 Jake P. Mann
Jake P. Mann Robert K. Semple
Robert K. Semple Matthew J. Armstrong
Matthew J. Armstrong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 16 November 2016
Sec. Systems Endocrinology
Volume 7 - 2016 | https://doi.org/10.3389/fendo.2016.00145
This article is part of the Research Topic Are Rodent Models Fit for Investigation of Human Obesity and Related Diseases? View all 17 articles
Improving understanding of the genetic basis of human non-alcoholic fatty liver disease (NAFLD) has the potential to facilitate risk stratification of affected patients, permit personalized treatment, and inform development of new therapeutic strategies. Animal models have been widely used to interrogate the pathophysiology of, and genetic predisposition to, NAFLD. Nevertheless, considerable interspecies differences in intermediary metabolism potentially limit the extent to which results can be extrapolated to humans. For example, human genome-wide association studies have identified polymorphisms in PNPLA3 and TM6SF2 as the two most prevalent determinants of susceptibility to NAFLD and its inflammatory component (NASH), but animal models of these mutations have had only variable success in recapitulating this link. In this review, we critically appraise selected murine monogenic models of NAFLD, NASH, and hepatocellular carcinoma (HCC) with a focus on how closely they mirror human disease.
Non-alcoholic fatty liver disease (NAFLD) is a pandemic disorder associated with premature morbidity and mortality. Its etiology is multifactorial, with both genetic predisposition and environmental factors playing important parts (1). Animal models have been central to recent major translational research efforts, serving as discovery tools to identify new candidate pathogenic mechanisms, as a means of testing hypotheses arising from human studies, and as pre-clinical models in which to assess potential therapeutic strategies (2). There are multiple different experimental perturbations known to trigger NAFLD in animal models, which have been used to generate all components of the NAFLD spectrum (3–10). Such perturbations include genetic manipulation (e.g., ob/ob mouse), pro-steatotic or pro-inflammatory diets [e.g., methionine–choline deficient (MCD) diet], and toxic insults (e.g., streptozotocin injection). Combinations of these may be used to reflect the multifactorial nature of the human disease (e.g., ob/ob mice fed a MCD diet); however, a detailed discussion of rodent diets is beyond the scope of this review.
While animal models do potentially give key insights into the disease process that cannot be obtained through any other means, it is necessary to temper appreciation of this utility with understanding of the limitations of each of the models used.
We, now, briefly review the current understanding of the genetic architecture of human NAFLD, and the major approaches adopted to modeling it in animals, while critically appraising the utility of these models.
Human NAFLD is defined by the presence of macrovesicular steatosis in more than 5% of hepatocytes in individuals with a history of less than 20 g/day ethanol intake (11). The differentiation of macrovesicular from microvesicular steatosis is a qualitative histological assessment, and the two may coexist in more advanced disease (12). NAFLD exists as a pathological spectrum ranging from non-alcoholic fatty liver (NAFL, also known as “simple” steatosis), which denotes hepatic steatosis in the absence of steatohepatitis, through to non-alcoholic steatohepatitis (NASH), in which there is histological evidence of inflammatory infiltrates, hepatocyte ballooning (featuring Mallory-Denk bodies), and commonly fibrosis of varying severity. Cirrhosis can develop at the most severe end of the spectrum, conferring increased risk of hepatocellular carcinoma (HCC), portal hypertension, and liver failure (13). Longitudinal studies have shown 10–20% of patients with each stage of disease to progress to the following stage in, while there is also an element of reversibility, particularly in NAFL and NASH (14, 15).
A “two-hit hypothesis” has been suggested to account for the pathogenesis of advanced NAFLD, whereby the first “hit” drives development of steatosis and the second triggers inflammation and its sequelae, critically including fibrosis (16).
In principle, hepatic steatosis may result from pre-hepatic derangements, intra-hepatic derangements, or both, as illustrated in simplified form in Figure 1. A comprehensive review of pathways leading to hepatic steatosis is beyond the scope of this article and is provided elsewhere (17–19). In brief, pre-hepatic pathogenic factors encompass increased substrate flux to the liver (e.g., non-esterified free fatty acids (NEFA), monosaccharides, or amino acids) and dysregulation of hormones that act directly on hepatocyte metabolism (e.g., insulin, glucagon and related peptides, and adipokines). Increased “preload” in the form of NEFA delivery generally results from failure of adipose tissue adequately to buffer positive energy balance. This may be a consequence of hyperphagic obesity, in which even normal adipose buffering capacity is overwhelmed, or lipodystrophy, in which adipose energy buffering capacity is pathologically constrained. In some situations, a mixture of these is at play, as in generalized lipodystrophy, where the harmful results of absent adipose tissue are potentiated by concomitant lack of leptin.
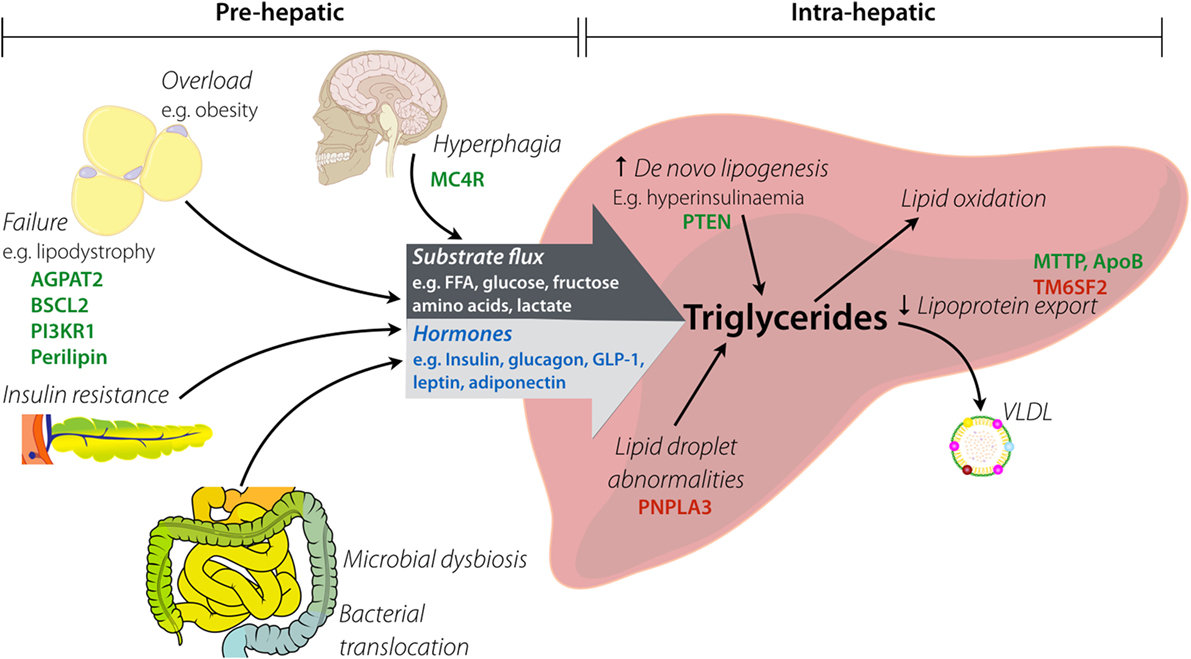
Figure 1. Factors implicated in hepatic steatosis. Pre-hepatic factors lead to increased substrate flux to the liver by both increased load (e.g., raised fatty acid delivery) and altered composition (e.g., increased fructose). Hyperphagia causes provision of excess macronutrients, which contributes to overload of white adipose tissue (via obesity). Lipodystrophy causes functional or anatomical failure of adipose, with the resulting spill over of substrates passing to the liver. Insulin resistance contributes to hormonal changes (e.g., raised insulin, low adiponectin) that alter intra-hepatic metabolism of lipids. Intestinal dysbiosis influences both substrate delivery to the liver and generation of gut-derived hormones (e.g., elevated GLP-1). Key: hormones are in blue, examples of genes involved in monogenic disorders are in green, and examples of genes with pro-steatotic common polymorphisms are in red.
Many pre-hepatic hormonal factors also influence propensity to NAFLD by acting on adipocytes to modulate lipolysis and/or through direct actions on hepatocytes (e.g., insulin, glucagon, glucagon-like peptides). There has been a particular focus on the ability of high levels of insulin, secondary to peripheral insulin resistance, to drive hepatic de novo lipogenesis. Another emerging influence on liver metabolism is the gut microbiome, which may affect gut hormone release and also signal directly through flux of bacterial metabolites such as acetate (20, 21).
Hepatocyte-autonomous (intra-hepatic) defects may also lead to triglyceride accumulation. Such defects may broadly be classified into: those increasing de novo synthesis of triglyceride; those perturbing lipid droplet dynamics, triglyceride mobilization and lipoprotein assembly or secretion; and those impairing catabolism of fatty acids by beta-oxidation. Although reduced ability to catabolize fatty acids via beta-oxidation (e.g., due to Mendelian disorders in key catabolic enzymes, or mitochondrial dysfunction) does result in hepatic steatosis, however, this is usually microvesicular in appearance and has a distinct clinical profile that often includes hypoglycemia, liver failure, and encephalopathy (22). These disorders will, thus, not be discussed further here.
Development of NASH is multifactorial; a comprehensive review of the inflammatory and fibrotic sequelae of hepatic lipid accumulation can be found elsewhere (23–26). Key elements of pathogenesis include oxidative stress (from lipid peroxidation and mitochondrial dysfunction) and activation of pro-inflammatory pathways (e.g., NF-κB) in hepatocytes, but other cellular pathways, including the endoplasmic reticulum stress response, have also been implicated (27). Coactivation of Kupffer cells, sinusoidal endothelium, and hepatic stellate cells gives rise to cytokines that augment inflammation [e.g., tumor necrosis factor alpha (TNFα), interleukin-1/-6] and drive fibrosis [e.g., transforming growth factor beta (TGFβ)] (19, 28, 29). These processes are also exacerbated by pre-hepatic factors, such as adipose inflammation/lipotoxicity, gut bacterial translocation, and endogenous alcohol production.
In the vast majority of patients, NAFLD is a multifactorial condition rooted in obesity and insulin resistance, based on strong clinical association and natural history studies in humans. Pandemic, idiopathic NAFLD is often referred to as “primary” NAFLD (30). Genetics can play a role in each stage of the pathophysiology of NAFLD, as illustrated both by rare monogenic conditions that feature severe NAFLD, and by the association of much more frequent single nucleotide polymorphisms (SNPs) with “common” NAFLD (31–33). The proliferation of recent human genetic findings puts their detailed treatment beyond the scope of this discussion; however, we select a series of mechanistically informative sentinel examples to appraise against rodent models.
Flux of substrates, such as free fatty acids, amino acids, and lactate, provide the building blocks for hepatocyte triglyceride accumulation as well as the energy required for activation of anabolic pathways (see Figure 1). Excess flux can thus be a potent driver for NAFLD. Most attention has been paid to flux of free fatty acids, the product either of lipolysis of triglyceride in adipose tissue or lipolysis of triglyceride in triglyceride-rich lipoproteins such as chylomicron remnants. Key determinants of free fatty acid flux to the liver are thus the dietary intake of fat and the efficiency of fatty acid trapping and storage in adipose tissue. Correspondingly, in states of hyperphagia or adipose tissue insufficiency, free fatty acid flux may be pathologically increased. Of note, “adipose tissue insufficiency” may arise from overload of adipose tissue that is normal or increased in amount, or may arise from frank anatomical deficiency of adipose tissue, or lipodystrophy.
Several human examples of monogenic hyperphagia exist. The first to be discovered was congenital leptin deficiency (34), with loss-of-function mutations in the leptin receptor gene discovered later (35). Leptin is an adipose-derived peptide adipokine that acts on the hypothalamus to signal replete energy stores and suppress appetite (36). It is expressed and secreted by white adipose tissue (WAT) in direct proportion to WAT volume, and serum levels fall commensurately with depletion of adipose stores during starvation. Primary leptin deficiency produces severe, hyperphagic obesity from a young age; however, liver fat accumulation has only rarely been commented on in published studies. In one study, severe NAFLD was identified in an obese leptin-deficient child that resolved quickly on introduction of recombinant leptin therapy (37). The long-term hepatic outcome of untreated human leptin deficiency or leptin receptor mutation is not known; however, leptin receptor polymorphisms have been associated with NASH and insulin resistance in patients with NAFLD (38, 39).
Several other forms of human monogenic obesity are now known. In contrast to genetic leptin or leptin receptor loss-of-function, which are extremely rare, heterozygous mutations in the melanocortin-4-receptor (MC4R) are the most common monogenic cause of obesity, accounting for around 6% of severe, early-onset obesity (40). There are no published reports of the impact of heterozygous MC4R mutations on hepatic steatosis in monogenic obesity, while polymorphisms in MC4R have been associated with alanine aminotransferase and BMI (41), but not with hepatic fat content in population-wide studies (42). Several other genes have been implicated in human monogenic obesity; however, in aggregate, these affect only a very small number of patients, and there is little information on the liver phenotype. Moreover, some of the gene products involved, such as prohormone convertase 1 and pro-opiomelanocortin have pleiotropic roles, not just in appetite control but also in the action of key peripheral hormones (peptide and steroid hormones respectively) which have independent effects on liver fat accumulation, so shall not be considered here.
The sparse attention paid in published human reports to the natural history of NAFLD in human monogenic obesity is likely to be attributable to the rarity of the diseases, to the fact that most patients identified are children in whom clinically overt liver NAFLD has not yet had time to develop and to that fact that, in leptin deficiency, curative therapy with recombinant human leptin is the standard of care. This means that the long-term natural history of NAFLD in leptin deficiency will be extremely difficult to document in future, although determining the long-term liver outcome of MC4R loss-of-function should be both tractable and informative.
In the face of severe hyperphagia, the ability even of “normal” adipose tissue to buffer chronic positive energy balance by trapping and sequestering lipid is finite and is eventually overwhelmed, leaving the liver exposed to excess free fatty acid flux. However, some rare humans have a congenital deficiency of adipose tissue, which we shall consider only in its most severe, generalized form. In this situation, there is effectively absolute adipose failure, and complete lack of energy buffering is compounded by secondary lack of leptin and consequent hyperphagia, as the hypothalamus erroneously interprets very low or absent leptin as an indication of starvation. Around 95% of cases in humans congenital generalized lipodystrophy (CGL) are accounted for by biallelic mutations in either AGPAT2, encoding an enzyme involved in triglyceride synthesis, or in BSCL2, encoding an endoplasmic reticulum protein involved in lipid droplet regulation and adipocyte differentiation (43, 44). A clinical hallmark of CGL is very severe NAFLD, with early development of inflammation, fibrosis, and HCC. Indeed, clinical experience and published case series suggest that complications of advanced liver disease are among the major sources of mortality in CGL (45, 46). Observations in CGL and other acquired and genetic lipodystrophies establish unequivocally that primary disorders of adipose tissue are sufficient to cause the full spectrum of NAFLD in humans (47). This does not prove that adipose dysfunction is the primary mechanism at play in pandemic NAFLD; however, current population-wide data do not rule this out.
Many hormones exert a major influence on lipid accumulation within hepatocytes, although in each case this is only possible in the context of adequate energy charge and substrate flux to the liver, so these cannot easily be teased apart. Hormones of proven importance in altering liver lipid accumulation include insulin, glucagon, and the incretin gut peptides (e.g., GLP-1). In humans, insulin has attracted particular attention as several lines of evidence suggest that insulin, acting through the insulin receptor, stimulates de novo lipogenesis sufficiently to make a major contribution to liver fat accumulation in NAFLD (48–50). Some negative evidence in support of this model comes from the observation that humans with loss-of-function mutations in the insulin receptor itself, although showing very severe insulin resistance, are protected from dyslipidemia and liver fat accumulation (51). Preliminary evidence suggests that this also holds for patients with lipodystrophy and loss-of-function mutations in PIK3R1, a component of phosphatidylinositol-3-kinase involved in insulin signaling (52).
Some genetic abnormalities can influence development of steatosis by hepatocyte-autonomous mechanisms. One of these mechanisms is impaired assembly and export of lipoproteins from hepatocytes, leading to intracellular accumulation of triglycerides. The best examples of this are heterozygous truncating mutations in APOB [encoding apolipoprotein B (ApoB)], causing hypobetalipoproteinaemia and biallelic mutations in MTTP (encoding microsomal triglyceride transfer protein), which cause abetalipoproteinaemia (53). These two conditions have a similar phenotype including NASH, low serum triglycerides, low LDL cholesterol, and neuropathy. Patients often develop progressive NASH with fibrosis even in the absence of diabetes, obesity, or other oxidative stresses (53, 54).
Further evidence in support of altered lipoprotein assembly and secretion as a significant player in NAFLD comes from GWAS of the commoner form of the disease. These have implicated a SNP in TM6SF2 (encoding transmembrane 6 superfamily member 2), which is needed for secretion of very-low density lipoproteins (VLDL), in NAFLD. The risk allele is found in around 1% patients with NAFLD (55) and correlates positively with serum aminotransferase levels (33), hepatic steatosis (31), NASH activity, and fibrosis (but not HCC) (56). Importantly, patients with this SNP have lower circulating LDL cholesterol, lower triglycerides, and reduced incidence of atherosclerotic disease, providing an example of dissociation of NAFLD from dyslipidemia and cardiovascular disease (57). Data relating to the association with insulin resistance and type 2 diabetes are currently inconclusive (58).
Population-wide human genetics has also implicated fundamental abnormalities in lipid droplets in NAFLD: for example, the p.Ile148Met SNP in PNPLA3, whose product is patatin-like phospholipase domain-containing 3, is associated with all stages of NAFLD, from simple steatosis, NASH, fibrosis, and development of HCC (59, 60). PNPLA3 (also known as adiponutrin) is a membrane-bound enzyme expressed at the surface of lipid droplets and on the smooth endoplasmic reticulum (61) and plays a role in lipid droplet dynamics. There has been conflicting evidence on whether this polymorphism is associated with insulin resistance, however.
Inflammation and fibrosis are the hallmarks of complicated NAFLD, and a genetic basis for the susceptibility to these complications is often mooted as the “second hit” needed to drive progression from simple steatosis to inflammatory and fibrotic sequelae. Unsurprisingly, no single gene examples of a primary inflammatory or fibrotic disorders leading to end stage NAFLD have been reported (62, 63); however, the GWAS approach has identified common genetic variants associated with progression of fibrosis (e.g., near the gene for platelet-derived growth factor alpha) or with the development of lobular inflammation (e.g., near genes encoding interleukin-6, or collagen type XIII alpha 1).
The ideal “one stop” animal model of NAFLD would recapitulate the full progression from simple steatosis through NASH to fibrosis, cirrhosis, and HCC. Ideally, the disease process would progress rapidly (in contrast to the human condition) to maximize experimental tractability of the model. However, a valid alternative would be to have a series of different models for different stages of the disease sequence, which may be primarily metabolic, inflammatory, or fibrotic. It has been suggested that the primary goal of rodent models should be to mimic the pathology of NAFLD, including the underlying histopathology (3). On this narrow basis, it has been argued that rodents do not accurately model human NAFLD because of differences in ballooning degeneration and distribution of inflammatory infiltrate in NASH (4).
Many animal models have been described in which different degrees of NAFLD are seen, and we shall consider selected models only as examples, classifying them into two groups according to the broad pathogenic mechanism at play: (1) models of “extrahepatic” NAFLD (Table 1) and (2) models of “intrahepatic” NAFLD (Table 2).
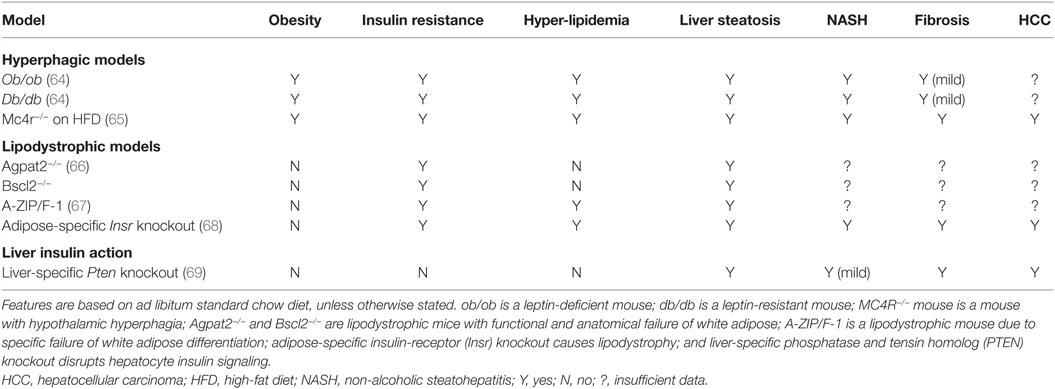
Table 1. Examples of murine genetic models relevant to “pre-hepatic” NAFLD.
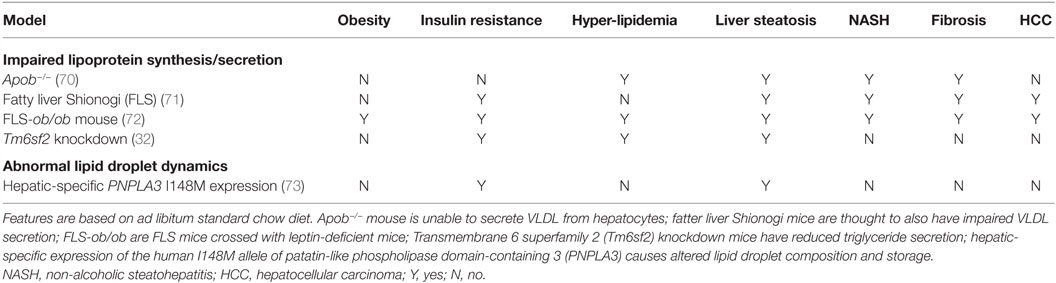
Table 2. Examples of murine genetic models relevant to “intrahepatic” NAFLD.
The severely obese ob/ob mouse strain arose spontaneously in 1949, and was eventually discovered in 1994 to harbor a loss-of-function mutation in the gene encoding leptin, thereby ushering in the modern era of investigation of the neuroscience of appetite control. Given an ad libitum diet, ob/ob mice develop obesity, insulin resistance, hyperglycemia, and hepatic steatosis (74), and they have been become a highly popular model for many aspects of obesity and related disorders, either being studied in isolation, or crossed with other strains of interest in order to determine the effect of severe hyperphagia on mice with different genetic perturbations. The severe obesity of ob/ob mice is attributable to hypothalamic hyperphagia; however, several studies have provided evidence for additional peripheral effects of leptin to modulate insulin sensitivity and metabolism directly in tissues, such as muscle and liver. Whether such peripheral effects are also seen in humans remains to be established beyond doubt (75–77).
The ob/ob mouse rapidly develops NAFL and at 20 weeks old early features of NASH are present (77, 78). NASH may been accelerated by a second “hit” of inflammatory/oxidative stress, using, for example, intraperitoneal injection of lipopolysaccharide (LPS) (79), dietary stressors such as a MCD diet (80), or crossing with another NASH-prone strain, such as the fatty liver Shionogi (FLS) mice, which are discussed later (81). Notably, however, leptin is required for normal immune function, and its deficiency dampens down both innate and acquired immune responses in humans (82, 83), though it seems to play a pro-inflammatory and pro-fibrogenic role in mice (78). This may explain why the inflammatory components of NAFLD appear relatively indolent in ob/ob mice than in more common NAFLD. On a related note, it has also be shown that the ob/ob mouse is relatively resistant to fibrosis (78, 80), partly due to reduced release of TNFα, which is necessary for activation of TGFβ, a key pro-fibrogenic molecule (84–86).
Collectively, these findings caution that, while the ob/ob mouse is a valuable model of primary hyperphagia-driven NAFLD, loss of specific actions of leptin on peripheral metabolism, coupled to some degree of immunosuppression, means that it is likely to deviate from pandemic NAFLD in key respects, especially related to the inflammatory and fibrotic end of the disease spectrum. Similar arguments may apply to leptin-resistant db/db mice, which harbor a splice site mutation abolishing expression of the long form of the leptin receptor; however, db/db mice are reported to exhibit more severe NAFLD than ob/ob mice (64, 80). In practice, a secondary stressor is also usually applied when using db/db mice to study NAFLD, with the MCD diet being widely used (87).
Mice in which Mc4r is genetically ablated have also been widely studied. Their primary defect lies within hypothalamic appetite control pathways, as the melanocortin 4 receptor responds to the neuropeptide α-MSH, which is generated in response to leptin action. Mc4r-null mice feature hyperphagic obesity without pathologically suppressed leptin levels, suggesting that they have the potential to model the extended spectrum of NAFLD more faithfully than ob/ob mice. In keeping with this, it has been reported that Mc4r-knockout mice, when exposed to a high-fat diet, develop not only steatosis but also exuberant NASH with established fibrosis after 20 weeks, progressing to HCC in all mice studied by 1 year (65).
Many different murine genetic models of lipodystrophy have been described. Where direct comparison is possible between mice and humans, it has been found that the common forms of CGL are well modeled in mice, while the situation for human genetic forms of partial lipodystrophy is more complex, and will not be discussed further here (45).
Both Agpat2- and Bscl2-knockout mice have been described (88, 89). Agpat2 encodes 1-acylglycerol-3-phosphate O-acyltransferase 2 and is needed for synthesis of triacylglycerols and glycerolphospholipids in white adipose. Many Agpat2−/− mice die in the first few weeks of life; however, the survivors accurately recapitulate the human condition of lipodystrophy with severe insulin resistance (66). Hepatic steatosis has also been reported to be a major feature of the mice before 16 weeks old, with liver weights twice normal, and modest inflammatory changes seen in addition to pronounced steatosis. More detailed time course studies of the liver disease in this model have not been reported, however.
Bscl2 encodes seipin, which is needed for both differentiation of white adipocytes and normal lipid droplet regulation, and deficiency has been suggested to shift the balance toward release of free fatty acids from adipocytes (89). Bscl2−/− mice display a lipodystrophic phenotype with near complete absence of WAT. Massive hepatic steatosis is a feature of all three Bscl2−/− models described. As in Agpat2 null mice, however, detailed natural history studies of liver pathology have not been reported: at 12 weeks, there is no evidence of NASH but there are no reported data beyond this age (90).
Although not directly modeling human disease, some other murine models have provided powerful evidence for the importance of adipose tissue in protection from NAFLD. One impactful model is the A-ZIP/F-1 (or “AZIP”) mouse, which is an adipose-deficient mouse generated by transgenic overexpression of an artificially engineered dominant negative protein that interferes with critical adipogenic transcription factors. This causes deficiency of white adipose, severe insulin resistance, and hepatic steatosis, although it was said not to feature inflammation at the relatively young age studied (67). It is not known whether these mice develop NAFLD-related fibrosis, in part because of the reduced survival of the mice, which are severely diabetic. A much more recent model featured knockout of the insulin receptor selectively in adipose tissue, knockout being mediated by cre recombinase driven by the adipose-specific adiponectin promoter (68). These mice developed severe lipodystrophy and fatty liver disease very early, and by 12 weeks old, livers demonstrated not only steatosis but also increased ROS, lipid peroxidation, ballooning degeneration of hepatocytes, and elevated serum transaminase levels. By 1 year old the liver accounted for 25% of body weight and showed highly dysplastic liver nodules in addition to worsened inflammation and fibrosis. High fat feeding worsened the liver injury.
Although it is impossible to mimic a primary increase in insulin without inducing complex and potentially confounding changes to systemic metabolism, liver-specific knockout of the insulin receptor does afford the opportunity to test the proposition that liver insulin action is necessary for the development of hepatic steatosis. Indeed, in 2008, it was reported that ablation of the insulin receptor in mouse liver, although inducing severe systemic insulin resistance, did not increase liver triglyceride or liver weight (91).
Conversely, liver-specific knockout of the phosphatase and tensin homolog (PTEN), which is a phosphatidylinositol-3,4,5-triphosphate (PIP3) phosphatase that serves to antagonize insulin’s metabolic actions, produces macrovesicular steatosis, NASH (including Mallory-Denk bodies), fibrosis, and HCC (69). Similarly, other modes of genetic activation of phosphatidylinositol-3-kinase pathway signaling also produce steatosis that progresses to HCC, but without such marked steatohepatitis (92, 93).
Assembly and secretion of VLDL represents a major route for the final disposition of intrahepatocyte triglyceride. Apolipoprotein B (ApoB) is a core component of VLDL particles, and genetic partial deficiency in humans causes familial hypobetalipoproteinaemia. Homozygous loss-of-function mutations of ApoB in mice cause embryonic lethality due to exencephalus. However, several different lines of mice heterozygous for mutated ApoB have been described, and these accurately recapitulate the hepatic steatosis, low serum triglycerides, and low HDL cholesterol of the human condition, although little fibrosis has been reported in the absence of additional pro-inflammatory stimuli (70, 94). Nevertheless, an important caveat is that natural history of liver inflammation and fibrosis has not been reported beyond 12 weeks of age. Similar to humans with hypobetalipoproteinaemia, mice with reduced ApoB function do not show marked insulin resistance, which differentiates them from most patients with NAFLD.
The FLS mouse is a further NAFLD model, which arose spontaneously as a result of inbreeding. FLS mice are non-obese and only mildly insulin resistant, but show marked accumulation of macrovesicular triglyceride with mononuclear inflammatory infiltrate and fibrosis, which eventually results in development of HCC by 13–16 months even without additional carcinogenic stimuli (95, 96). The precise underlying genetic defect is not yet known; however, a defect in microsomal triglyceride transfer protein (MTTP) (97) has been suggested. Precisely, why this model is so susceptible to HCC is not known. Crossing of FLS mice with ob/ob mice results in a model with the full metabolic syndrome and progressive fibrosis (81, 98); this model most closely recapitulates the whole human spectrum of NAFLD in a practical, experimental timeframe.
As discussed, human GWAS have shown a SNP in TM6SF2 to be associated with NAFLD (99). Reminiscent of hypobetalipoproteinaemia, however, carriers of the risk-conferring T-allele seem relatively protected from atherosclerotic disease despite increased lipid fat content. TM6SF2 is a membrane-bound protein located on the endoplasmic reticulum involved in the secretion of VLDL from hepatocytes (100). Germline knockout of Tm6sf− has not yet been described in mice; however, adeno-associated virus-mediated knockdown selectively in mouse liver resulted in increased hepatic triglyceride content and reduced VLDL secretion (32). Moreover transgenic hepatocyte-specific expression of human wild-type TM6SF2 in mice caused an increase in serum LDL cholesterol and lower HDL cholesterol (101); however, the full spectrum of NAFLD has not been demonstrated in this model.
The p.Ile148Met variant in human PNPLA3, found in around 20% of the population, is associated with NAFLD progression (55). Therefore, efforts have been made to generate a rodent model that recapitulates this genetic predisposing factor and to elucidate the role of PNPLA3 in NAFLD. PNPLA3 is expressed in many tissues in mice, including WAT and liver (102). Expression is suppressed upon fasting (103) and upregulated following a carbohydrate load (104), which is believed to be mediated by steroid regulatory element-binding protein-1c (SREBP-1c) and carbohydrate-response element-binding protein (ChREBP). PNPLA3 was initially thought to function as a lipase and be involved in the release of triglycerides from intracellular lipid droplets (105–107), so it was hypothesized that reduced activity would increase hepatocellular triglyceride content (108).
If reduced PNPLA3 activity was to accelerate NAFLD, Pnpla3 knockout mice would be expected to have severely fatty livers; however, in fact, they have no evidence of NAFLD (109). Indeed, there is no discernible difference between Pnpla3−/− and wild-type mice even when fed high-fat diet or crossed onto the Lepob/ob background. It should be noted, however, that mice show the highest Pnpla3 expression in WAT, unlike humans, in whom expression is highest in the liver.
In contrast to the knockout mice, mice with hepatic overexpression of human PNPLA3 p.Ile148Met show increased hepatic triglyceride content and fatty acid synthesis (73, 110). They develop steatosis only on a normal chow or sucrose diet, but not on a high-fat diet, which suggests that the mutant PNPLA3 acts upon de novo fatty acids, rather than re-absorbed circulating non-esterified fatty acids (111). This is consistent with epidemiological data that suggests fructose-rich diets are more harmful than high-fat diets (112).
The cumulative data from these mouse models suggest that PNPLA3 functions as a lysophosphatidic acid acyltransferase, and that the p.Ile148Met polymorphism increases this activity, stimulating development of hepatic steatosis (113, 114). This would have not been identified without the initially surprising finding in the knockout model. Although mice with hepatic expression of human PNPLA3 p.Ile148Met develop NAFLD, it is not known whether these mice develop HCC with age as reported studies extend to only 12 weeks, however.
The spectrum of human NAFLD disease arises from complex environmental influences allied to genetic predisposition. Human monogenic diseases demonstrate unequivocally that adipose tissue failure and primary genetic defects in intrahepatocyte lipid handling can give rise to NAFLD and its sequelae. Genetic perturbation in rodents may be combined with a pro-inflammatory or pro-steatotic diet to accelerate liver injury and mimic the multifactorial nature of human NAFLD. In general, murine genetic models closely mimic monogenic forms of NAFLD, where mouse and human liver phenotypes have been described in sufficient detail to draw conclusions; however, many gaps exist in descriptions of the natural history of NAFLD associated with several of the monogenic diseases in mice or humans. Findings in both species indicate that there is more than one possible pathogenic route to NAFLD, meaning that study of several different human conditions and their models will be important to tease out common mechanisms of liver damage. A critical advantage of mouse models over humans is that the tremendously powerful technologies available to perturb genes conditionally or in an organ-specific way permits isolation of only some parts of a highly interconnected system. So, while alignment of humans and their murine models could be further refined, murine models are a highly valuable tool in the study of NAFLD.
JM, RS, and MA were all involved in conceptualizing and writing the manuscript, in its critical review, and in approval of the final version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
RS is supported by the Wellcome Trust (grant number WT098498).
1. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol (2013) 10:627–36. doi:10.1038/nrgastro.2013.149
2. Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol (2006) 87:1–16. doi:10.1111/j.0959-9673.2006.00465.x
3. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol (2011) 8:35–44. doi:10.1038/nrgastro.2010.191
4. Imajo K, Yoneda M, Kessoku T, Ogawa Y, Maeda S, Sumida Y, et al. Rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Int J Mol Sci (2013) 14:21833–57. doi:10.3390/ijms141121833
5. Nagarajan P, Mahesh Kumar MJ, Venkatesan R, Majundar SS, Juyal RC. Genetically modified mouse models for the study of nonalcoholic fatty liver disease. World J Gastroenterol (2012) 18:1141–53. doi:10.3748/wjg.v18.i11.1141
6. Nakamura A, Terauchi Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int J Mol Sci (2013) 14:21240–57. doi:10.3390/ijms141121240
7. Riordan JD, Nadeau JH. Modeling progressive non-alcoholic fatty liver disease in the laboratory mouse. Mamm Genome (2014) 25:473–86. doi:10.1007/s00335-014-9521-3
8. Sanches SCL, Ramalho LNZ, Augusto MJ, da Silva DM, Ramalho FS. Nonalcoholic steatohepatitis: a search for factual animal models. Biomed Res Int (2015) 2015:1–13. doi:10.1155/2015/574832
9. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol (2012) 18:2300. doi:10.3748/wjg.v18.i19.2300
10. Willebrords J, Pereira IVA, Maes M, Crespo Yanguas S, Colle I, Van Den Bossche B, et al. Strategies, models and biomarkers in experimental non-alcoholic fatty liver disease research. Prog Lipid Res (2015) 59:106–25. doi:10.1016/j.plipres.2015.05.002
11. Hübscher SG. Histological assessment of non-alcoholic fatty liver disease. Histopathology (2006) 49:450–65. doi:10.1111/j.1365-2559.2006.02416.x
12. Tandra S, Yeh MM, Brunt EM, Vuppalanchi R, Cummings OW, Unalp-Arida A, et al. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol (2011) 55:654–9. doi:10.1016/j.jhep.2010.11.021
13. Brunt EM, Clouston AD. Histologic features of fatty liver disease. In: Bataller R, Caballeria J, editors. Nonalcoholic Steatohepatitis (NASH). Barcelona: Permanyer (2007). p. 95–110.
14. Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology (2015) 61:1547–54. doi:10.1002/hep.27368
15. Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis (2010) 28:155–61. doi:10.1159/000282080
16. Day CP, James OFW. Steatohepatitis: a tale of two “hits”? Gastroenterology (1998) 114:842–5. doi:10.1016/S0016-5085(98)70599-2
17. Berlanga A, Guiu-jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin end Exp Gastroenterol (2014) 7:221–39. doi:10.2147/CEG.S62831
18. Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med (2014) 371:1131–41. doi:10.1056/NEJMra1011035
19. Birkenfeld AL, Shulman GI. Non alcoholic fatty liver disease, hepatic insulin resistance and type 2 diabetes. Hepatology (2014) 59:713–23. doi:10.1002/hep.26672.Non
20. Marchesi JR, Adams DH, Fava F, Hermes GD, Hirschfield GM, Hold G, et al. The gut microbiota and host health: a new clinical frontier. Gut (2016) 65:330–9. doi:10.1136/gutjnl-2015-309990
21. Abu-Shanab A, Quigley EMM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol (2010) 7:691–701. doi:10.1038/nrgastro.2010.172
22. Hautekeete ML, Degott C, Benhamou JP. Microvesicular steatosis of the liver. Acta Clin Belg (1990) 45:311–26. doi:10.1080/17843286.1990.11718105
23. Nobili V, Svegliati-Baroni G, Alisi A, Miele L, Valenti L, Vajro P. A 360-degree overview of paediatric NAFLD: recent insights. J Hepatol (2013) 58:1218–29. doi:10.1016/j.jhep.2012.12.003
24. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol (2015) 12:387–400. doi:10.1038/nrgastro.2015.94
25. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology (2012) 142:711.e–25.e. doi:10.1053/j.gastro.2012.02.003
26. Noureddin M, Anstee QM, Loomba R. Review article: emerging anti-fibrotic therapies in the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther (2016) 43:1109–23. doi:10.1111/apt.13620
27. Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic Res (2015) 49:1405–18. doi:10.3109/10715762.2015.1078461
28. Than NN, Newsome PN. A concise review of non-alcoholic fatty liver disease. Atherosclerosis (2015) 239:192–202. doi:10.1016/j.atherosclerosis.2015.01.001
29. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol (2015) 62:S47–64. doi:10.1016/j.jhep.2014.12.012
30. Dongiovanni P, Valenti L. Genetics of nonalcoholic fatty liver disease. Metabolism (2016) 65(8):1026–37. doi:10.1016/j.metabol.2015.08.018
31. Speliotes EK, Yerges-armstrong LM, Wu J, Hernaez R, Lauren J, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet (2011) 7:e1001324. doi:10.1371/journal.pgen.1001324
32. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Heather H, Tybjaerg-hansen A, et al. Exone-wide association study identifies TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet (2014) 46:352–6. doi:10.1038/ng.2901.Exome-wide
33. Chambers JC, Zhang W, Sehmi J, Li X, Wass N, Van der Harst P, et al. Geno-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet (2012) 43:1131–8. doi:10.1038/ng.970.Genome-wide
34. Montague CT, Farooqi IS, Wareham NJ, Sewter CP, Cheetham CH, Earley AR, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature (1997) 387:903–8. doi:10.1038/43185
35. Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med (2007) 356:237–47. doi:10.1056/NEJMoa063988
36. Fujikawa T, Coppari R. Living without insulin: the role of leptin signaling in the hypothalamus. Front Neurosci (2015) 9:108. doi:10.3389/fnins.2015.00108
37. Von Schnurbein J, Heni M, Moss A, Nagel SA, MacHann J, Muehleder H, et al. Rapid improvement of hepatic steatosis after initiation of leptin substitution in a leptin-deficient girl. Horm Res Paediatr (2013) 79:310–7. doi:10.1159/000348541
38. Zain SM, Mohamed Z, Mahadeva S, Cheah PL, Rampal S, Chin KF, et al. Impact of leptin receptor gene variants on risk of non-alcoholic fatty liver disease and its interaction with adiponutrin gene. J Gastroenterol Hepatol (2013) 28:873–9. doi:10.1111/jgh.12104
39. Aller R, De Luis DA, Izaola O, González Sagrado M, Conde R, Pacheco D, et al. Lys656Asn polymorphism of leptin receptor, leptin levels and insulin resistance in patients with non alcoholic fatty liver disease. Eur Rev Med Pharmacol Sci (2012) 16:335–41. doi:10.3305/nh.2010.25.4.4484
40. Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med (2003) 348:1085–95. doi:10.1056/NEJMoa022050
41. Guan L, Shang X-R, Liu F-H, Song J-Y, Ma J, Wang H-J. Association of INSIG2 rs9308762 with ALT level independent of BMI. J Pediatr Gastroenterol Nutr (2014) 58:155–9. doi:10.1097/MPG.0b013e3182a87b71
42. Haupt A, Thamer C, Heni M, Tschritter O, Machann J, Schick F, et al. Impact of variation near MC4R on whole-body fat distribution, liver fat, and weight loss. Obesity (Silver Spring) (2009) 17:1942–5. doi:10.1038/oby.2009.233
43. Patni N, Garg A. Congenital generalized lipodystrophies – new insights into metabolic dysfunction. Nat Rev Endocrinol (2015) 11:522–34. doi:10.1038/nrendo.2015.123
44. Van Maldergem L, Magré J, Khallouf TE, Gedde-Dahl T, Delépine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet (2002) 39:722–33. doi:10.1136/jmg.39.10.722
45. Savage DB. Mouse models of inherited lipodystrophy. Dis Model Mech (2009) 2:554–62. doi:10.1242/dmm.002907
46. Parker VER, Semple RK. Genetics in endocrinology: genetic forms of severe insulin resistance: what endocrinologists should know. Eur J Endocrinol (2013) 169:R71–80. doi:10.1530/EJE-13-0327
47. Safar Zadeh E, Lungu AO, Cochran EK, Brown RJ, Ghany MG, Heller T, et al. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J Hepatol (2013) 59:131–7. doi:10.1016/j.jhep.2013.02.007
48. Savage DB, Semple RK. Recent insights into fatty liver, metabolic dyslipidaemia and their links to insulin resistance. Curr Opin Lipidol (2010) 21:329–36. doi:10.1097/MOL.0b013e32833b7782
49. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology (2014) 146:726–35. doi:10.1053/j.gastro.2013.11.049
50. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest (2005) 115:1343–51. doi:10.1172/JCI200523621
51. Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest (2009) 119:315–22. doi:10.1172/JCI37432
52. Huang-Doran I, Tomlinson P, Payne F, Gast A, Bottomley W, Harris J, et al. Insulin resistance uncoupled from dyslipidemia due to C-terminal 1 PIK3R1 mutations. JCI Insight (2016) 1(17):e88766. doi:10.1172/jci.insight.88766
53. Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol (2014) 61:891–902. doi:10.1016/j.jhep.2014.05.023
54. Amaro A, Fabbrini E, Kars M, Yue P, Schechtman K, Schonfeld G, et al. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology (2010) 139:149–53. doi:10.1053/j.gastro.2010.03.039
55. Anstee Q, Day CP. The genetics of nonalcoholic fatty liver disease: spotlight on PNPLA3 and TM6SF2. Semin Liver Dis (2015) 35:270–90. doi:10.1055/s-0035-1562947
56. Liu Y-L, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JBS, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun (2014) 5:4309. doi:10.1038/ncomms5309
57. Kahali B, Liu Y-L, Daly AK, Day CP, Anstee QM, Speliotes EK. TM6SF2: catch-22 in the fight against nonalcoholic fatty liver disease and cardiovascular disease? Gastroenterology (2015) 148:679–84. doi:10.1053/j.gastro.2015.01.038
58. Zhou Y, Llauradó G, Orešic M, Hyötyläinen T, Orho-melander M, Yki-järvinen H. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. J Hepatol (2015) 62:657–63. doi:10.1016/j.jhep.2014.10.010
59. Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology (2011) 53:1883–94. doi:10.1002/hep.24283
60. Romeo S, Kozlitina J, Zing C, Persemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet (2008) 40:1461–5. doi:10.1038/ng.257.Genetic
61. Baulande S, Lasnier F, Lucas M, Pairault J. Adiponutrin, a transmembrane protein corresponding to a novel dietary- and obesity-linked mRNA specifically expressed in the adipose lineage. J Biol Chem (2001) 276:33336–44. doi:10.1074/jbc.M105193200
62. Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol (2013) 10:645–55. doi:10.1038/nrgastro.2013.182
63. Macaluso FS, Maida M, Petta S. Genetic background in nonalcoholic fatty liver disease: a comprehensive review. World J Gastroenterol (2015) 21:11088–111. doi:10.3748/wjg.v21.i39.11088
64. Trak-Smayra V, Paradis V, Massart J, Nasser S, Jebara V, Fromenty B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int J Exp Pathol (2011) 92:413–21. doi:10.1111/j.1365-2613.2011.00793.x
65. Itoh M, Suganami T, Nakagawa N, Tanaka M, Yamamoto Y, Kamei Y, et al. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol (2011) 179:2454–63. doi:10.1016/j.ajpath.2011.07.014
66. Cortés V, Curtis DE, Sukumaran S, Shao X, Parameswara V, Rashid S, et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab (2009) 9:165–76. doi:10.1016/j.cmet.2009.01.002
67. Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, et al. Life without white fat: a transgenic mouse. Genes Dev (1998) 12:3168–81. doi:10.1101/gad.12.20.3168
68. Softic S, Boucher J, Solheim M, Fujisaka S, Haering M, Homan E, et al. Lipodystrophy due to adipose tissue specific insulin receptor knockout results in progressive NAFLD. Diabetes (2016) 65(8):2187–200. doi:10.2337/db16-0213
69. Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest (2004) 113:1774–83. doi:10.1172/JCI200420513
70. Farese RV, Ruland SL, Flynn LM, Stokowski RP, Young SG. Knockout of the mouse apolipoprotein B gene results in embryonic lethality in homozygotes and protection against diet-induced hypercholesterolemia in heterozygotes. Proc Natl Acad Sci U S A (1995) 92:1774–8. doi:10.1073/pnas.92.5.1774
71. Soga M, Kishimoto Y, Kawaguchi J, Nakai Y, Kawamura Y, Inagaki S, et al. The FLS mouse: a new inbred strain with spontaneous fatty liver. Lab Anim Sci (1999) 49:269–75.
72. Sugihara T, Koda M, Kishina M, Kato J, Tokunaga S, Matono T, et al. Fatty liver Shionogi-ob/ob mouse: a new candidate for a non-alcoholic steatohepatitis model. Hepatol Res (2013) 43:547–56. doi:10.1111/j.1872-034X.2012.01101.x
73. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3 I148M in mouse liver causes hepatic steatosis. J Clin Invest (2012) 122:4130. doi:10.1172/JCI65179DS1
74. Balland E, Cowley MA. New insights in leptin resistance mechanisms in mice. Front Neuroendocrinol (2015) 39:59–65. doi:10.1016/j.yfrne.2015.09.004
75. Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes (1996) 45:531–5. doi:10.2337/diabetes.45.4.531
76. Paz-Filho G, Mastronardi CA, Parker BJ, Khan A, Inserra A, Matthaei KI, et al. Molecular pathways involved in the improvement of non-alcoholic fatty liver disease. J Mol Endocrinol (2013) 51:167–79. doi:10.1530/JME-13-0072
77. Brix AE, Elgavish A, Nagy TR, Gower BA, Rhead WJ, Wood PA. Evaluation of liver fatty acid oxidation in the leptin-deficient obese mouse. Mol Genet Metab (2002) 75:219–26. doi:10.1006/mgme.2002.3298
78. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol (2002) 37:206–13. doi:10.1016/S0168-8278(02)00102-2
79. Bian Z, Peng Y, You Z, Wang Q, Miao Q, Liu Y, et al. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res (2013) 54:44–54. doi:10.1194/jlr.M026013
80. Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, et al. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol (2004) 287:G1035–43. doi:10.1152/ajpgi.00199.2004
81. Soga M, Hashimoto S, Kishimoto Y, Hirasawa T, Makino S, Inagaki S. Insulin resistance, steatohepatitis, and hepatocellular carcinoma in a new congenic strain of Fatty Liver Shionogi (FLS) mice with the Lep(ob) gene. Exp Anim (2010) 59:407–19. doi:10.1538/expanim.59.407
82. Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab (1999) 84:3686–95. doi:10.1210/jcem.84.10.5999
83. Procaccini C, Pucino V, Mantzoros CS, Matarese G. Leptin in autoimmune diseases. Metabolism (2015) 64:92–104. doi:10.1016/j.metabol.2014.10.014
84. Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, Nagarkatti M, et al. Micro RNA 21 inhibition of SMAD 7 enhances fibrogenesis via leptin mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol (2015) 308:G298–312. doi:10.1152/ajpgi.00346.2014
85. Wang J, Leclercq I, Brymora JM, Xu N, Ramezani-Moghadam M, London RM, et al. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology (2009) 137:713–23. doi:10.1053/j.gastro.2009.04.011.KUPFFER
86. Cayon A, Crespo J, Mayorga M, Guerra A, Pons-Romero F. Increased expression of Ob-Rb and its relationship with the overexpression of TGF-beta 1 and the stage of fibrosis in patients with nonalcoholic steatohepatitis. Liver Int (2006) 26:1065–71. doi:10.1111/J.1478-3231.2006.01337.X
87. Anstee QM, Concas D, Kudo H, Levene A, Pollard J, Charlton P, et al. Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J Hepatol (2010) 53:542–50. doi:10.1016/j.jhep.2010.03.016
88. Yang SJ, Iglayreger HB, Kadouh HC, Bodary PF. Inhibition of the chemokine (C-C motif) ligand 2/chemokine (C-C motif) receptor 2 pathway attenuates hyperglycaemia and inflammation in a mouse model of hepatic steatosis and lipoatrophy. Diabetologia (2009) 52:972–81. doi:10.1007/s00125-009-1309-8
89. Dollet L, Magré J, Cariou B, Prieur X. Function of seipin: new insights from Bscl2/seipin knockout mouse models. Biochimie (2014) 96:166–72. doi:10.1016/j.biochi.2013.06.022
90. Cui X, Wang Y, Tang Y, Liu Y, Zhao L, Deng J, et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet (2011) 20:3022–30. doi:10.1093/hmg/ddr205
91. Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Alemán JO, Suzuki R, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab (2008) 7:125–34. doi:10.1016/j.cmet.2007.11.013
92. Kudo Y, Tanaka Y, Tateishi K, Yamamoto K, Yamamoto S, Mohri D, et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J Hepatol (2011) 55:1400–8. doi:10.1016/j.jhep.2011.03.025
93. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology (2011) 140:1071–83. doi:10.1053/j.gastro.2010.12.006
94. Homanics GE, Smith TJ, Zhang SH, Lee D, Young SG, Maeda N. Targeted modification of the apolipoprotein B gene results in hypobetalipoproteinemia and developmental abnormalities in mice. Proc Natl Acad Sci U S A (1993) 90:2389–93. doi:10.1073/pnas.90.6.2389
95. Soga M, Kishimoto Y, Kawamura Y, Inagaki S, Makino S, Saibara T. Spontaneous development of hepatocellular carcinomas in the FLS mice with hereditary fatty liver. Cancer Lett (2003) 196:43–8. doi:10.1016/S0304-3835(03)00213-1
96. Oze-Fukai A, Fujisawa T, Sugimoto K, Nojima K, Shindo N, Shimoyoshi S, et al. A novel mouse model for type 2 diabetes and non-alcoholic fatty liver disease: spontaneous amelioration of diabetes by augmented beta cell mass. Endocr J (2009) 56:227–34. doi:10.1507/endocrj.K08E-315
97. Shindo N, Fujisawa T, Sugimoto K, Nojima K, Oze-Fukai A, Yoshikawa Y, et al. Involvement of microsomal triglyceride transfer protein in nonalcoholic steatohepatitis in novel spontaneous mouse model. J Hepatol (2010) 52:903–12. doi:10.1016/j.jhep.2009.12.033
98. Kato J, Koda M, Kishina M, Tokunaga S, Matono T, Sugihara T, et al. Therapeutic effects of angiotensin II type 1 receptor blocker, irbesartan, on non-alcoholic steatohepatitis using FLS-ob/ob male mice. Int J Mol Med (2012) 30:107–13. doi:10.3892/ijmm.2012.958
99. Roh Y, Loomba R, Seki E. The TM6SF2 variants, novel genetic predictors for nonalcoholic steatohepatitis. Gastroenterology (2013) 148:252–4. doi:10.1053/j.gastro.2014.11.014
100. Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A (2014) 111:8913–8. doi:10.1073/pnas.1323785111
101. Holmen O, Zhang H, Fan Y, Hovelson D, Schmidt E, Zhou W, et al. Systematic evaluation of coding variation identified a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet (2014) 46:345–51. doi:10.1038/ng.2926.Systematic
102. Lake AC, Sun Y, Li J-L, Kim JE, Johnson JW, Li D, et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res (2005) 46:2477–87. doi:10.1194/jlr.M500290-JLR200
103. Huang Y, He S, Li JZ, Seo Y-K, Osborne TF, Cohen JC, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A (2010) 107:7892–7. doi:10.1073/pnas.1003585107
104. Polson DA, Thompson MP. Adiponutrin mRNA expression in white adipose tissue is rapidly induced by meal-feeding a high-sucrose diet. Biochem Biophys Res Commun (2003) 301:261–6. doi:10.1016/S0006-291X(02)03027-9
105. Hoekstra M, Li Z, Kruijt JK, Van Eck M, Van Berkel TJC, Kuiper J. The expression level of non-alcoholic fatty liver disease-related gene PNPLA3 in hepatocytes is highly influenced by hepatic lipid status. J Hepatol (2010) 52:244–51. doi:10.1016/j.jhep.2009.11.004
106. Perttilä J, Huaman-Samanez C, Caron S, Tanhuanpää K, Staels B, Yki-Järvinen H, et al. PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis. Am J Physiol Endocrinol Metab (2012) 302:E1063–9. doi:10.1152/ajpendo.00125.2011
107. He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem (2010) 285:6706–15. doi:10.1074/jbc.M109.064501
108. Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Ståhlman M, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol (2012) 57:1276–82. doi:10.1016/j.jhep.2012.07.030
109. Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology (2010) 52:1134–42. doi:10.1002/hep.23812
110. Smagris E, BasuRay S, Li J, Huang Y, Lai KV, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology (2015) 61:108–18. doi:10.1002/hep.27242
111. Hao L, Ito K, Huang K, Sae-tan S, Lambert J, Ross A. Shifts in dietary carbohydrate-lipid exposure regulate expression of the non-alcoholic fatty liver disease-associated gene PNPLA3/adiponutrin in mouse liver and HepG2 human liver cells. Metabolism (2014) 63:1352–62. doi:10.1016/j.micinf.2011.07.011.Innate
112. Lim JS, Mietus-Snyder M, Valente A, Schwarz J-M, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol (2010) 7:251–64. doi:10.1038/nrgastro.2010.41
113. Kumari M, Schoiswohl G, Chitraju C, Paar M, Rangrez AY, Wongsiriroj N, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab (2012) 15:691–702. doi:10.1016/j.cmet.2012.04.008.Kumari
Keywords: steatosis, animal model, metabolic syndrome, steatohepatitis, genetic models
Citation: Mann JP, Semple RK and Armstrong MJ (2016) How Useful Are Monogenic Rodent Models for the Study of Human Non-Alcoholic Fatty Liver Disease? Front. Endocrinol. 7:145. doi: 10.3389/fendo.2016.00145
Received: 24 July 2016; Accepted: 01 November 2016;
Published: 16 November 2016
Edited by:
Nils P. Krone, University of Sheffield, UKReviewed by:
Rodolfo A. Rey, Consejo de Investigaciones Científicas y Técnicas (CONICET), ArgentinaCopyright: © 2016 Mann, Semple and Armstrong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert K. Semple, cmtzMTZAY2FtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.