 Karin Scheschowitsch
Karin Scheschowitsch Jacqueline Alves Leite
Jacqueline Alves Leite Jamil Assreuy
Jamil Assreuy
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 06 February 2017
Sec. Cellular Endocrinology
Volume 8 - 2017 | https://doi.org/10.3389/fendo.2017.00016
This article is part of the Research Topic Updates and new concepts in regulation of pro-inflammatory gene expression by steroid hormones View all 8 articles
The clinical use of classical glucocorticoids (GC) is narrowed by the many side effects it causes and the resistance to GC observed in some diseases. Since the great majority of GC effects depend on the activation of a glucocorticoid receptor (GR), many research groups had focused to better understand the signaling pathways involving those receptors. Transgenic animal models and genetic modifications of the receptor brought a huge insight into GR mechanisms of action. This in turn opened a new window for the search of selective GR modulators that ideally may have agonistic and antagonistic combined effects and activate one specific signaling pathway, inducing mostly transrepression or transactivation mechanisms. Another important research field concerns to posttranslational modifications that affect the GR and consequently also affect its signaling and function. In this mini review, we discuss many of those aspects of GR signaling, as well as findings like the ligand-independent activation of GR, which add another layer of complexity in GR signaling pathways. Although several recent data have been added to the GR field, much work has yet to be done, especially to find out the biological relevance of those alternative GR signaling pathways. Improving the knowledge about alternative GR signaling pathways and understanding how these pathways intercommunicate and in which situations they are relevant might help to develop new strategies to take benefit of it and to improve GC or other compounds efficacy causing minimal side effects.
Glucocorticoids (cortisol in humans and corticosterone in rodents) are steroid hormones [revised in Ref. (1)] involved in several physiological functions and in controlling inflammation (2–5). From their discovery (6, 7) until the present day, GC are considered the most effective anti-inflammatory drugs and one of the most widely prescribed drug classes worldwide (8–12).
Despite their potent anti-inflammatory effects, steroids cause relevant side effects when used for longer periods and at high doses (13–16), limiting their use and reducing adherence to treatment. Therefore, understanding the signaling mechanisms and pathways related to GC and their receptor [glucocorticoid receptor (GR)] is essential to provide the basis for the development of new selective glucocorticoid receptor modulators (SEGRMs) (17, 18). SEGRMs are expected to present the same or better efficacy compared to classical steroids, but causing minimal side effects (8, 14, 19–21). The present mini review will discuss advances in GR signaling pathways looking for a better comprehension about the beneficial and harmful effects of endogenous and exogenous GC, especially in inflammation.
The main actions of GC occur through the activation of GRs (NR3C1), which are transcription factors (TFs) belonging to the superfamily of nuclear receptors and are usually activated by ligands (6, 14, 22). GR is composed of three major functional domains, namely the N-terminal transactivation domain (NTD), the central DNA-binding domain (DBD), and the C-terminal ligand-binding domain (LBD) (23, 24). In the absence of ligands, GR is predominantly found in the cytoplasm complexed with accessory proteins (hsp90, hsp70, hsp56, p23, and immunophilins) and is kept in a conformation of high-binding affinity to GC (25, 26) (see Figure 1: 1). GR can actively shuttle between cytoplasm and nucleus, being the balance rate of nuclear import and export which determines the receptor cellular location (27–29). Increases in receptor density affect its conformation and location and may cause ligand-free dimerization that facilitates the subsequent binding of ligands, thus bypassing dimerization-dependent mechanisms of action (30).
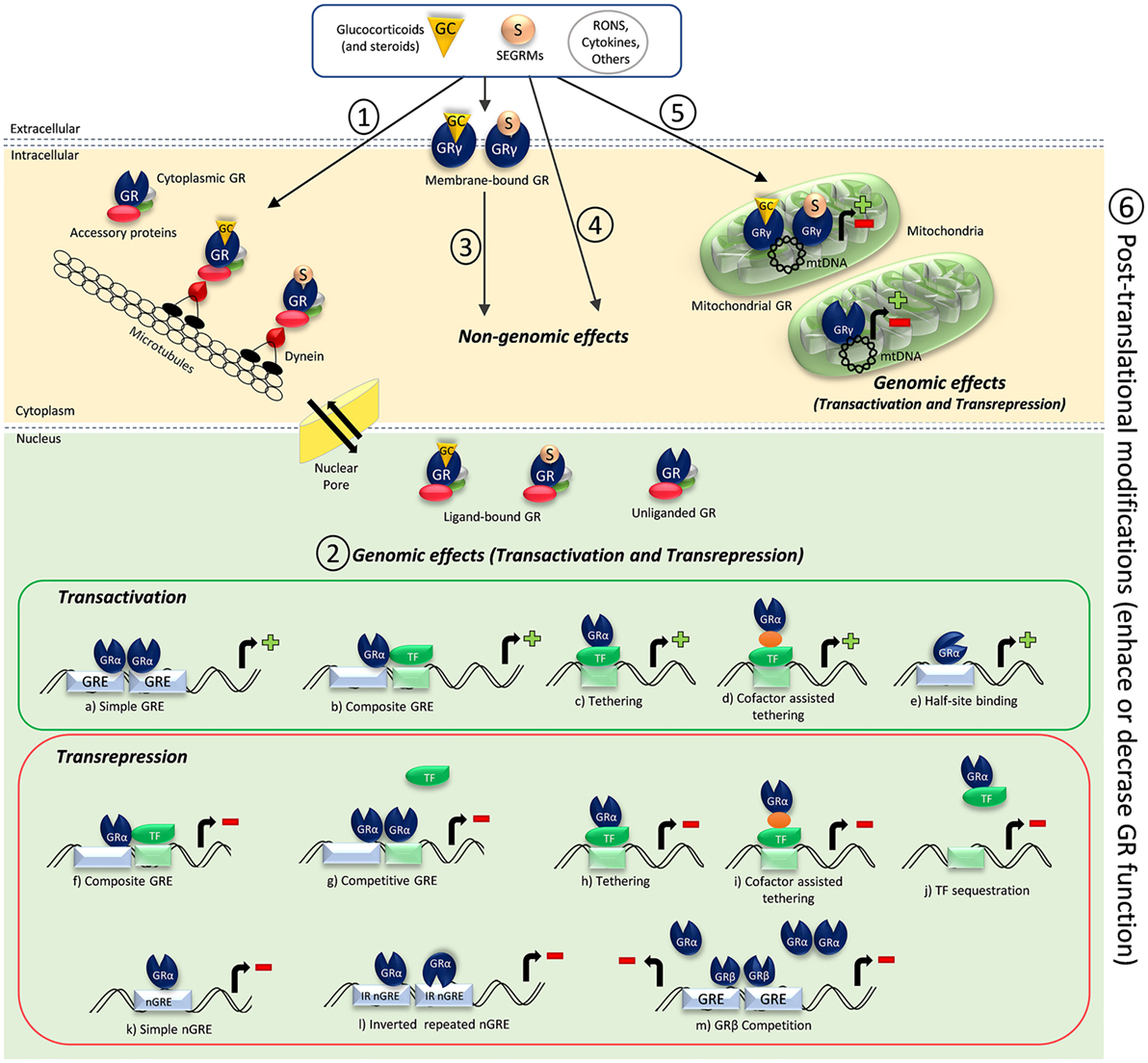
Figure 1. Schematic illustration of glucocorticoid receptor (GR) activation and GR-mediated mechanisms of action. (1) Cytoplasmic GR resides in the cytoplasm complexed with accessory proteins and present high affinity to ligands. Once ligands like glucocorticoids and other steroids or selective glucocorticoid receptor modulators (SEGRMs) bind to the cytoplasmic GR, the GR complex interacts with dynein and is transported along microtubules to a nuclear pore. Interaction with importins and nucleoporins of the nuclear pore allow the GR complex to enter the nucleus, dissociate from chaperones, and induce genomic effects. Dissociated chaperones and GR constantly shuttle between the nucleus and the cytoplasm through the nuclear pore. Reactive oxygen and nitrogen species (RONS), some cytokines, other substances, and conditions like shear stress can induce unliganded GR nuclear translocation, which seem to be cytoskeleton independent. However, unliganded GR nuclear translocation is still not completely understood. (2) Ligand-bound GR, and sometimes unliganded GR, can induce genomic effects through direct or indirect transactivation or transrepression mechanisms. GRα homodimers binding to glucocorticoid-responsive elements (GRE) (A), monomeric GRα DNA binding in a concerted manner with another transcription factor (TF) (B), direct (C) or indirect (D) binding of GRα onto a TF, and recently demonstrated monomeric GRα half-site binding (E) can result in promoter activation and gene expression. GR-negative regulation of gene transcription can occur by monomeric GRα DNA-binding crosstalk with another TF (F), GRα homodimers competition for an overlapping binding site (G), direct (H) or indirect (I) binding of GRα onto a TF, sequestration of a DNA-bound TF (J), direct binding of monomeric GRα onto a negative GRE (nGRE) (K), two monomeric GRα binding with inverted polarities to inverted repeated nGREs (L), or GRβ competition for an overlapping GRE, impairing GRα binding (M). (3) Ligands and other substances also can bind and interact with membrane-bound GR (claimed to be a GRγ isoform), causing fast non-genomic effects. (4) Ligands, particularly steroids in high concentrations, can induce non-genomic effects through GR-independent mechanisms of action. (5) Ligands and other substances can bind to mitochondrial GR, which is also suggested to be a GRγ isoform. Ligand-bound and unliganded mitochondrial GR induce genomic effects when bound to the mitochondrial DNA (mtDNA), and those effects are important to regulate mitochondrial functions and energy metabolism. (6) Posttranslational modifications can affect GR activation and function in all stages, enhancing or decreasing its function.
One of the first proposed signaling pathways for GR was that the binding of a ligand to the LBD of monomeric GR would induce its nuclear localization sequence (NLS) exposure. Then, accessory proteins would dissociate from the monomeric receptor, allowing it to dimerize and translocate along microtubules to the nucleus. There, GR dimers would bind to DNA through their DBD to exert their effects (27, 31–33).
Further evidence showed that ligand binding triggers NLS exposure due to the replacement of immunophilin FKBP51 by FKBP52, which interacts with dynein, carrying the whole monomeric GR complex along microtubules to a nuclear pore. FKBP52 also facilitates the entry of GR monomeric complex into the nucleus, as well as exporting the accessory proteins back to the cytoplasm via importins (25, 34–37) (see Figure 1: 1). Once in the nucleus, monomeric GR can assume different conformations depending on the glucocorticoid-responsive elements (GREs). GR monomer can recruit another monomer to form a GR homodimer on DNA, through distinct hydrophobic motifs of the LBD (38). Nevertheless, the subcellular compartment for GR dimerization is still in debate, and the GR binding regions used by specific GREs are still unclear (39). Therefore, more studies aiming to create predictive models for GR activity may help the development of new compounds.
Regarding GR nuclear translocation, it is suggested that different ligands can induce the exposure of one of the two so far described NLS for GR, influencing its nuclear translocation speed (27, 28, 34, 40). Classical GR ligands are suggested to induce NLS-1 exposure, which interacts with importins and nucleoporins, leading to rapid nuclear translocation (within 4–6 min). Shuttling of unliganded GR depends on NLS-1 interaction with importin-alpha. On the other hand, NLS-2 exposure is strictly ligand dependent and mediates slower GR nuclear translocation (45 min–1 h) (28, 36, 41).
Since GR was cloned (42), the development of animals and cells with full or partial GR depletion and with different GR mutations allowed the study of the role of GR. The first GR knockout mice (GRnull) presented severe lung developmental abnormalities and died shortly after birth (43). On the other hand, animals that overexpress GR are resistant to septic shock (44). Organ-specific GR depletion have shown that (a) hepatic GR is responsible for gluconeogenesis and production of postnatal insulin-like growth factor-1 (45, 46); (b) GR depletion in the central nervous system leads to numerous behavioral abnormalities (47); (c) mice with specific depletion of GR in macrophages, neutrophils (48–50), or endothelial cells (51) are more sensitive to pro-inflammatory stimuli, evidencing the important role for GR in these cells for the physiological control of inflammation.
Development of a mutation in the second zinc finger of DBD (52) (called GRdim) prevented GR homodimerization on most tandem GREs (53). Notwithstanding that this mutation strongly attenuates GR dimerization and impairs GR transcription activity from tandem GREs (21, 54, 55), it does not completely abrogate transactivation mechanisms, since it was recently discovered that it depends on promoter contexts (56). Furthermore, DNA motifs specify the genomic occupancy of monomeric GR and interfere with the availability of GR dimers binding sites (39). However, unlike GRnull mice, GRdim animals are viable and normal with respect to the major physiological GR-mediated functions (54), although more susceptible to inflammation (57). In a model of antigen-induced arthritis using GRdim mice, it was shown that GR dimerization is necessary for the anti-inflammatory effects of GC by suppressing Th1 and Th17 cells activity (58). Considering other GR mutations, it was observed that (a) heterozygous GR knockout mice, in which GR expression is reduced by half, were less sensitive to dexamethasone therapy in experimental autoimmune encephalomyelitis (59); (b) mice with selective GR depletion in T cells (GRlck-Cre) succumb to toxoplasma infection due to increased TNF and IFN-γ production by Th1 cells (60); and (c) mice with selective GR depletion in the thymocytes showed loss of the adaptive immune response and were immunocompromised (61).
Glucocorticoid effects usually depend on its interaction with GR in the cytoplasm to trigger a variety of cell responses that culminate in several changes in the whole body (62). Although most cells have similar GC response machineries, steroids exhibit varied effects in different tissues (63). This might be attributed to GR ubiquitous expression (except in red blood cells), to the numerous mechanisms that alter its function, and to the enormous assortment of GR-binding sites and their availability. Binding site availability depends on chromatin state, which is specific for each tissue and cell type. Also, GC concentration may determine which GREs are occupied by GR. Therefore, chromatin accessibility and GREs distinct sensitivity also help to explain why GC trigger different effects in different tissues (33). By using different chromatin immunoprecipitation sequencing, it was shown that C/EBPβ maintains chromatin accessibility to facilitate selective GR binding to GREs in liver tissue (64), another evidence of cell type-specific GR-induced transcription. Intracellular GR location, interaction with other proteins, binding capacity and sensitivity to GC and other modulators, receptor degradation rate, and intracellular density directly interfere with GC efficacy. Finally, the effects triggered by the GC-GR complex are tissue and cell type dependent and vary with the posttranslational modifications that will affect the receptor according to the host condition (19, 55).
The classical mechanism of action of nuclear receptors involves gene transcription (transactivation) or direct or indirect impairment of transcription of other TFs (transrepression) (Figure 1: 2). Currently, the most accepted transactivation model involves GR dimerization after binding of GC-GR monomeric complexes to GREs (see Figure 1A). This induces GR conformational changes to recruit cofactors, like histone acetyl transferases and C/EBPβ, that change the chromatin state facilitating gene expression (64–66). The type of dimer that will be formed and which cofactors will be recruited seem to be determined by the gene sequence in question (67, 68). Interestingly, it was recently shown that monomeric GR interacts with a half-site motif and drives transcription (see Figure 1E) in liver and primary macrophages, being more prevalent than homodimer binding under physiological conditions and being essential for those tissue-specific functions. Exogenous GC appear to favor gene expression by the binding of GR homodimers and disruption of GR monomeric binding from their half-site motifs (53). This discovery calls for a revision of transactivation mechanisms and other possible transrepression mechanisms involving monomeric GR binding to half-sites (Figures 1B–E).
Protein–protein interaction of GR with other TFs is an important mechanism of direct transrepression known as tethering mechanism. Monomeric GR can crosstalk with another TF (Figure 1F) or directly interact with critical points of TFs before they bind to its responsive element on DNA (Figure 1J). In this case, best exemplified for the nuclear transcription factor kappa-B (NF-κB), interaction may occur in the cytoplasm, preventing NF-κB activation and its nuclear translocation, or in the nucleus, impairing DNA NF-κB-binding capacity (62, 69). Nonetheless, GR monomers can also repress TFs even if they are already bound to DNA (70) (see Figures 1H,I) and can compete for an overlapping binding site (Figure 1G). Therefore, GR can regulate the expression of different TFs such as NF-κB, AP-1, STATs, Oct, NF-1, C/EBP, COUP-TFII, PPARs, and LXR (71–77).
Glucocorticoid receptor binding to DNA can also suppress gene expression by interacting with novel described negative GREs (nGREs). Recently, an extensive conserved family of negative palindromic GREs (IR nGREs) was found to form a repressing complex through association of SMRT/NCoR corepressors and HDACs, mediating transrepression by the direct binding of agonist-bound GR (78). Evidence showed that two GR monomers can bind to nGREs with inverted polarity if compared on how they bind to GREs (Figure 1L). This kind of monomer binding induce a unique conformational change that, together to the recruitment of corepressors, guarantee the presence of single monomers of GR bound to nGREs (79). As nGREs are near to responsive elements of other TFs, that binding can allosterically prevent the binding of other factors to DNA and recruit corepressors (62, 78) (see Figure 1K).
Indirect modes of reducing gene expression involve GC-induced expression of inhibitory proteins such as glucocorticoid-induced leucine zipper (GILZ) protein via modulation of MAPK pathways (16, 80). GILZ protein binds to the p65 subunit of NF-κB in T-cells and macrophages, impairing gene transcription and suppressing inflammation (81, 82). In addition, Lethe, a pseudogene non-coding RNAs induced by cytokines (IL-1β, TNF-α) and GC, might be involved in the negative feedback NF-κB signaling to control inflammation (83).
Since it was believed that GC side effects occur through transactivation mechanism, researchers became interested in identifying SGRMs to enhance GC therapeutic effects with fewer side effects (20, 21, 49, 84). Compound A (CpdA) is a non-steroidal ligand analog that binds GR with high affinity and induces its nuclear translocation. However, it mostly triggers transrepression tethering mechanisms with NF-κB (20, 85, 86) and/or binding to nGREs (79), while inhibiting GR dimerization and preventing dimerization-dependent side effects such as hyperglycemia (79, 85). GR unique conformation upon CpdA binding may impair receptor phosphorylation, which is the suggested mechanism why CpdA does not induce GC resistance and maintain its efficacy even after long periods of administration (86). Unfortunately, CpdA also present a limited therapeutic window due to its toxic effects in higher concentrations (84). In vitro studies with CpdX, a novel SEGRM, demonstrated that it is efficient in decreasing inflammation through tethering mechanism, albeit not inducing GRE transactivation and IR nGRE transrepression (87). In vivo studies are necessary to reveal the clinical value of CpdX. Compound C108297 presents agonistic and antagonistic properties in the rat brain (88) and can simultaneously prevent diet-induced obesity and inflammation (89). Although promising, it is still unclear whether C108297 is an antagonist or a partial agonist and if it induces a unique conformational of the GR-LBD or a two-state agonist conformation (88). Therefore, more studies are needed to better understand the signaling pathways involved in C108297 effects. Moreover, since new evidence suggests that those agonists or modulators do not necessarily need to be a GR ligand, SEGRMs concept is still in debate (90).
There are two major GR isoforms that differ only in their C-terminal regions, GRα and GRβ (subtypes of each isoform and other isoforms will not be discussed due to space limitation). The DBD is conserved across the nuclear receptor family and consists of two zinc fingers motifs important to GR dimerization and tandem GRE binding. GRα is the classic receptor responsible for GC actions, whereas GRβ does not bind GC and its biological relevance is still uncertain. It has been suggested that GRβ acts as a negative regulator of GRα isoform (see Figure 1M), because it can bind to GREs but does not induce its transcription (91–94). GRβ has an intrinsic transcriptional activity in non-GC-regulated genes (95). Recently, a pro-inflammatory role for GRβ was shown in the liver tissue of obese mice, suggesting that steatosis correlates to GRβ increased expression in adipose and liver tissues. The same study showed that GRβ antagonizes GC-induced signaling through GRα during fasting (96). So far, this is the first study showing a pro-inflammatory role for GRβ. More studies should focus in unveiling the biological relevance of GRβ in other tissues and cofactors of its singular signaling pathway.
Several posttranslational modifications also play an important role in enhancing or decreasing GR functionality to confer distinct biological functions (see Figure 1: 6). Examples include phosphorylation, acetylation, ubiquitination, methylation, nitrosylation, nitration, and SUMOylation [revised in Ref. (19, 97)]. Due to space limitations, we will focus on recent data about some of them. Nitrosylation of specific cysteine residues decreases GR binding capacity (98, 99) and increases resistance to GC action (100, 101), while tyrosine nitration residues favor nuclear translocation and receptor activity (102). However, exogenous NO seems to activate the endothelial cell GR (103). NO effects very much depend on its concentration and compartmentalization, and this is probably the reason for divergent results. Regarding to SUMOylation, it is related to stabilization, location, and transcriptional activity of GR, typically increasing it (104). However, recent reports demonstrated that SUMOylation is mandatory for GC-dependent transrepression mediated by IR nGREs. SUMOylation of GR lysine residues (mouse: K310 and human: K293) within the NTD is essential for the assembly of the repressive complex SMRT/NCoR-1-HDAC3 (105). The same SUMOylation site is essential for the tethering transrepression mechanism mediated by NF-κB/AP-1, which needs the formation of a GR small ubiquitin-related modifiers (SUMOs)–SMRT/NCoR1-HDAC3 repressing complex (87).
Beyond the consensus that steroidal or analog binding induces GR nuclear translocation, several evidences have shown that GR can be activated in the absence of ligands (30, 106–110). Sodium arsenite and dinitrophenol (31), some conditions such as elevated pH and temperature, and shear stress (111) can induce GR nuclear translocation in a ligand-independent manner (31, 111, 112) (Figure 1: 1). Interestingly, GR nuclear translocation induced by shear stress does not depend on ligands or intact cytoskeleton (113), but it is related to the nuclear lamina (114).
Unliganded GR acts as a positive regulator of the tumor suppressor gene BRCA1. This beneficial effect is lost upon addition of ligand, suggesting unliganded GR displacement from BRCA1 promoter in response to steroids decreases BRCA1 expression and increases the risk for breast cancer (109). That fact might be related to endogenous monomeric GR binding to half-site motifs, as already discussed. Transient transfection and GR overexpression in GR-deficient cells (COS-1) induced nuclear GR dimerization, GR binding to DNA, and transcription in the absence of exogenous GC (30) (see Figures 1: 1 and 2). Moreover, TNF-α can induce ligand-independent activation of GR pathways in COS-1 and epithelial cells, leading to decreased levels of IL-6 and IL-8 (108). However, the mechanisms of ligand-independent GR activation are not completely understood. One report suggests that GR phosphorylation at Ser-211 is enough for ligand-independent activation (107), whereas another report suggests that phosphorylation at Ser-134 also can induce ligand-independent GR nuclear translocation as part of the cellular stress pathway (115), but other mechanisms might be involved.
Several GC effects occur within a few seconds or minutes after administration, evidencing a non-genomic mechanism of action. This mechanism is particularly important in the vascular system and in dampening inflammation (62, 116) and usually involves activation of non-cytoplasmic GR or even GR-independent pathways (see Figures 1: 3 and 4). Nevertheless, activation of cytoplasmic GR cannot be excluded (33).
Membrane-bound receptors (mGR) have distinct properties if compared to cytoplasmic GR and are more related to intracellular signaling pathways mediated by G-protein-coupled receptors (117, 118). Although some studies suggest that mGR is a GRα isoform (117), others claim that it is in fact a GRγ isoform (119) (Figure 1: 3). Importantly, high doses of GC can act independently of GR by increasing second messengers, such as inositol-3-phosphate, cyclic adenosine monophosphate, and calcium ion (8, 20, 120, 121) (Figure 1: 4). The presence of GR in human mitochondria (122) and similar GRE sequences in mitochondrial DNA (123) were identified long time ago. Moreover, direct GC-stimulated mitochondrial transcription mediated by mitochondrial GR was also demonstrated in hepatocarcinoma cells (124). It was recently suggested that GRγ isoform resides in mitochondria and is related to cell energy metabolism regulation in a ligand-independent manner (119) (see Figure 1: 5).
Protein–protein interaction between ligand-activated GR and the regulatory subunit (p85α) of phosphoinositol-3-kinase in endothelial cells activates the protein kinase Akt, which phosphorylates and activates NO synthase-3. This mechanism would explain the rapid and transient cardiovascular protective effect of high GC doses in myocardial emergencies. A rapid decrease in peripheral resistance and blood pressure is observed minutes after GC administration, accompanied by an increase in coronary and cerebral blood flows (116). Since rapid vasoconstriction and changes in bronchial blood flow are observed few seconds after inhaled GC administration in asthmatic attacks, it suggests that non-genomic effects also may vary according to the tissue (125). Furthermore, endothelial cell stimulation with dexamethasone rapidly activates ERK and kinase c-Jun N-terminal MAPK to produce reactive oxygen species and activate TFs (126).
In summary, recent data have added new layers of complexity to GR signaling pathways. Clearly GR signaling does not depend only on ligand binding, and its classical mechanisms of action need further revision. Given the physiological and clinical importance of GC and their side effects, it is essential to further investigate alternative GR signaling pathways and their respective biological relevance. This might help to develop new strategies to take benefit of it as well as to improve GC or analogs efficacy with minimal side effects.
KS and JA discussed the structure of the manuscript. KS wrote the initial draft and created the figure. KS and JL worked to finish the revised manuscript, and JA did the final review prior to submission. KS, JL, and JA exchanged several revisions until the final manuscript was agreed upon all the authors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study received financial support from CNPq, CAPES, FINEP, FAPESP, and FAPESC.
1. Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM. Selective glucocorticoid receptor modulation: new directions with non-steroidal scaffolds. Pharmacol Ther (2015) 152:28–41. doi:10.1016/j.pharmthera.2015.05.001
2. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev (2000) 21:55–89. doi:10.1210/er.21.1.55
3. Meir Drexler S, Wolf OT. The role of glucocorticoids in emotional memory reconsolidation. Neurobiol Learn Mem (2016). doi:10.1016/j.nlm.2016.11.008
4. Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol (2010) 120:69–75. doi:10.1016/j.jsbmb.2010.03.058
5. Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci (2012) 1261:55–63. doi:10.1111/j.1749-6632.2012.06633.x
6. Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord (2007) 8:321–30. doi:10.1007/s11154-007-9059-8
7. Erhuma AM. Glucocorticoids: biochemical group that play key role in fetal programming of adult disease. In: Qian X, editor. Glucocorticoids – New Recognition of Our Familiar Friend. Rijeka: InTech (2012). p. 449–78.
8. De Bosscher K, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol (2009) 23:281–91. doi:10.1210/me.2008-0283
9. Bijlsma JWJ, Jacobs JWG, Buttgereit F. Glucocorticoids in the treatment of rheumatoid arthritis. Clin Exp Rheumatol (2015) 33(4 Suppl 92):S34–6.
10. Dibas A, Yorio T. Glucocorticoid therapy and ocular hypertension. Eur J Pharmacol (2016) 787:57–71. doi:10.1016/j.ejphar.2016.06.018
11. Hakim A, Usmani OS. Enhancing nuclear translocation: perspectives in inhaled corticosteroid therapy. Ther Deliv (2015) 6:443–51. doi:10.4155/tde.15.10
12. Normansell R, Kew KM, Mansour G. Different oral corticosteroid regimens for acute asthma. Cochrane Database Syst Rev (2016) (5):CD011801. doi:10.1002/14651858.CD011801.pub2
13. Ortsäter H, Sjöholm Å, Rafacho A. Regulation of glucocorticoid receptor signaling and the diabetogenic effects of glucocorticoid excess. State of the Art of Therapeutic Endocrinology. Rijeka: InTech (2012). p. 1–28.
14. Beck IM, De Bosscher K, Haegeman G. Glucocorticoid receptor mutants: man-made tools for functional research. Trends Endocrinol Metab (2011) 22:295–310. doi:10.1016/j.tem.2011.03.009
15. Huscher D, Thiele K, Gromnica-Ihle E, Hein G, Demary W, Dreher R, et al. Dose-related patterns of glucocorticoid-induced side effects. Ann Rheum Dis (2009) 68:1119–24. doi:10.1136/ard.2008.092163
16. Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J (2009) 23:3649–58. doi:10.1096/fj.09-134684
17. Rosen J, Miner JN. The search for safer glucocorticoid receptor ligands. Endocr Rev (2005) 26:452–64. doi:10.1210/er.2005-0002
18. De Bosscher K. Selective glucocorticoid receptor modulators. J Steroid Biochem Mol Biol (2010) 120:96–104. doi:10.1016/j.jsbmb.2010.02.027
19. Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol (2006) 102:11–21. doi:10.1016/j.jsbmb.2006.09.009
20. Löwenberg M, Stahn C, Hommes DW, Buttgereit F. Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids (2008) 73:1025–9. doi:10.1016/j.steroids.2007.12.002
21. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology (2013) 154:993–1007. doi:10.1210/en.2012-2045
22. Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev (2004) 125:697–706. doi:10.1016/j.mad.2004.06.010
23. Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol (2005) 94:383–94. doi:10.1016/j.jsbmb.2004.12.046
24. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids (2010) 75:1–12. doi:10.1016/j.steroids.2009.09.002
25. Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic (2012) 13:364–74. doi:10.1111/j.1600-0854.2011.01288.x
26. Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell (2014) 157:1685–97. doi:10.1016/j.cell.2014.04.038
27. Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Haché RJ, et al. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol (1999) 19:1025–37. doi:10.1128/MCB.19.2.1025
28. Haché RJ, Tse R, Reich T, Savory JG, Lefebvre YA. Nucleocytoplasmic trafficking of steroid-free glucocorticoid receptor. J Biol Chem (1999) 274:1432–9. doi:10.1074/jbc.274.3.1432
29. Carrigan A, Walther RF, Salem HA, Wu D, Atlas E, Lefebvre YA, et al. An active nuclear retention signal in the glucocorticoid receptor functions as a strong inducer of transcriptional activation. J Biol Chem (2007) 282:10963–71. doi:10.1074/jbc.M602931200
30. Robertson S, Rohwer JM, Hapgood JP, Louw A. Impact of glucocorticoid receptor density on ligand-independent dimerization, cooperative ligand-binding and basal priming of transactivation: a cell culture model. PLoS One (2013) 8:e64831. doi:10.1371/journal.pone.0064831
31. Sanchez ER. Heat shock induces translocation to the nucleus of the unliganded glucocorticoid receptor. J Biol Chem (1992) 267:17–20.
32. Robertson S, Hapgood JP, Louw A. Glucocorticoid receptor concentration and the ability to dimerize influence nuclear translocation and distribution. Steroids (2013) 78:182–94. doi:10.1016/j.steroids.2012.10.016
33. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol (2013) 132:1033–44. doi:10.1016/j.jaci.2013.09.007
34. Davies TH, Ning YM, Sánchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem (2002) 277:4597–600. doi:10.1074/jbc.C100531200
35. Storer CL, Dickey CA, Galigniana MD, Rein T, Cox MB. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol Metab (2011) 22:481–90. doi:10.1016/j.tem.2011.08.001
36. Echeverría PC, Mazaira G, Erlejman A, Gomez-Sanchez C, Piwien Pilipuk G, Galigniana MD. Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol Cell Biol (2009) 29:4788–97. doi:10.1128/MCB.00649-09
37. Galigniana MD, Harrell JM, O’Hagen HM, Ljungman M, Pratt WB. Hsp90-binding immunophilins link p53 to dynein during p53 transport to the nucleus. J Biol Chem (2004) 279:22483–9. doi:10.1074/jbc.M402223200
38. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell (2002) 110:93–105. doi:10.1016/S0092-8674(02)00817-6
39. Schiller BJ, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol (2014) 15:418. doi:10.1186/s13059-014-0418-y
40. Galigniana MD, Echeverría PC, Erlejman AG, Piwien-Pilipuk G. Role of molecular chaperones and TPR-domain proteins in the cytoplasmic transport of steroid receptors and their passage through the nuclear pore. Nucleus (2010) 1:299–308. doi:10.4161/nucl.1.4.11743
41. Hakim A, Barnes PJ, Adcock IM, Usmani OS. Importin-7 mediates glucocorticoid receptor nuclear import and is impaired by oxidative stress, leading to glucocorticoid insensitivity. FASEB J (2013) 27:4510–9. doi:10.1096/fj.12-222604
42. Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature (1985) 318:635–41. doi:10.1038/318635a0
43. Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev (1995) 9:1608–21. doi:10.1101/gad.9.13.1608
44. Reichardt HM, Umland T, Bauer A, Kretz O, Schütz G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol Cell Biol (2000) 20:9009–17. doi:10.1128/MCB.20.23.9009-9017.2000
45. Opherk C, Tronche F, Kellendonk C, Kohlmüller D, Schulze A, Schmid W, et al. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol (2004) 18:1346–53. doi:10.1210/me.2003-0283
46. Tronche F, Opherk C, Moriggl R, Kellendonk C, Reimann A, Schwake L, et al. Glucocorticoid receptor function in hepatocytes is essential to promote postnatal body growth. Genes Dev (2004) 18:492–7. doi:10.1101/gad.284704
47. Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, Muglia LJ. Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J Neurosci (2006) 26:1971–8. doi:10.1523/JNEUROSCI.2173-05.2006
48. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood (2007) 109:4313–9. doi:10.1182/blood-2006-10-048215
49. Kleiman A, Tuckermann JP. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol (2007) 275:98–108. doi:10.1016/j.mce.2007.05.009
50. Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest (2007) 117:1381–90. doi:10.1172/JCI28034
51. Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A (2013) 110(1):306–11. doi:10.1073/pnas.1210200110
52. Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J (1994) 13:4087–95.
53. Lim HW, Uhlenhaut NH, Rauch A, Weiner J, Hübner S, Hübner N, et al. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res (2015) 25:836–44. doi:10.1101/gr.188581.114
54. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell (1998) 93:531–41. doi:10.1016/S0092-8674(00)81183-6
55. Adcock IM, Caramori G, Ito K. New insights into the molecular mechanisms of corticosteroids actions. Curr Drug Targets (2006) 7:649–60. doi:10.2174/138945006777435344
56. Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol (2014) 12:e1001813. doi:10.1371/journal.pbio.1001813
57. Kleiman A, Hübner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J (2012) 26:722–9. doi:10.1096/fj.11-192112
58. Baschant U, Frappart L, Rauchhaus U, Bruns L, Reichardt HM, Kamradt T, et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc Natl Acad Sci U S A (2011) 108:19317–22. doi:10.1073/pnas.1105857108
59. Wüst S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol (2008) 180:8434–43. doi:10.4049/jimmunol.180.12.8434
60. Kugler DG, Mittelstadt PR, Ashwell JD, Sher A, Jankovic D. CD4+ T cells are trigger and target of the glucocorticoid response that prevents lethal immunopathology in toxoplasma infection. J Exp Med (2013) 210:1919–27. doi:10.1084/jem.20122300
61. Mittelstadt PR, Monteiro JP, Ashwell JD. Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J Clin Invest (2012) 122:2384–94. doi:10.1172/JCI63067
62. Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci (2009) 1179:167–78. doi:10.1111/j.1749-6632.2009.04986.x
63. Evans RM. The steroid and thyroid hormone receptor superfamily. Science (1988) 240:889–95. doi:10.1126/science.3283939
64. Grøntved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J (2013) 32:1568–83. doi:10.1038/emboj.2013.106
65. Hapgood JP, Avenant C, Moliki JM. Glucocorticoid-independent modulation of GR activity: implications for immunotherapy. Pharmacol Ther (2016) 165:93–113. doi:10.1016/j.pharmthera.2016.06.002
66. Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol (2010) 120:76–85. doi:10.1016/j.jsbmb.2010.02.018
67. Gronemeyer H, Bourguet W. Allosteric effects govern nuclear receptor action: DNA appears as a player. Sci Signal (2009) 2:e34. doi:10.1126/scisignal.273pe34
68. Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol (2013) 20:876–83. doi:10.1038/nsmb.2595
69. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol (2013) 380:41–54. doi:10.1016/j.mce.2012.12.014
70. Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol (2002) 71:9–15.
71. Lee HL, Archer TK. Nucleosome-mediated disruption of transcription factor-chromatin initiation complexes at the mouse mammary tumor virus long terminal repeat in vivo. Mol Cell Biol (1994) 14:32–41. doi:10.1128/MCB.14.1.32
72. De Martino MU, Bhattachryya N, Alesci S, Ichijo T, Chrousos GP, Kino T. The glucocorticoid receptor and the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II interact with and mutually affect each other’s transcriptional activities: implications for intermediary metabolism. Mol Endocrinol (2004) 18:820–33. doi:10.1210/me.2003-0341
73. Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell (2011) 43:145–55. doi:10.1016/j.molcel.2011.06.016
74. Patel R, Patel M, Tsai R, Lin V, Bookout AL, Zhang Y, et al. LXRbeta is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J Clin Invest (2011) 121:431–41. doi:10.1172/JCI41681
75. Rao NA, McCalman MT, Moulos P, Francoijs KJ, Chatziioannou A, Kolisis FN, et al. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res (2011) 21:1404–16. doi:10.1101/gr.118042.110
76. Siersbaek R, Nielsen R, John S, Sung MH, Baek S, Loft A, et al. Extensive chromatin remodelling and establishment of transcription factor “hotspots” during early adipogenesis. EMBO J (2011) 30:1459–72. doi:10.1038/emboj.2011.65
77. Langlais D, Couture C, Balsalobre A, Drouin J. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell (2012) 47:38–49. doi:10.1016/j.molcel.2012.04.021
78. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell (2011) 145:224–41. doi:10.1016/j.cell.2011.03.027
79. Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol (2013) 20:53–8. doi:10.1038/nsmb.2456
80. Clark AR, Lasa M. Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr Opin Pharmacol (2003) 3:404–11. doi:10.1016/S1471-4892(03)00073-0
81. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappa-B. Blood (2001) 98:743–53. doi:10.1182/blood.V98.3.743
82. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimège L, Mahieu T, et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med (2013) 5:456–70. doi:10.1002/emmm.201201683
83. Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge R-M, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and antiinflammatory therapeutics. Elife (2013) 2:1–16. doi:10.7554/eLife.00762
84. Reuter KC, Grunwitz CR, Kaminski BM, Steinhilber D, Radeke HH, Stein J. Selective glucocorticoid receptor agonists for the treatment of inflammatory bowel disease: studies in mice with acute trinitrobenzene sulfonic acid colitis. J Pharmacol Exp Ther (2012) 341:68–80. doi:10.1124/jpet.111.183947
85. De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, et al. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A (2005) 102:15827–32. doi:10.1073/pnas.0505554102
86. Gossye V, Elewaut D, Van Beneden K, Dewint P, Haegeman G, De Bosscher K. A plant-derived glucocorticoid receptor modulator attenuates inflammation without provoking ligand-induced resistance. Ann Rheum Dis (2010) 69:291–6. doi:10.1136/ard.2008.102871
87. Hua G, Ganti KP, Chambon P. Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proc Natl Acad Sci U S A (2015) 2015:201522821. doi:10.1073/pnas.1522826113
88. Zalachoras I, Houtman R, Atucha E, Devos R, Tijssen AM, Hu P, et al. Differential targeting of brain stress circuits with a selective glucocorticoid receptor modulator. Proc Natl Acad Sci U S A (2013) 110:7910–5. doi:10.1073/pnas.1219411110
89. Van Den Heuvel JK, Boon MR, Van Hengel I, Peschier-Van Der Put E, Van Beek L, Van Harmelen V, et al. Identification of a selective glucocorticoid receptor modulator that prevents both diet-induced obesity and inflammation. Br J Pharmacol (2016) 173:1793–804. doi:10.1111/bph.13477
90. De Bosscher K, Beck IM, Ratman D, Berghe WV, Libert C. Activation of the glucocorticoid receptor in acute inflammation: the SEDIGRAM concept. Trends Pharmacol Sci (2016) 37:4–16. doi:10.1016/j.tips.2015.09.002
91. Chrousos GP, Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE (2005) 2005:e48. doi:10.1126/stke.3042005pe48
92. Bamberger CM, Bamberger AM, De Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest (1995) 95:2435–41. doi:10.1172/JCI117943
93. de Castro M, Elliot S, Kino T, Bamberger C, Karl M, Webster E, et al. The non-ligand binding beta-isoform of the human glucocorticoid receptor (hGR beta): tissue levels, mechanism of action, and potential physiologic role. Mol Med (1996) 2:597–607.
94. Fruchter O, Kino T, Zoumakis E, Alesci S, De Martino M, Chrousos G, et al. The human glucocorticoid receptor (GR) isoform {beta} differentially suppresses GR{alpha}-induced transactivation stimulated by synthetic glucocorticoids. J Clin Endocrinol Metab (2005) 90:3505–9. doi:10.1210/jc.2004-1646
95. Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem Biophys Res Commun (2009) 381:671–5. doi:10.1016/j.bbrc.2009.02.110
96. Marino JS, Stechschulte LA, Stec DE, Nestor-Kalinoski A, Coleman S, Hinds TD. Glucocorticoid receptor β induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem (2016) 291:9383–94. doi:10.1074/jbc.M116.752311
97. Anbalagan M, Huderson B, Murphy L, Rowan BG. Post-translational modifications of nuclear receptors and human disease. Nucl Recept Signal (2012) 10:1–13. doi:10.1621/nrs.10001
98. Duma D, Silva-Santos JE, Assreuy J. Inhibition of glucocorticoid receptor binding by nitric oxide in endotoxemic rats. Crit Care Med (2004) 32:2304–10. doi:10.1097/01.CCM.0000145996.57901
99. Galigniana MD, Piwien-Pilipuk G, Assreuy J. Inhibition of glucocorticoid receptor binding by nitric oxide. Mol Pharmacol (1999) 55:317–23.
100. Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A (2008) 105:19183–7. doi:10.1073/pnas.0805514105
101. Malhotra D, Thimmulappa RK, Mercado N, Ito K, Kombairaju P, Kumar S, et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest (2011) 121:4289–302. doi:10.1172/JCI45144
102. Paul-Clark MJ, Roviezzo F, Flower RJ, Cirino G, Soldato PD, Adcock IM, et al. Glucocorticoid receptor nitration leads to enhanced anti-inflammatory effects of novel steroid ligands. J Immunol (2003) 171:3245–52. doi:10.4049/jimmunol.171.6.3245
103. Ji JY, Diamond SL. Exogenous nitric oxide activates the endothelial glucocorticoid receptor. Biochem Biophys Res Commun (2004) 318:192–7. doi:10.1016/j.bbrc.2004.04.008
104. Druker J, Liberman AC, Antunica-Noguerol M, Gerez J, Paez-Pereda M, Rein T, et al. RSUME enhances glucocorticoid receptor SUMOylation and transcriptional activity. Mol Cell Biol (2013) 33:2116–27. doi:10.1128/MCB.01470-12
105. Hua G, Paulen L, Chambon P. GR SUMOylation and formation of an SUMO-SMRT/NCoR1-HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc Natl Acad Sci U S A (2016) 113:E626–34. doi:10.1073/pnas.1522821113
106. Kotitschke A, Sadie-Van Gijsen H, Avenant C, Fernandes S, Hapgood JP. Genomic and nongenomic cross talk between the gonadotropin-releasing hormone receptor and glucocorticoid receptor signaling pathways. Mol Endocrinol (2009) 23:1726–45. doi:10.1210/me.2008-0462
107. Matthews L, Johnson J, Berry A, Trebble P, Cookson A, Spiller D, et al. Cell cycle phase regulates glucocorticoid receptor function. PLoS One (2011) 6:e22289. doi:10.1371/journal.pone.0022289
108. Verhoog NJ, Du Toit A, Avenant C, Hapgood JP. Glucocorticoid-independent repression of tumor necrosis factor (TNF) α-stimulated interleukin (IL)-6 expression by the glucocorticoid receptor: a potential mechanism for protection against an excessive inflammatory response. J Biol Chem (2011) 286:19297–310. doi:10.1074/jbc.M110.193672
109. Ritter HD, Antonova L, Mueller CR. The unliganded glucocorticoid receptor positively regulates the tumor suppressor gene BRCA1 through GABP beta. Mol Cancer Res (2012) 10:558–69. doi:10.1158/1541-7786.MCR-11-0423-T
110. Ritter HD, Mueller CR. Expression microarray identifies the unliganded glucocorticoid receptor as a regulator of gene expression in mammary epithelial cells. BMC Cancer (2014) 14:275. doi:10.1186/1471-2407-14-275
111. Ji JY, Jing H, Diamond SL. Shear stress causes nuclear localization of endothelial glucocorticoid receptor and expression from the GRE promoter. Circ Res (2003) 92:279–85. doi:10.1161/01.RES.0000057753.57106.0B
112. Vandevyver S, Dejager L, Van Bogaert T, Kleyman A, Liu Y, Tuckermann J, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest (2012) 122:2130–40. doi:10.1172/JCI60006
113. Nayebosadri A, Christopher L, Ji JY. Bayesian image analysis of dexamethasone and shear stress-induced glucocorticoid receptor intracellular movement. Ann Biomed Eng (2012) 40:1508–19. doi:10.1007/s10439-011-0499-7
114. Nayebosadri A, Ji JY. Endothelial nuclear lamina is not required for glucocorticoid receptor nuclear import but does affect receptor-mediated transcription activation. Am J Physiol Cell Physiol (2013) 305:C309–22. doi:10.1152/ajpcell.00293.2012
115. Galliher-Beckley AJ, Williams JG, Cidlowski JA. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol (2011) 31:4663–75. doi:10.1128/MCB.05866-11
116. Limbourg FP, Liao JK. Nontranscriptional actions of the glucocorticoid receptor. J Mol Med (Berl) (2003) 81:168–74. doi:10.1007/s00109-003-0418-y
117. Deng Q, Riquelme D, Trinh L, Low MJ, Tomic M, Stojilkovic S, et al. Rapid glucocorticoid feedback inhibition of ACTH secretion involves ligand-dependent membrane association of glucocorticoid receptors. Endocrinology (2015) 156:3215–27. doi:10.1210/EN.2015-1265
118. Strehl C, Buttgereit F. Unraveling the functions of the membrane-bound glucocorticoid receptors: first clues on origin and functional activity. Ann N Y Acad Sci (2014) 1318:1–6. doi:10.1111/nyas.12364
119. Morgan DJ, Poolman TM, Williamson AJ, Wang Z, Clark NR, Ma’ayan A, et al. Glucocorticoid receptor isoforms direct distinct mitochondrial programs to regulate ATP production. Sci Rep (2016) 6:26419. doi:10.1038/srep26419
120. Norman AW, Mizwicki MT, Norman DP. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov (2004) 3:27–41. doi:10.1038/nrd1283
121. Stahn C, Löwenberg M, Hommes DW, Buttgereit F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol (2007) 275:71–8. doi:10.1016/j.mce.2007.05.019
122. Scheller K, Sekeris CE, Krohne G, Hock R, Hansen IA, Scheer U. Localization of glucocorticoid hormone receptors in mitochondria of human cells. Eur J Cell Biol (2000) 79:299–307. doi:10.1078/S0171-9335(04)70033-3
123. Gruber CJ, Gruber DM, Gruber IM, Wieser F, Huber JC. Anatomy of the estrogen response element. Trends Endocrinol Metab (2004) 15:73–8. doi:10.1016/j.tem.2004.01.008
124. Psarra AM, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta (2011) 1813:1814–21. doi:10.1016/j.bbamcr.2011.05.014
125. Mendes ES, Pereira A, Danta I, Duncan RC, Wanner A. Comparative bronchial vasoconstrictive efficacy of inhaled glucocorticosteroids. Eur Respir J (2003) 21:989–93. doi:10.1183/09031936.03.00072402
Keywords: glucocorticoid receptor, glucocorticoids, selective glucocorticoid receptor modulators, signaling pathways, nuclear translocation
Citation: Scheschowitsch K, Leite JA and Assreuy J (2017) New Insights in Glucocorticoid Receptor Signaling—More Than Just a Ligand-Binding Receptor. Front. Endocrinol. 8:16. doi: 10.3389/fendo.2017.00016
Received: 21 July 2016; Accepted: 18 January 2017;
Published: 06 February 2017
Edited by:
Isaias Glezer, Federal University of São Paulo, BrazilReviewed by:
Jan Tuckermann, University of Ulm, GermanyCopyright: © 2017 Scheschowitsch, Leite and Assreuy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jamil Assreuy, amFtaWwuYXNzcmV1eUB1ZnNjLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.