BO; Immunology of Diabetes Society. Current advances and travails in islet transplantation. Diabetes (2009) 58:2175–84. doi:10.2337/db09-0476
14. Gala-Lopez BL, Pepper AR, Pawlick RL, O’gorman D, Kin T, Bruni A, et al. Antiaging glycopeptide protects human islets against tacrolimus-related injury and facilitates engraftment in mice. Diabetes (2016) 65:451–62. doi:10.2337/db15-0764
15. Akar Y, Yucel G, Durukan A, Yucel I, Arici G. Systemic toxicity of tacrolimus given by various routes and the response to dose reduction. Clin Exp Ophthalmol (2005) 33:53–9. doi:10.1111/j.1442-9071.2005.00942.x
16. Lee DY, Park SJ, Nam JH, Byun Y. A combination therapy of PEGylation and immunosuppressive agent for successful islet transplantation. J Control Release (2006) 110:290–5. doi:10.1016/j.jconrel.2005.12.011
17. Vaithilingam V, Tuch BE. Islet transplantation and encapsulation: an update on recent developments. Rev Diabet Stud (2011) 8:51–67. doi:10.1900/RDS.2011.8.51
18. Monti P, Scirpoli M, Maffi P, Ghidoli N, De Taddeo F, Bertuzzi F, et al. Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J Clin Invest (2008) 118:1806–14. doi:10.1172/JCI35197
19. Lombardi A, Gambardella J, Du XL, Sorriento D, Mauro M, Iaccarino G, et al. Sirolimus induces depletion of intracellular calcium stores and mitochondrial dysfunction in pancreatic beta cells. Sci Rep (2017) 7:15823. doi:10.1038/s41598-017-15283-y
20. Lombardi A, Trimarco B, Iaccarino G, Santulli G. Impaired mitochondrial calcium uptake caused by tacrolimus underlies beta-cell failure. Cell Commun Signal (2017) 15:47. doi:10.1186/s12964-017-0203-0
21. Kennedy ED, Rizzuto R, Theler JM, Pralong WF, Bastianutto C, Pozzan T, et al. Glucose-stimulated insulin secretion correlates with changes in mitochondrial and cytosolic Ca2+ in aequorin-expressing INS-1 cells. J Clin Invest (1996) 98:2524–38. doi:10.1172/JCI119071
22. Wiederkehr A, Wollheim CB. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta-cell. Cell Calcium (2008) 44:64–76. doi:10.1016/j.ceca.2007.11.004
23. Seleme MC, Lei W, Burg AR, Goh KY, Metz A, Steele C, et al. Dysregulated TLR3-dependent signaling and innate immune activation in superoxide-deficient macrophages from nonobese diabetic mice. Free Radic Biol Med (2012) 52:2047–56. doi:10.1016/j.freeradbiomed.2012.01.027
24. Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol (2010) 185:5247–58. doi:10.4049/jimmunol.1001472
25. Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, et al. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes (2011) 60:2144–51. doi:10.2337/db10-1222
26. Delmastro MM, Piganelli JD. Oxidative stress and redox modulation potential in type 1 diabetes. Clin Dev Immunol (2011) 2011:593863. doi:10.1155/2011/593863
27. Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol (2011) 7:504–11. doi:10.1038/nchembio.607
28. Ramkumar KM, Sekar TV, Bhakkiyalakshmi E, Foygel K, Rajaguru P, Berger F, et al. The impact of oxidative stress on islet transplantation and monitoring the graft survival by non-invasive imaging. Curr Med Chem (2013) 20:1127–46. doi:10.2174/0929867311320090003
29. Newsholme P, Cruzat V, Arfuso F, Keane K. Nutrient regulation of insulin secretion and action. J Endocrinol (2014) 221:R105–20. doi:10.1530/JOE-13-0616
30. Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol (1997) 159:591–8.
31. Sklavos MM, Tse HM, Piganelli JD. Redox modulation inhibits CD8 T cell effector function. Free Radic Biol Med (2008) 45:1477–86. doi:10.1016/j.freeradbiomed.2008.08.023
32. Padgett LE, Tse HM. NADPH oxidase-derived superoxide provides a third signal for CD4 T cell effector responses. J Immunol (2016) 197:1733–42. doi:10.4049/jimmunol.1502581
33. Chen J, Gusdon AM, Thayer TC, Mathews CE. Role of increased ROS dissipation in prevention of T1D. Ann N Y Acad Sci (2008) 1150:157–66. doi:10.1196/annals.1447.045
34. Kawasaki E, Abiru N, Eguchi K. Prevention of type 1 diabetes: from the view point of beta cell damage. Diabetes Res Clin Pract (2004) 66(Suppl 1):S27–32. doi:10.1016/j.diabres.2003.09.015
35. Mohseni Salehi Monfared SS, Larijani B, Abdollahi M. Islet transplantation and antioxidant management: a comprehensive review. World J Gastroenterol (2009) 15:1153–61. doi:10.3748/wjg.15.1153
36. Gould DS, Auchincloss H Jr. Direct and indirect recognition: the role of MHC antigens in graft rejection. Immunol Today (1999) 20:77–82. doi:10.1016/S0167-5699(98)01394-2
37. Lin CM, Gill RG. Direct and indirect allograft recognition: pathways dictating graft rejection mechanisms. Curr Opin Organ Transplant (2016) 21:40–4. doi:10.1097/MOT.0000000000000263
38. Brune B, Dehne N, Grossmann N, Jung M, Namgaladze D, Schmid T, et al. Redox control of inflammation in macrophages. Antioxid Redox Signal (2013) 19:595–637. doi:10.1089/ars.2012.4785
39. Kesarwani P, Murali AK, Al-Khami AA, Mehrotra S. Redox regulation of T-cell function: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal (2013) 18:1497–534. doi:10.1089/ars.2011.4073
40. Grazia TJ, Pietra BA, Johnson ZA, Kelly BP, Plenter RJ, Gill RG. A two-step model of acute CD4 T-cell mediated cardiac allograft rejection. J Immunol (2004) 172:7451–8. doi:10.4049/jimmunol.172.12.7451
41. Pietra BA, Wiseman A, Bolwerk A, Rizeq M, Gill RG. CD4 T cell-mediated cardiac allograft rejection requires donor but not host MHC class II. J Clin Invest (2000) 106:1003–10. doi:10.1172/JCI10467
42. Diamond AS, Gill RG. An essential contribution by IFN-gamma to CD8+ T cell-mediated rejection of pancreatic islet allografts. J Immunol (2000) 165:247–55. doi:10.4049/jimmunol.165.1.247
43. Sleater M, Diamond AS, Gill RG. Islet allograft rejection by contact-dependent CD8+ T cells: perforin and FasL play alternate but obligatory roles. Am J Transplant (2007) 7:1927–33. doi:10.1111/j.1600-6143.2007.01889.x
44. Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med (2001) 30:1191–212. doi:10.1016/S0891-5849(01)00480-4
45. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med (2010) 48:749–62. doi:10.1016/j.freeradbiomed.2009.12.022
46. Murata Y, Ohteki T, Koyasu S, Hamuro J. IFN-gamma and pro-inflammatory cytokine production by antigen-presenting cells is dictated by intracellular thiol redox status regulated by oxygen tension. Eur J Immunol (2002) 32:2866–73. doi:10.1002/1521-4141(2002010)32:10<2866::AID-IMMU2866>3.0.CO;2-V
47. Murata Y, Shimamura T, Hamuro J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int Immunol (2002) 14:201–12. doi:10.1093/intimm/14.2.201
48. Puddu P, Fantuzzi L, Borghi P, Varano B, Rainaldi G, Guillemard E, et al. IL-12 induces IFN-gamma expression and secretion in mouse peritoneal macrophages. J Immunol (1997) 159:3490–7.
49. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev (2011) 75:50–83. doi:10.1128/MMBR.00031-10
50. Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol (1999) 19:1–11. doi:10.1128/MCB.19.1.1
51. Pantano C, Shrivastava P, Mcelhinney B, Janssen-Heininger Y. Hydrogen peroxide signaling through tumor necrosis factor receptor 1 leads to selective activation of c-Jun N-terminal kinase. J Biol Chem (2003) 278:44091–6. doi:10.1074/jbc.M308487200
52. Eizirik DL, Mandrup-Poulsen T. A choice of death – the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia (2001) 44:2115–33. doi:10.1007/s001250100021
53. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell (1998) 94:491–501. doi:10.1016/S0092-8674(00)81590-1
54. Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta (2011) 1813:558–63. doi:10.1016/j.bbamcr.2011.01.026
55. Shi F, Zhang P, Mao Y, Wang C, Zheng M, Zhao Z. The nitroxide Tempo inhibits hydroxyl radical production from the Fenton-like reaction of iron(II)-citrate with hydrogen peroxide. Biochem Biophys Res Commun (2017) 483:159–64. doi:10.1016/j.bbrc.2016.12.174
56. Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology (2011) 283:65–87. doi:10.1016/j.tox.2011.03.001
57. Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol (2002) 3:1129–34. doi:10.1038/ni1202-1129
58. Fra AM, Fagioli C, Finazzi D, Sitia R, Alberini CM. Quality control of ER synthesized proteins: an exposed thiol group as a three-way switch mediating assembly, retention and degradation. EMBO J (1993) 12:4755–61.
59. Masciarelli S, Sitia R. Building and operating an antibody factory: redox control during B to plasma cell terminal differentiation. Biochim Biophys Acta (2008) 1783:578–88. doi:10.1016/j.bbamcr.2008.01.003
60. Bertolotti M, Yim SH, Garcia-Manteiga JM, Masciarelli S, Kim YJ, Kang MH, et al. B- to plasma-cell terminal differentiation entails oxidative stress and profound reshaping of the antioxidant responses. Antioxid Redox Signal (2010) 13:1133–44. doi:10.1089/ars.2009.3079
61. Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol (2003) 23:8786–94. doi:10.1128/MCB.23.23.8786-8794.2003
62. Akiyoshi T, Hirohashi T, Alessandrini A, Chase CM, Farkash EA, Neal Smith R, et al. Role of complement and NK cells in antibody mediated rejection. Hum Immunol (2012) 73:1226–32. doi:10.1016/j.humimm.2012.07.330
63. Lampropoulou V, Hoehlig K, Roch T, Neves P, Calderon Gomez E, Sweenie CH, et al. TLR-activated B cells suppress T cell-mediated autoimmunity. J Immunol (2008) 180:4763–73. doi:10.4049/jimmunol.180.7.4763
64. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med (2012) 209:1001–10. doi:10.1084/jem.20111675
65. Lee JR. Reactive oxygen species play roles on B cell surface receptor CD40-mediated proximal and distal signaling events: effects of an antioxidant, N-acetyl-L-cysteine treatment. Mol Cell Biochem (2003) 252:1–7. doi:10.1023/A:1025529704480
66. Lee JR, Koretzky GA. Production of reactive oxygen intermediates following CD40 ligation correlates with c-Jun N-terminal kinase activation and IL-6 secretion in murine B lymphocytes. Eur J Immunol (1998) 28:4188–97. doi:10.1002/(SICI)1521-4141(199812)28:12<4188::AID-IMMU4188>3.0.CO;2-B
67. Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene (2007) 26:6724–37. doi:10.1038/sj.onc.1210757
68. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta (2011) 1813:878–88. doi:10.1016/j.bbamcr.2011.01.034
69. Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res (2009) 69:1958–65. doi:10.1158/0008-5472.CAN-08-2944
70. Lin L, Fuchs J, Li C, Olson V, Bekaii-Saab T, Lin J. STAT3 signaling pathway is necessary for cell survival and tumorsphere forming capacity in ALDH(+)/CD133(+) stem cell-like human colon cancer cells. Biochem Biophys Res Commun (2011) 416:246–51. doi:10.1016/j.bbrc.2011.10.112
71. De Groot M, Schuurs TA, Keizer PPM, Fekken S, Leuvenink HGD, Van Schilfgaarde R. Response of encapsulated rat pancreatic islets to hypoxia. Cell Transplant (2003) 12:867–75. doi:10.3727/000000003771000219
73. Giuliani M, Moritz W, Bodmer E, Dindo D, Kugelmeier P, Lehmann R, et al. Central necrosis in isolated hypoxic human pancreatic islets: evidence for postisolation ischemia. Cell Transplant (2005) 14:67–76. doi:10.3727/000000005783983287
74. Barshes NR, Wyllie S, Goss JA. Inflammation-mediated dysfunction and apoptosis in pancreatic islet transplantation: implications for intrahepatic grafts. J Leukoc Biol (2005) 77:587–97. doi:10.1189/jlb.1104649
75. Bottino R, Fernandez LA, Ricordi C, Lehmann R, Tsan MF, Oliver R, et al. Transplantation of allogeneic islets of Langerhans in the rat liver: effects of macrophage depletion on graft survival and microenvironment activation. Diabetes (1998) 47:316–23. doi:10.2337/diabetes.47.3.316
76. Coffey LC, Berman DM, Willman MA, Kenyon NS. Immune cell populations in nonhuman primate islets. Cell Transplant (2009) 18:1213–22. doi:10.3727/096368909X12483162196728
77. Jansson L. Dissociation between pancreatic islet blood flow and insulin release in the rat. Acta Physiol Scand (1985) 124:223–8. doi:10.1111/j.1748-1716.1985.tb07655.x
78. Carlsson PO, Liss P, Andersson A, Jansson L. Measurements of oxygen tension in native and transplanted rat pancreatic islets. Diabetes (1998) 47:1027–32. doi:10.2337/diabetes.47.7.1027
79. Carlsson PO, Palm F, Andersson A, Liss P. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes (2001) 50:489–95. doi:10.2337/diabetes.50.3.489
80. Carlsson PO, Palm F, Andersson A, Liss P. Chronically decreased oxygen tension in rat pancreatic islets transplanted under the kidney capsule. Transplantation (2000) 69:761–6. doi:10.1097/00007890-200003150-00015
81. Biarnes M, Montolio M, Nacher V, Raurell M, Soler J, Montanya E. Beta-cell death and mass in syngeneically transplanted islets exposed to short- and long-term hyperglycemia. Diabetes (2002) 51:66–72. doi:10.2337/diabetes.51.1.66
82. Montana E, Bonner-Weir S, Weir GC. Beta cell mass and growth after syngeneic islet cell transplantation in normal and streptozocin diabetic C57BL/6 mice. J Clin Invest (1993) 91:780–7. doi:10.1172/JCI116297
83. Crutchlow MF, Yu M, Bae YS, Deng S, Stoffers DA. Exendin-4 does not promote beta-cell proliferation or survival during the early post-islet transplant period in mice. Transplant Proc (2008) 40:1650–7. doi:10.1016/j.transproceed.2008.03.161
84. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes (2003) 52:1–8. doi:10.2337/diabetes.52.1.1
85. Linn T, Schmitz J, Hauck-Schmalenberger I, Lai Y, Bretzel RG, Brandhorst H, et al. Ischaemia is linked to inflammation and induction of angiogenesis in pancreatic islets. Clin Exp Immunol (2006) 144:179–87. doi:10.1111/j.1365-2249.2006.03066.x
86. Olsson R, Carlsson PO. Oxygenation of cultured pancreatic islets. Adv Exp Med Biol (2006) 578:263–8. doi:10.1007/0-387-29540-2_42
87. Maechler P, De Andrade PB. Mitochondrial damages and the regulation of insulin secretion. Biochem Soc Trans (2006) 34:824–7. doi:10.1042/BST0340824
88. Kanak MA, Takita M, Itoh T, Sorelle JA, Murali S, Kunnathodi F, et al. Alleviation of instant blood-mediated inflammatory reaction in autologous conditions through treatment of human islets with NF-kappaB inhibitors. Transplantation (2014) 98:578–84. doi:10.1097/TP.0000000000000107
89. Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol (2014) 49:1–15. doi:10.3109/10409238.2013.838205
90. Linetsky E, Bottino R, Lehmann R, Alejandro R, Inverardi L, Ricordi C. Improved human islet isolation using a new enzyme blend, liberase. Diabetes (1997) 46:1120–3. doi:10.2337/diab.46.7.1120
91. Lakey JR, Warnock GL, Shapiro AM, Korbutt GS, Ao Z, Kneteman NM, et al. Intraductal collagenase delivery into the human pancreas using syringe loading or controlled perfusion. Cell Transplant (1999) 8:285–92. doi:10.1177/096368979900800309
92. Bottino R, Balamurugan AN, Bertera S, Pietropaolo M, Trucco M, Piganelli JD. Preservation of human islet cell functional mass by anti-oxidative action of a novel SOD mimic compound. Diabetes (2002) 51:2561–7. doi:10.2337/diabetes.51.8.2561
93. Bottino R, Balamurugan AN, Tse H, Thirunavukkarasu C, Ge X, Profozich J, et al. Response of human islets to isolation stress and the effect of antioxidant treatment. Diabetes (2004) 53:2559–68. doi:10.2337/diabetes.53.10.2559
94. Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic Biol Med (2004) 36:233–47. doi:10.1016/j.freeradbiomed.2003.10.029
95. Sklavos MM, Bertera S, Tse HM, Bottino R, He J, Beilke JN, et al. Redox modulation protects islets from transplant-related injury. Diabetes (2010) 59:1731–8. doi:10.2337/db09-0588
96. Zhang B, Kang M, Xie Q, Xu B, Sun C, Chen K, et al. Anthocyanins from Chinese bayberry extract protect beta cells from oxidative stress-mediated injury via HO-1 upregulation. J Agric Food Chem (2011) 59:537–45. doi:10.1021/jf1035405
97. Cai H, Yang B, Xu Z, Zhang B, Xu B, Li X, et al. Cyanidin-3-O-glucoside enhanced the function of syngeneic mouse islets transplanted under the kidney capsule or into the portal vein. Transplantation (2015) 99:508–14. doi:10.1097/TP.0000000000000628
98. Mysore TB, Shinkel TA, Collins J, Salvaris EJ, Fisicaro N, Murray-Segal LJ, et al. Overexpression of glutathione peroxidase with two isoforms of superoxide dismutase protects mouse islets from oxidative injury and improves islet graft function. Diabetes (2005) 54:2109–16. doi:10.2337/diabetes.54.7.2109
99. Bertera S, Crawford ML, Alexander AM, Papworth GD, Watkins SC, Robbins PD, et al. Gene transfer of manganese superoxide dismutase extends islet graft function in a mouse model of autoimmune diabetes. Diabetes (2003) 52:387–93. doi:10.2337/diabetes.52.2.387
100. Chou FC, Sytwu HK. Overexpression of thioredoxin in islets transduced by a lentiviral vector prolongs graft survival in autoimmune diabetic NOD mice. J Biomed Sci (2009) 16:71. doi:10.1186/1423-0127-16-71
101. Tran PO, Parker SM, Leroy E, Franklin CC, Kavanagh TJ, Zhang T, et al. Adenoviral overexpression of the glutamylcysteine ligase catalytic subunit protects pancreatic islets against oxidative stress. J Biol Chem (2004) 279:53988–93. doi:10.1074/jbc.M404809200
102. Moriscot C, Pattou F, Kerr-Conte J, Richard MJ, Lemarchand P, Benhamou PY. Contribution of adenoviral-mediated superoxide dismutase gene transfer to the reduction in nitric oxide-induced cytotoxicity on human islets and INS-1 insulin-secreting cells. Diabetologia (2000) 43:625–31. doi:10.1007/s001250051351
103. Fiorina P, Shapiro AM, Ricordi C, Secchi A. The clinical impact of islet transplantation. Am J Transplant (2008) 8:1990–7. doi:10.1111/j.1600-6143.2008.02353.x
104. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet (2006) 368:1696–705. doi:10.1016/S0140-6736(06)69705-5
105. Carlessi R, Lemos NE, Dias AL, Oliveira FS, Brondani LA, Canani LH, et al. Exendin-4 protects rat islets against loss of viability and function induced by brain death. Mol Cell Endocrinol (2015) 412:239–50. doi:10.1016/j.mce.2015.05.009
106. Padmasekar M, Lingwal N, Samikannu B, Chen C, Sauer H, Linn T. Exendin-4 protects hypoxic islets from oxidative stress and improves islet transplantation outcome. Endocrinology (2013) 154:1424–33. doi:10.1210/en.2012-1983
107. Davalli AM, Scaglia L, Zangen DH, Hollister J, Bonner-Weir S, Weir GC. Vulnerability of islets in the immediate posttransplantation period. Dynamic changes in structure and function. Diabetes (1996) 45:1161–7. doi:10.2337/diabetes.45.9.1161
108. Nilsson B, Ekdahl KN, Korsgren O. Control of instant blood-mediated inflammatory reaction to improve islets of Langerhans engraftment. Curr Opin Organ Transplant (2011) 16:620–6. doi:10.1097/MOT.0b013e32834c2393
109. Liuwantara D, Chew YV, Favaloro EJ, Hawkes JM, Burns HL, O’connell PJ, et al. Characterizing the mechanistic pathways of the instant blood-mediated inflammatory reaction in xenogeneic neonatal islet cell transplantation. Transplant Direct (2016) 2:e77. doi:10.1097/TXD.0000000000000590
110. Hardstedt M, Lindblom S, Karlsson-Parra A, Nilsson B, Korsgren O. Characterization of innate immunity in an extended whole blood model of human islet allotransplantation. Cell Transplant (2016) 25:503–15. doi:10.3727/096368915X688461
111. Moberg L, Johansson H, Lukinius A, Berne C, Foss A, Kallen R, et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet (2002) 360:2039–45. doi:10.1016/S0140-6736(02)12020-4
112. Johansson H, Lukinius A, Moberg L, Lundgren T, Berne C, Foss A, et al. Tissue factor produced by the endocrine cells of the islets of Langerhans is associated with a negative outcome of clinical islet transplantation. Diabetes (2005) 54:1755–62. doi:10.2337/diabetes.54.6.1755
113. Moberg L, Olsson A, Berne C, Felldin M, Foss A, Kallen R, et al. Nicotinamide inhibits tissue factor expression in isolated human pancreatic islets: implications for clinical islet transplantation. Transplantation (2003) 76:1285–8. doi:10.1097/01.TP.0000098905.86445.0F
114. Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J (1972) 128:617–30. doi:10.1042/bj1280617
115. Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep (1997) 17:3–8. doi:10.1023/A:1027374931887
116. Abdelli S, Ansite J, Roduit R, Borsello T, Matsumoto I, Sawada T, et al. Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes (2004) 53:2815–23. doi:10.2337/diabetes.53.11.2815
117. Liu Q, Ding JL. The molecular mechanisms of TLR-signaling cooperation in cytokine regulation. Immunol Cell Biol (2016) 94(6):538–42. doi:10.1038/icb.2016.18
118. Hultmark D. Macrophage differentiation marker MyD88 is a member of the toll/IL-1 receptor family. Biochem Biophys Res Commun (1994) 199:144–6. doi:10.1006/bbrc.1994.1206
119. Warner N, Nunez G. MyD88: a critical adaptor protein in innate immunity signal transduction. J Immunol (2013) 190:3–4. doi:10.4049/jimmunol.1203103
120. Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol (2009) 9:778–88. doi:10.1038/nri2655
121. Zhang X, Xing S, Li M, Zhang L, Xie L, He W, et al. Beyond knockout: a novel homodimerization-targeting MyD88 inhibitor prevents and cures type 1 diabetes in NOD mice. Metabolism (2016) 65:1267–77. doi:10.1016/j.metabol.2016.05.005
122. Wang J, Sun Z, Gou W, Adams DB, Cui W, Morgan KA, et al. Alpha-1 antitrypsin enhances islet engraftment by suppression of instant blood-mediated inflammatory reaction. Diabetes (2017) 66:970–80. doi:10.2337/db16-1036
123. Chavakis E, Choi EY, Chavakis T. Novel aspects in the regulation of the leukocyte adhesion cascade. Thromb Haemost (2009) 102:191–7. doi:10.1160/TH08-12-0844
124. Kourtzelis I, Kotlabova K, Lim JH, Mitroulis I, Ferreira A, Chen LS, et al. Developmental endothelial locus-1 modulates platelet-monocyte interactions and instant blood-mediated inflammatory reaction in islet transplantation. Thromb Haemost (2016) 115:781–8. doi:10.1160/TH15-05-0429
125. Miwa I, Ichimura N, Sugiura M, Hamada Y, Taniguchi S. Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology (2000) 141:2767–72. doi:10.1210/endo.141.8.7614
126. Lombardi A, Tomer Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J Autoimmun (2017) 80:48–55. doi:10.1016/j.jaut.2017.02.002
127. Chambers KT, Unverferth JA, Weber SM, Wek RC, Urano F, Corbett JA. The role of nitric oxide and the unfolded protein response in cytokine-induced beta-cell death. Diabetes (2008) 57:124–32. doi:10.2337/db07-0944
128. Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, et al. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci U S A (2001) 98:10845–50. doi:10.1073/pnas.191207498
129. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest (2005) 115:2656–64. doi:10.1172/JCI26373
130. Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes (2012) 61:818–27. doi:10.2337/db11-1293
131. Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, et al. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci Transl Med (2013) 5:211ra156. doi:10.1126/scitranslmed.3006534
132. Louvet C, Szot GL, Lang J, Lee MR, Martinier N, Bollag G, et al. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A (2008) 105:18895–900. doi:10.1073/pnas.0810246105
133. Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1alpha signaling spares ER-stressed pancreatic beta cells to reverse autoimmune diabetes. Cell Metab (2017) 25:1207. doi:10.1016/j.cmet.2017.03.018
134. Negi S, Park SH, Jetha A, Aikin R, Tremblay M, Paraskevas S. Evidence of endoplasmic reticulum stress mediating cell death in transplanted human islets. Cell Transplant (2012) 21:889–900. doi:10.3727/096368911X603639
135. Abadpour S, Gopel SO, Schive SW, Korsgren O, Foss A, Scholz H. Glial cell-line derived neurotrophic factor protects human islets from nutrient deprivation and endoplasmic reticulum stress induced apoptosis. Sci Rep (2017) 7:1575. doi:10.1038/s41598-017-01805-1
136. Korge P, Ping P, Weiss JN. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ Res (2008) 103:873–80. doi:10.1161/CIRCRESAHA.108.180869
137. Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol (2002) 282:C227–41. doi:10.1152/ajpcell.00112.2001
138. Li N, Brun T, Cnop M, Cunha DA, Eizirik DL, Maechler P. Transient oxidative stress damages mitochondrial machinery inducing persistent beta-cell dysfunction. J Biol Chem (2009) 284:23602–12. doi:10.1074/jbc.M109.024323
139. Lim S, Rashid MA, Jang M, Kim Y, Won H, Lee J, et al. Mitochondria-targeted antioxidants protect pancreatic beta-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cell Physiol Biochem (2011) 28:873–86. doi:10.1159/000335802
140. Lee MS, Gu D, Feng L, Curriden S, Arnush M, Krahl T, et al. Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-beta 1. Am J Pathol (1995) 147:42–52.
141. Cheng K, Fraga D, Zhang C, Kotb M, Gaber AO, Guntaka RV, et al. Adenovirus-based vascular endothelial growth factor gene delivery to human pancreatic islets. Gene Ther (2004) 11:1105–16. doi:10.1038/sj.gt.3302267
142. Narang AS, Cheng K, Henry J, Zhang C, Sabek O, Fraga D, et al. Vascular endothelial growth factor gene delivery for revascularization in transplanted human islets. Pharm Res (2004) 21:15–25. doi:10.1023/B:PHAM.0000012147.52900.b8
143. Jia X, Cheng K, Mahato RI. Coexpression of vascular endothelial growth factor and interleukin-1 receptor antagonist for improved human islet survival and function. Mol Pharm (2007) 4:199–207. doi:10.1021/mp060091s
144. Winter DT, Eich T, Jahr H, Brendel MD, Bretzel RG. Influence of antioxidant therapy on islet graft survival. Transplant Proc (2002) 34:2366–8. doi:10.1016/S0041-1345(02)03273-6
145. Xu B, Moritz JT, Epstein PN. Overexpression of catalase provides partial protection to transgenic mouse beta cells. Free Radic Biol Med (1999) 27:830–7. doi:10.1016/S0891-5849(99)00130-6
146. Ruttkay-Nedecky B, Nejdl L, Gumulec J, Zitka O, Masarik M, Eckschlager T, et al. The role of metallothionein in oxidative stress. Int J Mol Sci (2013) 14:6044–66. doi:10.3390/ijms14036044
147. Jung HS, Lim KS, Kim MJ, Hwang YH, Yoo C, Lee YK, et al. Hypoxic resistance of hypodermically transplanted pancreatic islets by using cell-absorbable antioxidant Tat-metallothionein. J Control Release (2013) 172:1092–101. doi:10.1016/j.jconrel.2013.09.031
148. Park L, Min D, Kim H, Park J, Choi S, Park Y. The combination of metallothionein and superoxide dismutase protects pancreatic beta cells from oxidative damage. Diabetes Metab Res Rev (2011) 27:802–8. doi:10.1002/dmrr.1254
149. Grover A, Shandilya A, Punetha A, Bisaria VS, Sundar D. Inhibition of the NEMO/IKKbeta association complex formation, a novel mechanism associated with the NF-kappaB activation suppression by Withania somnifera’s key metabolite withaferin A. BMC Genomics (2010) 11(Suppl 4):S25. doi:10.1186/1471-2164-11-S4-S25
150. Cucciolla V, Borriello A, Oliva A, Galletti P, Zappia V, Della Ragione F. Resveratrol: from basic science to the clinic. Cell Cycle (2007) 6:2495–510. doi:10.4161/cc.6.20.4815
151. Peng H, Olsen G, Tamura Y, Noguchi H, Matsumoto S, Levy MF, et al. Inhibition of inflammatory cytokine-induced response in human islet cells by withaferin A. Transplant Proc (2010) 42:2058–61. doi:10.1016/j.transproceed.2010.05.131
152. Breccia M, Muscaritoli M, Cannella L, Stefanizzi C, Frustaci A, Alimena G. Fasting glucose improvement under dasatinib treatment in an accelerated phase chronic myeloid leukemia patient unresponsive to imatinib and nilotinib. Leuk Res (2008) 32:1626–8. doi:10.1016/j.leukres.2008.01.015
153. Costa DB, Huberman MS. Improvement of type 2 diabetes in a lung cancer patient treated with erlotinib. Diabetes Care (2006) 29:1711. doi:10.2337/dc06-0558
154. Hagerkvist R, Jansson L, Welsh N. Imatinib mesylate improves insulin sensitivity and glucose disposal rates in rats fed a high-fat diet. Clin Sci (Lond) (2008) 114:65–71. doi:10.1042/CS20070122
155. Hagerkvist R, Makeeva N, Elliman S, Welsh N. Imatinib mesylate (Gleevec) protects against streptozotocin-induced diabetes and islet cell death in vitro. Cell Biol Int (2006) 30:1013–7. doi:10.1016/j.cellbi.2006.08.006
156. Hagerkvist R, Sandler S, Mokhtari D, Welsh N. Amelioration of diabetes by imatinib mesylate (Gleevec): role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J (2007) 21:618–28. doi:10.1096/fj.06-6910com
157. Mokhtari D, Li T, Lu T, Welsh N. Effects of imatinib mesylate (Gleevec) on human islet NF-kappaB activation and chemokine production in vitro. PLoS One (2011) 6:e24831. doi:10.1371/journal.pone.0024831
158. King AJ, Griffiths LA, Persaud SJ, Jones PM, Howell SL, Welsh N. Imatinib prevents beta cell death in vitro but does not improve islet transplantation outcome. Ups J Med Sci (2016) 121:140–5. doi:10.3109/03009734.2016.1151090
159. Kizilel S, Garfinkel M, Opara E. The bioartificial pancreas: progress and challenges. Diabetes Technol Ther (2005) 7:968–85. doi:10.1089/dia.2005.7.968
160. Qi M. Transplantation of encapsulated pancreatic islets as a treatment for patients with type 1 diabetes mellitus. Adv Med (2014) 2014:429710. doi:10.1155/2014/429710
161. Beck J, Angus R, Madsen B, Britt D, Vernon B, Nguyen KT. Islet encapsulation: strategies to enhance islet cell functions. Tissue Eng (2007) 13:589–99. doi:10.1089/ten.2006.0183
162. Tse HM, Kozlovskaya V, Kharlampieva E, Hunter CS. Minireview: directed differentiation and encapsulation of islet beta-cells-recent advances and future considerations. Mol Endocrinol (2015) 29:1388–99. doi:10.1210/me.2015-1085
163. Chick WL, Like AA, Lauris V. Beta cell culture on synthetic capillaries: an artificial endocrine pancreas. Science (1975) 187:847–9. doi:10.1126/science.187.4179.847
164. Scharp DW, Mason NS, Sparks RE. Islet immuno-isolation: the use of hybrid artificial organs to prevent islet tissue rejection. World J Surg (1984) 8:221–9. doi:10.1007/BF01655139
165. Leblond FA, Simard G, Henley N, Rocheleau B, Huet PM, Halle JP. Studies on smaller (approximately 315 microM) microcapsules: IV. Feasibility and safety of intrahepatic implantations of small alginate poly-L-lysine microcapsules. Cell Transplant (1999) 8:327–37. doi:10.1177/096368979900800303
166. Rafael E, Wernerson A, Arner P, Tibell A. In vivo studies on insulin permeability of an immunoisolation device intended for islet transplantation using the microdialysis technique. Eur Surg Res (1999) 31:249–58. doi:10.1159/000008700
167. Loudovaris T, Jacobs S, Young S, Maryanov D, Brauker J, Johnson RC. Correction of diabetic nod mice with insulinomas implanted within Baxter immunoisolation devices. J Mol Med (Berl) (1999) 77:219–22. doi:10.1007/s001090050340
168. Teramura Y, Oommen OP, Olerud J, Hilborn J, Nilsson B. Microencapsulation of cells, including islets, within stable ultra-thin membranes of maleimide-conjugated PEG-lipid with multifunctional crosslinkers. Biomaterials (2013) 34:2683–93. doi:10.1016/j.biomaterials.2013.01.015
169. Breger JC, Lyle DB, Shallcross JC, Langone JJ, Wang NS. Defining critical inflammatory parameters for endotoxin impurity in manufactured alginate microcapsules. J Biomed Mater Res B Appl Biomater (2009) 91:755–65. doi:10.1002/jbm.b.31452
170. Dusseault J, Tam SK, Menard M, Polizu S, Jourdan G, Yahia L, et al. Evaluation of alginate purification methods: effect on polyphenol, endotoxin, and protein contamination. J Biomed Mater Res A (2006) 76:243–51. doi:10.1002/jbm.a.30541
171. Zhu HT, Lu L, Liu XY, Yu L, Lyu Y, Wang B. Treatment of diabetes with encapsulated pig islets: an update on current developments. J Zhejiang Univ Sci B (2015) 16:329–43. doi:10.1631/jzus.B1400310
172. Sharma V, Hunckler M, Ramasubramanian MK, Opara EC, Katuri KC. Microfluidic approach to cell microencapsulation. Methods Mol Biol (2017) 1479:71–6. doi:10.1007/978-1-4939-6364-5_5
173. Leong W, Wang DA. Cell-laden polymeric microspheres for biomedical applications. Trends Biotechnol (2015) 33:653–66. doi:10.1016/j.tibtech.2015.09.003
174. Pathak S, Regmi S, Gupta B, Poudel BK, Pham TT, Kim JR, et al. Hybrid congregation of islet single cells and curcumin-loaded polymeric microspheres as an interventional strategy to overcome apoptosis associated with pancreatic islets transplantation. ACS Appl Mater Interfaces (2016) 8:25702–13. doi:10.1021/acsami.6b07897
175. Wilson JT, Cui W, Chaikof EL. Layer-by-layer assembly of a conformal nanothin PEG coating for intraportal islet transplantation. Nano Lett (2008) 8:1940–8. doi:10.1021/nl080694q
176. Hong S, Leroueil PR, Janus EK, Peters JL, Kober MM, Islam MT, et al. Interaction of polycationic polymers with supported lipid bilayers and cells: nanoscale hole formation and enhanced membrane permeability. Bioconjug Chem (2006) 17:728–34. doi:10.1021/bc060077y
177. Teramura Y, Kaneda Y, Iwata H. Islet-encapsulation in ultra-thin layer-by-layer membranes of poly(vinyl alcohol) anchored to poly(ethylene glycol)-lipids in the cell membrane. Biomaterials (2007) 28:4818–25. doi:10.1016/j.biomaterials.2007.07.050
178. Kozlovskaya V, Zavgorodnya O, Chen Y, Ellis K, Tse HM, Cui W, et al. Ultrathin polymeric coatings based on hydrogen-bonded polyphenol for protection of pancreatic islet cells. Adv Funct Mater (2012) 22:3389–98. doi:10.1002/adfm.201200138
179. Kozlovskaya V, Xue B, Lei W, Padgett LE, Tse HM, Kharlampieva E. Hydrogen-bonded multilayers of tannic acid as mediators of T-cell immunity. Adv Healthc Mater (2015) 4:686–94. doi:10.1002/adhm.201400657
180. Pham-Hua D, Padgett LE, Xue B, Anderson B, Zeiger M, Barra JM, et al. Islet encapsulation with polyphenol coatings decreases pro-inflammatory chemokine synthesis and T cell trafficking. Biomaterials (2017) 128:19–32. doi:10.1016/j.biomaterials.2017.03.002
181. Staels W, De Groef S, Heremans Y, Coppens V, Van Gassen N, Leuckx G, et al. Accessory cells for beta-cell transplantation. Diabetes Obes Metab (2016) 18:115–24. doi:10.1111/dom.12556
182. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol (2008) 8:726–36. doi:10.1038/nri2395
183. Berman DM, Willman MA, Han D, Kleiner G, Kenyon NM, Cabrera O, et al. Mesenchymal stem cells enhance allogeneic islet engraftment in nonhuman primates. Diabetes (2010) 59:2558–68. doi:10.2337/db10-0136
184. Johansson U, Rasmusson I, Niclou SP, Forslund N, Gustavsson L, Nilsson B, et al. Formation of composite endothelial cell-mesenchymal stem cell islets: a novel approach to promote islet revascularization. Diabetes (2008) 57:2393–401. doi:10.2337/db07-0981
185. Solari MG, Srinivasan S, Boumaza I, Unadkat J, Harb G, Garcia-Ocana A, et al. Marginal mass islet transplantation with autologous mesenchymal stem cells promotes long-term islet allograft survival and sustained normoglycemia. J Autoimmun (2009) 32:116–24. doi:10.1016/j.jaut.2009.01.003
186. Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, et al. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood (2007) 109:228–34. doi:10.1182/blood-2006-02-002246
187. Mohammadi Ayenehdeh J, Niknam B, Rasouli S, Hashemi SM, Rahavi H, Rezaei N, et al. Immunomodulatory and protective effects of adipose tissue-derived mesenchymal stem cells in an allograft islet composite transplantation for experimental autoimmune type 1 diabetes. Immunol Lett (2017) 188:21–31. doi:10.1016/j.imlet.2017.05.006
188. Kim HI, Yu JE, Lee SY, Sul AY, Jang MS, Rashid MA, et al. The effect of composite pig islet-human endothelial cell grafts on the instant blood-mediated inflammatory reaction. Cell Transplant (2009) 18:31–7. doi:10.3727/096368909788237113
189. Kim JH, Oh BJ, Lee HN, Park HS, Park SG, Park KS. Endothelial colony-forming cell coating of pig islets prevents xenogeneic instant blood-mediated inflammatory reaction. Cell Transplant (2011) 20:1805–15. doi:10.3727/096368911X566154
190. Johansson U, Elgue G, Nilsson B, Korsgren O. Composite islet-endothelial cell grafts: a novel approach to counteract innate immunity in islet transplantation. Am J Transplant (2005) 5:2632–9. doi:10.1111/j.1600-6143.2005.01076.x
191. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
192. Chou HS, Hsieh CC, Charles R, Wang L, Wagner T, Fung JJ, et al. Myeloid-derived suppressor cells protect islet transplants by B7-H1 mediated enhancement of T regulatory cells. Transplantation (2012) 93:272–82. doi:10.1097/TP.0b013e31823ffd39
193. Arakawa Y, Qin J, Chou HS, Bhatt S, Wang L, Stuehr D, et al. Cotransplantation with myeloid-derived suppressor cells protects cell transplants: a crucial role of inducible nitric oxide synthase. Transplantation (2014) 97:740–7. doi:10.1097/01.TP.0000442504.23885.f7
194. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets – results of one year follow-up. Clin Immunol (2014) 153:23–30. doi:10.1016/j.clim.2014.03.016
195. Marek-Trzonkowska N, Mysliwec M, Siebert J, Trzonkowski P. Clinical application of regulatory T cells in type 1 diabetes. Pediatr Diabetes (2013) 14:322–32. doi:10.1111/pedi.12029
196. Pawlick RL, Wink J, Pepper AR, Bruni A, Abualhassen N, Rafiei Y, et al. Reparixin, a CXCR1/2 inhibitor in islet allotransplantation. Islets (2016) 8:115–24. doi:10.1080/19382014.2016.1199303
197. Zhang W, Gorantla VS, Campbell PG, Li Y, Yang Y, Komatsu C, et al. Biopatterned CTLA4/Fc matrices facilitate local immunomodulation, engraftment, and glucose homeostasis after pancreatic islet transplantation. Diabetes (2016) 65:3660–6. doi:10.2337/db16-0320
Keywords: redox signaling, reactive oxygen species, type 1 diabetes, islet transplantation, immune rejection, immunology, encapsulation
Citation: Barra JM and Tse HM (2018) Redox-Dependent Inflammation in Islet Transplantation Rejection. Front. Endocrinol. 9:175. doi: 10.3389/fendo.2018.00175
Received: 24 January 2018; Accepted: 03 April 2018;
Published: 23 April 2018
Edited by:
David Taylor-Fishwick, Eastern Virginia Medical School, United StatesReviewed by:
Ankit Saxena, National Institutes of Health (NIH), United StatesAngela Lombardi, Albert Einstein College of Medicine, United States
Copyright: © 2018 Barra and Tse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hubert M. Tse, aHRzZUB1YWIuZWR1