 Sung Sik Choe
Sung Sik Choe Jae Bum Kim
Jae Bum Kim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 13 April 2016
Sec. Cellular Endocrinology
Volume 7 - 2016 | https://doi.org/10.3389/fendo.2016.00030
The adipose tissue is a central metabolic organ in the regulation of whole-body energy homeostasis. The white adipose tissue functions as a key energy reservoir for other organs, whereas the brown adipose tissue accumulates lipids for cold-induced adaptive thermogenesis. Adipose tissues secrete various hormones, cytokines, and metabolites (termed as adipokines) that control systemic energy balance by regulating appetitive signals from the central nerve system as well as metabolic activity in peripheral tissues. In response to changes in the nutritional status, the adipose tissue undergoes dynamic remodeling, including quantitative and qualitative alterations in adipose tissue-resident cells. A growing body of evidence indicates that adipose tissue remodeling in obesity is closely associated with adipose tissue function. Changes in the number and size of the adipocytes affect the microenvironment of expanded fat tissues, accompanied by alterations in adipokine secretion, adipocyte death, local hypoxia, and fatty acid fluxes. Concurrently, stromal vascular cells in the adipose tissue, including immune cells, are involved in numerous adaptive processes, such as dead adipocyte clearance, adipogenesis, and angiogenesis, all of which are dysregulated in obese adipose tissue remodeling. Chronic overnutrition triggers uncontrolled inflammatory responses, leading to systemic low-grade inflammation and metabolic disorders, such as insulin resistance. This review will discuss current mechanistic understandings of adipose tissue remodeling processes in adaptive energy homeostasis and pathological remodeling of adipose tissue in connection with immune response.
The adipose tissue is a critical regulator of systemic energy homeostasis by acting as a caloric reservoir. In excess nutrient conditions, the adipose tissue stores surplus nutrients in the form of neutral lipids, whereas in nutrient deficit conditions, it supplies nutrients to other tissues through lipolysis (1). In the past several decades, overnutrition and reduced daily activity have greatly increased obesity rates worldwide, creating a global health emergency due to concomitant increases in insulin resistance, type 2 diabetes, heart disease, atherosclerosis, hypertension, and many types of cancer (2–4). In response to alterations in the energy status, the adipose tissue is rapidly and dynamically remodeled through changes in the number and/or size of adipocytes. Simultaneously, various stromal vascular cells in the adipose tissue undergo numerical and/or functional changes, contributing to the maintenance of the adipose tissue function as an energy reservoir and endocrine organ. This series of events is called “adipose tissue remodeling.” However, under pathophysiological conditions, such as obesity, aberrant adipose tissue remodeling may induce dysregulation of adipose tissue-derived cytokines, hormones, and metabolites, leading to metabolic stresses and disorders in metabolic organs (2–4).
Living organisms need to consume energy from their environments to survive. In particular, storage of extra energy obtained during food abundance is an essential physiological activity that enhances survival during food scarcity periods. Multicellular organisms have evolved specialized cells or organs to store excess nutrients as lipids because lipids have higher calories than other nutrients. For example, Caenorhabditis elegans stores surplus energy in the form of lipids in intestinal cells (5), whereas sharks and Drosophila accumulate excess lipids in the liver and fat body, respectively (6, 7). In other organisms, particularly some fishes and higher vertebrates, the adipose tissue functions as a specialized energy reservoir (1). The adipose tissue is distributed throughout the body and is capable of expanding to accommodate excess energy in the form of accumulated lipids, characteristics distinguishing the adipose tissue from other organs and tissues (8). In humans, there are two major types of adipose tissues, white adipose tissue (WAT) and brown adipose tissue (BAT). Anatomically, WAT comprises two major depots, subcutaneous WAT (SAT) and visceral WAT (VAT) around internal organs. VAT, which is concentrated in the abdominal cavity, is further subdivided into mesenteric, omental, perirenal, and peritoneal depots (Figure 1) (8, 9). The key physiological functions of WAT are insulation and energy storage. In obesity, however, excess VAT is closely linked to metabolic complications, such as insulin resistance and type 2 diabetes (8, 9). Mesenteric and omental adipose tissues are particularly important for hepatic insulin resistance and steatosis because liver is directly exposed to releasing factors from these adipose tissues via the portal vein. As an animal model, rodents have gonadal adipose tissue that is considered part of VAT. However, because of lack of gonadal adipose tissue in humans, careful interpretation is required to extrapolate findings in rodents to human. BAT is a specialized form that participates in non-shivering thermogenesis through lipid oxidation (10, 11). The distinct brown color of BAT is attributed to its high mitochondrial density, which is critical for heat generation and lipid oxidation (10, 11). Although BAT is readily observed in both infant and adult rodents, it has been proposed that BAT in humans is limited to neonates and is gradually replaced by WAT with aging. However, recent positron emission tomography/computed tomography studies have shown that BAT is viable and functional in human adults (12, 13). Thus, various adipose tissues act as central regulators of energy homeostasis by storing excess energy as well as by controlling thermogenesis.
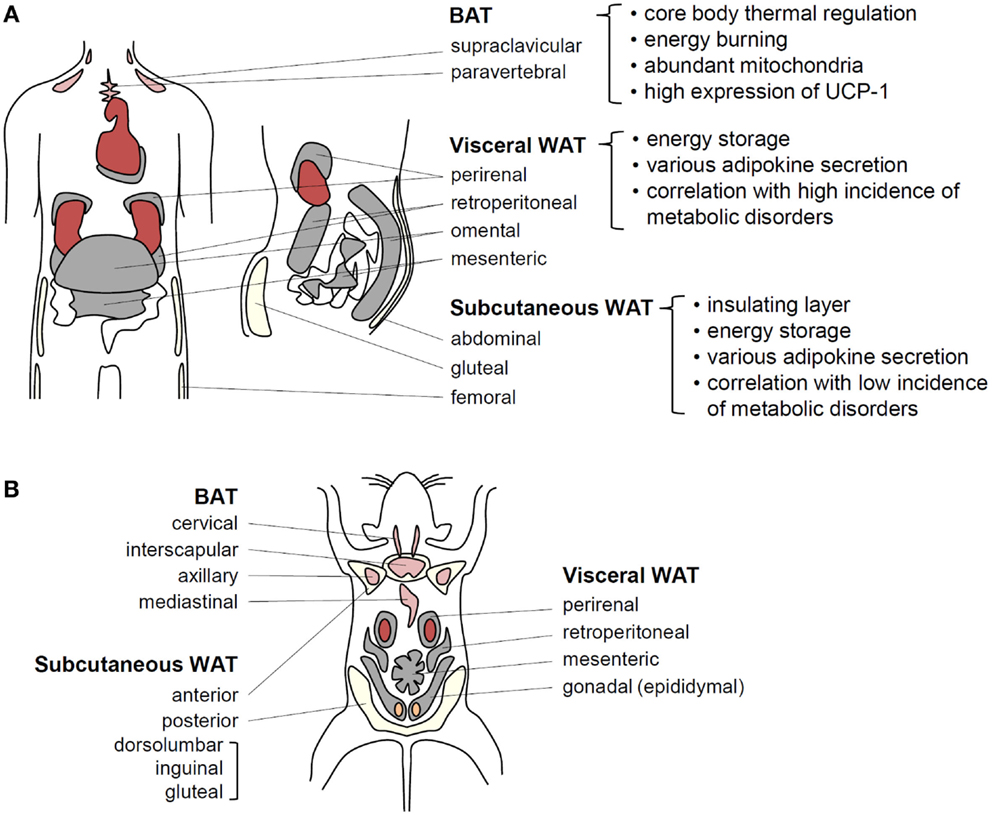
Figure 1. Adipose tissue functions in energy homeostasis and thermal regulation. (A) In humans, BAT localized around the shoulders and ribs contributes to heat generation. Brown adipocytes exhibit abundant mitochondria and UCP-1 expression related to thermogenesis. It has recently been speculated that BAT efficiency for fat-burning could be harnessed to reduce obesity. Visceral WAT (VAT) and subcutaneous WAT (SAT) possesses considerable capacities for energy storage. VAT surrounds intra-abdominal organs, whereas SAT spreads throughout the body beneath the skin. These fat tissues secrete various adipokines to regulate energy homeostasis. VAT is more strongly associated with obesity-induced metabolic disorders than SAT. (B) In adult mice, BAT is well developed and easily observed compared with that in adult humans. Among WAT depots within the abdominal cavity, the paired gonadal depots located around the ovaries in females and the testes in males are studied as a model of VAT. However, these depots do not exist in humans. The paired inguinal depots in the anterior to the upper part of the hind limbs are representative SATs in mice.
The adipose tissue has been historically considered merely an energy storage depot, but this concept was revised following the discovery of leptin, the first adipocyte-derived cytokine, by Friedman’s group in 1990. Leptin release in response to changes in nutritional status indicates that the adipose tissue acts as an endocrine organ involved in modulating energy homeostasis (14, 15). Since this discovery, additional cytokines, hormones, and peptides secreted by adipocytes, collectively termed as “adipokines,” have been identified and intensively investigated for roles in the control of energy homeostasis (16). Leptin, one of the most well studied adipokines, is secreted in response to food intake and inhibits appetite by regulating neural circuits located in the brain. Leptin acts on surface receptors expressed in AGRP neurons in the lateral hypothalamus and POMC neurons in the medial hypothalamus to inhibit appetite and stimulate satiety, respectively (17–19). Mice homozygous for the leptin mutation ob/ob or leptin receptor mutation db/db are massively obese because of uncontrolled appetite and ensuing excess food intake (20, 21). Leptin also promotes lipid oxidation and mitochondrial biogenesis and accelerates energy expenditure in peripheral tissues through both local signaling and regulation of brain-derived factors (22, 23). Circulating leptin is actually elevated in obesity, but hypothalamic leptin resistance aggravates obesity through inhibition of appetite control and lipid oxidation (17–19).
Adiponectin is another adipocyte-secreted adipokine that is abundant in the blood (24–26). Adiponectin exerts anti-obesity and antidiabetic effects and alleviates insulin resistance by stimulating lipid oxidation and anti-inflammatory responses (27–32). There are two major receptors for adiponectin AdipoR1 and AdipoR2. Both receptors stimulate AMP-activated protein kinase (AMPK), which is necessary for the anti-obesity and antidiabetic actions of adiponectin (29–31). There are two distinct forms of circulating plasma adiponectin, a low molecular weight (LMW) full-length trimer and a hexamer. The full-length trimer aggregates via disulfide bonding into high molecular weight (HMW) multimers of 12–18 monomers. It has been reported that these multimers are the primary bioactive forms and that each may exert different biological effects. In obesity, the circulating plasma levels of HMW and LMW adiponectin are reduced, and this altered adiponectin oligomeric profile has been suggested to be a reliable clinical indicator of metabolic disorders (33).
Recent studies have identified additional adipokines that promote obesity and associated morbidities (34–37). For instance, resistin, an adipokine mainly secreted from VAT, is elevated in obesity. In mice, administration of resistin impairs glucose tolerance and insulin action, implying that elevated levels of resistin may link obesity to diabetes (34). Given that the resistin receptor has not been identified, the molecular pathways through which resistin induces insulin resistance in obesity are still uncertain. Retinol-binding protein 4, another adipokine upregulated in serum of insulin-resistance mice and humans, impairs insulin signaling in liver and muscle (35). In contrast, omentin, identified in the human omental adipose depot, regulates blood glucose level by enhancing insulin action and is decreased in obesity (36, 37). The recently identified adipokine nesfatin, primarily derived from SAT, modulates appetite according to the nutrient status (38). In addition to peptide adipokines, lipid metabolites called “lipokines” have also been discovered. In the adipose tissue, C16:1n7-palmitoleate is synthesized de novo via stearoyl-CoA desaturase-1. Secreted C16:1n7-palmitoleate improves muscle insulin sensitivity and suppresses fat accumulation in the liver (hepatosteatosis) (39). The identification of multiple adipokines and lipokines with disparate cellular actions underscores the central role of adipose tissue depots as endocrine organs in the dynamic regulation of systemic energy homeostasis.
Compared with WAT, BAT is characterized by multilocular lipid droplets, rich vascularization, and abundant mitochondria. Unlike WAT, BAT functions prominently in thermoregulation through lipid oxidation-mediated heat generation (10, 11). Although both BAT and muscles harbor high levels of mitochondria, BAT is specialized for heat generation rather than for ATP synthesis by high expression of mitochondrial uncoupling protein 1 (UCP-1) (10, 11). The adipose tissues express high levels of β-adrenergic receptors that mediate cold-induced lipolysis (40). After cold exposure, it appears that large amounts of lipids from WAT flow into BAT (40). Concurrently, β-adrenergic signaling in BAT activates the expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), which stimulates the expression of UCP-1 and mitochondrial genes (41). In addition to UCP-1 expression, cold-induced BAT enhances lipid uptake for the efficient production of heat, accompanied by mitochondrial biogenesis (40). In accordance with this general model of homeostatic thermogenesis, it has been reported that cold exposure elevates the expression of pro-angiogenic factors, including vascular endothelial growth factor (VEGF), and impedes the expression of anti-angiogenic factors in BAT (42). VEGF receptor (VEGFR) 2 blockage abolishes cold-induced angiogenesis and impairs thermogenic capacity, indicating that angiogenic remodeling of BAT is one of important factors for thermoregulation (42).
Emerging evidence suggests that both BAT and SAT contribute to thermal regulation (43). Cold exposure induces “beiging” remodeling of SAT to the BAT phenotype (referred to as “adipose tissue browning”) (40, 43). Although beige/bright adipocytes and brown adipocytes share the ability for thermogenesis, there are several differences. First, myogenic factor five-negative beige adipocytes are derived from different developmental lineages compared with myogenic factor five-positive brown adipocytes (44). Second, even though PRD1-BF-RIZ1 homologous domain-containing protein-16 (PRDM16) has been recognized as a critical transcriptional coactivator for both brown and beige adipocytes, PRDM16 depletion impedes beige adipocyte development upon β-adrenergic stimulation but has no effect on brown adipocyte development or function (45, 46). To date, two hypotheses have been proposed for beige adipocyte differentiation: one is that beige adipocytes are derived by transdifferentiation of mature white adipocytes and the other is that beige adipocytes are differentiated from specific precursors (47–51). Permanent or inducible labeling of adipocyte lineages in mice demonstrated that the increased number of subcutaneous beige adipocytes after cold exposure resulted from de novo adipogenesis (51), indicating that there are distinct precursor cells capable of differentiating into beige adipocytes in SAT. It has also been reported that temperature can reverse this transformation in SAT. Beige adipocytes formed by cold stimulation revert to typical white adipocytes after warm adaptation (50). Moreover, subsequent cold stimulation reconverts these white-typical adipocytes to beige adipocytes. These results suggest the potential for repeated transdifferentiation of white adipocytes into beige adipocytes. Although it remains controversial whether mouse beige adipocytes have an equivalent cell population in human SAT, it has been demonstrated that human adult BAT consists of both classical brown adipocytes and beige adipocytes (52, 53). Moreover, recent marker gene analyses have suggested that the supraclavicular BAT of adult humans appears to consist of beige adipocytes rather than classical brown adipocytes (52–55).
Several cytokines are involved in the stimulation of beiging and/or browning. For instance, fibroblast growth factor 21 (FGF21) activates PGC-1α and accelerates the function of brown adipocytes (56–58). The newly identified adipokine irisin has been reported to link muscle-induced shivering thermogenesis to FGF21-induced brown (beige) adipose tissue-mediated non-shivering thermogenesis (59). Bone morphogenetic proteins (BMPs) 4 and 7 are also associated with the browning of adipocytes (60, 61). A better understanding of BAT activation and WAT browning may lead to the discovery of valuable therapeutic targets for the treatment of obesity and metabolic diseases (40, 43, 62). Despite these findings, more studies are needed to elucidate the molecular mechanisms of beiging/browning of adipocytes as well as their developmental origins.
Adipose tissue expansion in obesity could be mediated by hypertrophy (enlarged adipocytes), hyperplasia (increased numbers of adipocytes), or both (Figure 2). Adipocyte hypertrophy and hyperplasia are regulated by environmental and genetic factors (63). However, it is still uncertain how these two modes of adipose tissue expansion are controlled at the molecular level. In humans, SAT develops during weeks 14–24 of fetal gestation from the head and neck to the trunk and limbs through an increase in the number of adipocytes. By 28 weeks, major fat depots are organized (64). During the first year after birth, both adipocyte number and size increase in humans. The number of adipocytes then appears to remain stable until adolescence, when it increases once again (65, 66). In contrast to SAT development, very little is known about the developmental period of VAT. It has been shown that VAT is rarely formed before birth and that the total amount of VAT remains small until adolescence (67, 68).
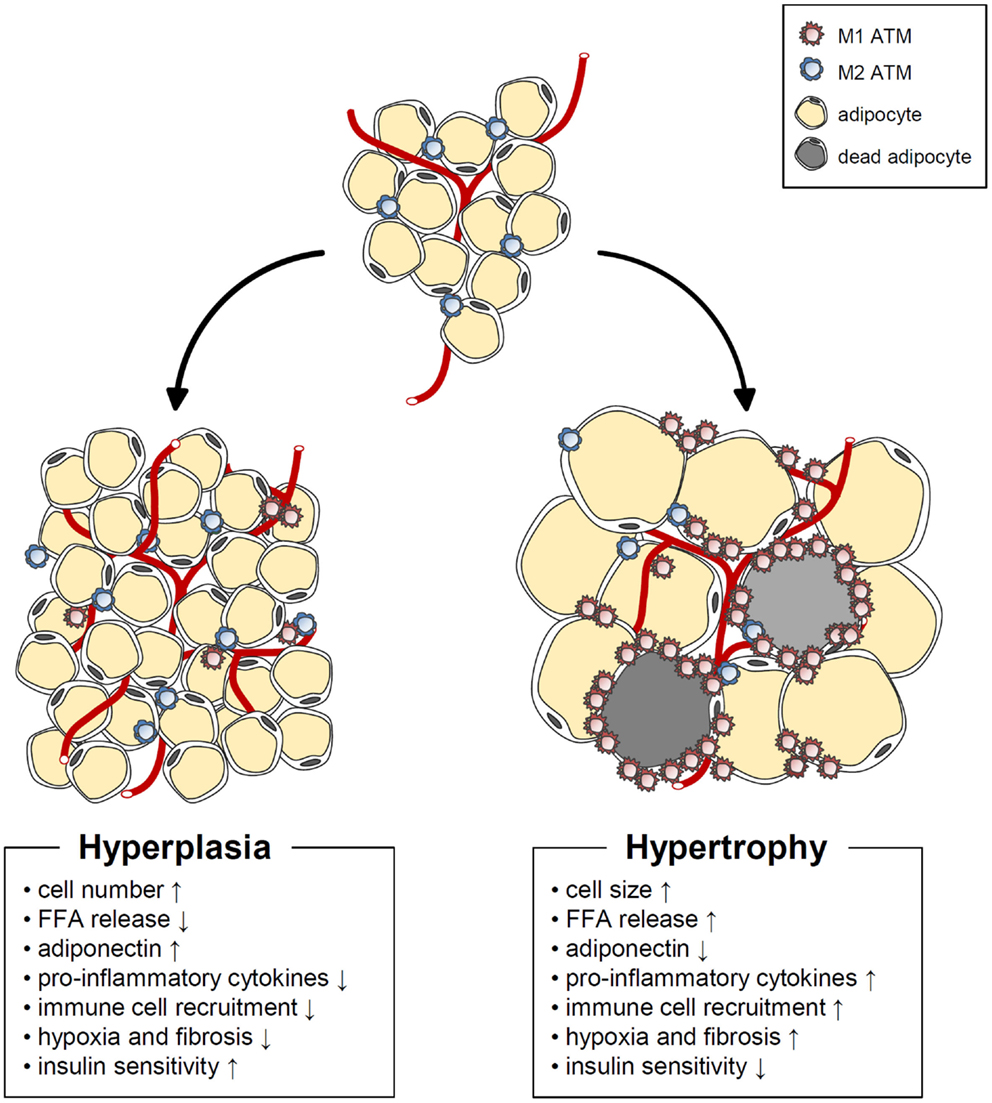
Figure 2. Characteristics of hypertrophic and hyperplasic adipocytes. In obesity, adipose tissue expansion occurs by two different mechanisms. Hypertrophic adipose expansion through increased adipocyte size is associated with such harmful phenomena as increased basal fatty acids release, pro-inflammatory cytokine release, immune cell recruitment, hypoxia, fibrosis, decreased adiponectin, and impaired insulin sensitivity. On the other hand, hyperplasic adipose expansion though increased adipocyte number is linked to beneficial phenomena, such as increased adiponectin, decreased basal fatty acids release, pro-inflammatory cytokine release, immune cell recruitment, hypoxia, fibrosis, and improved insulin sensitivity.
Both SAT and VAT masses are markedly increased in obese humans. In childhood obesity, WAT mass expansion is accompanied by adipocyte hyperplasia (69, 70). In obese animal models, it has been proposed that de novo adipogenesis begins when the adipocytes reach an upper volume limit (51, 71). Thereafter, formation of new adipocytes from precursor cells is required for further increases in energy storage capacity. In adult humans, however, the increase in WAT mass that triggers metabolic disorders is primarily due to adipocyte hypertrophy (72, 73). Spalding et al. reported that adipocyte numbers are roughly constant in lean and obese adults (74). In addition, neither the number nor the turnover rate of adipocytes during adulthood is altered by obesity or weight loss, implying that adipocyte hypertrophy is the predominant contributor to adult obesity.
In recent decades, many studies have suggested that adipocyte hypertrophy per se results in abnormal adipocyte function and leads to insulin resistance (75–79). The distinguishing features of hypertrophic adipocytes are as follows. First, in the obese adipose tissue, hypertrophic adipocytes show necrotic-like abnormalities (80–82). Necrosis-like adipocyte death is increased in hormone-sensitive lipase-deficient mice with adipocyte hypertrophy as well as in diet-induced obesity (DIO) mouse models (80–82). It has been proposed that an increase in dead adipocytes in obesity will impede adipose tissue function and induce inflammation. Second, hypertrophic adipocytes exhibit increased expression and secretion of pro-inflammatory cytokines, including tumor necrosis factor α (TNFα), interleukin (IL)-6, IL-8, and monocyte chemoattractant protein-1 (MCP-1) (83). This elevation of pro-inflammatory cytokines leads to serine phosphorylation of insulin receptor substrate-1 via nuclear factor κB and Jun N-terminal kinase signaling, resulting in the development of insulin resistance (84, 85). Pro-inflammatory cytokines also promote adipose tissue inflammation by recruiting various immune cells, including macrophages and T cells (2–4). Third, adipocyte hypertrophy induces local adipose tissue hypoxia because of a relative deficiency of vasculature (86–88). When hypoxia occurs in the adipose tissue, the expression levels of angiogenic factors and inflammatory response-associated genes are upregulated (86, 88, 89). Activation of hypoxia-inducible factor (HIF) 1α, a key transcription factor mediating hypoxic responses, accelerates adipose tissue fibrosis. Also, HIF1α augments the local inflammatory response in the obese adipose tissue, whereas HIF2α attenuates adipose tissue inflammation in obesity (87, 90, 91). Moreover, basal lipolysis is elevated in hypertrophic adipocytes (92, 93), increasing the leakage of free fatty acids (FFAs). Large amounts of FFAs released from the obese adipose tissue are taken up by other tissues, such as the liver and muscle, which can cause ectopic lipid accumulation and lipotoxicity (94, 95). Furthermore, saturated fatty acids, such as palmitic or stearic acids, activate the Toll-like receptor 4 signaling cascade, which plays an essential role in innate immunity, leading to chronic inflammation as well as insulin resistance (96–98). It was recently reported that adipocyte hypertrophy also impairs insulin-dependent glucose uptake because of a defect in GLUT4 trafficking (79). Upon chronic treatment with FFAs, 3T3-L1 adipocytes become enlarged with unilocular lipid droplets, mimicking in vivo hypertrophic adipocytes. These unilocular and hypertrophic adipocytes show disrupted cortical actin structures and impaired insulin-dependent GLUT4 trafficking to the plasma membrane (Figure 3). Thus, it is likely that adipocyte hypertrophy impairs adipocyte function though both inflammation-dependent and inflammation-independent mechanisms, thereby exacerbating insulin resistance and disrupting energy metabolism.
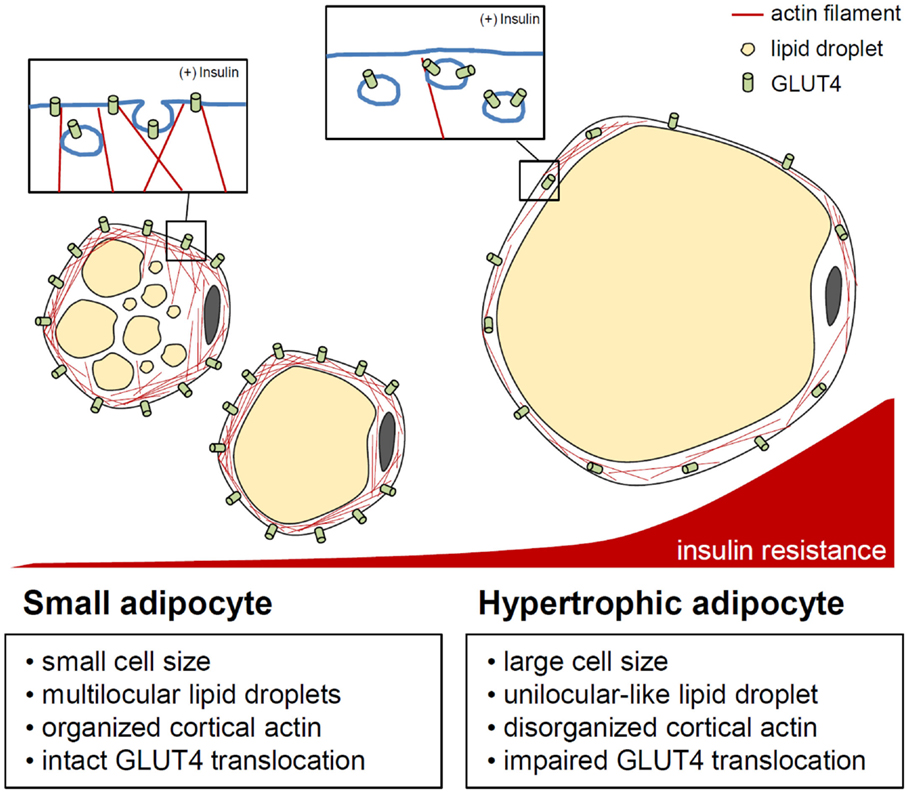
Figure 3. Actin cytoskeleton and insulin-stimulated GLUT4 translocation control in adipocytes. In adipocytes, cytosolic and cortical actin organization is involved in GLUT4 storing vesicle (GSV) transport by insulin stimulation. When adipocytes are hypertrophied, enlarged unilocular lipid droplets and expanded cell volume may impede cortical actin dynamics, resulting in improper/deficient translocation of GSVs. This indicates the importance of the adipocyte cytoskeleton in the regulation of adipocyte glucose metabolism in response to insulin.
Adipocyte hypertrophy occurs more readily in SAT than VAT until adolescence (65, 66, 71, 99). For hyperplasic expansion of the adipose tissue, adipocyte precursor cells must differentiate into adipocytes, a process regulated by various transcription factors and hormones. Peroxisome proliferator-activated receptor γ (PPARγ), a key regulator of adipogenesis, and CCAAT/enhancer binding protein families regulate the induction and maintenance of adipogenesis (100–102). Sterol regulatory element-binding transcription factor 1c (SREBP1c) is another key transcription factor that stimulates the expression of lipogenic genes, including acetyl-CoA carboxylase, fatty acid synthase, and saturated fatty acid dehydrogenase. It has been reported that activation of SREBP1c provides endogenous PPARγ ligands and consequently increases adipogenesis (103, 104). Other extracellular signals, including BMPs, insulin-like growth factors, and WNT, modulate adipocyte differentiation (105–108). However, the molecular mechanisms of in vivo adipogenesis in normal and obese conditions remain elusive. Recently, several ex vivo and in vivo studies have identified adipocyte precursor cell types (109–113). In human WAT, adipocyte precursor cells are abundant among CD31− and CD34+ stromal vascular fractions (109). In mice, more specific subpopulation of adipogenic precursor cells have been characterized, including highly pro-adipogenic subpopulations of CD45–, CD31–, Ter119–, CD29+, CD34+, Sca-1+, and CD24+ cells (110). Subsequent studies have shown that these subpopulations can form fully functional WAT deposits when transplanted into A/Zip lipodystrophic mice, implying that these adipocyte precursor cells can proliferate and differentiate into adipocytes in vivo. However, it remains unknown how and when adipocyte precursor cells can commit to adipocyte differentiation.
Many cohort studies have reported that several common parameters of obesity, including body mass index (BMI), insulin resistance, and cardiovascular diseases, are not always positively correlated with obesity. For instance, certain obese individuals with BMIs over 30 kg/m2 are insulin sensitive and exhibit normal metabolic parameters, including blood glucose and lipid levels, HOMA-IR, and plasma inflammatory markers. Such obese individuals are called “metabolically healthy obese” (MHO) (77, 114–117). A frequent feature of the MHO population is a lower number of immune cells and a higher proportion of relatively small adipocytes in adipose tissues (77, 114, 115, 117). Moreover, there are several mouse models that show similar MHO phenotypes (118, 119). Thiazolidinedione derivatives like rosiglitazone are antidiabetic agents that promote adipogenesis by acting as PPARγ ligands (120, 121). De novo small adipocytes are detected in obese db/db mice treated with rosiglitazone, and insulin-dependent glucose uptake is enhanced in rosiglitazone-induced newly differentiated small adipocytes (79, 122). These findings suggest that de novo differentiation of small adipocyte could ameliorate insulin resistance in obesity by providing additional capacity to store excess energy. Although the implications of adipocyte hyperplasia in the adipose tissue function are not fully understood, the regulation of hyperplasic adipocytes may exert beneficial effects against adipocyte hypertrophy and subsequent insulin resistance.
Inflammation is a biological defense response against harmful stimuli, such as pathogen invasion and cell damage (123). A close relationship between the inflammatory response and insulin resistance has been proposed on the basis of the suppression of insulin-dependent glucose uptake in sepsis patients (124, 125). Furthermore, the anti-inflammatory drug salicylate alleviates insulin resistance in diabetic subjects (126, 127). Expression of the inflammatory cytokine TNFα is increased in the obese adipose tissue, whereas blockade of TNFα/TNFα receptor signaling by TNFα neutralization improves insulin-dependent glucose uptake (128). Other pro-inflammatory cytokines, such as IL-1β, IL-6, and MCP-1, are also upregulated in the obese adipose tissue (129). These results indicate that inflammatory cytokines secreted from the adipose tissue contribute to the induction of insulin resistance.
Immune cells residing in the adipose tissue actively secrete numerous pro- and anti-inflammatory cytokines (2–4). Anti-inflammatory cytokines help in maintaining insulin sensitivity in the lean adipose tissue, while accumulation of pro-inflammatory cytokines in obesity leads to insulin resistance (2–4) (Figure 4). In addition, pro-inflammatory cytokines stimulate lipolysis in adipocytes, leading to lipotoxicity in other tissues (2, 3). In C57BL/6 mice, an inflammatory response was specifically induced in the adipose tissue but not in other metabolic tissues by a brief (1 week) high-fat diet (HFD) (130). During long-term HFD, however, pro-inflammatory responses were also greatly enhanced in other metabolic tissues, including the liver and muscles (130). This report proposes that inflammatory responses because of excess energy intake are primarily initiated in adipose tissues and that chronic inflammation of adipose tissues subsequently induces inflammation in other metabolic organs, such as the liver, muscle, and pancreas. Therefore, it is likely that inflammation is a causal factor for widespread systemic insulin resistance in obesity.
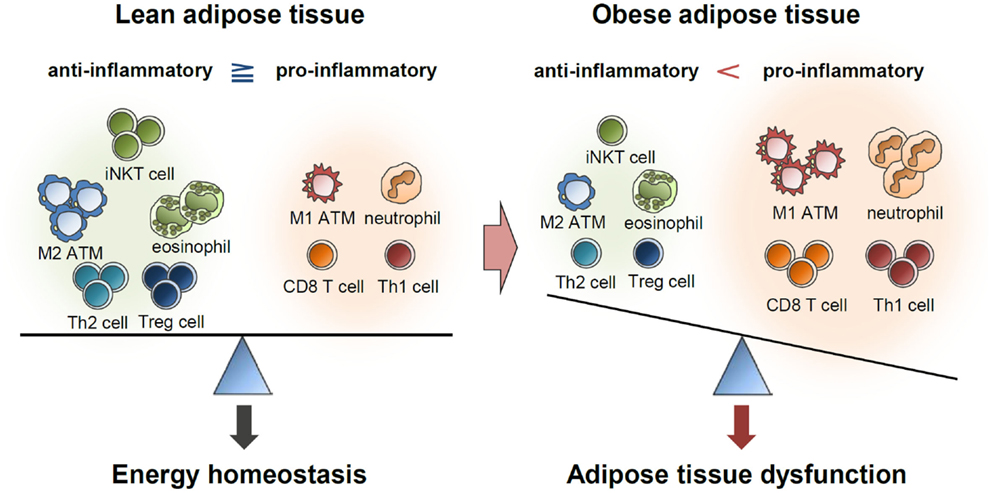
Figure 4. Balance of immune responses in the regulation of adipose tissue function. Lean adipose tissue harbors various anti-inflammatory immune cells, such as eosinophils, M2 macrophages, Th2 cells, iNKT cells, and Treg cells. These immune cells help in maintaining insulin sensitivity and store extra energy in the form of TGs. In obese adipose tissue, the numbers of pro-inflammatory immune cells, including neutrophils, M1 macrophages, mast cells, Th1 cells, and CD8 T cells, are greatly elevated. Simultaneously reduced number of anti-inflammatory immune cells accelerates pro-inflammatory response and adipose tissue dysfunction.
Adipose tissue remodeling is accompanied by reversible changes in immune cell composition as well as adipocyte size, altering numerous adipose tissue functions (2–4). The numbers of neutrophils and macrophages are rapidly increased in the adipose tissue upon HFD feeding (130–132). Neutrophils are the most abundant blood leukocyte type and rapidly react to inflammatory responses (133). It was reported that the expression levels of neutrophil-derived elastase and myeloperoxidase (MPO) were elevated after only 3 days of HFD feeding in mice and that neutrophils quickly accumulated in the obese adipose tissue (131, 132). Both HFD-induced adipose tissue inflammation and insulin resistance were attenuated in mice treated with a neutrophil elastase inhibitor and in neutrophil elastase knockout mice (132), implying that neutrophil elastase contributes to the pro-inflammatory response and insulin resistance in adipose tissues. However, one study reported that MPO-positive cells did not infiltration into adipose tissues of obese humans even though the levels of circulating MPO and neutrophils were elevated (134).
Macrophages are the most abundant cell type among adipose tissue leukocytes, constituting almost 50% of total immune cells (135, 136). The number of macrophages in adipose tissue prominently increases with obesity. Accumulated macrophages are frequently observed in a crown-like structure (CLS) surrounding lipid droplet-associated protein (perilipin)-negative adipocytes in obese subjects (80, 81, 137). MCP-1 is a well-known chemokine that attracts macrophages into the adipose tissue in obesity. Both MCP-1-deficient mice and MCP-1 receptor CCR2-deficient mice are protected against macrophage accumulation and inflammatory responses in adipose tissues and are resistant to DIO-induced insulin resistance (138, 139). Adipose tissue macrophages (ATMs) can be divided into M1 (classically activated) and M2 types (alternatively activated) (2, 140). The M1 type prominently express inducible nitric oxide synthase (iNOS), TNFα, IL-1β, and CD11c on the cell surface, whereas M2 macrophages express arginase, IL-10, and Ym-1. It has been reported that M1 ATMs are the major contributors to adipose tissue inflammation and insulin resistance in obesity. Furthermore, mice transplanted with bone marrow depleted of CD11c-positive cells show attenuated adipose tissue inflammation and insulin resistance during DIO (141). Although this simple dichotomous classification of ATMs has some experimental benefits, it appears that ATMs are composed of diverse cell populations not easily classified as M1 or M2.
The polarization of M1 macrophages can be induced by IFNγ or lipopolysaccharides (142). In the adipose tissue, IFNγ is predominantly expressed in T cells, particularly Th1 and CD8 T cells (142). The numbers of Th1 and CD8 T cells are elevated in adipose tissues of obese subjects (142–144). In mice, CD8 T cell depletion reduces M1 macrophage accumulation and TNFα expression in the adipose tissue and also improves insulin sensitivity (143). Coculture of CD8 T cells and adipose tissue from HFD-fed mice increases CD8 T cells proliferation and TNFα expression (143). These findings suggest that factors induced in the adipose tissue by HFD further activate CD8 T cells. The Th1 cell, a subtype of CD4 T cell, is also a major IFNγ-expressing cell that accumulates in obesity (144). IFNγ stimulates the expression of chemokines and pro-inflammatory cytokines in adipocytes as well as the M1 polarization of macrophages. Although HFD-fed IFNγ knockout mice show no difference in adipose tissue mass, adipose tissue inflammation, and glucose tolerance are ameliorated compared to wild-type mice (142). These findings suggest that pro-inflammatory immune cells amplify the inflammatory response via cell-to-cell interactions with adipocytes and other immune cells, which in turn induces chronic inflammation, adipose tissue dysfunction, and insulin resistance.
Other immune cell populations also protect against adipose tissue inflammatory responses. For instance, M2 macrophages strongly express arginase, which sequesters arginine from iNOS, resulting in inhibition of nitric oxide generation and suppression of M1 macrophage function (2). Moreover, M2 macrophages secrete the anti-inflammatory cytokine IL-10 (2). Treatment of adipocytes with IL-10 alleviates TNFα-induced insulin resistance (145). Activation of STAT6 signaling by Th2-type cytokines, such as IL-4, mediates M2 polarization (2). When IL-4 is administered to DIO mice, adipose tissue mass and inflammatory responses are alleviated, and insulin sensitivity is improved (146). IL-4 is primarily produced by eosinophils in murine adipose tissues (147), and compared with wild-type mice, eosinophil-deficient mice are more glucose-intolerant upon HFD feeding and show diminished numbers of M2 ATMs. In contrast, overexpression of IL-5, an eosinophil chemoattractant, induces glucose tolerance and increases eosinophil numbers in the adipose tissue (147). Thus, it has been proposed that eosinophils in the adipose tissue regulate insulin sensitivity by mediating M2 macrophage polarization.
Beneficial roles of invariant natural killer T (iNKT) cells in adipose tissue inflammation and insulin sensitivity have recently been identified, which are associated with secretion of anti-inflammatory cytokines, such as IL-4 and IL-10 (Figure 5) (148–152). These iNKT cells have unique functional characteristics. For example, iNKT cells recognize the lipid antigen loaded on CD1d molecules of antigen-presenting cells, whereas CD4 and CD8 T cells recognize the peptide antigens loaded on MHC molecules (153). In addition, iNKT cells can rapidly secrete large quantities of cytokines after activation (153). The number of iNKT cells in adipose tissues is reduced in obese subjects (149, 150, 152). In short-term HFD-fed mice, adipose iNKT cell numbers are rapidly reduced concomitant with enhanced expression of iNKT cell activation markers (152). These results suggest that iNKT cells are involved in the early inflammatory response to metabolic stress in the adipose tissue. In addition, it has been shown that iNKT cell-deficient mice are more susceptible to adipose tissue inflammation and insulin resistance than wild-type mice (150, 152). Moreover, when iNKT cells are supplemented by adoptive transfer or activated by α-galactosylceramide treatment in murine models, body weight gain is reduced; glucose tolerance is improved; and anti-inflammatory cytokines, such as IL-4 and IL-10, are elevated in DIO (149, 150). Under HFD, the expression of the arginase gene is increased in wild type but not in iNKT cell-depleted mice, suggesting a crucial role of iNKT cells in M2 polarization (148). These data suggest that iNKT cells suppress adipose tissue inflammation and insulin resistance in DIO mice. It was recently suggested that iNKT cells in the adipose tissue possess a unique tissue-resident property, unlike iNKT cells in other tissues. Moreover, iNKT cells can regulate the functions of regulatory T (Treg) cells via IL-2 production as well as those of M2 macrophages via IL-10 production in the adipose tissue (154). Further, CD1d molecules that present lipid antigens to iNKT cells are highly expressed in adipocytes that store large amounts of lipid metabolites (151, 152). Despite these recent findings, the roles of adipocyte CD1d under physiological and pathological conditions and the identity of the endogenous lipid antigens exclusively loaded on adipocyte CD1d remain to be elucidated.
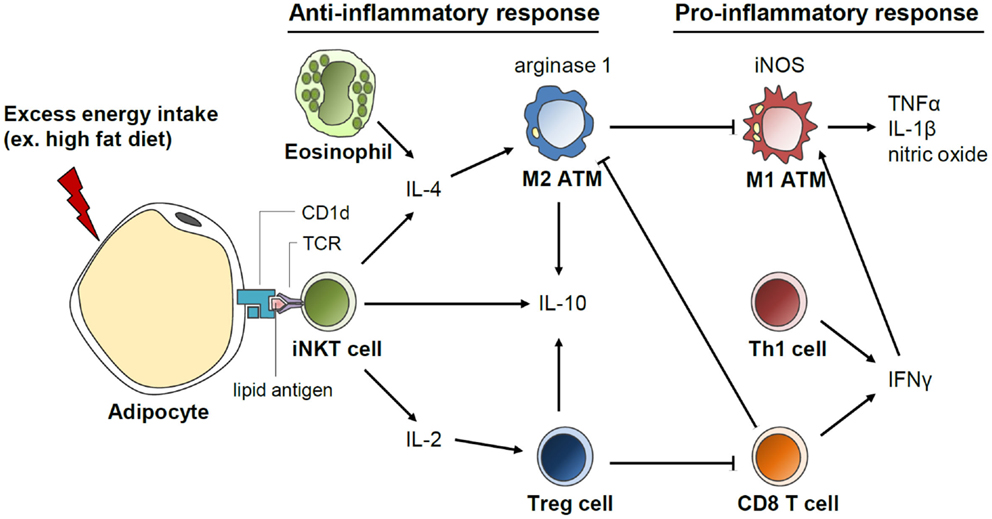
Figure 5. Invariant natural killer T (iNKT) cell-mediated regulation of anti-inflammatory response in the adipose tissue. Adipocytes secrete various inflammation-inducing factors including FFAs upon excess energy intake, such as HFD. In addition, antigen presentation by CD1d on adipocytes could activate iNKT cells, which rapidly secrete great quantities of cytokines, such as IL-4, IL-2, and IL-10. IL-4 produced by iNKT cells induces macrophage polarization into M2 type and arginase expression. IL-2 secretion by iNKT cells promotes Treg cell function in the adipose tissue. Activation of anti-inflammatory responses mediated by iNKT cells could play a crucial role in the suppression of excessive pro-inflammatory response in the adipose tissue upon HFD.
Treg cells protect the adipose tissue against severe inflammation through cell-to-cell interactions as well as by the secretion of anti-inflammatory cytokines, such as IL-10 and TGFβ. The number of Treg cells is reduced in adipose tissues of obese humans and mice models (155). In Treg cell-deficient mice, HFD-induced insulin resistance and expression of pro-inflammatory cytokines in the adipose tissue are exacerbated (155). Also, it has been reported that adipose tissue Treg cells have a 136-fold higher level of IL-10 transcripts than that in Treg cells in the lymph nodes (155).
Thus, it appears that diverse anti-inflammatory immune cells, including eosinophils, M2 macrophages, iNKT cells, and Treg cells, can cooperate to resolve inflammatory responses in adipose tissues during the early stages of obesity. Taken together, these results suggest that chronic excessive energy and an imbalanced ratio of pro-inflammatory to anti-inflammatory immune cells induce dysregulated immune responses in obese adipose tissues.
Several studies have also shown that the adipose tissue inflammatory response differs depending on tissue location (81, 137, 156–160). It is well known that VAT expansion is a stronger risk factor for the development of metabolic syndrome and insulin resistance than SAT expansion (70, 161). In addition, obesity-induced macrophage accumulation is greater in VAT than in SAT (137, 156, 158, 159). The mRNA levels of MCP-1, CCR2, and macrophage marker genes, such as CD14 and CD163, are consistently higher in human VAT than in SAT (156, 159). Moreover, IFNγ-secreting CD8 T cells and relative ratios of Th1 and Th2 cells are higher in VAT than in SAT (162). These differences in the inflammatory potential between fat depots need to be further elucidated to understand the numerical changes and roles of each immune cell in obese adipose tissues.
In the adipose tissue, the pro-inflammatory response causes insulin resistance, whereas the anti-inflammatory response preserves insulin sensitivity. The degree of inflammation in the adipose tissue is determined by dynamic interactions among various immune cells. For example, in mouse models, HFD feeding enhances M1 polarization to accelerate pro-inflammatory responses in adipose tissues. However, it is noteworthy that absolute number of M2 macrophages in fat tissue weight is also increased by HFD at the early stage of obesity (163, 164). It appears that immune homeostasis is determined by crosstalk between elevated M1 and M2 ATMs despite the change in nutritional status (165). On the other hand, each immune cell population in the adipose tissue exhibits a unique pattern of change under HFD feeding. In mouse models, neutrophils and macrophages are rapidly increased, whereas iNKT cells are decreased in the adipose tissue within 1 week during HFD (130, 132, 152). In contrast, the number of Treg cells remains stable for 10 weeks of HFD feeding (130, 143). These differences suggest that various adipose tissue immune cells have distinct roles in the adipose tissue immune response and may maintain immune balance to prevent uncontrolled pro-inflammatory responses. In short-term HFD-fed mice, changes in immune cell populations are not sufficient to affect glucose tolerance, possibly because of rebalancing of adipose tissue inflammation via crosstalk between local immune cell populations (130). However, under chronic HFD feeding, cumulative effects induced by long-term changes in immune cell populations induce metabolic dysregulation (132, 152). In other words, when obesity is prolonged, the net balance between pro- and anti-inflammation shifts to chronic inflammation. Without sustainable counteraction by anti-inflammatory responses, elevated pro-inflammatory immune cells appear to dysregulate adipose tissue functions with the progress of severe obesity, thereby disrupting glucose and lipid homeostasis.
Recent studies have revealed that adipose tissue remodeling due to inflammation is a reversible phenomenon closely associated with insulin sensitivity and adiposity. In both human obesity and mouse models, expression levels of pro-inflammatory markers and macrophage numbers increase in parallel with insulin resistance. However, after weight loss by calorie restriction, immune responses are re-balanced, and insulin sensitivity is restored by the reduction of M1 macrophages and induction of M2 macrophages (166–168). Furthermore, it has been reported that weight loss in obese and diabetic subjects after bariatric surgery results in reduced inflammatory gene expression and pro-inflammatory immune cell numbers (169–171). Thus, it is likely that rebalancing of pro- and anti-inflammatory immune cells could be driven by external stimuli, such as hormones, cytokines, or chemicals. Activation of M2 ATMs by Th2 cytokines, such as IL-4, attenuates the pro-inflammatory response in inflamed adipose tissues and ameliorates metabolic complications, including insulin resistance (146, 172). These results suggest that immune responses in adipose tissues are reversible and dynamically regulated by factors associated with changes in adiposity and immunostimulation, which eventually restore systemic insulin sensitivity.
In lean subjects, ATMs contribute to the regulation of adipose tissue function as well as adipose tissue remodeling (173). Multiple lines of evidence suggest that ATMs influence angiogenesis in adipose tissues (174–178). In mouse models, ATM depletion through the use of clodronate liposomes reduces blood vessel structures in adipose tissues (178). In addition, macrophages recruited into the tip of the gonadal adipose tissue promote angiogenesis during tissue outgrowth (174). In particular, lymphatic vessel endothelial receptor 1-positive macrophages secrete tissue remodeling factors, such as matrix metalloproteinase (MMP)-7, MMP-9, and MMP-12 that stimulate VEGF/VEGFR signaling for the formation of new vessels (174). It has been suggested that ATMs may be the major cell type that releases MMPs in the human adipose tissue (176). In Matrigel, ATMs isolated from the human adipose tissue promote the formation of endothelial cell tubes by secreting MMPs, such as MMP-9 (176). Extracellular matrix (ECM) remodeling, including the degradation of the connective tissue and basement membrane proteins, is necessary during the early stage of angiogenesis (179, 180). Thus, it has been suggested that ATMs contribute to angiogenic remodeling of the adipose tissue by modulating ECM.
Remodeling of ECM is also associated with the modulation of adipogenesis during adipose tissue expansion. Adipocyte differentiation is regulated by the deposition of collagen, a major protein component of ECM (181). In addition, it has been proposed that adipose tissue fibrosis in obese subjects caused by excess deposition of collagen, including type VI, leads to adipose tissue inflammation by triggering the infiltration of immune cells, such as macrophages, neutrophils, and lymphocytes (87, 182, 183). Pathological remodeling of ECM may also reduce the number of adipose tissue capillaries in obese and insulin-resistant subjects (184). Certain subtypes of collagen appear crucial for adipose tissue fibrosis-mediated inflammation and metabolic dysfunction in obesity (182, 183). Deletion of collagen VI in obese mice improves metabolic parameters and inflammatory profiles even though overall collagen content of the adipose tissue is not altered (182). However, it remains to be determined whether MMPs secreted from ATMs prevent collagen deposition linked to adipose tissue fibrosis.
Accumulation of ATMs is a key factor aggravating inflammatory responses in adipose tissues (2–4). However, recruited ATMs in obese adipose tissues may also play a protective role by enhancing angiogenesis in response to local hypoxia. Hypoxia is one of the most potent microenvironmental factors for macrophage recruitment into adipose tissues (185), and recruited macrophages are the predominant producers of pro-angiogenic factors in response to hypoxia (186). In mouse obese adipose tissues, ATM depletion decreases the expression level of platelet-derived growth factor (PDGF), an angiogenic factor that mediates endothelial cell tube formation and capillary maturation by pericyte recruitment (177). These results indicate that ATMs could be an important source of PDGF for angiogenesis. Furthermore, pro-inflammatory cytokines released from ATMs, such as TNFα, could act as angiogenic factors (187–189). Therefore, it has been speculated that ATMs may counteract hypoxia by promoting angiogenesis through production of angiogenic factors. However, recent elucidation of ATM-mediated angiogenesis in the obese adipose tissue still needs to be confirmed in vivo.
Another potential physiological role of ATMs in obesity is the clearance of dead or damaged adipocytes and fragmented cellular contents. In DIO mice, perilipin-negative adipocytes show features of necrosis-like death, such as plasma membrane rupture, dilated endoplasmic reticulum, and cell debris (80, 81). Most of the recruited ATMs in obese adipose tissues are present near perilipin-negative adipocytes, forming CLSs (80, 81). These CLS macrophages are multinucleated giant cells that surround residual adipocyte lipids, implying that CLS macrophages scavenge free lipids released from dead adipocytes to the interstitium (80). It was recently shown that depletion of mannose-binding lectin, which stimulates the phagocytic capacity of ATM, increases the numbers of CLSs in the adipose tissue of DIO mice, and decreases dead adipocyte clearance (190). Thus, it is likely that macrophage recruitment into the obese adipose tissue is involved in the clearance of dead adipocytes via phagocytosis. Furthermore, it has been suggested that ATMs could mediate the removal of extracellular lipids from adipose tissues (191). In both fasted and HFD-fed mice, ATMs absorb FFAs released from the adipocytes. This phenomenon could contribute to the prevention of lipotoxicity by buffering local extracellular FFA elevations in the adipose tissue. In obesity, however, lipid-loaded ATMs resembling foam cells have been suggested to have pro-inflammatory characteristics (192, 193) that contribute to insulin resistance. In both mice with adipocyte-targeted activation of caspase-8 and fat-transplanted mice, adipocyte death facilitates the recruitment of M1 and M2 macrophages (194, 195). It is possible that M1 macrophages phagocytize dead adipocytes, whereas M2 macrophages reconstruct ECM and resolve the activation of M1 macrophages after removal of dead adipocytes. These complementary functions of M1 and M2 macrophages may minimize tissue damage through tight regulation of phagocytosis and tissue repair. However, in obesity, the balance between M1 and M2 ATMs appears to be shifted by a greater increase in M1 macrophage number (2–4). Enhanced M1 polarization of ATMs in obese adipose tissues may interrupt the normal process of dead cell clearance, whereas further stimulating a pro-inflammatory response.
In response to cold exposure, β-adrenergic signaling modulates adaptive thermogenesis in BAT and SAT by promoting uncoupled respiration and thermogenic gene expression (40). Also, chronic cold exposure leads to the remodeling of BAT and SAT, resulting in a dramatic increase in the metabolic rate, mitochondrial biogenesis, and fatty acid oxidation (40). Emerging evidence suggests that M2 ATMs mediate thermogenic adipose tissue remodeling in response to cold exposure (196–198). Chawla’s group proposed that M2 ATMs induce thermogenic tissue remodeling by amplifying cold-induced β-adrenergic signaling via local catecholamine production (196, 198). In accordance with this notion, ATM depletion suppresses the cold-induced increase in thermogenic gene expression, lipolysis, and energy expenditure. In addition, blockage of M2 polarization by inhibition of IL-4 signaling diminishes adaptive thermogenesis and attenuates the generation of UCP-1-positive beige adipocytes in SAT. It has also been suggested that M2 ATMs promote the accumulation of PDGFRα-positive progenitor cells through osteopontin secretion and downstream β-adrenergic signaling (197). Recruited PDGFRα-positive progenitor cells can differentiate into UCP-1-positive adipocytes. Thus, it is likely that the interaction between M2 ATMs and PDGFRα-positive progenitor cells mediates the de novo differentiation of beige adipocytes in SAT during cold exposure. Together, these results indicate that ATM-associated adipose tissue remodeling is intimately involved in the modulation of adipose tissue function as well as adipose tissue growth and expansion in changing environments.
We have focused on the physiological and pathological roles of the adipose tissue and its remodeling in regulating whole-body energy metabolism by sensing the nutritional status and crosstalk between adipocytes and stromal vascular cells. During adipose tissue expansion, numerous cellular responses are dynamically altered. Modulation of ECM remodeling and angiogenesis may promote differentiation of de novo adipocytes, preventing the formation of hypertrophic adipocytes by providing additional capacity to store extra lipids. Recruited ATMs probably act to remove dead adipocytes and repair damaged adipose tissue for healthy adipose tissue remodeling. Adipose tissue remodeling is modulated by the inflammatory responses of multiple immune cell types, including macrophages and lymphocytes. However, in obesity, chronic excessive energy storage in the adipose tissue initiates pathological remodeling, which triggers pro-inflammatory responses of immune cells. Furthermore, tissue remodeling, including angiogenesis and tissue repair for healthy expansion of the adipose tissue, is hindered by inflammatory and metabolic stress. Thus, resolving uncontrolled pro-inflammatory responses during adipose tissue remodeling is important for maintaining a metabolically healthy status. Adipose tissue remodeling is a complex but well-orchestrated mechanism modulated by multiple adipose tissue cell types that allow adaptation to external environmental changes. A deeper mechanistic understanding of adipose tissue remodeling could facilitate the development of therapeutic approaches against obesity-induced metabolic diseases.
SSC, JYH, IJH, JIK, and JBK contributed to the writing of the manuscript under JBK’s supervision.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Yoon Jeong Park for critical reading and helpful comments. This work was supported by the National Creative Research Initiative Program (2011-0018312) funded by the Korean Ministry of Education, Science, and Technology.
1. Birsoy K, Festuccia WT, Laplante M. A comparative perspective on lipid storage in animals. J Cell Sci (2013) 126:1541–52. doi:10.1242/jcs.104992
2. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol (2011) 11:738–49. doi:10.1038/nri3071
3. Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm (2013) 2013:139239. doi:10.1155/2013/139239
4. Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells (2014) 37:365–71. doi:10.14348/molcells.2014.0074
5. McKay RM, McKay JP, Avery L, Graff JM. C. elegans: a model for exploring the genetics of fat storage. Dev Cell (2003) 4:131–42. doi:10.1016/S1534-5807(02)00411-2
6. Van Vleet ES, Candileri S, McNeillie J, Reinhardt SB, Conkright ME, Zwissler A. Neutral lipid components of eleven species of Caribbean sharks. Comp Biochem Physiol B (1984) 79:549–54.
7. Miller JM, Oligino T, Pazdera M, Lopez AJ, Hoshizaki DK. Identification of fat-cell enhancer regions in Drosophila melanogaster. Insect Mol Biol (2002) 11:67–77. doi:10.1046/j.0962-1075.2001.00310.x
8. Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell (2007) 131:242–56. doi:10.1016/j.cell.2007.10.004
9. Item F, Konrad D. Visceral fat and metabolic inflammation: the portal theory revisited. Obes Rev (2012) 13(Suppl 2):30–9. doi:10.1111/j.1467-789X.2012.01035.x
11. Richard D, Picard F. Brown fat biology and thermogenesis. Front Biosci (Landmark Ed) (2011) 16:1233–60. doi:10.2741/3786
12. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360:1509–17. doi:10.1056/Nejmoa0810780
13. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Brief report: functional brown adipose tissue in healthy adults. N Engl J Med (2009) 360:1518–25. doi:10.1056/Nejmoa0808949
14. Friedman JM, Leibel RL, Siegel DS, Walsh J, Bahary N. Molecular mapping of the mouse Ob mutation. Genomics (1991) 11:1054–62. doi:10.1016/0888-7543(91)90032-A
15. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi:10.1038/372425a0
16. Rondinone CM. Adipocyte-derived hormones, cytokines, and mediators. Endocrine (2006) 29:81–90. doi:10.1385/ENDO
17. Vaisse C, Halaas JL, Horvath CM, Darnell JE, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wildtype and ob/ob mice but not db/db mice. Nat Genet (1996) 14:95–7. doi:10.1038/Ng0996-95
18. Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology (2004) 145:4880–9. doi:10.1210/en.2004-0726
19. Myers MG, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab (2010) 21:643–51. doi:10.1016/j.tem.2010.08.002
20. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene-product on body-weight regulation in Ob/Ob mice. Science (1995) 269:540–3. doi:10.1126/science.7624776
21. Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell (1996) 84:491–5. doi:10.1016/S0092-8674(00)81294-5
22. Minokoshi Y, Kim YB, Peroni OD, Fryer LGD, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature (2002) 415:339–43. doi:10.1038/415339a
23. Guo ZH, Jiang HY, Xu XR, Duan WZ, Mattson MP. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J Biol Chem (2008) 283:1754–63. doi:10.1074/jbc.M703753200
24. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum-protein similar to C1q, produced exclusively in adipocytes. J Biol Chem (1995) 270:26746–9. doi:10.1074/jbc.270.45.26746
25. Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem (1996) 271:10697–703. doi:10.1074/jbc.271.18.10697
26. Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem Biophys Res Commun (1996) 221:286–9. doi:10.1006/bbrc.1996.0587
27. Berg AH, Combs TP, Du XL, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med (2001) 7:947–53. doi:10.1038/90992
28. Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med (2002) 8:731–7. doi:10.1038/nm724
29. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med (2002) 8:1288–95. doi:10.1038/nm788
30. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects (vol 423, pg 762-9, 2003). Nature (2004) 431:1123–1123. doi:10.1038/Nature03091
31. Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes (2006) 55:2562–70. doi:10.2337/db05-1322
32. Folco EJ, Rocha VZ, Lopez-Ilasaca M, Libby P. Adiponectin inhibits pro-inflammatory signaling in human macrophages independent of interleukin-10. J Biol Chem (2009) 284:25569–75. doi:10.1074/jbc.M109.019786
33. Lara-Castro C, Luo NL, Wallace P, Klein RL, Garvey WT. Adiponectin multimeric complexes and the metabolic syndrome trait cluster. Diabetes (2006) 55:249–59. doi:10.2337/diabetes.55.01.06.db05-1105
34. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature (2001) 409:307–12. doi:10.1038/35053000
35. Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature (2005) 436:356–62. doi:10.1038/nature03711
36. Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab (2006) 290:E1253–61. doi:10.1152/ajpendo.00572.2004
37. de Souza Batista CM, Yang RZ, Lee MJ, Glynn NM, Yu DZ, Pray J, et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes (2007) 56:1655–61. doi:10.2337/db06-1506
38. Ramanjaneya M, Chen J, Brown JE, Tripathi G, Hallschmid M, Patel S, et al. Identification of nesfatin-1 in human and murine adipose tissue: a novel depot-specific adipokine with increased levels in obesity. Endocrinology (2010) 151:3169–80. doi:10.1210/en.2009-1358
39. Cao HM, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell (2008) 134:933–44. doi:10.1016/j.cell.2008.07.048
40. Cohen P, Spiegelman BM. Brown and beige fat: molecular parts of a thermogenic machine. Diabetes (2015) 64:2346–51. doi:10.2337/db15-0318
41. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell (1998) 92:829–39. doi:10.1016/S0092-8674(00)81410-5
42. Xue Y, Petrovic N, Cao RH, Larsson O, Lim S, Chen S, et al. Hypoxia-independent angiogenesis in adipose tissues during cold acclimation. Cell Metab (2009) 9:99–109. doi:10.1016/j.cmet.2008.11.009
43. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med (2013) 19:1252–63. doi:10.1038/nm.3361
44. Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature (2008) 454:961–7. doi:10.1038/nature07182
45. Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest (2011) 121:96–105. doi:10.1172/JCI44271
46. Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell (2014) 156:304–16. doi:10.1016/j.cell.2013.12.021
47. Himms-Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, Cinti S. Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol (2000) 279:C670–81.
48. Schulz TJ, Huang TL, Tran TT, Zhang HB, Townsend KL, Shadrach JL, et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc Natl Acad Sci U S A (2011) 108:143–8. doi:10.1073/pnas.1010929108
49. Vitali A, Murano I, Zingaretti MC, Frontini A, Ricquier D, Cinti S. The adipose organ of obesity-prone C57BL/6J mice is composed of mixed white and brown adipocytes. J Lipid Res (2012) 53:619–29. doi:10.1194/jlr.M018846
50. Rosenwald M, Perdikari A, Rulicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol (2013) 15:659–67. doi:10.1038/ncb2740
51. Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med (2013) 19:1338–44. doi:10.1038/nm.3324
52. Sharp LZ, Shinoda K, Ohno H, Scheel DW, Tomoda E, Ruiz L, et al. Human BAT possesses molecular signatures that resemble beige/brite cells. PLoS One (2012) 7:e49452. doi:10.1371/journal.pone.0049452
53. Jespersen NZ, Larsen TJ, Peijs L, Daugaard S, Homoe P, Loft A, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab (2013) 17:798–805. doi:10.1016/j.cmet.2013.04.011
54. Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell (2012) 150:366–76. doi:10.1016/j.cell.2012.05.016
55. Shinoda K, Luijten IHN, Hasegawa Y, Hong H, Sonne SB, Kim M, et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat Med (2015) 21:389–94. doi:10.1038/nm.3819
56. Chartoumpekis DV, Habeos IG, Ziros PG, Psyrogiannis AI, Kyriazopoulou VE, Papavassiliou AG. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol Med (2011) 17:736–40. doi:10.2119/molmed.2011.00075
57. Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, et al. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J Biol Chem (2011) 286:12983–90. doi:10.1074/jbc.M110.215889
58. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 regulates PGC-1 alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev (2012) 26:271–81. doi:10.1101/gad.177857.111
59. Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab (2014) 19:302–9. doi:10.1016/j.cmet.2013.12.017
60. Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature (2008) 454:1000–U1044. doi:10.1038/nature07221
61. Xue RD, Wan Y, Zhang S, Zhang QY, Ye HY, Li YM. Role of bone morphogenetic protein 4 in the differentiation of brown fat-like adipocytes. Am J Physiol Endocrinol Metab (2014) 306:E363–72. doi:10.1152/ajpendo.00119.2013
62. Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol (2014) 10:24–36. doi:10.1038/nrendo.2013.204
63. Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell (2001) 104:531–43. doi:10.1016/S0092-8674(01)00240-9
64. Poissonnet CM, Burdi AR, Garn SM. The chronology of adipose tissue appearance and distribution in the human fetus. Early Hum Dev (1984) 10:1–11. doi:10.1016/0378-3782(84)90106-3
65. Knittle JL, Hirsch J. Effect of early nutrition on the development of rat epididymal fat pads: cellularity and metabolism. J Clin Invest (1968) 47:2091–8. doi:10.1172/JCI105894
66. Knittle JL, Timmers K, Ginsberg-Fellner F, Brown RE, Katz DP. The growth of adipose tissue in children and adolescents. Cross-sectional and longitudinal studies of adipose cell number and size. J Clin Invest (1979) 63:239–46. doi:10.1172/JCI109295
67. Fox K, Peters D, Armstrong N, Sharpe P, Bell M. Abdominal fat deposition in 11-year-old children. Int J Obes Relat Metab Disord (1993) 17:11–6.
68. Siegel MJ, Hildebolt CF, Bae KT, Hong C, White NH. Total and intraabdominal fat distribution in preadolescents and adolescents: measurement with MR imaging. Radiology (2007) 242:846–56. doi:10.1148/radiol.2423060111
69. Salans LB, Cushman SW, Weismann RE. Studies of human adipose tissue. Adipose cell size and number in nonobese and obese patients. J Clin Invest (1973) 52:929–41. doi:10.1172/JCI107258
70. Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev (2000) 21:697–738. doi:10.1210/edrv.21.6.0415
71. Joe AW, Yi L, Even Y, Vogl AW, Rossi FM. Depot-specific differences in adipogenic progenitor abundance and proliferative response to high-fat diet. Stem Cells (2009) 27:2563–70. doi:10.1002/stem.190
72. Hirsch J, Han PW. Cellularity of rat adipose tissue: effects of growth, starvation, and obesity. J Lipid Res (1969) 10:77–82.
73. Hirsch J, Knittle JL. Cellularity of obese and nonobese human adipose tissue. Fed Proc (1970) 29:1516–21.
74. Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, et al. Dynamics of fat cell turnover in humans. Nature (2008) 453:783–7. doi:10.1038/nature06902
75. Salans LB, Knittle JL, Hirsch J. The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. J Clin Invest (1968) 47:153–65. doi:10.1172/JCI105705
76. Stern JS, Batchelor BR, Hollander N, Cohn CK, Hirsch J. Adipose-cell size and immunoreactive insulin levels in obese and normal-weight adults. Lancet (1972) 2:948–51. doi:10.1016/S0140-6736(72)92474-9
77. Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab (2010) 299:E506–15. doi:10.1152/ajpendo.00586.2009
78. Cotillard A, Poitou C, Torcivia A, Bouillot JL, Dietrich A, Kloting N, et al. Adipocyte size threshold matters: link with risk of type 2 diabetes and improved insulin resistance after gastric bypass. J Clin Endocrinol Metab (2014) 99:E1466–70. doi:10.1210/jc.2014-1074
79. Kim JI, Huh JY, Sohn JH, Choe SS, Lee YS, Lim CY, et al. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol (2015) 35:1686–99. doi:10.1128/MCB.01321-14
80. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res (2005) 46:2347–55. doi:10.1194/jlr.M500294-JLR200
81. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW II, DeFuria J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes (2007) 56:2910–8. doi:10.2337/db07-0767
82. Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I, et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res (2013) 54:2423–36. doi:10.1194/jlr.M038638
83. Jernas M, Palming J, Sjoholm K, Jennische E, Svensson PA, Gabrielsson BG, et al. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J (2006) 20:1540–2. doi:10.1096/fj.05-5678fje
84. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science (2001) 293:1673–7. doi:10.1126/science.1061620
85. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature (2002) 420:333–6. doi:10.1038/nature01137
86. Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab (2007) 293:E1118–28. doi:10.1152/ajpendo.00435.2007
87. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol (2009) 29:4467–83. doi:10.1128/MCB.00192-09
88. Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev (2013) 93:1–21. doi:10.1152/physrev.00017.2012
89. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes (2007) 56:901–11. doi:10.2337/db06-0911
90. Choe SS, Shin KC, Ka S, Lee YK, Chun JS, Kim JB. Macrophage HIF-2alpha ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes (2014) 63:3359–71. doi:10.2337/db13-1965
91. Lee YS, Kim JW, Osborne O, Oh, da Y, Sasik R, et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell (2014) 157:1339–52. doi:10.1016/j.cell.2014.05.012
92. Berger JJ, Barnard RJ. Effect of diet on fat cell size and hormone-sensitive lipase activity. J Appl Physiol (1985) (1999) 87:227–32.
93. Wueest S, Rapold RA, Rytka JM, Schoenle EJ, Konrad D. Basal lipolysis, not the degree of insulin resistance, differentiates large from small isolated adipocytes in high-fat fed mice. Diabetologia (2009) 52:541–6. doi:10.1007/s00125-008-1223-5
94. Slawik M, Vidal-Puig AJ. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res Rev (2006) 5:144–64. doi:10.1016/j.arr.2006.03.004
95. Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol (2015) 208:501–12. doi:10.1083/jcb.201409063
96. Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, et al. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem (2003) 278:37041–51. doi:10.1074/jbc.M305213200
97. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116:3015–25. doi:10.1172/JCI28898
98. Song MJ, Kim KH, Yoon JM, Kim JB. Activation of toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun (2006) 346:739–45. doi:10.1016/j.bbrc.2006.05.170
99. Tchkonia T, Tchoukalova YD, Giorgadze N, Pirtskhalava T, Karagiannides I, Forse RA, et al. Abundance of two human preadipocyte subtypes with distinct capacities for replication, adipogenesis, and apoptosis varies among fat depots. Am J Physiol Endocrinol Metab (2005) 288:E267–77. doi:10.1152/ajpendo.00265.2004
100. Spiegelman BM, Hu E, Kim JB, Brun R. PPAR gamma and the control of adipogenesis. Biochimie (1997) 79:111–2. doi:10.1016/S0300-9084(97)81500-3
101. Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE, et al. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell (1999) 3:151–8. doi:10.1016/S1097-2765(00)80306-8
102. Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev (2002) 16:22–6. doi:10.1101/gad.948702
103. Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev (1996) 10:1096–107. doi:10.1101/gad.10.9.1096
104. Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci U S A (1998) 95:4333–7. doi:10.1073/pnas.95.8.4333
105. Smith PJ, Wise LS, Berkowitz R, Wan C, Rubin CS. Insulin-like growth factor-I is an essential regulator of the differentiation of 3T3-L1 adipocytes. J Biol Chem (1988) 263:9402–8.
106. Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science (2000) 289:950–3. doi:10.1126/science.289.5481.950
107. Huang H, Song TJ, Li X, Hu L, He Q, Liu M, et al. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A (2009) 106:12670–5. doi:10.1073/pnas.0906266106
108. Kim M, Kim JI, Kim JB, Choe S. The activin-betaA/BMP-2 chimera AB204 is a strong stimulator of adipogenesis. J Tissue Eng Regen Med (2015). doi:10.1002/term.2050
109. Sengenes C, Lolmede K, Zakaroff-Girard A, Busse R, Bouloumie A. Preadipocytes in the human subcutaneous adipose tissue display distinct features from the adult mesenchymal and hematopoietic stem cells. J Cell Physiol (2005) 205:114–22. doi:10.1002/jcp.20381
110. Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell (2008) 135:240–9. doi:10.1016/j.cell.2008.09.036
111. Berry R, Rodeheffer MS. Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol (2013) 15:302–8. doi:10.1038/ncb2696
112. Berry R, Jeffery E, Rodeheffer MS. Weighing in on adipocyte precursors. Cell Metab (2014) 19:8–20. doi:10.1016/j.cmet.2013.10.003
113. Church CD, Berry R, Rodeheffer MS. Isolation and study of adipocyte precursors. Methods Enzymol (2014) 537:31–46. doi:10.1016/B978-0-12-411619-1.00003-3
114. Sims EA. Are there persons who are obese, but metabolically healthy? Metabolism (2001) 50:1499–504. doi:10.1053/meta.2001.27213
115. O’Connell J, Lynch L, Cawood TJ, Kwasnik A, Nolan N, Geoghegan J, et al. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PLoS One (2010) 5:e9997. doi:10.1371/journal.pone.0009997
116. Pataky Z, Bobbioni-Harsch E, Golay A. Open questions about metabolically normal obesity. Int J Obes (Lond) (2010) 34(Suppl 2):S18–23. doi:10.1038/ijo.2010.235
117. Primeau V, Coderre L, Karelis AD, Brochu M, Lavoie ME, Messier V, et al. Characterizing the profile of obese patients who are metabolically healthy. Int J Obes (Lond) (2011) 35:971–81. doi:10.1038/ijo.2010.216
118. Yamauchi T, Kamon J, Waki H, Imai Y, Shimozawa N, Hioki K, et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem (2003) 278:2461–8. doi:10.1074/jbc.M209033200
119. Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med (2012) 18:1539–49. doi:10.1038/nm.2899
120. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem (1995) 270:12953–6. doi:10.1074/jbc.270.22.12953
121. Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes (1996) 45:1661–9. doi:10.2337/diabetes.45.12.1661
122. Koh YJ, Park BH, Park JH, Han J, Lee IK, Park JW, et al. Activation of PPAR gamma induces profound multilocularization of adipocytes in adult mouse white adipose tissues. Exp Mol Med (2009) 41:880–95. doi:10.3858/emm.2009.41.12.094
123. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest (2005) 115:1111–9. doi:10.1172/JCI25102
124. Clowes GH Jr, Martin H, Walji S, Hirsch E, Gazitua R, Goodfellow R. Blood insulin responses to blood glucose levels in high output sepsis and spetic shock. Am J Surg (1978) 135:577–83. doi:10.1016/0002-9610(78)90040-5
125. Wichterman KA, Chaudry IH, Baue AE. Studies of peripheral glucose uptake during sepsis. Arch Surg (1979) 114:740–5. doi:10.1001/archsurg.1979.01370300094020
126. Williamson RT. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. Br Med J (1901) 1:760–2. doi:10.1136/bmj.1.2100.760
128. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259:87–91. doi:10.1126/science.7678183
129. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi:10.1146/annurev-immunol-031210-101322
130. Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes (2011) 60:2474–83. doi:10.2337/db11-0194
131. Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res (2008) 49:1894–903. doi:10.1194/jlr.M800132-JLR200
132. Talukdar S, Oh, da Y, Bandyopadhyay G, Li D, Xu J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012) 18:1407–12. doi:10.1038/nm.2885
133. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol (2006) 6:541–50. doi:10.1038/nri1841
134. Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) (2009) 17:2014–8. doi:10.1038/oby.2009.113
135. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi:10.1172/JCI19246
136. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112:1821–30. doi:10.1172/JCI19451
137. Cancello R, Tordjman J, Poitou C, Guilhem G, Bouillot JL, Hugol D, et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes (2006) 55:1554–61. doi:10.2337/db06-0133
138. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116:1494–505. doi:10.1172/JCI26498
139. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest (2006) 116:115–24. doi:10.1172/JCI24335
140. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
141. Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab (2008) 8:301–9. doi:10.1016/j.cmet.2008.08.015
142. Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res (2008) 103:467–76. doi:10.1161/CIRCRESAHA.108.177105
143. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15:914–20. doi:10.1038/nm.1964
144. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med (2009) 15:921–9. doi:10.1038/nm.2001
145. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi:10.1172/JCI29881
146. Ricardo-Gonzalez RR, Red Eagle A, Odegaard JI, Jouihan H, Morel CR, Heredia JE, et al. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc Natl Acad Sci U S A (2010) 107:22617–22. doi:10.1073/pnas.1009152108
147. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332:243–7. doi:10.1126/science.1201475
148. Ji Y, Sun S, Xia S, Yang L, Li X, Qi L. Short-term high-fat-diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem (2012) 287(29):24378–86. doi:10.1074/jbc.M112.371807
149. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, et al. Activation of natural killer T cells promotes M2 macrophage polarization in adipose tissue and improves systemic glucose tolerance via the IL-4/STAT6 signaling axis in obesity. J Biol Chem (2012) 287(17):13561–71. doi:10.1074/jbc.M112.350066
150. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity (2012) 37:574–87. doi:10.1016/j.immuni.2012.06.016
151. Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest (2012) 122:3343–54. doi:10.1172/JCI62739
152. Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, et al. A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol Cell Biol (2013) 33:328–39. doi:10.1128/MCB.00552-12
153. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
154. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16:85–95. doi:10.1038/ni.3047
155. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15:930–9. doi:10.1038/nm.2002
156. Bruun JM, Lihn AS, Pedersen SB, Richelsen B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): implication of macrophages resident in the AT. J Clin Endocrinol Metab (2005) 90:2282–9. doi:10.1210/jc.2004-1696
157. Fain JN. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm (2006) 74:443–77. doi:10.1016/S0083-6729(06)74018-3
158. Yu R, Kim CS, Kwon BS, Kawada T. Mesenteric adipose tissue-derived monocyte chemoattractant protein-1 plays a crucial role in adipose tissue macrophage migration and activation in obese mice. Obesity (Silver Spring) (2006) 14:1353–62. doi:10.1038/oby.2006.153
159. Alvehus M, Buren J, Sjostrom M, Goedecke J, Olsson T. The human visceral fat depot has a unique inflammatory profile. Obesity (Silver Spring) (2010) 18:879–83. doi:10.1038/oby.2010.22
160. Bigornia SJ, Farb MG, Mott MM, Hess DT, Carmine B, Fiscale A, et al. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes (2012) 2:e30. doi:10.1038/nutd.2012.3
161. Fujioka S, Matsuzawa Y, Tokunaga K, Tarui S. Contribution of intra-abdominal fat accumulation to the impairment of glucose and lipid metabolism in human obesity. Metabolism (1987) 36:54–9. doi:10.1016/0026-0495(87)90063-1
162. McLaughlin T, Liu LF, Lamendola C, Shen L, Morton J, Rivas H, et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arterioscler Thromb Vasc Biol (2014) 34:2637–43. doi:10.1161/ATVBAHA.114.304636
163. Fujisaka S, Usui I, Bukhari A, Ikutani M, Oya T, Kanatani Y, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes (2009) 58:2574–82. doi:10.2337/db08-1475
164. Anderson EK, Gutierrez DA, Kennedy A, Hasty AH. Weight cycling increases T-cell accumulation in adipose tissue and impairs systemic glucose tolerance. Diabetes (2013) 62:3180–8. doi:10.2337/db12-1076
165. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi:10.1038/nature05894
166. Clement K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J (2004) 18:1657–69. doi:10.1096/fj.04-2204com
167. Li P, Lu M, Nguyen MT, Bae EJ, Chapman J, Feng D, et al. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J Biol Chem (2010) 285:15333–45. doi:10.1074/jbc.M110.100263
168. Mraz M, Lacinova Z, Drapalova J, Haluzikova D, Horinek A, Matoulek M, et al. The effect of very-low-calorie diet on mRNA expression of inflammation-related genes in subcutaneous adipose tissue and peripheral monocytes of obese patients with type 2 diabetes mellitus. J Clin Endocrinol Metab (2011) 96:E606–13. doi:10.1210/jc.2010-1858
169. Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes (2005) 54:2277–86. doi:10.2337/diabetes.54.8.2277
170. Trachta P, Dostalova I, Haluzikova D, Kasalicky M, Kavalkova P, Drapalova J, et al. Laparoscopic sleeve gastrectomy ameliorates mRNA expression of inflammation-related genes in subcutaneous adipose tissue but not in peripheral monocytes of obese patients. Mol Cell Endocrinol (2014) 383:96–102. doi:10.1016/j.mce.2013.11.013
171. Poitou C, Perret C, Mathieu F, Truong V, Blum Y, Durand H, et al. Bariatric surgery induces disruption in inflammatory signaling pathways mediated by immune cells in adipose tissue: a RNA-Seq study. PLoS One (2015) 10:e0125718. doi:10.1371/journal.pone.0125718
172. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab (2008) 7:496–507. doi:10.1016/j.cmet.2008.04.003
173. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol (2011) 6:275–97. doi:10.1146/annurev-pathol-011110-130138
174. Cho CH, Koh YJ, Han J, Sung HK, Jong Lee H, Morisada T, et al. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ Res (2007) 100:e47–57. doi:10.1161/01.RES.0000259564.92792.93
175. Nishimura S, Manabe I, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes (2007) 56:1517–26. doi:10.2337/db06-1749
176. Bourlier V, Zakaroff-Girard A, Miranville A, De Barros S, Maumus M, Sengenes C, et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation (2008) 117:806–15. doi:10.1161/CIRCULATIONAHA.107.724096
177. Pang C, Gao Z, Yin J, Zhang J, Jia W, Ye J. Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity. Am J Physiol Endocrinol Metab (2008) 295:E313–22. doi:10.1152/ajpendo.90296.2008
178. Xu F, Burk D, Gao Z, Yin J, Zhang X, Weng J, et al. Angiogenic deficiency and adipose tissue dysfunction are associated with macrophage malfunction in SIRT1-/- mice. Endocrinology (2012) 153:1706–16. doi:10.1210/en.2011-1667
179. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature (2000) 407:242–8. doi:10.1038/35025215
180. Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol (2001) 21:1104–17. doi:10.1161/hq0701.093685
181. Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell (2006) 125:577–91. doi:10.1016/j.cell.2006.02.050
182. Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol (2009) 29:1575–91. doi:10.1128/MCB.01300-08
183. Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, et al. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab (2009) 94:5155–62. doi:10.1210/jc.2009-0947
184. Spencer M, Unal R, Zhu B, Rasouli N, McGehee RE Jr, Peterson CA, et al. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J Clin Endocrinol Metab (2011) 96:E1990–8. doi:10.1210/jc.2011-1567
185. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest (2011) 121:2094–101. doi:10.1172/JCI45887
186. Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol (2005) 175:6257–63. doi:10.4049/jimmunol.175.10.6257
187. Cao Y. Angiogenesis modulates adipogenesis and obesity. J Clin Invest (2007) 117:2362–8. doi:10.1172/JCI32239
188. Fan Y, Ye J, Shen F, Zhu Y, Yeghiazarians Y, Zhu W, et al. Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J Cereb Blood Flow Metab (2008) 28:90–8. doi:10.1038/sj.jcbfm.9600509
189. Ligresti G, Aplin AC, Zorzi P, Morishita A, Nicosia RF. Macrophage-derived tumor necrosis factor-alpha is an early component of the molecular cascade leading to angiogenesis in response to aortic injury. Arterioscler Thromb Vasc Biol (2011) 31:1151–9. doi:10.1161/ATVBAHA.111.223917
190. Stienstra R, Dijk W, van Beek L, Jansen H, Heemskerk M, Houtkooper RH, et al. Mannose-binding lectin is required for the effective clearance of apoptotic cells by adipose tissue macrophages during obesity. Diabetes (2014) 63:4143–53. doi:10.2337/db14-0256
191. Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest (2010) 120:3466–79. doi:10.1172/JCI42845
192. Prieur X, Mok CY, Velagapudi VR, Nunez V, Fuentes L, Montaner D, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes (2011) 60:797–809. doi:10.2337/db10-0705
193. Shapiro H, Pecht T, Shaco-Levy R, Harman-Boehm I, Kirshtein B, Kuperman Y, et al. Adipose tissue foam cells are present in human obesity. J Clin Endocrinol Metab (2013) 98:1173–81. doi:10.1210/jc.2012-2745
194. Fischer-Posovszky P, Wang QA, Asterholm IW, Rutkowski JM, Scherer PE. Targeted deletion of adipocytes by apoptosis leads to adipose tissue recruitment of alternatively activated M2 macrophages. Endocrinology (2011) 152:3074–81. doi:10.1210/en.2011-1031
195. Kato H, Mineda K, Eto H, Doi K, Kuno S, Kinoshita K, et al. Degeneration, regeneration, and cicatrization after fat grafting: dynamic total tissue remodeling during the first 3 months. Plast Reconstr Surg (2014) 133:303e–13e. doi:10.1097/PRS.0000000000000066
196. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature (2011) 480:104–8. doi:10.1038/nature10653
197. Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab (2013) 18:355–67. doi:10.1016/j.cmet.2013.08.003
Keywords: adipose tissue, adipose tissue macrophage, hypertrophic adipocyte, inflammatory response, iNKT cell, metabolic disorder, obesity
Citation: Choe SS, Huh JY, Hwang IJ, Kim JI and Kim JB (2016) Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 7:30. doi: 10.3389/fendo.2016.00030
Received: 21 November 2015; Accepted: 31 March 2016;
Published: 13 April 2016
Edited by:
Gary Sweeney, York University, CanadaReviewed by:
Carol Huang, University of Calgary, CanadaCopyright: © 2016 Choe, Huh, Hwang, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jae Bum Kim, amFlYmtpbUBzbnUuYWMua3I=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.