 Andre Machado Xavier
Andre Machado Xavier Aparecida Kataryna Olimpio Anunciato
Aparecida Kataryna Olimpio Anunciato Tatiana Rosado Rosenstock
Tatiana Rosado Rosenstock Isaias Glezer
Isaias Glezer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 19 April 2016
Sec. Cellular Endocrinology
Volume 7 - 2016 | https://doi.org/10.3389/fendo.2016.00031
This article is part of the Research Topic Updates and new concepts in regulation of pro-inflammatory gene expression by steroid hormones View all 8 articles
Glucocorticoids (GCs) are potent anti-inflammatory compounds that have been extensively used in clinical practice for several decades. GC’s effects on inflammation are generally mediated through GC receptors (GRs). Signal transduction through these nuclear receptors leads to dramatic changes in gene expression programs in different cell types, typically due to GR binding to DNA or to transcription modulators. During the last decade, the view of GCs as exclusive anti-inflammatory molecules has been challenged. GR negative interference in pro-inflammatory gene expression was a landmark in terms of molecular mechanisms that suppress immune activity. In fact, GR can induce varied inhibitory molecules, including a negative regulator of Toll-like receptors pathway, or subject key transcription factors, such as NF-κB and AP-1, to a repressor mechanism. In contrast, the expression of some acute-phase proteins and other players of innate immunity generally requires GR signaling. Consequently, GRs must operate context-dependent inhibitory, permissive, or stimulatory effects on host defense signaling triggered by pathogens or tissue damage. This review aims to disclose how contradictory or comparable effects on inflammatory gene expression can depend on pharmacological approach (including selective GC receptor modulators; SEGRMs), cell culture, animal treatment, or transgenic strategies used as models. Although the current view of GR-signaling integrated many advances in the field, some answers to important questions remain elusive.
An inflammatory reaction relies on both fast triggering and tight control over intensity. Failure on fine-tuning immune cells activation and pro-inflammatory signaling can lead to unnecessary expended energy and tissue damage. Endogenous glucocorticoids (GCs), as cortisol in human and corticosterone in rodents, are key hormones produced by the adrenal cortex that regulate innate immune responses. Pioneering work showed that a hormone from adrenal cortex was necessary to keep adrenolectomized animals alive after bacterial challenge (1), while a specific steroidal corticoid reversed the effects associated with adrenolectomy (2). These early studies suggest either these hormones (i.e., GCs) are necessary to mount an efficient self-defense response or counteract the aggressive side effects of this crucial reaction. While the first hypothesis will be discussed later, the second statement received attention when Phillip Hench assumed that arthritis remission could be related to high GCs blood levels. The later was verified to be true, as demonstrated by the reduction of rheumatoid arthritis symptoms upon treatment with cortisone [reviewed in Ref. (3, 4)]. An important step toward a molecular mechanism was the involvement of gene expression description in GCs anti-inflammatory effects, specifically the synthesis of an inhibitory protein or peptide (5–7). During the subsequent years, the main anti-inflammatory mechanism associated with GCs was the synthesis of Lipocortin 1 (Annexin A1; ANXA1), a phospholipase-A2 inhibitory protein that prevents the production of downstream inflammatory mediators prostaglandins and leukotrienes [reviewed in Ref. (3)]. The molecular cloning of steroid receptors increased the knowledge regarding GC receptor (GR) binding to the DNA and transcriptional control through GC response elements (GREs). These DNA sequences can mediate transactivation, as described above for ANXA1, or repression as well [reviewed in Ref. (8)]. Different modalities of GR interference in inflammatory signaling were reported, but one particular mechanism was considered more relevant to the understanding of GR functions during inflammation. This pathway drived the development of the concept of repression through tethering, which involves GR inhibitory physical interactions with nuclear activators of pro-inflammatory genes transcription (9–11). More importantly, this alternative paradigm opened the door to include other regulatory models and explore novel ligands with dissociated effects called selective GR agonists (SEGRAs) (also called dissociated GR ligands, and selective GR modulators; SEGRMs – specially in case of non-steroidal molecules) (12). GCs are known to interfere with innate immune signaling that promotes gene expression through engagement of Toll-like receptors (TLRs) and cytokine receptors, which leads to activation of transcription factors: nuclear factor (NF)-κB, activator protein (AP)-1, signal transducers and activators of transcription (STATs), interferon regulatory factors (IRFs), and others (13–16) (Figure 1). Here, we will discuss the perspectives of GR-mediated transcriptional activation or repression through varied mechanisms during the course of innate immune responses. We also aim to contextualize the different models of transcriptional control associated with GCs, which do not only suppresses inflammatory signaling, and point key discrepant observed outcomes and their interpretations.
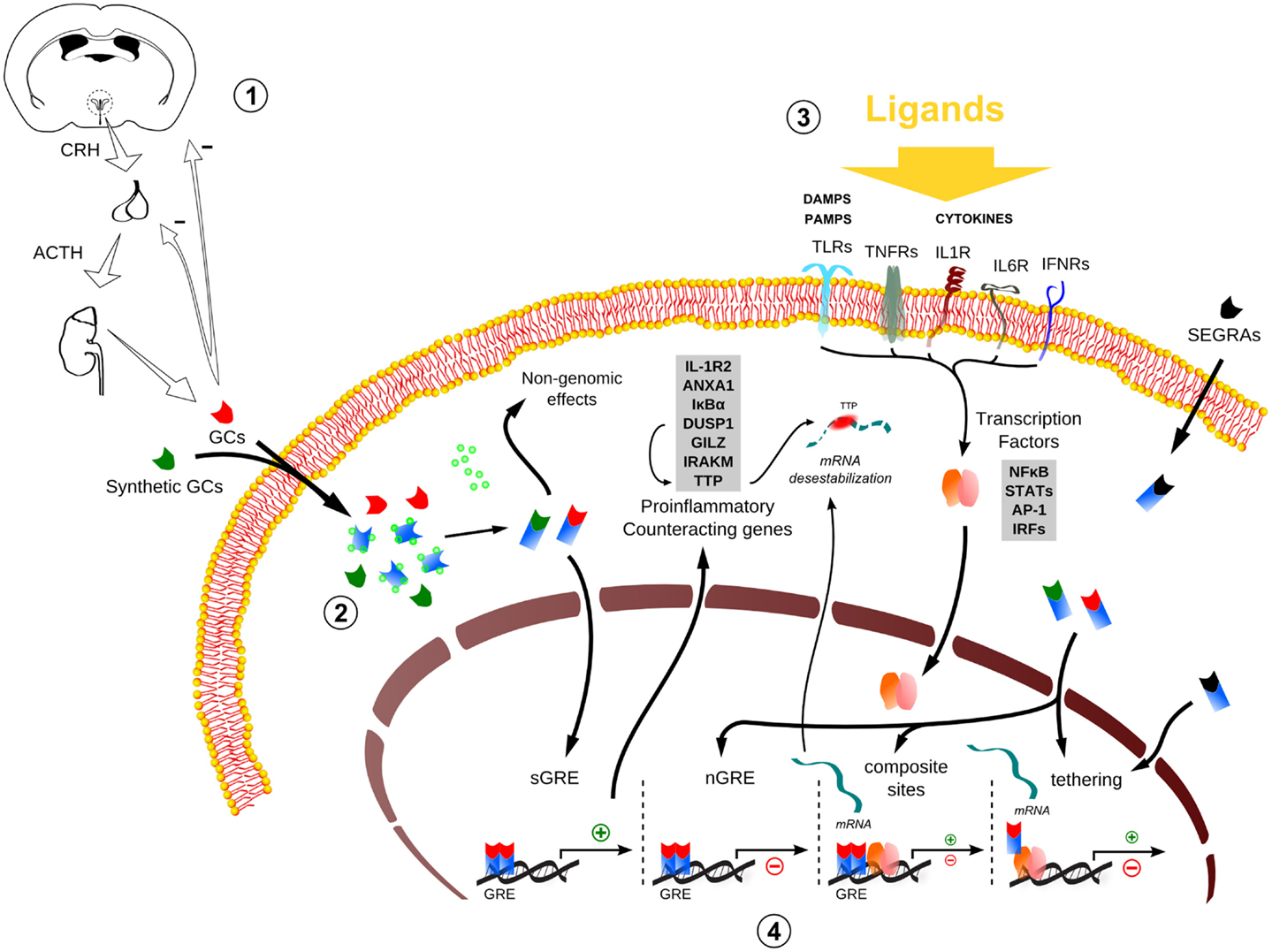
Figure 1. Global scheme of glucocorticoid signaling and transcriptional mechanism during inflammation. 1. Hypothalamus–pituitary–adrenal (HPA) signaling cascade upon stressors. CRH, corticotrophin-releasing hormone; ACTH, adrenocorticotropic hormone; GCs, glucocorticoids. 2. The endogenous/synthetic GCs bind to glucocorticoid receptor (GR) and can act in two ways: non-genomic effects in cytoplasm or translocation into the nucleus, resulting in the modulation of the transcriptional responses (for example, the transactivation of anti-inflammatory genes). Alternatively, selective glucocorticoid receptor agonists (SEGRAs) can act majorly through tethering mechanism. 3. In the context of an inflammatory scenario, cytokines, DAMPS, and PAMPs bind to their respective receptors and activate pro-inflammatory transcription factors (TFs). These TFs translocate to the nucleus and increases the activity at pro-inflammatory genes promoters by GC–GR complex (composite sites, tethering, or compete for DNA-binding sites – not shown). 4. The four main transcriptional mechanisms involved in the inflammatory response: sGRE, nGRE, binding to composite sites and tethering. In the first two modes (sGRE and nGRE), the GC–GR complex modulates the transcription in a GRE-dependent manner activating or repressing genes, if accessible. In the last two modes (composite site and tethering), the GC–GR complex is recruited to GRE sites modulating gene expression in conjunction with TFs (composite site) or interacting directly with TFs (tethering) or coactivators (not shown). Please refer to main text for more details.
The secretion of endogenous GCs results from hypothalamic–pituitary–adrenal (HPA) axis activation. Circadian, stress-related sensory information and inflammation trigger parvocellular corticotropin-releasing hormone (CRH)-secreting neurons in the paraventricular nucleus (PVN) of the hypothalamus. Once CRH reaches the anterior pituitary, responsive cells release adrenocorticotropic hormone into the bloodstream, stimulating the release of GCs from de adrenal cortex. GCs exert a negative feedback action at many levels of the HPA axis, keeping the corticoid at a physiological range (Figure 1) (17). Because steroids are water insoluble, they are transported through the blood to their target tissues mainly complexed with transcortin. GCs freely pass through cell membranes of their target cells, bind to their steroid receptors, and the complexes (steroid–receptor) translocate to nucleus, where they act as transcription factors [reviewed in Ref. (18)].
Glucocorticoid signaling depends largely on nuclear translocation and association of a hormone-bound GR dimer to 6-bp GREs (also called simple GREs), which are specific DNA sequence in the regulatory regions of target genes. The human (h) GR gene (NR3C1) is ubiquitously expressed and functions as a ligand-dependent transcription factor that regulates the expression of GC-responsive genes positively or negatively (18). There are two main highly homologous hGR isoforms: α and β, but others have been described as well. hGRα functions have been considerably detailed, including the complex diversity that results from alternative translation sites (19).
Glucocorticoid receptor is a modular protein, meaning that they have distinct domains: (1) the amino-terminal A/B region, also called immunogenic, functional, or N-terminal domain (NTD), and (2) the C, D, and E regions also known as structural domain, comprising the DNA-binding domain (DBD), the hinge region, and the ligand-binding domain (LBD), respectively. The NTD of the hGRα contains a major transactivation domain, named activation function (AF)-1, which plays an important role in the interaction of the receptor with molecules necessary for the initiation of transcription, such as coactivators, chromatin modulators, and basal transcription machinery. The DBD of the hGRα (region C) contains the ability to bind to GREs apart from sequences important for receptor dimerization and nuclear translocation. The LBD of the hGRα (region E) binds to GC and plays a critical role in the ligand-induced activation of hGRα. The LBD also contains a second transactivation domain, termed AF-2, which is ligand-dependent and has sequences important for receptor dimerization, nuclear translocation, and interaction with coactivators (20, 21).
Coactivators form a bridge between the DNA-bound hGRα and the transcription initiation complex and facilitate the transduction of the GC signal to RNA polymerase II activity. These include: (1) The p300 and the homologous cAMP-responsive element-binding protein (CREB)-binding protein (CBP), which also serve as macromolecular docking “platforms” for transcription factors from several signal transduction cascades, including nuclear receptors, CREB, AP-1, NF-κB, STATs, and others; (2) the p300/CBP-associated factor (p/CAF), which interacts with p300/CBP; and (3) the p160 family of coactivators (22, 23). These coactivators also have intrinsic histone acetyltransferase (HAT) activity, which promotes chromatin decondensation, and allows the transcription initiation complex of the RNA-polymerase II and its ancillary components to initiate and promote transcription (24–27).
The ligand-activated hGRα can modulate gene expression independently of binding to GREs, by interacting as a monomer with other transcription factors, such as AP-1, NF-κB, p53, and STATs (11, 28, 29). This GR nuclear action is independent of DNA-binding sites (DBSs) and involves modulation of transcriptional activity through direct protein–protein interaction (“tethering”) with inducible specific transcription factors by influencing their ability to stimulate or inhibit the transcription rates of the respective target genes (30). The negative regulation by tethering has been misleadingly conceived as the sole modality of “transrepression” during inflammation, which could be a general terminology for repression in trans, including those mediated by the recently characterized negative GREs (IR nGREs), a novel family of evolutionary-conserved cis-acting negative response elements that differs from simple GREs (31). The mechanism of repression by tethering has been the basis to revolutionize GR agonists pharmacological design (see SEGRAs/SEGRMs in Data Sheet S1 in Supplementary Material). In addition to that, GR and other transcription factors can compete for DBS, and there is also the model of composite regulation, in which GR interacts with other transcription factors at adjacent or overlapping DNA regulatory elements (32, 33). In sum, several modalities of GR interaction with DNA, coactivators/corepressors, and other transcription factors govern the complex transcriptional responses to GCs (Figure 1).
Post-translational modifications (PTMs) of GR are also relevant to GCs signaling. The hGR has several phosphorylation sites that typically occurs after binding to the ligand and may determine turnover, subcellular trafficking, target promoter specificity, cofactor interaction, strength and duration of receptor signaling, and receptor stability. Phosphorylation of the GR is a versatile mechanism for modulating and integrating multiple receptor functions (34, 35).
Other PTMs include ubiquitination, which also regulates the motility of GR inside the nucleus. After the binding of the ligand, the GR is destabilized and directed toward the proteasome pathway [reviewed in Ref. (18)]. Acetylation of the GR occurs after ligand-binding and prior to nuclear translocation. The acetylated GR is deacetylated by histone deacetylase 2 (HDAC2), and this deacetylation is necessary for the GR to be able to inhibit NF-κB activation (36). PTMs, including those not mentioned, offer an additional dimension of GR regulation that can be relevant depending on the inflammatory context.
Two basic approaches to interrogate GR functions include mimicking the signaling with agonists, or abolishing the signaling by inhibiting endogenous GCs synthesis or blocking the receptor with antagonists. The genetic counterparts of these strategies are transgene overexpression and genetic deletion/loss-of-function mutations at the level of GR or GCs metabolism. Revealing GR roles is a challenging task, since the outcomes can vary depending on timing (early or delayed), duration (acute or chronic), GCs levels (“physiological” or “supraphysiological”), or tissue/cell type and even species. We provide Data Sheet S1 in Supplementary Material to complement information for readers not familiar with GR agonists, antagonists, and transgenic mice, and briefly point out the limitations for each strategy.
Transgenic mice have already provided valuable information about GR signaling [reviewed in Ref. (37)], including the demonstration of cell-specific GCs signaling contribution during inflammation using promoter-driven Cre-lox P recombination, or the evaluation of GR signaling in dimerization deficient receptor knock-in mice (GRdim). Dissociated agonist (SEGRM) Compound A also have proved that targeting tethered transrepression efficiently promote anti-inflammatory effects (38), corroborating GRdim mice data. However, this simplified model have been questioned (39), and pharmacological investigation of GR-mediated gene transactivation showed that depending on the gene selected for analysis, a given steroid can behave as an antagonist, partial agonist, or full agonist (40). Due the complexity of GR DBSs, ligand-induced conformation changes, PTMs, and protein–protein interactions, a complete landscape of GCs transcriptional control during inflammation will require combined efforts and massive data acquisition to help define new cutting edge therapy rationale. In order to provide additional information about global gene expression in different immune cells, we provide a Table S1 in Supplementary Material comprising the main findings according to the indicated GR agonists (Table S1 in Supplementary Material).
Didactically, GR signaling may be divided into genomic (involving transcription regulation) and non-genomic. The later is faster and less characterized. Although probable, there are no strong evidences that GR engage relevant non-genomic signal transduction to repress pro-inflammatory signaling during innate immune responses (41, 42). GR genomic mechanisms that inhibit pro-inflammatory signaling include: (1) direct transcription of genes that will negatively interfere with pathways involved in the synthesis of inflammatory mediators; (2) direct repression of genes that contribute to immune cells activation; (3) negative interference in transcriptional events engaged by transcriptions factors that transduce pro-inflammatory signals; and (4) synergism between GR and other transcription factors activated during inflammation, ultimately promoting the induction of “anti-inflammatory” gene products. Examples of the first mechanism involves transcription of ANXA1, NFKBIA (IκBα), DUSP1 (MKP-1), GILZ, and ZFP36 (TPP). IκBα induction by GCs highjacks nuclear NF-κB, while MAP kinase phosphatase DUSP1 inactivates p38 kinase pro-inflammatory signaling, and tristetraproline (TPP) can destabilize many cytokine transcripts [reviewed in Ref. (43)]. A recent study showed that DUSP1 promotes activation of TPP destabilizing activity on Tnf, Il1b, and many other pro-inflammatory transcripts (44). For GILZ, we suggest a dedicated review (45). While direct repression (second mechanism) through nGREs during inflammation is not well characterized, some repressed genes associated with GCs anti-inflammatory effects through IR nGRE have been identified [C1qb, C3, Il6, etc., Ref. (31)]. However, an independent group was unable to recognize IR nGRE enrichment in their dataset (46). GR obstruction on the transactivation of pro-inflammatory genes activated by NF-κB/AP-1 (third mechanism) has been largely attributed to tethered transrepression, and to a lesser extent to composite sites or GR competition for DBS or limited coactivators (39, 43, 47). In the case of tethering, GR can associate with NF-κB and prevents the binding of IRF3 or positive transcription elongation factor b to the promoter. Conversely, GR recruits GRIP1 when tethered to AP-1, an event that may depend on nuclear thyroid hormone receptor interactor (48). We may expect that a “positive” GR tethering, recruiting coactivators, or activating the basal transcription machinery operates the expression of anti-inflammatory genes (fourth mechanism). It is not always clear if this is the case for some of the genes mentioned in the first mechanism. IRAK-M (IRAK3), which can block major TLRs pathways effectively, was recently described as a GR-induced gene through cooperation with NF-κB sites (49). Priming of chromatin state or presence and activity of coactivators/corepressor may impact greatly how GR modulates genes expression (50–53). We assume that all these transcriptional anti-inflammatory mechanisms prevail at late phases of an acute inflammatory reaction or when exogenous GR ligands are therapeutically delivered (Figure 2), which would be in agreement with time-dependent gene profiling assays (54).
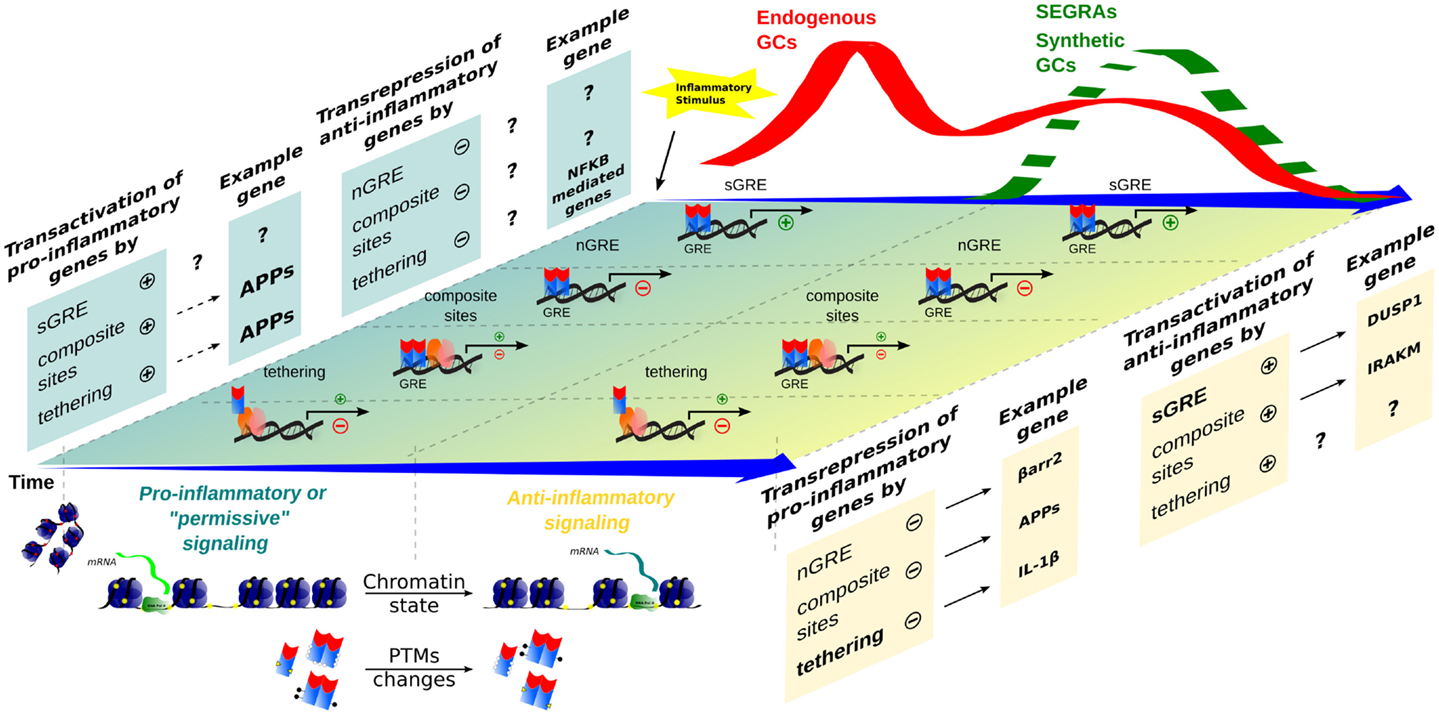
Figure 2. Time-dependent evolution of inflammatory responses as orchestrated by multiple GR-dependent mechanism. At the first moment, after an inflammatory stimulus, acute-phase proteins (APP) and other genes are transcribed through transactivation, contributing to a pro-inflammatory response that correlates with a peak of endogenous GCs levels (red wave); examples of the transcriptional modalities are still poorly described. In a second moment, a subsequent endogenous GCs wave, or administration of synthetic GCs and SEGRAs (green dashed wave), correlates with the prevalence of anti-inflammatory response governed by transactivation (anti-inflammatory genes)/transrepression (pro-inflammatory genes) mechanisms. A decreased expression of pro-inflammatory genes, for instance, IL-1β (tethering) and possible C1q (nGRE), reinforce the anti-inflammatory modality ruled by sGRE mode (DUSP1, GILZ, etc.). No positive tethering mechanism was described for anti-inflammatory genes. In contrast, IRAK-M induction through composite site with NF-κB has been reported (see main text). The chromatin remodeling and different PTMs are present in both phase of inflammatory response, offering the relevant protein interactions and DNA-binding sites for GRs/TFs.
In general, the effects of SEGRMs are less characterized; however, evidences suggest that these dissociated agonists of GR (e.g., Compound A) display an anti-inflammatory activity. In human peripheral blood mononuclear cells, Compound A inhibits the production of pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6 through transrepression (55). In addition, other described SEGRMs, like ZK 216348 and Org 214007-0, showed similar anti-inflammatory effects in comparison to synthetic GC prednisolone (56, 57). According to Table S1 in Supplementary Material, there is a general agreement on a GR-mediated anti-inflammatory effect considering individual immune cells and different agonists.
Although the in vivo response results from a complex interplay between different cells and organs that respond differently to GCs, it is reasonable that inflammation must proceed when endogenous GCs peaks at the beginning of the reaction. It is also understandable that GR agonists are a clinically relevant option after an inflammatory burst, unless chronic treatment is considered.
We pointed earlier the hypothesis that GCs contributes to the mounting of an efficient self-defense response. Sapolsky et al. reviewed this concept and referenced many important studies that have been historically neglected (58). When global gene expression assays became available, the fact that some genes escape GR suppression was not highlighted, since anti-inflammatory effects have always been expected. Several studies employing GR agonists or antagonists showed that various acute-phase proteins (APPs) such as serum amyloid A (SAAs), lipocalin 2 (LCN2), pentraxin 3 (PTX3), ceruloplasmin (CP), etc., are highly dependent on concomitant inflammatory stimulus and GR signaling (46, 59–62). Interestingly, induction of Lcn2 and Ptx3 genes by Gram-negative lipopolissacharyde (LPS) is increased by GCs and depends on IκBζ (63). Frequently, APPs pro- or anti-inflammatory nature is not clearly identified. The acute-phase response is defined as an acute inflammatory response involving non-antibody proteins whose concentration in the plasma increase in response to infection or injury of homeothermic animals (64). As part of inflammation, APPs are products of GR signaling and important players in innate immune responses (65). Fortunately, new studies have focused on the roles of some APPs, indicating that LNC2 may regulate myeloid cell polarization to pro-inflammatory (M1) phenotype (66), or contrarily, deactivate macrophages (67) and suppress cytokines production (68). PTX3 also presents ambiguous functions, since it reinforces complement function and reduces immune cells migration to sites of inflammation (69). Oncostatin M (OSM) and its receptor are potently induced by combined LPS and GR signaling in vivo and in vitro, as shown by the use of agonists and antagonists (70). The signal transduction of this neuropoietic cytokine via the cognate receptor can significantly synergize with pro-inflammatory cytokines (71). It is also interesting that GCs synergism with interferon signaling and STATs have been observed (59, 60), but usually the reported effect goes in the opposite direction (72). While the interpretation of synergism between GR and pro-inflammatory stimuli must be further elucidated regarding APPs, type I interferons, and non-canonical cytokines, other evidences suggests pro-inflammatory actions for GCs.
Busillo and Cidlowski proposed a molecular framework to explain how GR mediates anti-inflammatory and pro-inflammatory effects (73). The antagonistic effects were attributed to different target immune networks: pro-inflammation in innate immune response and anti-inflammatory in adaptive response. Based on several evidences that sensors NLRP3 (inflammasome component) and TLR2 are induced by GR signaling plus a pro-inflammatory trigger, the authors proposed that GCs prepares and reinforces the immune system to respond to pathogens and injury. The interpretation on NLRP3 and TLR2 inductions by GR/pro-inflammatory combination demands caution, because increased IL-1β levels and engagement of TLR signaling depends on multiple levels that can vary over the time. Their relevance must be evaluated by collectively checking if IL1B transcript is reduced (transcription and mRNA stability) or if toll-interacting protein (TOLLIP), a repressor of TLR signaling, is also induced [reported in Ref. (54)].
It has also been proposed by Busillo and Cidlowsky that the contrasting actions of GCs may rely in different signaling properties of target cells, chromatin state (availability of GR DBS), cellular binding partners, etc. Although we do agree with this model and recommend this article for the readers interested in specific cellular responses to GCs, we do not discard that pro- and anti-inflammatory resultants coexist in the same cells as a function of time (Figure 2), GCs levels (74), and interplay with other cell types. In fact, individual immune cells treated with synthetic GCs can present anti- or pro-inflammatory responses indirectly. As demonstrated by Hodrea et al., dexamethasone can promote enhanced phagocytosis by human dendritic cells through upregulation of genes related to this function, leading to subsequent increase in pro-inflammatory response (75). However, in other immune cell types, Dexamethasone exerted anti-inflammatory effects by enhancing apoptosis or by downregulating surface L-selectin in neutrophils (76, 77). Thus, GCs effects on inflammatory cells can be variable through different mechanisms. The literature regarding the effects of SEGRAs/SEGRMs in different immune cells remains quite scarce, but as demonstrated by Pazdrak et al., the responses may diverge in the same cell type depending on the agonist, as observed in eosinophils (78). We suggest that the current view on GR transcriptional modulation would benefit from models that consider additional factors not completely available in the literature yet.
Important molecular achievements have been made in terms of regulatory cis- and trans-components in GCs target genes, which are now recognized as highly heterogeneous sets. Strikingly, not all gene expression data fit in predicted models (54), pointing to unrecognized regulatory determinants. To better characterize how these genes are regulated by GR, important perspectives must be incorporated, such as evidences that this nuclear receptor occupies half-sites as monomers (79), and how different agonists provoke different PTMs on GRs and the respective consequences (12). It is also mysterious, at gene expression level, the ways GRs promote pro-inflammatory or anti-inflammatory effects. What are the exact context-dependent factors that determine predominance of well-known anti-inflammatory actions or complete resistance to GCs (80)? This field of research still awaits new breakthroughs.
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by research grants to IG from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP: 2007/53732-8) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 484869/2012-4). IG is member of the CEPID Redoxoma (FAPESP 2013/07937-8).
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fendo.2016.00031
1. Hartman FA, Scott WJ. Protection of adrenalectomized animals against bacterial intoxication by an extract of the adrenal cortex. J Exp Med (1932) 55:63–9. doi:10.1084/jem.55.1.63
2. Selye H. On the hormonal activity of a steroid compound. Science (1941) 94:94. doi:10.1126/science.94.2430.94
3. Flower RJ. Lipocortin and the mechanism of action of the glucocorticoids. Br J Pharmacol (1988) 94:987–1015. doi:10.1111/j.1476-5381.1988.tb11614.x
4. de Herder WW. Heroes in endocrinology: Nobel Prizes. Endocr Connect (2014) 3:R94–104. doi:10.1530/EC-14-0070
5. Flower RJ, Blackwell GJ. Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature (1979) 278:456–9. doi:10.1038/278456a0
6. Russo-Marie F, Paing M, Duval D. Involvement of glucocorticoid receptors in steroid-induced inhibition of prostaglandin secretion. J Biol Chem (1979) 254:8498–504.
7. Tsurufuji S, Sugio K, Takemasa F. The role of glucocorticoid receptor and gene expression in the anti-inflammatory action of dexamethasone. Nature (1979) 280:408–10. doi:10.1038/280408a0
8. Gustafsson J-A. Historical overview of nuclear receptors. J Steroid Biochem Mol Biol (2015) 157:3–6. doi:10.1016/j.jsbmb.2015.03.004
9. Lucibello FC, Slater EP, Jooss KU, Beato M, Müller R. Mutual transrepression of Fos and the glucocorticoid receptor: involvement of a functional domain in Fos which is absent in FosB. EMBO J (1990) 9:2827–34.
10. Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci U S A (1994) 91:752–6. doi:10.1073/pnas.91.2.752
11. Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol (1995) 15:943–53. doi:10.1128/MCB.15.2.943
12. Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM. Selective glucocorticoid receptor modulation: new directions with non-steroidal scaffolds. Pharmacol Ther (2015) 152:28–41. doi:10.1016/j.pharmthera.2015.05.001
13. Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol (2007) 82:237–43. doi:10.1189/jlb.1206763
14. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi:10.1038/nri3581
15. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol (2011) 12:695–708. doi:10.1038/ni.2065
16. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem (2007) 282:20059–63. doi:10.1074/jbc.R700016200
17. De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev (1998) 19:269–301. doi:10.1210/edrv.19.3.0331
18. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids (2010) 75:1–12. doi:10.1016/j.steroids.2009.09.002
19. Lu NZ, Cidlowski JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol (2006) 16:301–7. doi:10.1016/j.tcb.2006.04.005
20. Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol (2006) 102:11–21. doi:10.1016/j.jsbmb.2006.09.009
21. Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids (2005) 70:407–17. doi:10.1016/j.steroids.2005.02.006
22. Stallcup MR, Kim JH, Teyssier C, Lee Y-H, Ma H, Chen D. The roles of protein-protein interactions and protein methylation in transcriptional activation by nuclear receptors and their coactivators. J Steroid Biochem Mol Biol (2003) 85:139–45. doi:10.1016/S0960-0760(03)00222-X
23. York B, O’Malley BW. Steroid receptor coactivator (SRC) family: masters of systems biology. J Biol Chem (2010) 285:38743–50. doi:10.1074/jbc.R110.193367
24. Auboeuf D, Hönig A, Berget SM, O’Malley BW. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science (2002) 298:416–9. doi:10.1126/science.1073734
25. McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell (2002) 108:465–74. doi:10.1016/S0092-8674(02)00641-4
26. McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev (1999) 20:321–44. doi:10.1210/edrv.20.3.0366
27. McKenna NJ, Xu J, Nawaz Z, Tsai SY, Tsai MJ, O’Malley BW. Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J Steroid Biochem Mol Biol (1999) 69:3–12. doi:10.1016/S0960-0760(98)00144-7
28. Jonat C, Rahmsdorf HJ, Park KK, Cato AC, Gebel S, Ponta H, et al. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell (1990) 62:1189–204. doi:10.1016/0092-8674(90)90395-U
29. Kino T, Chrousos GP. Tissue-specific glucocorticoid resistance-hypersensitivity syndromes: multifactorial states of clinical importance. J Allergy Clin Immunol (2002) 109:609–13. doi:10.1067/mai.2002.123708
30. Göttlicher M, Heck S, Herrlich P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J Mol Med (Berl) (1998) 76:480–9. doi:10.1007/s001090050242
31. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell (2011) 145:224–41. doi:10.1016/j.cell.2011.03.027
32. Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science (1990) 249:1266–72. doi:10.1126/science.2119054
33. Hoeppner MA, Mordacq JC, Linzer DI. Role of the composite glucocorticoid response element in proliferin gene expression. Gene Expr (1995) 5:133–41.
34. Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci (2004) 1024:86–101. doi:10.1196/annals.1321.007
35. Orti E, Hu LM, Munck A. Kinetics of glucocorticoid receptor phosphorylation in intact cells. Evidence for hormone-induced hyperphosphorylation after activation and recycling of hyperphosphorylated receptors. J Biol Chem (1993) 268:7779–84.
36. Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med (2006) 203:7–13. doi:10.1084/jem.20050466
37. Kleiman A, Tuckermann JP. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol (2007) 275:98–108. doi:10.1016/j.mce.2007.05.009
38. De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, et al. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A (2005) 102:15827–32. doi:10.1073/pnas.0505554102
39. Newton R, Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol (2007) 72:799–809. doi:10.1124/mol.107.038794
40. Joshi T, Johnson M, Newton R, Giembycz M. An analysis of glucocorticoid receptor-mediated gene expression in BEAS-2B human airway epithelial cells identifies distinct, ligand-directed, transcription profiles with implications for asthma therapeutics. Br J Pharmacol (2015) 172:1360–78. doi:10.1111/bph.13014
41. Ayroldi E, Cannarile L, Migliorati G, Nocentini G, Delfino DV, Riccardi C. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB J (2012) 26:4805–20. doi:10.1096/fj.12-216382
42. Schoneveld JLM, Fritsch-Stork RD, Bijlsma JWJ. Nongenomic glucocorticoid signaling: new targets for immunosuppressive therapy? Arthritis Rheum (2011) 63:3665–7. doi:10.1002/art.30635
43. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci (2013) 34:518–30. doi:10.1016/j.tips.2013.07.003
44. Smallie T, Ross EA, Ammit AJ, Cunliffe HE, Tang T, Rosner DR, et al. Dual-specificity phosphatase 1 and tristetraprolin cooperate to regulate macrophage responses to lipopolysaccharide. J Immunol (2015) 195:277–88. doi:10.4049/jimmunol.1402830
45. Ronchetti S, Migliorati G, Riccardi C. GILZ as a mediator of the anti-inflammatory effects of glucocorticoids. Front Endocrinol (2015) 6:170. doi:10.3389/fendo.2015.00170
46. Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, et al. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell (2013) 49:158–71. doi:10.1016/j.molcel.2012.10.013
47. Chinenov Y, Gupte R, Rogatsky I. Nuclear receptors in inflammation control: repression by GR and beyond. Mol Cell Endocrinol (2013) 380:55–64. doi:10.1016/j.mce.2013.04.006
48. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol (2010) 10:365–76. doi:10.1038/nri2748
49. Miyata M, Lee J-Y, Susuki-Miyata S, Wang WY, Xu H, Kai H, et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat Commun (2015) 6:6062. doi:10.1038/ncomms7062
50. Beck IM, Vanden Berghe W, Vermeulen L, Bougarne N, Vander Cruyssen B, Haegeman G, et al. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-kappaB inhibition. EMBO J (2008) 27:1682–93. doi:10.1038/emboj.2008.95
51. Chinenov Y, Sacta MA, Cruz AR, Rogatsky I. GRIP1-associated SET-domain methyltransferase in glucocorticoid receptor target gene expression. Proc Natl Acad Sci U S A (2008) 105:20185–90. doi:10.1073/pnas.0810863105
52. Flammer JR, Dobrovolna J, Kennedy MA, Chinenov Y, Glass CK, Ivashkiv LB, et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol (2010) 30:4564–74. doi:10.1128/MCB.00146-10
53. Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J (2006) 25:108–17. doi:10.1038/sj.emboj.7600919
54. King EM, Chivers JE, Rider CF, Minnich A, Giembycz MA, Newton R. Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation- and transrepression-dependent mechanisms. PLoS One (2013) 8:e53936. doi:10.1371/journal.pone.0053936
55. Beck IM, Van Crombruggen K, Holtappels G, Daubeuf F, Frossard N, Bachert C, et al. Differential cytokine profiles upon comparing selective versus classic glucocorticoid receptor modulation in human peripheral blood mononuclear cells and inferior turbinate tissue. PLoS One (2015) 10:e0123068. doi:10.1371/journal.pone.0123068
56. Schäcke H, Schottelius A, Döcke W-D, Strehlke P, Jaroch S, Schmees N, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci U S A (2004) 101:227–32. doi:10.1073/pnas.0300372101
57. van Lierop MJC, Alkema W, Laskewitz AJ, Dijkema R, van der Maaden HM, Smit MJ, et al. Org 214007-0: a novel non-steroidal selective glucocorticoid receptor modulator with full anti-inflammatory properties and improved therapeutic index. PLoS One (2012) 7:e48385. doi:10.1371/journal.pone.0048385
58. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev (2000) 21:55–89. doi:10.1210/er.21.1.55
59. Glezer I, Chernomoretz A, David S, Plante MM, Rivest S. Genes involved in the balance between neuronal survival and death during inflammation. PLoS One (2007) 2:e310. doi:10.1371/journal.pone.0000310
60. Langlais D, Couture C, Balsalobre A, Drouin J. Regulatory network analyses reveal genome-wide potentiation of LIF signaling by glucocorticoids and define an innate cell defense response. PLoS Genet (2008) 4:e1000224. doi:10.1371/journal.pgen.1000224
61. Lannan EA, Galliher-Beckley AJ, Scoltock AB, Cidlowski JA. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology (2012) 153:3701–12. doi:10.1210/en.2012-1020
62. Vizzardelli C, Pavelka N, Luchini A, Zanoni I, Bendickson L, Pelizzola M, et al. Effects of dexamethazone on LPS-induced activationand migration of mouse dendritic cells revealed by a genome-wide transcriptional analysis. Eur J Immunol (2006) 36:1504–15. doi:10.1002/eji.200535488
63. Yamazaki S, Akira S, Sumimoto H. Glucocorticoid augments lipopolysaccharide-induced activation of the IkappaBzeta-dependent genes encoding the anti-microbial glycoproteins lipocalin 2 and pentraxin 3. J Biochem (2015) 157:399–410. doi:10.1093/jb/mvu086
64. Smith AD, Datta SP, Smith GH, Campbell PN, Bentley R, McKenzie H, et al. Oxford Dictionary of Biochemistry and Molecular Biology. Oxford: Oxford University Press (1997).
65. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med (1999) 340:448–54. doi:10.1056/NEJM199902113400607
66. Jang E, Lee S, Kim JH, Kim JH, Seo JW, Lee WH, et al. Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J (2013) 27:1176–90. doi:10.1096/fj.12-222257
67. Warszawska JM, Gawish R, Sharif O, Sigel S, Doninger B, Lakovits K, et al. Lipocalin 2 deactivates macrophages and worsens pneumococcal pneumonia outcomes. J Clin Invest (2013) 123:3363–72. doi:10.1172/Jci67911
68. Zhang J, Wu Y, Zhang Y, Leroith D, Bernlohr DA, Chen X. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol Endocrinol (2008) 22:1416–26. doi:10.1210/me.2007-0420
69. Vilahur G, Badimon L. Biological actions of pentraxins. Vascul Pharmacol (2015) 73:38–44. doi:10.1016/j.vph.2015.05.001
70. Glezer I, Rivest S. Oncostatin M is a novel glucocorticoid-dependent neuroinflammatory factor that enhances oligodendrocyte precursor cell activity in demyelinated sites. Brain Behav Immun (2010) 24:695–704. doi:10.1016/j.bbi.2010.01.005
71. Le Goff B, Singbrant S, Tonkin BA, Martin TJ, Romas E, Sims NA, et al. Onostatin M acting via OSMR, augments the actions of IL-1 and TNF in synovial fibroblasts. Cytokine (2014) 68:101–9. doi:10.1016/j.cyto.2014.04.001
72. Flammer JR, Rogatsky I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol (2011) 25:1075–86. doi:10.1210/me.2011-0068
73. Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab (2013) 24:109–19. doi:10.1016/j.tem.2012.11.005
74. Lim H-Y, Müller N, Herold MJ, Van Den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology (2007) 122:47–53. doi:10.1111/j.1365-2567.2007.02611.x
75. Hodrea J, Majai G, Doró Z, Zahuczky G, Pap A, Rajnavölgyi É, et al. The glucocorticoid dexamethasone programs human dendritic cells for enhanced phagocytosis of apoptotic neutrophils and inflammatory response. J Leukoc Biol (2012) 91:127–36. doi:10.1189/jlb.0511243
76. Vago JP, Nogueira CRC, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol (2012) 92:249–58. doi:10.1189/jlb.0112008
77. Weber PSD. Mechanisms of glucocorticoid-induced down-regulation of neutrophil L-selectin in cattle: evidence for effects at the gene-expression level and primarily on blood neutrophils. J Leukoc Biol (2003) 75:815–27. doi:10.1189/jlb.1003505
78. Pazdrak K, Moon Y, Straub C, Stafford S, Kurosky A. Eosinophil resistance to glucocorticoid-induced apoptosis is mediated by the transcription factor NFIL3. Apoptosis (2016) 21:421–31. doi:10.1007/s10495-016-1226-5
79. Schiller BJ, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol (2014) 15:418. doi:10.1186/PREACCEPT-2066059439130185
Keywords: acute-phase response, cortisol, gene expression, inflammatory diseases, innate immune response, GRE, SEGRAs, transrepression
Citation: Xavier AM, Anunciato AKO, Rosenstock TR and Glezer I (2016) Gene Expression Control by Glucocorticoid Receptors during Innate Immune Responses. Front. Endocrinol. 7:31. doi: 10.3389/fendo.2016.00031
Received: 19 December 2015; Accepted: 04 April 2016;
Published: 19 April 2016
Edited by:
Sandhya Srikant Visweswariah, Indian Institute of Science, IndiaReviewed by:
Norifumi Iijima, Yale University School of Medicine, USACopyright: © 2016 Xavier, Anunciato, Rosenstock and Glezer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isaias Glezer, aWdsZXplckB1bmlmZXNwLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.