 Waldemar Kanczkowski
Waldemar Kanczkowski Mariko Sue1
Mariko Sue1 Stefan R. Bornstein
Stefan R. Bornstein- 1Department of Internal Medicine III, Technische Universität Dresden, Dresden, Germany
- 2Department of Endocrinology and Diabetes, King’s College London, London, UK
Survival of all living organisms depends on maintenance of a steady state of homeostasis, which process relies on its ability to react and adapt to various physical and emotional threats. The defense against stress is executed by the hypothalamic–pituitary–adrenal axis and the sympathetic–adrenal medullary system. Adrenal gland is a major effector organ of stress system. During stress, adrenal gland rapidly responds with increased secretion of glucocorticoids (GCs) and catecholamines into circulation, which hormones, in turn, affect metabolism, to provide acutely energy, vasculature to increase blood pressure, and the immune system to prevent it from extensive activation. Sepsis resulting from microbial infections is a sustained and extreme example of stress situation. In many critical ill patients, levels of both corticotropin-releasing hormone and adrenocorticotropin, the two major regulators of adrenal hormone production, are suppressed. Levels of GCs, however, remain normal or are elevated in these patients, suggesting a shift from central to local intra-adrenal regulation of adrenal stress response. Among many mechanisms potentially involved in this process, reduced GC metabolism and activation of intra-adrenal cellular systems composed of adrenocortical and adrenomedullary cells, endothelial cells, and resident and recruited immune cells play a key role. Hence, dysregulated function of any of these cells and cellular compartments can ultimately affect adrenal stress response. The purpose of this mini review is to highlight recent insights into our understanding of the adrenal gland microenvironment and its role in coordination of stress-induced hormone secretion.
Introduction
In order to survive all, living organisms must maintain a steady state of homeostasis. This unique capacity to react and adapt to various physical and emotional threats in higher vertebrates is mediated by a coordinated action of the nervous, endocrine, and immune systems, known as stress system (1). Activation of the stress system, which is composed of the hypothalamic–pituitary–adrenal (HPA) axis and the sympathetic–adrenal medullary system, leads to increased synthesis and release of glucocorticoids (GCs) and catecholamines (CAs) from the adrenal cortex and medulla, respectively (2–4). The main function of these hormones is maintenance and restoration of basal and stress-related body homeostasis. This protective action of adrenal hormones is mostly accomplished by their metabolic, cardioprotective, and anti-inflammatory actions. In particular, both GCs and CAs are known to acutely enhance plasma glucose levels and promote an increased cardiac output and high blood pressure, while protecting against excessive inflammation (5–8).
Secretion of adrenal hormones during stress-free conditions is regulated centrally and is characterized by ultradian rhythms (9). An early morning release of adrenocorticotropic hormone (ACTH) into circulation is enhanced by increased concentrations of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) in the hypophyseal portal system of anterior pituitary (10, 11). Consequently, elevated plasma levels of ACTH stimulate adrenocortical cells to produce and release GC hormones. In turn, as a part of negative feedback, GCs act directly on the pituitary gland to reduce ACTH secretion and on hypothalamic neurons to reduce CRH release (11, 12). Systemic rise in CA levels is in turn initiated by activation of the sympathetic nervous system and splenic nerves innervating the adrenal medulla (13, 14).
One of the extreme examples of sustained and severe physical stress is sepsis syndrome. It is characterized by abnormal host response to infection, resulting in systemic inflammation that frequently culminates in a life-threatening dysfunction of multiple organs (15, 16). Severe sepsis remains the leading cause of mortality worldwide, and its incidence is increasing (17, 18). An intact function of the stress system, and in particular the activation of GC and CA production, is critical to survive this adverse condition (19, 20). During sepsis, this homeostatic function of the stress system is frequently impaired with mechanisms remaining largely unknown (21, 22). Consequently, many critically ill patients demonstrate suppressed ACTH and CRH levels while having normal or elevated cortisol levels (22). Hence, a key involvement of pituitary-independent factors was proposed (2). Among these, reduced GC metabolism and activation of local adrenal microenvironment seem to play crucial role (23, 24) (Figure 1).
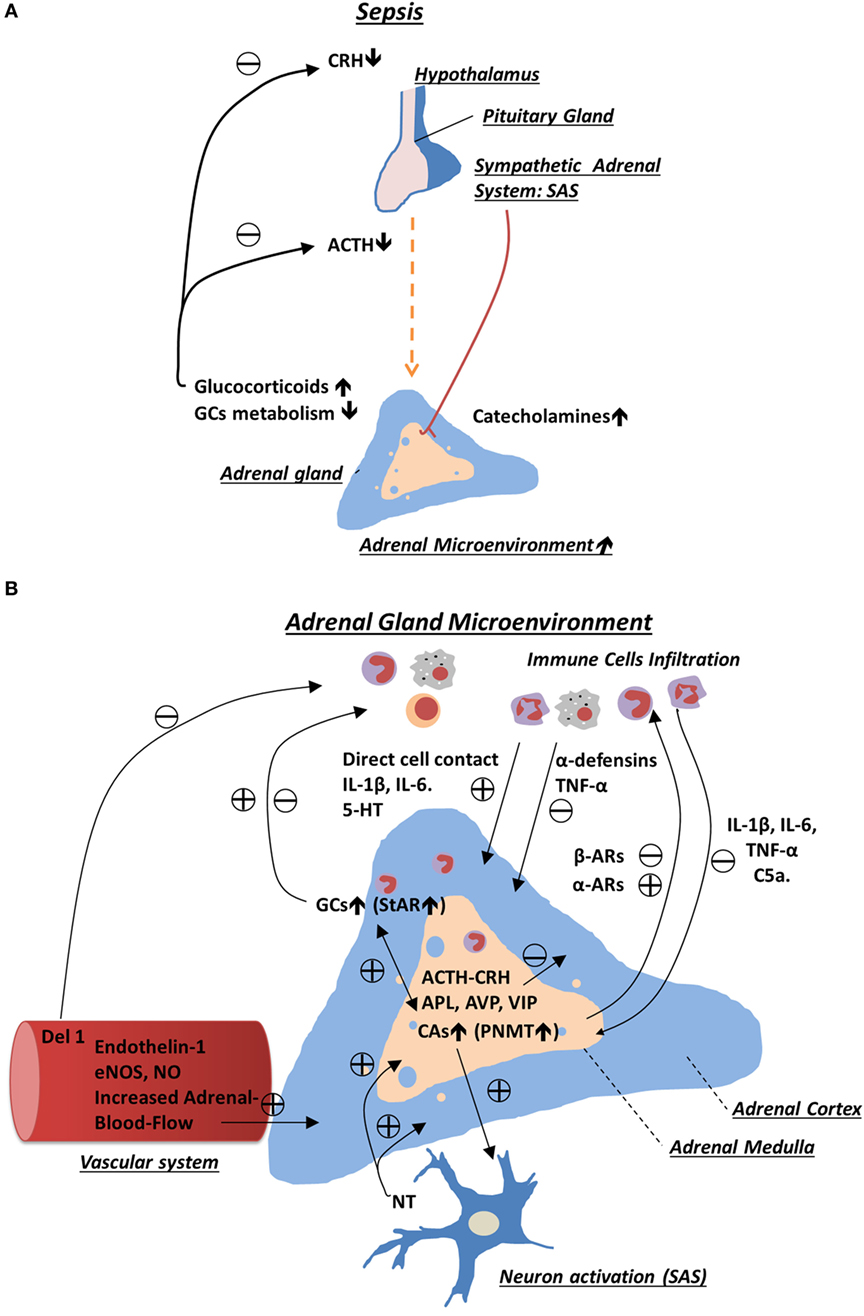
Figure 1. Schematic representation of regulatory mechanisms involved in adrenal gland stress response during sepsis. (A) Sepsis activates the hypothalamic–pituitary–adrenal axis and sympathetic–adrenal medullary system, which in turn enhances production of glucocorticoids (GCs) and catecholamines from the adrenal gland. During chronic phase of sepsis, production of corticotropin-releasing hormone and adrenocorticotropic hormone (ACTH) is inhibited as a result of increased negative feedback from the elevated levels of GCs. High level of adrenal GCs in plasma is sustained due to stimulatory effects provided by intra-adrenal microenvironment and reduced GCs metabolism. (B) In the adrenal gland, many cell types including resident and infiltrating immune cells, sympathetic neurons, endothelial cells, and adrenal chromaffin and adrenocortical cells interact with each other. This interaction occurs through a direct cell–cell contact and in a paracrine way, which ultimately leads to sustained production of adrenal hormones. Among many mediators produced in adrenal gland, which are reported to modulate adrenal hormone production, are adrenocortical steroids, catecholemines, and various cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor-α, serotonin (5-HT), neurotransmitters, and neurohormones (e.g., APL, ACTH, and vasoactive intestinal peptide).
As regulation of the stress system and importance of impaired metabolism of GCs during sepsis were recently presented and discussed in many review articles (25–27), the main purpose of this mini review is to highlight recent insights into our understanding of the role local adrenal gland microenvironment in the regulation of stress-induced hormone secretion during sepsis.
Adrenal Gland Microenvironment
Within adrenal gland, two embryonically different tissues coexist: mesodermally derived, steroid-producing cortex and ectodermally derived, CA-producing medulla. Within these two environmental niches, interplay between various cells takes place including adrenocortical and chromaffin cells, neuronal cells, immune cells, endothelial cells, and glia cells (28). The adrenal gland is a source of many bioactive substances including steroid hormones, CAs, cytokines, neurotransmitters, and neuropeptides (Table 1). These substances are known to interact with various cell types within adrenal gland itself, thereby influencing its function during stress conditions and disease (24, 28).

Table 1. Intra-adrenal interaction involved in a regulation of adrenal stress response.
Adrenal Cortex and Medulla Interactions during Sepsis
Ample evidence exists suggesting a bidirectional interaction between adrenal cortex and the medulla (28). Despite a classical view on the adrenal gland anatomy, demonstrating a clear separation of the cortex and medulla, it has been well documented that these two endocrine tissues are morphologically intermingled (29, 30). This close localization, in turn, enables direct cell–cell and paracrine interactions between adrenocortical and chromaffin cells involving their secretory products.
Consequently, the adrenal cortex-derived GCs were found to enhance synthesis of CAs both in vitro (31) and in vivo (32–34). GCs were found to execute these effects by upregulating expression of phenylethanolamine N-methyltransferase (PNMT), which gene encodes for an enzyme that catalyzes the synthesis of epinephrine from norepinephrine. Furthermore, studies using various knockout mice, which were deficient in steroidogenic factor-1, glucocorticoid receptor, or corticotropin-releasing hormone type 1 receptor, clearly demonstrated that lack of one of the these key steroidogenic regulators impair PNMT gene expression and thus basal and stress-induced epinephrine production (35–38). Similarly, an intact function of adrenal medulla is important to maintain adrenocortical function. It has been shown that coculture of bovine adrenocortical cells with chromaffin cells enhanced a basal GC secretion by 10-fold (39). Furthermore, CAs were found to enhance production of various steroids including cortisol, aldosterone, and androstenedione, through mechanism involving upregulation of steroidogenic acute regulatory protein (28, 40, 41).
During sepsis, an enhanced production of epinephrine and norepinephrine (42–44) and increased secretory capacity of adrenal chromaffin cells were reported (45). Unlike expression of tyrosine hydroxylase, which enzyme is involved in first step of CA biosynthesis, upregulation of PNMT gene expression in adrenal medulla was found to be regulated independently of splanchnic nerve stimulation (28). Instead, an extremely high concentration of GCs was reported to be involved in regulation of PNMT expression during sepsis (46). Consequently, stress-induced expression of PNMT is strongly attenuated in CRH-deficient mice (47).
In addition, adrenal chromaffin cells release many active neuropeptides and transmitters, including neuropeptide Y (NPY), vasoactive intestinal peptide (VIP), or substance P, with known stimulatory action on GCs production (48, 49). For example, VIP, which levels increase during experimental endotoxemia in adrenal medulla, was found to stimulate DHEA, testosterone, androstenedione, cortisol, and aldosterone secretion in adrenocortical cells (48–52). Among other mediators found in adrenal medulla, which may influence GC production during sepsis, are angiotensin, AVP, CRH, ACTH, and apelin (53–55). Although direct stimulatory effects of angiotensin 2, CRH, and ACTH on adrenal hormone production are well described, the degree to which each of these peptides contributes to hormone production during sepsis conditions has not been fully elucidated yet (2, 56). Another interesting peptide found recently to be expressed in the adrenal medulla is apelin. Apelin is a neurohormone, which acts through its receptor APJ expressed in various organs including hypothalamic neurons, anterior pituitary, and adrenal gland. It was recently found to modulate neuroendocrine response to stress through stimulation of secretion of both ACTH and corticosterone directly and via AVP- and CRH-dependent pathways (54, 55, 57).
Recently, a population of nestin-positive glia-like multipotent stem cells was identified in adrenal medulla of nestin-GFP transgenic mice (58). Interestingly, when subjected to chronic stress, this population was able to give rise to new chromaffin cells, suggesting a direct involvement of these cells in the adrenal stress adaptation (58–60). It is therefore very interesting to investigate the fate of this cells and their potential interaction with adrenocortical cells during sepsis conditions.
In summary, an intact function of both adrenal cortex and adrenal medulla is of pivotal importance in the regulation of adrenal stress response. Consequently, any disorders or medications that can attenuate adrenal cortex function will also ultimately affect CAs production by chromaffin cells and vice versa potentially impacting the outcome of many stress-related disorders (19).
Immune–Adrenal Crosstalk
In the last 40 years, it has become evident that immune and endocrine systems interact with each other at the level of adrenal gland and that this interaction is crucially involved in the regulation of adrenal gland function during normal and stress conditions (61). This immune–endocrine crosstalk is mostly executed by bidirectional action of paracrine factors, such as steroid hormones, CAs, and various vasoactive or proteolytic enzymes, as well as by direct cell–cell contacts and activation of toll-like receptors (TLRs) (28, 62).
Immune–Adrenal Interaction at the Level of Adrenal Cortex
During non-stress conditions, many immune cells can be found in the innermost zone of adrenal cortex known as zona reticularis; these are particularly tissue macrophages (63), dendritic cells (64), mast cells (65), and lymphocytes (66). However, some of them, especially macrophages and mast cells, were also reported in other parts of the adrenal gland including subcapsular region and in the adrenal medulla (64). In non-stress conditions, adrenal-resident immune cells play important homeostatic functions by sensing pathogens, removing apoptotic cells, and promoting tissue remodeling through, e.g., secretion of growth factors (24, 67). Recently, a new population of adrenal macrophages was identified based on the high expression of a complement receptor immunoglobulin, CRIg, which molecule is known to mediate phagocytosis of pathogens and apoptotic cells (68). During systemic inflammation induced either by LPS injection or CLP, highly dynamic changes within adrenal immune cell populations can be observed. For instance, it has been shown that already within first 3 h after administration of LPS or induction of peritonitis in rodents, a rapid infiltration of neutrophils takes place, while the number of local dendritic cells and macrophages declines (69–71).
Using electron microscopy, close cell–cell localization between local immune cells and surrounding adrenocortical, chromaffin, or endothelial cells was demonstrated (64, 66). This in turn enables various bidirectional and direct interactions between each cell type. For example, a functional crosstalk between adrenocortical cells, expressing major histocompatibility complex class II molecules (MHC/HLA), and leukocytes was described. Indeed, a co-incubation of T cells with primary cultures of human adrenocortical cells promoted adrenal androgen and cortisol secretion (66). Besides direct interaction, various immune cells were found to regulate adrenal hormone production in a paracrine way through secretion of cytokines, such as interleukins (IL) 1 and 6 (72), and pro-opiomelanocortin (POMC)-derived peptides, e.g., ACTH and biogenic amines (28, 73).
The ample experimental evidence exists demonstrating that cytokines are critically involved in ACTH-independent activation of GC production during sepsis (72). For example, injection of IL-1β enhanced GC production in hypophysectomized rats (74). Furthermore, an impaired synthesis of GCs after LPS injection was observed in mice receiving sera against tumor necrosis factor (TNF)-α and IL-6 and, to a lesser extent, IL-1β- or in IL-6-deficient mice (75, 76). However, it has been demonstrated that cytokine levels found normally in plasma are far too low to induce hormone production from adrenocortical cells. Therefore, the main source of cytokines must come from the adrenal gland itself (28). Sustained production of cytokines in highly anti-inflammatory environment of the adrenal gland during systemic inflammation was found to be enabled by increased expression of migration inhibitory factor (77). During sepsis, various cell types present within adrenal gland microenvironment may be a potential source of pro-inflammatory cytokines. For example, adrenocortical cells were found to express several TLRs and secrete several cytokines in response to bacterial ligands (78, 79). However, in a recent study, inactivation of myeloid but not adrenocortical TLR signaling resulted in significant reduction of intra-adrenal cytokine levels and activation of the HPA axis after LPS administration (80). The latter observation suggests that immune cells are the key sources of cytokines in the adrenal gland during sepsis. Due to high expression of both cytokine and GC receptors in adrenocortical and immune cells, respectively, cytokines and adrenal hormones are known to regulate production in each other. For example, IL-1β or IL-6 was found to increase adrenal hormone production, whereas TNF stimulation demonstrated rather opposite effect (81–83). In turn, adrenal steroid hormones are known to influence immune cells function. In particular, it has been demonstrated that in naïve immune cells, GCs can activate several inflammatory-related genes, e.g., TLRs, yet in cells treated with LPS, they inhibit inflammation (6). In a recent study, a chronic exposure to GCs was found to cause a shift in the innate–adaptive balance of the immune response, particularly, influencing the chemokine–chemokine receptor networks (84). Besides cytokines, other biologically active substances, such as serotonin (5-HT) or histamine, which are stored by mast cells show stimulatory effects on adrenal steroidogenesis (85, 86).
During prolonged sepsis, the immune–adrenal interaction may also result in the suppression of adrenal hormone production. Despite their important role in host defenses against bacterial infections limiting bacterial spread in circulation (87), a prolonged exposure of adrenal cells to neutrophil-derived antimicrobial agents, such as ROS and proteolytic enzymes secreted during formation of neutrophil extracellular traps, results in tissue damage (88). In addition, neutrophils were found to secrete corticostatins, e.g., α-defensins, which substances are known to interfere with ACTH-mediated increase in adrenocortical hormone production (89). In a recent study using developmental endothelial locus 1 (Del-1)-deficient mice (90), an association between higher amount of infiltrating neutrophils and impaired adrenal corticosterone production after systemic LPS administration was found (91).
Immune–Adrenal Interaction at the Level of Adrenal Medulla
Activation of the sympathetic nerve system during sepsis results in enhanced production and secretion of CAs from the adrenal chromaffin cells (44) through a mechanism involving an increased release of Ca2+ from endothelial reticulum (45). An early increase in endogenous CA level plays an important protective role as its absence due to either pharmacological or surgical intervention resulted in induction of hypotension and increased mortality of rats during experimental endotoxemia (20, 43). One of the mechanisms involved in the protective role of CAs, besides well-known vasopressor function, is the anti-inflammatory action on various immune cells. The immunomodulatory effects of CAs are mediated via their direct interactions with β-adrenergic receptors (ARs) expressed by a variety of immune cells. In particular, it has been shown that CAs promote IL-10 secretion while decreasing production of pro-inflammatory TNF cytokine in LPS-treated macrophages (92). Furthermore, incubation of neutrophils with CAs decreased NO production and ROS generation (93). However, these immune-suppressive effects of CAs should be carefully interpreted as they may correspond predominantly to the leukocyte populations or be restricted to β-AR activation. In fact, recently both epinephrine and norepinephrine were shown to increase IL-6 production in endothelial cell line: HMEC-1 and in human skin microvascular endothelial cells (94). It has been also found that activation of α-ARs in macrophages by CA may potentiate inflammation by increasing TNF-α production (95). Recently, phagocytes were shown to be able not only to secrete CAs in response to LPS stimulation but also to possess all major CA-generating and -degrading enzymes required for their production and inactivation (96). This discovery adds additional complexity to already complex immune–chromaffin cell interaction. During sepsis, the adrenal medulla shows high degree of immune infiltration (69). This suggests that overactivation of immune cells could potentially impair production and secretion of adrenomedullary hormones or induce structural damage. Indeed, during experimental sepsis induced by CLP, systemic inflammation resulted in a strong apoptosis in the adrenal chromaffin cells, which process required activation of receptors for complement C5a (97). The latter observation suggests that during progression of sepsis, an increased activation of immune system promoting apoptosis of chromaffin cells may impact stress-induced CA production.
Adrenal Vasculature
Vascular system plays a key role in the proper functioning of many vital organs during sepsis. Besides enabling the secretion of steroid hormones and CAs into circulation, adrenal vasculature was found to control leukocyte infiltration through expression of adhesion molecules and Del-1 protein (91).
Adrenal cortex is characterized by high density of endothelial cells that stay in close vicinity with steroid-producing cells. Consequently, both adrenal hormones and endothelial cell-derived products were shown to influence function of adrenal vasculature and steroid-producing cells, respectively. For example, human adrenocortical cells increase production of aldosterone and cortisol once incubated with endothelial cell-conditioned media. Although the exact composition of the latter media was not determined, this stimulatory effect was found to require cAMP, but not PKA, pathway (98). Endothelial cells are known to produce several factors that, e.g., nitric oxide (NO) or endothelin-1, could potentially influence adrenal hormone production. In particular, in rat and human adrenocortical cells, it has been found that endothelin-1 can directly promote GC synthesis and potentiate angiotensin II and ACTH-induced aldosterone production (99). Besides endothelin-1, nitric oxide (NO) was found to exert stimulatory effect on adrenal steroidogenesis by mediating the acute response to ACTH (28). In fact, blocking of NO production either by using NO synthase inhibitor or NO scavengers resulted in decreased ACTH-mediated corticosterone release (100). However, in other studies using perfused adrenal glands administration of l-arginine, the substrate for nitric oxide synthesis did not change corticosterone response to ACTH, suggesting an indirect modulatory action of NO (101).
During sepsis, in many critically ill patients, an increased size of adrenal gland resulting from the hypervascularization and increased blood flow and adrenal hyperplasia were found to positively correlate with survival rate (102). Although an intact activity of splanchnic nerve is crucial in regulation of adrenal blood flow, other factors such as VIP, NPY, or neurotensin were found also to be involved (103, 104). Furthermore, a positive correlation between an increased blood flow and enhanced adrenal steroidogenesis was demonstrated (105). Indeed, an enhanced adrenal blood flow was found to participate in angiotensin II- and ACTH-induced aldosterone and cortisol production. This stimulatory action on adrenal vascular response to angiotensin II and ACTH was found to be mediated by vasoactive substances released from granules of mast cells (106). However, more recently, additional involvement of the cytochrome 450 in controlling the adrenal blood flow after AII and ACTH was demonstrated in rats (24, 107).
Summary
Sepsis and septic shock strongly impacts body homeostasis, which if not sufficiently counteracted by activated stress system results in increased mortality of critically ill patients (56, 108, 109). During chronic phase of sepsis, elevated adrenal hormone secretion was reported to be mediated by pituitary-independent mechanisms including activation of intra-adrenal microenvironment (22, 23). An increasing amount of experimental studies support the key involvement of several cell types and cellular systems present within adrenal gland microenvironment in the sustained production of adrenal hormones during sepsis. Consequently, any impairment in function of these systems can ultimately affect adrenal stress response (2, 24).
Author Contributions
WK, MS, and SB have all made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer OL and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Funding
This work was supported by grants of the Deutsche Forschungs Gemeinschaft (DFG) to WK (KA 3013/2-2) and SB (BO 1141/11-2).
References
1. Nicolaides NC, Kyratzi E, Lamprokostopoulou A, Chrousos GP, Charmandari E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation (2015) 22:6–19. doi:10.1159/000362736
2. Bornstein SR, Chrousos GP. Clinical review 104: adrenocorticotropin (ACTH)- and non-ACTH-mediated regulation of the adrenal cortex: neural and immune inputs. J Clin Endocrinol Metab (1999) 84:1729–36. doi:10.1210/jcem.84.5.5631
3. Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med (1995) 332:1351–62. doi:10.1056/NEJM199505183322008
4. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol (2009) 5:374–81. doi:10.1038/nrendo.2009.106
5. Bornstein SR, Briegel J. A new role for glucocorticoids in septic shock: balancing the immune response. Am J Respir Crit Care Med (2003) 167:485–6. doi:10.1164/rccm.2211010
6. Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J (2002) 16:61–71. doi:10.1096/fj.01-0245com
7. Ensinger H, Geisser W, Brinkmann A, Wachter U, Vogt J, Radermacher P, et al. Metabolic effects of norepinephrine and dobutamine in healthy volunteers. Shock (2002) 18:495–500. doi:10.1097/00024382-200212000-00002
8. Trager K, Radermacher P. Catecholamines in the treatment of septic shock: effects beyond perfusion. Crit Care Resusc (2003) 5:270–6.
9. Spiga F, Walker JJ, Terry JR, Lightman SL. HPA axis-rhythms. Compr Physiol (2014) 4:1273–98. doi:10.1002/cphy.c140003
10. Gillies GE, Linton EA, Lowry PJ. Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature (1982) 299:355–7. doi:10.1038/299355a0
11. Hinz B, Hirschelmann R. Rapid non-genomic feedback effects of glucocorticoids on CRF-induced ACTH secretion in rats. Pharm Res (2000) 17:1273–7. doi:10.1023/A:1026499604848
12. Evans AN, Liu Y, Macgregor R, Huang V, Aguilera G. Regulation of hypothalamic corticotropin-releasing hormone transcription by elevated glucocorticoids. Mol Endocrinol (2013) 27:1796–807. doi:10.1210/me.2013-1095
13. Winkler H, Apps DK, Fischer-Colbrie R. The molecular function of adrenal chromaffin granules: established facts and unresolved topics. Neuroscience (1986) 18:261–90. doi:10.1016/0306-4522(86)90154-5
14. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev (2000) 52:595–638.
15. Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315:775–87. doi:10.1001/jama.2016.0289
16. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315:801–10. doi:10.1001/jama.2016.0287
17. Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med (2013) 41:1167–74. doi:10.1097/CCM.0b013e31827c09f8
18. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence (2014) 5:4–11. doi:10.4161/viru.27372
19. Chan CM, Mitchell AL, Shorr AF. Etomidate is associated with mortality and adrenal insufficiency in sepsis: a meta-analysis. Crit Care Med (2012) 40:2945–53. doi:10.1097/CCM.0b013e31825fec26
20. McKechnie K, Dean HG, Furman BL, Parratt JR. Plasma catecholamines during endotoxin infusion in conscious unrestrained rats: effects of adrenal demedullation and/or guanethidine treatment. Circ Shock (1985) 17:85–94.
21. Annane D, Maxime V, Ibrahim F, Alvarez JC, Abe E, Boudou P. Diagnosis of adrenal insufficiency in severe sepsis and septic shock. Am J Respir Crit Care Med (2006) 174:1319–26. doi:10.1164/rccm.200509-1369OC
22. Bornstein SR, Engeland WC, Ehrhart-Bornstein M, Herman JP. Dissociation of ACTH and glucocorticoids. Trends Endocrinol Metab (2008) 19:175–80. doi:10.1016/j.tem.2008.01.009
23. Boonen E, Bornstein SR, Van den Berghe G. New insights into the controversy of adrenal function during critical illness. Lancet Diabetes Endocrinol (2015) 3:805–15. doi:10.1016/S2213-8587(15)00224-7
24. Kanczkowski W, Sue M, Zacharowski K, Reincke M, Bornstein SR. The role of adrenal gland microenvironment in the HPA axis function and dysfunction during sepsis. Mol Cell Endocrinol (2015) 408:241–8. doi:10.1016/j.mce.2014.12.019
25. Turrin NP, Rivest S. Unraveling the molecular details involved in the intimate link between the immune and neuroendocrine systems. Exp Biol Med (Maywood) (2004) 229:996–1006.
26. Bellavance MA, Rivest S. The HPA-immune axis and the immunomodulatory actions of glucocorticoids in the brain. Front Immunol (2014) 5:136. doi:10.3389/fimmu.2014.00136
27. Boonen E, Van den Berghe G. Endocrine responses to critical illness: novel insights and therapeutic implications. J Clin Endocrinol Metab (2014) 99:1569–82. doi:10.1210/jc.2013-4115
28. Ehrhart-Bornstein M, Hinson JP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocr Rev (1998) 19:101–43. doi:10.1210/edrv.19.2.0326
29. Bornstein SR, Gonzalez-Hernandez JA, Ehrhart-Bornstein M, Adler G, Scherbaum WA. Intimate contact of chromaffin and cortical cells within the human adrenal gland forms the cellular basis for important intraadrenal interactions. J Clin Endocrinol Metab (1994) 78:225–32. doi:10.1210/jcem.78.1.7507122
30. Gallo-Payet N, Pothier P, Isler H. On the presence of chromaffin cells in the adrenal cortex: their possible role in adrenocortical function. Biochem Cell Biol (1987) 65:588–92. doi:10.1139/o87-076
31. Wurtman RJ. Stress and the adrenocortical control of epinephrine synthesis. Metabolism (2002) 51:11–4. doi:10.1053/meta.2002.33185
32. Wong DL, Bildstein CL, Siddall B, Lesage A, Yoo YS. Neural regulation of phenylethanolamine N-methyltransferase in vivo: transcriptional and translational changes. Brain Res Mol Brain Res (1993) 18:107–14. doi:10.1016/0169-328X(93)90178-R
34. Sharara-Chami RI, Joachim M, Pacak K, Majzoub JA. Glucocorticoid treatment – effect on adrenal medullary catecholamine production. Shock (2010) 33:213–7. doi:10.1097/SHK.0b013e3181af0633
35. Gut P, Huber K, Lohr J, Bruhl B, Oberle S, Treier M, et al. Lack of an adrenal cortex in Sf1 mutant mice is compatible with the generation and differentiation of chromaffin cells. Development (2005) 132:4611–9. doi:10.1242/dev.02052
36. Yoshida-Hiroi M, Bradbury MJ, Eisenhofer G, Hiroi N, Vale WW, Novotny GE, et al. Chromaffin cell function and structure is impaired in corticotropin-releasing hormone receptor type 1-null mice. Mol Psychiatry (2002) 7:967–74. doi:10.1038/sj.mp.4001143
37. Finotto S, Krieglstein K, Schober A, Deimling F, Lindner K, Bruhl B, et al. Analysis of mice carrying targeted mutations of the glucocorticoid receptor gene argues against an essential role of glucocorticoid signalling for generating adrenal chromaffin cells. Development (1999) 126:2935–44.
38. Jeong KH, Jacobson L, Pacak K, Widmaier EP, Goldstein DS, Majzoub JA. Impaired basal and restraint-induced epinephrine secretion in corticotropin-releasing hormone-deficient mice. Endocrinology (2000) 141:1142–50. doi:10.1210/en.141.3.1142
39. Haidan A, Bornstein SR, Glasow A, Uhlmann K, Lubke C, Ehrhart-Bornstein M. Basal steroidogenic activity of adrenocortical cells is increased 10-fold by coculture with chromaffin cells. Endocrinology (1998) 139:772–80. doi:10.1210/en.139.2.772
40. Haidan A, Bornstein SR, Liu Z, Walsh LP, Stocco DM, Ehrhart-Bornstein M. Expression of adrenocortical steroidogenic acute regulatory (StAR) protein is influenced by chromaffin cells. Mol Cell Endocrinol (2000) 165:25–32. doi:10.1016/S0303-7207(00)00267-7
41. Guse-Behling H, Ehrhart-Bornstein M, Bornstein SR, Waterman MR, Scherbaum WA, Adler G. Regulation of adrenal steroidogenesis by adrenaline: expression of cytochrome P450 genes. J Endocrinol (1992) 135:229–37. doi:10.1677/joe.0.1350229
42. Lukewich MK, Rogers RC, Lomax AE. Divergent neuroendocrine responses to localized and systemic inflammation. Semin Immunol (2014) 26:402–6. doi:10.1016/j.smim.2014.01.004
43. Jones SB, Kotsonis P, Majewski H. Endotoxin enhances norepinephrine release in the rat by peripheral mechanisms. Shock (1994) 2:370–5. doi:10.1097/00024382-199411000-00012
44. Hahn PY, Wang P, Tait SM, Ba ZF, Reich SS, Chaudry IH. Sustained elevation in circulating catecholamine levels during polymicrobial sepsis. Shock (1995) 4:269–73. doi:10.1097/00024382-199510000-00007
45. Lukewich MK, Lomax AE. Endotoxemia enhances catecholamine secretion from male mouse adrenal chromaffin cells through an increase in Ca(2+) release from the endoplasmic reticulum. Endocrinology (2014) 155:180–92. doi:10.1210/en.2013-1623
46. Wurtman RJ, Axelrod J. Adrenaline synthesis: control by the pituitary gland and adrenal glucocorticoids. Science (1965) 150:1464–5. doi:10.1126/science.150.3702.1464
47. Kvetnansky R, Kubovcakova L, Tillinger A, Micutkova L, Krizanova O, Sabban EL. Gene expression of phenylethanolamine N-methyltransferase in corticotropin-releasing hormone knockout mice during stress exposure. Cell Mol Neurobiol (2006) 26:735–54. doi:10.1007/s10571-006-9063-7
48. Ehrhart-Bornstein M, Bornstein SR, Scherbaum WA, Pfeiffer EF, Holst JJ. Role of the vasoactive intestinal peptide in a neuroendocrine regulation of the adrenal cortex. Neuroendocrinology (1991) 54:623–8. doi:10.1159/000125969
49. Bornstein SR, Haidan A, Ehrhart-Bornstein M. Cellular communication in the neuro-adrenocortical axis: role of vasoactive intestinal polypeptide (VIP). Endocr Res (1996) 22:819–29. doi:10.1080/07435809609043781
50. Cobb VJ, Williams BC, Mason JI, Walker SW. Direct stimulation of cortisol secretion from the human NCI H295 adrenocortical cell line by vasoactive intestinal polypeptide. J Hypertens (1997) 15:1735–8. doi:10.1097/00004872-199715120-00081
51. Ait-Ali D, Stroth N, Sen JM, Eiden LE. PACAP-cytokine interactions govern adrenal neuropeptide biosynthesis after systemic administration of LPS. Neuropharmacology (2010) 58:208–14. doi:10.1016/j.neuropharm.2010.03.009
52. Haase M, Willenberg HS, Bornstein SR. Update on the corticomedullary interaction in the adrenal gland. Endocr Dev (2011) 20:28–37. doi:10.1159/000321211
53. Pope GR, Roberts EM, Lolait SJ, O’Carroll AM. Central and peripheral apelin receptor distribution in the mouse: species differences with rat. Peptides (2012) 33(1):139–48. doi:10.1016/j.peptides.2011.12.005
54. Reaux-Le GA, Alvear-Perez R, Zizzari P, Epelbaum J, Bluet-Pajot MT, Llorens-Cortes C. Cellular localization of apelin and its receptor in the anterior pituitary: evidence for a direct stimulatory action of apelin on ACTH release. Am J Physiol Endocrinol Metab (2007) 292:E7–15. doi:10.1152/ajpendo.00521.2005
55. Gallo-Payet N, Roussy JF, Chagnon F, Roberge C, Lesur O. Hypothalamic-pituitary-adrenal axis and multiple and organ dysfunction syndrome in critical illness: a special focus on arginine-vasopressin and apelin. J Organ Dysfunct (2008) 4:216–29. doi:10.1080/17471060802339711
56. Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med (2009) 360:2328–39. doi:10.1056/NEJMra0804635
57. Newson MJ, Roberts EM, Pope GR, Lolait SJ, O’Carroll AM. The effects of apelin on hypothalamic-pituitary-adrenal axis neuroendocrine function are mediated through corticotrophin-releasing factor- and vasopressin-dependent mechanisms. J Endocrinol (2009) 202:123–9. doi:10.1677/JOE-09-0093
58. Rubin de Celis MF, Bornstein SR, Androutsellis-Theotokis A, Andoniadou CL, Licinio J, Wong ML, et al. The effects of stress on brain and adrenal stem cells. Mol Psychiatry (2016) 21:590–3. doi:10.1038/mp.2015.230
59. Steenblock C, Rubin de Celis MF, Androutsellis-Theotokis A, Sue M, Delgadillo Silva LF, Eisenhofer G, et al. Adrenal cortical and chromaffin stem cells: is there a common progeny related to stress adaptation? Mol Cell Endocrinol (2016). doi:10.1016/j.mce.2016.09.011
60. Rubin de Celis MF, Garcia-Martin R, Wittig D, Valencia GD, Enikolopov G, Funk RH, et al. Multipotent glia-like stem cells mediate stress adaptation. Stem Cells (2015) 33:2037–51. doi:10.1002/stem.2002
61. Besedovsky H, Sorkin E. Network of immune-neuroendocrine interactions. Clin Exp Immunol (1977) 27:1–12.
62. Bornstein SR, Ziegler CG, Krug AW, Kanczkowski W, Rettori V, McCann SM, et al. The role of toll-like receptors in the immune-adrenal crosstalk. Ann N Y Acad Sci (2006) 1088:307–18. doi:10.1196/annals.1366.027
63. Schober A, Huber K, Fey J, Unsicker K. Distinct populations of macrophages in the adult rat adrenal gland: a subpopulation with neurotrophin-4-like immunoreactivity. Cell Tissue Res (1998) 291:365–73. doi:10.1007/s004410051006
64. Gonzalez-Hernandez JA, Bornstein SR, Ehrhart-Bornstein M, Geschwend JE, Adler G, Scherbaum WA. Macrophages within the human adrenal gland. Cell Tissue Res (1994) 278:201–5. doi:10.1007/BF00414161
65. Naccache A, Louiset E, Duparc C, Laquerriere A, Patrier S, Renouf S, et al. Temporal and spatial distribution of mast cells and steroidogenic enzymes in the human fetal adrenal. Mol Cell Endocrinol (2016) 434:69–80. doi:10.1016/j.mce.2016.06.015
66. Wolkersdorfer GW, Lohmann T, Marx C, Schroder S, Pfeiffer R, Stahl HD, et al. Lymphocytes stimulate dehydroepiandrosterone production through direct cellular contact with adrenal zona reticularis cells: a novel mechanism of immune-endocrine interaction. J Clin Endocrinol Metab (1999) 84:4220–7. doi:10.1210/jcem.84.11.6110
67. Bornstein SR, Rutkowski H, Vrezas I. Cytokines and steroidogenesis. Mol Cell Endocrinol (2004) 215:135–41. doi:10.1016/j.mce.2003.11.022
68. Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124:915–27. doi:10.1016/j.cell.2005.12.039
69. Jennewein C, Tran N, Kanczkowski W, Heerdegen L, Kantharajah A, Drose S, et al. Mortality of septic mice strongly correlates with adrenal gland inflammation. Crit Care Med (2016) 44:e190–9. doi:10.1097/CCM.0000000000001373
70. Kanczkowski W, Chatzigeorgiou A, Samus M, Tran N, Zacharowski K, Chavakis T, et al. Characterization of the LPS-induced inflammation of the adrenal gland in mice. Mol Cell Endocrinol (2013) 371:228–35. doi:10.1016/j.mce.2012.12.020
71. Engstrom L, Rosen K, Angel A, Fyrberg A, Mackerlova L, Pieter KJ, et al. Systemic immune challenge activates an intrinsically regulated local inflammatory circuit in the adrenal gland. Endocrinology (2008) 149:1436–50. doi:10.1210/en.2007-1456
72. Bethin KE, Vogt SK, Muglia LJ. Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc Natl Acad Sci U S A (2000) 97:9317–22. doi:10.1073/pnas.97.16.9317
73. Oates EL, Allaway GP, Armstrong GR, Boyajian RA, Kehrl JH, Prabhakar BS. Human lymphocytes produce pro-opiomelanocortin gene-related transcripts. Effects of lymphotropic viruses. J Biol Chem (1988) 263:10041–4.
74. Andreis PG, Neri G, Belloni AS, Mazzocchi G, Kasprzak A, Nussdorfer GG. Interleukin-1 beta enhances corticosterone secretion by acting directly on the rat adrenal gland. Endocrinology (1991) 129:53–7. doi:10.1210/endo-129-1-53
75. Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M, et al. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med (1994) 180:1243–50. doi:10.1084/jem.180.4.1243
76. Hadid R, Spinedi E, Chautard T, Giacomini M, Gaillard RC. Role of several mediators of inflammation on the mouse hypothalamo-pituitary-adrenal axis response during acute endotoxemia. Neuroimmunomodulation (1999) 6:336–43. doi:10.1159/000026393
77. Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, et al. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol (1997) 150:235–46.
78. Gonzalez-Hernandez JA, Ehrhart-Bornstein M, Spath-Schwalbe E, Scherbaum WA, Bornstein SR. Human adrenal cells express tumor necrosis factor-alpha messenger ribonucleic acid: evidence for paracrine control of adrenal function. J Clin Endocrinol Metab (1996) 81:807–13. doi:10.1210/jc.81.2.807
79. Kanczkowski W, Zacharowski K, Wirth MP, Ehrhart-Bornstein M, Bornstein SR. Differential expression and action of toll-like receptors in human adrenocortical cells. Mol Cell Endocrinol (2009) 300:57–65. doi:10.1016/j.mce.2008.10.028
80. Kanczkowski W, Alexaki VI, Tran N, Grossklaus S, Zacharowski K, Martinez A, et al. Hypothalamo-pituitary and immune-dependent adrenal regulation during systemic inflammation. Proc Natl Acad Sci U S A (2013) 110:14801–6. doi:10.1073/pnas.1313945110
81. Path G, Bornstein SR, Spath-Schwalbe E, Scherbaum WA. Direct effects of interleukin-6 on human adrenal cells. Endocr Res (1996) 22:867–73. doi:10.1080/07435809609043788
82. Gonzalez-Hernandez JA, Bornstein SR, Ehrhart-Bornstein M, Gschwend JE, Gwosdow A, Jirikowski G, et al. IL-1 is expressed in human adrenal gland in vivo. Possible role in a local immune-adrenal axis. Clin Exp Immunol (1995) 99:137–41. doi:10.1111/j.1365-2249.1995.tb03484.x
83. Judd AM, Call GB, Barney M, McIlmoil CJ, Balls AG, Adams A, et al. Possible function of IL-6 and TNF as intraadrenal factors in the regulation of adrenal steroid secretion. Ann N Y Acad Sci (2000) 917:628–37. doi:10.1111/j.1749-6632.2000.tb05428.x
84. van de Garde MD, Martinez FO, Melgert BN, Hylkema MN, Jonkers RE, Hamann J. Chronic exposure to glucocorticoids shapes gene expression and modulates innate and adaptive activation pathways in macrophages with distinct changes in leukocyte attraction. J Immunol (2014) 192:1196–208. doi:10.4049/jimmunol.1302138
85. Lefebvre H, Contesse V, Delarue C, Feuilloley M, Hery F, Grise P, et al. Serotonin-induced stimulation of cortisol secretion from human adrenocortical tissue is mediated through activation of a serotonin4 receptor subtype. Neuroscience (1992) 47:999–1007. doi:10.1016/0306-4522(92)90047-6
86. Lefebvre H, Prevost G, Louiset E. Autocrine/paracrine regulatory mechanisms in adrenocortical neoplasms responsible for primary adrenal hypercorticism. Eur J Endocrinol (2013) 169:R115–38. doi:10.1530/EJE-13-0308
87. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi:10.1038/nm1565
88. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209:819–35. doi:10.1084/jem.20112322
89. Tominaga T, Fukata J, Naito Y, Nakai Y, Funakoshi S, Fujii N, et al. Effects of corticostatin-I on rat adrenal cells in vitro. J Endocrinol (1990) 125:287–92. doi:10.1677/joe.0.1250287
90. Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science (2008) 322:1101–4. doi:10.1126/science.1165218
91. Kanczkowski W, Chatzigeorgiou A, Grossklaus S, Sprott D, Bornstein SR, Chavakis T. Role of the endothelial-derived endogenous anti-inflammatory factor Del-1 in inflammation-mediated adrenal gland dysfunction. Endocrinology (2013) 154:1181–9. doi:10.1210/en.2012-1617
92. Spengler RN, Chensue SW, Giacherio DA, Blenk N, Kunkel SL. Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J Immunol (1994) 152:3024–31.
93. Barnett CC Jr, Moore EE, Partrick DA, Silliman CC. Beta-adrenergic stimulation down-regulates neutrophil priming for superoxide generation, but not elastase release. J Surg Res (1997) 70:166–70. doi:10.1006/jsre.1997.5118
94. Gornikiewicz A, Sautner T, Brostjan C, Schmierer B, Fugger R, Roth E, et al. Catecholamines up-regulate lipopolysaccharide-induced IL-6 production in human microvascular endothelial cells. FASEB J (2000) 14:1093–100.
95. Spengler RN, Allen RM, Remick DG, Strieter RM, Kunkel SL. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol (1990) 145:1430–4.
96. Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature (2007) 449:721–5. doi:10.1038/nature06185
97. Flierl MA, Rittirsch D, Gao H, Hoesel LM, Nadeau BA, Day DE, et al. Adverse functions of IL-17A in experimental sepsis. FASEB J (2008) 22:2198–205. doi:10.1096/fj.07-105221
98. Ansurudeen I, Willenberg HS, Kopprasch S, Krug AW, Ehrhart-Bornstein M, Bornstein SR. Endothelial factors mediate aldosterone release via PKA-independent pathways. Mol Cell Endocrinol (2009) 300:66–70. doi:10.1016/j.mce.2008.11.020
99. Hinson JP, Vinson GP, Kapas S, Teja R. The role of endothelin in the control of adrenocortical function: stimulation of endothelin release by ACTH and the effects of endothelin-1 and endothelin-3 on steroidogenesis in rat and human adrenocortical cells. J Endocrinol (1991) 128(2):275–80. doi:10.1677/joe.0.1280275
100. Mohn CE, Fernandez-Solari J, De LA, Prestifilippo JP, de la Cal C, Funk R, et al. The rapid release of corticosterone from the adrenal induced by ACTH is mediated by nitric oxide acting by prostaglandin E2. Proc Natl Acad Sci U S A (2005) 102:6213–8. doi:10.1073/pnas.0502136102
101. Cameron LA, Hinson JP. The role of nitric oxide derived from l-arginine in the control of steroidogenesis, and perfusion medium flow rate in the isolated perfused rat adrenal gland. J Endocrinol (1993) 139:415–23. doi:10.1677/joe.0.1390415
102. Jung B, Nougaret S, Chanques G, Mercier G, Cisse M, Aufort S, et al. The absence of adrenal gland enlargement during septic shock predicts mortality: a computed tomography study of 239 patients. Anesthesiology (2011) 115:334–43. doi:10.1097/ALN.0b013e318225cfd7
103. Breslow MJ. Regulation of adrenal medullary and cortical blood flow. Am J Physiol (1992) 262:H1317–30.
104. Engeland WC, Gann DS. Splanchnic nerve stimulation modulates steroid secretion in hypophysectomized dogs. Neuroendocrinology (1989) 50:124–31. doi:10.1159/000125211
105. Jones CT, Edwards AV, Bloom SR. The effect of changes in adrenal blood flow on adrenal cortical responses to adrenocorticotrophin in conscious calves. J Physiol (1990) 429:377–86. doi:10.1113/jphysiol.1990.sp018262
106. Hinson JP, Vinson GP, Pudney J, Whitehouse BJ. Adrenal mast cells modulate vascular and secretory responses in the intact adrenal gland of the rat. J Endocrinol (1989) 121:253–60. doi:10.1677/joe.0.1210253
107. Ansurudeen I, Kopf PG, Gauthier KM, Bornstein SR, Cowley AW Jr, Campbell WB. Aldosterone secretagogues increase adrenal blood flow in male rats. Endocrinology (2014) 155:127–32. doi:10.1210/en.2013-1532
108. Cohen J, Vincent JL, Adhikari NK, Machado FR, Angus DC, Calandra T, et al. Sepsis: a roadmap for future research. Lancet Infect Dis (2015) 15:581–614. doi:10.1016/S1473-3099(15)70112-X
Keywords: stress system, hypothalamic–pituitary–adrenal axis, immune–adrenal crosstalk, ACTH, glucocorticoids
Citation: Kanczkowski W, Sue M and Bornstein SR (2016) Adrenal Gland Microenvironment and Its Involvement in the Regulation of Stress-Induced Hormone Secretion during Sepsis. Front. Endocrinol. 7:156. doi: 10.3389/fendo.2016.00156
Received: 16 September 2016; Accepted: 29 November 2016;
Published: 14 December 2016
Edited by:
Nicole Gallo-Payet, Université de Sherbrooke, CanadaReviewed by:
Tamas Kozicz, Radboud University Nijmegen, NetherlandsOlivier Lesur, Université de Sherbrooke, Canada
Copyright: © 2016 Kanczkowski, Sue and Bornstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Waldemar Kanczkowski, d2FsZGVtYXIua2FuY3prb3dza2lAdW5pa2xpbmlrdW0tZHJlc2Rlbi5kZQ==