 Matthew L. Bettini
Matthew L. Bettini Maria Bettini
Maria Bettini- Pediatric Diabetes and Endocrinology, Baylor College of Medicine, Texas Children’s Hospital, McNair Medical Institute, Houston, TX, United States
The strongest susceptibility allele for Type 1 Diabetes (T1D) is human leukocyte antigen (HLA), which supports a central role for T cells as the drivers of autoimmunity. However, the precise mechanisms that allow thymic escape and peripheral activation of beta cell antigen-specific T cells are still largely unknown. Studies performed with the non-obese diabetic (NOD) mouse have challenged several immunological dogmas, and have made the NOD mouse a key experimental system to study the steps of immunodysregulation that lead to autoimmune diabetes. The structural similarities between the NOD I-Ag7 and HLA-DQ8 have revealed the stability of the T cell receptor (TCR)/HLA/peptide tri-molecular complex as an important parameter in the development of autoimmune T cells, as well as afforded insights into the key antigens targeted in T1D. In this review, we will provide a summary of the current understanding with regard to autoimmune T cell development, the significance of the antigens targeted in T1D, and the relationship between TCR affinity and immune regulation.
Introduction
Autoimmunity is generally associated with polygenetic susceptibility, while the initial precipitating event is likely triggered by an environmental stressor (1–4). The major alleles associated with most autoimmune disorders are the human leukocyte antigen (HLA), and several alleles are shared among autoimmune conditions (5–8). This suggests that a common T cell-dependent mechanism is the underlying cause of tissue-specific autoimmunity irrespective of the organ or tissue being targeted. Although several hypotheses have been put forth to explain the HLA-mediated susceptibility, the exact mechanisms are still largely unknown. HLA structure selects for a particular peptide sequence motif and can affect the stability of the peptide:HLA complex (9). It is likely that autoimmune epitopes are not efficiently presented within the susceptible HLA molecules during thymic selection, or alternatively are presented with increased stability or at a higher concentration in the target tissue (10). Clearly, HLA allele structure is not the only parameter that might affect the stability of the tri-molecular complex [T cell receptor (TCR)/HLA/peptide], and not all individuals with T1D possess susceptible HLA alleles. Lower level of tissue antigen expression in the thymus, the relative abundance of self-antigen at the tissue site, an increase in immunogenicity of self-peptides either via post-translational modifications (PTMs) or molecular mimicry could all influence the stimulatory capacity of peptide:HLA complexes in periphery (Figure 1). How these changes in epitope immunogenicity could affect disease development will be discussed in this review.
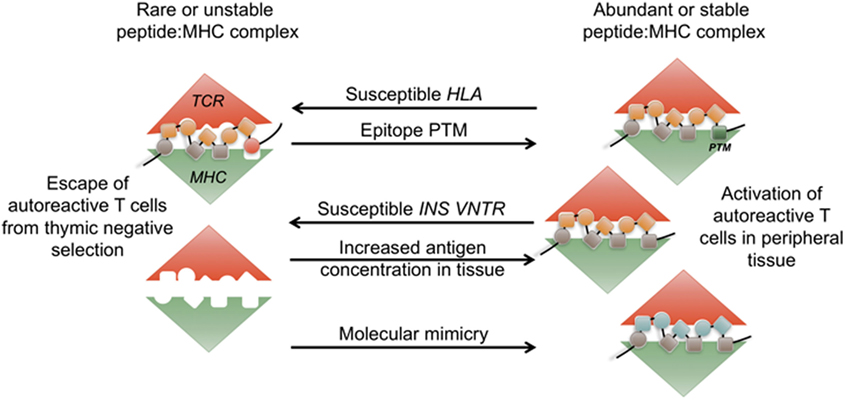
Figure 1. Abundance and stability of the tri-molecular complex at the interface of tolerance and autoimmunity. During thymic development, rare or unstable self-peptide: major histocompatibility (MHC) complexes can lead to escape of autoimmune T cells. Human leukocyte antigen (HLA)-DQ8 and H2-IAg7 susceptible alleles form unstable complexes with insulin epitope B:9-23. INS-VNTR susceptible allele results in lower level of insulin presentation in the thymus. Post-translational modifications (PTM) of self-epitopes can lead to more stable complexes in periphery. Increase in antigen availability in periphery or presence of structurally similar peptides in the context of infection (molecular mimicry) leads to priming of autoimmune T cells.
The spontaneously diabetic non-obese diabetic (NOD) mouse model has been a useful system for identification of the key mechanisms important in the development of autoimmunity due to its significant similarity to human T1D (11, 12). Nearly 6 years after HLA was first associated with T1D in humans (13, 14), the spontaneously generated NOD diabetic strain was obtained by the Jackson Laboratory from CLEA Japan, where it quickly became an invaluable tool in the etiology of T1D (11, 15). The importance of the major histocompatibility (MHC) locus was originally traced by congenic approach, where MHC locus was introgressed onto the NOD background (16, 17). Further analysis of mice that received a non-NOD MHC class II transgene confirmed the important contribution of I-Ag7 to diabetes susceptibility (18). Although MHC II confers most of the susceptibility, there are over 50 genetic loci that make up the NOD diabetic phenotype (19). The polygenetic susceptibility of the NOD mouse strain mirrors human disease, and further underlies the complexity of T1D (20). Importantly, the I-Ag7 MHC II variant has structural similarities with human susceptible DQ8 (DQA1*0301/DQB1*0302) (9, 21, 22). Moreover, many of the antigens targeted in autoimmune diabetes are shared between the two species (19). The similarities of the shallow and positively charged peptide-binding groove characteristic of both human DQ8 and mouse I-Ag7, and significant concordance in antigenic targets have made it possible to uncover sequence characteristics of autoimmune epitopes that are relevant to human disease (23, 24). Nevertheless, the precipitating events that lead to T cell priming and beta cell destruction remain unclear (4, 25). While the NOD mouse model has been a prolific tool for mechanistic insight into the many facets of T1D pathogenesis, recent expansion of HLA-humanized mouse models now allow direct interrogation of human autoimmune tri-molecular complex (TCR/HLA/peptide) and its role in loss of self-tolerance.
Evidence for T Cell-Mediated T1D
A large body of evidence accumulated over several decades has implicated beta cell-specific immune response and, in particular, beta cell-specific T cells as the main drivers of autoimmune tissue damage and development of T1D (12, 26, 27). Progression to disease in humans is associated with islet antigen-specific antibody responses, and T cells specific to islet antigens are found at higher frequencies in T1D patients (28–31). Importantly, both CD4 and CD8 T cells were observed directly in the pancreatic lesions, and islet antigen-specific T cells have been cloned from pancreatic islets of T1D organ donors (32–38). HLA, being the major risk allele, implies that inherent structural differences in HLA and, consequently, TCRs selected on those HLA alleles lead to erroneous T cell reactivity to self (5, 39, 40). While class II HLA alleles confer the majority of the genetic susceptibility, certain class I alleles have been shown to impose a separate risk (41). Multiple antigens are targeted by both CD4 and CD8 T cells in T1D. Beta cell-specific antigens presented by Class II molecules include preproinsulin (PPI), insulinoma-associated antigen (I-A2), glutamic acid decarboxylase (GAD) 65, heat shock protein (HSP)-60, HSP-70, islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), and zinc transporter (ZnT8) (42–44). While MHC class I responses display similar wide range of antigenic targets, including PPI signal peptide, IA2, ZNT8, human islet amyloid polypeptide (IAPP), IGRP, and GAD65 (45). The progression to T1D in humans is associated with accumulation of islet antigen antibody reactivity to IAA, GAD65, IA-2, and ZnT8, which mirrors the intra- and inter-molecular “antigenic spread” of T cell responses (46, 47). Other non-HLA allelic risk variants are associated with pathways involved in T cell development, activation, and function, further highlighting the importance of T cells are the key drivers of autoimmunity (19).
HLA Mechanisms of Autoimmunity
While the precise mechanisms that lead to loss of tolerance are multifaceted, HLA-DQ8 susceptibility implies that the stability of the tri-molecular complex is an important aspect that underlies autoimmune T cell responses (Figure 1). The inbred NOD mouse model that possesses a single susceptible MHC class II allele I-Ag7 (I-Adα/I-Ag7β) has played a vital role in uncovering the mechanisms involved in the development of T1D. The structural similarities characterized by the shallow peptide-binding groove and the positive charge in the p9 peptide-binding pocket present in both I-Ag7 and human HLA-DQ8 point to a similar mechanism of autoimmune susceptibility (22). The potential mechanisms include altered thymic selection due to peptide:MHC instability, and/or preferential binding and presentation of beta cell neo-antigens formed via post-transnational modifications in the periphery (Figure 1). Biochemical analyses revealed a propensity for both DQ8 and I-Ag7 to bind peptides with negatively charged C-terminus (48). In the case of celiac disease, which is also associated with DQ8 susceptibility, gluten peptides targeted in disease have a negative charge at the C-terminus, which results in their stable binding to DQ8 (49). Although this observation suggests that key epitopes targeted in T1D should similarly contain negatively charged residues at p9, most beta cell antigenic epitopes lack this trait. Moreover, the dominant insulin epitope B:9-23 has a positively charged arginine at the C-terminus. Nevertheless, in support of this hypothesis, a mutation of InsB9–23 at presumptive p9 to a negatively charged glutamic acid increased the immunogenicity of the epitope and augmented the activation of insulin-specific T cells (50). In addition, a recent study has identified IAPP and Chromogranin A (ChgA) epitopes in beta cells that have been modified by peptide fusion to acquire a negative charge at the C-terminus (35). The modified peptides were significantly more immunogenic compared to unmodified wild-type epitopes. This groundbreaking finding offered a potential explanation for lack of efficient thymic selection under conditions of unstable tri-molecular complex formation in the thymus, followed by priming and activation of autoreactive T cells in response to modified and stable peptide:MHC complexes in peripheral tissue.
Thymic Development of Autoreactive T Cells—What is the Evidence for Altered Thymic Selection in Autoimmunity?
A body of evidence suggests an important role for altered thymic selection in the development of autoimmunity. Negative selection of autoimmune lymphocytes depends on sufficient amount of self-antigen available for presentation in the thymus, which is regulated by intra-thymic and extra-thymic sources, genetic variation in tissue antigen promoters, and effective antigen presentation on certain HLA alleles (Figures 1 and 2A). Normally, tissue-specific antigens are presented by Autoimmune regulator (Aire) and Fezf2 expressing thymic medullary epithelial cells (mTECs) to aide in the deletion of self-reactive thymocytes (51, 52). MTECs can also transfer antigens, including beta cell antigens, to thymic resident dendritic cells (DCs), which in turn delete self-reactive T cells (53). Both DCs and Aire expressing mTECs are also essential in generating thymically derived Foxp3+ regulatory T cells (Tregs), a critical population for the establishment and maintenance of self-tolerance (54, 55). Indeed, there appears to be a correlation between a reduction in DC numbers and residual β cell function in T1D subjects (56), while the NOD mouse exhibits an overall reduction in DCs (57, 58). These observations suggest a relationship between self-tolerance and the absolute number of DCs present in the thymus and periphery. However, not all peripheral antigens are expressed by mTECs and, therefore, negative selection must also rely on peripheral antigen retrieval and delivery to the thymus by DCs. Importantly, studies have shown that the generation of thymic regulatory T cells by antigen-presenting mTECs and DCs early in life (neonatal) is critical in maintaining tolerance to self (55, 59). The idea of peripheral antigen exposure generating tissue-specific Tregs was elegantly demonstrated by Scharschmidt et. al., where skin colonization of S. epidermidis allowed for the development and trafficking of microflora-specific Tregs to the skin. Using sphingosine-1-phosphate receptor antagonist, FTY720, the authors blocked Treg egress and pinpointed the thymus as the main source for Treg development (59).
There is no direct evidence for thymic selection deficiencies in individuals with T1D; however, several key observations suggest that there is a role for altered selection in the development of autoimmune responses to insulin. The level of thymic insulin expression in humans is controlled in part by the polymorphic variable number of tandem nucleotide repeats found in the region proximal to the promoter region of the insulin gene (INS–VNTR) (60). It has been shown that VNTR I alleles express 26–63 tandem repeats while the VNTR III carries 141–209 repeats. This difference translates into higher thymic transcript levels for the VNTR III individuals and a threefold to fourfold relative protection from T1D (61, 62). It appears that the number of repeats affects AIRE binding to the insulin promoter region, thus controlling transcriptional regulation of insulin in the thymus (51, 62, 63). In support of alterations in thymic selection, analysis of human peripheral blood from T1D patients and healthy controls revealed that subjects expressing the INS-VNTR I (T1D-predisposing) allele displayed elevated frequencies of high affinity proinsulin-specific T cells compared to INS–VNTR I HLA–DR4 subjects (64). INS-VNTR allelic expression appears to determine insulin reactivity rather than the total number of insulin-reactive T cells, as both VNTR I and VNTR III groups displayed similar total number of insulin-reactive T cells in peripheral blood (64). However, it has only been hypothesized that the differences in thymic insulin expression between VNTR I and VNTR III subjects influence positive and negative selections of insulin-reactive T cells, but this has never been formally demonstrated in vivo due to a lack of VNTR mouse models.
The role of thymic insulin expression in the establishment of central tolerance has been addressed in the NOD mouse model by both deletion and overexpression of insulin in the thymus. Deletion of insulin specifically in thymic Aire expressing mTECs enhanced diabetes development in both male and female mice (65). In addition, transgenic overexpression of proinsulin, but not GAD65 (66) or IGRP (67), significantly delayed (68) or prevented (69) diabetes progression in NOD mice. However, in these studies overexpression of insulin was targeted to all MHC class II expressing APCs and, therefore, the relative role of central compared to peripheral tolerance was not determined (68, 69). A more recent set of experiments determined that a narrow window of ectopic proinsulin expression in APCs (from birth until weaning) could prevent the development of diabetes in NOD mice (70). This timeframe fits with a previous study that showed organ specific autoreactive T cell escape from the thymus is greatest during the first 10 days of life in NOD mice (71). In the former study by Jhala et. al., protection was due in part to the deletion of insulin-specific T cells, but also the inability of the remaining insulin-specific T cells to respond to cognate antigen in periphery (70). In our recent study, we tested two TCRs (4-8 and 12-4.1, Table 1) with defined affinities for InsB9–23 for their ability to escape negative selection in the presence of ectopic overexpression of insulin. Surprisingly, we did not observe any increase in thymic deletion of the relatively high (4-8) or low (12-4.1) TCRs, although the increase in insulin expression did protect mice from developing autoimmune diabetes. Protection from disease appeared to be due to an increase in Treg development with a significant increase in thymic, splenic, and pancreas-residing insulin-specific Tregs (72). These findings pose an intriguing possibility that the amount or stability of self-peptide:MHC complexes during thymic selection is more important for Treg development rather than deletion of self-reactive T cells (Figure 2A).
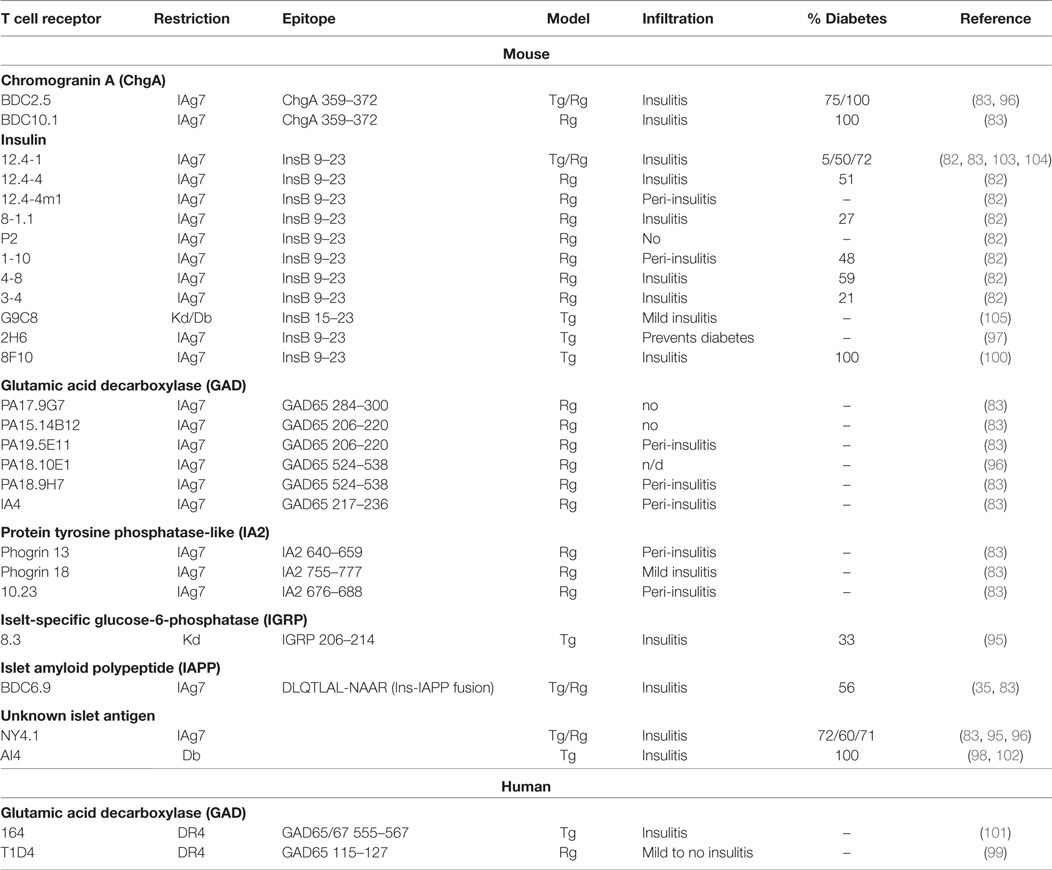
Table 1. Pathogenicity of beta antigen-reactive T cells.
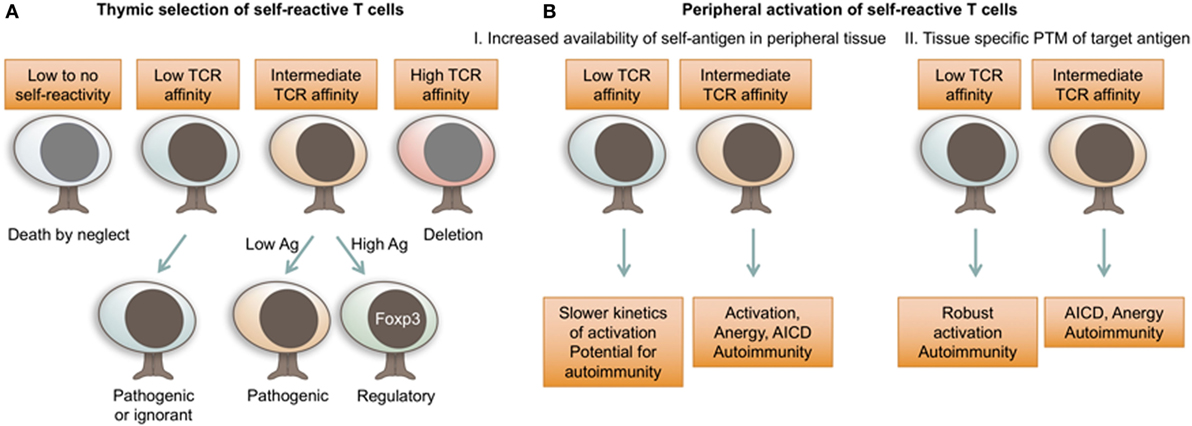
Figure 2. T cell receptor (TCR) affinity for self dictates autoimmune T cell fate decisions. (A) TCR affinity for self-ligands and antigen availability dictate thymocyte fate choices during thymic selection. While autoimmune T cells can be selected with a range of TCR affinities, increased antigen availability and relatively stronger self-reactivity will preferentially result in the development of regulatory Foxp3+ T cells. (B) In peripheral tissues, self-reactive T cells are activated in response to increased concentrations of tissue antigen or highly immunogenic PTM antigens. While autoimmune T cells can possess a range of TCR affinities for self-antigen, lower affinity TCRs are less susceptible to peripheral mechanisms of tolerance. Ag, antigen; AICD, activation induced cell death; PTM, post-translational modification.
Chromogranin A is the only other currently known beta cell antigen necessary for the initiation of autoimmune diabetes in NOD mice; however, expression of ChgA in the thymus has not yet been detected (73). Therefore, tolerance to ChgA may rely in part on transport of antigen by peripheral DCs to the thymus. Whether islet-derived antigens are carried to the thymus to promote islet-specific Treg development has not been explored; nevertheless, the divergent TCR repertoire between islet-infiltrating effector and regulatory T cells suggests a lack of local Treg conversion in favor of thymic lineage being the predominant Treg population in the pancreas (74). The thymic Treg niche was thought to be highly specialized and restricted (75); however, a recent study has demonstrated that the manipulation of either the number of antigen-presenting cells or an increase in antigen exposure within the thymus can expand the Treg niche (54).
While highly self-reactive T cells are removed from the T cell repertoire by negative selection, the quality and the quantity of self-reactive Tregs that develop from the moderately self-reactive thymocyte pool is a critical component of peripheral self-tolerance (Figure 2A). This idea is consistent with the observation that healthy individuals possess significant frequencies of self-reactive T cells, but are free from autoimmunity (76, 77). The escape of self-reactive T cells in itself is not just a byproduct of Treg development, but seems to serve an important immunological purpose, since some level of self-reactivity is associated with enhanced responsiveness to foreign pathogens (78–80). It is likely that the fine balance between beneficial self-reactivity and self-tolerance is uniquely perturbed in individuals with a susceptible genetic background. A slight change in thymic antigen expression or the overall stability of the tri-molecular complex could shift the T cell development spectrum toward Treg insufficiency, rather than escape of higher affinity cells. Therefore, the ratio of beta cell antigen-reactive Tregs vs. effector or memory T cells might be a better predictive biomarker of autoimmunity than the overall frequencies of self-reactive cells.
TCR Parameters of T Cell Pathogenicity
Autoimmune T cell responses in general, as well as, the T cell population that infiltrates the NOD pancreatic islets, are composed of cells with different T cell lineages, diabetogenic or regulatory capabilities, antigenic specificities, and TCR affinities (10, 81–83). All of these parameters are directly influenced by the TCR (84). Therefore, TCR sequence, specificity, and affinity hold the key to understanding the dynamics of diabetogenic T cell responses during chronic progressive autoimmune disorders, such as T1D. The antigenic specificity of each TCR is dictated by the highly variable CDR3 region found within the α and β chains of the TCR heterodimer. The variability is the result of random genetic recombination events that bring together one of many variable (V) genetic segments with a joining (J) region. The large number of TCR sequences infiltrating an organ, their variability among individuals, and the heterodimeric structure of the TCR has been a significant roadblock in a comprehensive functional analysis of TCRs. In this section, we will summarize the studies that have investigated beta cell-specific TCR parameters for their ability to predict T cell pathogenic potential.
TCR Sequence As a Biomarker of Pathogenicity
One of the main hurdles in the identification and functional analysis of beta cell-reactive T cells in humans with T1D is the breadth of antigens and epitopes that are targeted among affected individuals. Peptide/MHC tetrameric reagents have been the most effective approach to identify T cells with autoimmune potential; however, beta cell-reactive cells comprise a small population of peripheral blood, which makes such approaches technically challenging. Moreover, tetramers detect only the highest affinity subpopulation of T cells specific for a particular epitope, while the majority of autoimmune responder T cells are often overlooked, as was effectively demonstrated in the mouse model of multiple sclerosis (85). As such, the field is currently lacking sufficient approaches to perform in depth tracking of antigen-specific T cells over time. Recent technological advances in high-throughput sequencing have opened new avenues for tracking self-reactive T cells and could be easily applicable to studies of human tissue-infiltrating T cells (86). A promising biomarker approach could be based on high-throughput TCR sequencing with focus on TCR motifs known to be associated with a specific target epitope. While human CD4 responses have proven to be highly diverse (87, 88), CD8 T cells are generally more clonotypic (86). A recent study was able to identify a public CDR3 motif associated with IGRP265–273 specific memory T cells in antibody-positive subjects and individuals diagnosed with T1D (89). Their findings suggest that dominant clonotypes persist in the same individual over time, and some TCR sequences could be shared among individuals. Interestingly, the public TCR motif was also identified in healthy controls, although it was restricted to the naïve T cell compartment. While promising as a potential biomarker, such deep sequencing approaches necessitate knowledge of multiple TCR sequences associated with reactivity to several beta cell antigens.
Antigen Specificity of Pathogenic TCRs
Although, T cells of multiple antigenic reactivities have been isolated from pancreatic islets of T1D donors (35–38), it does not necessitate that these cells are equally pathogenic or are actively involved in beta cell destruction. In order to identify potentially important initiating antigens in T1D, multiple beta cell proteins have been mutated on the NOD background, including IAPP, GAD65, insulin, IGRP, and islet Ag-2 (90–94). Interestingly, only the mutation of insulin and chromogranin resulted in protection against diabetes (73, 94). This suggests that insulin and chromogranin-reactive T cells are either critical for the initiation of autoimmunity, or are necessary for further propagation of the disease and the ultimate destruction of beta cells.
Over the years, pathogenic potential of T cells reactive to various islet antigens was directly assessed in single TCR systems. Multiple mouse and a few human TCRs reactive against various beta cell proteins have been expressed in mice utilizing both transgenic and retrogenic approaches (82, 83, 95–105) (Table 1). Importantly, the observed tissue infiltration and spontaneous disease development were highly variable among the antigenic specificities (Table 1). Single TCR mice expressing either insulin, chromogranin, or IGRP reactive mouse TCRs developed spontaneous diabetes, supporting the important pathogenic role for these reactivities in autoimmune diabetes. The majority of phogrin (IA2b) and I-A2 reactive mouse TCRs can induce islet infiltration, albeit without overt diabetes. Reactivity to multiple GAD epitopes, however, results in no disease and very limited infiltration for both human and mouse TCRs (Table 1). Based on these observations, it is likely that TCRs with select beta cell antigenic specificities are central to disease pathogenesis. Moreover, certain specificities might be important at different stages of disease, while others might not have a pathogenic but rather a regulatory effect, as was observed for GAD-reactive mouse T cells (106–109). Nevertheless, our ability to effectively extrapolate contribution of T cell specificities to disease in a polyclonal multi-antigen specific environment by analyzing their behavior in single TCR systems is limited. NOD mouse models exhibit a single MHC II restriction; therefore, pathogenic responses to antigens presented in alternative susceptible HLA class II or class I alleles might be overlooked. Alternatively, it is possible that inflammation induced by T cells specific for the initiating antigen results in exposure or modification of secondary antigens, leading to pathogenic activation of a distinct repertoire of T cells specific to the newly displayed epitopes.
The molecular determinants of pathogenic TCRs in autoimmunity are still largely unknown. Antigen availability, immunogenic modification of T cell epitopes, and TCR avidity could all shape the responses of beta cell-specific T cells (Figure 2). While it is still unclear whether antigen reactivity is an absolute pre-requisite for tissue entry, several experimental approaches have shown that T cell accumulation in NOD pancreatic islets is driven by antigen recognition (110–112). The difference in antigen availability could explain relative importance of T cell specificities in the development and progression of autoimmunity. For example, reduced pathogenicity of GAD65-reactive T cells in NOD mouse model might be due to insufficient antigen availability in the pancreas. T cell reactivity to GAD65 and GAD67 can be observed early in NOD mice (113, 114), and antibodies specific for GAD are associated with progression to T1D in humans (115), which suggests a role for GAD reactivity in T1D. However, relative to other beta cell antigens, GAD T cells exhibit reduced pathogenicity in mouse models compared to other antigens (Table 1), with only one study showing diabetogenic activity of GAD65-reactive T cells (116). The rather mild pathogenic potential of GAD-reactive T cells in NOD model could be attributed to relatively low levels of both GAD65 and GAD67 expressed in the mouse islets compared to rat or human pancreas (117). In support of this, overexpression of GAD65 under the rat insulin promoter enhanced pancreatic infiltration of GAD-reactive T cells (110). Although, this observation serves as a proof of principle for the importance of antigen availability for islet infiltration, overexpression of GAD65 in polyclonal NOD mice does not result in enhanced insulitis or diabetes (118). Therefore, other parameters in addition to islet antigen availability must regulate T cell pathogenic potential.
TCR Affinity of Pathogenic T Cells
It is logical to assume that TCR affinity for antigen is associated with increased pathogenicity; however, that is not always the case, as we have shown for insulin-reactive TCRs. When eight NOD CD4 T cell-derived TCRs with variable affinity for insulin InsB9–23 epitope were compared for their ability to drive spontaneous diabetes, high- and low-affinity T cells were similarly pathogenic (82) (Table 1). This is consistent with observations that a polyclonal autoimmune T cell response can encompass a wide range of TCR affinities, and low-affinity T cells are important contributors to the immune response (85, 119). However, it appears that there are certain functional distinctions between high- and low-affinity insulin-reactive T cells. Compared to high-affinity TCRs, low-affinity TCRs were less sensitive to thymic negative selection pressures, exhibited lower frequencies of Foxp3+ T cell development, and had a reduction in negative regulators of T cell activation (82). Their inability to reach the threshold for engagement of regulatory elements could allow the low-affinity cells to exert effector functions and induce beta cell damage even under relatively low level of TCR stimulation (Figure 2B).
Peripheral Priming of Autoimmune T Cells
Molecular Mimicry
The mechanisms behind self-reactive T cell priming and ensuing loss of self-tolerance are complex and poorly understood. Autoimmune T cells exhibit a level of reactivity for self-antigens, but are somehow able to escape negative selection in the thymus. In the periphery, these cells encounter cognate self-antigen with enough affinity and in the right context to become activated and cause tissue damage. In the case of T1D, studies have implicated molecular mimicry as a potential trigger, where beta cell-reactive T cells could undergo initial priming and activation in response to structurally similar microbial epitopes (120) (Figure 1). While the direct evidence for molecular mimicry as a cause for autoimmunity is lacking, recent work exposing the previously unrecognized propensity of T cells for cross-reactivity reinforces molecular mimicry as a valid hypothesis (87, 121, 122). Islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP)-reactive CD8 T cells were shown to recognize a transporter protein peptide of Fusobacteria. Importantly, activation of IGRP-specific NY8.3 T cells by Fusobacteria contributed to enhanced diabetes development (123). Microflora composition in general has been implicated in both human and mouse T1D. In mice, gender hormones influence microbiota and subsequent T1D development (124, 125), while autoantibody-positive children have distinct microbiota signatures (126). It has yet to be seen whether specific microbiota species drive activation of islet-reactive T cells leading to beta cell destruction.
Unusual Orientation of the Tri-Molecular Complex
It is hard to reconcile exceedingly lower reactivity of autoimmune T cells to their cognate antigen, compared to non-self-reactive TCRs, with their capacity to exert significant tissue damage. For example, insulin-reactive TCR 12-4.1 isolated from pancreatic islets of NOD mice exhibits barely detectable reactivity to insulin in vitro (82), but causes spontaneous diabetes in 50–80% of mice (82, 103) (Table 1). As we alluded to earlier, lower affinity self-reactive TCRs are to some degree resistant to central and peripheral tolerance mechanisms, which might explain their ability to persist in an activated state (82). However, it is still unclear how self-reactive T cells with very low affinity for antigen are capable of causing beta cell destruction and highly penetrant diabetes. It is possible that the inherent unusual TCR structural and signaling characteristics are potential contributing factors that lead to unique responsiveness of autoimmune T cells. Crystal structures of autoimmune TCR:pMHC complexes have uncovered an unconventional docking of self-reactive TCRs on pMHC (127–129). Moreover, self-reactive human and mouse TCRs form unusual disorganized T cell synapses, exhibit slower kinetics of TCR signaling pathways, and yet they are still able to undergo activation and exert effector functions (130, 131). Conceivably, these characteristics allow autoimmune T cell escape from thymic selection, while in the target tissue high level of antigen is sufficient to elicit effector response.
Tissue-Specific PTM of Target Epitopes
In the case of autoimmune T1D, beta cell fragility characterized by increased susceptibility to oxidative and ER stress may be a critical factor in loss of self-tolerance. A consequence of the cellular stress is the altered processing and changes in PTM of proteins. The changes in beta cell epitopes can lead to the generation of tissue-specific neo-antigens that are not expressed in the thymus. T cells specific for neo-antigens can evade mechanisms of central tolerance and initiate an autoimmune response once exposed to PTM antigens in periphery (Figure 1). Interestingly, insulin containing granules are highly immunogenic compared to artificially synthesized protein, which suggests some manner of PTM takes place within the NOD beta cell granules (10). In the case of the dominant insulin epitope targeted in the NOD mice (InsB9–23), the modification likely affects the MHC-binding residue of the peptide, resulting in stable binding of peptide in a register that is normally unstable and very likely presented at low levels in the thymus (10, 50, 132). In support of this idea, studies have shown that a mimotope of the InsB9–23 insulin peptide with a change in the MHC anchor residue (R22E) was highly stimulatory for insulin-reactive T cells, and R22E peptide:MHC tetramers identified insulin-reactive cells within the islet-infiltrating T cell population (50, 133). Just in the last few years, it has been demonstrated that neo-antigenic PTM epitopes can form by fusion of either ChgA or IAPP peptide with a pro-insulin peptide (35). These fusion peptides were highly stimulatory to IAPP- and ChgA-reactive diabetogenic NOD T cell clones, as well as CD4 T cells isolated from the islets of T1D donors (35, 37). While the fusion peptides were identified in beta cells, it is unknown whether their formation is increased during inflammation or ER stress. More recent work has identified immunogenic peptides generated from an alternate insulin reading frame, the translation of which was further increased under ER stress (134). CD8 T cell clones isolated from peripheral blood of T1D subjects and specific for these defective ribosomal products (DRiPs) were able to cause direct beta cell damage in vitro, supporting a potentially critical role for DRiPs in T1D. This is yet another PTM mechanism within a mounting evidence for connection between beta cell ER stress and generation of immunogenic PTMs. Nevertheless, it is still unknown exactly to what extent PMT antigen-specific T cells contribute to T1D.
At the moment, we have very little insight into the functional concentration of PTM antigens vs. wild type epitopes presented in the inflamed tissue, the relative frequency of PTM-reactive T cells vs. T cells that recognize the wild-type epitopes, or how these parameters change over the course of chronic autoimmune tissue damage. It is likely that some T cells have a restricted specificity to either PTM or wild-type antigens, while others respond to both with different levels of activation. Addressing these questions will lead to our better understanding of the triggers that induce autoimmune response, as well as identification of the initiating antigens and the key pathogenic T cell populations. It is currently unknown whether tissue-specific PTM antigens are transported and expressed in the thymus. In order to model how the presence of post-translationally modified peptides in the thymus could alter the selection of insulin-reactive TCRs (4-8 and 12-4.1) that normally escape negative selection, we ectopically expressed the R22E insulin mimetope in bone marrow-derived APCs (72). In the presence of R22E, the high-affinity 4-8 TCR bearing thymocytes were efficiently deleted, while the low affinity 12-4.1 population was affected to a lesser degree, albeit still showing an increase in negative selection based on Annexin V staining. Nevertheless, the ectopic expression of R22E significantly reduced peripheral T cells and halted any islet infiltration in both the 4-8 and 12-4.1 retrogenic mice. These results suggest that unlike expression of wild-type antigen, expression of PTM epitopes in the thymus results in efficient deletion of autoimmune T cells.
Accumulating evidence indicates PTMs as the key to our understanding of autoimmune disease development (35, 135–140). Importantly, T cells specific for PTM GAD65 and ChgA epitopes have been identified in individuals diagnosed with T1D (35, 139). Although the evidence so far is limited, PTM epitope expression is likely restricted to peripheral tissue and is absent from the thymus. While wild-type self-proteins presented in the thymus successfully limit development of high-affinity self-reactive T cells, lower affinity T cells evade central tolerance to be able to respond to PTM antigens in periphery (Figures 1 and 2B). Moreover, it is conceivable that the lack of PTM antigen expression in the thymus could lead to holes in the Treg repertoire. While multiple studies have shown that modification of beta cell epitopes increases their immunogenicity, it is unclear what proportion of antigens in the pancreas has been modified. Presumably, relatively low concentrations of immunogenic PTM epitopes are sufficient to prime autoimmune T cells, while presence of wild-type epitope is adequate for propagation of chronic autoimmune response. Further biochemical analyses of the pancreatic beta cells are necessary to identify the predominant PTM epitopes and the stress conditions that lead to their development.
HLA-Humanized Mice to Model T1D Antigen Responses
While we have learned a great deal from the NOD mouse, there are certain limitations to the conclusions and parallels we can draw to human T1D. In order to improve the model, several HLA transgenic mouse strains expressing susceptible or protective alleles have been generated, some of these on the NOD background. Surprisingly, NOD mice expressing susceptible DQ8 or DR4 alleles do not develop spontaneous diabetes (141–143). However, HLA-DQ8 humanized mice do develop spontaneous autoimmune cardiomyopathy (144). Still, both DR4 and DQ8 alleles support the development of beta cell-reactive autoimmune T cells but require an additional trigger to initiate beta cell targeted autoimmunity. When DR4 and DQ8 mice were crossed with transgenic mice expressing B7.1 co-stimulatory molecule on beta cells, both HLA-humanized strains developed spontaneous diabetes (141). The main utility for HLA-humanized mice has been realized by performing systematic identification of the key antigenic epitopes presented on human HLAs (24). Future studies should be extended to assess the in vivo functional potential of human autoimmune TCRs specific for key immunogenic epitopes. To date only one beta cell antigen-reactive human TCR transgenic mouse with specificity for GAD65 has been described (101). In vivo functional analysis of TCRs, and human TCRs in particular, has been hindered due to limited access to patient samples, labor, and time involved in generating TCR transgenic mice. We have overcome the limitation of TCR transgenic system by utilizing a TCR retrogenic approach that allows rapid functional analysis of multiple TCRs through retroviral gene delivery (110, 145, 146). Using this approach, we have expressed a GAD65115–127 reactive TCR isolated from peripheral blood of an individual diagnosed with T1D (99). Although we observed robust development of GAD-reactive T cells in this system, similar to the transgenic expression, we detected a low level of islet infiltration. Future analyses should be expanded to other beta cell protein epitopes targeted in human T1D, including PTM epitopes. The humanized TCR retrogenic approach will allow efficient and relatively high-throughput analysis of autoimmune antigens important in human disease, and can be utilized as a platform for development of antigen-specific immunotherapies. It is likely that many questions pertinent to our understanding of autoimmune T cell development and pathogenicity will be eventually addressed in the context of human susceptible HLA alleles and human TCRs.
Conclusion
The biology of low-affinity autoimmune T cells has been perplexing due to the seeming contradiction between suboptimal in vitro responses and robust in vivo pathogenicity. In many cases, self-reactive autoimmune T cells do not follow the dogma prescribed by studies performed with T cells specific for infectious or model antigens. In addition to unusual TCR:pMHC interactions and downstream signaling, autoimmune antigens themselves can have atypical characteristics. Over the years, it has become clear that antigens targeted in autoimmunity, and particularly in T1D, are often modified versions of self-peptides that are presented during thymic selection. These exceptions to the rule characteristic of autoimmune T cell responses are often centered on the stability of the tri-molecular complex as a master switch from tolerance to autoimmunity.
Author Contributions
MB and MLB developed the concept, prepared the figures, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the NIH (AI125301-01A1 to MB and K22AI104761 to MLB), ADA (1-17-JDF-013 to MLB and 7-14-JF-07 to MB), and The Robert and Janice McNair Foundation.
References
1. van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev (2011) 91(1):79–118. doi:10.1152/physrev.00003.2010
2. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe (2015) 17(2):260–73. doi:10.1016/j.chom.2015.01.001
3. Bonifacio E, Warncke K, Winkler C, Wallner M, Ziegler AG. Cesarean section and interferon-induced helicase gene polymorphisms combine to increase childhood type 1 diabetes risk. Diabetes (2011) 60(12):3300–6. doi:10.2337/db11-0729
4. Bian X, Wallstrom G, Davis A, Wang J, Park J, Throop A, et al. Immunoproteomic profiling of antiviral antibodies in new-onset type 1 diabetes using protein arrays. Diabetes (2016) 65(1):285–96. doi:10.2337/db15-0179
5. Tsai S, Santamaria P. MHC Class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol (2013) 4:321. doi:10.3389/fimmu.2013.00321
6. Hammer J, Gallazzi F, Bono E, Karr RW, Guenot J, Valsasnini P, et al. Peptide binding specificity of HLA-DR4 molecules: correlation with rheumatoid arthritis association. J Exp Med (1995) 181(5):1847–55. doi:10.1084/jem.181.5.1847
7. Menconi F, Monti MC, Greenberg DA, Oashi T, Osman R, Davies TF, et al. Molecular amino acid signatures in the MHC class II peptide-binding pocket predispose to autoimmune thyroiditis in humans and in mice. Proc Natl Acad Sci U S A (2008) 105(37):14034–9. doi:10.1073/pnas.0806584105
8. Smith KJ, Pyrdol J, Gauthier L, Wiley DC, Wucherpfennig KW. Crystal structure of HLA-DR2 (DRA*0101, DRB1*1501) complexed with a peptide from human myelin basic protein. J Exp Med (1998) 188(8):1511–20. doi:10.1084/jem.188.8.1511
9. Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity (2000) 12(6):699–710. doi:10.1016/S1074-7613(00)80220-4
10. Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol (2010) 11(4):350–4. doi:10.1038/ni.1850
11. Leiter EH, Prochazka M, Coleman DL. The non-obese diabetic (NOD) mouse. Am J Pathol (1987) 128(2):380–3.
12. Bettini M, Vignali DA. T cell-driven initiation and propagation of autoimmune diabetes. Curr Opin Immunol (2011) 23(6):754–60. doi:10.1016/j.coi.2011.10.002
13. Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, et al. HL-A antigens and diabetes mellitus. Lancet (1974) 2(7885):864–6. doi:10.1016/S0140-6736(74)91201-X
14. Cudworth AG, Woodrow JC. Letter: HL-A antigens and diabetes mellitus. Lancet (1974) 2(7889):1153. doi:10.1016/S0140-6736(74)90930-1
15. Prochazka M, Leiter EH, Serreze DV, Coleman DL. Three recessive loci required for insulin-dependent diabetes in nonobese diabetic mice. Science (1987) 237(4812):286–9. doi:10.1126/science.2885918
16. Wicker LS, Appel MC, Dotta F, Pressey A, Miller BJ, DeLarato NH, et al. Autoimmune syndromes in major histocompatibility complex (MHC) congenic strains of nonobese diabetic (NOD) mice. The NOD MHC is dominant for insulitis and cyclophosphamide-induced diabetes. J Exp Med (1992) 176(1):67–77. doi:10.1084/jem.176.1.67
17. Prochazka M, Serreze DV, Worthen SM, Leiter EH. Genetic control of diabetogenesis in NOD/Lt mice. Development and analysis of congenic stocks. Diabetes (1989) 38(11):1446–55. doi:10.2337/diab.38.11.1446
18. Yamamura K, Miyazaki T, Uno M, Toyonaga T, Miyazaki J. Non-obese diabetic transgenic mouse. Springer Semin Immunopathol (1992) 14(2):115–25. doi:10.1007/BF00195289
19. Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol (2011) 33(1):67–87. doi:10.1007/s00281-010-0204-1
20. Thayer TC, Wilson SB, Mathews CE. Use of nonobese diabetic mice to understand human type 1 diabetes. Endocrinol Metab Clin North Am (2010) 39(3):541–61. doi:10.1016/j.ecl.2010.05.001
21. Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science (2000) 288(5465):505–11. doi:10.1126/science.288.5465.505
22. Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol (2001) 2(6):501–7. doi:10.1038/88694
23. Suri A, Unanue ER. The murine diabetogenic class II histocompatibility molecule I-Ag7: structural and functional properties and specificity of peptide selection. Adv Immunol (2005) 88:235–65. doi:10.1016/S0065-2776(05)88007-1
24. Babad J, Geliebter A, DiLorenzo TP. T-cell autoantigens in the non-obese diabetic mouse model of autoimmune diabetes. Immunology (2010) 131(4):459–65. doi:10.1111/j.1365-2567.2010.03362.x
25. Skog O, Korsgren S, Melhus A, Korsgren O. Revisiting the notion of type 1 diabetes being a T-cell-mediated autoimmune disease. Curr Opin Endocrinol Diabetes Obes (2013) 20(2):118–23. doi:10.1097/MED.0b013e32835edb89
26. Luo X, Herold KC, Miller SD. Immunotherapy of type 1 diabetes: where are we and where should we be going? Immunity (2010) 32(4):488–99. doi:10.1016/j.immuni.2010.04.002
27. Gorodezky C, Alaez C, Murguia A, Rodriguez A, Balladares S, Vazquez M, et al. HLA and autoimmune diseases: type 1 diabetes (T1D) as an example. Autoimmun Rev (2006) 5(3):187–94. doi:10.1016/j.autrev.2005.06.002
28. Reijonen H, Novak EJ, Kochik S, Heninger A, Liu AW, Kwok WW, et al. Detection of GAD65-specific T-cells by major histocompatibility complex class II tetramers in type 1 diabetic patients and at-risk subjects. Diabetes (2002) 51(5):1375–82. doi:10.2337/diabetes.51.5.1375
29. Danke NA, Yang J, Greenbaum C, Kwok WW. Comparative study of GAD65-specific CD4+ T cells in healthy and type 1 diabetic subjects. J Autoimmun (2005) 25(4):303–11. doi:10.1016/j.jaut.2005.08.007
30. Oling V, Marttila J, Ilonen J, Kwok WW, Nepom G, Knip M, et al. GAD65- and proinsulin-specific CD4+ T-cells detected by MHC class II tetramers in peripheral blood of type 1 diabetes patients and at-risk subjects. J Autoimmun (2005) 25(3):235–43. doi:10.1016/j.jaut.2005.09.018
31. Kent SC, Chen Y, Bregoli L, Clemmings SM, Kenyon NS, Ricordi C, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature (2005) 435(7039):224–8. doi:10.1038/nature03625
32. In’t Veld P. Insulitis in human type 1 diabetes: the quest for an elusive lesion. Islets (2011) 3(4):131–8. doi:10.4161/isl.3.4.15728
33. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155(2):173–81. doi:10.1111/j.1365-2249.2008.03860.x
34. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209(1):51–60. doi:10.1084/jem.20111187
35. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351(6274):711–4. doi:10.1126/science.aad2791
36. Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PT, et al. Proinsulin-specific, HLA-DQ8, and HLA-DQ8-transdimer-restricted CD4+ T cells infiltrate islets in type 1 diabetes. Diabetes (2015) 64(1):172–82. doi:10.2337/db14-0858
37. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22(12):1482–7. doi:10.1038/nm.4203
38. Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, et al. Islet-derived CD4 T cells targeting proinsulin in human autoimmune diabetes. Diabetes (2017) 66(3):722–34. doi:10.2337/db16-1025
39. Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med (2009) 360(16):1646–54. doi:10.1056/NEJMra0808284
40. Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes (2008) 57(4):1084–92. doi:10.2337/db07-1331
41. Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature (2007) 450(7171):887–92. doi:10.1038/nature06406
42. Wenzlau JM, Liu Y, Yu L, Moua O, Fowler KT, Rangasamy S, et al. A common nonsynonymous single nucleotide polymorphism in the SLC30A8 gene determines ZnT8 autoantibody specificity in type 1 diabetes. Diabetes (2008) 57(10):2693–7. doi:10.2337/db08-0522
43. Pietropaolo M, Surhigh JM, Nelson PW, Eisenbarth GS. Primer: immunity and autoimmunity. Diabetes (2008) 57(11):2872–82. doi:10.2337/db07-1691
44. Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol (2007) 148(1):1–16. doi:10.1111/j.1365-2249.2006.03244.x
45. Skowera A, Ellis RJ, Varela-Calvino R, Arif S, Huang GC, Van-Krinks C, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest (2008) 118(10):3390–402. doi:10.1172/JCI35449
46. Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity (2010) 32(4):468–78. doi:10.1016/j.immuni.2010.03.018
47. Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol (2001) 166(8):5265–70. doi:10.4049/jimmunol.166.8.5265
48. Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity. J Clin Invest (2005) 115(8):2268–76. doi:10.1172/JCI25350
49. Tollefsen S, Arentz-Hansen H, Fleckenstein B, Molberg O, Raki M, Kwok WW, et al. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J Clin Invest (2006) 116(8):2226–36. doi:10.1172/JCI27620
50. Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A (2010) 107(24):10978–83. doi:10.1073/pnas.1006545107
51. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298(5597):1395–401. doi:10.1126/science.1075958
52. Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, et al. Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell (2015) 163(4):975–87. doi:10.1016/j.cell.2015.10.013
53. Gardner JM, Fletcher AL, Anderson MS, Turley SJ. AIRE in the thymus and beyond. Curr Opin Immunol (2009) 21(6):582–9. doi:10.1016/j.coi.2009.08.007
54. Lin J, Yang L, Silva HM, Trzeciak A, Choi Y, Schwab SR, et al. Increased generation of Foxp3(+) regulatory T cells by manipulating antigen presentation in the thymus. Nat Commun (2016) 7:10562. doi:10.1038/ncomms10562
55. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science (2015) 348(6234):589–94. doi:10.1126/science.aaa7017
56. Galgani M, Nugnes R, Bruzzese D, Perna F, De Rosa V, Procaccini C, et al. Meta-immunological profiling of children with type 1 diabetes identifies new biomarkers to monitor disease progression. Diabetes (2013) 62(7):2481–91. doi:10.2337/db12-1273
57. Prasad SJ, Goodnow CC. Cell-intrinsic effects of non-MHC NOD genes on dendritic cell generation in vivo. Int Immunol (2002) 14(6):677–84. doi:10.1093/intimm/dxf034
58. Feili-Hariri M, Morel PA. Phenotypic and functional characteristics of BM-derived DC from NOD and non-diabetes-prone strains. Clin Immunol (2001) 98(1):133–42. doi:10.1006/clim.2000.4959
59. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity (2015) 43(5):1011–21. doi:10.1016/j.immuni.2015.10.016
60. Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet (1995) 9(3):284–92. doi:10.1038/ng0395-284
61. Sabater L, Ferrer-Francesch X, Sospedra M, Caro P, Juan M, Pujol-Borrell R. Insulin alleles and autoimmune regulator (AIRE) gene expression both influence insulin expression in the thymus. J Autoimmun (2005) 25(4):312–8. doi:10.1016/j.jaut.2005.08.006
62. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet (1997) 15(3):289–92. doi:10.1038/ng0397-289
63. Pugliese A, Zeller M, Fernandez A Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet (1997) 15(3):293–7. doi:10.1038/ng0397-293
64. Durinovic-Bello I, Wu RP, Gersuk VH, Sanda S, Shilling HG, Nepom GT. Insulin gene VNTR genotype associates with frequency and phenotype of the autoimmune response to proinsulin. Genes Immun (2010) 11(2):188–93. doi:10.1038/gene.2009.108
65. Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J (2009) 28(18):2812–24. doi:10.1038/emboj.2009.212
66. Jaeckel E, Klein L, Martin-Orozco N, von Boehmer H. Normal incidence of diabetes in NOD mice tolerant to glutamic acid decarboxylase. J Exp Med (2003) 197(12):1635–44. doi:10.1084/jem.20030215
67. Krishnamurthy B, Dudek NL, McKenzie MD, Purcell AW, Brooks AG, Gellert S, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest (2006) 116(12):3258–65. doi:10.1172/JCI29602
68. Jaeckel E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat Immunol (2004) 5(10):1028–35. doi:10.1038/ni1120
69. French MB, Allison J, Cram DS, Thomas HE, Dempsey-Collier M, Silva A, et al. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes (1997) 46(1):34–9. doi:10.2337/diab.46.1.34
70. Jhala G, Chee J, Trivedi PM, Selck C, Gurzov EN, Graham KL, et al. Perinatal tolerance to proinsulin is sufficient to prevent autoimmune diabetes. JCI Insight (2016) 1(10):e86065. doi:10.1172/jci.insight.86065
71. He Q, Morillon YM II, Spidale NA, Kroger CJ, Liu B, Sartor RB, et al. Thymic development of autoreactive T cells in NOD mice is regulated in an age-dependent manner. J Immunol (2013) 191(12):5858–66. doi:10.4049/jimmunol.1302273
72. Lee T, Sprouse ML, Banerjee P, Bettini M, Bettini ML. Ectopic expression of self-antigen drives regulatory T cell development and not deletion of autoimmune T cells. J Immunol (2017) 199(7):2270–8. doi:10.4049/jimmunol.1700207
73. Baker RL, Bradley B, Wiles TA, Lindsay RS, Barbour G, Delong T, et al. Cutting edge: nonobese diabetic mice deficient in chromogranin A are protected from autoimmune diabetes. J Immunol (2016) 196(1):39–43. doi:10.4049/jimmunol.1501190
74. Wong J, Mathis D, Benoist C. TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J Exp Med (2007) 204(9):2039–45. doi:10.1084/jem.20070822
75. Leung MW, Shen S, Lafaille JJ. TCR-dependent differentiation of thymic Foxp3+ cells is limited to small clonal sizes. J Exp Med (2009) 206(10):2121–30. doi:10.1084/jem.20091033
76. Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature (1990) 346(6280):183–7. doi:10.1038/346183a0
77. Ito Y, Hashimoto M, Hirota K, Ohkura N, Morikawa H, Nishikawa H, et al. Detection of T cell responses to a ubiquitous cellular protein in autoimmune disease. Science (2014) 346(6207):363–8. doi:10.1126/science.1259077
78. Mandl JN, Monteiro JP, Vrisekoop N, Germain RN. T cell-positive selection uses self-ligand binding strength to optimize repertoire recognition of foreign antigens. Immunity (2013) 38(2):263–74. doi:10.1016/j.immuni.2012.09.011
79. Persaud SP, Parker CR, Lo WL, Weber KS, Allen PM. Intrinsic CD4+ T cell sensitivity and response to a pathogen are set and sustained by avidity for thymic and peripheral complexes of self peptide and MHC. Nat Immunol (2014) 15(3):266–74. doi:10.1038/ni.2822
80. Yu W, Jiang N, Ebert PJ, Kidd BA, Muller S, Lund PJ, et al. Clonal deletion prunes but does not eliminate self-specific alphabeta CD8(+) T lymphocytes. Immunity (2015) 42(5):929–41. doi:10.1016/j.immuni.2015.05.001
81. Haskins K, Cooke A. CD4 T cells and their antigens in the pathogenesis of autoimmune diabetes. Curr Opin Immunol (2011) 23(6):739–45. doi:10.1016/j.coi.2011.08.004
82. Bettini M, Blanchfield L, Castellaw A, Zhang Q, Nakayama M, Smeltzer MP, et al. TCR affinity and tolerance mechanisms converge to shape T cell diabetogenic potential. J Immunol (2014) 193(2):571–9. doi:10.4049/jimmunol.1400043
83. Burton AR, Vincent E, Arnold PY, Lennon GP, Smeltzer M, Li CS, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes (2008) 57(5):1321–30. doi:10.2337/db07-1129
84. Hogquist KA, Jameson SC. The self-obsession of T cells: how TCR signaling thresholds affect fate ‘decisions’ and effector function. Nat Immunol (2014) 15(9):815–23. doi:10.1038/ni.2938
85. Sabatino JJ Jr, Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med (2011) 208(1):81–90. doi:10.1084/jem.20101574
86. Seay HR, Yusko E, Rothweiler SJ, Zhang L, Posgai AL, Campbell-Thompson M, et al. Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight (2016) 1(20):e88242. doi:10.1172/jci.insight.88242
87. Eugster A, Lindner A, Catani M, Heninger AK, Dahl A, Klemroth S, et al. High diversity in the TCR repertoire of GAD65 autoantigen-specific human CD4+ T cells. J Immunol (2015) 194(6):2531–8. doi:10.4049/jimmunol.1403031
88. Cerosaletti K, Barahmand-Pour-Whitman F, Yang J, DeBerg HA, Dufort MJ, Murray SA, et al. Single-cell RNA sequencing reveals expanded clones of islet antigen-reactive CD4+ T cells in peripheral blood of subjects with type 1 diabetes. J Immunol (2017) 199(1):323–35. doi:10.4049/jimmunol.1700172
89. Fuchs YF, Eugster A, Dietz S, Sebelefsky C, Kuhn D, Wilhelm C, et al. CD8+ T cells specific for the islet autoantigen IGRP are restricted in their T cell receptor chain usage. Sci Rep (2017) 7:44661. doi:10.1038/srep44661
90. Baker RL, Delong T, Barbour G, Bradley B, Nakayama M, Haskins K. Cutting edge: CD4 T cells reactive to an islet amyloid polypeptide peptide accumulate in the pancreas and contribute to disease pathogenesis in nonobese diabetic mice. J Immunol (2013) 191(8):3990–4. doi:10.4049/jimmunol.1301480
91. Yamamoto T, Yamato E, Tashiro F, Sato T, Noso S, Ikegami H, et al. Development of autoimmune diabetes in glutamic acid decarboxylase 65 (GAD65) knockout NOD mice. Diabetologia (2004) 47(2):221–4. doi:10.1007/s00125-003-1296-0
92. Kubosaki A, Gross S, Miura J, Saeki K, Zhu M, Nakamura S, et al. Targeted disruption of the IA-2beta gene causes glucose intolerance and impairs insulin secretion but does not prevent the development of diabetes in NOD mice. Diabetes (2004) 53(7):1684–91. doi:10.2337/diabetes.53.7.1684
93. Alkemade GM, Clemente-Casares X, Yu Z, Xu BY, Wang J, Tsai S, et al. Local autoantigen expression as essential gatekeeper of memory T-cell recruitment to islet grafts in diabetic hosts. Diabetes (2013) 62(3):905–11. doi:10.2337/db12-0600
94. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature (2005) 435(7039):220–3. doi:10.1038/nature03523
95. Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med (1997) 186(10):1663–76. doi:10.1084/jem.186.10.1663
96. Arnold PY, Burton AR, Vignali DA. Diabetes incidence is unaltered in glutamate decarboxylase 65-specific TCR retrogenic nonobese diabetic mice: generation by retroviral-mediated stem cell gene transfer. J Immunol (2004) 173(5):3103–11. doi:10.4049/jimmunol.173.5.3103
97. Du W, Wong FS, Li MO, Peng J, Qi H, Flavell RA, et al. TGF-beta signaling is required for the function of insulin-reactive T regulatory cells. J Clin Invest (2006) 116(5):1360–70. doi:10.1172/JCI27030C1
98. Chaparro RJ, Burton AR, Serreze DV, Vignali DA, DiLorenzo TP. Rapid identification of MHC class I-restricted antigens relevant to autoimmune diabetes using retrogenic T cells. J Immunol Methods (2008) 335(1–2):106–15. doi:10.1016/j.jim.2008.03.007
99. Sprouse ML, Blahnik G, Lee T, Tully N, Banerjee P, James EA, et al. Rapid identification and expression of human TCRs in retrogenic mice. J Immunol Methods (2016) 439:29–36. doi:10.1016/j.jim.2016.08.010
100. Mohan JF, Calderon B, Anderson MS, Unanue ER. Pathogenic CD4(+) T cells recognizing an unstable peptide of insulin are directly recruited into islets bypassing local lymph nodes. J Exp Med (2013) 210(11):2403–14. doi:10.1084/jem.20130582
101. Gebe JA, Unrath KA, Yue BB, Miyake T, Falk BA, Nepom GT. Autoreactive human T-cell receptor initiates insulitis and impaired glucose tolerance in HLA DR4 transgenic mice. J Autoimmun (2008) 30(4):197–206. doi:10.1016/j.jaut.2007.08.001
102. Graser RT, DiLorenzo TP, Wang F, Christianson GJ, Chapman HD, Roopenian DC, et al. Identification of a CD8 T cell that can independently mediate autoimmune diabetes development in the complete absence of CD4 T cell helper functions. J Immunol (2000) 164(7):3913–8. doi:10.4049/jimmunol.164.7.3913
103. Jasinski JM, Yu L, Nakayama M, Li MM, Lipes MA, Eisenbarth GS, et al. Transgenic insulin (B:9-23) T-cell receptor mice develop autoimmune diabetes dependent upon RAG genotype, H-2g7 homozygosity, and insulin 2 gene knockout. Diabetes (2006) 55(7):1978–84. doi:10.2337/db06-0058
104. Kobayashi M, Jasinski J, Liu E, Li M, Miao D, Zhang L, et al. Conserved T cell receptor alpha-chain induces insulin autoantibodies. Proc Natl Acad Sci U S A (2008) 105(29):10090–4. doi:10.1073/pnas.0801648105
105. Wong FS, Siew LK, Scott G, Thomas IJ, Chapman S, Viret C, et al. Activation of insulin-reactive CD8 T-cells for development of autoimmune diabetes. Diabetes (2009) 58(5):1156–64. doi:10.2337/db08-0800
106. Quinn A, McInerney B, Reich EP, Kim O, Jensen KP, Sercarz EE. Regulatory and effector CD4 T cells in nonobese diabetic mice recognize overlapping determinants on glutamic acid decarboxylase and use distinct V beta genes. J Immunol (2001) 166(5):2982–91. doi:10.4049/jimmunol.166.5.2982
107. Tisch R, Wang B, Atkinson MA, Serreze DV, Friedline R. A glutamic acid decarboxylase 65-specific Th2 cell clone immunoregulates autoimmune diabetes in nonobese diabetic mice. J Immunol (2001) 166(11):6925–36. doi:10.4049/jimmunol.166.11.6925
108. You S, Chen C, Lee WH, Brusko T, Atkinson M, Liu CP. Presence of diabetes-inhibiting, glutamic acid decarboxylase-specific, IL-10-dependent, regulatory T cells in naive nonobese diabetic mice. J Immunol (2004) 173(11):6777–85. doi:10.4049/jimmunol.173.11.6777
109. Chen G, Han G, Feng J, Wang J, Wang R, Xu R, et al. Glutamic acid decarboxylase-derived epitopes with specific domains expand CD4(+)CD25(+) regulatory T cells. PLoS One (2009) 4(9):e7034. doi:10.1371/journal.pone.0007034
110. Lennon GP, Bettini M, Burton AR, Vincent E, Arnold PY, Santamaria P, et al. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity (2009) 31(4):643–53. doi:10.1016/j.immuni.2009.07.008
111. Wang J, Tsai S, Shameli A, Yamanouchi J, Alkemade G, Santamaria P. In situ recognition of autoantigen as an essential gatekeeper in autoimmune CD8+ T cell inflammation. Proc Natl Acad Sci U S A (2010) 107(20):9317–22. doi:10.1073/pnas.0913835107
112. Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci U S A (2011) 108(4):1561–6. doi:10.1073/pnas.1018973108
113. Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature (1993) 366(6450):72–5. doi:10.1038/366072a0
114. Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GS, Robinson P, et al. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature (1993) 366(6450):69–72. doi:10.1038/366069a0
115. Achenbach P, Warncke K, Reiter J, Naserke HE, Williams AJ, Bingley PJ, et al. Stratification of type 1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes (2004) 53(2):384–92. doi:10.2337/diabetes.53.2.384
116. Zekzer D, Wong FS, Ayalon O, Millet I, Altieri M, Shintani S, et al. GAD-reactive CD4+ Th1 cells induce diabetes in NOD/SCID mice. J Clin Invest (1998) 101(1):68–73. doi:10.1172/JCI119878
117. Kim J, Richter W, Aanstoot HJ, Shi Y, Fu Q, Rajotte R, et al. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes (1993) 42(12):1799–808. doi:10.2337/diabetes.42.12.1799
118. Bridgett M, Cetkovic-Cvrlje M, O’Rourke R, Shi Y, Narayanswami S, Lambert J, et al. Differential protection in two transgenic lines of NOD/Lt mice hyperexpressing the autoantigen GAD65 in pancreatic beta-cells. Diabetes (1998) 47(12):1848–56. doi:10.2337/diabetes.47.12.1848
119. Martinez RJ, Andargachew R, Martinez HA, Evavold BD. Low-affinity CD4+ T cells are major responders in the primary immune response. Nat Commun (2016) 7:13848. doi:10.1038/ncomms13848
120. Cole DK, Bulek AM, Dolton G, Schauenberg AJ, Szomolay B, Rittase W, et al. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J Clin Invest (2016) 126(9):3626. doi:10.1172/JCI89919
121. Nelson RW, Beisang D, Tubo NJ, Dileepan T, Wiesner DL, Nielsen K, et al. T cell receptor cross-reactivity between similar foreign and self peptides influences naive cell population size and autoimmunity. Immunity (2015) 42(1):95–107. doi:10.1016/j.immuni.2014.12.022
122. Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem (2012) 287(2):1168–77. doi:10.1074/jbc.M111.289488
123. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice. J Exp Med (2016) 213(10):2129–46. doi:10.1084/jem.20160526
124. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity (2013) 39(2):400–12. doi:10.1016/j.immuni.2013.08.013
125. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339(6123):1084–8. doi:10.1126/science.1233521
126. de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes (2013) 62(4):1238–44. doi:10.2337/db12-0526
127. Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol (2005) 6(5):490–6. doi:10.1038/ni1187
128. Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J (2005) 24(17):2968–79. doi:10.1038/sj.emboj.7600771
129. Sethi DK, Schubert DA, Anders AK, Heroux A, Bonsor DA, Thomas CP, et al. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med (2011) 208(1):91–102. doi:10.1084/jem.20100725
130. Schubert DA, Gordo S, Sabatino JJ Jr, Vardhana S, Gagnon E, Sethi DK, et al. Self-reactive human CD4 T cell clones form unusual immunological synapses. J Exp Med (2012) 209(2):335–52. doi:10.1084/jem.20111485
131. Rosenthal KM, Edwards LJ, Sabatino JJ Jr, Hood JD, Wasserman HA, Zhu C, et al. Low 2-dimensional CD4 T cell receptor affinity for myelin sets in motion delayed response kinetics. PLoS One (2012) 7(3):e32562. doi:10.1371/journal.pone.0032562
132. Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med (2011) 208(12):2375–83. doi:10.1084/jem.20111502
133. Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A (2011) 108(40):16729–34. doi:10.1073/pnas.1113954108
134. Unger WW, Pearson T, Abreu JR, Laban S, van der Slik AR, der Kracht SM, et al. Islet-specific CTL cloned from a type 1 diabetes patient cause beta-cell destruction after engraftment into HLA-A2 transgenic NOD/scid/IL2RG null mice. PLoS One (2012) 7(11):e49213. doi:10.1371/journal.pone.0049213
135. Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med (2005) 202(9):1191–7. doi:10.1084/jem.20051251
136. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes (2012) 61(12):3239–46. doi:10.2337/db12-0112
137. Gottlieb PA, Delong T, Baker RL, Fitzgerald-Miller L, Wagner R, Cook G, et al. Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun (2014) 50:38–41. doi:10.1016/j.jaut.2013.10.003
138. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes (2014) 63(1):237–47. doi:10.2337/db12-1214
139. McGinty JW, Chow IT, Greenbaum C, Odegard J, Kwok WW, James EA. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes (2014) 63(9):3033–40. doi:10.2337/db13-1952
140. Marre ML, Profozich JL, Coneybeer JT, Geng X, Bertera S, Ford MJ, et al. Inherent ER stress in pancreatic islet beta cells causes self-recognition by autoreactive T cells in type 1 diabetes. J Autoimmun (2016) 72:33–46. doi:10.1016/j.jaut.2016.04.009
141. Wen L, Wong FS, Tang J, Chen NY, Altieri M, David C, et al. In vivo evidence for the contribution of human histocompatibility leukocyte antigen (HLA)-DQ molecules to the development of diabetes. J Exp Med (2000) 191(1):97–104. doi:10.1084/jem.191.1.97
142. Kudva YC, Rajagopalan G, Raju R, Abraham RS, Smart M, Hanson J, et al. Modulation of insulitis and type 1 diabetes by transgenic HLA-DR3 and DQ8 in NOD mice lacking endogenous MHC class II. Hum Immunol (2002) 63(11):987–99. doi:10.1016/S0198-8859(02)00435-4
143. Elliott JF, Liu J, Yuan ZN, Bautista-Lopez N, Wallbank SL, Suzuki K, et al. Autoimmune cardiomyopathy and heart block develop spontaneously in HLA-DQ8 transgenic IAbeta knockout NOD mice. Proc Natl Acad Sci U S A (2003) 100(23):13447–52. doi:10.1073/pnas.2235552100
144. Taneja V, David CS. Spontaneous autoimmune myocarditis and cardiomyopathy in HLA-DQ8.NODAbo transgenic mice. J Autoimmun (2009) 33(3–4):260–9. doi:10.1016/j.jaut.2009.09.005
145. Bettini ML, Bettini M, Vignali DA. T-cell receptor retrogenic mice: a rapid, flexible alternative to T-cell receptor transgenic mice. Immunology (2012) 136(3):265–72. doi:10.1111/j.1365-2567.2012.03574.x
Keywords: T cell, autoimmunity, type 1 diabetes, human leukocyte antigen, regulatory T cell, thymic selection
Citation: Bettini ML and Bettini M (2017) Understanding Autoimmune Diabetes through the Prism of the Tri-Molecular Complex. Front. Endocrinol. 8:351. doi: 10.3389/fendo.2017.00351
Received: 31 July 2017; Accepted: 30 November 2017;
Published: 14 December 2017
Edited by:
Marc S. Horwitz, University of British Columbia, CanadaReviewed by:
Hubert M. Tse, University of Alabama at Birmingham, United StatesClayton E. Mathews, University of Florida, United States
David H. Wagner, University of Colorado Denver, United States
Copyright: © 2017 Bettini and Bettini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew L. Bettini, bWF0dGhldy5iZXR0aW5pQGJjbS5lZHU=;
Maria Bettini, bWFyaWEuYmV0dGluaUBiY20uZWR1