Abstract
Brown adipose tissue (BAT) is considered an interesting target organ for the treatment of metabolic disease due to its high metabolic capacity. Non-shivering thermogenesis, once activated, can lead to enhanced partitioning and oxidation of fuels in adipose tissues, and reduce the burden of glucose and lipids on other metabolic organs such as liver, pancreas, and skeletal muscle. Sustained long-term activation of BAT may also lead to meaningful bodyweight loss. In this review, we discuss three different drug classes [the thiazolidinedione (TZD) class of PPARγ agonists, β3-adrenergic receptor agonists, and fibroblast growth factor 21 (FGF21) analogs] that have been proposed to regulate BAT and beige recruitment or activation, or both, and which have been tested in both rodent and human. The learnings from these classes suggest that restoration of functional BAT and beige mass as well as improved activation might be required to fully realize the metabolic potential of these tissues. Whether this can be achieved without the undesired cardiovascular side effects exhibited by the TZD PPARγ agonists and β3-adrenergic receptor agonists remains to be resolved.
Introduction
According to the International Diabetes Federation (IDF), 8.3% of adults worldwide – 370 million people – have type-2 diabetes (T2D), and the number of people with the disease is set to rise beyond 592 million in under 25 years (1). Although there are many drugs available for diabetes, none of them safely and durably prevent or reverse disease progress and its associated comorbidities. Poor diet, sedentary lifestyle, and obesity are considered major risk factors for diabetes. Inappropriate fuel handling by adipose tissue, liver, and skeletal muscle, combined with ectopic lipid deposition in key metabolic organs (such as liver, pancreas, muscle, and heart) have been hypothesized to play a significant role in the development of insulin resistance. Insulin resistance increases the overall burden on β-cells, which over time leads to β-cell failure and development of T2D.
Adipose tissue can be grossly divided into two major depots, white adipose tissue (WAT) and brown adipose tissue (BAT). WAT stores excess energy as triglycerides (TGs), which can be mobilized by lipolysis to generate free fatty acids (FFAs) for use by other tissues. BAT, on the other hand, is the main site of non-shivering thermogenesis (NST), which requires a brown adipocyte-specific protein called uncoupling protein 1 (UCP1).
Non-shivering thermogenesis by BAT is an interesting target for the treatment of metabolic disease due to the high metabolic capacity of BAT. BAT is highly vascularized and richly innervated by sympathetic nerves, and its activation is predominantly regulated by the sympathetic nerve system via β-adrenergic receptors (β-ARs). Enhancing energy expenditure (EE) through activation of NST by β3-adrenergic receptor (β3-AR) agonists has been investigated as an alternative to inhibition of food intake for bodyweight loss. This has, however, been unsuccessful in human clinical trials. This lack of effect on EE was partly attributed to negligible BAT function in adult humans compared to the situation in rodents.
The rediscovery of BAT in the adult human in 2007, and the subsequent demonstration of functional involvement of human BAT in NST have revitalized this area (2–5). In addition, the presence of brown-like adipocytes in WAT [referred to as beige or brown-in-white (brite) adipocytes] further increased the interest in brown adipocyte biology, as WAT mass is relatively large and any increase in cellular energetics in this tissue may have a significant impact on whole-body metabolism and EE. The beige nomenclature will be used for this review. Utilization of FFAs during NST could lead to depletion of brown and or beige adipocytes’ lipid stores, which may result in redistribution of fuels [including glucose and non-esterified fatty acids (NEFA)] toward brown and beige adipocytes. In turn, this could lead to a reduced fuel over-supply to other metabolic organs (heart, skeletal muscle, and liver) and, thus, improved insulin sensitivity (Figure 1).
Figure 1
Transcriptional and hormonal regulation of the “browning” program of adipose stem cells and characterization of the molecular signature have been reviewed extensively elsewhere (6–12). This paper focuses on the metabolic potential of BAT and beige adipocytes, how these systems can be manipulated by pharmacological means, and how to assess if a brown adipocyte phenotype has been achieved by pharmacological intervention. Finally, we discuss the challenges of drug discovery in this area by reviewing three classes of clinically investigated pharmacological agents that regulate various aspects of BAT and beige adipocyte function: thiazolidinedione (TZD) PPARγ agonists, β3-AR agonists, and fibroblast growth factor 21 (FGF21) analogs.
Brown Adipose Tissue in Adult Humans
Typically, BAT is located in the interscapular (iBAT), cervical, auxiliary, perirenal and paraaortic areas of animals or human infants (13–15). In adult humans, BAT depots have a diffuse anatomic distribution, with mixtures of white and beige adipocytes, seeming to coexist in close proximity. Beige adipocytes have also been reported to arise in what are normally considered WAT depots (such as inguinal adipose in rodents) in response to various stimuli such as cold, TZDs, and β-AR agonists (9). In humans, beige adipocytes were found in the WAT of pheochromocytoma patients due to the presence of catecholamine-secreting tumors (16, 17) and in the subcutaneous adipose of severely burned patients where heat loss is increased and who experience prolonged adrenergic stress (18). Both lineage-tracing studies and transcriptional profiling of classical brown and beige adipocytes indicate that these two cell types seem to originate developmentally from distinct cell lineages. Classical brown adipocytes in interscapular BAT arise from precursors that are myf5+, a gene known to be also expressed in committed skeletal muscle precursors (19, 20). The developmental origin of the beige adipocyte remains to be elucidated. The whole-body NST will have contributions from both classical brown and beige adipocytes.
It is now beyond doubt that BAT is present in adult humans and plays a role in NST (3). BAT activity can be detected by 18F-fluorodeoxyglucose (18F-FDG) uptake using PET–CT (2, 4, 5). Retrospective analysis of populations that have undergone PET–CT examination indicates that the prevalence of BAT varies between 1 and 5%. For example, one study on 4011 asymptomatic individuals (<5% obese subjects) showed that BAT prevalence is 5% in female and 1.3% in male (21). The BAT-positive subjects had lower body mass index (BMI), less visceral and subcutaneous fat areas, lower fasting glucose and TG levels, and increased HDL cholesterol concentrations compared to the BAT-negative subjects. Similar results were reported for 56 healthy volunteers (22), and for cancer patients (2, 23). The inverse correlation between BAT activity and BMI was further confirmed in non-diabetic subjects over a wide range of body compositions (BMI ranging from 22 to 48 kg/m2) (4, 24–27). A recent retrospective analysis of 18F-FDG uptake data (analysis of the neck regions of two relatively large cohorts of individuals) reports that the average body weight of BAT-positive individuals is approximately 5 kg lower compared to that of BAT-negative subjects (21, 23).
Using a combination of MRI and molecular analysis, Enerbäck’s group clearly demonstrated that iBAT in human infants consists of classical brown adipocytes (15). However, the molecular signature of brown adipocytes isolated from the neck regions of adult humans resembles that of the rodent beige phenotype rather than classical brown adipocytes (15, 28–31). What causes the BAT phenotype transformation between infant and adult humans is not understood. It is conceptually important to unravel the function, regulation, and differentiation of beige and classical brown adipocytes in order to be able to pharmacologically enhance thermogenesis in humans. For example, if the adipocytes with various white and beige appearances in adult human neck region are, in fact, simply dormant brown adipocytes, they may be readily re-activated by cold or sympathomimetics. In addition, it is important to understand the translational aspects of BAT biology, i.e., whether the same or different pharmacological agent(s) will show desirable effects in preclinical animal species and man?
Bat and Metabolic Significance
The contribution of BAT to whole-body metabolism in rodents has recently been examined using tools such as radioactive tracers and PET–CT imaging. Bartelt et al. showed that BAT is the major site of triglyceride-rich lipoprotein (TRL) clearance during acute cold exposure (32). Cold exposure also dramatically increased the glucose disposal to BAT tissue. The remarkable capacity of BAT to take up substrates is illustrated by the ratio of BAT mass to the total glucose and TG uptake by BAT compared to that of other major organs in mice under cold challenge. Labbe et al. extended this observation through PET–CT analysis of the rate of substrate flux and oxidation in the iBAT of both warm- and cold-adapted rats (33). The rate of glucose uptake into iBAT was relatively low at 27°C but increased 10-fold upon acute cold exposure and increased 46-fold after cold acclimation at 10°C. Similarly, NEFA levels rose 6-fold upon acute cold exposure, and ~100-fold after cold acclimation. The metabolic activity of the iBAT reached levels similar to that of heart and liver after 6 h of cold exposure. In spite of these results, it should be kept in mind that although BAT glucose uptake per unit volume of tissue is important, the bulk of glucose turnover during cold exposure is mediated by skeletal muscle metabolic activation even when shivering is minimized (7).
It is more challenging to determine the specific contribution of beige adipocytes to whole-body metabolism. Bartelt et al. showed that acute cold exposure also increases TRL uptake in inguinal WAT (iWAT), but to a smaller extent compared to iBAT (32, 34). Seale’s group reported an aP2-PRDM16 transgenic mouse that exhibited a highly favorable metabolic phenotype, in which iBAT remained unchanged but with extensive browning in iWAT. This suggests that beige adipocytes may contribute to the overall metabolic phenotype observed in this mouse (35).
On the whole-body level, Reitman’s group dissected the relative contributions of cold-induced, diet-induced, and physical activity-associated EE in mice in relation to the basal metabolic rate (BMR) at various temperatures (36). This work clearly illustrated that at 22°C, the temperature at which most reported metabolic studies have been conducted, mice expend a relatively large amount of energy to generate heat (120% of BMR). Adult humans, on the other hand, live in or near their thermoneutral zone, with a relatively small contribution from adaptive thermogenesis to EE (5% of BMR) (11). Interestingly, in mice housed at thermoneutrality (30–32°C), the relative contributions of BMR, diet, physical activity, and adaptive thermogenesis in mice to overall EE are reported to be ~60, 12, 25, and 0%, respectively (36). These figures are very similar to the relative contributions seen in humans with low activity levels, and might represent experimental conditions more suitable for translational research in this field. The EE increase in relation to external temperature is conceptually depicted in Figure 2 (not scaled to real data).
Figure 2
Numerous studies that address the role of human BAT activation and its quantitative impact on whole-body metabolism have been published. Based on the heat production capacity of mouse BAT, Rothwell and Stock proposed in the 1980s that 40–50 g of BAT, if maximally activated, could account for 20% of daily EE in human (37). More recently PET–CT measurements estimated the average active BAT volume in healthy humans to be 137 cm3, corresponding to a conservative estimate of around 50 g BAT mass (11). Virtanen et al. estimated the EE of human BAT to be 55 W/kg (5) based on the rate of glucose uptake during cold exposure, as measured by dynamic PET–CT. These conservative estimates suggest that the EE of fully activated BAT could amount to 2–5% of BMR. Recent cold exposure experiments confirmed that the cold-induced NST-associated increase in EE accounts for 0–15% of BMR (11). Using a human body-composition model (38), we further extrapolated that a 4% increase of BMR could lead to a 3% bodyweight reduction per year, assuming that the effects are sustained. A key assumption in such an extrapolation is that functional tolerance can be avoided (39). In support of this assumption, some pieces of evidence suggest that increased thermogenesis is not always fully compensated for and negated by an increase in food intake, for reasons that are not fully understood (40).
Cypess et al. recently reported a 13% increase in BMR as a result of BAT activation upon administration of an acute dose of the β3-AR agonist mirabegron (41). Again, assuming that this BMR increase could be sustained upon chronic treatment, modeling suggests that this could potentially lead to an 8% bodyweight loss per year in BAT-positive healthy men. Besides potential functional tolerance, it remains uncertain if chronic β-AR-mediated stimulation of BAT is possible without encountering the side effects often associated with β-AR agonists.
Bodyweight reduction has been shown to have positive effects not only on preventing the progression of pre-diabetes to diabetes, but also leads to a reduction of hemoglobin A1c (HbA1c) (42). More recently, Hanssen et al. showed that 10-day cold acclimation in obese T2D patients could increase BAT activity, which was in turn associated with a reduction of BAT TG content and increased EE (43). One very encouraging observation is the 43% improvement of glucose infusion rate during a clamp study. Importantly, the improvement in insulin sensitivity of both adipose and skeletal muscle appeared before any bodyweight change could be seen (43). As indicated by Hanssen et al., the cold-induced improvement in insulin sensitivity exceeds that which was observed after long-term exercise training. To place this in a pharmacological context, this improvement in insulin sensitivity is similar in extent to that seen after 2-week treatment with dapagliflozin (Farxiga/Forxiga), which affords an 18% improvement in tissue glucose disposal (44).
Functional Brown/Beige Adipocytes
The remarkable metabolic capacity of classical brown adipocytes in the activated state has been well characterized using rodent brown adipocytes. Functional characterization of beige adipocytes from the inguinal adipose tissue of mice or differentiated beige adipocytes from human adipose stem cells suggests that these cells are functionally similar to classical brown adipocytes (45, 46). The developmental origin of brown and beige adipocytes is currently an area of intense research and has been extensively reviewed elsewhere (47, 48).
Uncoupling of the mitochondrial transmembrane proton gradient in brown adipocytes in order to generate heat in response to cold is a complicated but well-orchestrated event. The key metabolic and signaling pathways in the brown or beige adipocyte are summarized in Figure 3. To be able to carry out this function, brown adipocytes need to be equipped with a complex machinery that is able to (i) signal through norepinephrine (NE) or other catecholamines via β-ARs, (ii) generate intracellular FFAs through hydrolysis of TGs from lipid droplets (lipolysis), and (iii) uncouple the ATP-generating process via UCP1 activation (13, 49). Chronically, cold challenge leads to a recruitment process in which cell proliferation, mitochondrial biogenesis, and angiogenesis are enhanced. Given the broad range of cellular processes that are involved in these events, it is not surprising that large numbers of genes/factors have been described to regulate the differentiation and function of brown and beige adipocytes (9, 34).
Figure 3
The β3-AR has emerged as a leading molecular target for the activation of brown or beige adipocytes (18, 41). Rodent and human adipose tissue display different β-AR expression profiles. (50–53). In mouse, the β3-AR is highly expressed in both BAT and WAT with several-fold higher abundance than the β1-AR, whereas in the adult human, the β3-AR is only expressed in BAT. In human WAT, β1-AR has been reported to be 50-fold more abundant than β3-AR (52). The β3-AR subtypes also differ in their potency toward various ligands, G-protein coupling, and desensitization. The binding affinity of NE to β3-AR was reported to be in the low micromolar range, while the potency for cAMP accumulation is in the low nanomolar range when measured in intact cells, suggesting that the coupling efficacy to adenylyl cyclase of β3-AR was higher than that of β1-AR (52, 54). In addition, β1-AR desensitizes more rapidly than β3-AR upon exposure to agonists. This led to the hypothesis that β1-AR may mediate the NE response to low levels of sympathetic stimulation. On the other hand, the activation of β3-AR may require higher levels of sympathetic stimulation, but once activated this receptor is likely to deliver a more sustained effect (52). It is important to note that circulating NE levels are generally in the low nanomolar concentration range, and it is likely that NE concentrations at the synaptic clefts will be much higher during cold response, providing sufficiently high local concentrations in BAT to enable local activation of β3-AR in spite of the low plasma concentration. Recently, several studies have shown that systemic administration of adrenergic activators, such as isoproterenol (ISO) and ephedrine, fail to elicit BAT activation in man (55–57). To properly interpret these clinical data, it is crucial to understand if the plasma concentrations of the adrenergic agonists used reached a sufficiently high concentration to enable activation of lipolysis and thermogenesis in human BAT.
Norepinephrine-mediated β-AR activation results in an increased intracellular cAMP concentration, which in turn stimulates lipolysis in brown adipocytes via activation of the protein kinase A pathway. Lipolysis is a stepwise process with different enzymes acting at each step: TGs are hydrolyzed by desnutrin/adipose triglyceride lipase (ATGL) to form diacylglycerol (DAG). DAG is then hydrolyzed by hormone-sensitive lipase (HSL) to monoacylglycerol and, subsequently glycerol, with a fatty acid released at each stage. Intracellular FFAs are the direct activators of UCP1 (58). In humans, the BAT radiodensity (which is indicative of intracellular TG stores) is inversely correlated with NST, strongly suggesting that depletion of intracellular TG occurs during cold exposure. In rats, inhibition of lipolysis by nicotinic acid-mediated GPR109a agonism significantly reduced the oxidative capacity of iBAT in response to cold, again suggesting a key role for lipolysis in UCP1-mediated thermogenesis (33). Additionally, adipose tissue-specific knockout of ATGL led to the formation of “whitened” brown adipocytes and resulted in impaired lipolysis and defective thermogenesis in BAT (59–62).
The brown adipocytes’ cold-depleted energy stores are replenished by de novo lipogenesis and glycogen synthesis, which requires cellular uptake of circulating glucose and FFAs (derived either from TRLs or from lipolysis in WAT). In brown adipocytes, glucose uptake is mostly mediated by glucose transporter 1 (GLUT1) and glucose transporter 4 (GLUT4) and subsequently stored as glycogen or converted to lactate through anaerobic glycolysis. As shown in Figure 3, FFAs are transported into the cell by cluster of differentiation 36 (CD36) and TGs are subsequently synthesized through re-esterification by a series of enzymes, including glycerol-3-phosphate acyltransferase (GPAT) and diacylglycerol O-acyltransferases (DGATs) (49). The process of recruitment of new brown or beige adipocytes and the maintenance of mature adipocyte function during acute and chronic cold acclimation are subject to complex transcriptional and hormonal regulation, for example, by PPARγ, bone morphogenic proteins (BMPs), the thyroid axis, and FGF21. These regulatory mechanisms have been extensively reviewed elsewhere (8, 47, 48, 63).
The functional activity of BAT is reduced by chronic warm acclimation, old age, obesity, and diabetes. Rodent models of genetic deficiency of leptin (ob/ob, db/db, and obese fa/fa rats) are cold sensitive, and their brown adipocytes have a white adipocyte appearance with reduced expression of UCP1 protein (64, 65). Old age and lack of cold challenge also reduce the thermogenic activity of the brown adipocytes (66). Brown adipocytes isolated from guinea pigs housed at 30°C appear unilocular, although these cells appear to retain their brown adipocyte identity and are able to respond to NE with a robust increase of thermogenesis, which is not the case for adipocytes isolated from WAT (67). In humans, BAT activity is inversely correlated with age, fat mass, and BMI. Insulin-stimulated glucose uptake is also compromised in the BAT of diabetic individuals (68). Recent reports that both chronic cold acclimation and weight loss can enhance BAT activity in humans are encouraging (27, 69, 70), indicating that reduced BAT function may be restored. In this respect, adipocytes or adipose precursor cells are highly plastic and able to adapt to the functional needs, as has been shown in rodent using lineage tracing experiments (71).
Thiazolidinediones
The TZDs [represented by rosiglitazone (Avandia) and pioglitazone (Actos)] are a chemical class of PPARγ agonists used as insulin sensitizers for the treatment of T2D (72). The primary site of action of the TZDs is adipose tissue, where they improve several functional aspects, including the uptake and storage of plasma NEFA. They also increase FFA mobilization under fasting conditions and enhance postprandial suppression of FFA mobilization by insulin (73).
Thiazolidinediones have been shown to increase UCP1 expression and BAT mass in rodents. Rosiglitazone has been shown in vitro by several laboratories to induce UCP1 in rodent brown adipocytes and differentiated adipose stem cells (74–77). In addition, chronic treatment of human subcutaneous adipose stem cells with rosiglitazone upregulated several components of the mitochondrial electron transport chain, which is consistent with what has been observed in human (78). The UCP1 protein in human adipocytes differentiated in vitro in the presence of rosiglitazone is only functional when cells have been allowed to differentiate for a longer time than is typically reported in mouse studies (45). Although TZDs have the ability to recruit the “browning program” in both mouse and human adipocytes, the thermogenic capacity of UCP1-expressing cells cannot be unleashed without subsequent activation (e.g., by β-AR agonists). An increase in the oxygen consumption rate (OCR) of human adipocytes differentiated in vitro in the presence of rosiglitazone could only be observed when the cells were stimulated with isoproterenol or in the presence of exogenously provided FFAs. The increase of OCR in these beige cells is completely UCP1 dependent, as UCP1 knock-down abolishes the effect (45).
Various in vivo rodent models of insulin resistance have been used to show that the TZDs increase UCP1 mRNA in iBAT and overall iBAT weight. However, this is not associated with a subsequent increase in thermogenesis or whole-body EE. In addition, TZD treatment leads to the brown adipocytes becoming lipid filled (79–81). This led to the hypothesis that obese and diabetic animals or humans could first be primed by a PPARγ agonist to expand the BAT capacity, followed by its activation via a β-AR agonist. This concept was tested pre-clinically in ob/ob mice by pre-treating with a PPARγ agonist (COOH, a non-TZD PPARγ agonist) followed by treatment with the β3-AR agonist CL-316,243. Synergistic effects on EE and bodyweight reduction were indeed observed in this single study (82). However, a later study using rosiglitazone followed by acute cold exposure failed to reproduce these effects (83). A possible explanation for the lack of increased EE in spite of increased TG storage, UCP1 expression and BAT mass induced upon PPARγ activation by rosiglitazone (84) may be the downregulation of β3-AR and iodothyronine deiodinase type II (DIO2) expression caused by TZD treatment (85, 86). In addition, as COOH belongs to a different structural class of PPARγ agonists to the TZDs, it is not unlikely that different outcomes could be observed between the two compounds. In humans, 12 weeks of pioglitazone treatment has been shown to generate a small increase in UCP1 mRNA levels in subcutaneous adipose tissue (87). However, combined pioglitazone and ephedrine treatment for 12 weeks in obese human subjects failed to deliver significant bodyweight reduction (88).
When comparing the observed outcomes of studies employing PPARγ agonists, it must be kept in mind that even structurally closely related compounds may exhibit very different PPARγ-dependent pharmacodynamic profiles. This is, in part, due to ligand-dependent modulation of the PPARγ protein that leads to recruitment of different coactivator and corepressor proteins, in turn resulting in a unique transcriptional profile for each compound (89). Since the identification of PPARγ as the molecular target of the TZDs (90), significant effort has been invested by the pharmaceutical industry in PPARγ agonist drug discovery (91–96) albeit largely without commercial success (97). The availability and high throughput of PPARγ LBD-based ligand binding assays and chimeric PPARγ–GAL4 reporter gene/transactivation assays enabled the generation of many highly potent, structurally diverse, selective PPARγ full and partial agonists. Despite this, since the launch of pioglitazone and rosiglitazone, no new selective PPARγ agonists have survived clinical testing and remain on the market, mainly due to preclinical and clinical safety issues (98–101). The central role of PPARγ in adipose biology has, however, not diminished due to these failures and PPARγ remains an attractive but challenging drug target.
The lack of translation between in vitro receptor binding and the functional (both in vitro and in vivo) effects of the compounds and inter-species differences in PPARγ biology are considered major obstacles in this field. This is due to a number of factors, including the combinatorial nature of the activation of the PPARγ:RXR heterodimer (102, 103), subtle differences in coactivator/corepressor recruitment between superficially related compounds (104–107), ligand effects on the extent of posttranslational modifications (108, 109), and non-transcriptional effects of the ligands (72). This suggests that a reductionist approach to PPARγ agonist discovery based on the use of isolated protein domains and chimeric reporter gene assays is unlikely to provide compounds with the desired functional or clinical outcome.
Since the “traditional” approach of optimizing receptor binding and agonist potency has not borne fruit, a radical change in the preclinical approach to PPARγ drug discovery is needed in order for the functional potential (for example, browning of WAT) of small-molecule PPARγ activation to be realized. For example, the application of phenotypic screening in relevant cell systems [i.e. primary human cells (110)] is one approach to frontload the functional assessment of compounds, with traditional in vitro assessment of PPARγ activity included in a secondary wave of assays. In addition, the recent widespread availability of omics techniques (such as RNAomics and proteomics) makes the preclinical identification of PPARγ agonists with a desirable functional profile a realistic prospect. A combination of such approaches avoids focus on a single receptor-dependent pathway or mechanism and allows pleiotropy to be accounted for. However, whether the perceived target-related risk associated with PPARγ agonism is considered acceptable in proportion to the potential commercial viability of a safe PPARγ agonist remains to be seen.
Beta-3 Adrenergic Receptor
Several sympathomimetic β3-AR agonists that selectively stimulate rodent brown and white adipocyte lipolysis were discovered from the mid-1980s onward (e.g., BRL-37344, CL-316,243, and CGP-12177A) (50, 51, 111). Early optimization of these compounds was mostly performed in rodent tissue or cell models, as the human β3-AR was not cloned until 1989 (112). The compounds showed potent anti-obesity and anti-diabetic effects in rodent models of obesity and diabetes, but none of these compounds advanced beyond the clinical phase II due to lack of efficacy. Specifically, compounds optimized using rodent β3-AR did not effectively translate to human. Several β3-AR agonists were synthesized and evaluated after the cloning of human β3-AR cDNA (113). A summary of studies investigating the effect of β3-AR agonism on EE in man is given in Table 1. Note that many binding and adenylyl cyclase activity assays were performed using isolated membrane preparations, returning β3-AR binding affinities in the micromolar range for the compounds tested. Lower potencies (in the nanomolar range) have been reported for the same compounds when measured in whole-cell assays (cAMP, lipolysis, or respiration) (52), suggesting a G-protein coupling efficiency for the β3-AR that is only captured in a whole-cell context.
Table 1
| PHYSICAL AND PHARMACOKINETIC PROPERTIES | ||||||
|---|---|---|---|---|---|---|
| Compound name(s) and chemical structure | CL-316,243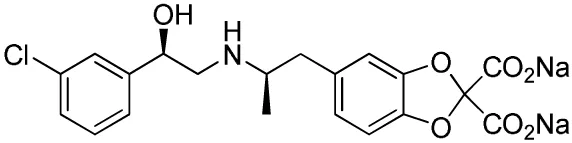 | ZD7114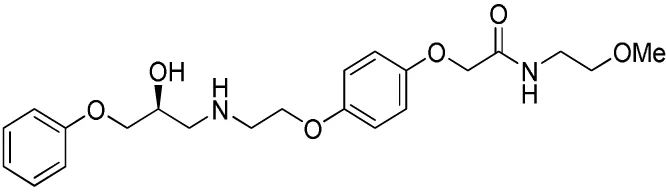 | ZD2079 (Talibegron)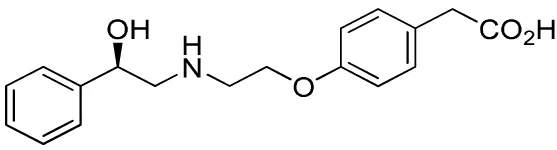 | L-796568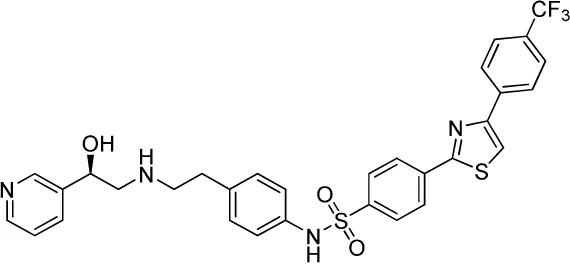 | TAK-677 (Rafabegron)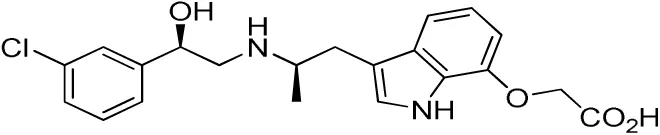 | Mirabegron (YM-178)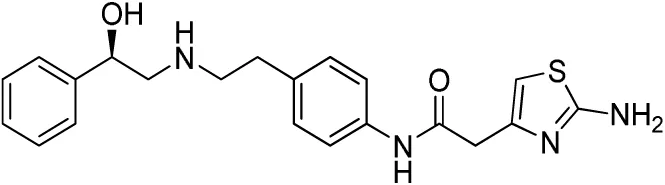 |
| MW (g/mol) | 466.8 (Na2 salt), 421.8 (free acid) | 418.5 | 315.4 | 624.7 | 402.9 (free acid) | 396.5 |
| clogP | −1.4 | 2.1 | −0.3 | 5.3 | 0.7 | 1.3 |
| Human β3 potency/TE | EC50 1.15 μM/0.63a (114) | Kact 20 nM (for ZD201651, the major active metabolite) (115) | Kact 191 nM/0.91a (116)b | 3.6 nM/0.94a (117) | EC50 0.062 nM/1.16a (118) | 22 nM/0.8a (119) |
| Rodent β3 potency/TE | Rat Ki 1 μM, Kact 0.71 nM/1a in β3 overexpressing CHO cells (111) | – | – | – | 0.016 nM/1.1a (rat cAMP) | – |
| Fold selectivity β3:β2:β1 | 1:228:96 Based on CHO data (114). Reported as an antagonist @ β1 and β2 (120) | – | – | 1:667 (partial):1333 (partial) | 1:209 (partial):1032 (partial) | 1:>446 (partial):>446 (partial) |
| T1/2/%F | 16 h/10% (human)c | – | – | >8 h/17% (rat) | – | – |
| PHARMACODYNAMICS | ||||||
| Population | Healthy young lean males (124) | Obese men and women with BMI 27–39 kg/m2 (115) | Obese men and women with BMI 27–39 kg/m2 (115) | Healthy overweight to obese men (121) | Obese men and women, mean BMI 33.9 kg/m2 (122) | Healthy male subjects with detectable BAT (41) |
| Administration | 1.5 g/day for 8 weeks (n = 10) | 150 mg/day for 2 weeks (n = 5) | 1.2 g/day for 2 weeks (n = 9) | 375 mg/day for 28 days (n = 10) | 0.5 mg BID for 29 days (n = 22) | 200 mg acutely (n = 12; crossover study) |
| Placebo (n = 4) | 300 mg/day for 2 weeks (n = 8) Placebo (n = 22) | Placebo (n = 8) | Placebo (n = 10) | Placebo (n = 4) | ||
| Exposure | 30 ± 11 nM (steady-state Cmin) | – | – | 77 ± 30 nM (steady-state Cmin) | 24 ± 13 nM (at t = 2 h) | 781 ± 184 nM (Cmax) |
| Fold in vitro EC50 | <0.1 | – | – | >20 | >1000 | >30 |
| BAT activity | – | – | – | – | – | Significant increase in BAT glucose uptake, from 1 to 130 mL × SUVmean × g/mL |
| Energy expenditure | 24-h EE after 8 weeks did not differ from baseline | No effect on 24 h EE | Trend for stimulatory effect on 24-h EE (2.4%) | Mean change in 24-h EE upon treatment did not differ significantly between treated and placebo | Slight increase (~50 kcal/day) in 24-h EE at the highest dose | Increased resting metabolic rate by 203 ± 40 kcal/day (+13%; p = 0.001) |
| Fasting plasma insulin | Trend toward decrease | – | – | No significant change | No significant change | Significant increase from 5.06 (3.56–6.19) to 7.61 (6.90–8.66) (μU/mL) |
| Other observations | No change in body weight or body composition. Increase in insulin action, decrease in 24-h RQ implying 23% increase in fat oxidation. No β1 or β2 side effects (bp/heart rate) | Acid metabolite also a β3 partial agonist. Increases UCP1 expression in dogs (2-week study 10 mg/kg BID) (123) | Trend for increase in spontaneous physical activity | No major lipolytic or thermogenic effect but lowered triacylglycerol concentrations. No β1/β2 side effects (heart rate or tremor) | No effect on 24-h RQ or fat oxidation | BAT metabolic activity was a significant predictor of the changes in RMR. Heart rate and systolic BP increased |
In vitro properties of β3-adrenergic receptor agonists that have been tested in humans, and their effect on energy expenditure in man.
MW, molecular weight; TE, target engagement; EE, energy expenditure; RQ, respiratory quotient.
aIsoproterenol response defined as 100%.
bPrimary reference refers to an, at that time, yet unpublished paper from ICI.
cClaimed to be a full agonist, but only 80% TE in in vitro assay.
Most early human trials of β3-AR agonists used bodyweight reduction as a clinical endpoint, to be achieved through increased EE. Data on BAT activation and metabolic parameters are scarce, with the exception being for the CL-316,243 study (124). In this study, treatment of lean healthy men for 4 weeks did not increase EE, but resulted in a 45% increase of insulin-mediated glucose disposal and a reduced 24-h respiratory quotient (24-h RQ), indicating enhanced fat oxidation. Intriguingly, CL-316,243 significantly increased fasting FFA levels in parallel with improved insulin action. These data are in line with recently published improved glucose infusion rates in T2D patients after 10 days of mild cold exposure (43). In both cases, improved insulin action preceded any significant weight loss. This improved action of insulin may be due to partitioning of FFAs toward BAT, which in turn reduces the fatty acid burden on other metabolic tissues (see Figure 1). Indeed, improved glucose uptake was seen in the skeletal muscle of T2D patients after cold exposure (43).
Similarly to CL-316,243 treatment, chronic dosing of the β3-AR agonists ZD7114 and ZD2079 did not significantly increase EE, although the latter compound showed a tendency toward a non-statistically significant increase of 2% (115).
Data have been reported for both acute and chronic dosing of β3-AR agonist L-796568 in overweight to obese men. Acutely, van Baak et al. (125) reported a significant increase (8%) in EE after 4 h for the highest dose used. However, no average increase was observed over the period of observation, 0–4 h. Chronic dosing of L-796568 for 4 weeks did not result in any significant change in EE (121). The authors discuss possible reasons for this lack of effect, e.g., declining plasma exposure levels over time, insufficient agonist-induced BAT proliferative capability of β3-AR-responsive tissues in humans during chronic stimulation, potential downregulation of β3-AR receptors, or other functional feedback. We believe that the small effect on EE observed in the acute study is another reason.
A chronic β3-AR agonism study (using TAK-677) in obese humans with no observed effect on EE or diabetic parameters was reported by Redman et al. (122). Whether BAT activation truly occurred after administration of compound was not carefully assessed in this study, which makes data interpretation challenging.
In contrast to the above-mentioned β3-AR agonism studies, Cypess et al. recently reported a ~13% increase in resting metabolic rate (RMR) upon acute treatment with high doses of β3-AR agonist mirabegron (41). In this study, healthy young male subjects with detectable BAT were selected in order to provide proof of concept (PoC) for β3-AR agonist-mediated BAT activation. Importantly, it was found that BAT metabolic activity was a significant predictor of the changes in RMR. This study also reports a weak correlation between cold- and drug-induced detectable BAT activities. Generally, such data from the same individuals are important to help put the large quantity of cold-induced BAT activation data into a drug-discovery context. It remains to be investigated if the reported energy-expenditure effect is sustained upon chronic dosing and if the efficacy of mirabegron will persist in females and other patient subpopulations, such as those with different ages and BMIs.
The failure of β3-AR agonists to show clinical effects on weight loss decreased interest in the mechanism as a means of treating the metabolic syndrome. However, the recent rediscovery of BAT in adult humans as well as the demonstration of functional activation of BAT by a β3-AR agonist may lead to a resurgent interest in β3-AR agonists for the treatment of metabolic disorders. The observation that improved insulin action preceded any significant weight loss upon β3-AR agonist treatment is particularly encouraging.
Clearly, important questions remain unanswered with respect to the role of the β3-AR and the clinical profile of β3-AR agonists. In order to make significant progress in β3-AR drug discovery, the lack of translation from rodent to human (i.e., receptor expression and functional differences) and from in vitro to in vivo for the human setting needs to be resolved. The clinically assessed β3-AR agonists exhibit structural and physicochemical property diversity; however, clinical plasma exposures relative to EC50 are similar for the chronic and acute studies (Table 1), and activation of BAT would therefore be expected in the chronic studies. The lack of effect on EE observed in the chronic studies may either be due to not measuring a mechanism-relevant clinical endpoint or that the patients recruited lacked sufficient BAT to lead to an effect on overall EE. The activation of pre-existing BAT by β3-AR agonists may require a personalized healthcare approach, unless combined with a compound or mechanism to expand the BAT depot.
Finally, cardiovascular side effects have been associated with β3-AR agonist treatment, usually attributed to insufficient selectivity toward the β1- and β2-ARs. However, the in vitro functional activity of many of the clinical compounds assessed does not explain the increase in heart rate observed – several of the compounds are functional β1-AR antagonists (β1-AR blockers), and as such could be expected to have the opposite effect to that clinically observed. The complex nature of AR biology needs to be further investigated if this mechanism is to be reconsidered as a means of activating BAT.
Fibroblast Growth Factor 21
Fibroblast growth factor 21 (FGF21) is a member of the endocrine FGF 19/21/23 family. FGF21 protein is expressed in liver, pancreas, and adipose tissue and is regulated by fasting, ketogenic diet, low protein diet, PPARγ, and PPARα activation as well as glucagon action (126). It acts through binding to a cell-surface receptor complex composed of conventional FGF receptors (FGFR1c/2c/3c) and the co-factor β-Klotho, leading to activation of FGF receptor substrate 2α and ERK1/2 phosphorylation. While FGF receptors are ubiquitously expressed, β-Klotho expression is restricted to a few tissues, including BAT, WAT, and liver (127, 128), which are the main sites of FGF21 action. When administered pharmacologically, FGF21 enhances EE and insulin sensitivity, reduces bodyweight, glucose, and lipids, and thus has the potential to be used for treatment of the metabolic syndrome via multiple mechanisms (129–131).
Functional enhancement of existing BAT and recruitment of beige adipocytes have been hypothesized to be the mechanism behind the EE increase, bodyweight loss, and improved glucose and lipid homeostasis induced by FGF21. FGF21 expression is increased in BAT upon cold challenge (132, 133). Adipose-derived FGF21 acts in an autocrine/paracrine manner to increase expression of UCP1 and other thermogenic genes, such as PGC1α, PRDM16, BMP8b, and DIO2 in iBAT and iWAT (134, 135). In neonates, FGF21 expression in liver is increased by suckling, which occurs via activation of PPARα, leading to induced BAT thermogenesis (136).
Attempts have been made to directly assess the contribution of BAT and beige adipocytes to FGF21-mediated effects. Surgical resection of iBAT in two different mouse models showed that FGF21-mediated EE increase and bodyweight reduction were retained (137, 138). However, one of the studies showed that several beige adipocyte markers, including PPARγ, PRDM16, PGC1α, and CIDEA tended toward upregulation in subcutaneous adipose tissue, and perigonadal adipose weight also was reduced (138). The authors propose that residual BAT and beige adipocytes may have compensated for the loss of iBAT. Recently, the role of UCP1 in mediating the pharmacological effects of FGF21 was assessed by treating UCP1 knockout (UCP1 KO) mice with either recombinant FGF21 or FGF21 fused with fragment crystallizable region (Fc-FGF21) (139, 140). Surprisingly, several FGF21-mediated effects were largely retained in the absence of UCP1. Again, compensatory mechanisms seem to be activated in UCP1 KO mice treated with recombinant FGF21. In FGF21-treated UCP1 KO mice, reduced food intake offset the decrease in EE and resulted in a similar bodyweight reduction to that observed in FGF21-treated wild-type mice. In addition, genes regulating fatty acid metabolism were upregulated in liver and epididymal adipose tissue, suggesting that FGF21 recruits UCP1-independent pathways in these tissues to compensate for the lack of UCP1. An intriguing observation was a two- to threefold increase in FGF21 secretion in iBAT when UCP1 KO mice were challenged by cold, suggesting that FGF21 may be one of the factors that recruit alternative thermogenic mechanisms when iBAT/UCP1 fails to generate heat. It should be noted that many previous publications have showed remodeling of the WAT in UCP1 KO mice, and that alternative thermogenic mechanisms have been discussed (141, 142).
In spite of the attractive metabolic effects of FGF21, development of FGF21-based therapeutics has encountered several technical challenges, including short in vivo half-life and poor biophysical properties of FGF21 protein (e.g., it is prone to aggregation). To date, three different analogs of FGF21 have advanced to concept testing in human: (130, 143, 144) (i) LY2405319 (an aggregation-resistant FGF21 analog), (ii) PF-05231023 (FGF21 linked to a Fab fragment of a scaffold antibody), and (iii) ARX-618 (PEGylated FGF21). For both LY2405319 and PF-05231023, the effects on TG (close to 50% reduction in 28 days), LDL cholesterol, and HDL cholesterol were substantial and bodyweight reduction was significant. However, the failure of FGF21 analogs to achieve clinically meaningful glucose-lowering effects was unexpected, and the mechanism behind this remains to be understood. Recently, Genentech reported a bispecific monoclonal antibody agonist that binds to both FGFR1c and β-Klotho, with sustained effects on NST, EE, and bodyweight over a 35-day period after a single administration to diet-induced obese mice (145). The effects on glucose and lipids were similar to those observed after administration of recombinant FGF21, and mostly attributed to peripheral FGF21 action as CNS exposure of the antibody was reported to be minimal. Together, these data further support the importance of BAT function in FGF21 action. As for the β3-AR agonist story, it may be important for future clinical concept testing to assess BAT recruitment/activation in order to understand if sufficiently high FGF21 levels have been achieved in patients. In addition, it is critical to constantly monitor potential safety concerns of this treatment principle in all studies, including bone density, growth hormone resistance, and female fertility.
Perspectives
The understanding of BAT physiology has increased rapidly in recent years. Data generated in both rodents and humans in cold/warm acclimation studies suggest that both BAT and beige adipocytes demonstrate recruitability and plasticity, which opens the door to pharmacological intervention. As obese and T2D patients are likely to have reduced BAT function, in order to realize the full potential of NST, it may be necessary to first restore the function of “whitened” brown or beige adipocytes before activation (Figure 2). In this respect, future studies using rodent models require consideration of external temperature in order to more closely mimic human physiology. Cold acclimation for a short duration in obese and diabetic humans indicates that it is possible to enhance BAT activity and achieve an improved metabolic profile before significant bodyweight reduction occurs. The improved insulin action in the absence of bodyweight change seen in the 4-week treatment of healthy individuals by CL-316,243 (124) suggests that restoration of BAT function may at least lead to improved metabolic control in patients with T2D. Acute treatment of BAT-positive human subjects with the β3-AR agonist mirabegron resulted in a significant increase in EE. If this effect is sustained upon chronic dosing and without eliciting cardiovascular side effects, this mechanism may finally lead to significant weight loss.
PPARγ remains an interesting target for the recruitment of BAT or beige adipocytes, although discovery of a compound that does not impair the adrenergic sensitivity or thyroid function of the adipocyte is extremely challenging. Phenotypic screening, for example a screen based on human brown adipocyte function, may however facilitate the discovery of such a compound.
Finally, if an FGF21 analog with appropriate half-life and potency could be developed, it would enable PoC testing in humans in order to understand if the potential beneficial metabolic effects of this hormone can be realized.
Statements
Author contributions
XP: conception, structure and content of the text, including the figures. PG: mathematical modeling of the literature data and critical examination and interpretation of pharmacokinetic and pharmacodynamic data on the β3-AR agonists in clinical studies. Major contribution to the Table 1. GO’M critical review and interpretation of PPARγ and β3-AR agonists structure, physiochemical properties and assays used for generating the data of all compounds included in Table 1. SB: critical review of bioenergetic of cell biology, general contribution of bioscience-related text, and critical reading and editing of review.
Acknowledgments
The authors would like to thank Nick Oakes (AstraZeneca) for his contribution to Figure 1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
IDF DIABETES ATLASSixth edition (2013). Available from: https://www.idf.org/sites/default/files/EN_6E_Atlas_Full_0.pdf
2
CypessAMLehmanSWilliamsGTalIRodmanDGoldfineABet alIdentification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360:1509–17.10.1056/NEJMoa0810780
3
NedergaardJBengtssonTCannonB. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab (2007) 293:E444–52.10.1152/ajpendo.00691.2006
4
van Marken LichtenbeltWDVanhommerigJWSmuldersNMDrossaertsJMKemerinkGJBouvyNDet alCold-activated brown adipose tissue in healthy men. N Engl J Med (2009) 360:1500–8.10.1056/NEJMoa0808718
5
VirtanenKALidellMEOravaJHeglindMWestergrenRNiemiTet alFunctional brown adipose tissue in healthy adults. N Engl J Med (2009) 360:1518–25.10.1056/NEJMoa0808949
6
BetzMJEnerbackS. Human brown adipose tissue: what we have learned so far. Diabetes (2015) 64:2352–60.10.2337/db15-0146
7
BlondinDPLabbeSMPhoenixSGuerinBTurcotteEERichardDet alContributions of white and brown adipose tissues and skeletal muscles to acute cold-induced metabolic responses in healthy men. J Physiol (2015) 593:701–14.10.1113/jphysiol.2014.283598
8
ChechiKCarpentierACRichardD. Understanding the brown adipocyte as a contributor to energy homeostasis. Trends Endocrinol Metab (2013) 24:408–20.10.1016/j.tem.2013.04.002
9
HarmsMSealeP. Brown and beige fat: development, function and therapeutic potential. Nat Med (2013) 19:1252–63.10.1038/nm.3361
10
NedergaardJCannonB. The browning of white adipose tissue: some burning issues. Cell Metab (2014) 20:396–407.10.1016/j.cmet.2014.07.005
11
van Marken LichtenbeltWDSchrauwenP. Implications of nonshivering thermogenesis for energy balance regulation in humans. Am J Physiol Regul Integr Comp Physiol (2011) 301:R285–96.10.1152/ajpregu.00652.2010
12
VosselmanMJvan Marken LichtenbeltWDSchrauwenP. Energy dissipation in brown adipose tissue: from mice to men. Mol Cell Endocrinol (2013) 379:43–50.10.1016/j.mce.2013.04.017
13
CannonBNedergaardJ. Brown adipose tissue: function and physiological significance. Physiol Rev (2004) 84:277–359.10.1152/physrev.00015.2003
14
EnerbackS. Human brown adipose tissue. Cell Metab (2010) 11:248–52.10.1016/j.cmet.2010.03.008
15
LidellMEBetzMJDahlqvist LeinhardOHeglindMElanderLSlawikMet alEvidence for two types of brown adipose tissue in humans. Nat Med (2013) 19:631–4.10.1038/nm.3017
16
FrontiniAVitaliAPeruginiJMuranoIRomitiCRicquierDet alWhite-to-brown transdifferentiation of omental adipocytes in patients affected by pheochromocytoma. Biochim Biophys Acta (2013) 1831:950–9.10.1016/j.bbalip.2013.02.005
17
RicquierDNechadMMoryG. Ultrastructural and biochemical characterization of human brown adipose tissue in pheochromocytoma. J Clin Endocrinol Metab (1982) 54:803–7.
18
SidossisLSPorterCSarafMKBorsheimERadhakrishnanRSChaoTet alBrowning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab (2015) 22:219–27.10.1016/j.cmet.2015.06.022
19
SealePBjorkBYangWKajimuraSChinSKuangSet alPRDM16 controls a brown fat/skeletal muscle switch. Nature (2008) 454:961–7.10.1038/nature07182
20
TimmonsJAWennmalmKLarssonOWaldenTBLassmannTPetrovicNet alMyogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc Natl Acad Sci U S A (2007) 104:4401–6.10.1073/pnas.0610615104
21
WangQZhangMXuMGuWXiYQiLet alBrown adipose tissue activation is inversely related to central obesity and metabolic parameters in adult human. PLoS One (2015) 10:e0123795.10.1371/journal.pone.0123795
22
SaitoMOkamatsu-OguraYMatsushitaMWatanabeKYoneshiroTNio-KobayashiJet alHigh incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes (2009) 58:1526–31.10.2337/db09-0530
23
PersichettiASciutoRReaSBascianiSLubranoCMarianiSet alPrevalence, mass, and glucose-uptake activity of (1)(8)F-FDG-detected brown adipose tissue in humans living in a temperate zone of Italy. PLoS One (2013) 8:e63391.10.1371/journal.pone.0063391
24
LeePGreenfieldJRHoKKFulhamMJ. A critical appraisal of the prevalence and metabolic significance of brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab (2010) 299:E601–6.10.1152/ajpendo.00298.2010
25
LeePSwarbrickMMZhaoJTHoKK. Inducible brown adipogenesis of supraclavicular fat in adult humans. Endocrinology (2011) 152:3597–602.10.1210/en.2011-1349
26
OuelletVLabbeSMBlondinDPPhoenixSGuerinBHamanFet alBrown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest (2012) 122:545–52.10.1172/JCI60433
27
VijgenGHBouvyNDTeuleGJBransBHoeksJSchrauwenPet alIncrease in brown adipose tissue activity after weight loss in morbidly obese subjects. J Clin Endocrinol Metab (2012) 97:E1229–33.10.1210/jc.2012-1289
28
CypessAMWhiteAPVernochetCSchulzTJXueRSassCAet alAnatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med (2013) 19:635–9.10.1038/nm.3112
29
JespersenNZLarsenTJPeijsLDaugaardSHomoePLoftAet alA classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab (2013) 17:798–805.10.1016/j.cmet.2013.04.011
30
SharpLZShinodaKOhnoHScheelDWTomodaERuizLet alHuman BAT possesses molecular signatures that resemble beige/brite cells. PLoS One (2012) 7:e49452.10.1371/journal.pone.0049452
31
WuJBostromPSparksLMYeLChoiJHGiangAHet alBeige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell (2012) 150:366–76.10.1016/j.cell.2012.05.016
32
BarteltABrunsOTReimerRHohenbergHIttrichHPeldschusKet alBrown adipose tissue activity controls triglyceride clearance. Nat Med (2011) 17:200–5.10.1038/nm.2297
33
LabbeSMCaronABakanILaplanteMCarpentierACLecomteRet alIn vivo measurement of energy substrate contribution to cold-induced brown adipose tissue thermogenesis. FASEB J (2015) 29:2046–58.10.1096/fj.14-266247
34
BarteltAHeerenJ. Adipose tissue browning and metabolic health. Nat Rev Endocrinol (2014) 10:24–36.10.1038/nrendo.2013.204
35
SealePConroeHMEstallJKajimuraSFrontiniAIshibashiJet alPrdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest (2011) 121:96–105.10.1172/JCI44271
36
Abreu-VieiraGXiaoCGavrilovaOReitmanML. Integration of body temperature into the analysis of energy expenditure in the mouse. Mol Metab (2015) 4:461–70.10.1016/j.molmet.2015.03.001
37
RothwellNJStockMJ. Whither brown fat?Biosci Rep (1986) 6:3–18.10.1007/BF01145174
38
HallKDSacksGChandramohanDChowCCWangYCGortmakerSLet alQuantification of the effect of energy imbalance on bodyweight. Lancet (2011) 378:826–37.10.1016/S0140-6736(11)60812-X
39
GennemarkPHjorthSGabrielssonJ. Modeling energy intake by adding homeostatic feedback and drug intervention. J Pharmacokinet Pharmacodyn (2015) 42:79–96.10.1007/s10928-014-9399-4
40
CannonBNedergaardJ. Thermogenesis challenges the adipostat hypothesis for body-weight control. Proc Nutr Soc (2009) 68:401–7.10.1017/S0029665109990255
41
CypessAMWeinerLSRoberts-TolerCFranquet EliaEKesslerSHKahnPAet alActivation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab (2015) 21:33–8.10.1016/j.cmet.2014.12.009
42
McAdam-MarxCMukherjeeJBellowsBKUnniSYeXIloejeUet alEvaluation of the relationship between weight change and glycemic control after initiation of antidiabetic therapy in patients with type 2 diabetes using electronic medical record data. Diabetes Res Clin Pract (2014) 103:402–11.10.1016/j.diabres.2013.12.038
43
HanssenMJHoeksJBransBvan der LansAASchaartGvan den DriesscheJJet alShort-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med (2015) 21:863–5.10.1038/nm.3891
44
MerovciASolis-HerreraCDanieleGEldorRFiorentinoTVTripathyDet alDapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest (2014) 124:509–14.10.1172/JCI70704
45
BartesaghiSHallenSHuangLSvenssonPAMomoRAWallinSet alThermogenic activity of UCP1 in human white fat-derived beige adipocytes. Mol Endocrinol (2015) 29:130–9.10.1210/me.2014-1295
46
ShabalinaIGPetrovicNde JongJMKalinovichAVCannonBNedergaardJ. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep (2013) 5:1196–203.10.1016/j.celrep.2013.10.044
47
SealeP. Transcriptional regulatory circuits controlling brown fat development and activation. Diabetes (2015) 64:2369–75.10.2337/db15-0203
48
WuJJunHMcDermottJR. Formation and activation of thermogenic fat. Trends Genet (2015) 31:232–8.10.1016/j.tig.2015.03.003
49
FestucciaWTBlanchardPGDeshaiesY. Control of brown adipose tissue glucose and lipid metabolism by PPARgamma. Front Endocrinol (2011) 2:84.10.3389/fendo.2011.00084
50
ArchJR. The discovery of drugs for obesity, the metabolic effects of leptin and variable receptor pharmacology: perspectives from beta3-adrenoceptor agonists. Naunyn Schmiedebergs Arch Pharmacol (2008) 378:225–40.10.1007/s00210-008-0271-1
51
AtgieCD’AllaireFBukowieckiLJ. Role of beta1- and beta3-adrenoceptors in the regulation of lipolysis and thermogenesis in rat brown adipocytes. Am J Physiol (1997) 273:C1136–42.
52
GrannemanJG. Why do adipocytes make the beta 3 adrenergic receptor?Cell Signal (1995) 7:9–15.10.1016/0898-6568(94)00066-K
53
LowellBBFlierJS. Brown adipose tissue, beta 3-adrenergic receptors, and obesity. Annu Rev Med (1997) 48:307–16.10.1146/annurev.med.48.1.307
54
MuzzinPRevelliJPKuhneFGocayneJDMcCombieWRVenterJCet alAn adipose tissue-specific beta-adrenergic receptor. Molecular cloning and down-regulation in obesity. J Biol Chem (1991) 266:24053–8.
55
CareyALFormosaMFVan EveryBBertovicDEikelisNLambertGWet alEphedrine activates brown adipose tissue in lean but not obese humans. Diabetologia (2013) 56:147–55.10.1007/s00125-012-2748-1
56
CypessAMChenYCSzeCWangKEnglishJChanOet alCold but not sympathomimetics activates human brown adipose tissue in vivo. Proc Natl Acad Sci U S A (2012) 109:10001–5.10.1073/pnas.1207911109
57
VosselmanMJvan der LansAABransBWiertsRvan BaakMASchrauwenPet alSystemic beta-adrenergic stimulation of thermogenesis is not accompanied by brown adipose tissue activity in humans. Diabetes (2012) 61:3106–13.10.2337/db12-0288
58
NichollsDG. A history of UCP1. Biochem Soc Trans (2001) 29:751–5.10.1042/bst0290751
59
AhmadianMAbbottMJTangTHudakCSKimYBrussMet alDesnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab (2011) 13:739–48.10.1016/j.cmet.2011.05.002
60
LiYFrommeTSchweizerSSchottlTKlingensporM. Taking control over intracellular fatty acid levels is essential for the analysis of thermogenic function in cultured primary brown and brite/beige adipocytes. EMBO Rep (2014) 15:1069–76.10.15252/embr.201438775
61
MottilloEPBlochAELeffTGrannemanJG. Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) alpha and delta in brown adipocytes to match fatty acid oxidation with supply. J Biol Chem (2012) 287:25038–48.10.1074/jbc.M112.374041
62
ZechnerR. FLUX FAT: enzymes, regulators, and pathophysiology of intracellular lipolysis. EMBO Mol Med (2015) 7:359–62.10.15252/emmm.201404846
63
KajimuraSSaitoM. A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu Rev Physiol (2014) 76:225–49.10.1146/annurev-physiol-021113-170252
64
CollinsSDanielKWRohlfsEMRamkumarVTaylorILGettysTW. Impaired expression and functional activity of the beta 3- and beta 1-adrenergic receptors in adipose tissue of congenitally obese (C57BL/6J ob/ob) mice. Mol Endocrinol (1994) 8:518–27.10.1210/me.8.4.518
65
TriandafillouJHimms-HagenJ. Brown adipose tissue in genetically obese (fa/fa) rats: response to cold and diet. Am J Physiol (1983) 244:E145–50.
66
RogersNHLandaAParkSSmithRG. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell (2012) 11:1074–83.10.1111/acel.12010
67
RafaelJFesserWNichollsDG. Cold adaptation in guinea pig at level of isolated brown adipocyte. Am J Physiol (1986) 250:C228–35.
68
BlondinDPLabbeSMNollCKunachMPhoenixSGuerinBet alSelective impairment of glucose but not fatty acid or oxidative metabolism in brown adipose tissue of subjects with type 2 diabetes. Diabetes (2015) 64:2388–97.10.2337/db14-1651
69
BlondinDPLabbeSMTingelstadHCNollCKunachMPhoenixSet alIncreased brown adipose tissue oxidative capacity in cold-acclimated humans. J Clin Endocrinol Metab (2014) 99:E438–46.10.1210/jc.2013-3901
70
LeePSmithSLindermanJCourvilleABBrychtaRJDieckmannWet alTemperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes (2014) 63:3686–98.10.2337/db14-0513
71
RosenwaldMPerdikariARulickeTWolfrumC. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol (2013) 15:659–67.10.1038/ncb2740
72
SoccioREChenERLazarMA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab (2014) 20:573–91.10.1016/j.cmet.2014.08.005
73
OakesNDThalenPGJacintoSMLjungB. Thiazolidinediones increase plasma-adipose tissue FFA exchange capacity and enhance insulin-mediated control of systemic FFA availability. Diabetes (2001) 50:1158–65.10.2337/diabetes.50.5.1158
74
DigbyJEMontagueCTSewterCPSandersLWilkisonWOO’RahillySet alThiazolidinedione exposure increases the expression of uncoupling protein 1 in cultured human preadipocytes. Diabetes (1998) 47:138–41.10.2337/diab.47.1.138
75
ElabdCChielliniCCarmonaMGalitzkyJCochetOPetersenRet alHuman multipotent adipose-derived stem cells differentiate into functional brown adipocytes. Stem Cells (2009) 27:2753–60.10.1002/stem.200
76
OhnoHShinodaKSpiegelmanBMKajimuraS. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab (2012) 15:395–404.10.1016/j.cmet.2012.01.019
77
PetrovicNShabalinaIGTimmonsJACannonBNedergaardJ. Thermogenically competent nonadrenergic recruitment in brown preadipocytes by a PPARgamma agonist. Am J Physiol Endocrinol Metab (2008) 295:E287–96.10.1152/ajpendo.00035.2008
78
SearsDDHsiaoGHsiaoAYuJGCourtneyCHOfrecioJMet alMechanisms of human insulin resistance and thiazolidinedione-mediated insulin sensitization. Proc Natl Acad Sci U S A (2009) 106:18745–50.10.1073/pnas.0903032106
79
BurkeyBFDongMGagenKEckhardtMDragonasNChenWet alEffects of pioglitazone on promoting energy storage, not expenditure, in brown adipose tissue of obese fa/fa Zucker rats: comparison to CL 316,243. Metabolism (2000) 49:1301–8.10.1053/meta.2000.9524
80
KellyLJVicarioPPThompsonGMCandeloreMRDoebberTWVentreJet alPeroxisome proliferator-activated receptors gamma and alpha mediate in vivo regulation of uncoupling protein (UCP-1, UCP-2, UCP-3) gene expression. Endocrinology (1998) 139:4920–7.10.1210/en.139.12.4920
81
SmithSRDe JongeLVolaufovaJLiYXieHBrayGA. Effect of pioglitazone on body composition and energy expenditure: a randomized controlled trial. Metabolism (2005) 54:24–32.10.1016/j.metabol.2004.07.008
82
SellHBergerJPSamsonPCastriotaGLalondeJDeshaiesYet alPeroxisome proliferator-activated receptor gamma agonism increases the capacity for sympathetically mediated thermogenesis in lean and ob/ob mice. Endocrinology (2004) 145:3925–34.10.1210/en.2004-0321
83
FestucciaWTBlanchardPGOliveiraTBMagdalonJPaschoalVARichardDet alPPARgamma activation attenuates cold-induced upregulation of thyroid status and brown adipose tissue PGC-1alpha and D2. Am J Physiol Regul Integr Comp Physiol (2012) 303:R1277–85.10.1152/ajpregu.00299.2012
84
FestucciaWTBlanchardPGTurcotteVLaplanteMSariahmetogluMBrindleyDNet alThe PPARgamma agonist rosiglitazone enhances rat brown adipose tissue lipogenesis from glucose without altering glucose uptake. Am J Physiol Regul Integr Comp Physiol (2009) 296:R1327–35.10.1152/ajpregu.91012.2008
85
BakopanosESilvaJE. Thiazolidinediones inhibit the expression of beta3-adrenergic receptors at a transcriptional level. Diabetes (2000) 49:2108–15.10.2337/diabetes.49.12.2108
86
FestucciaWTOztezcanSLaplanteMBerthiaumeMMichelCDohguSet alPeroxisome proliferator-activated receptor-gamma-mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology (2008) 149:2121–30.10.1210/en.2007-1553
87
BogackaIUkropcovaBMcNeilMGimbleJMSmithSR. Structural and functional consequences of mitochondrial biogenesis in human adipocytes in vitro. J Clin Endocrinol Metab (2005) 90:6650–6.10.1210/jc.2005-1024
88
BogackaIGettysTWde JongeLNguyenTSmithJMXieHet alThe effect of beta-adrenergic and peroxisome proliferator-activated receptor-gamma stimulation on target genes related to lipid metabolism in human subcutaneous adipose tissue. Diabetes Care (2007) 30:1179–86.10.2337/dc06-1962
89
NettlesKWGreeneGL. Ligand control of coregulator recruitment to nuclear receptors. Annu Rev Physiol (2005) 67:309–33.10.1146/annurev.physiol.66.032802.154710
90
LehmannJMMooreLBSmith-OliverTAWilkisonWOWillsonTMKliewerSA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem (1995) 270:12953–6.10.1074/jbc.270.22.12953
91
ChoNMomoseY. Peroxisome proliferator-activated receptor gamma agonists as insulin sensitizers: from the discovery to recent progress. Curr Top Med Chem (2008) 8:1483–507.10.2174/156802608786413474
92
HenkeBR. 1. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands and their therapeutic utility. Prog Med Chem (2004) 42:1–53.10.1016/S0079-6468(04)42001-3
93
NevinDKLloydDGFayneD. Rational targeting of peroxisome proliferating activated receptor subtypes. Curr Med Chem (2011) 18:5598–623.10.2174/092986711798347243
94
SavkurRSMillerAR. Investigational PPAR-gamma agonists for the treatment of Type 2 diabetes. Expert Opin Investig Drugs (2006) 15:763–78.10.1517/13543784.15.7.763
95
TakadaIMakishimaM. PPARgamma ligands and their therapeutic applications: a patent review (2008-2014). Expert Opin Ther Pat (2015) 25:175–91.10.1517/13543776.2014.985206
96
WillsonTMBrownPJSternbachDDHenkeBR. The PPARs: from orphan receptors to drug discovery. J Med Chem (2000) 43:527–50.10.1021/jm990554g
97
Garcia-VallveSGuaschLTomas-HernandezSDel BasJMOllendorffVArolaLet alPeroxisome proliferator-activated receptor gamma (PPARgamma) and ligand choreography: newcomers take the stage. J Med Chem (2015) 58:5381–94.10.1021/jm501155f
98
AbbasABlandonJRudeJElfarAMukherjeeD. PPAR-gamma agonist in treatment of diabetes: cardiovascular safety considerations. Cardiovasc Hematol Agents Med Chem (2012) 10:124–34.10.2174/187152512800388948
99
BortoliniMWrightMBBopstMBalasB. Examining the safety of PPAR agonists – current trends and future prospects. Expert Opin Drug Saf (2013) 12:65–79.10.1517/14740338.2013.741585
100
RubenstrunkAHanfRHumDWFruchartJCStaelsB. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta (2007) 1771:1065–81.10.1016/j.bbalip.2007.02.003
101
TangWHMarooA. PPARgamma agonists: safety issues in heart failure. Diabetes Obes Metab (2007) 9:447–54.10.1111/j.1463-1326.2006.00616.x
102
GermainPIyerJZechelCGronemeyerH. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature (2002) 415:187–92.10.1038/415187a
103
HamzaMSPottSVegaVBThomsenJSKandhadayarGSNgPWet alDe-novo identification of PPARgamma/RXR binding sites and direct targets during adipogenesis. PLoS One (2009) 4:e4907.10.1371/journal.pone.0004907
104
BulynkoYAO’MalleyBW. Nuclear receptor coactivators: structural and functional biochemistry. Biochemistry (2011) 50:313–28.10.1021/bi101762x
105
LonardDMO’MalleyBW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell (2007) 27:691–700.10.1016/j.molcel.2007.08.012
106
MillardCJWatsonPJFairallLSchwabeJW. An evolving understanding of nuclear receptor coregulator proteins. J Mol Endocrinol (2013) 51:T23–36.10.1530/JME-13-0227
107
NolteRTWiselyGBWestinSCobbJELambertMHKurokawaRet alLigand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature (1998) 395:137–43.10.1038/25931
108
AhmadianMSuhJMHahNLiddleCAtkinsARDownesMet alPPARgamma signaling and metabolism: the good, the bad and the future. Nat Med (2013) 19:557–66.10.1038/nm.3159
109
BanksASMcAllisterFECamporezJPZushinPJJurczakMJLaznik-BogoslavskiDet alAn ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature (2015) 517:391–5.10.1038/nature13887
110
MoisanALeeYKZhangJDHudakCSMeyerCAPrummerMet alWhite-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nat Cell Biol (2015) 17:57–67.10.1038/ncb3075
111
DolanJAMuenkelHABurnsMGPellegrinoSMFraserCMPietriFet alBeta-3 adrenoceptor selectivity of the dioxolane dicarboxylate phenethanolamines. J Pharmacol Exp Ther (1994) 269:1000–6.
112
EmorineLJMarulloSBriend-SutrenMMPateyGTateKDelavier-KlutchkoCet alMolecular characterization of the human beta 3-adrenergic receptor. Science (1989) 245:1118–21.10.1126/science.2570461
113
LeliasJMKaghadMRodriguezMChalonPBonninJDupreIet alMolecular cloning of a human beta 3-adrenergic receptor cDNA. FEBS Lett (1993) 324:127–30.10.1016/0014-5793(93)81377-C
114
HuBEllingboeJHanSLargisELimKMalamasMet alNovel (4-piperidin-1-yl)-phenyl sulfonamides as potent and selective human beta(3) agonists. Bioorg Med Chem (2001) 9:2045–59.10.1016/S0968-0896(01)00114-6
115
BuemannBToubroSAstrupA. Effects of the two beta3-agonists, ZD7114 and ZD2079 on 24 hour energy expenditure and respiratory quotient in obese subjects. Int J Obes Relat Metab Disord (2000) 24:1553–60.10.1038/sj.ijo.0801452
116
Pietri-RouxelFStrosbergAD. Pharmacological characteristics and species-related variations of beta 3-adrenergic receptors. Fundam Clin Pharmacol (1995) 9:211–8.10.1111/j.1472-8206.1995.tb00288.x
117
MathvinkRJTolmanJSChittyDCandeloreMRCascieriMAColwellLFJret alDiscovery of a potent, orally bioavailable beta(3) adrenergic receptor agonist, (R)-N-[4-[2-[[2-hydroxy-2-(3-pyridinyl)ethyl]amino]ethyl]phenyl]-4-[4 -[4-(trifluoromethyl)phenyl]thiazol-2-yl]benzenesulfonamide. J Med Chem (2000) 43:3832–6.10.1021/jm000286i
118
HaradaHHirokawaYSuzukiKHiyamaYOueMKawashimaHet alDiscovery of a novel and potent human and rat beta3-adrenergic receptor agonist, [3-[(2R)-[[(2R)-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]-1H-indol-7-yloxy]acetic acid. Chem Pharm Bull (2005) 53:184–98.10.1248/cpb.53.184
119
TakasuTUkaiMSatoSMatsuiTNagaseIMaruyamaTet alEffect of (R)-2-(2-aminothiazol-4-yl)-4’-{2-[(2-hydroxy-2-phenylethyl)amino]ethyl} acetanilide (YM178), a novel selective beta3-adrenoceptor agonist, on bladder function. J Pharmacol Exp Ther (2007) 321:642–7.10.1124/jpet.106.115840
120
BlinNNahmiasCDrumareMFStrosbergAD. Mediation of most atypical effects by species homologues of the beta 3-adrenoceptor. Br J Pharmacol (1994) 112:911–9.10.1111/j.1476-5381.1994.tb13167.x
121
LarsenTMToubroSvan BaakMAGottesdienerKMLarsonPSarisWHet alEffect of a 28-d treatment with L-796568, a novel beta(3)-adrenergic receptor agonist, on energy expenditure and body composition in obese men. Am J Clin Nutr (2002) 76:780–8.
122
RedmanLMde JongeLFangXGamlinBReckerDGreenwayFLet alLack of an effect of a novel beta3-adrenoceptor agonist, TAK-677, on energy metabolism in obese individuals: a double-blind, placebo-controlled randomized study. J Clin Endocrinol Metab (2007) 92:527–31.10.1210/jc.2006-1740
123
ChampignyORicquierDBlondelOMayersRMBriscoeMGHollowayBR. Beta 3-adrenergic receptor stimulation restores message and expression of brown-fat mitochondrial uncoupling protein in adult dogs. Proc Natl Acad Sci U S A (1991) 88:10774–7.10.1073/pnas.88.23.10774
124
WeyerCTataranniPASnitkerSDanforthEJrRavussinE. Increase in insulin action and fat oxidation after treatment with CL 316,243, a highly selective beta3-adrenoceptor agonist in humans. Diabetes (1998) 47:1555–61.10.2337/diabetes.47.10.1555
125
van BaakMAHulGBToubroSAstrupAGottesdienerKMDeSmetMet alAcute effect of L-796568, a novel beta 3-adrenergic receptor agonist, on energy expenditure in obese men. Clin Pharmacol Ther (2002) 71:272–9.10.1067/mcp.2002.122527
126
LaegerTHenaganTMAlbaradoDCRedmanLMBrayGANolandRCet alFGF21 is an endocrine signal of protein restriction. J Clin Invest (2014) 124:3913–22.10.1172/JCI74915
127
AdamsACChengCCCoskunTKharitonenkovA. FGF21 requires betaklotho to act in vivo. PLoS One (2012) 7:e49977.10.1371/journal.pone.0049977
128
DingXBoney-MontoyaJOwenBMBookoutALCoateKCMangelsdorfDJet albetaKlotho is required for fibroblast growth factor 21 effects on growth and metabolism. Cell Metab (2012) 16:387–93.10.1016/j.cmet.2012.08.002
129
AngelinBLarssonTERudlingM. Circulating fibroblast growth factors as metabolic regulators – a critical appraisal. Cell Metab (2012) 16:693–705.10.1016/j.cmet.2012.11.001
130
GimenoREMollerDE. FGF21-based pharmacotherapy – potential utility for metabolic disorders. Trends Endocrinol Metab (2014) 25:303–11.10.1016/j.tem.2014.03.001
131
OwenBMDingXMorganDACoateKCBookoutALRahmouniKet alFGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab (2014) 20:670–7.10.1016/j.cmet.2014.07.012
132
ChartoumpekisDVHabeosIGZirosPGPsyrogiannisAIKyriazopoulouVEPapavassiliouAG. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol Med (2011) 17:736–40.10.2119/molmed.2011.00075
133
HanssenMJBroedersESammsRJVosselmanMJvan der LansAAChengCCet alSerum FGF21 levels are associated with brown adipose tissue activity in humans. Sci Rep (2015) 5:10275.10.1038/srep10275
134
FisherFMKleinerSDourisNFoxECMepaniRJVerdeguerFet alFGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev (2012) 26:271–81.10.1101/gad.177857.111
135
LeePLindermanJDSmithSBrychtaRJWangJIdelsonCet alIrisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab (2014) 19:302–9.10.1016/j.cmet.2013.12.017
136
HondaresEGallego-EscuredoJMFlachsPFrontiniACereijoRGodayAet alFibroblast growth factor-21 is expressed in neonatal and pheochromocytoma-induced adult human brown adipose tissue. Metabolism (2014) 63:312–7.10.1016/j.metabol.2013.11.014
137
CamporezJPJornayvazFRPetersenMCPestaDGuigniBASerrJet alCellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology (2013) 154:3099–109.10.1210/en.2013-1191
138
EmanuelliBVienbergSGSmythGChengCStanfordKIArumugamMet alInterplay between FGF21 and insulin action in the liver regulates metabolism. J Clin Invest (2015) 125:458.10.1172/JCI80223
139
SammsRJSmithDPChengCCAntonellisPPPerfieldJWIIKharitonenkovAet alDiscrete aspects of FGF21 in vivo pharmacology do not require UCP1. Cell Rep (2015) 11:991–9.10.1016/j.celrep.2015.04.046
140
VeniantMMSivitsGHelmeringJKomorowskiRLeeJFanWet alPharmacologic effects of FGF21 are independent of the “browning” of white adipose tissue. Cell Metab (2015) 21:731–8.10.1016/j.cmet.2015.04.019
141
KeipertSKutschkeMLampDBrachthauserLNeffFMeyerCWet alGenetic disruption of uncoupling protein 1 in mice renders brown adipose tissue a significant source of FGF21 secretion. Mol Metab (2015) 4:537–42.10.1016/j.molmet.2015.04.006
142
UkropecJAnunciadoRPRavussinYHulverMWKozakLP. UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1-/- mice. J Biol Chem (2006) 281:31894–908.10.1074/jbc.M606114200
143
DongJQRossulekMSomayajiVRBaltrukonisDLiangYHudsonKet alPharmacokinetics and pharmacodynamics of PF-05231023, a novel long-acting FGF21 mimetic, in a first-in-human study. Br J Clin Pharmacol (2015) 80(5):1051–63.10.1111/bcp.12676
144
GaichGChienJYFuHGlassLCDeegMAHollandWLet alThe effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab (2013) 18:333–40.10.1016/j.cmet.2013.08.005
145
KolumamGChenMZTongRZavala-SolorioJKatesLvan BruggenNet alSustained brown fat stimulation and insulin sensitization by a humanized bispecific antibody agonist for fibroblast growth factor receptor 1/betaKlotho complex. EBioMedicine (2015) 2:730–43.10.1016/j.ebiom.2015.05.028
Summary
Keywords
brown adipose tissue, thermogenesis, uncoupling protein 1, drug discovery, PPARγ agonists, thiazolidinediones, β3-adrenergic receptor agonists, FGF21 analogs
Citation
Peng X-R, Gennemark P, O’Mahony G and Bartesaghi S (2015) Unlock the Thermogenic Potential of Adipose Tissue: Pharmacological Modulation and Implications for Treatment of Diabetes and Obesity. Front. Endocrinol. 6:174. doi: 10.3389/fendo.2015.00174
Received
26 September 2015
Accepted
28 October 2015
Published
19 November 2015
Volume
6 - 2015
Edited by
Jun Wu, University of Michigan, USA
Reviewed by
Bin Xu, Virginia Tech, USA; Qin Yang, University of California Irvine, USA
Updates
Copyright
© 2015 Peng, Gennemark, O’Mahony and Bartesaghi.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Rong Peng, xiao-rong.peng@astrazeneca.com
Specialty section: This article was submitted to Cellular Endocrinology, a section of the journal Frontiers in Endocrinology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.