 Mina Thon
Mina Thon Toru Hosoi
Toru Hosoi- Department of Pharmacotherapy, Graduate School of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan
Obesity has emerged as one of the most burdensome conditions in modern society. In this context, understanding the mechanisms controlling food intake is critical. At present, the adipocyte-derived hormone leptin and the pancreatic β-cell-derived hormone insulin are considered the principal anorexigenic hormones. Although leptin and insulin signal transduction pathways are distinct, their regulation of body weight maintenance is concerted. Resistance to the central actions of leptin or insulin is linked to the emergence of obesity and diabetes mellitus. A growing body of evidence suggests a convergence of leptin and insulin intracellular signaling at the insulin–receptor–substrate–phosphatidylinositol-3-kinase level. Moreover, numerous factors mediating the pathophysiology of leptin resistance, a hallmark of obesity, such as endoplasmic reticulum stress, protein tyrosine phosphatase 1B, and suppressor of cytokine signaling 3 also contribute to insulin resistance. Recent studies have also indicated that insulin potentiates leptin-induced signaling. Thus, a greater understanding of the overlapping functions of leptin and insulin in the central nervous system is vital to understand the associated physiological and pathophysiological states. This mini-review focuses on the cross talk and integrative signaling of leptin and insulin in the regulation of energy homeostasis in the brain.
Leptin
Since its discovery in 1994 (1), the 16 kDa adipocyte-derived hormone leptin has attracted interest in the field of obesity research due to its role in the regulation of energy balance (1–3). Leptin, secreted from adipose tissues in proportion to the fat store (4), acts in the hypothalamus to regulate feeding behavior (1–3). Leptin receptors (ObR) exist as six different isoforms (ObRa–ObRf), classified as short (ObRa, ObRc, OBRd, and ObRf), long (ObRb), and secreted (ObRe) isoforms (5, 6) (Figure 1). ObRa, ObRb, ObRc, ObRd, and ObRe were reported to be expressed in mice (6) and ObRa, ObRb, ObRc, ObRf (7), and ObRe (8) in rats. In humans, expression of ObRa, ObRb, ObRc (9), and ObRe (10) has been described. ObRa, ObRb, ObRc, ObRd, and ObRf isoforms are transmembrane receptors that share the characteristic of possessing a box 1 motif-binding Janus kinase 2 (JAK2). ObRe is the only isoform of the leptin receptor lacking a transmembrane domain (10–12). ObRb, the only isoform featuring a full-length intracellular domain for interaction with other proteins functioning as intracellular signal transducers, is considered the main functional receptor of leptin (13, 14), while the roles of the other isoforms remain to be elucidated (10, 15). ObRb expression is very high in the hypothalamus, where its role is well known in mediating body weight regulation. ObRb is a member of the class I cytokine receptor family, which mediates the Janus kinase signal transducer and activator of transcription (JAK-STAT) pathway (16). By binding to ObRb, leptin activates multiple signaling cascades, such as JAK2–STAT3 (14), mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) (17, 18), and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathways (19). The major anti-obesity effects of leptin are initiated by the phosphorylation of JAK2. The activation of JAK2 permits STAT3 phosphorylation and nuclear translocation. Nuclear phospho-STAT3 acts as a transcription factor for STAT3-targeted genes (20, 21).
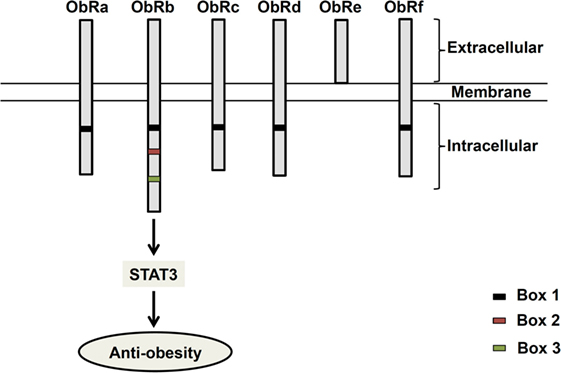
Figure 1. Structure of leptin receptor isoforms. Six different spliced isoforms of the leptin receptors (ObR) have been documented as ObRa–ObRf. All the isoforms share identical extracellular binding domain. ObRb possesses the longest intracellular domain, which is important for leptin signaling.
Although leptin has been considered a candidate for combating obesity, leptin insensitivity represents the barrier to its proper function in obese subjects. Consequently, identifying the mechanisms by which leptin resistance develops is critical. Impairment of leptin signaling is thought to be one such mechanism. Suppressor of cytokine signaling 3 (SOCS3), a mediator of negative feedback to STAT3, is known to attenuate leptin-induced signaling, hence SOCS3-deficient mice displayed increased leptin-induced STAT3 phosphorylation in the hypothalamus (22). Similarly, protein tyrosine phosphatase 1B (PTP1B) dephosphorylates JAK2, thereby inhibiting leptin activity. Consistent with this action, deactivation of PTP1B results in a decrease in body weight and adiposity, and an increase in energy expenditure in mice (23). Thus, SOCS3 and PTP1B are molecular mediators of leptin resistance.
Insulin
The pancreatic hormone insulin is widely known to reduce blood glucose levels via stimulation of glucose uptake by peripheral tissues, such as fat, the liver, and skeletal muscle. Insulin signaling is initiated through its binding with and mediation of protein kinase activity in the beta subunit of the insulin receptor (IR) (24). This stimulation permits phosphorylation of the insulin receptor substrate (IRS) to promote the activation of the PI3K–Akt pathway, which is a major metabolic pathway of insulin (25).
In addition to its peripheral actions, insulin enters the brain from the circulation (26). Insulin in the central nervous system (CNS) affects feeding behavior and energy homeostasis (27–29). Several entry pathways of peripheral insulin into the brain have been reported (30, 31). These include the transport of insulin by brain micovascular endothelial cells from peripheral vessels and the delivery of insulin to cerebrospinal fluid (CSF) via choroid plexus (30–33). In vivo studies have shown the injection of insulin (34) or an insulin-mimetic compound (35) intracerebroventricularly (icv) to reduce food intake in rats. In a similar way of its expression in periphery, IR is expressed in the brain (36). The hypothalamic signaling pathway of insulin activates IRS–PI3K, resulting in the activation of its downstream target protein Akt. Insulin-induced Akt activation elicits Akt’s phosphorylation of the transcription factor forkhead box protein 1 (FoxO1) to suppress the expression of orexigenic neuropeptides (37). This insulin-activated PI3K–Akt pathway may be linked to anorexia, as the administration of PI3K inhibitors has been shown to hinder the effect of insulin on lowering food intake (38).
The Mechanisms of Leptin and Insulin Resistance
A number of mechanisms have been proposed to explain leptin and insulin resistance. These include alteration of leptin and insulin transport across the blood–brain barrier (BBB) (39, 40), alteration of their intracellular signal transduction [e.g., SOCS3, PTP1B, and endoplasmic reticulum (ER) stress] (22, 23, 41–45), and other such abnormalities. In this part, we will focus on the mechanisms-mediated disruption of leptin and insulin signal transduction.
Endoplasmic reticulum stress is one of the mechanisms involved in defective action of leptin and insulin signaling. The ER, an organelle fulfilling diverse cellular functions, plays critical roles in the folding and quality control of proteins. Accumulation of unfolded or misfolded proteins in the ER disrupts ER homeostasis, which in turn causes ER stress. In reaction to this ER stress, cells trigger an adaptive response termed the “unfolded protein response” (UPR). To restore normalcy in ER function, UPR serves to downregulate protein translation, upregulate several chaperone proteins, and activate degradation pathways to clear the unfolded or misfolded protein from the ER (46–49). ER stress is implicated in a wide range of diseases, including metabolic diseases (50), neurodegenerative diseases (51), and cancers (52). Obesity is associated with the activation of inflammatory pathways and stress response signaling. In the obesity model, fat secretes various cytokines (53) and free fatty acids (FFAs) (54). These factors were suggested to cause ER stress (55). Supporting this theory, the pro-inflammatory cytokines TNFα (56), IL-1β (57), and interferon-γ (58) were shown to induce ER stress. Current evidence suggests that overnutrition may contribute to the development of ER stress and the activation of the UPR signaling pathway (59, 60). For instance, excess dietary saturated fatty acids (SFAs) consumption induces ER stress markers (61). Sensitivity to leptin and insulin is reduced in obese rats (62, 63). Others and we have reported that ER stress is an underlying mechanism mediating leptin resistance (43–45). Insulin resistance is also a hallmark of obesity and type 2 diabetes, and ER stress is known to induce insulin resistance by impairing IR signaling (64). Furthermore, ER stress is known to induce beta cell death, consequently, compromising insulin release (60). ER stress may therefore play a key role in leptin and insulin resistance.
Another mediator implicated in the attenuation of leptin and insulin signaling is PTP1B. PTP1B is involved in the negative regulation of both leptin and insulin signaling (65, 66). PTP1B inhibits leptin and insulin activities via dephosphorylation of JAK2 (67) and the activated insulin receptor (42), respectively. In this regard, PTP1B knockout mice increases sensitivity to leptin and insulin, and are resistant to a high-fat diet-induced obesity (42, 68). Therefore, development of potent and specific inhibitors for PTP1B has become interest in the treatment of type 2 diabetes and obesity (69). Over the last decades, diverse PTP1B inhibitors have been developed (70–72).
Besides PTP1B, SOCS3 is another negative regulator of leptin and insulin signaling. SOCS3 inhibits leptin- and insulin-induced signal transduction (41, 73, 74). Deletion of SOCS3 in hypothalamic neurons enhances leptin sensitivity, reduces appetite, and protects from diet-induced obesity (75); while overexpression of SOCS3 in proopiomelanocortin (POMC) neurons leads to hyperphagia and obesity (76, 77). In addition, SOCS3 knockout mice show improvement in glucose tolerance and insulin sensitivity (78). Based on these evidences, molecules that intervene SOCS3 actions would represent a potential therapeutic target in the treatment of obesity and type 2 diabetes.
Possible Interaction of Leptin and Insulin Activity in the CNS
Leptin and insulin induce the JAK–STAT3 and PI3K–Akt pathways, respectively, in hypothalamic neurons. Although leptin and insulin mediate distinct and common signaling pathways, they are both documented as major regulators of energy homeostasis and adiposity. It has been indicated that the disruption of IRs in the brain may cause obesity, insulin resistance, hyperphagia, and hyperleptinemia in mice (79). Moreover, an increase in food intake and obesity was observed in IRS-2-deficient mice despite their high circulating levels of leptin (80). These findings highlight the probable role of insulin in the control of food intake.
Leptin and insulin receptors (IRs) expressed in the CNS mediate the anorexigenic effects of these hormones (81–83). The hypothalamus is the principal site where leptin and insulin exert their regulatory effects on the maintenance of energy homeostasis (Figure 2). Leptin and insulin suppress the activity of the orexigenic neuropeptide Y (NPY)/agouti-related protein (AgRP) neurons, while they stimulate the anorexigenic POMC/cocaine- and amphetamine-related transcript (CART) neurons (84, 85) (Figure 2). Additionally, recent studies have elucidated the effects of leptin and insulin on each other’s actions in the body. For instance, leptin resistance can lead to the inhibition of insulin signaling, while insulin resistance can alter leptin signaling in a hypothalamic cell line (86). Conversely, our study revealed that insulin can potentiate leptin-induced STAT3, a transcription factor critical to a major signaling pathway exerting anti-obesity effects of leptin (87). Thus, it is possible that leptin and insulin may act synergistically to reduce body weight and food intake. If this is the case, cross talk between leptin and insulin would be crucial to the regulation of whole body energy homeostasis.
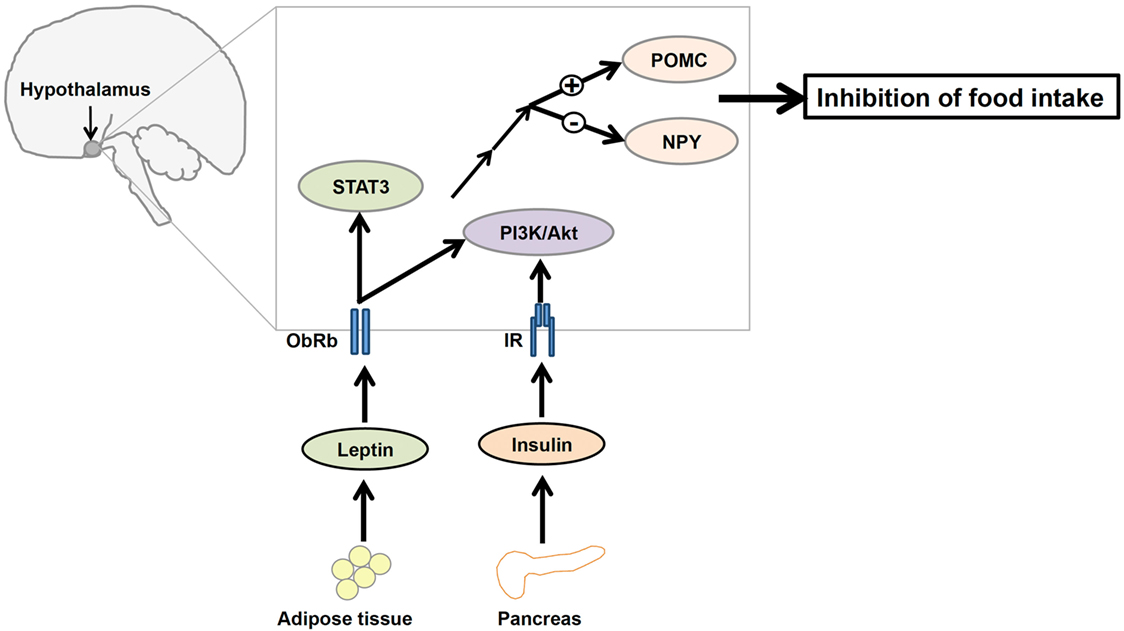
Figure 2. A diagrammatic representation of the integration of leptin and insulin signals in the regulation of energy homeostasis in the central nervous system. Adipocyte-derived hormone leptin and pancreatic hormone insulin bind to leptin receptors (ObRb) and insulin receptors (IRs), respectively, in the hypothalamus. Leptin and insulin regulate the expression of proopiomelanocortin (POMC) and neuropeptide Y (NPY) neurons in the hypothalamus via the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathway to suppress food intake.
ObRb and IR share an aspect of their signaling pathways, namely the involvement of PI3K, suggesting that perhaps the IRS–PI3K interaction is a mechanism by which the regulatory effects of both leptin and insulin on the reduction of food intake are integrated (88, 89) (Figure 2). Although the role of leptin-induced PI3K signaling is not well understood, evidence suggests that icv infusion of a PI3K inhibitor interferes with leptin-mediated reduction of food intake (19). Therefore, understanding this molecular mediator that links leptin and insulin signaling in the hypothalamus may be critical to understanding the regulation of energy homeostasis.
Recent literatures have focused on the role of FoxO1 in the regulation of food intake and energy expenditure. FoxO1 stimulates expression of transcription of orexigenic NPY and AgRP, suppresses the transcription of anorexigenic POMC, and blocks STAT3 action in POMC and AgRP neurons. Mechanistically, the activation of PI3K/Akt signaling pathway by insulin leads to phosphorylation of its downstream mediator, FoxO1. Inactivation of FoxO1 by its phosphorylation results in its translocation from the nucleus to cytoplasm, allowing STAT3 to bind to POMC or AgRP promoter (90, 91). IRS/PI3K/Akt axis is crucial for both leptin and insulin in the CNS (38, 92). In addition, the deletion of FoxO1 in POMC neuron results in weight loss and increases leptin sensitivity (93). Based on these evidences, FoxO1 might be a mediator of the potential cross talk between leptin and insulin in the regulation of food intake.
As mentioned above, SOCS3 and PTP1B are molecules that inhibit leptin signaling. Interestingly, SOCS3 and PTP1B also serve as regulators of insulin signaling. It has been suggested that SOCS3 suppresses IR signaling (41). Conversely, mice lacking PTP1B show enhanced insulin sensitivity (42). Therefore, SOCS3 (94) and/or PTP1B (95, 96) appear to share the suppressive effects on leptin and insulin signaling that are commonly seen in obesity.
Endoplasmic reticulum stress contributes to both leptin and insulin resistance in obesity. Thus, interventions that alleviate ER stress, by, for instance, improving protein folding via increasing chaperone capacity, would offer a potential therapeutic approach for the amelioration of obesity and ER-stress-related diseases. Recent literature proposes the involvement of 78 kDa glucose-regulated protein (GRP78) in the regulation of whole-body insulin sensitivity (97), glucose homeostasis (98), and protection against ER stress (99). Moreover, 4-phenylbutyrate (4-BPA), a chemical chaperone that enhances protein folding (100), was shown to reverse ER-stress-induced leptin resistance (43, 44). Furthermore, BPA alleviated FFAs-induced insulin resistance and beta cell dysfunction (101). Additionally, ob/ob mice, given a chemical chaperone, showed a reduction in ER stress markers, and improved insulin sensitivity and glucose homeostasis (99). Of note, GRP78 levels can be upregulated by insulin (102) and leptin (103) themselves. Based on a growing body of supporting evidence, it seems possible that GRP78 might be a candidate for therapeutic application, working by contributing to the actions of leptin, and insulin in the maintenance of energy homeostasis. On the other hand, the small GTPase Rap1 in the CNS has recently been identified as a key component in development of high-fat diet-induced obesity through ER stress (104). The suppression of Rap1 protect against obesity and metabolic disorders through the regulation of food intake and maintaining leptin and insulin signaling (104). Thus, manipulation of neuronal Rap1 would represent a potential therapeutic target for obesity.
In conclusion, the central signaling of leptin and insulin may be vital in controlling energy homeostasis via feeding suppression. Resistance to the actions of leptin or insulin is associated with the pathophysiology of obesity and type 2 diabetes. An advanced understanding of the physiological and pathophysiological actions of leptin and insulin in the CNS will shed light on potential therapeutic interventions for obesity.
Author Contributions
MT and TH wrote the manuscript. KO checked the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was supported by the Kobayashi International Scholarship Foundation and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; Takeda Science Foundation.
References
1. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372(6505):425–32. doi:10.1038/372425a0
2. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science (1995) 269(5223):543–6. doi:10.1126/science.7624777
3. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science (1995) 269(5223):540–3. doi:10.1126/science.7624776
4. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med (1996) 334(5):292–5. doi:10.1056/NEJM199602013340503
5. Iida M, Murakami T, Ishida K, Mizuno A, Kuwajima M, Shima K. Phenotype-linked amino acid alteration in leptin receptor cDNA from Zucker fatty (fa/fa) rat. Biochem Biophys Res Commun (1996) 222(1):19–26. doi:10.1006/bbrc.1996.0691
6. Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature (1996) 379(6566):632–5. doi:10.1038/379632a0
7. Porzionato A, Rucinski M, Macchi V, Stecco C, Castagliuolo I, Malendowicz LK, et al. Expression of leptin and leptin receptor isoforms in the rat and human carotid body. Brain Res (2011) 1385:56–67. doi:10.1016/j.brainres.2011.02.028
8. Smith JT, Mark PJ, Waddell BJ. Developmental increases in plasma leptin binding activity and tissue Ob-Re mRNA expression in the rat. J Endocrinol (2005) 184(3):535–41. doi:10.1677/joe.1.06045
9. Chua SC Jr, Koutras IK, Han L, Liu SM, Kay J, Young SJ, et al. Fine structure of the murine leptin receptor gene: splice site suppression is required to form two alternatively spliced transcripts. Genomics (1997) 45(2):264–70. doi:10.1006/geno.1997.4962
10. Ge H, Huang L, Pourbahrami T, Li C. Generation of soluble leptin receptor by ectodomain shedding of membrane-spanning receptors in vitro and in vivo. J Biol Chem (2002) 277(48):45898–903. doi:10.1074/jbc.M205825200
11. Lee G, Li C, Montez J, Halaas J, Darvishzadeh J, Friedman JM. Leptin receptor mutations in 129 db3J/db3J mice and NIH facp/facp rats. Mamm Genome (1997) 8(6):445–7. doi:10.1007/s003359900466
12. Li C, Ioffe E, Fidahusein N, Connolly E, Friedman JM. Absence of soluble leptin receptor in plasma from dbPas/dbPas and other db/db mice. J Biol Chem (1998) 273(16):10078–82. doi:10.1074/jbc.273.16.10078
13. Bjørbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem (1997) 272(51):32686–95. doi:10.1074/jbc.272.51.32686
14. Vaisse C, Halaas JL, Horvath CM, Darnell JE Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet (1996) 14(1):95–7. doi:10.1038/ng0996-95
15. Uotani S, Bjørbaek C, Tornøe J, Flier JS. Functional properties of leptin receptor isoforms: internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes (1999) 48(2):279–86. doi:10.2337/diabetes.48.2.279
16. Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, et al. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A (1997) 94(13):7001–5. doi:10.1073/pnas.94.13.7001
17. Takahashi Y, Okimura Y, Mizuno I, Iida K, Takahashi T, Kaji H, et al. Leptin induces mitogen-activated protein kinase-dependent proliferation of C3H10T1/2 cells. J Biol Chem (1997) 272(20):12897–900. doi:10.1074/jbc.272.20.12897
18. Cui H, Cai F, Belsham DD. Leptin signaling in neurotensin neurons involves STAT, MAP kinases ERK1/2, and p38 through c-Fos and ATF1. FASEB J (2006) 20(14):2654–6. doi:10.1096/fj.06-5989fje
19. Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature (2001) 413(6858):794–5. doi:10.1038/35101657
20. Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature (2003) 421(6925):856–9. doi:10.1038/nature01388
21. Piper ML, Unger EK, Myers MG Jr, Xu AW. Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol Endocrinol (2008) 22(3):751–9. doi:10.1210/me.2007-0389
22. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med (2004) 10(7):739–43. doi:10.1038/nm1071
23. Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med (2006) 12(8):917–24. doi:10.1038/nm1435
24. Hendricks SA, Agardh CD, Taylor SI, Roth J. Unique features of the insulin receptor in rat brain. J Neurochem (1984) 43(5):1302–9. doi:10.1111/j.1471-4159.1984.tb05387.x
25. Kanai F, Ito K, Todaka M, Hayashi H, Kamohara S, Ishii K, et al. Insulin-stimulated GLUT4 translocation is relevant to the phosphorylation of IRS-1 and the activity of PI3-kinase. Biochem Biophys Res Commun (1993) 195(2):762–8. doi:10.1006/bbrc.1993.2111
26. Baura GD, Foster DM, Porte D Jr, Kahn SE, Bergman RN, Cobelli C, et al. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest (1993) 92(4):1824–30. doi:10.1172/JCI116773
27. Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature (1979) 282(5738):503–5. doi:10.1038/282503a0
28. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci (2002) 5(6):566–72. doi:10.1038/nn861
29. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med (2002) 8(12):1376–82. doi:10.1038/nm798
30. Gray SM, Meijer RI, Barrett EJ. Insulin regulates brain function, but how does it get there? Diabetes (2014) 63(12):3992–7. doi:10.2337/db14-0340
31. Meijer RI, Gray S, Aylor K, Barrett EJ. Pathways for insulin access to the brain: the role of the microvascular endothelial cell. Am J Physiol Heart Circ Physiol (2016). doi:10.1152/ajpheart.00081.2016
32. Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, et al. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest (2013) 123(3):1299–309. doi:10.1172/JCI67677
33. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med (2012) 4(147):147ra111. doi:10.1126/scitranslmed.3003748
34. Sipols AJ, Baskin DG, Schwartz MW. Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes (1995) 44(2):147–51. doi:10.2337/diab.44.2.147
35. Air EL, Strowski MZ, Benoit SC, Conarello SL, Salituro GM, Guan XM, et al. Small molecule insulin mimetics reduce food intake and body weight and prevent development of obesity. Nat Med (2002) 8(2):179–83. doi:10.1038/nm0202-179
36. Wozniak M, Rydzewski B, Baker SP, Raizada MK. The cellular and physiological actions of insulin in the central nervous system. Neurochem Int (1993) 22(1):1–10. doi:10.1016/0197-0186(93)90062-A
37. Kitamura T, Feng Y, Kitamura YI, Chua SC Jr, Xu AW, Barsh GS, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med (2006) 12(5):534–40. doi:10.1038/nm1392
38. Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes (2003) 52(2):227–31. doi:10.2337/diabetes.52.2.227
39. Golden PL, Maccagnan TJ, Pardridge WM. Human blood-brain barrier leptin receptor. Binding and endocytosis in isolated human brain microvessels. J Clin Invest (1997) 99(1):14–8. doi:10.1172/JCI119125
40. Neumann KF, Rojo L, Navarrete LP, Farías G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links & clinical implications. Curr Alzheimer Res (2008) 5(5):438–47. doi:10.2174/156720508785908919
41. Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem (2000) 275(21):15985–91. doi:10.1074/jbc.275.21.15985
42. Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science (1999) 283(5407):1544–8. doi:10.1126/science.283.5407.1544
43. Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M, et al. Endoplasmic reticulum stress induces leptin resistance. Mol Pharmacol (2008) 4(6):1610–9. doi:10.1124/mol.108.050070
44. Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab (2009) 9(1):35–51. doi:10.1016/j.cmet.2008.12.004
45. Won JC, Jang PG, Namkoong C, Koh EH, Kim SK, Park JY, et al. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity (Silver Spring) (2009) 17(10):1861–5. doi:10.1038/oby.2009.194
46. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature (1999) 397(6716):271–4. doi:10.1038/16729
47. Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell (2000) 101(5):451–4. doi:10.1016/S0092-8674(00)80855-7
48. Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol (2000) 2(7):379–84. doi:10.1038/35017001
49. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol (2007) 8(7):519–29. doi:10.1038/nrm2199
50. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev (2006) 86(4):1133–49. doi:10.1152/physrev.00015.2006
51. Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ (2006) 13(3):385–92. doi:10.1038/sj.cdd.4401778
52. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer (2004) 4(12):966–77. doi:10.1038/nrc1505
53. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol (2004) 25(1):4–7. doi:10.1016/j.it.2003.10.013
54. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am (2008) 37(3):635–46. doi:10.1016/j.ecl.2008.06.007
55. Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology (2006) 147(7):3398–407. doi:10.1210/en.2005-1494
56. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol (2006) 26(8):3071–84. doi:10.1128/MCB.26.8.3071-3084.2006
57. Verma G, Datta M. IL-1beta induces ER stress in a JNK dependent manner that determines cell death in human pancreatic epithelial MIA PaCa-2 cells. Apoptosis (2010) 15(7):864–76. doi:10.1007/s10495-010-0498-4
58. Pirot P, Eizirik DL, Cardozo AK. Interferon-gamma potentiates endoplasmic reticulum stress-induced death by reducing pancreatic beta cell defence mechanisms. Diabetologia (2006) 49(6):1229–36. doi:10.1007/s00125-006-0214-7
59. Kaufman RJ, Scheuner D, Schröder M, Shen X, Lee K, Liu CY, et al. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol (2002) 3(6):411–21. doi:10.1038/nrm829
60. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science (2004) 306(5695):457–61. doi:10.1126/science.1103160
61. Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KL, Shenoy MK, et al. Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep (2016) 14(11):2611–23. doi:10.1016/j.celrep.2016.02.053
62. Cusin I, Rohner-Jeanrenaud F, Stricker-Krongrad A, Jeanrenaud B. The weight-reducing effect of an intracerebroventricular bolus injection of leptin in genetically obese fa/fa rats. Reduced sensitivity compared with lean animals. Diabetes (1996) 45(10):1446–50. doi:10.2337/diab.45.10.1446
63. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146(10):4192–9. doi:10.1210/en.2004-1520
64. Liang L, Chen J, Zhan L, Lu X, Sun X, Sui H, et al. Endoplasmic reticulum stress impairs insulin receptor signaling in the brains of obese rats. PLoS One (2015) 10(5):e0126384. doi:10.1371/journal.pone.0126384
65. Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell (2002) 2(4):497–503. doi:10.1016/S1534-5807(02)00149-1
66. Kenner KA, Anyanwu E, Olefsky JM, Kusari J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling. J Biol Chem (1996) 271(33):19810–6. doi:10.1074/jbc.271.33.19810
67. Kaszubska W, Falls HD, Schaefer VG, Haasch D, Frost L, Hessler P, et al. Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol Cell Endocrinol (2002) 195(1–2):109–18. doi:10.1016/S0303-7207(02)00178-8
68. Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol (2000) 20(15):5479–89. doi:10.1128/MCB.20.15.5479-5489
69. Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat Rev Drug Discov (2002) 1(9):696–709. doi:10.1038/nrd895
70. He R, Zeng LF, He Y, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J (2013) 280(2):731–50. doi:10.1111/j.1742-4658.2012.08718.x
71. Tamrakar AK, Maurya CK, Rai AK. PTP1B inhibitors for type 2 diabetes treatment: a patent review (2011 – 2014). Expert Opin Ther Pat (2014) 24(10):1101–15. doi:10.1517/13543776.2014.947268
72. Liu P, Du Y, Song L, Shen J, Li Q. Discovery of novel, high potent, ABC type PTP1B inhibitors with TCPTP selectivity and cellular activity. Eur J Med Chem (2016) 118:27–33. doi:10.1016/j.ejmech.2016.04.014
73. Bjørbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell (1998) 1(4):619–25. doi:10.1016/S1097-2765(00)80062-3
74. Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem (2002) 277(44):42394–8. doi:10.1074/jbc.C200444200
75. Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, et al. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab (2006) 4(2):123–32. doi:10.1016/j.cmet.2006.06.010
76. Reed AS, Unger EK, Olofsson LE, Piper ML, Myers MG Jr, Xu AW. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes (2010) 59(4):894–906. doi:10.2337/db09-1024
77. Ernst MB, Wunderlich CM, Hess S, Paehler M, Mesaros A, Koralov SB, et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci (2009) 29(37):11582–93. doi:10.1523/JNEUROSCI.5712-08.2009
78. Pedroso JA, Buonfiglio DC, Cardinali LI, Furigo IC, Ramos-Lobo AM, Tirapegui J, et al. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Mol Metab (2014) 3(6):608–18. doi:10.1016/j.molmet.2014.06.001
79. Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science (2000) 289(5487):2122–5. doi:10.1126/science.289.5487.2122
80. Burks DJ, Font de Mora J, Schubert M, Withers DJ, Myers MG, Towery HH, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature (2000) 407(6802):377–82. doi:10.1038/35030105
81. Baskin DG, Breininger JF, Schwartz MW. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes (1999) 48(4):828–33. doi:10.2337/diabetes.48.4.828
82. Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology (1997) 138(10):4489–92. doi:10.1210/endo.138.10.5570
83. Baskin DG, Wilcox BJ, Figlewicz DP, Dorsa DM. Insulin and insulin-like growth factors in the CNS. Trends Neurosci (1988) 11(3):107–11. doi:10.1016/0166-2236(88)90155-5
84. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature (2006) 443(7109):289–95. doi:10.1038/nature05026
85. Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature (2000) 404(6778):661–71. doi:10.1038/35007534
86. Nazarians-Armavil A, Menchella JA, Belsham DD. Cellular insulin resistance disrupts leptin-mediated control of neuronal signaling and transcription. Mol Endocrinol (2013) 27(6):990–1003. doi:10.1210/me.2012-1338
87. Thon M, Hosoi T, Ozawa K. Insulin enhanced leptin-induced STAT3 signaling by inducing GRP78. Sci Rep (2016) 6:34312. doi:10.1038/srep34312
88. Niswender KD, Schwartz MW. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol (2003) 24(1):1–10. doi:10.1016/S0091-3022(02)00105-X
89. Benomar Y, Roy AF, Aubourg A, Djiane J, Taouis M. Cross down-regulation of leptin and insulin receptor expression and signalling in a human neuronal cell line. Biochem J (2005) 388(Pt 3):929–39. doi:10.1042/BJ20041621
90. Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci (2006) 9(7):901–6. doi:10.1038/nn1731
91. Morrison CD, Morton GJ, Niswender KD, Gelling RW, Schwartz MW. Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. Am J Physiol Endocrinol Metab (2005) 289(6):E1051–7. doi:10.1152/ajpendo.00094.2005
92. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest (2005) 115(4):951–8. doi:10.1172/JCI24301
93. Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, et al. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med (2009) 15(10):1195–201. doi:10.1038/nm.2026
94. Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab (2006) 17(9):365–71. doi:10.1016/j.tem.2006.09.007
95. Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell (2002) 2(4):489–95. doi:10.1016/S1534-5807(02)00148-X
96. Elchebly M, Cheng A, Tremblay ML. Modulation of insulin signaling by protein tyrosine phosphatases. J Mol Med (Berl) (2000) 78(9):473–82. doi:10.1007/s001090000141
97. Ye R, Jung DY, Jun JY, Li J, Luo S, Ko HJ, et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes (2010) 59(1):6–16. doi:10.2337/db09-0755
98. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science (2006) 313(5790):1137–40. doi:10.1126/science.1128294
99. Suyama K, Watanabe M, Sakabe K, Okada Y, Matsuyama D, Kuroiwa M, et al. Overexpression of GRP78 protects glial cells from endoplasmic reticulum stress. Neurosci Lett (2011) 504(3):271–6. doi:10.1016/j.neulet.2011.09.045
100. de Almeida SF, Picarote G, Fleming JV, Carmo-Fonseca M, Azevedo JE, de Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem (2007) 282(38):27905–12. doi:10.1074/jbc.M702672200
101. Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes (2011) 60(3):918–24. doi:10.2337/db10-1433
102. Inageda K. Insulin modulates induction of glucose-regulated protein 78 during endoplasmic reticulum stress via augmentation of ATF4 expression in human neuroblastoma cells. FEBS Lett (2010) 584(16):3649–54. doi:10.1016/j.febslet.2010.07.040
103. Thon M, Hosoi T, Yoshii M, Ozawa K. Leptin induced GRP78 expression through the PI3K-mTOR pathway in neuronal cells. Sci Rep (2014) 4:7096. doi:10.1038/srep07096
Keywords: leptin, insulin, GRP78, leptin resistance, endoplasmic reticulum stress, food intake
Citation: Thon M, Hosoi T and Ozawa K (2016) Possible Integrative Actions of Leptin and Insulin Signaling in the Hypothalamus Targeting Energy Homeostasis. Front. Endocrinol. 7:138. doi: 10.3389/fendo.2016.00138
Received: 19 August 2016; Accepted: 07 October 2016;
Published: 20 October 2016
Edited by:
Jae B. Kim, Seoul National University, South KoreaReviewed by:
Sana Siddiqui, University of California San Francisco, USAKi Woo Kim, Yonsei University, South Korea
Copyright: © 2016 Thon, Hosoi and Ozawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toru Hosoi, dG9ydWhAaGlyb3NoaW1hLXUuYWMuanA=;
Koichiro Ozawa, b3phd2FrQGhpcm9zaGltYS11LmFjLmpw