 Meghan L. Marré
Meghan L. Marré Jon D. Piganelli
Jon D. Piganelli- Division of Pediatric Surgery, Department of Surgery, Children’s Hospital of Pittsburgh, University of Pittsburgh, Pittsburgh, PA, United States
Type 1 diabetes (T1D) is an autoimmune disease in which immune-mediated targeting and destruction of insulin-producing pancreatic islet β cells leads to chronic hyperglycemia. There are many β cell proteins that are targeted by autoreactive T cells in their native state. However, recent studies have demonstrated that many β cell proteins are recognized as neo-antigens following posttranslational modification (PTM). Although modified neo-antigens are well-established targets of pathology in other autoimmune diseases, the effects of neo-antigens in T1D progression and the mechanisms by which they are generated are not well understood. We have demonstrated that PTM occurs during endoplasmic reticulum (ER) stress, a process to which β cells are uniquely susceptible due to the high rate of insulin production in response to dynamic glucose sensing. In the context of genetic susceptibility to autoimmunity, presentation of these modified neo-antigens may activate autoreactive T cells and cause pathology. However, inherent β cell ER stress and protein PTM do not cause T1D in every genetically susceptible individual, suggesting the contribution of additional factors. Indeed, many environmental factors, such as viral infection, chemicals, or inflammatory cytokines, are associated with T1D onset, but the mechanisms by which these factors lead to disease onset remain unknown. Since these environmental factors also cause ER stress, exposure to these factors may enhance production of neo-antigens, therefore boosting β cell recognition by autoreactive T cells and exacerbating T1D pathogenesis. Therefore, the combined effects of physiological ER stress and the stress that is induced by environmental factors may lead to breaks in peripheral tolerance, contribute to antigen spread, and hasten disease onset. This Hypothesis and Theory article summarizes what is currently known about ER stress and protein PTM in autoimmune diseases including T1D and proposes a role for environmental factors in breaking immune tolerance to β cell antigens through neo-antigen formation.
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease in which insulin-producing pancreatic islet β cells are targeted and destroyed by autoreactive immune cells. Autoimmune recognition of β cell antigens leads to decreased β cell mass and to the subsequent decline of insulin-mediated regulation of glucose levels in the blood. Eventually, too few β cells remain to meet the demand for insulin to maintain normal blood glucose levels. This insufficient insulin secretion leads to chronic hyperglycemia and T1D.
Type 1 diabetes is strongly associated with a genetic predisposition to autoimmunity that is conferred by single-nucleotide polymorphisms (SNPs) and gene variants found at many genetic loci. In particular, SNPs and variants in genes associated with both the innate and adaptive branches of the immune system cause failures of central and peripheral tolerance that eventually lead to autoimmune targeting of β cells. Of these loci, polymorphisms in the major histocompatibility complex (MHC) locus are most strongly associated with T1D onset (1–3). MHC proteins are crucial to central tolerance, because the antigens they present during T cell development in the thymus determine which T cells survive selection. This process directly shapes the mature adaptive immune repertoire. Strongly autoreactive T cells should be deleted upon encountering self-antigen presented by MHC during selection (4), but in individuals expressing MHC polymorphisms associated with autoimmunity, autoreactive T cells successfully mature and exit the thymus (5, 6). If peripheral tolerance mechanisms also fail, these autoreactive T cells become activated when they encounter β cell antigens in pancreatic lymph nodes. This autoimmune response destroys pancreatic β cells and ultimately causes T1D.
To better understand the processes by which the autoimmune response leads to T1D, and to identify the β cell proteins that are targeted by autoreactive T cells, researchers have studied the non-obese diabetic (NOD) mouse. These mice develop a spontaneous autoimmune diabetes that is similar in many ways to the human disease. These similarities include genetic susceptibility at the MHC locus and other immune-related loci, intra-islet infiltration of autoreactive immune cells as disease progresses, and ultimate β cell destruction (7–9). The β cell autoantigens identified using this murine model include preproinsulin (10), glutamic acid decarboxylase 65 (GAD65) (11), islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) (12), chromogranin A (CHgA) (13), islet amyloid polypeptide (14), zinc transporter 8 (15), and 78 kDa glucose-regulated protein (GRP78) (16) (Table 1). Subsequent studies confirmed the relevance of these autoantigens to human T1D (17–23) (Table 1). In addition, several additional autoantigens have been identified in humans but not yet confirmed in NOD mice, including tyrosine phosphatase-like insulinoma antigen 2 and IA-2β (also known as phosphatase homolog of granules from rat insulinomas) (24, 25), and islet cell autoantigen 69 (26) (Table 1).
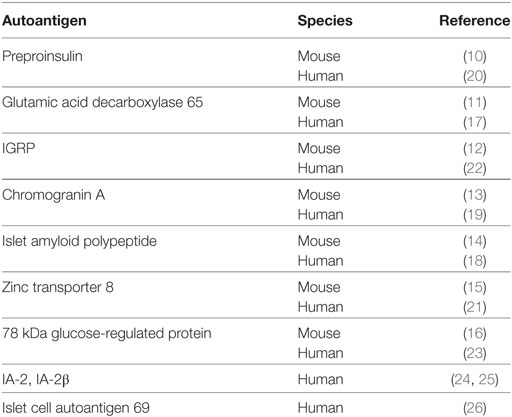
Table 1. β Cell autoantigens identified in murine and human T1D.
The immunogenicity of these β cell autoantigens has long been attributed to failures in the mechanisms that govern immune tolerance to self-peptides. While this likely remains true, seminal studies conducted by several laboratories demonstrated that many of these β cell peptides undergo posttranslational modification (PTM). These studies propose that aberrant PTM of these β cell proteins generates so called “neo-antigens” that are then recognized as non-self by immune cells (16, 19, 23, 27–32), hastening the break in tolerance and exacerbating immune targeting and destruction of β cells. However, most of these studies did not explore the cellular processes that lead to PTM of these proteins in the context of β cell function and biology.
To address this question, our laboratory demonstrated that endoplasmic reticulum (ER) stress in the β cell leads to the activation of PTM enzymes and the modification of β cell proteins, which in turn leads to increased recognition of these β cells by diabetogenic T cells (32). ER stress in the β cell originates from various sources. For instance, the normal function of β cells (to produce and secrete insulin) causes ER stress (32–42). We demonstrated that this inherent physiological ER stress is sufficient to activate PTM enzymes and to generate β cell immunogenicity (32) (Figure 1). In addition, many of the environmental factors associated with T1D onset such as viral infection (43–48), chemicals (32, 49–51), reactive oxygen species (ROS) (52–55), dysglycemia (56), and inflammation (57–59) may cause β cell ER stress (Figure 1). Therefore, any of these environmental factors has the potential to enhance autoimmune targeting of β cells through the generation of ER stress- and PTM-dependent neo-antigens (32, 60, 61). However, the mechanisms by which these factors hasten T1D onset, and whether the ER stress they cause cooperates with that caused by β cell physiology, remain unknown (Figure 1).
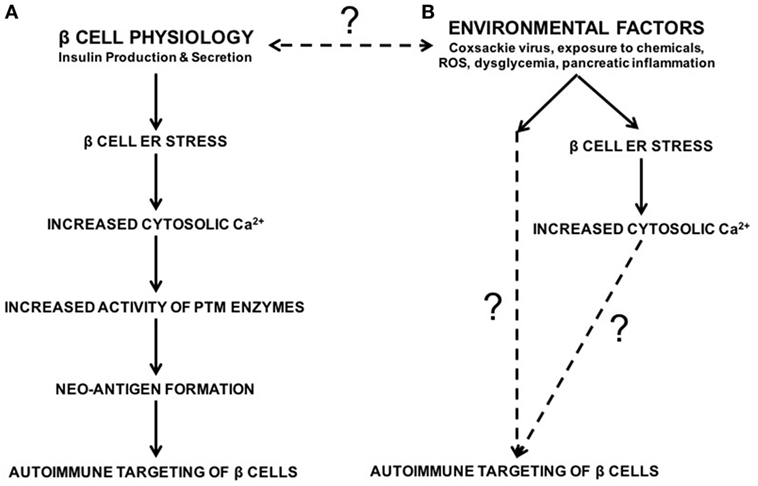
Figure 1. The roles of β cell physiology and environmental favors in the autoimmune targeting of β cells in type 1 diabetes (T1D). (A) Normal β cell secretory physiology causes inherent endoplasmic reticulum (ER) stress, which in turn results in a release of Ca2+ from the ER into the cytosol. We have previously demonstrated that ER stress and its Ca2+ efflux lead to increased activity of Ca2+-dependent posttranslational modification enzymes, formation of neo-antigens, and β cell immunogenicity (32). (B) In addition, many environmental factors are associated with T1D onset, such as viral infection, exposure to chemicals and reactive oxygen species, dysglycemia, and pancreatic inflammation. Although the mechanisms by which these factors lead to autoimmune targeting of β cells remain unknown, these environmental factors all cause β cell ER stress and Ca2+ efflux. Whether the ER stress and Ca2+ efflux caused by these environmental factors contributes to T1D onset, and whether this ER stress cooperates with physiological ER stress to generate neo-antigens, remain unknown.
Here, we review what is known about β cell ER stress, neo-antigen formation, and the progression to pathology in T1D. We also review the role that the environmental factors associated with T1D may play in exacerbating β cell ER stress. Finally, we discuss the evidence supporting our novel hypothesis that environmental factors converge with β cell physiology to increase ER stress above a putative threshold. According to our “threshold hypothesis,” ER stress must be sufficiently severe or prolonged to allow for the generation of PTM-dependent neo-antigens. We hypothesize that the convergence between β cell physiology and exposure to environmental factors increases ER stress above this threshold, leading to neo-antigen formation, β cell immunogenicity, and ultimately to the onset of T1D.
ER Stress and the Unfolded Protein Response (UPR)
The ER is primarily responsible for the proper folding and modification of proteins that are membrane bound or destined for secretion. Therefore, the ER lumen contains the molecular chaperones and the environment necessary for protein folding and PTM, including sufficient levels of adenosine triphosphate, an oxidizing environment to support disulfide bond formation, and millimolar concentrations of calcium (Ca2+) (62). Proteins that are folded and modified properly exit the ER and are shuttled to their intended intra- or extracellular locations. However, proteins that become misfolded cannot exit the ER lumen. The accumulation of misfolded or aberrantly modified proteins causes ER stress.
Endoplasmic reticulum stress induces the UPR, which functions in two main modes: the adaptive UPR and the terminal UPR (63, 64). The adaptive UPR occurs early in ER stress and functions largely to alleviate ER stress and restore normal cellular homeostasis through three signaling cascades, each of which begins with the activation of protein sensors of stress in the ER membrane (65). First, protein kinase RNA (PKR)-like ER kinase (PERK) activates a signaling cascade that inhibits mRNA translation and reduces the protein burden in the lumen of the ER (66, 67). Second, activating transcription factor 6 signaling leads to increased production of new molecular chaperones to aid with the folding of accumulated misfolded proteins (68). And third, the signaling pathway initiated by inositol-requiring protein 1 increases expression of chaperones for protein folding and of proteins involved in lipid synthesis to increase ER volume (69, 70). Together, these branches of the UPR work to facilitate the proper folding of proteins that have accumulated, and also reduce the entrance of additional non-chaperone proteins into the ER lumen. In these ways, the adaptive UPR acts to allow the ER to return to normal homeostasis.
Although the adaptive UPR aims to protect the cell from the negative effects of ER stress, ER dysfunction that is excessive or extended may overcome these cytoprotective mechanisms. Under these conditions, the terminal UPR activates proapoptotic processes (71–73) leading to death of the affected cell. However, long before apoptosis pathways are activated, even temporary ER stress and the adaptive UPR may have important consequences for cellular function and physiology.
All cells undergo periods of increased protein production, which increases the ER burden, leading to misfolding or aberrant modification of nascent proteins, and ultimately to ER stress and UPR activation. However, secretory cells, due to their normal physiology, are uniquely susceptible to ER stress. These cells must produce not only the proteins necessary for normal cellular maintenance, but also the proteins to be secreted and the proteins that comprise the secretory pathway itself. Even with a larger ER volume and greater numbers of chaperones to account for this increased demand (74), the secretory function of these cells leads to significantly increased ER burden and stress.
Like other secretory cells, β cells undergo naturally high levels of ER stress due to their normal physiological role of insulin production and secretion (32–42). Indeed, increased ER stress, and its consequences for protein folding, occurs as a direct consequence of glucose sensing (37, 38). In response to increased glucose concentrations, β cells upregulate the translation of preproinsulin by 50-fold, reaching a production rate of one million molecules of preproinsulin per minute (75). These one million molecules of preproinsulin inundate the ER lumen for folding and the formation of three disulfide bonds per molecule, causing tremendous ER stress. Under these conditions, many of the insulin molecules produced by β cells become misfolded or incorrectly modified (75) (Figure 2). In addition to this inherent ER stress due to normal physiology, β cell ER stress may rise due to exposure to the environmental factors that are associated with T1D onset (32, 43–61) (Figure 1). Under these circumstances, β cell ER stress may rise above physiological levels.
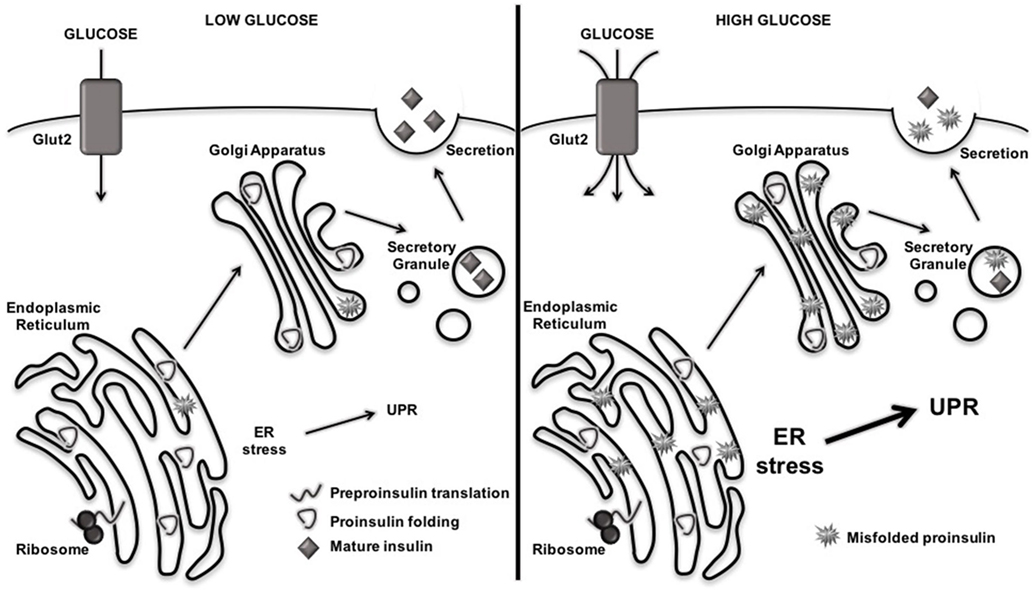
Figure 2. Rising blood glucose increases misfolding of proinsulin and endoplasmic reticulum (ER) stress levels. When blood glucose levels are low, preproinsulin is translated, properly folded and modified in the ER, and secreted as mature insulin into the extracellular space. When blood glucose levels rise, β cells increase production of preproinsulin, flooding the ER lumen with one million molecules per minute that require folding and disulfide bond formation. This increased protein burden in the ER leads to misfolding of proteins and aberrant posttranslational modification, which further exacerbates ER dysfunction and activates the unfolded protein response (UPR).
Heightened β cell ER stress does not necessarily activate the terminal UPR or suggest β cell exhaustion or impending death as observed in some models (63, 76–80). Rather, β cells exhibit naturally high ER stress very early and activate the adaptive UPR long before β cell death. In a study using a reporter mouse in which green fluorescent protein is expressed with the activation of the UPR, the pancreas exhibited the highest ER stress of all tissues examined, and did so as early as day 16 of life (81). In spite of the observed ER stress and UPR activation, these mice (on the C57Bl/6 background) never succumbed to loss of β cell mass and diabetes (81). These data confirm that high levels of β cell ER stress does not necessarily activate the terminal UPR and lead to β cell failure and death. Indeed, the β cells in most individuals resolve ER stress through the proper function of the adaptive UPR and therefore maintain healthy and functional β cell mass throughout their lifetimes. Therefore, β cell death is not the only consequence of ER stress. We hypothesize that lower and more transient ER stress, and the activation of the adaptive UPR, may have consequences for β cell function and for the autoimmune targeting of β cells much earlier.
ER Stress Affects Ca2+-Dependent Cellular Functions
In addition to its role in the folding and modification of new proteins, the ER contains the largest store of intracellular Ca2+ and is an important organelle for regulating Ca2+ concentrations, and therefore Ca2+-dependent processes, throughout the cell (82). One consequence of ER stress is the release of Ca2+ from the ER lumen into the cytosol. This Ca2+ efflux has important consequences for cellular physiology.
In the ER, millimolar concentrations of Ca2+ are necessary for proper protein folding and modification (62). Indeed, molecular chaperones that assist in protein folding and protein disulfide isomerases that facilitate the formation of disulfide bonds depend on these high Ca2+ concentrations (83, 84). These high concentrations of Ca2+ are maintained by sarco/endoplasmic reticulum Ca2+ ATPases (SERCA) pumps in the ER membrane that actively transport Ca2+ from the cytosol into the ER. When Ca2+ leaves the ER lumen during ER stress, the function of these proteins also decreases, further inhibiting protein folding and modification and contributing to greater ER dysfunction (85).
In the cytosol, Ca2+ is required for the regulation of normal cellular processes such as metabolism, vesicular trafficking, protein secretion, mRNA transcription, and apoptosis (86). To achieve the necessary cytosolic concentrations, Ca2+ is released from the ER by the ryanodine receptor and inositol 1,4,5-trisphosphate receptor channels. Under conditions of ER stress, the efflux of Ca2+ from the ER lumen increases cytosolic concentrations above normal physiological levels. This increased cytosolic Ca2+ can be deleterious for cellular function. For example, increased cytosolic Ca2+ can initiate apoptosis through activation of caspase-dependent cell death pathways (87, 88) or mitochondria-dependent pathways (89–92).
It is clear, then, that ER stress greatly affects Ca2+-dependent cellular functions. While the adaptive UPR works to relieve ER stress, cytosolic Ca2+ still increases before ER homeostasis is regained. β cells, which are particularly susceptible to ER stress, are therefore also prone to the dysregulation of cellular processes following even a temporary efflux of Ca2+ from the ER. In addition, the environmental factors associated with T1D onset also lead to increased cytosolic Ca2+ (32, 43–61) (Figure 1). Therefore, we propose that the combination of physiological ER stress and that derived from environmental factors, even if transient, may have consequences for β cell health and function.
ER Stress Activates Cytosolic PTM Enzymes
Transient ER stress and increased cytosolic Ca2+ concentrations can activate cytosolic Ca2+-dependent enzymes, including those that mediate PTM. Activation of these PTM enzymes can have significant implications for proteins being folded in the ER. In particular, two such PTM enzymes reside in the cytosol and are activated during the ER stress Ca2+ flux: tissue transglutaminase 2 (Tgase2) and peptidylarginine deiminase 2 (PAD2).
Tissue transglutaminase 2 is ubiquitously expressed and resides in the cytosol (93). When activated, Tgase2 translocates to several intracellular compartments (94), including the ER (95–97) and secretory granules (98) to modify proteins through two mechanisms (99): first, Tgase2 crosslinks proteins through the formation of ε(γ-glutamyl) isopeptide bonds between glutamine and lysine residues, and second, Tgase2 mediates the deamidation of glutamine residues. Tgase2 plays important roles in the regulation of apoptosis (100, 101), gene expression (93, 102, 103), and cellular adhesion and wound healing (104–107). Of relevance to T1D, Tgase2 is expressed in and functions in β cells (32, 60).
Of the five mammalian PAD isoforms, PAD2 is the most widely expressed and is the isoform expressed in the pancreas (108). PAD2 also resides in the cytosol (109), and, similar to Tgase2, activated PAD2 is recruited to various subcellular compartments for the modification of proteins (110). PAD2 mediates the conversion of arginine to citrulline. This amino acid conversion alters the overall charge and hydrophobicity of the protein (111), causing changes in protein folding and conformation (112). PAD2 plays roles in many cellular functions, including the negative regulation of nuclear factor kappa-light-chain-enhancer of activated B cells activation (113), cytoskeleton disassembly (114), and in the formation of neutrophil extracellular traps (115).
While Ca2+-dependent activation of these enzymes is necessary for normal cellular function, these enzymes also contribute to pathology in many diseases.
PTM Generates Neo-Antigens in Autoimmune Diseases
Protein PTM is necessary for cellular viability and function. However, autoantigens in many different autoimmune diseases such as celiac disease (116), collagen-induced arthritis (117), multiple sclerosis/experimental autoimmune encephalomyelitis (118–121), rheumatoid arthritis (122–127), and systemic lupus erythematosus (128–131) contain PTM, suggesting that these modifications may contribute to breaks in tolerance that exacerbate disease.
Central tolerance is established during T cell development in the thymus. In the thymus, medullary thymic epithelial cells (mTECs) express peptides normally found in peripheral tissues through the function of autoimmune regulator (132–134). When these peptides are presented to developing T cells in the context of MHC molecules, T cells that respond too strongly to these self-peptides are deleted and are thus absent from the mature T cell population (4, 135–137). However, if self-proteins undergo PTM in peripheral tissues, as in the autoimmune diseases listed above, these proteins may be processed and presented differently by peripheral antigen-presenting cells (APCs) than by the mTECs (138). If such modified epitopes were not expressed and presented by mTECs, T cells that recognize these modified epitopes escape negative selection and are present in circulation as mature T cells. When these T cells encounter these neo-antigens in peripheral tissues, they become activated and lead the autoimmune targeting of peripheral tissues.
As with all peripheral tissues, peptides from β cell proteins, including insulin and insulin-like growth factor 2, are presented by mTECs to developing T cells in the thymus (5, 6, 139–142). However, the presence of T cells in the periphery that recognize islet proteins and target β cells suggests the failure of crucial tolerance mechanisms. This failure in central tolerance mechanisms may be explained by the growing body of literature that abnormal PTM increases the immunogenicity of β cell peptides in both murine and human models of T1D (Table 2). These studies have demonstrated that some β cell proteins undergo various modifications including oxidation (28, 143), Tgase2-mediated crosslinking by isopeptide bond (19, 29), Tgase2-mediated deamidation (30–32), PAD2-mediated citrullination (16, 23, 31), the formation of hybrid peptides (144, 145), and the formation of a defective ribosomal insulin gene product (146). Furthermore, the neo-antigens formed by these PTM elicit stronger immune responses than the native proteins (16, 19, 23, 28–31, 143, 145), suggesting an important role for these neo-antigens in precipitating disease onset. These findings have been of great importance to the understanding of T1D pathogenesis, because these studies identified novel autoantigens that are targeted in T1D. However, the mechanisms by which these neo-antigens arise in β cells was not examined.
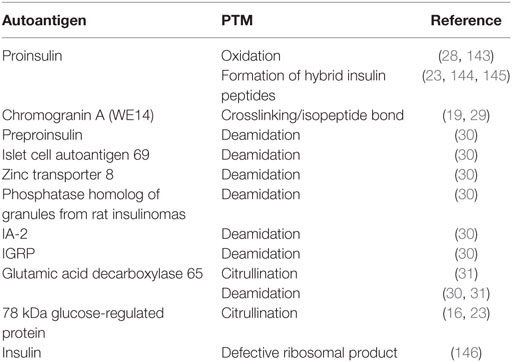
Table 2. Posttranslational modification (PTM)-mediated neo-antigen formation in type 1 diabetes.
β Cell Neo-Antigens Arise During ER Stress
To begin to elucidate how PTM neo-antigens might arise in β cells, our laboratory examined the consequences of β cell ER stress for β cell immunogenicity, since β cells inherently undergo high levels of ER stress (32–42, 60, 81). To do so, we used a model system of β cell recognition by diabetogenic BDC2.5 CD4+ T cells. These particular T cells were chosen because they recognize a Tgase2-modified peptide of CHgA (29) and secrete interferon gamma (IFNγ) when they encounter their PTM-dependent antigen.
Our studies demonstrated that, in primary murine islets, ER stress induced by thapsigargin [a widely accepted chemical inducer of ER stress (96, 147, 148)] contributed to heightened cytosolic Ca2+ concentrations, increased Tgase2 activity, and increased β cell immunogenicity (32). In fact, murine islets undergoing ER stress elicited greater IFNγ secretion from BDC2.5 T cells (32) and by all other β cell antigen-specific T cells examined (Figure 3), suggesting a role for Ca2+-dependent PTM in immunogenicity of many other β cell antigens. This increased immunogenicity was dependent upon both Ca2+ and Tgase2-mediated PTM, since chelation of cytosolic Ca2+ or decreased expression of Tgase2 reduced this consequence of ER stress (32). These data show that β cell ER stress leads to β cell immunogenicity through Ca2+-dependent PTM of endogenous proteins.
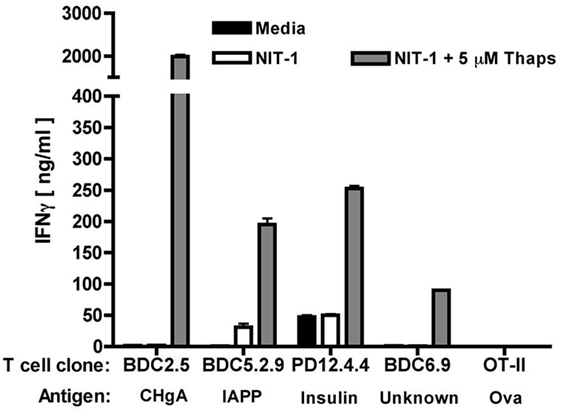
Figure 3. Endoplasmic reticulum (ER) stress increases the immunogenicity of several β cell autoantigens. The immunogenicity of NIT-1 insulinoma cells treated with 5 µM thapsigargin or control for 1 h was assessed by T cell assay. Briefly, T cells (2 × 104), NOD. scid splenocytes as antigen-presenting cells (4 × 105), and NIT-1 cells as antigen (1 × 103) were combined in 200 µl in triplicate in 96-well flat-bottom tissue culture plates and incubated at 37°C for 72 h. TH1 effector function was determined by measuring interferon gamma (IFNγ) secretion by enzyme-linked immunosorbent assay. Data are mean IFNγ secretion ± SD and are from one representative experiment of three independent experiments. For all specificities examined, NIT-1 cells undergoing ER stress elicited higher effector responses from the T cells, suggesting that ER stress contributes to the modification and greater immunogenicity of each of these proteins.
Since ER stress is inherent to β cell physiology and function (32–42, 60), we hypothesized that ER stress induced by normal physiology [e.g., dynamic glucose sensing and secretory function (33–42, 60)] may be sufficient to cause Ca2+- and PTM-dependent β cell immunogenicity. Indeed, a murine insulinoma (NIT-1) that exhibited low ER stress and immunogenicity was exposed to physiological milieu by transplantation into NOD. scid mice. After transplant, these cells exhibited insulin secretion, ER stress, Tgase2 activity, and immunogenicity (32). These data confirm that β cell physiology and insulin secretion contributes to the autoimmune targeting of β cells (60).
Many groups have demonstrated an increase in β cell ER stress long before β cell death and T1D onset (79, 81, 149, 150). In fact, relief of ER stress has been proposed as therapeutic opportunity for preventing β cell death and maintaining euglycemia (63, 80, 151, 152). However, most researchers conclude that ER stress leads to β cell death through the terminal UPR and activation of apoptosis pathways (76, 77, 80). Ours was the first study to demonstrate that normal, physiological β cell ER stress and the adaptive UPR contribute to T1D through the formation of β cell neo-antigens. In doing so, we became the first to propose a mechanism by which β cell neo-antigens (Table 2) may occur (Figure 4).
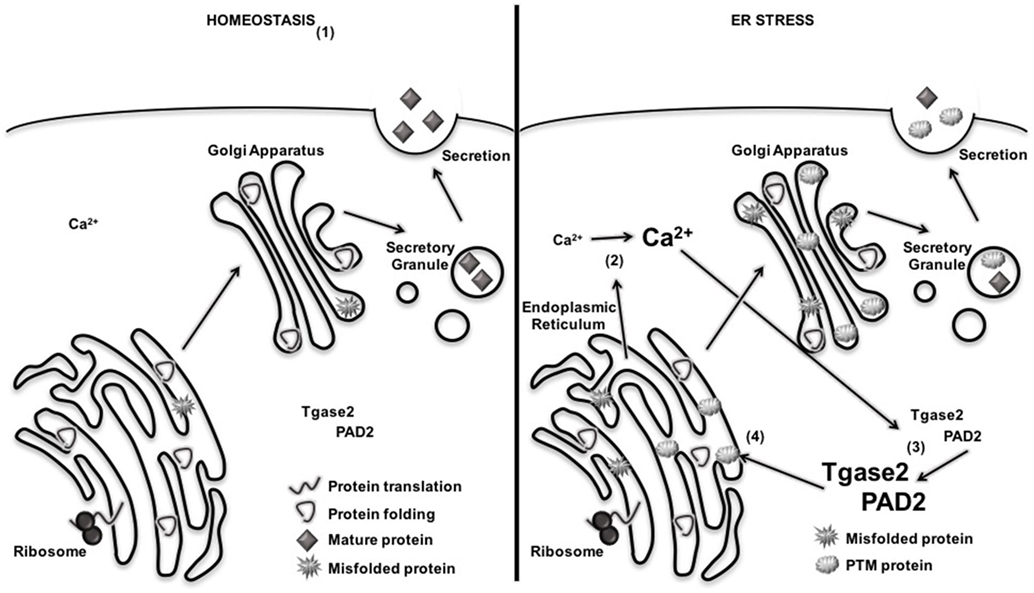
Figure 4. Endoplasmic reticulum (ER) stress increases the activation of Ca2+-dependent posttranslational modification (PTM) enzymes and the formation of PTM-dependent β cell neo-antigens. (1) Under homeostatic conditions, proteins are translated, folded, and packaged into secretory granules. Cytosolic Ca2+ and PTM enzyme activity remain low. (2) During β cell ER stress, Ca2+ stores are released from the ER, increasing cytosolic Ca2+. (3) Increased Ca2+ concentrations activated Ca2+-dependent enzymes tissue transglutaminase 2 (Tgase2) and peptidylarginine deiminase 2 (PAD2). (4) Active PTM enzymes modify nascent proteins. If presented to autoreactive T cells by antigen-presenting cell, modified β cell proteins break tolerance and facilitate immune recognition of β cells.
β Cell Immunogenicity Requires a Threshold of ER Stress
Endoplasmic reticulum stress occurs along a gradient. The burden of unfolded proteins in the ER lumen can vary from mild to severe, resulting in varying degrees of ER dysfunction and stress. This variance in levels of ER stress has important implications for the cellular consequences of ER stress. As discussed earlier, the strength and duration of ER stress-induced UPR signaling is a major factor in determining whether the adaptive UPR or terminal UPR is initiated (63, 64). One explanation may be that the severity and duration of ER stress affects the strength of the Ca2+ efflux from the ER lumen and determines whether cytosolic Ca2+ concentrations cross a putative threshold. Differences in cytosolic Ca2+ concentrations may significantly alter PTM enzyme activity, neo-antigen generation, and β cell immunogenicity.
This “threshold hypothesis” is further supported by literature that demonstrates that Tgase2 and PAD2 remain largely inactive in the cytosol, and activation requires significantly increased concentrations of cytosolic Ca2+. In fact, the activation of both enzymes requires Ca2+ concentrations up to 100-fold higher than what is necessary for normal cellular physiology and function. Therefore, these enzymes generally become activated only under conditions of cellular distress or dysfunction, such as ER stress (96, 97, 108, 109, 147, 153, 154). Since these PTM enzymes require particular levels of cytosolic Ca2+ to become activated, it follows that a particular level of ER stress must be achieved before PTM-dependent neo-antigen formation can occur.
Previous work in our laboratory examined whether varying levels of ER stress lead to different degrees of β cell PTM-dependent immunogenicity. NIT-1 cells were incubated with increasing doses of thapsigargin, which increases ER stress and cytosolic Ca2+ by inhibiting the SERCA pumps that transport Ca2+ from the cytosol into the ER. As expected, thapsigargin induced ER stress and UPR activity in a dose-dependent manner (Figure 5A). The immunogenicity of these cells was examined by the BDC2.5 T cell clone, and T cell effector function was measured by IFNγ as previously described (32). Only the highest dose of thapsigargin elicited detectable IFNγ secretion from the T cells (Figure 5B). Therefore, although lower doses of thapsigargin induced ER stress, the stress (and consequences thereof) at these lower doses was not sufficient to result in β cell immunogenicity.
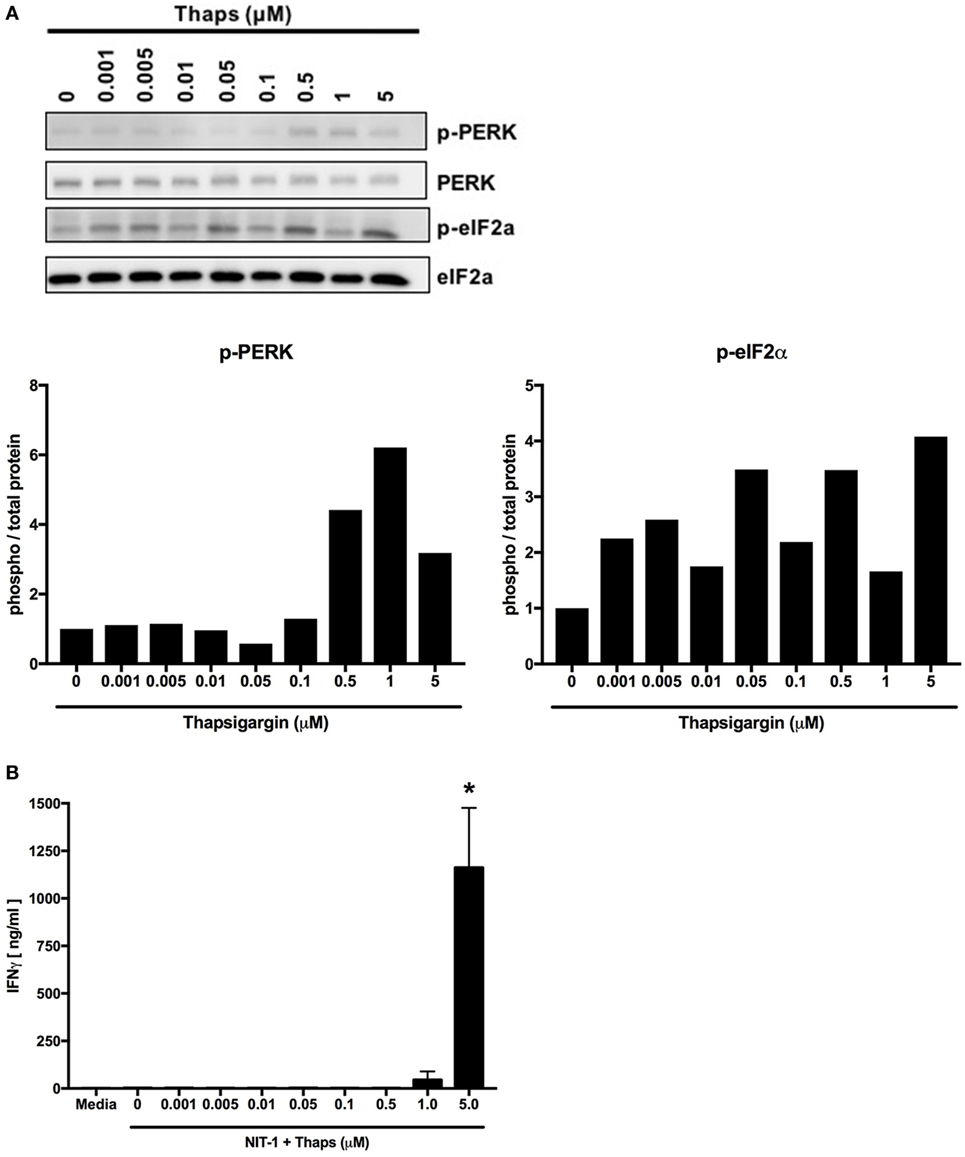
Figure 5. Endoplasmic reticulum stress must increase above a threshold to induce posttranslational modification-dependent immunogenicity. (A) NIT-1 insulinoma cells were incubated with increasing concentrations of thapsigargin for 1 h and washed extensively. Cell lysates were analyzed for the phosphorylation of UPR proteins—protein kinase RNA (PKR)-like ER kinase (PERK) and eIF2α. Data are representative of two independent experiments. Densitometry data are phosphorylation levels normalized by total protein and relative to that in control (0 µM) treated cells. (B) The immunogenicity of NIT-1 cells treated with increasing concentrations of thapsigargin for 1 h was measured by BDC2.5 T cell assay. Data are mean interferon gamma (IFNγ) secretion ± SEM. *p < 0.05.
Tunicamycin is another chemical inducer of ER stress that blocks the initial steps of glycoprotein synthesis in the ER and thus increases the burden of unfolded proteins in the ER lumen (148). Increasing doses of tunicamycin increased ER stress in NIT-1 cells (Figure 6A), but to lesser degrees compared with thapsigargin (Figure 5A). Also, as with lower doses of thapsigargin, the lower ER stress induced by tunicamycin was not sufficient to elicit effector responses from BDC2.5 T cells (Figure 6B). Together, these data serve as further evidence that a particular threshold of ER stress must be reached to achieve PTM-dependent β cell immunogenicity.
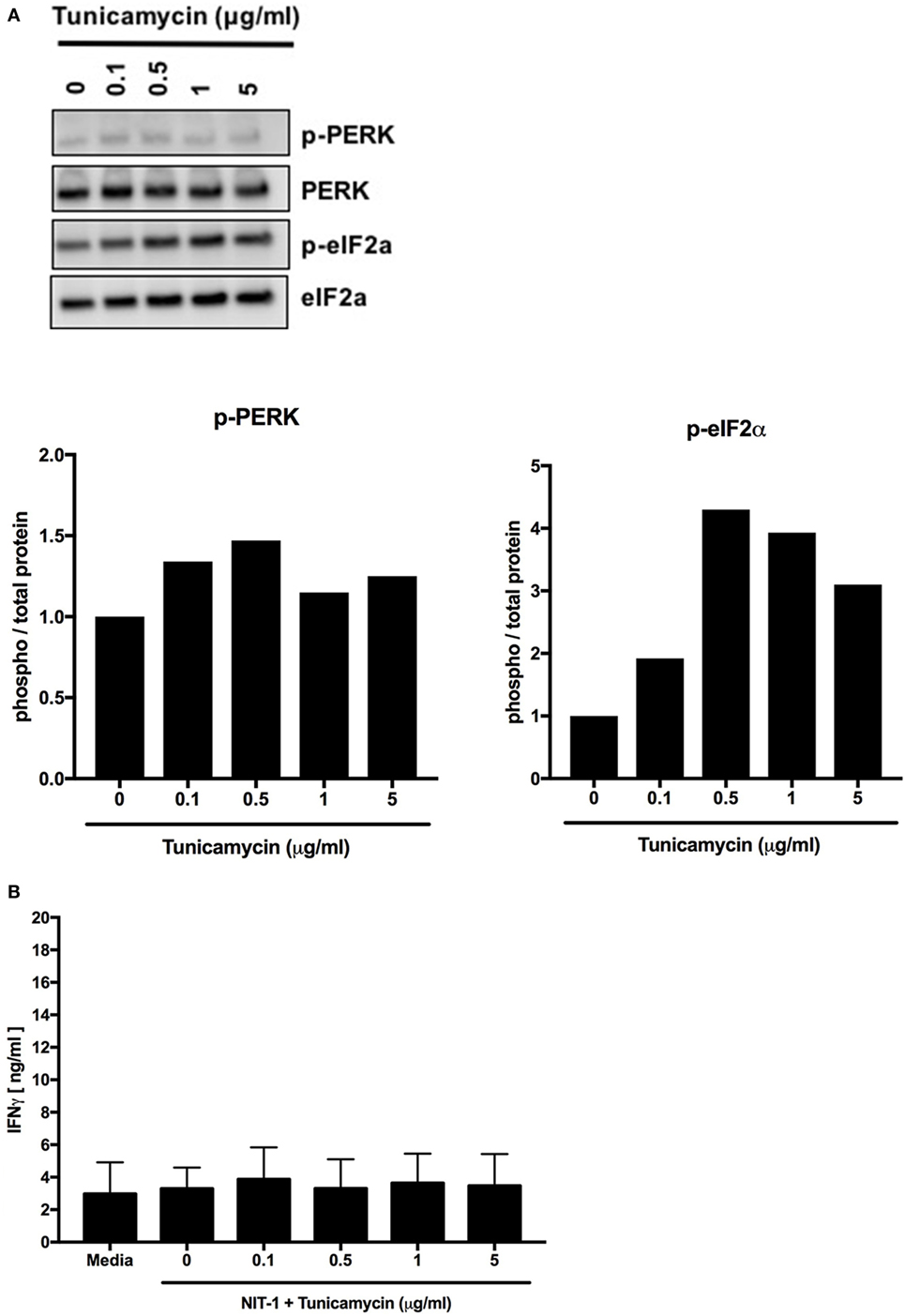
Figure 6. Endoplasmic reticulum stress below a threshold does not induce posttranslational modification-dependent immunogenicity. (A) NIT-1 insulinoma cells were incubated with increasing concentrations of tunicamycin for 4 h and washed extensively. Cell lysates were analyzed for the phosphorylation of UPR proteins—protein kinase RNA (PKR)-like ER kinase (PERK) and eIF2α. Data are representative of two independent experiments. Densitometry data are phosphorylation levels normalized by total protein and relative to that in control (0 µg/ml) treated cells. (B) The immunogenicity of NIT-1 cells treated with increasing concentrations of tunicamycin for 4 h was measured by BDC2.5 T cell assay. Data are mean interferon gamma (IFNγ) secretion ± SEM.
Environmental Factors Associated with T1D Induce Heightened β Cell ER Stress
Every pancreas undergoes ER stress (32, 81), but this stress does not lead to T1D in every individual. In fact, even in those with a genetic predisposition to autoimmunity, T1D may never occur (155) or may occur much later than expected (156, 157). These observations suggest that environmental factors may precipitate disease onset. Indeed, T1D onset is associated with several environmental factors such as viral infection (43–48), chemicals (49–51), ROS (52–55), dysglycemia (56), and inflammation (57–59). Although these environmental factors are thought to exacerbate the autoimmune targeting of β cells and hasten disease onset, the mechanisms by which these environmental factors advance pathology, and whether these factors contribute to PTM-mediated neo-antigen formation, remain unknown (Figure 1).
As discussed earlier, β cell ER stress and Ca2+ flux into the cytosol must cross a threshold before Tgase2 and PAD2 can modify β cell proteins to generate neo-antigens and elicit effector responses from diabetogenic T cells. While β cell physiology causes ER stress (32–42) and this ER stress can, under some circumstances generate neo-antigens and immunogenicity (32, 60) (Figure 1), the discrepancy in disease onset in those genetically predisposed to autoimmunity (155–157) suggests that this physiological stress alone may not be sufficient to generate neo-antigens. Interestingly, each of the environmental factors associated with T1D also lead to an increase in β cell ER stress and cytosolic Ca2+.
Coxsackie Virus
Coxsackie virus infection is associated with T1D onset. Recent onset T1D patients have viral RNA in their pancreas and higher titers of antibodies against Coxsackie virus (158, 159). Also, Coxsackie virus infection accelerates disease onset in NOD mice with established insulitis (46, 160–162), suggesting a role for Coxsackie virus in breaking immune tolerance. Studies with BDC2.5 TCR transgenic NOD mice attributed this acceleration to activation of bystander immune cells (46). These data provide a strong link between pancreatic viral infection and broken tolerance. Since BDC2.5 T cells do not recognize a viral protein (29) but rather modified CHgA, activation of BDC2.5 T cells in these mice suggests that Coxsackie virus infection may lead to PTM of endogenous β cell proteins and neo-antigen formation. Indeed, viruses cause neo-antigen generation and exacerbate pathology in other models of autoimmunity (163).
Moreover, Coxsackie virus protein 2B disrupts the ER membrane (164–166), releasing Ca2+ from the ER into the cytosol and causing ER stress. We have shown that β cell ER stress contributes to neo-antigen formation and immunogenicity (32). Therefore, it is plausible that Coxsackie virus may raise β cell ER stress and cytosolic Ca2+ concentrations above the levels attributed to normal physiology, increasing neo-antigen production through Ca2+-dependent PTM.
Exposure to Chemicals
Exposure of β cells to chemicals such as alloxan and streptozotocin cause the loss of insulin secretion and β cell death (167). For each of these chemicals, β cells experience DNA damage, protein ADP ribosylation (168), and ROS generation (169–171), all of which ultimately lead to apoptosis and significant loss of β cell death. However, before apoptosis pathways are activated, ADP ribosylation and ROS cause misfolding and accumulation of nascent proteins in the ER lumen. As discussed earlier, the accumulation of misfolded and abnormally modified proteins leads to ER stress and release of Ca2+ into the cytosol (172, 173).
Reactive Oxygen Species
Reactive oxygen species, which have the potential to cause irreversible damage to cellular proteins and organelles (174–176), are generated both during normal β cell function (52) and when β cells are exposed to other insults such as Coxsackie virus (177–179). Although antioxidant defenses work to prevent ROS-mediated damage, β cell mitochondria express very low levels of antioxidant enzymes (180–182), making these cells particularly susceptible to ROS-mediated damage. When ROS exceeds the capacity of the cell to scavenge these species, oxidative stress leads to β cell death (183, 184) and ultimately to T1D (52, 54, 180, 185–190). However, before the loss of β cell mass, ROS leads to oxidative modification of proteins and lipids (191), and to the release of Ca2+ from the ER into the cytosol (192–194). Therefore, ER stress and Ca2+ efflux caused by ROS may lead to protein PTM and the formation for neo-antigens in β cells.
Dysglycemia
As discussed earlier, increased glucose sensing by β cells during times of dysglycemia increases insulin production and secretion (75). Normal insulin secretion raises β cell ER stress (32–42), but when blood glucose rises too high, or the hyperglycemia is too prolonged, so called “glucotoxicity” further enhances β cell ER stress. At later stages of T1D, ER stress induced by glucotoxicity is thought to be a major contributor to β cell death through the terminal UPR. However, fluctuation in blood glucose levels as β cell mass is gradually lost may also induce the adaptive UPR. In this way, glucotoxicity may, long before β cell death, contribute to Ca2+- and PTM-dependent neo-antigen formation and therefore to autoimmune targeting of β cells.
Inflammation
As autoreactive immune cells infiltrate the islets to target their antigens, these activated immune cells secrete pro-inflammatory cytokines. In addition, β cells themselves release additional pro-inflammatory cytokines during viral infection (195), and cellular stress (196). These inflammatory mediators initiate signaling cascades in the β cells. For example, pro-inflammatory cytokines activate NF-kB in β cells, which inhibits the expression of other transcription factors necessary for normal β cell function (197). Also, inflammatory cytokines activate c-jun N-terminal mitogen-activated protein kinase signaling, which is associated with ER stress and Ca2+ release (198, 199). Finally, inflammatory cytokines reduce SERCA expression, effectively preventing the return of Ca2+ from the cytosol to the ER and further exacerbating ER stress (197, 200). Therefore, pancreatic inflammation may lead to β cell neo-antigen formation and exacerbate autoimmune targeting of β cells.
Therefore, we hypothesize that the ER stress generated by these environmental factors may converge with the stress caused by normal physiology to allow cytosolic Ca2+ to cross the necessary threshold to activate PTM enzymes and generate neo-antigens long before the terminal UPR initiates apoptosis pathways. In this way, ER stress-mediated neo-antigen formation may be a common mechanism by which these environmental factors augment autoimmune targeting of β cells and hasten T1D onset.
Conclusion
Type 1 diabetes is caused by the autoimmune targeting and destruction of pancreatic β cells. The autoreactive immune cells target many β cell proteins (Table 1) when central and peripheral tolerance fail. The mechanisms by which tolerance fails are still being elucidated, but a growing body of literature demonstrates that β cell peptides modified by Ca2+-dependent PTM elicit stronger responses from autoreactive T cells than their native counterparts (16, 19, 23, 28–31, 143, 145). However, the mechanisms by which these β cell peptides become modified during β cell physiology is only beginning to be explored (32, 60).
We have previously demonstrated that β cell ER stress leads to PTM-dependent immunogenicity (32). Although this ER stress may be derived from the natural secretory physiology of the β cell (32–42), inherent, physiological ER stress alone may not sufficient to precipitate T1D onset even in those individuals harboring a genetic predisposition to autoimmunity (155–157). We therefore propose a model in which β cell ER stress leads to neo-antigen formation and immunogenicity of β cells when this ER stress reaches a critical threshold. The ER stress induced by the environmental factors associated with T1D may combine with physiological ER stress to raise cytosolic Ca2+ above this putative threshold, allowing for the activation of PTM enzymes and the generation of PTM-dependent neo-antigens (Figure 7). This convergence of with physiological stress may explain how environmental factors hasten T1D onset.
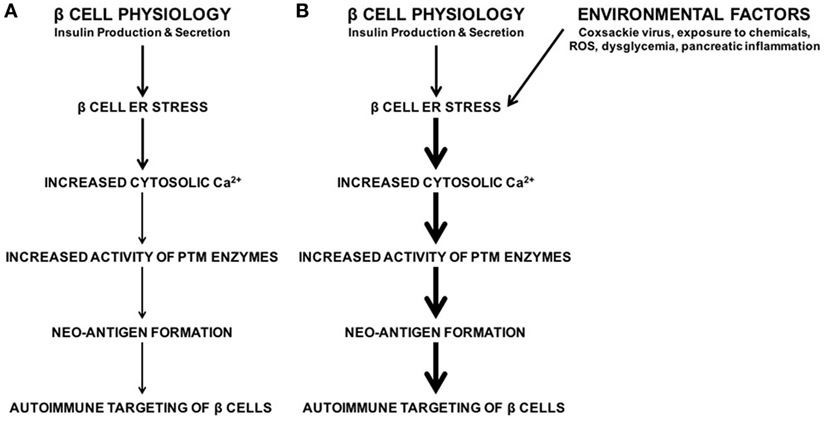
Figure 7. Model. (A) Normal β cell secretory physiology causes inherent endoplasmic reticulum (ER) stress, which in turn results in a release of Ca2+ from the ER into the cytosol. This ER stress and Ca2+ efflux lead to increased activity of Ca2+-dependent posttranslational modification (PTM) enzymes, formation of neo-antigens, and β cell immunogenicity (32). However, neo-antigen formation and immunogenicity due to inherent physiological ER stress may not be enough to cause type 1 diabetes (T1D). (B) The environmental factors associated with T1D onset cause β cell ER stress and Ca2+ efflux. The ER stress induced by these environmental factors cooperates with the physiological ER stress to raise cytosolic Ca2+ concentrations above a threshold to activate PTM enzymes, generate neo-antigens, cause autoimmune targeting of β cells, and precipitate T1D onset.
It is important to note that, although physiological and environmental factor-derived ER stress likely occurs in the β cells of all individuals, autoimmunity predominantly occurs in the context of genetic predisposition to autoimmunity. For patients who express the MHC molecules that predispose them to autoimmunity, β cell neo-antigens generated during ER stress are presented by these MHC molecules and activate the T cells that escaped negative selection during development. The activation of these T cells ultimately leads to the autoimmune destruction of the β cells and to T1D onset. However, in those without this MHC predisposition, β cell ER stress may still result in the modification of β cell proteins without leading to disease. In these patients, these neo-antigens may not be presented by APC or may not be recognized if autoreactive T cells are correctly deleted from the repertoire during negative selection in the thymus. Therefore, β cell ER stress and the subsequent neo-antigen formation likely still require genetic predisposition to autoimmunity to lead to T1D.
Our model proposes a “threshold hypothesis” according to which cytosolic Ca2+ must cross a particular threshold to allow for the generation of PTM-dependent β cell neo-antigens. Additional studies are necessary to confirm the cooperation between physiological ER stress and that derived from exposure to environmental factors to reach this threshold. These studies will further advance our understanding both of how neo-antigens are formed in the β cell and the mechanisms by which environmental factors hasten disease onset. Such studies may reveal novel opportunities for therapeutic intervention to prevent or delay T1D onset in at-risk patients.
Materials and Methods
Mice
Mice were bred and housed under specific pathogen-free conditions at Rangos Research Center of Children’s Hospital of Pittsburgh of University of Pittsburgh Medical Center. All experiments were approved by Institutional Animal Care and Use Committee of the University of Pittsburgh.
Cell Culture
The NIT-1 insulinoma cell line was a gift from Clayton Mathews (University of Florida) and were maintained at 37°C in a 5% CO2 humid air incubator, in DMEM (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Mediatech), 10 mM HEPES buffer (Gibco), 4 mM l-glutamine (Gibco), 200 µM nonessential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), 61.5 µM β-mercaptoethanol (Sigma-Aldrich), and 100 µg/ml gentamicin (Gibco).
CD4+, MHC class II-restricted BDC2.5, BDC5.2.9, PD12.4.4, and BDC6.9 T cells were a gift from Kathryn Haskins (University of Colorado). T cell clones were maintained in supplemented DMEM as described previously (32, 201–203).
OT-II splenocytes were harvested and prepared in supplemented DMEM as described previously (204–208).
Induction of ER Stress
NIT-1 cells were cultured in 25 cm2 tissue culture flasks (Greiner Bio-One) with various concentrations of thapsigargin or control for 1 h at 37°C or with various concentrations of tunicamycin or control for 4 h at 37°C. Before downstream analysis, the cells were washed extensively (50,000× original volume) to remove residual thapsigargin or tunicamycin, and removed from the flask with 0.05% trypsin–EDTA (Gibco).
T Cell Assays
T cells (2 × 104), NOD. scid splenocytes as APC (4 × 105), and antigen (1 × 103 dispersed NIT-1 cells) were combined in 200 µl supplemented DMEM in triplicate in 96-well flat-bottom tissue culture plates (Greiner Bio-One) and incubated at 37°C for 72 h. TH1 effector function was determined by measuring IFNγ secretion by enzyme-linked immunosorbent assay (ELISA).
Splenocyte Assay
OT-II splenocytes (1 × 106) were combined with antigen (1 × 103 dispersed NIT-1 cells) in 200 µl supplemented DMEM in triplicate in 96-well flat-bottom tissue culture plates (Greiner Bio-One) and incubated at 37°C for 72 h as described previously (204–208). TH1 effector function was determined by measuring IFNγ secretion by ELISA.
Enzyme-Linked Immunosorbent Assay
Interferon gamma from T cell assays was measured with murine IFNγ ELISA antibody pairs (BD Biosciences) as described previously (32, 202–204, 208). Absorbance was measured at 450 nm with a SpectraMax M2 microplate reader (Molecular Devices). Data were analyzed with SoftMax Pro (Molecular Devices).
Preparation of Cell Lysates
Cells were lysed by sonication in 50 mM Tris pH 8.0, 137 mM NaCl, 10% glycerol, 1% NP-40, 1 mM NaF, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 2 mM Na3VO4, and 1 mM PMSF. Protein concentration was determined by bicinchoninic acid protein assay (Thermo Fisher Scientific).
Western Blotting
Lysates were separated by SDS-PAGE with 10% polyacrylamide gels and transferred to PVDF membranes. Membranes were blocked in 5% BSA in TBST for 1 h, and probed with antibodies to phosphorylated PERK (Cell Signaling Technology; 1:200), phosphorylated eIF2α (Cell Signaling Technology; 1:1,000), total PERK (Cell Signaling Technology; 1:1,000), and total eIF2α (Cell Signaling Technology; 1:1,000) overnight at 4°C. Membranes were washed and incubated with HRP-conjugated goat anti-rabbit (Cell Signaling Technology; 1:2,000) for 1 h. Chemiluminescence was detected with Luminata Crescendo Western HRP Substrate (Millipore) and analyzed with Fujifilm LAS-4000 imager and Multi Gage Software (Fujifilm Life Science).
Statistical Analysis
For ELISA, data are mean IFNγ secretion ± SD or SEM (as indicated). For Western blotting, data are representative of two experiments. Densitometry data are phosphorylation levels normalized by total and relative to that in control-treated cells. Statistical significance was determined by Student’s t-test, and statistically significant differences are shown for *p < 0.05.
Ethics Statement
This study was carried out in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Author Contributions
MM and JP contributed to the composition of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank members of the Piganelli Laboratory for the review of this manuscript. In particular, the authors thank Christina P. Martins, Ethan Bassin, Dr. Gina M. Coudriet, and Dr. Dana M. Previte.
Funding
This work was supported in part by the Juvenile Diabetes Research Foundation (3-PDF-2014-213-A-N to MM and 2-SRA-2014-296-Q-R to JP) and Children’s Hospital of Pittsburgh of the UPMC Health System (Cochrane-Weber Endowed Fund to JP).
Abbreviations
Aire, autoimmune regulator; APC, antigen-presenting cell; ATF6, activating transcription factor 6; ATP, adenosine triphosphate; Ca2+, calcium; CHgA, chromogranin A; DRiP, defective ribosomal product; EAE, experimental autoimmune encephalomyelitis; ER, endoplasmic reticulum; GAD65, glutamic acid decarboxylase 65; GFP, green fluorescent protein; GRP78, 78 kDa glucose-regulated protein; IA-2, tyrosine phosphatase-like insulinoma antigen 2; IAPP, islet amyloid polypeptide; ICA69, islet cell autoantigen 69; IGF-2, insulin-like growth factor 2; IGRP, islet-specific glucose-6-phosphatase catalytic subunit-related protein; IFNγ, interferon gamma; IP3R, inositol 1,4,5-trisphosphate receptor; IRE1, inositol-requiring protein 1; JNK, c-jun N-terminal kinase; MAP, mitogen-activated protein kinase; MHC, major histocompatibility complex; mTEC, medullary thymic epithelial cell; NET, neutrophil extracellular traps; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOD, non-obese diabetic mouse; PAD2, peptidylarginine deiminase 2; PDI, protein disulfide isomerases; PERK, protein kinase RNA (PKR)-like ER kinase; Phogrin, phosphatase homolog of granules from rat insulinomas; PTM, posttranslational modification; ROS, reactive oxygen species; RyR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+ ATPases; SNP, single-nucleotide polymorphisms; T1D, type 1 diabetes; Tgase2, tissue transglutaminase 2; UPR, unfolded protein response; ZnT8, zinc transporter 8.
References
1. Dorman JS, LaPorte RE, Stone RA, Trucco M. Worldwide differences in the incidence of type I diabetes are associated with amino acid variation at position 57 of the HLA-DQ beta chain. Proc Natl Acad Sci U S A (1990) 87(19):7370–4. doi:10.1073/pnas.87.19.7370
2. Luca D, Ringquist S, Klei L, Lee AB, Gieger C, Wichmann HE, et al. On the use of general control samples for genome-wide association studies: genetic matching highlights causal variants. Am J Hum Genet (2008) 82(2):453–63. doi:10.1016/j.ajhg.2007.11.003
3. Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature (1987) 329(6140):599–604. doi:10.1038/329599a0
4. Hogquist KA, Jameson SC. The self-obsession of T cells: how TCR signaling thresholds affect fate ‘decisions’ and effector function. Nat Immunol (2014) 15(9):815–23. doi:10.1038/ni.2938
5. Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J (2009) 28(18):2812–24. doi:10.1038/emboj.2009.212
6. Geenen V. Thymus and type 1 diabetes: an update. Diabetes Res Clin Pract (2012) 98(1):26–32. doi:10.1016/j.diabres.2012.05.023
7. Driver JP, Chen YG, Mathews CE. Comparative genetics: synergizing human and NOD mouse studies for identifying genetic causation of type 1 diabetes. Rev Diabet Stud (2012) 9(4):169–87. doi:10.1900/RDS.2012.9.169
10. Wegmann DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol (1994) 24(8):1853–7. doi:10.1002/eji.1830240820
11. Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature (1993) 366(6450):72–5. doi:10.1038/366072a0
12. Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A (2003) 100(14):8384–8. doi:10.1073/pnas.0932778100
13. Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol (2010) 11(3):225–31. doi:10.1038/ni.1844
14. Delong T, Baker RL, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Islet amyloid polypeptide is a target antigen for diabetogenic CD4+ T cells. Diabetes (2011) 60:2325–30. doi:10.2337/db11-0288
15. Nayak DK, Calderon B, Vomund AN, Unanue ER. ZnT8-reactive T cells are weakly pathogenic in NOD mice but can participate in diabetes under inflammatory conditions. Diabetes (2014) 63(10):3438–48. doi:10.2337/db13-1882
16. Rondas D, Crevecoeur I, D’Hertog W, Ferreira GB, Staes A, Garg AD, et al. Citrullinated glucose-regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes (2015) 64(2):573–86. doi:10.2337/db14-0621
17. Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature (1990) 347(6289):151–6. doi:10.1038/347151a0
18. Gorus FK, Sodoyez JC, Pipeleers DG, Keymeulen B, Foriers A, Van Schravendijk CF. Detection of autoantibodies against islet amyloid polypeptide in human serum. Lack of association with type 1 (insulin-dependent) diabetes mellitus, or with conditions favouring amyloid deposition in islets. The Belgian Diabetes Registry. Diabetologia (1992) 35(11):1080–6. doi:10.1007/BF02221685
19. Gottlieb PA, Delong T, Baker RL, Fitzgerald-Miller L, Wagner R, Cook G, et al. Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun (2014) 50:38–41. doi:10.1016/j.jaut.2013.10.003
20. Keller RJ. Cellular immunity to human insulin in individuals at high risk for the development of type I diabetes mellitus. J Autoimmun (1990) 3(3):321–7. doi:10.1016/0896-8411(90)90150-Q
21. Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A (2007) 104(43):17040–5. doi:10.1073/pnas.0705894104
22. Yang J, Danke NA, Berger D, Reichstetter S, Reijonen H, Greenbaum C, et al. Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J Immunol (2006) 176(5):2781–9. doi:10.4049/jimmunol.176.5.2781
23. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22(12):1482–7. doi:10.1038/nm.4203
24. Lan MS, Wasserfall C, Maclaren NK, Notkins AL. IA-2, a transmembrane protein of the protein tyrosine phosphatase family, is a major autoantigen in insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A (1996) 93(13):6367–70. doi:10.1073/pnas.93.13.6367
25. Bonifacio E, Lampasona V, Genovese S, Ferrari M, Bosi E. Identification of protein tyrosine phosphatase-like IA2 (islet cell antigen 512) as the insulin-dependent diabetes-related 37/40K autoantigen and a target of islet-cell antibodies. J Immunol (1995) 155(11):5419–26.
26. Pietropaolo M, Castano L, Babu S, Buelow R, Kuo YL, Martin S, et al. Islet cell autoantigen 69 kD (ICA69). Molecular cloning and characterization of a novel diabetes-associated autoantigen. J Clin Invest (1993) 92(1):359–71. doi:10.1172/JCI116574
27. Dunne JL, Overbergh L, Purcell AW, Mathieu C. Posttranslational modifications of proteins in type 1 diabetes: the next step in finding the cure? Diabetes (2012) 61(8):1907–14. doi:10.2337/db11-1675
28. Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med (2005) 202(9):1191–7. doi:10.1084/jem.20051251
29. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-Cell clones recognize an altered peptide of chromogranin A. Diabetes (2012) 61:3239–46. doi:10.2337/db12-0112
30. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes (2014) 63(1):237–47. doi:10.2337/db12-1214
31. McGinty JW, Chow IT, Greenbaum C, Odegard J, Kwok WW, James EA. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes (2014) 63(9):3033–40. doi:10.2337/db13-1952
32. Marre ML, Profozich JL, Coneybeer JT, Geng X, Bertera S, Ford MJ, et al. Inherent ER stress in pancreatic islet beta cells causes self-recognition by autoreactive T cells in type 1 diabetes. J Autoimmun (2016) 72:33–46. doi:10.1016/j.jaut.2016.04.009
33. Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes mellitus. Int Med (2003) 42(1):7–14. doi:10.2169/internalmedicine.42.7
34. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev (2008) 29(1):42–61. doi:10.1210/er.2007-0015
35. Fonseca SG, Lipson KL, Urano F. Endoplasmic reticulum stress signaling in pancreatic beta-cells. Antioxid Redox Sig (2007) 9(12):2335–44. doi:10.1089/ars.2007.1790
36. Kim MK, Kim HS, Lee IK, Park KG. Endoplasmic reticulum stress and insulin biosynthesis: a review. Exp Diabetes Res (2012) 2012:509437. doi:10.1155/2012/509437
37. Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab (2006) 4(3):245–54. doi:10.1016/j.cmet.2006.07.007
38. Lipson KL, Fonseca SG, Urano F. Endoplasmic reticulum stress-induced apoptosis and auto-immunity in diabetes. Curr Mol Med (2006) 6(1):71–7. doi:10.2174/156652406775574613
39. Ortsater H, Sjoholm A. A busy cell – endoplasmic reticulum stress in the pancreatic beta-cell. Mol Cell Endocrinol (2007) 277(1–2):1–5. doi:10.1016/j.mce.2007.06.006
40. Teodoro T, Odisho T, Sidorova E, Volchuk A. Pancreatic beta-cells depend on basal expression of active ATF6alpha-p50 for cell survival even under non-stress conditions. Am J Physiol Cell Physiol (2012) 302(7):C992–1003. doi:10.1152/ajpcell.00160.2011
41. Volchuk A, Ron D. The endoplasmic reticulum stress response in the pancreatic beta-cell. Diabetes Obes Metab (2010) 12(Suppl 2):48–57. doi:10.1111/j.1463-1326.2010.01271.x
42. Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ (2006) 13(3):374–84. doi:10.1038/sj.cdd.4401840
43. Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and Coxsackie virus in insulin-dependent diabetes. J Clin Invest (1994) 94(5):2125–9. doi:10.1172/JCI117567
44. Harkonen T, Lankinen H, Davydova B, Hovi T, Roivainen M. Enterovirus infection can induce immune responses that cross-react with beta-cell autoantigen tyrosine phosphatase IA-2/IAR. J Med Virol (2002) 66(3):340–50. doi:10.1002/jmv.2151
45. Hiemstra HS, Schloot NC, van Veelen PA, Willemen SJ, Franken KL, van Rood JJ, et al. Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Pro Natl Acad Sci U S A (2001) 98(7):3988–91. doi:10.1073/pnas.071050898
46. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med (1998) 4(7):781–5. doi:10.1038/nm0798-781
47. Horwitz MS, Ilic A, Fine C, Balasa B, Sarvetnick N. Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin Immunol (2004) 110(2):134–44. doi:10.1016/j.clim.2003.09.014
48. Schulte BM, Kramer M, Ansems M, Lanke KH, van Doremalen N, Piganelli JD, et al. Phagocytosis of enterovirus-infected pancreatic beta-cells triggers innate immune responses in human dendritic cells. Diabetes (2010) 59:1182–91. doi:10.2337/db09-1071
49. Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science (1976) 193(4251):415–7. doi:10.1126/science.180605
50. Rossini AA, Appel MC, Williams RM, Like AA. Genetic influence of the streptozotocin-induced insulitis and hyperglycemia. Diabetes (1977) 26(10):916–20. doi:10.2337/diab.26.10.916
51. Takasu N, Asawa T, Komiya I, Nagasawa Y, Yamada T. Alloxan-induced DNA strand breaks in pancreatic islets. Evidence for H2O2 as an intermediate. J Biol Chem (1991) 266(4):2112–4.
52. Delmastro MM, Piganelli JD. Oxidative stress and redox modulation potential in type 1 diabetes. Clin Dev Immunol (2011) 2011:593863. doi:10.1155/2011/593863
53. Delmastro-Greenwood MM, Tse HM, Piganelli JD. Effects of metalloporphyrins on reducing inflammation and autoimmunity. Antioxid Redox Signal (2014) 20(15):2465–77. doi:10.1089/ars.2013.5257
54. Piganelli JD, Flores SC, Cruz C, Koepp J, Batinic-Haberle I, Crapo J, et al. A metalloporphyrin-based superoxide dismutase mimic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T-cell clone. Diabetes (2002) 51:347–55. doi:10.2337/diabetes.51.2.347
55. Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol (2010) 185:5247–58. doi:10.4049/jimmunol.1001472
56. Sosenko JM, Palmer JP, Rafkin-Mervis L, Krischer JP, Cuthbertson D, Mahon J, et al. Incident dysglycemia and progression to type 1 diabetes among participants in the diabetes prevention trial-type 1. Diabetes Care (2009) 32(9):1603–7. doi:10.2337/dc08-2140
57. Held W, MacDonald HR, Weissman IL, Hess MW, Mueller C. Genes encoding tumor necrosis factor alpha and granzyme A are expressed during development of autoimmune diabetes. Proc Natl Acad Sci U S A (1990) 87(6):2239–43. doi:10.1073/pnas.87.6.2239
58. Jiang Z, Woda BA. Cytokine gene expression in the islets of the diabetic biobreeding/Worcester rat. J Immunol (1991) 146(9):2990–4.
59. Mandrup-Poulsen T, Spinas GA, Prowse SJ, Hansen BS, Jorgensen DW, Bendtzen K, et al. Islet cytotoxicity of interleukin 1. Influence of culture conditions and islet donor characteristics. Diabetes (1987) 36(5):641–7. doi:10.2337/diabetes.36.5.641
60. Marre ML, James EA, Piganelli JD. Beta cell ER stress and the implications for immunogenicity in type 1 diabetes. Front Cell Dev Biol (2015) 3:67. doi:10.3389/fcell.2015.00067
61. McGinty JW, Marre ML, Bajzik V, Piganelli JD, James EA. T cell epitopes and post-translationally modified epitopes in type 1 diabetes. Curr Diab Rep (2015) 15(11):90. doi:10.1007/s11892-015-0657-7
62. Gething MJ, Sambrook J. Protein folding in the cell. Nature (1992) 355(6355):33–45. doi:10.1038/355033a0
63. Papa FR. Endoplasmic reticulum stress, pancreatic beta-cell degeneration, and diabetes. Cold Spring Harb Perspect Med (2012) 2(9):a007666. doi:10.1101/cshperspect.a007666
64. Eizirik DL, Cnop M. ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci Signal (2010) 3(110):e7. doi:10.1126/scisignal.3110pe7
65. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13(2):89–102. doi:10.1038/nrm3270
66. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell (2000) 6(5):1099–108. doi:10.1016/S1097-2765(00)00108-8
67. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell (2000) 5(5):897–904. doi:10.1016/S1097-2765(00)80330-5
68. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell (1999) 10(11):3787–99. doi:10.1091/mbc.10.11.3787
69. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol (2003) 23(21):7448–59. doi:10.1128/MCB.23.21.7448-7459.2003
70. Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol (2004) 167(1):35–41. doi:10.1083/jcb.200406136
71. Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol (1996) 16(8):4273–80. doi:10.1128/MCB.16.8.4273
72. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol (2001) 21(4):1249–59. doi:10.1128/MCB.21.4.1249-1259.2001
73. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev (1998) 12(7):982–95. doi:10.1101/gad.12.7.982
74. Shimizu Y, Hendershot LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signal (2009) 11(9):2317–31. doi:10.1089/ARS.2009.2501
75. Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev (2008) 29(3):317–33. doi:10.1210/er.2007-0039
76. Araki E, Oyadomari S, Mori M. Impact of endoplasmic reticulum stress pathway on pancreatic beta-cells and diabetes mellitus. Exp Biol Med (2003) 228(10):1213–7. doi:10.1177/153537020322801018
77. Nozaki J, Kubota H, Yoshida H, Naitoh M, Goji J, Yoshinaga T, et al. The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic beta cells. Genes Cells (2004) 9(3):261–70. doi:10.1111/j.1356-9597.2004.00721.x
78. Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest (2002) 109(4):525–32. doi:10.1172/JCI14550
79. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ (2004) 11(4):381–9. doi:10.1038/sj.cdd.4401373
80. Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest (2002) 109(4):443–5. doi:10.1172/JCI15020
81. Iwawaki T, Akai R, Kohno K, Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med (2004) 10(1):98–102. doi:10.1038/nm970
82. Meldolesi J, Pozzan T. The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem Sci (1998) 23(1):10–4. doi:10.1016/S0968-0004(97)01143-2
83. Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. J Chem Neuroanat (2004) 28(1–2):51–65. doi:10.1016/j.jchemneu.2003.08.007
84. Nigam SK, Goldberg AL, Ho S, Rohde MF, Bush KT, Sherman MY. A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca(2+)-binding proteins and members of the thioredoxin superfamily. J Biol Chem (1994) 269(3):1744–9.
85. Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol (2011) 3(6):a004317. doi:10.1101/cshperspect.a004317
86. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol (2000) 1(1):11–21. doi:10.1038/35036035
87. Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol (2004) 165(3):347–56. doi:10.1083/jcb.200310015
88. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature (2000) 403(6765):98–103. doi:10.1038/47513
89. Gil-Parrado S, Fernandez-Montalvan A, Assfalg-Machleidt I, Popp O, Bestvater F, Holloschi A, et al. Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J Biol Chem (2002) 277(30):27217–26. doi:10.1074/jbc.M202945200
90. Hajnoczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun (2003) 304(3):445–54. doi:10.1016/S0006-291X(03)00616-8
91. Kim MJ, Jo DG, Hong GS, Kim BJ, Lai M, Cho DH, et al. Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc Natl Acad Sci U S A (2002) 99(15):9870–5. doi:10.1073/pnas.152336999
92. Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol (2000) 150(4):887–94. doi:10.1083/jcb.150.4.887
93. Lesort M, Attanavanich K, Zhang J, Johnson GV. Distinct nuclear localization and activity of tissue transglutaminase. J Biol Chem (1998) 273(20):11991–4. doi:10.1074/jbc.273.20.11991
94. Park D, Choi SS, Ha KS. Transglutaminase 2: a multi-functional protein in multiple subcellular compartments. Amino Acids (2010) 39(3):619–31. doi:10.1007/s00726-010-0500-z
95. Orru S, Caputo I, D’Amato A, Ruoppolo M, Esposito C. Proteomics identification of acyl-acceptor and acyl-donor substrates for transglutaminase in a human intestinal epithelial cell line. Implications for celiac disease. J Biol Chem (2003) 278(34):31766–73. doi:10.1074/jbc.M305080200
96. Verhaar R, Drukarch B, Bol JG, Jongenelen CA, Musters RJ, Wilhelmus MM. Increase in endoplasmic reticulum-associated tissue transglutaminase and enzymatic activation in a cellular model of Parkinson’s disease. Neurobiol Dis (2012) 45(3):839–50. doi:10.1016/j.nbd.2011.10.012
97. Wilhelmus MM, Verhaar R, Andringa G, Bol JG, Cras P, Shan L, et al. Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in Parkinson’s disease brain. Brain Pathol (2011) 21(2):130–9. doi:10.1111/j.1750-3639.2010.00429.x
98. Russo L, Marsella C, Nardo G, Massignan T, Alessio M, Piermarini E, et al. Transglutaminase 2 transamidation activity during first-phase insulin secretion: natural substrates in INS-1E. Acta Diabetol (2013) 50(1):61–72. doi:10.1007/s00592-012-0381-6
99. Facchiano F, Facchiano A, Facchiano AM. The role of transglutaminase-2 and its substrates in human diseases. Front Biosci (2006) 11:1758–73. doi:10.2741/1921
100. Yamaguchi H, Wang HG. Tissue transglutaminase serves as an inhibitor of apoptosis by cross-linking caspase 3 in thapsigargin-treated cells. Mol Cell Biol (2006) 26(2):569–79. doi:10.1128/MCB.26.2.569-579.2006
101. Fok JY, Mehta K. Tissue transglutaminase induces the release of apoptosis inducing factor and results in apoptotic death of pancreatic cancer cells. Apoptosis (2007) 12(8):1455–63. doi:10.1007/s10495-007-0079-3
102. Ballestar E, Abad C, Franco L. Core histones are glutaminyl substrates for tissue transglutaminase. J Biol Chem (1996) 271(31):18817–24. doi:10.1074/jbc.271.31.18817
103. Han JA, Park SC. Transglutaminase-dependent modulation of transcription factor Sp1 activity. Mol Cells (2000) 10(6):612–8. doi:10.1007/s10059-000-0612-5
104. Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol (2000) 148(4):825–38. doi:10.1083/jcb.148.4.825
105. Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J (1999) 13(13):1787–95.
106. Stephens P, Grenard P, Aeschlimann P, Langley M, Blain E, Errington R, et al. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J Cell Sci (2004) 117(Pt 15):3389–403. doi:10.1242/jcs.01188
107. Verderio EA, Johnson T, Griffin M. Tissue transglutaminase in normal and abnormal wound healing: review article. Amino Acids (2004) 26(4):387–404. doi:10.1007/s00726-004-0094-4
108. Takahara H, Tsuchida M, Kusubata M, Akutsu K, Tagami S, Sugawara K. Peptidylarginine deiminase of the mouse. Distribution, properties, and immunocytochemical localization. J Biol Chem (1989) 264(22):13361–8.
109. Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays (2003) 25(11):1106–18. doi:10.1002/bies.10357
110. Jang B, Shin HY, Choi JK, Nguyen du PT, Jeong BH, Ishigami A, et al. Subcellular localization of peptidylarginine deiminase 2 and citrullinated proteins in brains of scrapie-infected mice: nuclear localization of PAD2 and membrane fraction-enriched citrullinated proteins. J Neuropathol Exp Neurol (2011) 70(2):116–24. doi:10.1097/NEN.0b013e318207559e
111. Rogers GE, Harding HW, Llewellyn-Smith IJ. The origin of citrulline-containing proteins in the hair follicle and the chemical nature of trichohyalin, an intracellular precursor. Biochim Biophys Acta (1977) 495(1):159–75. doi:10.1016/0005-2795(77)90250-1
112. Tarcsa E, Marekov LN, Mei G, Melino G, Lee SC, Steinert PM. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J Biol Chem (1996) 271(48):30709–16. doi:10.1074/jbc.271.48.30709
113. Lee HJ, Joo M, Abdolrasulnia R, Young DG, Choi I, Ware LB, et al. Peptidylarginine deiminase 2 suppresses inhibitory {kappa}B kinase activity in lipopolysaccharide-stimulated RAW 264.7 macrophages. J Biol Chem (2010) 285(51):39655–62. doi:10.1074/jbc.M110.170290
114. Inagaki M, Takahara H, Nishi Y, Sugawara K, Sato C. Ca2+-dependent deimination-induced disassembly of intermediate filaments involves specific modification of the amino-terminal head domain. J Biol Chem (1989) 264(30):18119–27.
115. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207(9):1853–62. doi:10.1084/jem.20100239
116. Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med (1998) 4(6):713–7. doi:10.1038/nm0698-713
117. Corthay A, Backlund J, Broddefalk J, Michaelsson E, Goldschmidt TJ, Kihlberg J, et al. Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur J Immunol (1998) 28(8):2580–90. doi:10.1002/(SICI)1521-4141(199808)28:08<2580::AID-IMMU2580>3.0.CO;2-X
118. Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L, Rothbard JB. T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature (1986) 324(6094):258–60. doi:10.1038/324258a0
119. Martin R, Whitaker JN, Rhame L, Goodin RR, McFarland HF. Citrulline-containing myelin basic protein is recognized by T-cell lines derived from multiple sclerosis patients and healthy individuals. Neurology (1994) 44(1):123–9. doi:10.1212/WNL.44.1.123
120. Wallberg M, Bergquist J, Achour A, Breij E, Harris RA. Malondialdehyde modification of myelin oligodendrocyte glycoprotein leads to increased immunogenicity and encephalitogenicity. Eur J Immunol (2007) 37(7):1986–95. doi:10.1002/eji.200636912
121. van Stipdonk MJ, Willems AA, Amor S, Persoon-Deen C, Travers PJ, Boog CJ, et al. T cells discriminate between differentially phosphorylated forms of alphaB-crystallin, a major central nervous system myelin antigen. Int Immunol (1998) 10(7):943–50. doi:10.1093/intimm/10.7.943
122. Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific auto-antibodies. J Clin Invest (1998) 101(1):273–81. doi:10.1172/JCI1316
123. Masson-Bessiere C, Sebbag M, Girbal-Neuhauser E, Nogueira L, Vincent C, Senshu T, et al. The major synovial targets of the rheumatoid arthritis-specific anti-filaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J Immunol (2001) 166(6):4177–84. doi:10.4049/jimmunol.166.6.4177
124. Vossenaar ER, Despres N, Lapointe E, van der Heijden A, Lora M, Senshu T, et al. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res Ther (2004) 6(2):R142–50. doi:10.1186/ar1149
125. Vossenaar ER, Nijenhuis S, Helsen MM, van der Heijden A, Senshu T, van den Berg WB, et al. Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum (2003) 48(9):2489–500. doi:10.1002/art.11229
126. Burkhardt H, Sehnert B, Bockermann R, Engstrom A, Kalden JR, Holmdahl R. Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur J Immunol (2005) 35(5):1643–52. doi:10.1002/eji.200526000
127. Kinloch A, Tatzer V, Wait R, Peston D, Lundberg K, Donatien P, et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther (2005) 7(6):R1421–9. doi:10.1186/ar1541
128. Mamula MJ, Gee RJ, Elliott JI, Sette A, Southwood S, Jones PJ, et al. Isoaspartyl post-translational modification triggers autoimmune responses to self-proteins. J Biol Chem (1999) 274(32):22321–7. doi:10.1074/jbc.274.32.22321
129. Monneaux F, Lozano JM, Patarroyo ME, Briand JP, Muller S. T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70K snRNP protein administered in MR/lpr mice. Eur J Immunol (2003) 33(2):287–96. doi:10.1002/immu.200310002
130. Coudevylle N, Rokas D, Sakarellos-Daitsiotis M, Krikorian D, Panou-Pomonis E, Sakarellos C, et al. Phosphorylated and nonphosphorylated epitopes of the La/SSB autoantigen: comparison of their antigenic and conformational characteristics. Biopolymers (2006) 84(4):368–82. doi:10.1002/bip.20458
131. Brahms H, Raymackers J, Union A, de Keyser F, Meheus L, Luhrmann R. The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for anti-Sm autoantibodies. J Biol Chem (2000) 275(22):17122–9. doi:10.1074/jbc.M000300200
132. Abramson J, Husebye ES. Autoimmune regulator and self-tolerance – molecular and clinical aspects. Immunol Rev (2016) 271(1):127–40. doi:10.1111/imr.12419
133. Chan AY, Anderson MS. Central tolerance to self revealed by the autoimmune regulator. Ann N Y Acad Sci (2015) 1356:80–9. doi:10.1111/nyas.12960
134. Ucar O, Rattay K. Promiscuous gene expression in the thymus: a matter of epigenetics, miRNA, and more? Front Immunol (2015) 6:93. doi:10.3389/fimmu.2015.00093
135. Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol (2003) 21:139–76. doi:10.1146/annurev.immunol.21.120601.141107
136. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14(6):377–91. doi:10.1038/nri3667
137. Palmer E. Negative selection – clearing out the bad apples from the T-cell repertoire. Nat Rev Immunol (2003) 3(5):383–91. doi:10.1038/nri1085
138. Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol (2012) 24(1):112–8. doi:10.1016/j.coi.2011.12.003
139. Geenen V, Lefebvre PJ. The intrathymic expression of insulin-related genes: implications for pathophysiology and prevention of type 1 diabetes. Diabetes Metab Rev (1998) 14(1):95–103. doi:10.1002/(SICI)1099-0895(199803)14:1<95::AID-DMR200>3.0.CO;2-W
140. Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes (2002) 51(5):1383–90. doi:10.2337/diabetes.51.5.1383
141. Geenen V, Achour I, Robert F, Vandersmissen E, Sodoyez JC, Defresne MP, et al. Evidence that insulin-like growth factor 2 (IGF2) is the dominant thymic peptide of the insulin superfamily. Thymus (1993) 21(2):115–27.
142. Jolicoeur C, Hanahan D, Smith KM. T-cell tolerance toward a transgenic beta-cell antigen and transcription of endogenous pancreatic genes in thymus. Proc Natl Acad Sci U S A (1994) 91(14):6707–11. doi:10.1073/pnas.91.14.6707
143. Strollo R, Vinci C, Arshad MH, Perrett D, Tiberti C, Chiarelli F, et al. Antibodies to post-translationally modified insulin in type 1 diabetes. Diabetologia (2015) 58(12):2851–60. doi:10.1007/s00125-015-3746-x
144. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351(6274):711–4. doi:10.1126/science.aad2791
145. Wiles TA, Delong T, Baker RL, Bradley B, Barbour G, Powell RL, et al. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J Autoimmun (2017) 78:11–8. doi:10.1016/j.jaut.2016.10.007
146. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med (2017) 23(4):501–7. doi:10.1038/nm.4289
147. Ientile R, Caccamo D, Griffin M. Tissue transglutaminase and the stress response. Amino Acids (2007) 33:385–94. doi:10.1007/s00726-007-0517-0
148. Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol (2011) 490:71–92. doi:10.1016/B978-0-12-385114-7.00004-0
149. Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes (2012) 61(4):818–27. doi:10.2337/db11-1293
150. Zhong J, Rao X, Xu JF, Yang P, Wang CY. The role of endoplasmic reticulum stress in autoimmune-mediated beta-cell destruction in type 1 diabetes. Exp Diabetes Res (2012) 2012:238980. doi:10.1155/2012/238980
151. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science (2006) 313(5790):1137–40. doi:10.1126/science.1128294
152. O’Sullivan-Murphy B, Urano F. ER stress as a trigger for beta-cell dysfunction and autoimmunity in type 1 diabetes. Diabetes (2012) 61(4):780–1. doi:10.2337/db12-0091
153. Kojima S, Kuo TF, Tatsukawa H, Hirose S. Induction of cross-linking and silencing of Sp1 by transglutaminase during liver injury in ASH and NASH via different ER stress pathways. Dig Dis (2010) 28(6):715–21. doi:10.1159/000324278
154. Kuo TF, Tatsukawa H, Matsuura T, Nagatsuma K, Hirose S, Kojima S. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J Cell Physiol (2012) 227(3):1130–7. doi:10.1002/jcp.22833
155. Knip M, Veijola R, Virtanen SM, Hyoty H, Vaarala O, Akerblom HK. Environmental triggers and determinants of type 1 diabetes. Diabetes (2005) 54(Suppl 2):S125–36. doi:10.2337/diabetes.54.suppl_2.S125
156. Barnett AH, Eff C, Leslie RD, Pyke DA. Diabetes in identical twins. A study of 200 pairs. Diabetologia (1981) 20(2):87–93. doi:10.1007/BF00262007
157. Verge CF, Gianani R, Yu L, Pietropaolo M, Smith T, Jackson RA, et al. Late progression to diabetes and evidence for chronic beta-cell autoimmunity in identical twins of patients with type I diabetes. Diabetes (1995) 44(10):1176–9. doi:10.2337/diab.44.10.1176
158. Banatvala JE, Bryant J, Schernthaner G, Borkenstein M, Schober E, Brown D, et al. Coxsackie B, mumps, rubella, and cytomegalovirus specific IgM responses in patients with juvenile-onset insulin-dependent diabetes mellitus in Britain, Austria, and Australia. Lancet (1985) 1(8443):1409–12. doi:10.1016/S0140-6736(85)91843-4
159. Clements GB, Galbraith DN, Taylor KW. Coxsackie B virus infection and onset of childhood diabetes. Lancet (1995) 346(8969):221–3. doi:10.1016/S0140-6736(95)91270-3
160. Horwitz MS, Fine C, Ilic A, Sarvetnick N. Requirements for viral-mediated autoimmune diabetes: beta-cell damage and immune infiltration. J Autoimmun (2001) 16(3):211–7. doi:10.1006/jaut.2000.0486
161. Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology (2004) 329(2):381–94. doi:10.1016/j.virol.2004.06.049
162. Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes (2000) 49(5):708–11. doi:10.2337/diabetes.49.5.708
163. Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Ann N Y Acad Sci (2005) 1050:1–9. doi:10.1196/annals.1313.001
164. Campanella M, de Jong AS, Lanke KW, Melchers WJ, Willems PH, Pinton P, et al. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J Biol Chem (2004) 279(18):18440–50. doi:10.1074/jbc.M309494200
165. van Kuppeveld FJ, de Jong AS, Melchers WJ, Willems PH. Enterovirus protein 2B po(u)res out the calcium: a viral strategy to survive? Trends Microbiol (2005) 13(2):41–4. doi:10.1016/j.tim.2004.12.005
166. van Kuppeveld FJ, Hoenderop JG, Smeets RL, Willems PH, Dijkman HB, Galama JM, et al. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J (1997) 16(12):3519–32. doi:10.1093/emboj/16.12.3519
167. Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia (2008) 51(2):216–26. doi:10.1007/s00125-007-0886-7
168. Sandler S, Swenne I. Streptozotocin, but not alloxan, induces DNA repair synthesis in mouse pancreatic islets in vitro. Diabetologia (1983) 25(5):444–7. doi:10.1007/BF00282526
169. Bedoya FJ, Solano F, Lucas M. N-monomethyl-arginine and nicotinamide prevent streptozotocin-induced double strand DNA break formation in pancreatic rat islets. Experientia (1996) 52(4):344–7. doi:10.1007/BF01919538
170. Heikkila RE, Winston B, Cohen G. Alloxan-induced diabetes-evidence for hydroxyl radical as a cytotoxic intermediate. Biochem Pharmacol (1976) 25(9):1085–92. doi:10.1016/0006-2952(76)90502-5
171. Takasu N, Komiya I, Asawa T, Nagasawa Y, Yamada T. Streptozocin- and alloxan-induced H2O2 generation and DNA fragmentation in pancreatic islets. H2O2 as mediator for DNA fragmentation. Diabetes (1991) 40(9):1141–5. doi:10.2337/diabetes.40.9.1141
172. Kim HR, Rho HW, Park BH, Park JW, Kim JS, Kim UH, et al. Role of Ca2+ in alloxan-induced pancreatic beta-cell damage. Biochim Biophys Acta (1994) 1227(1–2):87–91. doi:10.1016/0925-4439(94)90111-2
173. Park BH, Rho HW, Park JW, Cho CG, Kim JS, Chung HT, et al. Protective mechanism of glucose against alloxan-induced pancreatic beta-cell damage. Biochem Biophys Res Commun (1995) 210(1):1–6. doi:10.1006/bbrc.1995.1619
174. Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J (1972) 128(3):617–30. doi:10.1042/bj1280617
175. Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep (1997) 17(1):3–8. doi:10.1023/A:1027374931887
176. Johnson FB, Sinclair DA, Guarente L. Molecular biology of aging. Cell (1999) 96(2):291–302. doi:10.1016/S0092-8674(00)80567-X
177. Yoon JW, Onodera T, Notkins AL. Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with Coxsackie virus B4. J Exp Med (1978) 148(4):1068–80. doi:10.1084/jem.148.4.1068
178. Xie B, Zhou JF, Lu Q, Li CJ, Chen P. Oxidative stress in patients with acute Coxsackie virus myocarditis. Biomed Environ Sci (2002) 15(1):48–57.
179. Peterhans E, Grob M, Burge T, Zanoni R. Virus-induced formation of reactive oxygen intermediates in phagocytic cells. Free Radic Res Commun (1987) 3(1–5):39–46. doi:10.3109/10715768709069768
180. Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans (2008) 36(Pt 3):343–7. doi:10.1042/BST0360343
181. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med (1996) 20(3):463–6. doi:10.1016/0891-5849(96)02051-5
182. Grankvist K, Marklund SL, Taljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J (1981) 199(2):393–8. doi:10.1042/bj1990393
183. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature (2000) 404(6779):787–90. doi:10.1038/35008121
184. Welsh N, Hellerstrom C. In vitro restoration of insulin production in islets from adult rats treated neonatally with streptozotocin. Endocrinology (1990) 126(4):1842–8. doi:10.1210/endo-126-4-1842
185. Chen J, Gusdon AM, Thayer TC, Mathews CE. Role of increased ROS dissipation in prevention of T1D. Ann N Y Acad Sci (2008) 1150:157–66. doi:10.1196/annals.1447.045
186. Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal (2010) 12(4):537–77. doi:10.1089/ars.2009.2531
187. West IC. Radicals and oxidative stress in diabetes. Diabet Med (2000) 17(3):171–80. doi:10.1046/j.1464-5491.2000.00259.x
188. Horio F, Fukuda M, Katoh H, Petruzzelli M, Yano N, Rittershaus C, et al. Reactive oxygen intermediates in autoimmune islet cell destruction of the NOD mouse induced by peritoneal exudate cells (rich in macrophages) but not T cells. Diabetologia (1994) 37(1):22–31. doi:10.1007/BF00428773
189. Nerup J, Mandrup-Poulsen T, Molvig J, Helqvist S, Wogensen L, Egeberg J. Mechanisms of pancreatic beta-cell destruction in type I diabetes. Diabetes Care (1988) 11(Suppl 1):16–23.
190. Suarez-Pinzon WL, Szabo C, Rabinovitch A. Development of autoimmune diabetes in NOD mice is associated with the formation of peroxynitrite in pancreatic islet beta-cells. Diabetes (1997) 46(5):907–11. doi:10.2337/diab.46.5.907
191. Santini SA, Marra G, Giardina B, Cotroneo P, Mordente A, Martorana GE, et al. Defective plasma antioxidant defenses and enhanced susceptibility to lipid peroxidation in uncomplicated IDDM. Diabetes (1997) 46(11):1853–8. doi:10.2337/diab.46.11.1853
192. Favero TG, Zable AC, Abramson JJ. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J Biol Chem (1995) 270(43):25557–63. doi:10.1074/jbc.270.43.25557
193. Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal (2006) 8(9–10):1391–418. doi:10.1089/ars.2006.8.1391
194. Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science (1998) 279(5348):234–7. doi:10.1126/science.279.5348.234
195. Schulte BM, Lanke KH, Piganelli JD, Kers-Rebel ED, Bottino R, Trucco M, et al. Cytokine and chemokine production by human pancreatic islets upon enterovirus infection. Diabetes (2012) 61(8):2030–6. doi:10.2337/db11-1547
196. Bottino R, Balamurugan AN, Tse H, Thirunavukkarasu C, Ge X, Profozich J, et al. Response of human islets to isolation stress and the effect of antioxidant treatment. Diabetes (2004) 53:2559–68. doi:10.2337/diabetes.53.10.2559
197. Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes (2005) 54(Suppl 2):S97–107. doi:10.2337/diabetes.54.suppl_2.S97
198. Lee H, Park MT, Choi BH, Oh ET, Song MJ, Lee J, et al. Endoplasmic reticulum stress-induced JNK activation is a critical event leading to mitochondria-mediated cell death caused by beta-lapachone treatment. PLoS One (2011) 6(6):e21533. doi:10.1371/journal.pone.0021533
199. Wang Q, Zhang H, Zhao B, Fei H. IL-1beta caused pancreatic beta-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Mol Cell Biochem (2009) 324(1–2):183–90. doi:10.1007/s11010-008-9997-9
200. Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes (2005) 54(2):452–61. doi:10.2337/diabetes.54.2.452
201. Haskins K, Portas M, Bradley B, Wegmann D, Lafferty K. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes (1988) 37(10):1444–8. doi:10.2337/diab.37.10.1444
202. Batinic-Haberle I, Spasojevic I, Tse HM, Tovmasyan A, Rajic Z, Clair DK, et al. Erratum to: design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids (2012) 42(1):95–113. doi:10.1007/s00726-010-0603-6
203. Milton MJ, Poulin M, Mathews C, Piganelli JD. Generation, maintenance, and adoptive transfer of diabetogenic T-cell lines/clones from the nonobese diabetic mouse. Methods Mol Med (2004) 102:213–25. doi:10.1385/1-59259-805-6:213
204. Delmastro MM, Styche AJ, Trucco MM, Workman CJ, Vignali DA, Piganelli JD. Modulation of redox balance leaves murine diabetogenic TH1 T cells “LAG-3-ing” behind. Diabetes (2012) 61(7):1760–8. doi:10.2337/db11-1591
205. Previte DM, O’Connor EC, Novak EA, Martins CP, Mollen KP, Piganelli JD. Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS One (2017) 12(4):e0175549. doi:10.1371/journal.pone.0175549
206. Sklavos MM, Coudriet GM, Delmastro M, Bertera S, Coneybeer JT, He J, et al. Administration of a negative vaccination induces hyporesponsiveness to islet allografts. Cell Transplant (2013) 22(7):1147–55. doi:10.3727/096368912X657233
207. Sklavos MM, Tse HM, Piganelli JD. Redox modulation inhibits CD8 T cell effector function. Free Radic Biol Med (2008) 45:1477–86. doi:10.1016/j.freeradbiomed.2008.08.023
Keywords: type 1 diabetes, β cell, environmental factors, endoplasmic reticulum stress, posttranslation modification, neo-antigen, autoimmunity
Citation: Marré ML and Piganelli JD (2017) Environmental Factors Contribute to β Cell Endoplasmic Reticulum Stress and Neo-Antigen Formation in Type 1 Diabetes. Front. Endocrinol. 8:262. doi: 10.3389/fendo.2017.00262
Received: 09 August 2017; Accepted: 20 September 2017;
Published: 29 September 2017
Edited by:
Marc S. Horwitz, University of British Columbia, CanadaReviewed by:
Margaret A. Morris, Eastern Virginia Medical School, United StatesDavid H. Wagner, University of Colorado Denver, United States
Copyright: © 2017 Marré and Piganelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jon D. Piganelli, amRwNTFAcGl0dC5lZHU=