 Fumihiro Sanada1*
Fumihiro Sanada1* Yoshiaki Taniyama1,2Jun Muratsu1,2Rei Otsu1
Yoshiaki Taniyama1,2Jun Muratsu1,2Rei Otsu1 Hideo Shimizu1Hiromi Rakugi2
Hideo Shimizu1Hiromi Rakugi2 Ryuichi Morishita1*
Ryuichi Morishita1*
- 1Department of Clinical Gene Therapy, Osaka University Graduate School of Medicine, Suita, Osaka, Japan
- 2Department of Geriatric and General Medicine, Osaka University Graduate School of Medicine, Suita, Osaka, Japan
Cellular senescence is the complex process of deterioration that drives the aging of an organism, resulting in the progressive loss of organ function and eventually phenotypic aging. Senescent cells undergo irreversible growth arrest, usually by inducing telomere shortening. Alternatively, senescence may also occur prematurely in response to various stress stimuli, such as oxidative stress, DNA damage, or activated oncogenes. Recently, it has been shown that IGF binding protein-5 (IGFBP-5) with the induction of the tumor suppressor p53 is upregulated during cellular senescence. This mechanism mediates interleukin-6/gp130-induced premature senescence in human fibroblasts, irradiation-induced premature senescence in human endothelial cells (ECs), and replicative senescence in human ECs independent of insulin-like growth factor I (IGF-I) and IGF-II. Additionally, a link between IGFBP-5, hyper-coagulation, and inflammation, which occur with age, has been implicated. Thus, IGFBP-5 seems to play decisive roles in controlling cell senescence and cell inflammation. In this review, we describe the accumulating evidence for this role of IGFBP-5 including our new finding.
Introduction
Insulin-like growth factor I (IGF-I) and II (IGF-II) are insulin superfamily members and are ubiquitously distributed in several organs (1, 2). Six high-affinity IGF binding proteins (IGFBPs) interact with IGFs, regulating IGF-I/II bioavailability, distribution, and signaling. IGFBPs are secreted and bind to IGFs in the circulation and extracellular environment (3). In addition to IGF-dependent action, IGF-independent functions of IGFBPs, many of which occur intracellularly, have recently been reported (4, 5). For example, IGFBP-1 and -2 are associated with cancer cell proliferation, adhesion, and migration through the specific binding of IGFBP-1 and -2 to alpha 5 beta1 integrin, followed by alterations in the phosphorylation status of downstream signaling molecules (6, 7). By regulating enzymes involved in sphingolipid metabolism, IGFBP-3 and -5 affect the balance between growth inhibitory lipids and growth stimulatory lipids (8, 9). Additional evidence has implicated that IGF binding protein-5 (IGFBP-5) is upregulation in the irradiation-induced premature senescence and replicative senescence of umbilical vein endothelial cells (HUVECs) (10). Knockdown of IGFBP-5 in aged HUVECs partially reversed the process of senescence, whereas the application of exogenous IGFBP-5 or IGFBP-5 overexpression induced premature senescence in HUVECs in vitro (11), indicating a decisive role for IGFBP-5 in controlling cell senescence and proliferation. The insulin/IGF signaling pathway has been implicated in the aging of many organisms, ranging from nematodes to mammals. The observation that IGFBPs modulate the availability or the distribution of IGF-1 adds further support to the hypothesis that IGFBPs have a vital role in the aging process (12). Many changes in the immune system, hemostasis, and vasculature, including alterations in inflammation, coagulation, and vascular senescence, occur during aging. However, its mechanism is not fully understood.
In this review, we first overview the mechanism of chronic inflammation during aging and later possible mechanism linking between cell senescence and senescence-inducing stimuli via IGFBP-5 is discussed.
Mechanism of Chronic Inflammation in Aging
Inflammation is required for an acute, transient immune response to invading pathogens or acute traumatic injury. This process is essential for facilitating the tissue repair by increasing division and migration of cells. In contrast, chronic inflammation causes low-grade and persistent inflammation leading to tissue degeneration rather than the solution to infection, injury, or disease (13). Many aged tissues are chronically inflamed, which is the common pathological mechanism for age-associated diseases, such as cardiovascular disease, diabetes, cancer, and Alzheimer’s disease (14). Interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) counteracts anabolic signaling, including insulin and erythropoietin cascades. Thus, chronic low-grade inflammation is now recognized as an important causative factor for insulin resistance and sarcopenia (15). Several sources of chronic inflammation during aging termed “inflammaging” have been described (Figure 1) (14). (i) Debris from cells or immunoglobulin accumulation due to increased cell death or inappropriate cell elimination systems in aging activates the innate immune system, resulting in chronic inflammation in aged organs (16). According to Zhang et al. (17), circulating mitochondrial damage-associated molecular patterns cause inflammation in response to injury. Mitochondrial damage-associated molecular patterns released from damaged cells are evolutionally conserved with bacterial pathogen-associated molecular patterns activating innate immunity (18). Thus, age-related failing of mitochondria quality control is associated with inflammaging. (ii) The ability of the oral and gut mucosa to protect against bacterial invasion declines with age, leading to persist low-grade inflammation (19). Periodontal disease has been also demonstrated to increase the inflammatory response with age (20). Additionally, the abundance of anti-inflammatory microbiota decreases with age and is inversely correlated with serum level of inflammatory cytokines, such as TNF-α and IL-1β (21). (iii) The increased number of senescent cells in tissue secretes various inflammatory cytokines, which play a causal role in age-related diseases. Senescent cells have been identified in age-related diseases including atherosclerosis, cancer, and diabetes (22–24). Senescence-associated secretory phenotype (SASP) is considered to be the main mechanism by which persistent prolonged inflammation occurs even after the initial stimulus has been removed. (iv) Age-related changes in the immune system termed “immunosenescence” increase the susceptibility to infections, malignancy, and autoimmunity, decrease the response to vaccinations, and impair wound healing (25, 26). These changes in the innate and adaptive immune responses associated with increasing age cause inappropriate inflammation. (v) Increased activity of the coagulation and fibrinolysis systems during aging has recently been reported to increase inflammation through the protease-activated receptors (PARs) (27, 28). The plasma concentrations of coagulation factors V, VII, VIII, and IX increase in healthy humans in parallel with the physiological processes of aging. In addition, fibrinogen levels, which have emerged in several trials as a primary risk factor for thrombotic disorders, have been shown to increase with advancing age (29). Thus, uncontrolled coagulation activity and the subsequent activation of the fibrinolysis system contribute to the pathophysiology of aging and several age-related chronic inflammatory diseases, such as atherosclerosis and lung fibrosis (30).
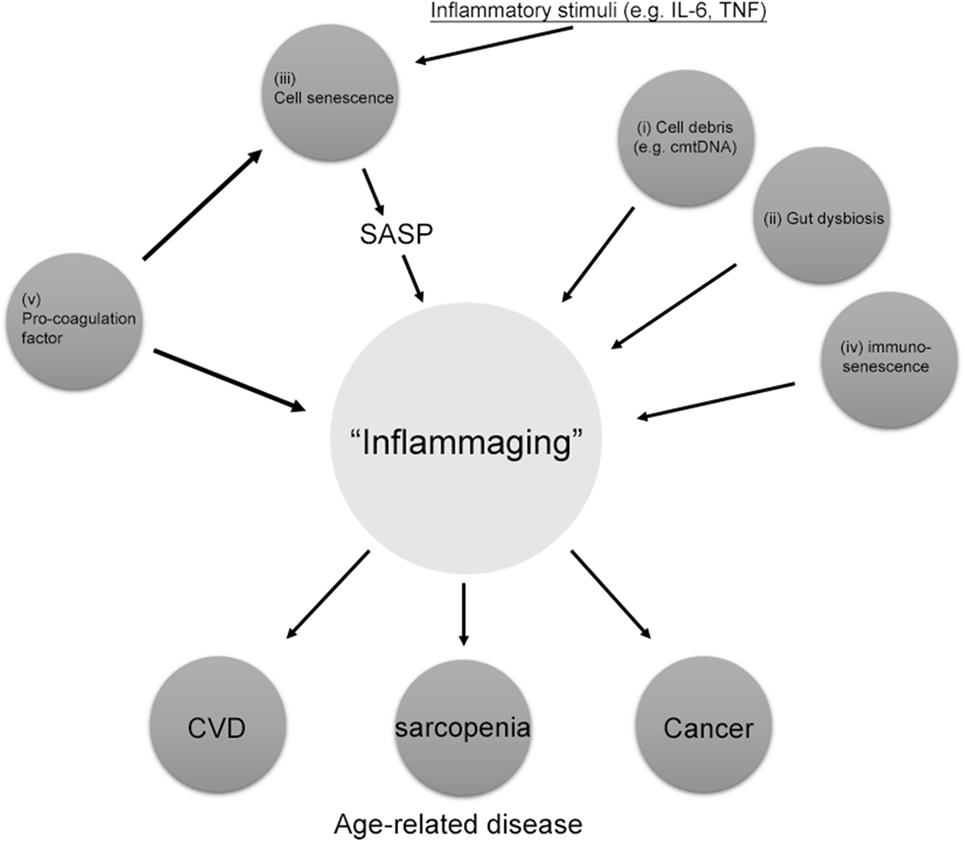
Figure 1. Source of “inflammaging.” Among the main causes of inflammaging, the accumulation of pro-coagulation factors, cell senescence, cell debris such as circulating mitochondrial DNA (cmtDNA), gut dysbiosis, and immune senescence are the main causes of inflammaging. Pro-coagulation factors also cause cell senescence. Inflammaging can also be influenced by many other factors, including age, reactive oxygen species, and those not directly related to inflammation, such as microRNAs (miRs) and agalactosylated N-glycans. SASP, senescence-associated secretory phenotype.
IGFBP-5 and Cell Senescence
Increasing evidence has implied that the clearance of senescent cells in animal models attenuates the progression of age-related disorders, including osteoarthritis and atherosclerosis (31–33). This evidence strongly supports the hypothesis that senescent cell clearance or the modulation of pro-inflammatory pathways related to the acquisition of SASP might be potential therapeutic strategies for combating age-related diseases and expanding the health span of humans. The IGF/IGFBPs system has been implicated to be a potential target of age-related disease. Of the six IGFBPs, IGFBP-5 plays a critical role in the process of replicative and premature cell senescence (10, 11). PAR-1/2 signaling induced by coagulation factor Xa (FXa) and the fibrinolytic factor plasmin has been shown to increase IGFBP-5 expression in endothelial cells (ECs) and smooth muscle cells (SMCs) (34–36). FXa stimulation of ECs and SMCs increased inflammatory cytokine secretion via enhancing cellular senescence through the early growth response 1–IGFBP-5–p53 pathway (34, 37). Interestingly, the FXa- and IGFBP-5-positive areas within the atherosclerotic plaques of human were similarly distributed (37). Kojima et al. have demonstrated that IGFBP-5, as a major signal transducer and activator of transcription 3 mediator, regulates IL-6-induced reactive oxygen species (ROS) production, as well as the subsequent DNA damage response and senescence of TIG3 fibroblast cells (38). They also discovered that IGFBP-5 itself had senescence-inducing activity in TIG cells with increased ROS production. Knocking down of IGFBP-5 significantly reduced IL-6/IL-6R-induced ROS increase and premature senescence. Together, all of these data support the hypothesis that IGFBP-5, which is produced in p53-dependent manner, plays an important role in FXa- or IL-6-induced premature senescence of ECs, SMCs, and fibroblast. IGFBP-5 plays a multifunctional role, possessing growth inhibitory and growth promoting functions (39). IGFBP-5 in breast cancer cells enhances cell proliferation (40). In contrast, IGFBP-5 transgenic mice show retarded growth and reduced litter size (41). Additionally, IGFBP-5 directly regulates apoptosis by interfering with the IGF signaling cascade (42). Moreover, cytoplasmic accumulation of IGFBP-5 in breast cancer cells interacted with sphingosine kinase and protein kinase C, stimulating antiapoptotic effects (9, 43). Thus, IGFBP-5 seems to exert its multifunction depending on cell type, pattern of its distribution in cells and tissue, and IGF-I/II bioavailability.
IGFBP-3, -4, and -6 are also associated with the process of cell senescence. Through Akt, p53, FOXO3a, IGFBP-3 promotes ECs and fibroblast senescence (44, 45). Senescent mesenchymal stem cells secrete IGFBP-4, and it promotes their senescence (39). Senescence induced by pro-oxidative stimuli increases IGFBP-6 levels and IGFBP-6 enhances cell senescence in fibroblast (46, 47), although some experiments demonstrated contradictory results (48). Structurally, six IGFBPs have highly conserved N- and C-terminal domains (49) and different protein sequence in the linker domains (3). Considering their tissue distribution pattern (50), the six IGFBPs might have similar effect on cell senescence in different tissues. Alternatively diverse posttranslational modification in the linker domain of IGFBPs during aging might modify their function on cell growth and senescence. Additional work is required for the elucidation of their function in cell senescence.
Conclusion
IGF binding protein-5 has decisive roles in controlling cell senescence and subsequent cell inflammation independent of IGF-I and -II. IGFBP-5 expression was recently shown to be increased following stimulation with coagulation fXa, plasmin, IL-6, and irradiation, leading to cell senescence (Figure 2). Additionally, IGFBP-5 induces fibroblast activation and the inflammatory response, contributing to tissue fibrosis. Currently, information on the roles of IGFBPs in the aging of different cells and tissues and the molecules related to IGFBPs signaling is limited. Therefore, the molecular mechanisms underlying the effect of the IGFBP system on aging requires further research. Therapies targeting the coagulation and fibrinolysis cascades might represent new options for the treatment of chronic inflammatory diseases.
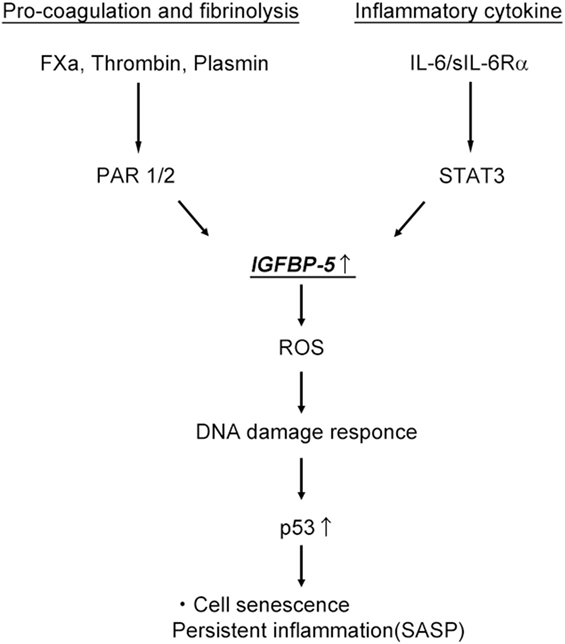
Figure 2. Activation of the pro-coagulation cascade and interleukin-6 (IL-6)/signal transducer and activator of transcription (STAT3) pathway induce cell senescence and persistent inflammation through IGF binding protein-5 (IGFBP-5). ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype.
Author Contributions
FS and YT organized, performed experiment, and wrote manuscript. JM, RO, and HS collected data. HR and RM supervised experiment.
Conflict of Interest Statement
RM received research funding from Bayer Yakuhin, Ltd. Other authors have no conflicts of interests.
Acknowledgments
We would like to thank Blure May Kanako and Kana Shibata for providing technical support and all members of the Clinical Gene Therapy group at the Osaka University Graduate School of Medicine.
Funding
This work was partially supported by a Grant-in-Aid from the Organization for Pharmaceutical Safety and Research, a Grant-in-Aid from the Ministry of Public Health and Welfare, a Grant-in-Aid from Japan Promotion of Science, special coordination funds from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese government, and research funding from Bayer Yakuhin, Ltd.
References
1. Bartke A. Impact of reduced insulin-like growth factor-1/insulin signaling on aging in mammals: novel findings. Aging Cell (2008) 7(3):285–90. doi:10.1111/j.1474-9726.2008.00387.x
2. Ewald CY, Landis JN, Porter Abate J, Murphy CT, Blackwell TK. Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature (2015) 519(7541):97–101. doi:10.1038/nature14021
3. Forbes BE, McCarthy P, Norton RS. Insulin-like growth factor binding proteins: a structural perspective. Front Endocrinol (2012) 3:38. doi:10.3389/fendo.2012.00038
4. Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer (2014) 14(5):329–41. doi:10.1038/nrc3720
5. Li P, Sun X, Cai G, Chen X. Insulin-like growth factor system and aging. J Aging Sci (2017) 5:171. doi:10.4172/2329-8847.1000171
6. Perks CM, Newcomb PV, Norman MR, Holly JM. Effect of insulin-like growth factor binding protein-1 on integrin signalling and the induction of apoptosis in human breast cancer cells. J Mol Endocrinol (1999) 22(2):141–50. doi:10.1677/jme.0.0220141
7. Wang GK, Hu L, Fuller GN, Zhang W. An interaction between insulin-like growth factor-binding protein 2 (IGFBP-2) and integrin alpha5 is essential for IGFBP2-induced cell mobility. J Biol Chem (2006) 281(20):14085–91. doi:10.1074/jbc.M513686200
8. Granata R, Trovato L, Garbarino G, Taliano M, Ponti R, Sala G, et al. Dual effects of IGFBP-3 on endothelial cell apoptosis and survival: involvement of the sphingolipid signaling pathways. FASEB J (2004) 18(12):1456–8. doi:10.1096/fj.04-1618fje
9. McCaig C, Perks CM, Holly JM. Signaling pathways involved in the direct effects of IGFBP-5 on breast epithelial cell attachment and survival. J Cell Biochem (2002) 84:784–94. doi:10.1002/jcb.10093
10. Rombouts C, Aerts A, Quintens R, Baselet B, El-Saghire H, Harms-Ringdahl M, et al. Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int J Radiat Biol (2014) 90(7):560–74. doi:10.3109/09553002.2014.905724
11. Kim KS, Seu YB, Baek SH, Kim MJ, Kim KJ, Kim JH, et al. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol Biol Cell (2007) 18(11):4543–52. doi:10.1091/mbc.E07-03-0280
12. Anisimov VN, Bartke A. The key role of growth hormone-insulin-IGF-1 signaling in aging and cancer. Crit Rev Oncol Hematol (2013) 87(3):201–23. doi:10.1016/j.critrevonc.2013.01.005
13. Straub RH, Schradin C. Chronic inflammatory systemic diseases: an evolutionary trade-off between acutely beneficial but chronically harmful programs. Evol Med Public Health (2016) 2016(1):37–51. doi:10.1093/emph/eow001
14. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci (2014) 69(Suppl 1):S4–9. doi:10.1093/gerona/glu057
15. Beyer I, Mets T, Bautmans I. Chronic low-grade inflammation and age-related sarcopenia. Curr Opin Clin Nutr Metab Care (2012) 15(1):12–22. doi:10.1097/MCO.0b013e32834dd297
16. Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol (2008) 152(3):448–55. doi:10.1111/j.1365-2249.2008.03658.x
17. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
18. Dall’Olio F, Vanhooren V, Chen CC, Slagboom PE, Wuhrer M, Franceschi C. N-glycomic biomarkers of biological aging and longevity: a link with inflammaging. Ageing Res Rev (2013) 12(2):685–98. doi:10.1016/j.arr.2012.02.002
19. Biagi E, Candela M, Franceschi C, Brigidi P. The aging gut microbiota: new perspectives. Ageing Res Rev (2011) 10(4):428–9. doi:10.1016/j.arr.2011.03.004
20. Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell (2006) 126(5):855–67. doi:10.1016/j.cell.2006.08.019
21. Toward R, Montandon S, Walton G, Gibson GR. Effect of prebiotics on the human gut microbiota of elderly persons. Gut Microbes (2012) 3:57–60. doi:10.4161/gmic.19411
22. Sanada F, Taniyama Y, Azuma J, Iekushi K, Dosaka N, Yokoi T, et al. Hepatocyte growth factor, but not vascular endothelial growth factor, attenuates angiotensin II-induced endothelial progenitor cell senescence. Hypertension (2009) 53(1):77–82. doi:10.1161/HYPERTENSIONAHA.108.120725
23. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest (2013) 123(3):966–72. doi:10.1172/JCI64098
24. He S, Sharpless NE. Senescence in health and disease. Cell (2017) 169(6):1000–11. doi:10.1016/j.cell.2017.05.015
25. Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology (2007) 120(4):435–46. doi:10.1111/j.1365-2567.2007.02555.x
26. Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol (2007) 211(2):144–56. doi:10.1002/path.2104
27. Chu AJ. Blood coagulation as an intrinsic pathway for proinflammation: a mini review. Inflamm Allergy Drug Targets (2010) 9(1):32–44. doi:10.2174/187152810791292890
28. Hess K, Grant PJ. Inflammation and thrombosis in diabetes. Thromb Haemost (2011) 105(Suppl 1):S43–54. doi:10.1160/THS10-11-0739
29. Favaloro EJ, Franchini M, Lippi G. Aging hemostasis: changes to laboratory markers of hemostasis as we age – a narrative review. Semin Thromb Hemost (2014) 40(6):621–33. doi:10.1055/s-0034-1384631
30. Chu AJ. Tissue factor, blood coagulation, and beyond: an overview. Int J Inflam (2011) 2011:367284. doi:10.4061/2011/367284
31. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (2016) 354(6311):472–7. doi:10.1126/science.aaf6659
32. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med (2017) 23(6):775–81. doi:10.1038/nm.4324
33. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature (2011) 479(7372):232–6. doi:10.1038/nature10600
34. Sanada F, Taniyama Y, Muratsu J, Otsu R, Iwabayashi M, Carracedo M, et al. Activated factor X induces endothelial cell senescence through IGFBP-5. Sci Rep (2016) 6:35580. doi:10.1038/srep35580
35. Kamio N, Hashizume H, Nakao S, Matsushima K, Sugiya H. Plasmin is involved in inflammation via protease-activated receptor-1 activation in human dental pulp. Biochem Pharmacol (2008) 75(10):1974–80. doi:10.1016/j.bcp.2008.02.018
36. Carmo AA, Costa BR, Vago JP, de Oliveira LC, Tavares LP, Nogueira CR, et al. Plasmin induces in vivo monocyte recruitment through protease-activated receptor-1-, MEK/ERK-, and CCR2-mediated signaling. J Immunol (2014) 193(7):3654–63. doi:10.4049/jimmunol.1400334
37. Sanada F, Muratsu J, Otsu R, Shimizu H, Koibuchi N, Uchida K, et al. Local production of activated factor X in atherosclerotic plaque induced vascular smooth muscle cell senescence. Sci Rep (2017) 7:17172. doi:10.1038/s41598-017-17508-6
38. Kojima H, Kunimoto H, Inoue T, Nakajima K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle (2012) 11(4):730–9. doi:10.4161/cc.11.4.19172
39. Severino V, Alessio N, Farina A, Sandomenico A, Cipollaro M, Peluso G, et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis (2013) 4(11):e911. doi:10.1038/cddis.2013.445
40. Akkiprik M, Hu L, Sahin A, Hao X, Zhang W. The subcellular localization of IGFBP5 affects its cell growth and migration functions in breast cancer. BMC Cancer (2009) 9:103. doi:10.1186/1471-2407-9-103
41. Salih DA, Tripathi G, Holding C, Szestak TA, Gonzalez MI, Carter EJ, et al. Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. Proc Natl Acad Sci U S A (2004) 101:4314–9. doi:10.1073/pnas.0400230101
42. Marshman E, Streuli CH. Insulin-like growth factors and insulin-like growth factor binding proteins in mammary gland function. Breast Cancer Res (2002) 6:231–9. doi:10.1186/bcr535
43. Akkiprik M, Feng Y, Wang H, Chen K, Hu L, Sahin A, et al. Multifunctional roles of insulin-like growth factor binding protein 5 in breast cancer. Breast Cancer Res (2008) 10:212. doi:10.1186/bcr2116
44. Kim KS, Kim M-S, Seu YB, Chung HY, Kim JH, Kim J-R. Regulation of replicative senescence by insulin-like growth factor-binding protein 3 in human umbilical vein endothelial cells. Aging Cell (2007) 6(4):535–45. doi:10.1111/j.1474-9726.2007.00315.x
45. Elzi DJ, Lai Y, Song M, Hakala K, Weintraub ST, Shiio Y. Plasminogen activator inhibitor 1-insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci U S A (2012) 109(30):12052–7. doi:10.1073/pnas.1120437109
46. Coppe J-P, Patil CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One (2010) 5:e9188. doi:10.1371/journal.pone.0009188
47. Qiu J, Ma XL, Wang X, Chen H, Huang BR. Insulin-like growth factor binding protein-6 interacts with the thyroid hormone receptor alpha1 and modulates the thyroid hormone-response in osteoblastic differentiation. Mol Cell Biochem (2012) 361:197–208. doi:10.1007/s11010-011-1104-y
48. Bach LA. Recent insights into the actions of IGFBP-6. J Cell Commun Signal (2015) 9(2):189–200. doi:10.1007/s12079-015-0288-4
49. Kim HS, Nagalla SR, Oh Y, Wilson E, Roberts CT Jr, Rosenfeld RG. Identification of a family of low-affinity insulin-like growth factor binding proteins (IGFBPs): characterization of connective tissue growth factor as a member of the IGFBP superfamily. Proc Natl Acad Sci U S A (1997) 94(24):12981–6. doi:10.1073/pnas.94.24.12981
Keywords: IGF binding protein-5, cell senescence, inflammation, coagulation system, age-related disease
Citation: Sanada F, Taniyama Y, Muratsu J, Otsu R, Shimizu H, Rakugi H and Morishita R (2018) IGF Binding Protein-5 Induces Cell Senescence. Front. Endocrinol. 9:53. doi: 10.3389/fendo.2018.00053
Received: 08 December 2017; Accepted: 05 February 2018;
Published: 20 February 2018
Edited by:
Briony Forbes, Flinders University, AustraliaReviewed by:
Anica Dricu, University of Medicine and Pharmacy of Craiova, RomaniaFumihiko Hakuno, The University of Tokyo, Japan
Copyright: © 2018 Sanada, Taniyama, Muratsu, Otsu, Shimizu, Rakugi and Morishita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fumihiro Sanada, c2FuYWRhQGNndC5tZWQub3Nha2EtdS5hYy5qcA==;
Ryuichi Morishita, bW9yaXNoaXRAY2d0Lm1lZC5vc2FrYS11LmFjLmpw